
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 29 September 2020
Sec. Primary Immunodeficiencies
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.557521
 Kazuki Nemoto1
Kazuki Nemoto1 Toshinori Kawanami1
Toshinori Kawanami1 Takayuki Hoshina2*
Takayuki Hoshina2* Masataka Ishimura3
Masataka Ishimura3 Kei Yamasaki1
Kei Yamasaki1 Satoshi Okada4
Satoshi Okada4 Hirokazu Kanegane5
Hirokazu Kanegane5 Kazuhiro Yatera1
Kazuhiro Yatera1 Koichi Kusuhara2
Koichi Kusuhara2Hypogammaglobulinemia is a rare complication of STAT1 gain-of-function (GOF) mutations. We report an adult patient diagnosed with hypogammaglobulinemia caused by B-cell depletion during the treatment of disseminated cryptococcosis. The patient carried the STAT1 GOF mutation (c.820C>T, p.R274W). The flow cytometric analysis of his bone marrow revealed that B-cell differentiation was blocked in the stages between pre-B1b and pre-B2 cells. On the other hand, his brother who carried the same mutation displayed normal B-cell counts, thereby indicating that the unrecognized variants in same or other gene might be associated with abnormal B-cell differentiation in the patients. In conclusion, impaired B-cell differentiation in the bone marrow can cause hypogammaglobulinemia in patients with STAT1 GOF mutations.
Gain-of-function (GOF) mutations in the signal transducer and activator of transcription 1 (STAT1) gene are recognized as genetic abnormalities that cause chronic mucocutaneous candidiasis disease (CMCD) (1, 2). This autosomal dominant immunodeficiency is characterized by recurrent or persistent mucocutaneous lesions, such as on the nails, skin, and oral or genital mucosae, caused by the Candida species, and some patients with this disorder develop invasive fungal infections (3). Low B-cell count (19%) and low immunoglobulin (Ig) G levels (3%) have been observed as rare immunological abnormalities in patients with STAT1 GOF mutations (3). Although the mechanisms underlying B-cell lymphopenia and hypogammaglobulinemia have previously been investigated (4), they are still somewhat elusive.
We herein report an adult patient diagnosed with STAT 1 GOF mutation-induced hypogammaglobulinemia caused by B-cell depletion during the treatment of disseminated cryptococcosis. To the best of our knowledge, this is the first cases report that demonstrated the impairment of B-cell differentiation in bone marrow of such a patient.
A 30-year-old Japanese man was admitted to our hospital because of high-grade fever with transient consciousness disorder and double vision. He had no medical history throughout his childhood and adolescence but continued to have repeated fever and productive cough since the age of 25. He also presented with stomatitis and dental caries. His mother had a history of disseminated cryptococcosis and died from chronic hepatitis C virus infection. His younger brother had been clinically diagnosed with CMCD for having recurrent and refractory cutaneous candidiasis although no genetic analysis has been performed on his dissent.
Physical examination on the admission revealed that the majority of his teeth were corroded, and that he had white moss throughout his mouth. A nuchal stiffness was noted with right abductor paralysis. His chest computed tomography (CT) showed bronchiectasis and multiple granular shadows randomly distributed in the bilateral lower and middle lobes, thin-walled cysts predominantly in both upper lobes, and consolidation in the lower left lobe (Figure 1). Cryptococcus neoformans was isolated from his blood and cerebrospinal fluid samples and was also detected in liver and lung biopsies. Accordingly, he was diagnosed with disseminated cryptococcosis with meningitis and oral and cutaneous candidiasis.
