 Bhagirath Chaurasia
Bhagirath Chaurasia Chad Lamar Talbot2
Chad Lamar Talbot2 Scott A. Summers
Scott A. Summers
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 23 September 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.576347
This article is part of the Research Topic The Immunomodulatory Roles of Adipocytes View all 11 articles
Adipose depots are heterogeneous tissues that store and sense fuel levels. Through the secretion of lipids, cytokines, and protein hormones (adipokines), they communicate with other organ systems, informing them of the organism's nutritional status. The adipose tissues include diverse types of adipocytes (white, beige, and brown) distinguished by the number/size of lipid droplets, mitochondrial density, and thermogenic capacity. Moreover, they include a spectrum of immune cells that modulate metabolic activity and tissue remodeling. The unique characteristics and interplay of these cells control the production of ceramides, a class of nutrient signals derived from fat and protein metabolism that modulate adipocyte function to regulate glucose and lipid metabolism. The excessive accumulation of ceramides contributes to the adipose tissue inflammation and dysfunction that underlies cardiometabolic disease. Herein we review findings on this important class of lipid species and discuss their role at the convergence point that links overnutrition/inflammation to key features of the metabolic syndrome.
Obesity increases one's risk for metabolic diseases such as diabetes, coronary artery disease, non-alcoholic steatohepatitis, and heart failure. The condition promotes (a) the accumulation of deleterious lipid metabolites in non-adipose tissues (i.e., lipotoxicity) and (b) chronic low-grade inflammation, which in turn produces the tissue dysfunction that fuels these disorders. The lipotoxicity is secondary to adipose dysfunction, such that excessive lipids are delivered to peripheral tissues rather than being safely stored as triglycerides within the healthy adipocyte (1–5). The inflammation results from the increased recruitment of pro-inflammatory macrophages into the expanded adipose depots, leading to increased secretion of inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukins (IL), and chemokines (6–8). Together, these lipotoxic and inflammatory pathways account for virtually all of the features of the metabolic syndrome including insulin resistance, dyslipidemia, and hypertension.
Lipids, in addition to being major fuel reservoirs (e.g., triglycerides), have important roles in the regulation of nutrient storage. In particular, sphingolipids such as ceramides are metabolic signals that accumulate in obesity and trigger evolutionarily conserved cellular responses to lipid overload (9). Such mechanisms include inhibiting the uptake of glucose and amino acids, leading to the preferential utilization of free fatty acids (FFAs) for energy; slowing rates of triglyceride lipolysis; and impairing mitochondrial respiration (9). At higher concentrations, ceramides induce apoptosis (9). These sphingolipid actions contribute to the tissue dysfunction that underlies non-alcoholic steatohepatitis, diabetes, and heart disease. Inflammatory cytokines, including TNF-α and IL-1, reinforce this signal by accelerating ceramide production (10). Ceramides thus function at the nexus of lipid metabolism and inflammation.
Studies in mice reveal that inhibition of ceramide synthesis resolves hepatic steatosis and improves insulin-stimulated glucose disposal to slow the progression of cardiometabolic diseases (11). These ceramide-lowering interventions also alter adipose tissue metabolism and morphology, enhancing glucose utilization, and energy expenditure. These manipulations also decrease adipose tissue inflammation and alter macrophage polarization, converting them from pro-inflammatory M1-macrophages into anti-inflammatory M2-macrophages (12). Herein we will review the synergy between the free fatty acids (FFAs) and ceramides that accumulate in obesity and inflammation that accompanies adipose tissue expansion for the development of cardiometabolic diseases. In addition, we will discuss the potential therapeutic approaches for targeting ceramides to reduce inflammation and improve adipose health.
Elevations in circulating FFA resulting from increased nutrient consumption or unchecked lipolysis have been implicated in metabolic disorders including insulin resistance, type 2 diabetes, and cardiovascular disease (13). Emerging studies suggest that these fatty acids fuel production of deleterious lipid metabolites such as ceramides while inducing chronic inflammation (3, 6, 14, 15). To this end, FFA, particularly saturated fatty acids such as palmitate which is a key substrate for ceramide production while also modulating innate immune cells to elicit a proinflammatory response, have important roles at the origin of metabolic disease (16, 17).
Ceramides are precursors of complex sphingolipids (e.g., sphingomyelin) that are integral components of cell membranes. The sphingolipid content of the adipose depots is influenced by nutrient availability (e.g., increased levels of sphingolipid precursors such as serine and palmitate), inflammatory signals, adiponectin, and other factors that control global stress responses. Ceramides can thus serve as metabolic messengers that integrate input from a variety of factors associated with obesity and metabolic disease. Their cellular levels are determined by three enzymatic pathways: de novo synthesis, sphingomyelin hydrolysis, and the salvage pathway (Figure 1) (18, 19).
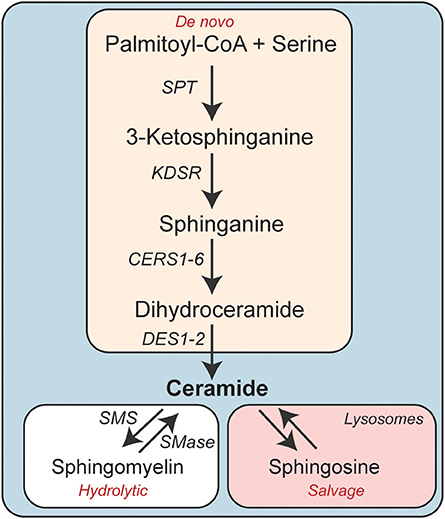
Figure 1. Schematic depicting the enzymatic pathways involved in cellular ceramide synthesis. CERS, Ceramide synthase; DES, Dihydroceramide desaturase; KDSR, 3-ketodihydrosphinganine reductase; SMS, Sphingomyelin synthase; SMase, Sphingomyelinase.
The de novo synthesis pathway comprises four sequential enzymatic steps (20). Serine palmitoyltransferase (SPT) catalyzes the first reaction, condensing palmitoyl-CoA (CoA) and serine to produce 3-ketosphinganine. This transient intermediate doesn't accumulate in cells, as it is rapidly converted to sphinganine by 3-ketosphinganine reductase (3Ksn). Ceramide synthases (CERS1-6) then add a fatty acid, ranging in chain length from 14-carbon to 34-carbon atoms, to sphinganine to produce dihydroceramides. The CERS enzymes have variable substrate specificity and unique tissue distributions and account for much of the diversity in sphingolipids (21). In the fourth and final step, dihydroceramide desaturase (Degs1 and 2) introduces a critical double-bond into dihydroceramide, generating ceramides (22).
The second pathway involves the hydrolysis of sphingomyelin by neutral or acid sphingomyelinase to produce phosphocholine and re-form ceramide (23).
