 Francesca Alessandrini1
Francesca Alessandrini1 Frank Blanco-Pérez
Frank Blanco-Pérez Melanie Albrecht
Melanie Albrecht- 1Center of Allergy & Environment (ZAUM), Technical University of Munich (TUM) and Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany
- 2Molecular Allergology/Vice President's Research Group, Paul-Ehrlich-Institut, Langen, Germany
Asthma is a heterogeneous disease with increasing prevalence worldwide characterized by chronic airway inflammation, increased mucus secretion and bronchial hyperresponsiveness. The phenotypic heterogeneity among asthmatic patients is accompanied by different endotypes, mainly Type 2 or non-Type 2. To investigate the pathomechanism of this complex disease many animal models have been developed, each trying to mimic specific aspects of the human disease. Rodents have classically been employed in animal models of asthma. The present review provides an overview of currently used Type 2 vs. non-Type 2 rodent asthma models, both acute and chronic. It further assesses the methods used to simulate disease development and exacerbations as well as to quantify allergic airway inflammation, including lung physiologic, cellular and molecular immunologic responses. Furthermore, the employment of genetically modified animals, which provide an in-depth understanding of the role of a variety of molecules, signaling pathways and receptors implicated in the development of this disease as well as humanized models of allergic inflammation, which have been recently developed to overcome differences between the rodent and human immune systems, are discussed. Nevertheless, differences between mice and humans should be carefully considered and limits of extrapolation should be wisely taken into account when translating experimental results into clinical use.
Introduction
Asthma is a heterogeneous disease which affects around 300 million individuals of all age groups and its prevalence is increasing worldwide. Its impact is considered similar to other major chronic diseases such as diabetes or Alzheimer disease (1, 2). Asthma is defined by a history of respiratory symptoms such a wheeze, shortness of breath, chest tightness, cough and variable expiratory airflow limitation (3). A chronic airway inflammation leads to airway remodeling with hyperplasia of goblet cells and mucus hypersecretion, hypertrophy and hyperplasia of smooth muscle cells and lung fibrosis. Different asthma phenotypes have been described, which drove the development of the concept of asthma endotypes, where each endotype is a subtype of a disease condition and defined by a distinct pathophysiological mechanism, in contrast to the disease phenotype, which comprises the observable characteristics of the disease (4, 5). Generally, asthma can be separated in two main endotypes, a so-called Type 2 endotype, characterized by T-helper Type 2-high inflammatory response and a non-Type 2 endotype, whereby also mixed endotypes are not rare (6, 7). While asthmatic reactions can also be induced without exogenous triggers (8, 9), only antigen-driven asthma models will be discussed here.
Airway Type 2 immune responses are mainly mediated by eosinophils, mast cells, basophils, T2 cells, group 2 innate lymphoid cells (ILC2s) and IgE-producing B cells (10, 11). The whole inflammatory process starts with the activation of epithelial cells and release of cytokines such as IL-25, IL-31, IL-33, and TSLP which contribute to downstream T cells- and innate lymphoid cells-mediated T2 immune responses. These are characterized by the release of cytokines such as IL-4, IL-5, IL-9, and IL-13, consequent production of allergen-specific IgE, recruitment of eosinophils and other inflammatory cells, production of mucus and smooth muscle hyperreactivity (12). Non-Type 2 asthma, instead, is characterized by airway inflammation in the absence of eosinophils and is often associated with environmental and/or host hazards, such as cigarette smoke, pollution, work-related agents, infections, and obesity. These risk factors, alone or in conjunction, can activate specific cellular and molecular pathways leading to non-type 2 pulmonary inflammation (13). Growing evidence supports two major characteristic features of non-Type 2 asthma, namely a neutrophilic-driven inflammation and an IL-6-driven activation of the IL-17-dependent pathway (14, 15). To allow for a detailed investigation of molecular pathways critical for this complex disease or for a specific endotype in a functioning immune and respiratory system, many animal models of asthma have been developed, each of them trying to reproduce specific aspects of the human disease. Because of their low cost, high breeding efficiency and the large availability of transgenic models, rodents, and especially mice have classically been employed in asthma research, although considerations have been made regarding their limitations in mimicking human asthma (16, 17). In this review we will focus on antigen-driven asthma models and methods used for the elicitation and quantification of allergic airway inflammation, including lung physiologic, cellular and molecular immunologic responses. Furthermore, approaches to study exacerbations, chronicity and non-allergic airway inflammation as well as the value of humanized models will be discussed. Nevertheless, differences between mice and humans should be carefully considered and limits of extrapolation should be wisely taken into account when translating experimental results into clinical use.
Eliciting Allergic Airway Inflammation
Historically, experimental asthma research was performed sensitizing rodents intraperitoneally with chicken ovalbumin (OVA) in combination with the pro-T2 adjuvant aluminum hydroxide (alum), followed by repetitive OVA exposures via the airways in order to elicit a Th-2 skewed adaptive immune response leading to eosinophilia, goblet cell hyperplasia and airway hyperresposiveness (18–20). Alum plays an important role in boosting the adaptive immune system via the inflammasome (21). The benefits of OVA lie on the fact that this substance is efficient, inexpensive and has well-characterized MHCI and MHCII epitopes and moreover OT1 and OT2 T-cell receptor transgenic mice are available, which allow monitoring of OVA-specific immune responses in the airways (22, 23), making OVA a very good option for unraveling underlying mechanisms of the disease. However, OVA is not allergenic upon inhalation, therefore it has been more and more replaced by naturally occurring allergens which possess higher clinical relevance.
Allergens frequently used in sensitization protocols include the house dust mites (HDM) Dermatopagoides pteronyssinus and farinae, the fungus Alternaria alternata, cockroach and pollen extracts. The principle of sensitization and challenge remained the same as it was for OVA, but here the use of the adjuvant became dispensable. Adjuvant-free models have been established using several intranasal instillations of these allergens, mimicking the natural exposure to airborne allergens via the nasal mucosa and airway tract (24–27). Some of these allergen complexes like HDM are characterized by an intrinsic protease activity which favors the initiation of the allergic response, stimulating the production of interleukin-25 (IL-25), IL-33, and thymic stromal lymphopoietin (TSLP) from airway epithelial cells, which in turn activate both dendritic cells, promoting T2 responses (28), and local ILC2, leading to the increased release of IL-5, IL-9, IL-13, and amphiregulin (26). Some others, like birch, grass or ragweed pollen grains, do not only release allergens, but also proinflammatory and immunomodulatory lipids and adenosine, which act as critical co-factors in the development of lung allergic inflammation (24, 29).
Whereas models using allergen sensitization/provocation via the airways is reminiscent of the standard route of sensitization in asthma and hay fever, there is also compelling data on the relevance of cutaneous exposure in the development of pulmonary allergy along the lines of the so-called “atopic march” in which eczema precedes food allergies, asthma or hay fever (30). Mouse models have confirmed that repeated epicutaneous sensitization to protein (aero)-allergens leads to phenotypes of atopic dermatitis and to increased risk of allergic rhinitis, lung inflammation and airway hyperresponsiveness, where skin barrier dysfunction and TSLP expression from keratinocytes play essential roles (31–35).