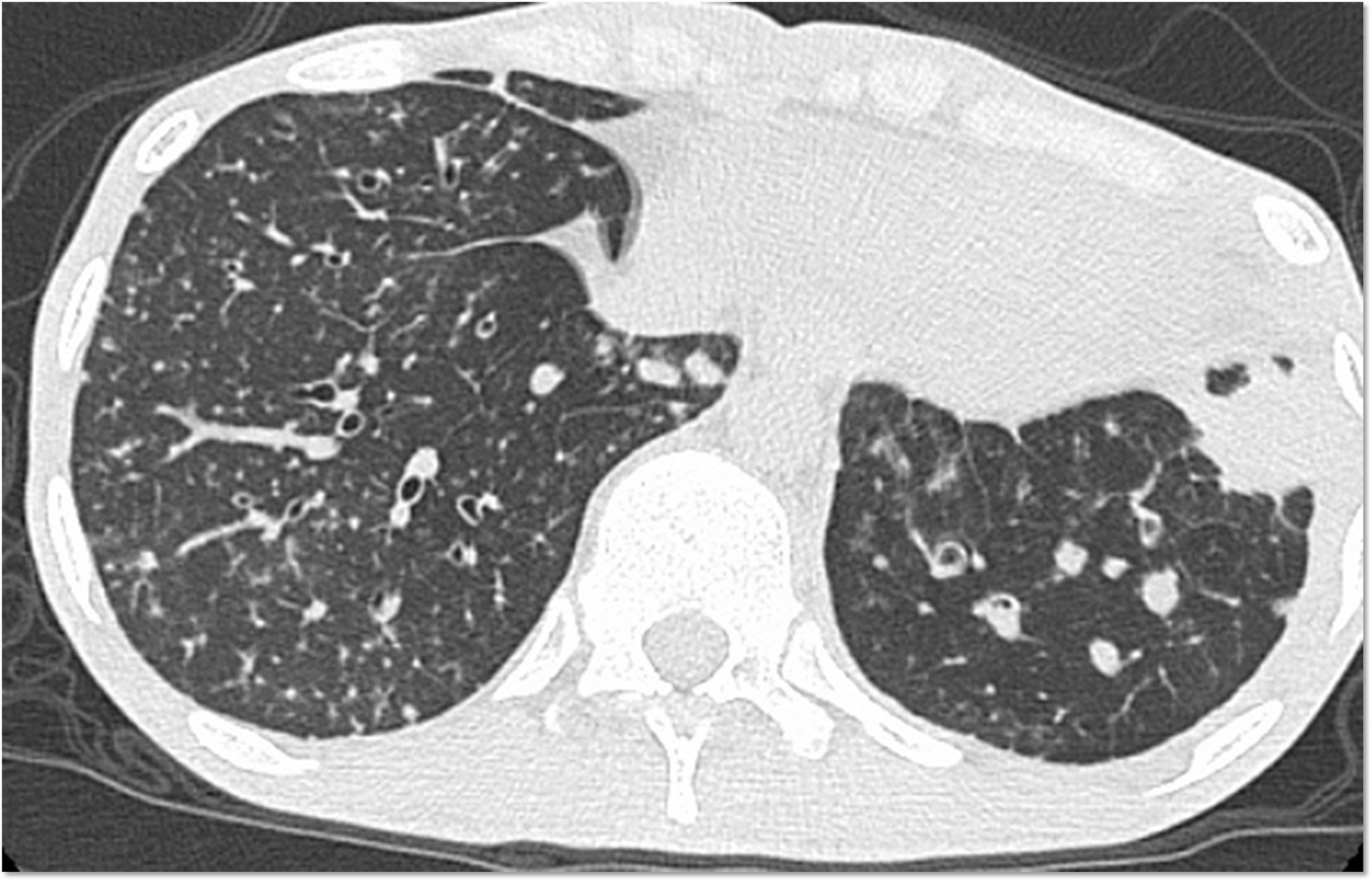
Figure 1 Chest computed tomography scan. Bronchiectasis and multiple granular shadows are randomly distributed in the bilateral lobes, and consolidation in the lower left lobe is present.