The third pathway, termed the salvage pathway, allows for the reformation of ceramides from sphingolipids after they are degraded in late endosomes or lysosomes (24). The liberated sphingoid base can be re-acylated by the aforementioned CERS enzymes, re-synthesizing ceramides.
Within the last decade, advances in mass-spectrometry have allowed researchers to confidently assess whether plasma and tissue ceramide levels correlate with indices of metabolic diseases. Numerous groups have found that circulating ceramides and FFAs are elevated in subjects with insulin resistance, type 2-diabetes, non-alcoholic fatty liver diseases, chronic kidney diseases and major adverse cardiovascular events including mortality (25–35). In parallel, researchers have shown that circulating inflammatory cytokines also positively associate with these metabolic outcomes (36, 37). Interestingly, circulating FFAs, ceramides and inflammatory cytokines also correlate with one another in human subjects with coronary artery disease, hepatic steatosis, or insulin resistance (10, 33, 38–40). These studies suggest that they may have interrelated roles in the etiology of metabolic disorders.
Adipose tissue ceramide content and inflammation have also been evaluated in subjects with obesity, insulin resistance and/or diabetes. One such study, by the Yki-Jävinen group, demonstrated that ceramide levels are elevated in the adipose tissues of individuals with insulin resistance, independent of obesity (41). In this study, the tissue also showed increases in inflammatory markers. The Brüning laboratory also found that ceramides, particularly C16-ceramides, were elevated in individuals with obesity (42). They also observed dramatically increased expression of ceramide synthase 6 (CERS6), the enzyme that is responsible for generating C16-ceramides. Additionally, CERS6 expression positively correlated with insulin resistance.
The oversupply of precursors such as palmitate and serine undoubtedly account for much of the ceramide accumulation that occurs in obesity. Indeed, a small number of dietary studies have shown that dietary fat intake influences ceramide synthesis and accumulation (43, 44). As outlined below, inflammatory modulators also influence the rate of ceramide production.
In obesity, the recruitment of macrophages to the expanding adipose depots can induce an inflammatory state characterized by increased expression and secretion of inflammatory cytokines such as TNF-α, IL-6, and IL-1β (6, 15, 45–50). Some of these cytokines have been shown to produce ceramides (51–53). In particular, serum and adipose TNF-α are often elevated in individuals with obesity and/or type 2 diabetes and correlate with the severity of insulin resistance (54–56) and with levels of ceramides (33). In cultured cells, the cytokine stimulates ceramide accumulation by inducing expression of ceramide synthesis genes [e.g., serine palmitoyltransferase (SPT)] and increasing expression and activity of sphingomyelin hydrolyzing enzymes (e.g., sphingomyelinase) (51, 57–62). Similar effects on ceramide synthesis have been demonstrated with certain cytokines such as the TNF-α in vivo (63), which antagonize insulin-stimulated glucose disposal in rats and humans (64, 65). In cultured adipocytes and myeloid cells, researchers confirmed that it inhibits insulin signaling and action via receptor-mediated activation of sphingomyelinase (66).
In mice, genetic manipulations to ablate TNF-α or its receptors ameliorate obesity-induced insulin resistance (46, 67). However, clinical trials targeting TNF-α have generally shown little or no beneficial effect on systemic insulin sensitivity (68, 69), indicating that TNF-α lowering is insufficient to combat insulin resistance in humans.
The lipotoxic environment in obesity increases the supply of saturated fatty acids that either directly or indirectly activate toll-like receptor (TLR)-4 (70–74). These pattern recognition receptors, which are typically involved in innate immune responses, have been implicated in inflammation and insulin resistance that accompanies obesity and underlies metabolic disease. For example, Flier et al. found that mice lacking TLR-4 were protected from lipid or high fat diet-induced insulin resistance (17, 75). They also found that long-chain fatty acids signal via TLR-4 to induce transcription of inflammatory cytokines (e.g., TNF-α and IL-6), thus reinforcing and enhancing the inflammatory state. Using similar approaches with various loss-of-function TLR-4 mouse models, four other laboratories described essential roles for TLR-4 in obesity and/or insulin resistance (76–79). Curiously, Shulman et al. found the opposite result, concluding that TLR-4 was not required for lipid-induced insulin resistance (80).
Activation of toll-like receptor (TLR)-4, via lipopolysaccharides (LPS) or a more specific ligand Kdo(2)-lipid A, induces ceramide accumulation by increasing the expression of several ceramide synthesis enzymes (77, 81–84). In cultured myotubes, nuclear factor kappa B (NFκB) was found to be an obligate intermediate in these TLR-4 mediated effects on ceramide production (77). In contrast, ablation of TLR-4 in mice reduces ceramides, and even prevents their synthesis in models of lipid oversupply (i.e., mice fed a high fat diet or infused with lipid cocktails) (77). These findings indicate that TLR-4 enhances ceramide production and reveal the interplay between TLR-4 and ceramides in the metabolic dysfunction that accompanies obesity.
The mechanisms controlling TLR-4 activation in obesity have been controversial. Though saturated fatty acids were initially speculated to be TLR-4 ligands (70–73), some have argued that fatty acids signal through indirect signaling mechanisms (74). Others have argued that this observation is an artifact, likely due to contamination of the saturated fatty acid preparations with lipopolysaccharide (85). In an elegant study, Lancaster et al. found that saturated fatty acids do not bind directly to the TLR-4 receptors, but rather prime TLR-4 to induce lipid-mediated inflammatory signaling (74). These authors found that activating TLR-4 led to a marked upregulation of ceramides and ceramide-synthesizing genes (74).
Inflammasomes are large, multiprotein complexes that form in response to endogenous stress signals, initiating a wide range of cellular activities that include production of the pro-inflammatory cytokines (e.g., IL-1β). The best characterized inflammasome is termed NLRP3 because of the presence of NOD-, LRR-, and pyrin domain-containing protein 3 within the complex. Other components include the adapter ASC and pro-caspase-1. Saturated FFAs were recently found to induce inflammasome activation in macrophages, prompting speculation that lipotoxic intermediates such as ceramides might drive inflammasome activation (49). In both macrophages and adipocytes, ceramides activate the NLRP3 inflammasome, promoting cleavage of caspase-1 and subsequent stimulation of cytokine secretion (86). Subsequent studies found roles for inflammasomes as a downstream ceramide effector in other cell types (87–89), Within adipocytes, this ceramide interaction with the NLRP3 inflammasome may contribute to the adipose inflammation that contributes to insulin resistance. Interestingly, inhibiting de novo ceramide biosynthesis in macrophages did not influence the inflammasome (90), nor did it impact glucose tolerance (11, 12, 90). Moreover, palmitate has been shown to elicit activation of inflammasome by modulating the AMPK-ROS-autophagy pathway, suggesting alternative mechanisms link FFAs to this immune complex (49).