Besides the pulmonary inflammation upon allergen exposure, exacerbations induced by other factors like viral and bacterial infections are a characteristic feature in the course of disease (36, 37). Here murine models of asthma have been especially useful to identify possible effects of infections with the development of the pathology. Particularly, influenza (38–40), rhinovirus (41, 42) and respiratory syncytial virus (38, 43) are important pathogens in the childhood that have been associated with exacerbations in asthma.
Haptens are also broadly used in rodent models to investigate exacerbation in airway inflammation. Studies with toluene diisocyanate (TDI), trimellitic anhydride (TMA), dinitrofluorobenzene (DNFB), and picryl chloride (PCL), allowed dissecting the hapten-induced allergy as well as the similarities and differences between the different compounds (44–47). Rodent models of DNFB, a powerful sensitizer of non-atopic asthma (47), have recently shown increased numbers of macrophages in bronchoalveolar lavage fluid (BALF), tracheal hyper-reactivity and a strong neutrophilic-based lung inflammation that could reflect characteristic features of non-atopic asthma in humans (46, 48).
Quantifying Allergic Airway Inflammation
The physiological characteristics of asthma are mediated by a complex interaction between multiple effector cells and mediators.
The increased infiltration of inflammatory cells is determined by total and differential cell counts as well as measurement of inflammatory mediator content in the BALF or lung tissue (24, 49, 50). Upon allergen provocation, especially the role of eosinophils is shown to be indispensable for the development of allergic airway inflammation by mediating influx of T cell subsets [reviewed in (51)] into the lung (52, 53). For their release of pro-inflammatory mediators these cells are important contributors to pathophysiological changes, including airway epithelial cell damage, mucus hypersecretion and goblet cell hyperplasia which can be observed and quantified in histological staining of lung tissue (20, 54). In this context, eosinophils can be quantified by cell surface markers and by direct counting of stained cells in histological specimen (55).
Regarding the measurement of inflammatory mediators, tissue-based ex vivo cultures are another way to examine which cytokines are regulated in the development of airway inflammation and asthma and which cell type plays a decisive role in the concerned organs [reviewed in (56)]. As an alternative to the determination of cytokines in the supernatant of lung homogenates, stimulation of cells isolated from lung tissue or draining lymph nodes, by adding e.g., the allergen is used to evaluate the distinct cytokine patterns and to examine cell type specific responses more precisely (57, 58), allowing initial mechanistic conclusions about the observed phenotype.
As a hallmark of T2-driven allergic asthma, allergen-specific IgE responses are quantified in murine sera e.g., by means of ELISA (enzyme-linked immunosorbent assay) or functional cellular assays (59). Another factor to be taken into account in this context is IgG (and its subclasses), which are known to modulate inflammation via its receptors (FcγR) (60, 61). For example, antigen-specific IgG has been shown to improve allergic airway inflammation when signaling via FcγRIIB on DCs (62) and triggering different FcgR via certain IgG subclasses engage different pathways in murine IgE-independent anaphylaxis (63). Interestingly, similar mechanisms are discussed to take place in humans as well (64).
Airway hyperresponsiveness (AHR), defined as the predisposition of the airways to react excessively to bronchoconstrictor agents or to noxious stimuli, is an essential component of the asthma phenotype. The degree of AHR usually correlates with disease severity (65), and can be employed clinically for therapy management (66). AHR may not replace measurements of lung function such as FEV1, however it has been proposed to be included with other indices of lung function for asthma control (67). Similarly to spirometry in cooperative humans, lung function testing has been developed for rodents. Analysis of AHR in animal models is usually performed using one cholinergic agonist (methacholine, carbachol, histamine, serotonine), which act on the muscarinic receptor transduction pathway coupling to airway smooth muscle contraction (68). Measurement of AHR is usually performed shortly (24–48 h) after allergen challenge either in whole body chambers in conscious animals (body plethysmography) or in tracheostomized animals, using systems such as the Buxco® or the Flexivent®, with the agonist being either injected or aerosolized (24, 50, 69). Whilst the measurement of Penh (enhanced pause) using body plethysmography has lost acceptance in the scientific community (70), measurement of respiratory system resistance (RL) and dynamic compliance (DC) together with other physiologic parameters under mechanical ventilation in tracheostomized animals is often employed in asthma research (50, 71, 72). An increase in RL reflects both narrowing of the conducting airways and alterations in the lung periphery (distal airways and parenchyma). On the contrary, decreases in DC reflect only events occurring in the lung periphery. Therefore, if the response to an intervention is limited largely to RL, then a relatively proximal location is implicated for the effect, whereas a distinctive effect on DC is indicative of a more distal site of action. (73).
The limitation of this technique is based on the fact that it is only applicable in terminal experiments. This has been overcome by the use of oro-tracheal intubation technique, allowing for repetitive measurements in the same animals, which can be of advantage in longitudinal studies (74, 75).
Non-Allergic Asthma Models
Since the non-allergic asthmatic phenotype occurs also in patients with severe, steroid resistant asthma and management of asthma evolves into precision medicine with therapies directed toward specific phenotypes/endotypes (76–78), proper models of these conditions are needed to facilitate research on adequate therapeutic options (79). In this regard, it was shown that a Th17-driven non-eosinophilic lung inflammation is insensitive to several treatment options including steroids, by using adoptive transfer of in vitro polarized antigen specific Th17 cells with subsequent pulmonary allergen application (80, 81). Manni et al. could create a mixed phenotype by adoptive transfer of T2 and Th17 cells enabling them to dissect contributions of the different cytokine pathways to distinct features of airway disease like mucus metaplasia or tissue inflammation (82). Microbial components like bacterial lipopolysaccharides (LPS) used as adjuvants in airway application of allergen have been proven to elicit a non-eosinophilic airway inflammation by triggering pathogen recognition receptors (PRR). Kim et al. could demonstrate that in such models the dose of LPS during sensitization plays a decisive role in shaping the resulting lung inflammation either toward eosinophilic (low dose LPS) or neutrophilic (high dose LPS) inflammation (83). Comparing this airway sensitization model to intraperitoneal allergen application (with alum) Wilson et al. could illustrate how different sensitization regimes lead to different molecular and phenotypical pattern in the resulting airway disease identifying a prominent role for Th17 in neutrophilic airway inflammation and AHR (84). Hadebe et al. demonstrated the importance of microbial triggers in airway immune responses via initiation of a non-allergic steroid-refractory airway inflammation by combining two different agents (LPS and beta-glucan) (85). A more sophisticated approach uses biolistic transfection of a plasmid containing the genetic information of the allergen via gene-gun, with targeted expression in dendritic cells ensured by a specific promotor, leading to a Th1/Tc1 driven inflammation depending on IFNγ that is sensitive to steroid treatment (86). Application of Poly I/C, a dsRNA analog mimicking a viral infection, in combination with an allergen results in a Th1-driven airway inflammation as well, offering the possibility to study the pathomechanism underlying virus-induced airway inflammation (87). Taking advantage of the possibility to shape the resulting airway inflammation by means of different sensitization regimes (using the same allergen: house dust mite), Tan et al. were able to directly compare transcriptomic lung profiles of eosinophilic, neutrophilic and mixed phenotypes enabling identification of molecular pattern that are linked to distinct inflammatory phenotypes (88).