The patient was treated with a combination therapy of liposomal amphotericin B and flucytosine for 8 weeks. Consequently, his respiratory symptoms and neurological abnormalities resolved. The cryptococcal antigen titers in his cerebrospinal fluid decreased from 1,024 times to 32. His chest CT on day 44 of the treatment showed that the granular shadows and consolidation entirely improved, but the bronchiectases in his right middle lobe and left lingular segment remained. The antifungal treatment was then switched to oral fluconazole, and the disease did not recur for one year.
The patient was suspected of having primary immunodeficiency because of the present clinical course and his family history. We sequenced the patient’s CMCD-related genes and found a previously recognized heterozygous missense mutation in the coiled-coil domain of the STAT1 gene (c.820C>T, p.R274W) (5, 6). The same mutation was also detected in his younger brother. The monocytes of the patient showed hyper-phosphorylation of STAT1 after stimulation with interferon-γ (Figure 2). Based on these results, the patient was diagnosed with STAT1 GOF.
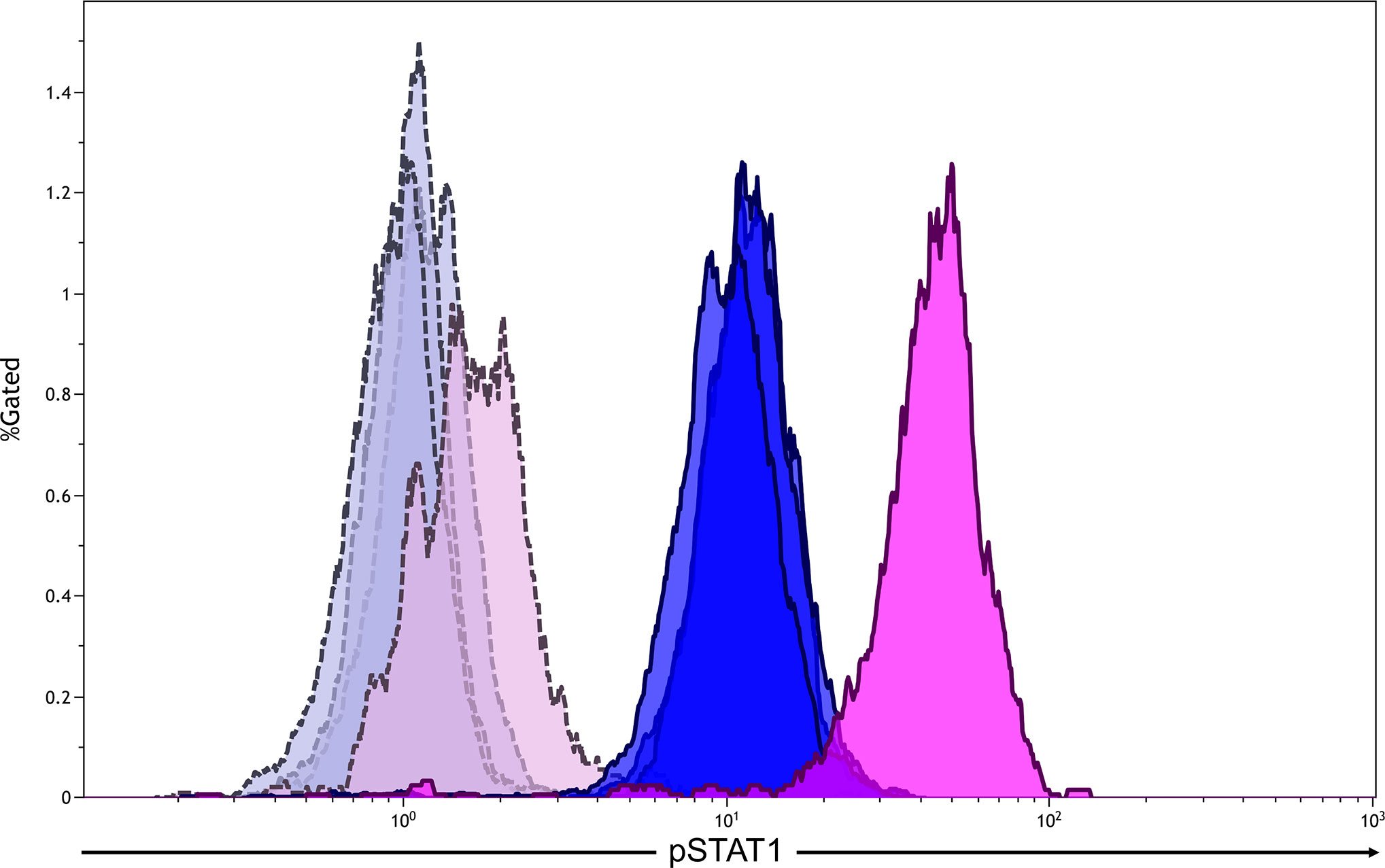
Figure 2 Flow cytometric analysis of phosphorylationed STAT1 (pSTAT1) in monocytes after the stimulation with interferon-γ (500 U/mL). The analysis gate was set in CD14+ cells. Blue areas indicate healthy adults (n = 3), whereas pink areas indicate the patient. Gray and purple areas indicate pSTAT1 in monocytes without interferon-γ stimulation in healthy adults and the patient, respectively.