Plasminogen activator inhibitor-1 (PAI-1) is a glycoprotein that is synthesized in endothelial cells, liver, adipose tissue, and other tissue types. It inhibits the serine proteases that covert plasminogen into the active fibrinolytic enzyme plasmin (91, 92). Plasma PAI-1 concentrations are elevated in obesity and diabetes and correlate with the severity of insulin resistance (93–95). Pharmacological inhibition or genetic ablation of PAI-1 in mice protects them from both obesity and insulin resistance while improving adipocyte health and decreasing adipose inflammation (96–99). PAI-1 ablation ensures this protection, at least in part, by reducing accumulation of ceramides in adipocytes, which it accomplishes by decreasing expression of ceramide synthesis genes (96). Conversely, ceramides were reported to induce PAI-1 expression in adipocytes (100), revealing bidirectional interplay between PAI-1 and ceramides that modulates adipose tissue inflammation and function.
The adipokine adiponectin attenuates many features of diabetes and heart disease, including insulin resistance, dyslipidemia, inflammation and cardiomyocyte, endothelial cell and beta-cell apoptosis (101–106). Holland, Scherer et al. were intrigued by the fact that adiponectin and ceramides have such oppositional roles in biology. Moreover, they observed a sequence similarity between adiponectin receptors (AdipoRs) and a family of ceramidases. They thus tested the provocative idea that adiponectin elicited its broad spectrum of actions by reducing (via diacylation) ceramides. They confirmed that the receptor had ceramidase activity that is activated by ligand binding (105). In mice, the cardioprotective and anti-diabetic actions of adiponectin were accompanied by reductions in ceramides (105). Moreover, they identified key residues in AdipoRs that were required for ceramidase activity and for all of adiponectin's downstream actions (105). These findings were then validated by Vasiliauskaite-Brooks et al. who crystalized the AdipoRs in presence of short-chain ceramide analogs, discovering it bound to the liberated sphingoid base (107, 108). They also confirmed that the purified receptors possess ceramidase activity (107, 108). These studies suggest yet another key regulatory mechanism that controls cellular ceramides in order to modulate inflammation and other features of the metabolic syndrome.
Insulin resistance is a defining attribute of the metabolic syndrome that increases one's risk for diabetes and heart disease. As noted above, numerous studies have described correlational relationships between insulin resistance, circulating cytokines, and ceramides in clinical populations (9, 21, 22). Studies in rodents further indicate that ceramides play causative roles in insulin resistance, often linking inflammatory agonists to their deleterious effects on glucose uptake and utilization.
The earliest studies evaluating the role of ceramides in insulin resistance analyzed their effects in 3T3-L1 adipocytes, a murine cell line that shows many of the hallmark metabolic attributes of human adipose tissue. Those studies revealed that ceramides inhibit glucose uptake by inhibiting activation of Akt/PKB (109), a serine/threonine kinase that is an obligate intermediate in insulin-stimulated glucose transporter GLUT4 translocation, as well as glycogen and protein synthesis and protection from apoptosis. Curiously, ceramides did not inhibit the signaling events that precede Akt/PKB activation, such as the activation of PI3-kinase or generation of its product, 3'-polyphosphoinositides (110). Moreover, they blocked activation of the enzyme by numerous other stimuli, including those that don't utilize the signaling scaffold insulin receptor substrate-1 (110), which had recently been identified as a putative site of insulin resistance (111). This observation prompted a flurry of studies seeking to elucidate the signaling mechanisms that linked elevations in ceramides to the inhibition of this important enzyme. These studies revealed that ceramides inhibit Akt/PKB by two known mechanisms, which impact different portions of the enzyme (112). Ceramides dephosphorylate key activating residues through protein phosphatase 2A(PP2A) (112), which is an established ceramide effector (113). Through an alternate mechanism, ceramide blocks the translocation of Akt/PKB to the plasma membrane (112). Studies by the Hundal laboratory subsequently revealed that the translocation effect was due to ceramide actions on atypical protein kinase C (PKCζ), which phosphorylates a key residue in the pleckstrin homology domain of Akt/PKB to block its recruitment to the plasma membrane (114–117). These disparate ceramide mechanisms are clearly separable, as they impact different protein domains and are responsive to distinct inhibitors (112). They also vary by cell type, seeming to be contingent on the relative quantity of caveolar membranes. Adipocytes that have a high abundance of caveolae favor the PKCζ-Akt/PKB axis rather than the PP2A-Akt/PKB axis (118) (Figure 2).
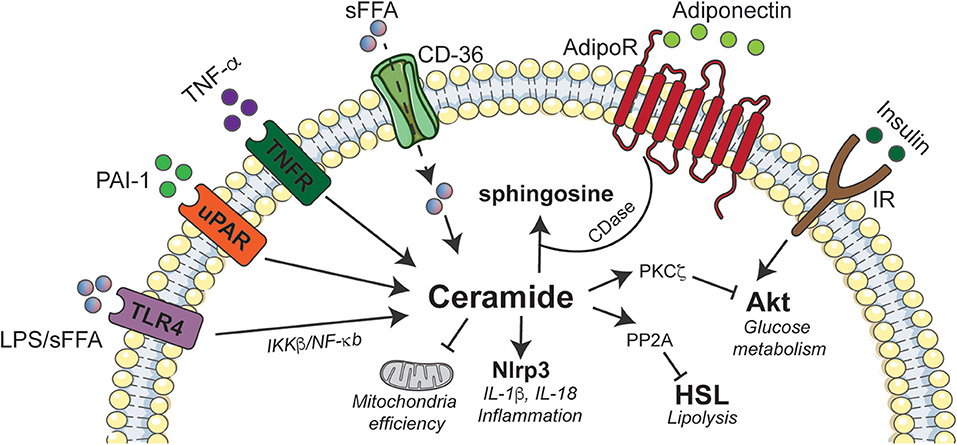
Figure 2. Schematic depicting interactions between ceramides and inflammatory agonists in adipose tissue. Ceramide accumulation elicits deleterious effects on adipose tissue function by activating Nlrp3 inflammasome that induces inflammation, inhibition of Akt via PKCζ to abrogate insulin signaling, and promoting excessive lipid storage by inhibiting HSL. The immunomodulatory adiponectin exhibits some of its beneficial effects by stimulating ceramidase activity that converts ceramides to sphingosine. Akt, Protein Kinase B; CD-36, cluster of differentiation 36; AdipoR, Adiponectin receptor; CDase, Ceramidase; IKK, Ikappa kinase; IL, interleukin; IR, insulin receptor; LPS, lipopolysaccharide; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; Nlrp3, NLR family, pyrin domain containing 3; PAI-1, Plasminogen activator inhibitor 1; PKC, protein kinase C; PP2A, Protein phosphatase 2A; sFFA, Saturated fatty acids; TLR4, Toll like receptor-4; TNF-α, Tumor necrosis factor alpha; TNFR, Tumor necrosis factor alpha receptor; uPAR, Urokinase-type plasminogen activator receptor.