Aspirin-exacerbated respiratory disease (AERD) is a common, severe variant of asthma, which affects 7–10% of all asthmatics and is associated with overproduction of cysteinyl leukotrienes (cysLTs) and respiratory reactions to drugs that block cyclooxygenase 1 (89). The pathophysiology has not been fully solved yet, but in order to model this disease deficiency or overexpression genetic animal models have been used presenting severe eosinophilic respiratory mucosal inflammation (90, 91).
Chronicity and Remodeling
Most of the above-mentioned models focus on the development of symptoms after a short period of antigen exposure. While this has provided a broad range of information on causal and mechanistic effects on asthma, it usually cannot mimic characteristics like chronic inflammation of the airway wall, mucus production and remodeling (92–95).
To compensate that limitation, several methods applying allergen for a longer period of time have been established. This causes a protracted experimental window up to several months and in some cases, due to the continuous exposure to the allergen, leads to tolerance in the mice (96–101). The transgenic technology allowed the generation of mice with characteristics of chronic asthma and airway remodeling (102, 103). Furthermore, transgenic models allowed the identification of an important migration factor of DCs to the lung (104) and the role of IL-33 receptor suppressor of tumorigenicity 2 (ST2) in development of chronic asthma in mice by regulating ILC2s, mast cells, IL9 and IL-13 in the lungs (105). In addition, recent gene modification in mice allowed to identify for example the role of the potassium channels Kca3.1 in airway remodeling (106), and the regulatory role of semaphorin 3E (Sema3E) in inflammatory and remodeling responses in chronic asthma (107).
Recently, CRISPR/Cas 9, a gene disruption technology, allowed to knock-out/down several genes in associated with exacerbation, inflammation and remodeling in asthmatic diseases, identifying roles for these molecules in some pathophysiological features of asthma. For example, using the CRISPR/Cas 9 technology the transient receptor potential (TRP) 1, an ion channel was successfully knocked-out by Reese et al. They could demonstrate its role in the protection from airway inflammation in rats as well as in mice, suggesting TRP1A as a therapeutic target in asthma (108). In another study depletion of long non-coding RNAs (lncRNAs), particularly AK085865, led to reduction of the inflammatory response in a murine model of asthma, by modulating differentiation of innate lymphoid cells progenitor (ILCP) into ILC2s (109). CRISPR/Cas 9, because of its high target specificity, is a tool that could be of high importance in the understanding of the pathomechanisms of asthma and identification of novel therapeutic targets.
Humanized Mouse Models
Despite the widespread use of mouse models for the evaluation of asthmatic diseases, there are restrictions when comparing components of the murine biology (e.g., the immune system) with those of the human biology (110). Humanized mouse models, that are immunodeficient mice engrafted with functional human (immune) cells, help to overcome some of these discrepancies. They have become an important pre-clinical tool for biomedical research, but to date only a small number of humanized mouse models are available in the research field of asthma.
Currently immunodeficient mouse strains for this purpose are often based on IL2rgnull mice, which lack a functional common gamma chain (γc) of the IL-2 receptor. This chain is not only part of the receptor complex for IL-2, but assembles with other chains to form receptors for IL-4, IL-7, IL-9, IL-15, and IL-21 as well, which are expressed on several cells of the immune system and signaling via these receptors is essential for homeostasis of these immune cells [reviewed in (111)]. Thus, the lack of γc results in absence of functional T, B, and NK cells.
The three most commonly used strains in humanized models are: the NSG mouse, the NOG mouse and the BRG or BALB/c-Rag2null IL2rgnull mouse. BRG and NSG mice have no gamma chain while NOG mice have a truncated cytoplasmic domain of the gamma chain, preventing signal transmission (112, 113). All three models allow for efficient engraftment with human immune cells, due to a severe impairment in development of T and B as well as NK cells. These new models have enabled a multitude of new findings in the field of asthma research such as the interaction of allergen immunotherapy, clinical tolerance and cellular response, as well as new therapeutic options through the induction of peripheral tolerance by sGARP (glycoprotein A repetitions predominant) (114, 115). New mechanistic relationships were also clarified, such as the influence of the IL-33/IL-13 axis on the asthmatic airway inflammation or the anti-inflammatory effect of IL-35 in asthmatic diseases (116). Based on the immunodeficient IL2rgnull mouse, further mouse models emerged, including the Hu-SRC-SCID mouse and the BLT mouse as well as the Hu-PBL-SCID mouse providing further insight into our understanding of the development of AHR as a characteristic feature of allergic asthma (117) and discovery of new therapeutics, such as the use of TIM-1 antagonists as a possible treatment strategy for asthma (118).
Limits of Extrapolation
Taken together, recent developments in asthma research led to a shift from solely applying allergic T2-driven eosinophilic airway inflammation models to a broader variety of airway inflammation models following the demand for precision medicine based on phenotype/endotypes in asthma management. However, it is important to be aware that, while the main hallmarks of asthmatic airway inflammation can be mimicked in such models, there are certain differences between mice and men which are reviewed in detail elsewhere (119, 120), that might limit translational impact of results obtained in mouse models. Some of these differences include immunological features (121, 122), which might be overcome by using humanized models, whereas others like anatomical discrepancies [e.g., lung anatomy, cell composition in the airways (123, 124)] will still differ in humanized mice. Moreover, the course of disease and treatment can often not be mimicked: asthma often begins in childhood when the lung is not fully developed yet, whereas experiments are mostly done in mice which do not spontaneously develop asthma, using adult animals with fully developed lung structure. Since the immunological response is shaped not only by the route, but also the amount and frequency of allergen exposure (23, 125, 126), a model that efficiently results in allergic airway inflammation might not necessarily mimic allergen exposure as it is experienced by the patients. Direct extrapolation of efficacy for therapeutic interventions obtained in mouse models is hampered by the fact that mouse models are conducted under highly controlled conditions (e.g., under specific pathogen-free conditions) which substantially affects the diversity of intrinsic and acquired immune responsiveness and may cause substantial immunological differences between these models and human (127, 128). Moreover, experiments are usually performed in genetically similar animals, which do not reflect the heterogeneity of asthmatic patients. To sum this up there is not the “one asthma model” mimicking human disease, but there is a huge variety of different approaches that allow to closely reproduce certain aspects of this complex syndrome with certain advantages and disadvantages (Table 1), enabling researchers to examine a scientific question from several different angels in order to contribute mosaic pieces for better understanding asthma.