Additionally, the patient presented with low serum IgG and IgA levels [IgG: 2.36 g/L, normal range (NR): 8.61–17.47 g/L; IgA: 0.05 g/L, NR: 0.93–3.93; IgM: 0.92 g/L, NR: 0.33–1.83]. Surface marker analyses revealed that 72.3% (NR: 54.3–81.9%) of his peripheral lymphocytes comprised CD3+ cells with a CD4/CD8 ratio of 32.8 (NR: 24.3–49.7%)/36.2 (NR: 18.4–49.0%), and that 0.1% (NR: 2.9–20.1%) comprised CD19+ cells (Table 1). Peripheral blood lymphocyte responsiveness to phytohemagglutinin was normal (Stimulation Index: 281, NR: 254–388). The level of T-cell receptor excision circles was 2.06 x 102 copies/μgDNA [NR: 3.4 ± 3.6 x 102 copies/μg DNA (7)], whereas both those of the coding joint and signal joint kappa-deleting recombination excision circles were undetermined. We then investigated the cause of hypogammaglobulinemia and B-cell lymphopenia. The B-cell differentiation was blocked in the stages between VpreB+IgMlow (pre-B1b) cells and VpreB−IgMlow (pre-B2) cells in the bone marrow sample obtained three months after the onset of cryptococcosis (Figure 3). Human immunodeficiency virus-RNA and Epstein-Barr virus- and cytomegalovirus-DNA were undetectable by polymerase chain reaction. No pathogenic variants in antibody deficiency-causing genes listed in the 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity were detected using the target panel sequence (8). The reduction in the peripheral B-cell counts had persisted even after the patient’s recovery from cryptococcosis (Table 1). His prolonged respiratory symptoms did not recur upon regular subcutaneous immunoglobulin infusion to maintain a trough level of >7.0 g/L.

Table 1 The analysis of lymphocyte subsets in the acute phase of cryptococcosis and the non-infectious state.