These studies suggested that ceramides, induced by either the oversupply of fatty acid substrates or the inflammation-induced upregulation or activation of ceramide-producing enzymes, might drive insulin resistance in vivo. Data in rodents support this hypothesis. For example, a pharmacological inhibitor of SPT (i.e., myriocin) prevents and/or reverses insulin resistance in high fat diet fed mice (12, 77, 119–121), lipid-infused rats (121), fructose-fed hamsters (122), and leptin-deficient mice and rats (i.e., Zucker fa/fa rats and ob/ob mice) (121). It also resolves steatosis, decreases adipocyte size, and enhances recruitment of M2 macrophages into subcutaneous adipose tissue (12). Similar findings were obtained with pharmacological (i.e., fenretinide) or genetic (i.e., gene knockout) inhibition of DES1 (11, 123, 124). Many of these actions could be explained by ceramide actions within the adipocyte. Adipocyte-specific depletion of SPTLC2, a critical subunit within the SPT complex, or DES1 improved insulin sensitivity, resolved hepatic steatosis, and decreased inflammation of the adipose beds (12). A comparable spectrum of effects was obtained using adipose-specific over-expression of acid ceramidase (125).
While the mechanisms that allow ceramide to modulate lipid and inflammation-induced insulin resistance are fairly clear, the means by which adipocyte ceramides induce the recruitment of macrophages are not. Of note, most of the protective actions of ceramide depletion are unlikely to be driven by ceramides within the macrophage, as depleting SPTLC2 or DES1 from myeloid cells did not influence glucose homeostasis (11, 12, 90).
In mice, myriocin also increases energy expenditure via a mechanism that involves changes to the adipose depot. The SPT inhibitor increased the allotment of adipocytes that express uncoupling protein 1 (UCP1) (12), a mitochondrial protein that dissipates the proton gradient generated by the electron transport chain. This uncoupling reduces mitochondrial membrane potential and leads to high rates of substrate oxidation, heat production and energy expenditure (126). Similar observations were obtained following the adipocyte-specific depletion of the SPTLC2 subunit (12).
Myriocin also caused a shift in macrophage polarization from M1 to M2, which has been shown to induce adipose “browning” characterized by the upregulation of UCP1. Given these data, we profiled macrophage content in adipose tissue following a myriocin intervention. This revealed a recruitment of M2-macrophages in the adipose tissue that was associated with a reduction in expression of key pro-inflammatory cytokines (e.g., IL-6, MCP-1, and TNF-α) and an induction of a crucial anti-inflammatory cytokine IL-10 (11). To resolve whether these improvements were due to cell-autonomous ceramide actions within the adipocytes or macrophages, we depleted the Sptlc2 gene from both adipocytes and macrophages. Adipocyte-specific depletion recapitulated the effects of myriocin and increased the recruitment of M2-macrophages and expression of thermogenic genes (e.g., Ucp1, Pgc1a, and Prdm16). These data indicated that adipocyte sphingolipids likely drove the cellular responses that increased energy expenditure. By comparison, depleting Sptlc2 from macrophages failed to impact energy expenditure. Moreover, ectopic ceramides were also shown to inhibit mitochondrial respiration and block activation of hormone-sensitive lipase by β-adrenergic agonists. The effects on lipolysis were mediated by the aforementioned ceramide effector PP2A (Figure 2).
Beyond the effects on UCP1 and HSL, ceramides seem to slow energy expenditure by inhibiting mitochondrial respiration. Indeed, addition of ceramides to cells is sufficient to inhibit mitochondrial activity (12). Hammerschmidt et al. (127) elucidated one mechanism that underlies this effect, determining that ceramides bind to mitochondrial fission factor (MFF) to alter mitochondrial morphology and reduce respiratory capacity (127). This effect is specific for the C16-ceramides produced by CERS6 (127).
Two other studies have evaluated the effects of reducing ceramides in white adipocytes. Curiously, while these studies did find that depleting ceramides from adipose tissue influenced glucose and lipid homeostasis, neither intervention induced adipose browning. One was the aforementioned study evaluating the consequence of acid ceramidase expression, while the other was our study involving DES1 depletion (11). While these interventions affected mitochondrial respiration, they did not induce UCP1 expression. We thus conclude that the effect on UCP1 is not due to direct ceramide actions, but rather to another intermediate in the pathway. One attractive hypothesis is that the browning effects are mediated by the CERS enzymes (128, 129), which have been shown to be transcriptional repressors that move to the nucleus and regulate lipase expression following encounters with fatty acids. By comparison, we conclude that the effects on mitochondrial fission and lipolysis (i.e., HSL) are due to direct actions of the sphingolipid analogs on MFF and PP2A, respectively. The effects on mitochondrial morphology/respiration and HSL were observed in all of the interventional studies described.
Inflammation has long been known to be a hallmark of obesity, owing to the recruitment of macrophages to adipose depots and the enhancement of TLR-4 signaling by saturated fatty acids. Herein we discussed how the impact of chronic inflammation on host metabolism are linked to ceramide-driven lipotoxicity. Ceramides, which are universally upregulated by inflammatory stimuli, inhibit insulin-stimulated glucose disposal and mitochondrial respiration. They thus provide a convergence point that links overnutrition/dyslipidemia and inflammation to drive many of the key features of the metabolic syndrome. Curiously, manipulating ceramides in adipose tissue also influences the inflammatory state of the organ, suggesting the existence of feedback mechanisms that involve ceramide-dependent, adipocyte autonomous signals that control the immune cell population (e.g., via the NLRP3 inflammasome). Additional research on ceramides and their inflammatory regulators thus holds great promise as a means to combat metabolic disease and improve adipose tissue health.
Some of the figures presented in this manuscript were prepared using Servier Art.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors received research support from the National Institutes of Health (DK115824, DK116888, and DK116450 to SS; DK115824 and DK124326 to BC), the Juvenile Diabetes Research Foundation (JDRF 3-SRA-2019-768-A-B to SS), the American Diabetes Association (to SS), the American Heart Association (to SS), the American Heart Association Career Development Award (to BC), the Margolis Foundation (to SS), and the USDA (2019-67018-29250 to BC).
