
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 December 2020
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.553715
 Beom K. Choi1
Beom K. Choi1 Hyeon-Woo Lee2*
Hyeon-Woo Lee2*CD137, a member of the TNFR family, is a costimulatory receptor, and CD137L, a member of the TNF family, is its ligand. Studies using CD137- and CD137L-deficient mice and antibodies against CD137 and CD137L have revealed the diverse and paradoxical effects of these two proteins in various cancers, autoimmunity, infections, and inflammation. Both their cellular diversity and their spatiotemporal expression patterns indicate that they mediate complex immune responses. This intricacy is further enhanced by the bidirectional signal transduction events that occur when these two proteins interact in various types of immune cells. Here, we review the biology of murine CD137/CD137L, particularly, the complexity of their proximal signaling pathways, and speculate on their roles in immune responses.
The mammalian immune system is a highly evolved complex of cells and proteins that defends against intruders (1). It comprises adaptive and innate immune responses carried out by various specific cells (1, 2). Communication between immune cells is critical for initiating, modulating, and maintaining immunity against infections and controlling dysregulated immune responses (3–5). Cytokines produced by immune cells and cell-cell contact are the main means of communication (5, 6). Cell-cell contact via adhesion between extracellular proteins and receptor/ligand interactions plays key roles in information flow (3–5). A major example of cell-cell contact in the immune system that we focus on here is the interaction of T lymphocytes with antigen-presenting cells (APCs) (7–9). APCs deliver antigenic information to T lymphocytes. In addition to major histocompatibility complex (MHC)/T-cell receptor (TCR) interaction, costimulatory receptors must associate with their ligands for APCs to achieve antigen-dependent activation of T cells. Upon activation of pattern recognition receptors such as Toll-like receptors (TLRs) by pathogen-associated molecular patterns such as lipopolysaccharide (LPS), APCs mature into functional immune cells that present antigens to T cells. This maturation process involves the expression of molecules such as MHCs, adhesion proteins, ligands for costimulatory receptors, and chemokines (7, 10).
Twenty-nine members of the tumor necrosis factor receptor (TNFR) superfamily (TNFRSF) and 19 members of the TNF ligand superfamily (TNFSF) have been identified. They include OX40/OX40 ligand (L), CD27/CD70, glucocorticoid-induced TNFR-related protein (GITR)/GITRL, and CD137/CD137L pairs, which are all costimulatory receptors and their ligands expressed on T lymphocytes and APCs, respectively (11–13). Ligation of TNFRSF members by the corresponding TNFSF members induces signaling via dedicated signal transduction pathways (11, 14). At the same time, TNFSF ligation activates ligand-dependent signal transduction pathways, so-called “reverse signals,” that elicit reverse signal-induced cellular responses (15–17). CD137 (4-1BB, TNFRSF9), a 30-kDa glycoprotein, is an inducible type I TNFRSF transmembrane protein expressed on murine T lymphocytes following antigen recognition by the TCR (12, 18). CD137 ligation evokes survival signals in CD8+ T lymphocytes (11, 14). CD137 is expressed in various cells including natural killer (NK) cells, NKT cells, regulatory T cells (Tregs), dendritic cells (DCs), follicular DCs, mast cells, differentiating myeloid-lineage cells, eosinophils, neutrophils, and monocytes (19–23). CD137L, a 34-kDa type II transmembrane glycoprotein, was originally identified as a binding partner to soluble forms of CD137 (sCD137) (24). It is expressed on monocytes, macrophages, B cells, DCs, T cells, differentiating hematopoietic cells, bone marrow cells, and tumor cells (25).
Crosslinking of CD137 with CD137L induces various immune responses involving both adaptive and innate immunity (11, 25–28). In addition, CD137L reverse signals increase the antigen-presenting capacity and inflammation of APCs (29–31). Because binding between CD137 and CD137L on various cells can trigger bidirectional signals, it is difficult to dissect the individual effects of CD137 and CD137L using current experimental tools such as knockout mice and antibodies (Abs) against the two molecules in animal models. Table 1 summarizes CD137/CD137L-mediated immune responses in mouse models. Although it is accepted that CD137 provides T cells with strong survival signals, it also inhibits CD4+ T-cell responses in autoimmune disease models and B-cell responses in vivo (44, 48, 64). CD137 knockout results in hyperimmune responses in mice and in hyperproliferation of T cells and myeloid progenitors in vitro (27, 40, 41, 65). In contrast, CD137L deficiency lowers CD8+ T-cell responses in virus infection models (66, 67) and reduces cytotoxic T lymphocyte activity against vesicular stomatitis virus (27). Reverse signaling via CD137L, elicited by treating myeloid cells with recombinant CD137-Fc protein (rCD137-Fc), enhanced myelopoiesis during inflammation (25, 65). In contrast, CD137 deficiency increased the number of myeloid cells in vitro, and this effect was abrogated by treatment with rCD137-Fc, implying that CD137L provides negative signals to myeloid cells (27, 65). Even in cell culture, treatment with CD137Ab or rCD137-Fc can influence CD137-mediated cellular responses because certain types of cells express both CD137 and CD137L. Therefore, CD137/CD137L-mediated immune responses appear paradoxical and puzzling. Although plausible explanations have been suggested for the complex effects of mouse CD137/CD137L (mCD137/CD137L), none of the explanations are definitive yet. In this review, we discuss the major factors contributing to the complexity of mCD137/CD137L signals and recent findings that hint at links between the mCD137/CD137L axis and other key immune processes.