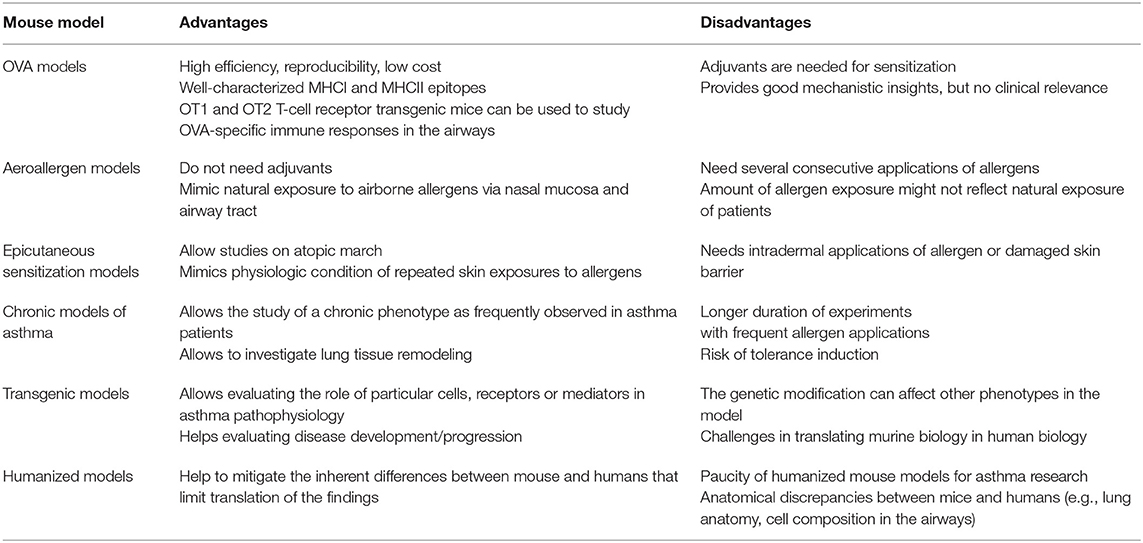
Table 1. Advantages and disadvantages of T2-driven asthma mouse models.
Author Contributions
FA and MA conceived topic and structure of this mini review. FA, SM, ES, FB-P, and MA wrote the review. FA, FB-P, and MA reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Croisant S. Epidemiology of asthma: prevalence and burden of disease. Adv Exp Med Biol. (2014) 795:17–29. doi: 10.1007/978-1-4614-8603-9_2
2. Boulet LP, Reddel HK, Bateman E, Pedersen S, FitzGerald JM, O'Byrne PM. The global initiative for asthma (GINA): 25 years later. Eur Respir J. (2019) 54:1900598. doi: 10.1183/13993003.00598-2019
3. Reddel HK, Bateman ED, Becker A, Boulet LP, Cruz AA, Drazen JM, et al. A summary of the new GINA strategy: a roadmap to asthma control. Eur Respir J. (2015) 46:622–39. doi: 10.1183/13993003.00853-2015
4. Wenzel S. Severe asthma: from characteristics to phenotypes to endotypes. Clin Exp Allergy. (2012) 42:650–8. doi: 10.1111/j.1365-2222.2011.03929.x
5. Agache I, Akdis C, Jutel M, Virchow JC. Untangling asthma phenotypes and endotypes. Allergy. (2012) 67:835–46. doi: 10.1111/j.1398-9995.2012.02832.x
6. Fahy JV. Type 2 inflammation in asthma–present in most, absent in many. Nat Rev Immunol. (2015) 15:57–65. doi: 10.1038/nri3786
7. Kuo CS, Pavlidis S, Loza M, Baribaud F, Rowe A, Pandis I, et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J. (2017) 49:1602135. doi: 10.1183/13993003.02135-2016
9. Pite H, Aguiar L, Morello J, Monteiro EC, Alves AC, Bourbon M, et al. Metabolic dysfunction and asthma: current perspectives. J Asthma Allergy. (2020) 13:237–47. doi: 10.2147/JAA.S208823
10. Locksley RM. Asthma and allergic inflammation. Cell. (2010) 140:777–83. doi: 10.1016/j.cell.2010.03.004
11. Agache I, Sugita K, Morita H, Akdis M, Akdis CA. The complex type 2 endotype in allergy and asthma: from laboratory to bedside. Curr Allergy Asthma Rep. (2015) 15:29. doi: 10.1007/s11882-015-0529-x
12. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. (2015) 16:45–56. doi: 10.1038/ni.3049
13. Esteban-Gorgojo I, Antolin-Amerigo D, Dominguez-Ortega J, Quirce S. Non-eosinophilic asthma: current perspectives. J Asthma Allergy. (2018) 11:267–81. doi: 10.2147/JAA.S153097
14. Furukawa T, Sakagami T, Koya T, Hasegawa T, Kawakami H, Kimura Y, et al. Characteristics of eosinophilic and non-eosinophilic asthma during treatment with inhaled corticosteroids. J Asthma. (2015) 52:417–22. doi: 10.3109/02770903.2014.975357
15. Ullah MA, Revez JA, Loh Z, Simpson J, Zhang V, Bain L, et al. Allergen-induced IL-6 trans-signaling activates gammadelta T cells to promote type 2 and type 17 airway inflammation. J Allergy Clin Immunol. (2015) 136:1065–73. doi: 10.1016/j.jaci.2015.02.032
16. Aun MV, Bonamichi-Santos R, Arantes-Costa FM, Kalil J, Giavina-Bianchi P. Animal models of asthma: utility and limitations. J Asthma Allergy. (2017) 10:293–301. doi: 10.2147/JAA.S121092
17. Sagar S, Akbarshahi H, Uller L. Translational value of animal models of asthma: challenges and promises. Eur J Pharmacol. (2015) 759:272–7. doi: 10.1016/j.ejphar.2015.03.037
18. Alessandrini F, Weichenmeier I, van Miert E, Takenaka S, Karg E, Blume C, et al. Effects of ultrafine particles-induced oxidative stress on Clara cells in allergic lung inflammation. Part Fibre Toxicol. (2010) 7:11. doi: 10.1186/1743-8977-7-11
19. Debeuf N, Haspeslagh E, van Helden M, Hammad H, Lambrecht BN. Mouse models of asthma. Curr Protoc Mouse Biol. (2016) 6:169–84. doi: 10.1002/cpmo.4
20. Alessandrini F, Schulz H, Takenaka S, Lentner B, Karg E, Behrendt H, et al. Effects of ultrafine carbon particle inhalation on allergic inflammation of the lung. J Allergy Clin Immunol. (2006) 117:824–30. doi: 10.1016/j.jaci.2005.11.046
21. Kool M, Petrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. (2008) 181:3755–9. doi: 10.4049/jimmunol.181.6.3755
22. Daniels NJ, Hyde E, Ghosh S, Seo K, Price KM, Hoshino K, et al. Antigen-specific cytotoxic T lymphocytes target airway CD103+ and CD11b+ dendritic cells to suppress allergic inflammation. Mucosal Immunol. (2016) 9:229–39. doi: 10.1038/mi.2015.55
23. Aguilar-Pimentel JA, Alessandrini F, Huster KM, Jakob T, Schulz H, Behrendt H, et al. Specific CD8 T cells in IgE-mediated allergy correlate with allergen dose and allergic phenotype. Am J Respir Crit Care Med. (2010) 181:7–16. doi: 10.1164/rccm.200902-0190OC
24. Wimmer M, Alessandrini F, Gilles S, Frank U, Oeder S, Hauser M, et al. Pollen-derived adenosine is a necessary cofactor for ragweed allergy. Allergy. (2015) 70:944–54. doi: 10.1111/all.12642
25. Yee MC, Nichols HL, Polley D, Saifeddine M, Pal K, Lee K, et al. Protease-activated receptor-2 signaling through beta-arrestin-2 mediates Alternaria alkaline serine protease-induced airway inflammation. Am J Physiol Lung Cell Mol Physiol. (2018) 315:L1042–57. doi: 10.1152/ajplung.00196.2018
26. Yasuda Y, Nagano T, Kobayashi K, Nishimura Y. Group 2 innate lymphoid cells and the house dust mite-induced asthma mouse model. Cells. (2020) 9:1178. doi: 10.3390/cells9051178
27. Arizmendi NG, Abel M, Puttagunta L, Asaduzzaman M, Davidson C, Karimi K, et al. Mucosal exposure to cockroach extract induces allergic sensitization and allergic airway inflammation. Allergy Asthma Clin Immunol. (2011) 7:22. doi: 10.1186/1710-1492-7-22
28. Tjota MY, Hrusch CL, Blaine KM, Williams JW, Barrett NA, Sperling AI. Signaling through FcRgamma-associated receptors on dendritic cells drives IL-33-dependent TH2-type responses. J Allergy Clin Immunol. (2014) 134:706–13 e8. doi: 10.1016/j.jaci.2014.06.013