SS is founder and consultant for Centaurus Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Unger RH. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol Metab. (2003) 14:398–403. doi: 10.1016/j.tem.2003.09.008
2. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. (2005) 115:1111–9. doi: 10.1172/JCI25102
3. Chaurasia B, Summers SA. Ceramides - Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol Metab. (2015) 26:538–50. doi: 10.1016/j.tem.2015.07.006
4. Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDGenetic M, clinical implications. Diabetes. (1995) 44:863–70. doi: 10.2337/diabetes.44.8.863
5. Unger RH, Clark GO, Scherer PE, Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta. (2010) 1801:209–14. doi: 10.1016/j.bbalip.2009.10.006
6. Hotamisligil GS. Inflammation and metabolic disorders. Nature. (2006) 444:860–7. doi: 10.1038/nature05485
7. Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest. (1995) 95:2111–9. doi: 10.1172/JCI117899
8. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/JCI200319246
9. Summers SA, Chaurasia B, Holland WL. Metabolic messengers: ceramides. Nat Metab. (2019) 1:1051–8. doi: 10.1038/s42255-019-0134-8
10. Gill JM, Sattar N. Ceramides a new player in the inflammation-insulin resistance paradigm? Diabetologia. (2009) 52:2475–7. doi: 10.1007/s00125-009-1546-x
11. Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L, et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science. (2019) 365:386–92. doi: 10.1126/science.aav3722
12. Chaurasia B, Kaddai VA, Lancaster GI, Henstridge DC, Sriram S, Galam DL, et al. Adipocyte ceramides regulate subcutaneous adipose browning, inflammation, and metabolism. Cell Metab. (2016) 24:820–34. doi: 10.1016/j.cmet.2016.10.002
13. Sears B, Perry M. The role of fatty acids in insulin resistance. Lipids Health Dis. (2015) 14:121. doi: 10.1186/s12944-015-0123-1
14. Brookheart RT, Michel CI, Schaffer JE. As a matter of fat. Cell Metab. (2009) 10:9–12. doi: 10.1016/j.cmet.2009.03.011
15. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85. doi: 10.1038/nature21363
16. Boden G. Interaction between free fatty acids and glucose metabolism. Curr Opin Clin Nutr Metab Care. (2002) 5:545–9. doi: 10.1097/00075197-200209000-00014
17. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. (2006) 116:3015–25. doi: 10.1172/JCI28898
18. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. (2018) 19:175–91. doi: 10.1038/nrm.2017.107
19. Gault CR, Obeid LM, Hannun YA. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol. (2010) 688:1–23. doi: 10.1007/978-1-4419-6741-1_1
20. Merrill AH Jr. De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J Biol Chem. (2002) 277:25843–6. doi: 10.1074/jbc.R200009200
21. Zelnik ID, Rozman B, Rosenfeld-Gur E, Ben-Dor S, Futerman AH. A stroll down the cers lane. Adv Exp Med Biol. (2019) 1159:49–63. doi: 10.1007/978-3-030-21162-2_4
22. Siddique MM, Li Y, Chaurasia B, Kaddai VA, Summers SA. Dihydroceramides. From bit players to lead actors. J Biol Chem. (2015) 290:15371–9. doi: 10.1074/jbc.R115.653204
23. Bienias K, Fiedorowicz A, Sadowska A, Prokopiuk S, Car H. Regulation of sphingomyelin metabolism. Pharmacol Rep. (2016) 68:570–81. doi: 10.1016/j.pharep.2015.12.008
24. Kitatani K, Idkowiak-Baldys J, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. (2008) 20:1010–8. doi: 10.1016/j.cellsig.2007.12.006
25. Huynh K, Barlow CK, Jayawardana KS, Weir JM, Mellett NA, Cinel M, et al. High-throughput plasma lipidomics: detailed mapping of the associations with cardiometabolic risk factors. Cell Chem Biol. (2019) 26:71–84.e4. doi: 10.1016/j.chembiol.2018.10.008
26. Lemaitre RN, Jensen PN, Hoofnagle A, McKnight B, Fretts AM, King IB, et al. Plasma ceramides and sphingomyelins in relation to heart failure risk. Circ Heart Fail. (2019) 12:e005708. doi: 10.1161/CIRCHEARTFAILURE.118.005708
27. Jensen PN, Fretts AM, Yu C, Hoofnagle AN, Umans JG, Howard BV, et al. Circulating sphingolipids, fasting glucose, and impaired fasting glucose: the strong heart family study. EBioMedicine. (2019) 41:44–49. doi: 10.1016/j.ebiom.2018.12.046
28. Poss AM, Maschek JA, Cox JE, Hauner BJ, Hopkins PN, Hunt SC, et al. Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. J Clin Invest. (2020) 130:1363–76. doi: 10.1172/JCI131838
29. Wigger L, Cruciani-Guglielmacci C, Nicolas A, Denom J, Fernandez N, Fumeron F, et al. Plasma dihydroceramides are diabetes susceptibility biomarker candidates in mice and humans. Cell Rep. (2017) 18:2269–79. doi: 10.1016/j.celrep.2017.02.019
30. Mantovani A, Bonapace S, Lunardi G, Salgarello M, Dugo C, Gori S, et al. Association of plasma ceramides with myocardial perfusion in patients with coronary artery disease undergoing stress myocardial perfusion scintigraphy. Arterioscler Thromb Vasc Biol. (2018) 38:2854–61. doi: 10.1161/ATVBAHA.118.311927
31. Havulinna AS, Sysi-Aho M, Hilvo M, Kauhanen D, Hurme R, Ekroos K, et al. Circulating ceramides predict cardiovascular outcomes in the population-based FINRISK 2002 cohort. Arterioscler Thromb Vasc Biol. (2016) 36:2424–30. doi: 10.1161/ATVBAHA.116.307497
32. Laaksonen R, Ekroos K, Sysi-Aho M, Hilvo M, Vihervaara T, Kauhanen D, et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur Heart J. (2016) 37:1967–76. doi: 10.1093/eurheartj/ehw148
33. Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, et al. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes. (2009) 58:337–43. doi: 10.2337/db08-1228
34. Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am. (2008) 37:635–46. doi: 10.1016/j.ecl.2008.06.007
35. Capurso C, Capurso A. From excess adiposity to insulin resistance: the role of free fatty acids. Vascul Pharmacol. (2012) 57:91–7. doi: 10.1016/j.vph.2012.05.003
36. Marques-Vidal P, Bastardot F, von Kanel R, Paccaud F, Preisig M, Waeber G, et al. Association between circulating cytokine levels, diabetes and insulin resistance in a population-based sample (CoLaus study). Clin Endocrinol. (2013) 78:232–41. doi: 10.1111/j.1365-2265.2012.04384.x