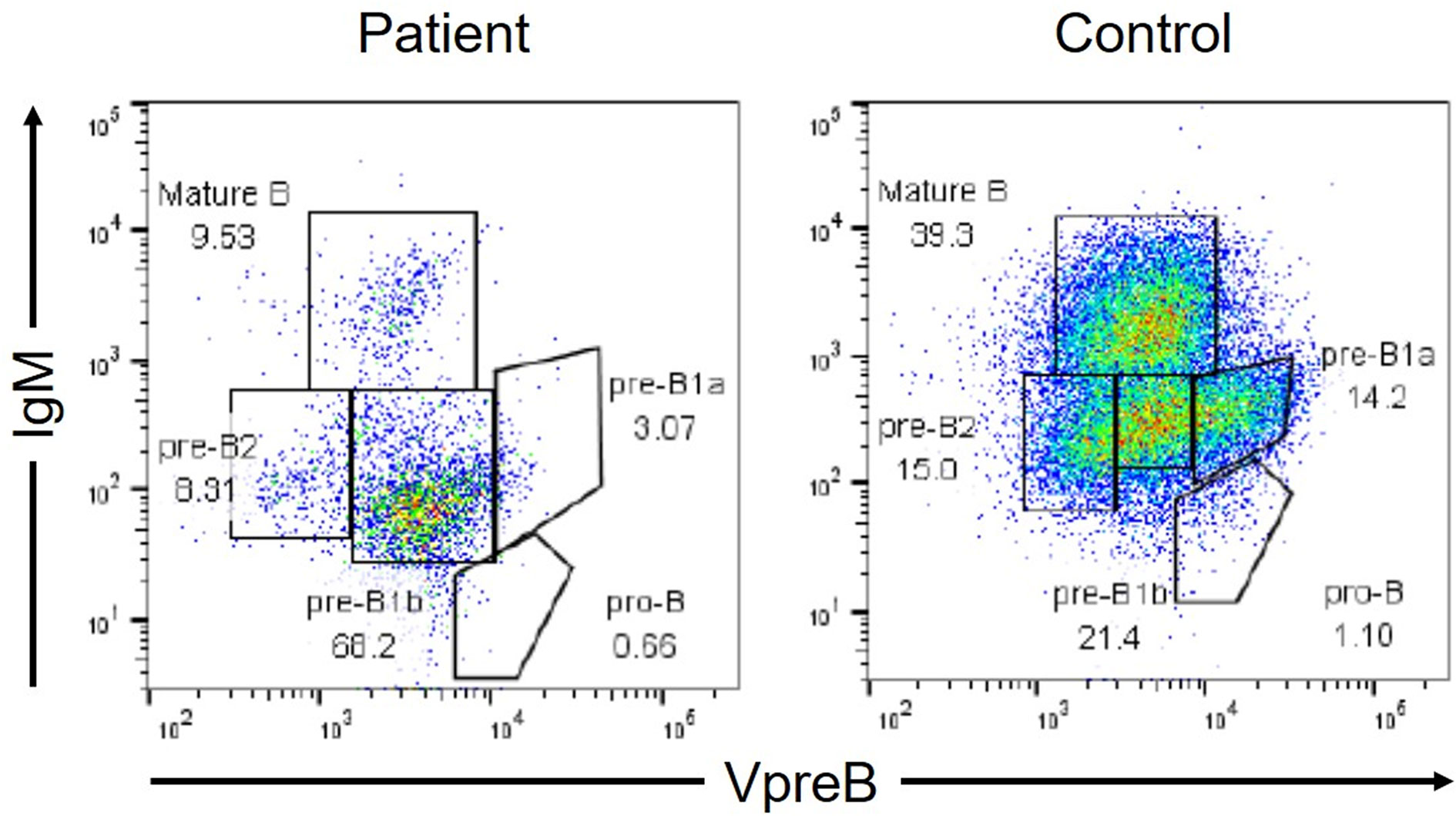
Figure 3 Flow cytometric analysis of B-cell differentiation in the bone marrow (BM). VpreB is a component of the surrogate light chains. The analysis gate was set in CD19+ and/or VpreB+ cells. The BM cells were fixed and permeabilized to simultaneously analyze the cell surface antigens (IgM and CD19) and the intracellular molecule (VpreB).
Disseminated cryptococcosis is a fungal infection that frequently develops in patients with STAT1 GOF mutations (3). Th17 cells are thought to play an important role in the protection against cryptococcal infections (9, 10). The STAT1 signaling is overactive in patients with STAT1 GOF mutations. Increased STAT1 function inversely leads to the impaired production of STAT3-dependent cytokines, including interleukin (IL)-17A and IL-17F, in T cells. The present patients developed disseminated cryptococcosis as the initial manifestation of STAT1 GOF mutation, suggesting that this mutation should be considered for differential diagnosis, even if a previously-healthy adult develops disseminated cryptococcosis, because this genetic disorder may, although rarely, develop in adulthood (3, 5).