29. Oeder S Alessandrini F Wirz OF Braun A Wimmer M Frank U . Pollen-derived nonallergenic substances enhance Th2-induced IgE production in B cells. Allergy. (2015) 70:1450–60. doi: 10.1111/all.12707
30. Davidson WF, Leung DYM, Beck LA, Berin CM, Boguniewicz M, Busse WW, et al. Report from the National Institute of Allergy and Infectious Diseases workshop on “Atopic dermatitis and the atopic march: Mechanisms and interventions”. J Allergy Clin Immunol. (2019) 143:894–913. doi: 10.1016/j.jaci.2019.01.003
31. He R, Kim HY, Yoon J, Oyoshi MK, MacGinnitie A, Goya S, et al. Exaggerated IL-17 response to epicutaneous sensitization mediates airway inflammation in the absence of IL-4 and IL-13. J Allergy Clin Immunol. (2009) 124:761–70 e1. doi: 10.1016/j.jaci.2009.07.040
32. Akei HS, Brandt EB, Mishra A, Strait RT, Finkelman FD, Warrier MR, et al. Epicutaneous aeroallergen exposure induces systemic TH2 immunity that predisposes to allergic nasal responses. J Allergy Clin Immunol. (2006) 118:62–9. doi: 10.1016/j.jaci.2006.04.046
33. Strid J, Hourihane J, Kimber I, Callard R, Strobel S. Disruption of the stratum corneum allows potent epicutaneous immunization with protein antigens resulting in a dominant systemic Th2 response. Eur J Immunol. (2004) 34:2100–9. doi: 10.1002/eji.200425196
34. Demehri S, Morimoto M, Holtzman MJ, Kopan R. Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol. (2009) 7:e1000067. doi: 10.1371/journal.pbio.1000067
35. Han H, Xu W, Headley MB, Jessup HK, Lee KS, Omori M, et al. Thymic stromal lymphopoietin (TSLP)-mediated dermal inflammation aggravates experimental asthma. Mucosal Immunol. (2012) 5:342–51. doi: 10.1038/mi.2012.14
36. Hansbro NG, Horvat JC, Wark PA, Hansbro PM. Understanding the mechanisms of viral induced asthma: new therapeutic directions. Pharmacol Ther. (2008) 117:313–53. doi: 10.1016/j.pharmthera.2007.11.002
37. Iikura M, Hojo M, Koketsu R, Watanabe S, Sato A, Chino H, et al. The importance of bacterial and viral infections associated with adult asthma exacerbations in clinical practice. PLoS ONE. (2015) 10:e0123584. doi: 10.1371/journal.pone.0123584
38. Barends M, de Rond LG, Dormans J, van Oosten M, Boelen A, Neijens HJ, et al. Respiratory syncytial virus, pneumonia virus of mice, and influenza A virus differently affect respiratory allergy in mice. Clin Exp Allergy. (2004) 34:488–96. doi: 10.1111/j.1365-2222.2004.01906.x
39. Ravanetti L, Dijkhuis A, Sabogal Pineros YS, Bal SM, Dierdorp BS, Dekker T, et al. An early innate response underlies severe influenza-induced exacerbations of asthma in a novel steroid-insensitive and anti-IL-5-responsive mouse model. Allergy. (2017) 72:737–53. doi: 10.1111/all.13057
40. Doorley LA, LeMessurier KS, Iverson AR, Palipane M, Samarasinghe AE. Humoral immune responses during asthma and influenza co-morbidity in mice. Immunobiology. (2017) 222:1064–73. doi: 10.1016/j.imbio.2017.08.002
41. Mahmutovic Persson I, Menzel M, Ramu S, Cerps S, Akbarshahi H, Uller L. IL-1beta mediates lung neutrophilia and IL-33 expression in a mouse model of viral-induced asthma exacerbation. Respir Res. (2018) 19:16. doi: 10.1186/s12931-018-0725-z
42. Kantor DB, Stenquist N, McDonald MC, Schultz BJ, Hauptman M, Smallwood CD, et al. Rhinovirus and serum IgE are associated with acute asthma exacerbation severity in children. J Allergy Clin Immunol. (2016) 138:1467–471 e9. doi: 10.1016/j.jaci.2016.04.044
43. Hu X, Li X, Hu C, Qin L, He R, Luo L, et al. Respiratory syncytial virus exacerbates OVA-mediated asthma in mice through C5a-C5aR regulating CD4(+)T cells immune responses. Sci Rep. (2017) 7:15207. doi: 10.1038/s41598-017-15471-w
44. Vanoirbeek JA, Tarkowski M, De Vooght V, Nemery B, Hoet PH. Immunological determinants in a mouse model of chemical-induced asthma after multiple exposures. Scand J Immunol. (2009) 70:25–33. doi: 10.1111/j.1365-3083.2009.02263.x
45. Russjan E, Kaczynska K. Murine models of hapten-induced asthma. Toxicology. (2018) 410:41–8. doi: 10.1016/j.tox.2018.09.001
46. Russjan E, Kaczynska K. Beneficial effects of neurotensin in murine model of hapten-induced asthma. Int J Mol Sci. (2019) 20:5025. doi: 10.3390/ijms20205025
47. van Houwelingen AH, Kraneveld AD, Nijkamp FP. Hapten-induced hypersensitivity reactions in the airways: atopic versus non-atopic. Environ Toxicol Pharmacol. (2002) 11:197–205. doi: 10.1016/S1382-6689(02)00007-8
48. Bozkurt TE, Kaya Y, Durlu-Kandilci NT, Onder S, Sahin-Erdemli I. The effect of cannabinoids on dinitrofluorobenzene-induced experimental asthma in mice. Respir Physiol Neurobiol. (2016) 231:7–13. doi: 10.1016/j.resp.2016.05.012
49. Alessandrini F, Beck-Speier I, Krappmann D, Weichenmeier I, Takenaka S, Karg E, et al. Role of oxidative stress in ultrafine particle-induced exacerbation of allergic lung inflammation. Am J Respir Crit Care Med. (2009) 179:984–91. doi: 10.1164/rccm.200807-1061OC
50. Marzaioli V, Aguilar-Pimentel JA, Weichenmeier I, Luxenhofer G, Wiemann M, Landsiedel R, et al. Surface modifications of silica nanoparticles are crucial for their inert versus proinflammatory and immunomodulatory properties. Int J Nanomed. (2014) 9:2815–32. doi: 10.2147/IJN.S57396
51. Gelfand EW, Joetham A, Wang M, Takeda K, Schedel M. Spectrum of T-lymphocyte activities regulating allergic lung inflammation. Immunol Rev. (2017) 278:63–86. doi: 10.1111/imr.12561
52. Jacobsen EA, Ochkur SI, Pero RS, Taranova AG, Protheroe CA, Colbert DC, et al. Allergic pulmonary inflammation in mice is dependent on eosinophil-induced recruitment of effector T cells. J Exp Med. (2008) 205:699–710. doi: 10.1084/jem.20071840