37. Rubin DA, McMurray RG, Harrell JS, Hackney AC, Thorpe DE, Haqq AM. The association between insulin resistance and cytokines in adolescents: the role of weight status and exercise. Metabolism. (2008) 57:683–90. doi: 10.1016/j.metabol.2008.01.005
38. de Mello VD, Lankinen M, Schwab U, Kolehmainen M, Lehto S, Seppanen-Laakso T, et al. Link between plasma ceramides, inflammation and insulin resistance: association with serum IL-6 concentration in patients with coronary heart disease. Diabetologia. (2009) 52:2612–5. doi: 10.1007/s00125-009-1482-9
39. Rosqvist F, Kullberg J, Stahlman M, Cedernaes J, Heurling K, Johansson HE, et al. Overeating saturated fat promotes fatty liver and ceramides compared with polyunsaturated fat: a randomized trial. J Clin Endocrinol Metab. (2019) 104:6207–19. doi: 10.1210/jc.2019-00160
40. Zabielski P, Blachnio-Zabielska AU, Wojcik B, Chabowski A, Gorski J. Effect of plasma free fatty acid supply on the rate of ceramide synthesis in different muscle types in the rat. PLoS ONE. (2017) 12:e0187136. doi: 10.1371/journal.pone.0187136
41. Kolak M, Westerbacka J, Velagapudi VR, Wagsater D, Yetukuri L, Makkonen J, et al. Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity. Diabetes. (2007) 56:1960–8. doi: 10.2337/db07-0111
42. Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. (2014) 20:678–86. doi: 10.1016/j.cmet.2014.08.002
43. Kien CL, Bunn JY, Poynter ME, Stevens R, Bain J, Ikayeva O, et al. A lipidomics analysis of the relationship between dietary fatty acid composition and insulin sensitivity in young adults. Diabetes. (2013) 62:1054–63. doi: 10.2337/db12-0363
44. Luukkonen PK, Sadevirta S, Zhou Y, Kayser B, Ali A, Ahonen L, et al. Saturated fat is more metabolically harmful for the human liver than unsaturated fat or simple sugars. Diabetes Care. (2018) 41:1732–39. doi: 10.2337/dc18-0071
45. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. (1993) 259:87–91. doi: 10.1126/science.7678183
46. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. (1997) 389:610–4. doi: 10.1038/3933
47. Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. (2010) 12:593–605. doi: 10.1016/j.cmet.2010.11.011
48. Tack CJ, Stienstra R, Joosten LA, Netea MG. Inflammation links excess fat to insulin resistance: the role of the interleukin-1 family. Immunol Rev. (2012) 249:239–52. doi: 10.1111/j.1600-065X.2012.01145.x
49. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12:408–15. doi: 10.1038/ni.2022
50. Han MS, White A, Perry RJ, Camporez JP, Hidalgo J, Shulman GI, et al. Regulation of adipose tissue inflammation by interleukin 6. Proc Natl Acad Sci USA. (2020) 117:2751–60. doi: 10.1073/pnas.1920004117
51. Xu J, Yeh CH, Chen S, He L, Sensi SL, Canzoniero LM, et al. Involvement of de novo ceramide biosynthesis in tumor necrosis factor-alpha/cycloheximide-induced cerebral endothelial cell death. J Biol Chem. (1998) 273:16521–6. doi: 10.1074/jbc.273.26.16521
52. Tong L, Balazs R, Soiampornkul R, Thangnipon W, Cotman CW. Interleukin-1 beta impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol Aging. (2008) 29:1380–93. doi: 10.1016/j.neurobiolaging.2007.02.027
53. Homaidan FR, El-Sabban ME, Chakroun I, El-Sibai M, Dbaibo GS. IL-1 stimulates ceramide accumulation without inducing apoptosis in intestinal epithelial cells. Mediators Inflamm. (2002) 11:39–45. doi: 10.1080/09629350210313
54. Plomgaard P, Nielsen AR, Fischer CP, Mortensen OH, Broholm C, Penkowa M, et al. Associations between insulin resistance and TNF-alpha in plasma, skeletal muscle and adipose tissue in humans with and without type 2 diabetes. Diabetologia. (2007) 50:2562–71. doi: 10.1007/s00125-007-0834-6
55. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. (1995) 95:2409–15. doi: 10.1172/JCI117936
56. Olson NC, Callas PW, Hanley AJ, Festa A, Haffner SM, Wagenknecht LE, et al. Circulating levels of TNF-alpha are associated with impaired glucose tolerance, increased insulin resistance, and ethnicity: the insulin resistance atherosclerosis study. J Clin Endocrinol Metab. (2012) 97:1032–40. doi: 10.1210/jc.2011-2155
57. Kolesnick RN, Haimovitz-Friedman A, Fuks Z. The sphingomyelin signal transduction pathway mediates apoptosis for tumor necrosis factor. Fas, ionizing radiation. Biochem Cell Biol. (1994) 72:471–4. doi: 10.1139/o94-063
58. Rivas CI, Golde DW, Vera JC, Kolesnick RN. Involvement of the sphingomyelin pathway in autocrine tumor necrosis factor signaling for human immunodeficiency virus production in chronically infected HL-60 cells. Blood. (1994) 83:2191–7. doi: 10.1182/blood.V83.8.2191.bloodjournal8382191
59. Kolesnick R. Signal transduction through the sphingomyelin pathway. Mol Chem Neuropathol. (1994) 21:287–97. doi: 10.1007/BF02815356
60. Jarvis WD, Kolesnick RN, Fornari FA, Traylor RS, Gewirtz DA, Grant S. Induction of apoptotic DNA damage and cell death by activation of the sphingomyelin pathway. Proc Natl Acad Sci USA. (1994) 91:73–7. doi: 10.1073/pnas.91.1.73
61. Brindley DN, Wang CN, Mei J, Xu J, Hanna AN. Tumor necrosis factor-alpha and ceramides in insulin resistance. Lipids. (1999) 34:S85–8. doi: 10.1007/BF02562240
62. Meyer SG, de Groot H. Cycloserine and threo-dihydrosphingosine inhibit TNF-alpha-induced cytotoxicity: evidence for the importance of de novo ceramide synthesis in TNF-alpha signaling. Biochim Biophys Acta. (2003) 1643:1–4. doi: 10.1016/j.bbamcr.2003.10.002
63. Mallampalli RK, Peterson EJ, Carter AB, Salome RG, Mathur SN, Koretzky GA. TNF-alpha increases ceramide without inducing apoptosis in alveolar type II epithelial cells. Am J Physiol. (1999) 276:L481–90. doi: 10.1152/ajplung.1999.276.3.L481
64. Krogh-Madsen R, Plomgaard P, Moller K, Mittendorfer B, Pedersen BK. Influence of TNF-alpha and IL-6 infusions on insulin sensitivity and expression of IL-18 in humans. Am J Physiol Endocrinol Metab. (2006) 291:E108–14. doi: 10.1152/ajpendo.00471.2005
65. Lang CH, Dobrescu C, Bagby GJ. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology. (1992) 130:43–52. doi: 10.1210/endo.130.1.1727716
66. Peraldi P, Hotamisligil GS, Buurman WA, White MF, Spiegelman BM. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem. (1996) 271:13018–22. doi: 10.1074/jbc.271.22.13018