The novelty of this case report lies in the analysis of B-cell differentiation of the bone marrow sample of a patient with STAT1 GOF mutation. The mechanistic details of such impaired Th17 responses are accordingly somewhat understood, whereas those of impaired humoral immunity caused by B-cell lymphopenia in patients with STAT1 GOF mutations are still elusive because of the small number of patients with humoral immunodeficiency (3, 5). A previous study analyzed the peripheral mature B-cells and consequently suggested that the accelerated B-cell apoptosis resulting from increased caspase activity might cause B-cell lymphopenia (4). However, no study has investigated B-cell differentiation in the bone marrow of patients with the STAT1 GOF mutations accompanying B-cell lymphopenia. It has been speculated that Btk mutations in X-linked agammaglobulinemia (XLA) may interrupt the proliferation and survival of the earliest pre-B cells (pre-B1a cells) (11). STAT3 whose functions are inhibited in patients with the STAT1 GOF mutations is also associated with early B-cell development (earlier stage of pro-B cells) in the bone marrow (12). We observed that B-cell differentiation in such a patient was blocked in the stages between the pre-B1b and pre-B2 cells of the bone marrow, which was later than in patients with XLA. Furthermore, the STAT1-dependent B-cell differentiation is affected by infection. In mice, STAT1 plays an important role in the promotion, but not the impairment, of B-cell differentiation in the bone marrow and marginal zone (13, 14). In our patient, the reduction in the peripheral B-cell counts persisted even after his recovery from cryptococcosis, thereby indicating that the impaired B-cell differentiation was not caused by infection. Although the molecular biological analysis to elucidate the mechanism of the impaired B-cell differentiation was performed, we believe that B-cell differentiation in patients with STAT1 GOF mutations is interrupted by a pathway other than the BTK- and STAT3-associated pathways.
The decreased number of B cells is only observed in some the patients with such mutations. In fact, the B-cell counts and the immunoglobulin levels of the patient’s younger brother were within normal ranges (data not shown). Although no pathogenic variants in previously recognized antibody deficiency-causing genes (8) have been detected in our patient, unrecognized variants in same or other gene may have caused the impaired B-cell differentiation in the bone marrow. Indeed, a recent study has suggested that the interplay between rare and common genetic variants may provide the phenotypic heterogeneity of primary immunodeficiency (15). Whole-genome sequencing is a future prospective to elucidate genetic difference between the siblings, thus providing a better understanding of the molecular mechanisms underlying impaired B-cell differentiation. Alternatively, the reduction in the B-cell counts observed in patients with the STAT1 GOF mutations may depend on age. Our patient might have developed hypogammaglobulinemia in adulthood because his respiratory symptoms appeared 5 years before the present episode. Further studies are needed to elucidate the details of the mechanism underlying the impaired B-cell differentiation in this disorder.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Written informed consent was obtained from the patient for the publication of any potentially identifiable images or data included in this article.
KN, TK, and TH were involved in the treatment of the patient, conceptualized the study, carried out the initial analysis of data for work, and drafted the initial manuscript. KeY and KaY were involved in the treatment of the patient. MI, HK, and SO carried out the acquisition and analysis of data for the work. KK coordinated and supervised data collection. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Naomi Terada (Tokyo Medical and Dental University) for performing the flow cytometric analysis of the bone marrow sample, and Tamami Tanaka and Motoshi Sonoda (Kyushu University) for performing the flow cytometric analysis of peripheral blood samples. We would like to thank Editage (www.editage.com) for English language editing.
1. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med (2011) 365:54–61. doi: 10.1056/NEJMoa1100102
2. Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med (2011) 208:1635–48. doi: 10.1084/jem.20110958
3. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood (2016) 127:3154–64. doi: 10.1182/blood-2015-11-679902
4. Romberg N, Morbach H, Lawrence MG, Kim S, Kang I, Holland SM, et al. Gain-of-function STAT1 mutations are associated with PD-L1 overexpression and a defect in B-cell survival. J Allergy Clin Immunol (2013) 131:1691–3. doi: 10.1016/j.jaci.2013.01.004
5. Depner M, Fuchs S, Raabe J, Frede N, Glocker C, Doffinger R, et al. The extended clinical phenotype of 26 patients with chronic mucocutaneous candidiasis due to gain-of-function mutations in STAT1. J Clin Immunol (2016) 36:73–84. doi: 10.1007/s10875-015-0214-9
6. Kagawa R, Fujiki R, Tsumura M, Sakata S, Nishimura S, Itan Y, et al. Alanine-scanning mutagenesis of human signal transducer and activator of transcription 1 to estimate loss- or gain-of-function variants. J Allergy Clin Immunol (2017) 140:232–41. doi: 10.1016/j.jaci.2016.09.035
7. Morinishi Y, Imai K, Nakagawa N, Sato H, Horiuchi K, Ohtsuka Y, et al. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal guthrie cards. J Pediatr (2009) 155:829–33. doi: 10.1016/j.jpeds.2009.05.026
8. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
9. Valdez PA, Vithayathil PJ, Janelsins BM, Shaffer AL, Williamson PR, Datta SK. Prostaglandin E2 suppresses antifungal immunity by inhibiting interferon regulatory factor 4 function and interleukin-17 expression in T cells. Immunity (2012) 36:668–79. doi: 10.1016/j.immuni.2012.02.013
10. Odio CD, Milligan KL, McGowan K, Rudman Spergel AK, Bishop R, Boris L, et al. Endemic mycoses in patients with STAT3-mutated hyper-IgE (Job) syndrome. J Allergy Clin Immunol (2015) 136:1411–3.e1-2. doi: 10.1016/j.jaci.2015.07.003
11. Nomura K, Kanegane H, Karasuyama H, Tsukada S, Agematsu K, Murakami G, et al. Genetic defect in human X-linked agammaglobulinemia impedes a maturational evolution of pro-B cells into a later stage of pre-B cells in the B-cell differentiation pathway. Blood (2000) 96:610–7.
12. Chou WC, Levy DE, Lee CK. STAT3 positively regulates an early step in B-cell development. Blood (2006) 108:3005–11. doi: 10.1182/blood-2006-05-024430
13. Chen TT, Tsai MH, Kung JT, Lin KI, Decker T, Lee CK. STAT1 regulates marginal zone B cell differentiation in response to inflammation and infection with blood-borne bacteria. J Exp Med (2016) 213:3025–39. doi: 10.1084/jem.20151620
14. Hsu CC, Meeker SM, Escobar S, Brabb TL, Paik J, Park H, et al. Murine norovirus inhibits B cell development in the bone marrow of STAT1-deficient mice. Virology (2018) 515:123–33. doi: 10.1016/j.virol.2017.12.013
Keywords: STAT1 gain-of-function mutation, disseminated cryptococcosis, hypogammaglobulinemia, impaired B-cell differentiation, bone marrow (BM)
Citation: Nemoto K, Kawanami T, Hoshina T, Ishimura M, Yamasaki K, Okada S, Kanegane H, Yatera K and Kusuhara K (2020) Impaired B-Cell Differentiation in a Patient With STAT1 Gain-of-Function Mutation. Front. Immunol. 11:557521. doi: 10.3389/fimmu.2020.557521
Received: 30 April 2020; Accepted: 14 September 2020;
Published: 29 September 2020.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Lisa Renee Forbes, Baylor College of Medicine, United StatesCopyright © 2020 Nemoto, Kawanami, Hoshina, Ishimura, Yamasaki, Okada, Kanegane, Yatera and Kusuhara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takayuki Hoshina, aG9zaGluYUBtZWQudW9laC11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.