53. Mattes J, Yang M, Mahalingam S, Kuehr J, Webb DC, Simson L, et al. Intrinsic defect in T cell production of interleukin (IL)-13 in the absence of both IL-5 and eotaxin precludes the development of eosinophilia and airways hyperreactivity in experimental asthma. J Exp Med. (2002) 195:1433–44. doi: 10.1084/jem.20020009
54. Sussan TE, Gajghate S, Chatterjee S, Mandke P, McCormick S, Sudini K, et al. Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L27–36. doi: 10.1152/ajplung.00398.2014
55. Dyer KD, Garcia-Crespo KE, Killoran KE, Rosenberg HF. Antigen profiles for the quantitative assessment of eosinophils in mouse tissues by flow cytometry. J Immunol Methods. (2011) 369:91–7. doi: 10.1016/j.jim.2011.04.009
56. Lambrecht BN, Hammad H, Fahy JV. The cytokines of asthma. Immunity. (2019) 50:975–91. doi: 10.1016/j.immuni.2019.03.018
57. Turner DL, Goldklang M, Cvetkovski F, Paik D, Trischler J, Barahona J, et al. Biased generation and in situ activation of lung tissue-resident memory CD4 T cells in the pathogenesis of allergic asthma. J Immunol. (2018) 200:1561–9. doi: 10.4049/jimmunol.1700257
58. Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, et al. Interleukin-2-dependent allergen-specific tissue-resident memory cells drive asthma. Immunity. (2016) 44:155–66. doi: 10.1016/j.immuni.2015.11.004
59. Ward MDW, Copeland LB. Evaluating antigen-specific IgE using the rat basophil leukemia cell (RBL) assay. Methods Mol Biol. (2018) 1803:371–81. doi: 10.1007/978-1-4939-8549-4_22
60. Cassard L, Jonsson F, Arnaud S, Daeron M. Fcgamma receptors inhibit mouse and human basophil activation. J Immunol. (2012) 189:2995–3006. doi: 10.4049/jimmunol.1200968
61. Nimmerjahn F, Ravetch JV. Fc-receptors as regulators of immunity. Adv Immunol. (2007) 96:179–204. doi: 10.1016/S0065-2776(07)96005-8
62. Ishikawa Y, Kobayashi K, Yamamoto M, Nakata K, Takagawa T, Funada Y, et al. Antigen-Specific IgG ameliorates allergic airway inflammation via Fcgamma receptor IIB on dendritic cells. Respir Res. (2011) 12:42. doi: 10.1186/1465-9921-12-42
63. Beutier H, Gillis CM, Iannascoli B, Godon O, England P, Sibilano R, et al. IgG subclasses determine pathways of anaphylaxis in mice. J Allergy Clin Immunol. (2017) 139:269–80 e7. doi: 10.1016/j.jaci.2016.03.028
64. Finkelman FD, Khodoun MV, Strait R. Human IgE-independent systemic anaphylaxis. J Allergy Clin Immunol. (2016) 137:1674–80. doi: 10.1016/j.jaci.2016.02.015
65. Fowler SJ, Dempsey OJ, Sims EJ, Lipworth BJ. Screening for bronchial hyperresponsiveness using methacholine and adenosine monophosphate. Relationship to asthma severity and beta(2)-receptor genotype. Am J Respir Crit Care Med. (2000) 162(4 Pt 1):1318–22. doi: 10.1164/ajrccm.162.4.9912103
66. Leuppi JD, Salome CM, Jenkins CR, Anderson SD, Xuan W, Marks GB, et al. Predictive markers of asthma exacerbation during stepwise dose reduction of inhaled corticosteroids. Am J Respir Crit Care Med. (2001) 163:406–12. doi: 10.1164/ajrccm.163.2.9912091
67. Reddel HK, Taylor DR, Bateman ED, Boulet LP, Boushey HA, Busse WW, et al. An official American Thoracic Society/European Respiratory Society statement: asthma control and exacerbations: standardizing endpoints for clinical asthma trials and clinical practice. Am J Respir Crit Care Med. (2009) 180:59–99. doi: 10.1164/rccm.200801-060ST
68. Fernandez-Rodriguez S, Broadley KJ, Ford WR, Kidd EJ. Increased muscarinic receptor activity of airway smooth muscle isolated from a mouse model of allergic asthma. Pulm Pharmacol Ther. (2010) 23:300–7. doi: 10.1016/j.pupt.2010.03.001
69. Glaab T, Mitzner W, Braun A, Ernst H, Korolewitz R, Hohlfeld JM, et al. Repetitive measurements of pulmonary mechanics to inhaled cholinergic challenge in spontaneously breathing mice. J Appl Physiol (1985). (2004) 97:1104–11. doi: 10.1152/japplphysiol.01182.2003
70. Adler A, Cieslewicz G, Irvin CG. Unrestrained plethysmography is an unreliable measure of airway responsiveness in BALB/c and C57BL/6 mice. J Appl Physiol (1985). (2004) 97:286–92. doi: 10.1152/japplphysiol.00821.2003
71. McGovern TK, Robichaud A, Fereydoonzad L, Schuessler TF, Martin JG. Evaluation of respiratory system mechanics in mice using the forced oscillation technique. J Vis Exp. (2013) 75:e50172. doi: 10.3791/50172
72. Spacova I, Petrova MI, Fremau A, Pollaris L, Vanoirbeek J, Ceuppens JL, et al. Intranasal administration of probiotic Lactobacillus rhamnosus GG prevents birch pollen-induced allergic asthma in a murine model. Allergy. (2019) 74:100–10. doi: 10.1111/all.13502
73. Kanehiro A, Takeda K, Joetham A, Tomkinson A, Ikemura T, Irvin CG, et al. Timing of administration of anti-VLA-4 differentiates airway hyperresponsiveness in the central and peripheral airways in mice. Am J Respir Crit Care Med. (2000) 162(3 Pt 1):1132–9. doi: 10.1164/ajrccm.162.3.9910100
74. Bonnardel E, Prevel R, Campagnac M, Dubreuil M, Marthan R, Berger P, et al. Determination of reliable lung function parameters in intubated mice. Respir Res. (2019) 20:211. doi: 10.1186/s12931-019-1177-9
75. De Vleeschauwer SI, Rinaldi M, De Vooght V, Vanoirbeek JA, Vanaudenaerde BM, Verbeken EK, et al. Repeated invasive lung function measurements in intubated mice: an approach for longitudinal lung research. Lab Anim. (2011) 45:81–9. doi: 10.1258/la.2010.010111
76. Assaf SM, Hanania NA. Biological treatments for severe asthma. Curr Opin Allergy Clin Immunol. (2019) 19:379–86. doi: 10.1097/ACI.0000000000000549
77. Chung KF. Precision medicine in asthma: linking phenotypes to targeted treatments. Curr Opin Pulm Med. (2018) 24:4–10. doi: 10.1097/MCP.0000000000000434
78. Lang DM. Severe asthma: epidemiology, burden of illness, and heterogeneity. Allergy Asthma Proc. (2015) 36:418–24. doi: 10.2500/aap.2015.36.3908