67. Ventre J, Doebber T, Wu M, MacNaul K, Stevens K, Pasparakis M, et al. Targeted disruption of the tumor necrosis factor-alpha gene: metabolic consequences in obese and nonobese mice. Diabetes. (1997) 46:1526–31. doi: 10.2337/diabetes.46.9.1526
68. Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab. (2000) 85:1316–9. doi: 10.1210/jcem.85.3.6417
69. Wascher TC, Lindeman JH, Sourij H, Kooistra T, Pacini G, Roden M. Chronic TNF-alpha neutralization does not improve insulin resistance or endothelial function in “healthy” men with metabolic syndrome. Mol Med. (2011) 17:189–93. doi: 10.2119/molmed.2010.00221
70. Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, et al. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem. (2004) 279:16971–9. doi: 10.1074/jbc.M312990200
71. Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, et al. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. (2003) 278:37041–51. doi: 10.1074/jbc.M305213200
72. Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, et al. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res. (2003) 44:479–86. doi: 10.1194/jlr.M200361-JLR200
73. Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. (2001) 276:16683–9. doi: 10.1074/jbc.M011695200
74. Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. (2018) 27:1096–110.e5. doi: 10.1016/j.cmet.2018.03.014
75. Kim JK. Fat uses a TOLL-road to connect inflammation and diabetes. Cell Metab. (2006) 4:417–9. doi: 10.1016/j.cmet.2006.11.008
76. Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring). (2008) 16:1248–55. doi: 10.1038/oby.2008.210
77. Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. (2011) 121:1858–70. doi: 10.1172/JCI43378
78. Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, et al. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res. (2007) 100:1589–96. doi: 10.1161/CIRCRESAHA.106.142851
79. Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. (2007) 56:1986–98. doi: 10.2337/db06-1595
80. Galbo T, Perry RJ, Jurczak MJ, Camporez JP, Alves TC, Kahn M, et al. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. Proc Natl Acad Sci USA. (2013) 110:12780–5. doi: 10.1073/pnas.1311176110
81. Memon RA, Holleran WM, Uchida Y, Moser AH, Grunfeld C, Feingold KR. Regulation of sphingolipid and glycosphingolipid metabolism in extrahepatic tissues by endotoxin. J Lipid Res. (2001) 42:452–9.
82. Memon RA, Holleran WM, Moser AH, Seki T, Uchida Y, Fuller J, et al. Endotoxin and cytokines increase hepatic sphingolipid biosynthesis and produce lipoproteins enriched in ceramides and sphingomyelin. Arterioscler Thromb Vasc Biol. (1998) 18:1257–65. doi: 10.1161/01.ATV.18.8.1257
83. Schilling JD, Machkovech HM, He L, Sidhu R, Fujiwara H, Weber K, et al. Palmitate and lipopolysaccharide trigger synergistic ceramide production in primary macrophages. J Biol Chem. (2013) 288:2923–32. doi: 10.1074/jbc.M112.419978
84. Sims K, Haynes CA, Kelly S, Allegood JC, Wang E, Momin A Jr, et al. Kdo2-lipid A, a TLR4-specific agonist, induces de novo sphingolipid biosynthesis in RAW264.7 macrophages, which is essential for induction of autophagy. J Biol Chem. (2010) 285:38568–79. doi: 10.1074/jbc.M110.170621
85. Erridge C, Samani NJ. Saturated fatty acids do not directly stimulate toll-like receptor signaling. Arterioscler Thromb Vasc Biol. (2009) 29:1944–9. doi: 10.1161/ATVBAHA.109.194050
86. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. (2011) 17:179–88. doi: 10.1038/nm.2279
87. Kursawe R, Dixit VD, Scherer PE, Santoro N, Narayan D, Gordillo R, et al. A role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes. (2016) 65:610–8. doi: 10.2337/db15-1478
88. Youm YH, Kanneganti TD, Vandanmagsar B, Zhu X, Ravussin A, Adijiang A, et al. The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. (2012) 1:56–68. doi: 10.1016/j.celrep.2011.11.005
89. Youm YH, Adijiang A, Vandanmagsar B, Burk D, Ravussin A, Dixit VD. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology. (2011) 152:4039–45. doi: 10.1210/en.2011-1326
90. Camell CD, Nguyen KY, Jurczak MJ, Christian BE, Shulman GI, Shadel GS, et al. Macrophage-specific de novo synthesis of ceramide is dispensable for inflammasome-driven inflammation and insulin resistance in obesity. J Biol Chem. (2015) 290:29402–13. doi: 10.1074/jbc.M115.680199
91. Alessi MC, Poggi M, Juhan-Vague I. Plasminogen activator inhibitor-1, adipose tissue and insulin resistance. Curr Opin Lipidol. (2007) 18:240–5. doi: 10.1097/MOL.0b013e32814e6d29
92. Kaji H. Adipose tissue-derived plasminogen activator inhibitor-1 function and regulation. Compr Physiol. (2016) 6:1873–96. doi: 10.1002/cphy.c160004
93. Festa A, Williams K, Tracy RP, Wagenknecht LE, Haffner SM. Progression of plasminogen activator inhibitor-1 and fibrinogen levels in relation to incident type 2 diabetes. Circulation. (2006) 113:1753–9. doi: 10.1161/CIRCULATIONAHA.106.616177
94. Hanley AJ, Festa A, D'Agostino RB Jr, Wagenknecht LE, Savage PJ, et al. Metabolic and inflammation variable clusters and prediction of type 2 diabetes: factor analysis using directly measured insulin sensitivity. Diabetes. (2004) 53:1773–81. doi: 10.2337/diabetes.53.7.1773
95. D'Agostino RB Jr, Hamman RF, Karter AJ, Mykkanen L, Wagenknecht LE, Haffner SM, et al. Insulin resistance atherosclerosis study. Cardiovascular disease risk factors predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes Care. (2004) 27:2234–40. doi: 10.2337/diacare.27.9.2234
96. Shah C, Yang G, Lee I, Bielawski J, Hannun YA, Samad F. Protection from high fat diet-induced increase in ceramide in mice lacking plasminogen activator inhibitor 1. J Biol Chem. (2008) 283:13538–48. doi: 10.1074/jbc.M709950200
97. Ma LJ, Mao SL, Taylor KL, Kanjanabuch T, Guan Y, Zhang Y, et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes. (2004) 53:336–46. doi: 10.2337/diabetes.53.2.336
98. Tamura Y, Kawao N, Yano M, Okada K, Matsuo O, Kaji H. Plasminogen activator inhibitor-1 deficiency ameliorates insulin resistance and hyperlipidemia but not bone loss in obese female mice. Endocrinology. (2014) 155:1708–17. doi: 10.1210/en.2013-1888
99. Wang L, Chen L, Liu Z, Liu Y, Luo M, Chen N, et al. PAI-1 exacerbates white adipose tissue dysfunction and metabolic dysregulation in high fat diet-induced obesity. Front Pharmacol. (2018) 9:1087. doi: 10.3389/fphar.2018.01087
100. Samad F, Hester KD, Yang G, Hannun YA, Bielawski J. Altered adipose and plasma sphingolipid metabolism in obesity: a potential mechanism for cardiovascular and metabolic risk. Diabetes. (2006) 55:2579–87. doi: 10.2337/db06-0330
101. Wang ZV, Scherer PE. Adiponectin, the past two decades. J Mol Cell Biol. (2016) 8:93–100. doi: 10.1093/jmcb/mjw011
102. Holland WL, Xia JY, Johnson JA, Sun K, Pearson MJ, Sharma AX, et al. Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Mol Metab. (2017) 6:267–75. doi: 10.1016/j.molmet.2017.01.002
103. Ye R, Holland WL, Gordillo R, Wang M, Wang QA, Shao M, et al. Adiponectin is essential for lipid homeostasis and survival under insulin deficiency and promotes beta-cell regeneration. Elife. (2014) 3:e03851. doi: 10.7554/eLife.03851
104. Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. (2013) 17:790–7. doi: 10.1016/j.cmet.2013.03.019
105. Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. (2011) 17:55–63. doi: 10.1038/nm.2277
106. Kobayashi H, Ouchi N, Kihara S, Walsh K, Kumada M, Abe Y, et al. Selective suppression of endothelial cell apoptosis by the high molecular weight form of adiponectin. Circ Res. (2004) 94:e27–31. doi: 10.1161/01.RES.0000119921.86460.37
107. Vasiliauskaite-Brooks I, Healey RD, Granier S. 7TM proteins are not necessarily GPCRs. Mol Cell Endocrinol. (2019) 491:110397. doi: 10.1016/j.mce.2019.02.009
108. Vasiliauskaite-Brooks I, Sounier R, Rochaix P, Bellot G, Fortier M, Hoh F, et al. Structural insights into adiponectin receptors suggest ceramidase activity. Nature. (2017) 544:120–3. doi: 10.1038/nature21714
109. Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol. (1998) 18:5457–64. doi: 10.1128/MCB.18.9.5457
110. Stratford S, DeWald DB, Summers SA. Ceramide dissociates 3'-phosphoinositide production from pleckstrin homology domain translocation. Biochem J. (2001) 354:359–68. doi: 10.1042/bj3540359
111. White MF. The IRS-signalling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem. (1998) 182:3–11. doi: 10.1007/978-1-4615-5647-3_1
112. Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem. (2004) 279:36608–15. doi: 10.1074/jbc.M406499200
113. Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem. (1993) 268:15523–30.
114. Stretton C, Evans A, Hundal HS. Cellular depletion of atypical PKC{lambda} is associated with enhanced insulin sensitivity and glucose uptake in L6 rat skeletal muscle cells. Am J Physiol Endocrinol Metab. (2010) 299:E402–12. doi: 10.1152/ajpendo.00171.2010
115. Hajduch E, Turban S, Le Liepvre X, Le Lay S, Lipina C, Dimopoulos N, et al. Targeting of PKCzeta and PKB to caveolin-enriched microdomains represents a crucial step underpinning the disruption in PKB-directed signalling by ceramide. Biochem J. (2008) 410:369–79. doi: 10.1042/BJ20070936
116. Powell DJ, Turban S, Gray A, Hajduch E, Hundal HS. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J. (2004) 382:619–29. doi: 10.1042/BJ20040139
117. Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol. (2003) 23:7794–808. doi: 10.1128/MCB.23.21.7794-7808.2003
118. Blouin CM, Prado C, Takane KK, Lasnier F, Garcia-Ocana A, Ferre P, et al. Plasma membrane subdomain compartmentalization contributes to distinct mechanisms of ceramide action on insulin signaling. Diabetes. (2010) 59:600–10. doi: 10.2337/db09-0897
119. Ussher JR, Koves TR, Cadete VJ, Zhang L, Jaswal JS, Swyrd SJ, et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes. (2010) 59:2453–64. doi: 10.2337/db09-1293
120. Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, et al. CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. (2014) 20:687–95. doi: 10.1016/j.cmet.2014.09.015
121. Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. (2007) 5:167–79. doi: 10.1016/j.cmet.2007.01.002
122. Dekker MJ, Baker C, Naples M, Samsoondar J, Zhang R, Qiu W, et al. Inhibition of sphingolipid synthesis improves dyslipidemia in the diet-induced hamster model of insulin resistance: evidence for the role of sphingosine and sphinganine in hepatic VLDL-apoB100 overproduction. Atherosclerosis. (2013) 228:98–109. doi: 10.1016/j.atherosclerosis.2013.01.041
123. Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. (2005) 436:356–62. doi: 10.1038/nature03711
124. Bikman BT, Guan Y, Shui G, Siddique MM, Holland WL, Kim JY, et al. Fenretinide prevents lipid-induced insulin resistance by blocking ceramide biosynthesis. J Biol Chem. (2012) 287:17426–37. doi: 10.1074/jbc.M112.359950
125. Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, et al. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab. (2015) 22:266–78. doi: 10.1016/j.cmet.2015.06.007
126. Kajimura S, Spiegelman BM, Seale P. Brown and beige fat: physiological roles beyond heat generation. Cell Metabolism. (2015) 22:546–59. doi: 10.1016/j.cmet.2015.09.007
127. Hammerschmidt P, Ostkotte D, Nolte H, Gerl MJ, Jais A, Brunner HL, et al. CerS6-derived sphingolipids interact with Mff and promote mitochondrial fragmentation in obesity. Cell. (2019) 177:1536–52.e23. doi: 10.1016/j.cell.2019.05.008
128. Sociale M, Wulf AL, Breiden B, Klee K, Thielisch M, Eckardt F, et al. Ceramide synthase schlank is a transcriptional regulator adapting gene expression to energy requirements. Cell Rep. (2018) 22:967–78. doi: 10.1016/j.celrep.2017.12.090
Keywords: ceramide, inflammation, insulin, diabetes, adipocyte
Citation: Chaurasia B, Talbot CL and Summers SA (2020) Adipocyte Ceramides—The Nexus of Inflammation and Metabolic Disease. Front. Immunol. 11:576347. doi: 10.3389/fimmu.2020.576347
Received: 25 June 2020; Accepted: 20 August 2020;
Published: 23 September 2020.
Edited by:
Aimin Xu, The University of Hong Kong, Hong KongReviewed by:
Oreste Gualillo, Servicio Gallego de Salud, SpainCopyright © 2020 Chaurasia, Talbot and Summers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bhagirath Chaurasia, YmhhZ2lyYXRoLWNoYXVyYXNpYUB1aW93YS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.