79. Martin RA, Hodgkins SR, Dixon AE, Poynter ME. Aligning mouse models of asthma to human endotypes of disease. Respirology. (2014) 19:823–33. doi: 10.1111/resp.12315
80. McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. (2008) 181:4089–97. doi: 10.4049/jimmunol.181.6.4089
81. Joean O, Hueber A, Feller F, Jirmo AC, Lochner M, Dittrich AM, et al. Suppression of Th17-polarized airway inflammation by rapamycin. Sci Rep. (2017) 7:15336. doi: 10.1038/s41598-017-15750-6
82. Manni ML, Mandalapu S, McHugh KJ, Elloso MM, Dudas PL, Alcorn JF. Molecular mechanisms of airway hyperresponsiveness in a murine model of steroid-resistant airway inflammation. J Immunol. (2016) 196:963–77. doi: 10.4049/jimmunol.1501531
83. Kim YK, Oh SY, Jeon SG, Park HW, Lee SY, Chun EY, et al. Airway exposure levels of lipopolysaccharide determine type 1 versus type 2 experimental asthma. J Immunol. (2007) 178:5375–82. doi: 10.4049/jimmunol.178.8.5375
84. Wilson RH, Whitehead GS, Nakano H, Free ME, Kolls JK, Cook DN. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med. (2009) 180:720–30. doi: 10.1164/rccm.200904-0573OC
85. Hadebe S, Kirstein F, Fierens K, Chen K, Drummond RA, Vautier S, et al. Microbial ligand costimulation drives neutrophilic steroid-refractory asthma. PLoS ONE. (2015) 10:e0134219. doi: 10.1371/journal.pone.0134219
86. Stein J, Maxeiner JH, Montermann E, Hohn Y, Raker V, Taube C, et al. Non-eosinophilic airway hyper-reactivity in mice, induced by IFN-gamma producing CD4(+) and CD8(+) lung T cells, is responsive to steroid treatment. Scand J Immunol. (2014) 80:327–38. doi: 10.1111/sji.12217
87. Jeon SG, Moon HG, Kim YS, Choi JP, Shin TS, Hong SW, et al. 15-lipoxygenase metabolites play an important role in the development of a T-helper type 1 allergic inflammation induced by double-stranded RNA. Clin Exp Allergy. (2009) 39:908–17. doi: 10.1111/j.1365-2222.2009.03211.x
88. Tan HT, Hagner S, Ruchti F, Radzikowska U, Tan G, Altunbulakli C, et al. Tight junction, mucin, and inflammasome-related molecules are differentially expressed in eosinophilic, mixed, and neutrophilic experimental asthma in mice. Allergy. (2019) 74:294–307. doi: 10.1111/all.13619
89. Rajan JP, Wineinger NE, Stevenson DD, White AA. Prevalence of aspirin-exacerbated respiratory disease among asthmatic patients: a meta-analysis of the literature. J Allergy Clin Immunol. (2015) 135:676–81 e1. doi: 10.1016/j.jaci.2014.08.020
90. Liu T, Laidlaw TM, Katz HR, Boyce JA. Prostaglandin E2 deficiency causes a phenotype of aspirin sensitivity that depends on platelets and cysteinyl leukotrienes. Proc Natl Acad Sci USA. (2013) 110:16987–92. doi: 10.1073/pnas.1313185110
91. Liu T, Kanaoka Y, Barrett NA, Feng C, Garofalo D, Lai J, et al. Aspirin-exacerbated respiratory disease involves a cysteinyl leukotriene-driven IL-33-mediated mast cell activation pathway. J Immunol. (2015) 195:3537–45. doi: 10.4049/jimmunol.1500905
92. Lloyd CM. Building better mouse models of asthma. Curr Allergy Asthma Rep. (2007) 7:231–6. doi: 10.1007/s11882-007-0077-0
93. Van Hove CL, Maes T, Cataldo DD, Gueders MM, Palmans E, Joos GF, et al. Comparison of acute inflammatory and chronic structural asthma-like responses between C57BL/6 and BALB/c mice. Int Arch Allergy Immunol. (2009) 149:195–207. doi: 10.1159/000199715
94. Fehrenbach H, Wagner C, Wegmann M. Airway remodeling in asthma: what really matters. Cell Tissue Res. (2017) 367:551–69. doi: 10.1007/s00441-016-2566-8
95. Agache I, Bilo M, Braunstahl GJ, Delgado L, Demoly P, Eigenmann P, et al. In vivo diagnosis of allergic diseases–allergen provocation tests. Allergy. (2015) 70:355–65. doi: 10.1111/all.12586
96. Yiamouyiannis CA, Schramm CM, Puddington L, Stengel P, Baradaran-Hosseini E, Wolyniec WW, et al. Shifts in lung lymphocyte profiles correlate with the sequential development of acute allergic and chronic tolerant stages in a murine asthma model. Am J Pathol. (1999) 154:1911–21. doi: 10.1016/S0002-9440(10)65449-1
97. Lee HY, Kim IK, Yoon HK, Kwon SS, Rhee CK, Lee SY. Inhibitory effects of resveratrol on airway remodeling by transforming growth factor-beta/smad signaling pathway in chronic asthma model. Allergy Asthma Immunol Res. (2017) 9:25–34. doi: 10.4168/aair.2017.9.1.25
98. Schramm CM, Puddington L, Wu C, Guernsey L, Gharaee-Kermani M, Phan SH, et al. Chronic inhaled ovalbumin exposure induces antigen-dependent but not antigen-specific inhalational tolerance in a murine model of allergic airway disease. Am J Pathol. (2004) 164:295–304. doi: 10.1016/S0002-9440(10)63119-7
99. Coltherd JC, Rodgers DT, Lawrie RE, Al-Riyami L, Suckling CJ, Harnett W, et al. The parasitic worm-derived immunomodulator, ES-62 and its drug-like small molecule analogues exhibit therapeutic potential in a model of chronic asthma. Sci Rep. (2016) 6:19224. doi: 10.1038/srep19224
100. Hove CLV, Maes T, Joos GF, Tournoy KG. Prolonged inhaled allergen exposure can induce persistent tolerance. Am J Respir Cell Mol Biol. (2007). 36:573–84. doi: 10.1165/rcmb.2006-0385OC
101. Bracken SJ, Adami AJ, Szczepanek SM, Ehsan M, Natarajan P, Guernsey LA, et al. Long-term exposure to house dust mite leads to the suppression of allergic airway disease despite persistent lung inflammation. Int Arch Allergy Immunol. (2015) 166:243–58. doi: 10.1159/000381058
102. Elias J. The relationship between asthma and COPD. Lessons from transgenic mice. Chest. (2004) 126(2 Suppl.):111S−6S; discussion 159S−61S. doi: 10.1378/chest.126.2_suppl_1.111S
103. Sun X, Hou T, Cheung E, Iu TN, Tam VW, Chu IM, et al. Anti-inflammatory mechanisms of the novel cytokine interleukin-38 in allergic asthma. Cell Mol Immunol. (2020) 17:631–46. doi: 10.1038/s41423-019-0300-7
104. Echeverri Tirado LC, Ghonim MA, Wang J, Al-Khami AA, Wyczechowska D, Luu HH, et al. PARP-1 is critical for recruitment of dendritic cells to the lung in a mouse model of asthma but dispensable for their differentiation and function. Mediat Inflamm. (2019) 2019:1656484. doi: 10.1155/2019/1656484
105. Verma M, Liu S, Michalec L, Sripada A, Gorska MM, Alam R. Experimental asthma persists in IL-33 receptor knockout mice because of the emergence of thymic stromal lymphopoietin-driven IL-9(+) and IL-13(+) type 2 innate lymphoid cell subpopulations. J Allergy Clin Immunol. (2018) 142:793–803 e8. doi: 10.1016/j.jaci.2017.10.020
106. Yu Z, Wang Y, Qin L, Chen H. Functional cooperation between KCa3.1 and TRPV4 channels in bronchial smooth muscle cell proliferation associated with chronic asthma. Front Pharmacol. (2017) 8:559. doi: 10.3389/fphar.2017.00559
107. Movassagh H, Shan L, Duke-Cohan JS, Chakir J, Halayko AJ, Koussih L, et al. Downregulation of semaphorin 3E promotes hallmarks of experimental chronic allergic asthma. Oncotarget. (2017) 8:98953–63. doi: 10.18632/oncotarget.22144
108. Reese RM, Dourado M, Anderson K, Warming S, Stark KL, Balestrini A, et al. Behavioral characterization of a CRISPR-generated TRPA1 knockout rat in models of pain, itch, and asthma. Sci Rep. (2020) 10:979. doi: 10.1038/s41598-020-57936-5
109. Pei W, Zhang Y, Li X, Luo M, Chen T, Zhang M, et al. LncRNA AK085865 depletion ameliorates asthmatic airway inflammation by modulating macrophage polarization. Int Immunopharmacol. (2020) 83:106450. doi: 10.1016/j.intimp.2020.106450
110. Zschaler J, Schlorke D, Arnhold J. Differences in innate immune response between man and mouse. Crit Rev Immunol. (2014) 34:433–54. doi: 10.1615/CritRevImmunol.2014011600
111. Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. (2009) 9:480–90. doi: 10.1038/nri2580
112. Kenney LL, Shultz LD, Greiner DL, Brehm MA. Humanized mouse models for transplant immunology. Am J Transplant. (2016) 16:389–97. doi: 10.1111/ajt.13520
113. Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. (2012) 12:786–98. doi: 10.1038/nri3311
114. Meyer-Martin H, Hahn SA, Beckert H, Belz C, Heinz A, Jonuleit H, et al. GARP inhibits allergic airway inflammation in a humanized mouse model. Allergy. (2016) 71:1274–83. doi: 10.1111/all.12883
115. Vizzardelli C, Gindl M, Roos S, Mobs C, Nagl B, Zimmann F, et al. Blocking antibodies induced by allergen-specific immunotherapy ameliorate allergic airway disease in a human/mouse chimeric model. Allergy. (2018) 73:851–61. doi: 10.1111/all.13363
116. Ito R, Maruoka S, Soda K, Katano I, Kawai K, Yagoto M, et al. A humanized mouse model to study asthmatic airway inflammation via the human IL-33/IL-13 axis. JCI Insight. (2018) 3:121580. doi: 10.1172/jci.insight.121580
117. Duez C, Kips J, Pestel J, Tournoy K, Tonnel AB, Pauwels R. House dust mite-induced airway changes in hu-SCID mice. Am J Respir Crit Care Med. (2000) 161:200–6. doi: 10.1164/ajrccm.161.1.9806026
118. Sonar SS, Hsu YM, Conrad ML, Majeau GR, Kilic A, Garber E, et al. Antagonism of TIM-1 blocks the development of disease in a humanized mouse model of allergic asthma. J Clin Invest. (2010) 120:2767–81. doi: 10.1172/JCI39543
119. Wenzel S, Holgate ST. The mouse trap: it still yields few answers in asthma. Am J Respir Crit Care Med. (2006) 174:1173–6; discussion 1176–8. doi: 10.1164/rccm.2609002
120. Graham MT, Nadeau KC. Lessons learned from mice and man: mimicking human allergy through mouse models. Clin Immunol. (2014) 155:1–16. doi: 10.1016/j.clim.2014.08.002
121. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
122. Gould HJ, Ramadani F. IgE responses in mouse and man and the persistence of IgE memory. Trends Immunol. (2015) 36:40–8. doi: 10.1016/j.it.2014.11.002
123. Boers JE, Ambergen AW, Thunnissen FB. Number and proliferation of clara cells in normal human airway epithelium. Am J Respir Crit Care Med. (1999) 159(5 Pt 1):1585–91. doi: 10.1164/ajrccm.159.5.9806044
124. Hyde DM, Hamid Q, Irvin CG. Anatomy, pathology, and physiology of the tracheobronchial tree: emphasis on the distal airways. J Allergy Clin Immunol. (2009) 124(6 Suppl.):S72–7. doi: 10.1016/j.jaci.2009.08.048
125. Furuhashi K, Chua YL, Wong KHS, Zhou Q, Lee DCP, Liong KH, et al. Priming with high and low respiratory allergen dose induces differential CD4(+) T helper type 2 cells and IgE/IgG1 antibody responses in mice. Immunology. (2017) 151:227–38. doi: 10.1111/imm.12726
126. Nelde A, Teufel M, Hahn C, Duschl A, Sebald W, Brocker EB, et al. The impact of the route and frequency of antigen exposure on the IgE response in allergy. Int Arch Allergy Immunol. (2001) 124:461–9. doi: 10.1159/000053781
127. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. (2016) 532:512–6. doi: 10.1038/nature17655
Keywords: mouse model, asthma, T2 airway inflammation, non-T2 airway inflammation, endotypes
Citation: Alessandrini F, Musiol S, Schneider E, Blanco-Pérez F and Albrecht M (2020) Mimicking Antigen-Driven Asthma in Rodent Models—How Close Can We Get? Front. Immunol. 11:575936. doi: 10.3389/fimmu.2020.575936
Received: 24 June 2020; Accepted: 31 August 2020;
Published: 30 September 2020.
Edited by:
Christiane Hilger, Luxembourg Institute of Health, LuxembourgReviewed by:
Matthew Poynter, University of Vermont, United StatesTatiana Michel, Luxembourg Institute of Health, Luxembourg
Copyright © 2020 Alessandrini, Musiol, Schneider, Blanco-Pérez and Albrecht. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melanie Albrecht, bWVsYW5pZS5hbGJyZWNodEBwZWkuZGU=