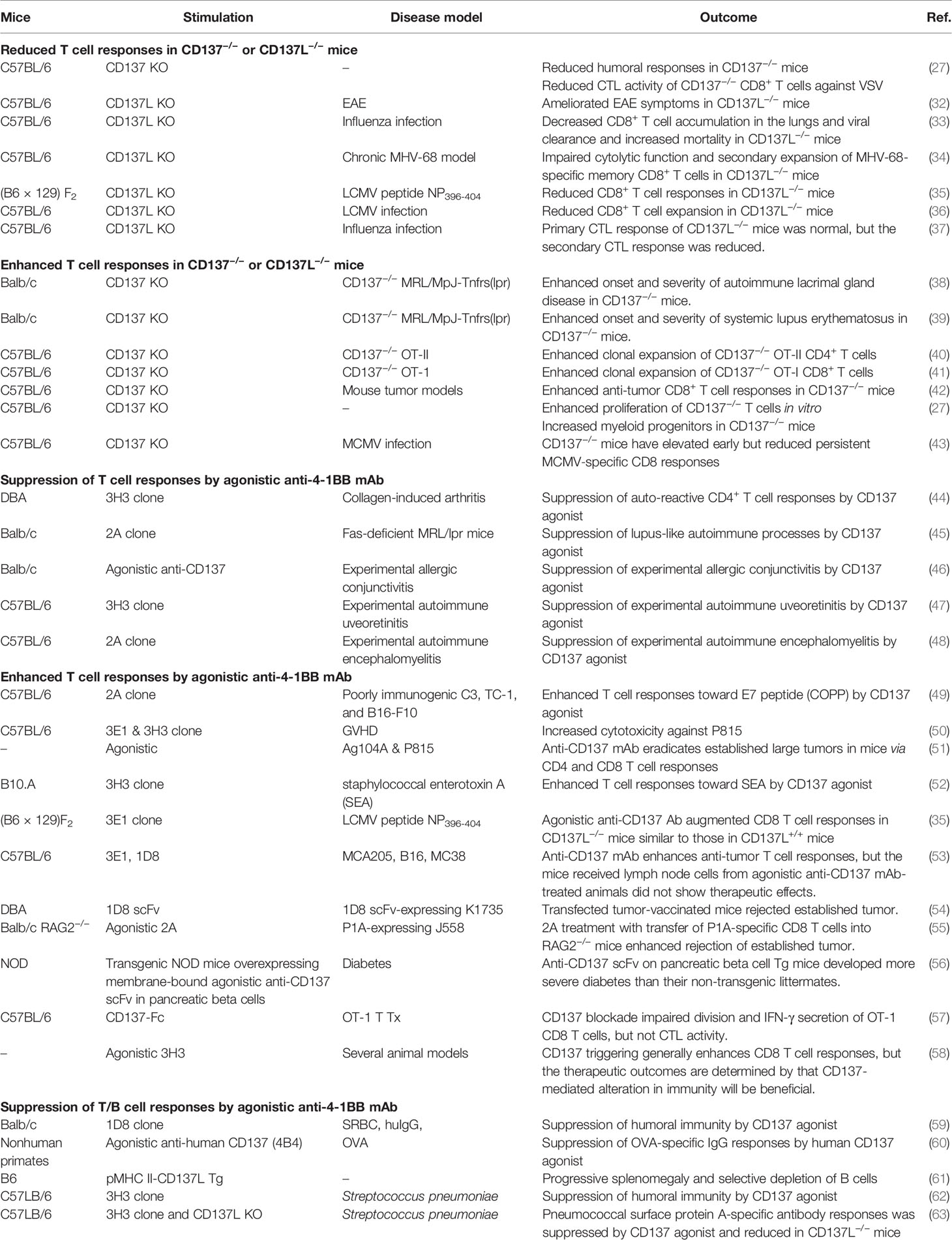
Table 1 Immune responses generated by CD137/CD137L in animal models.
Members of the TNF/TNFR superfamilies, such as CD137L/CD137, evoke dual cellular responses by initiating bidirectional signals (11, 14–17). The interaction between CD137L and CD137 can give rise to a great variety of cellular responses because their expression on both innate and/or adaptive immune cells is induced or enhanced by primary cell activation, such as TCR activation for CD137 and TLR4 activation for CD137L (18, 19, 66–68). For example, resting T cells express little, if any, CD137 (14, 19); however, TCR ligation by antigenic epitopes/MHCs on APCs induces or upregulates CD137 expression on T cells (69, 70). Differences in the affinity and/or avidity of antigenic epitopes for the TCR can influence the level, time of onset, and duration of CD137 expression (Figure 1). However, the expression of CD137L on APCs including DCs can be enhanced via TLR4 or other receptors in response to stimuli such as LPS (66–68) (Figure 1).
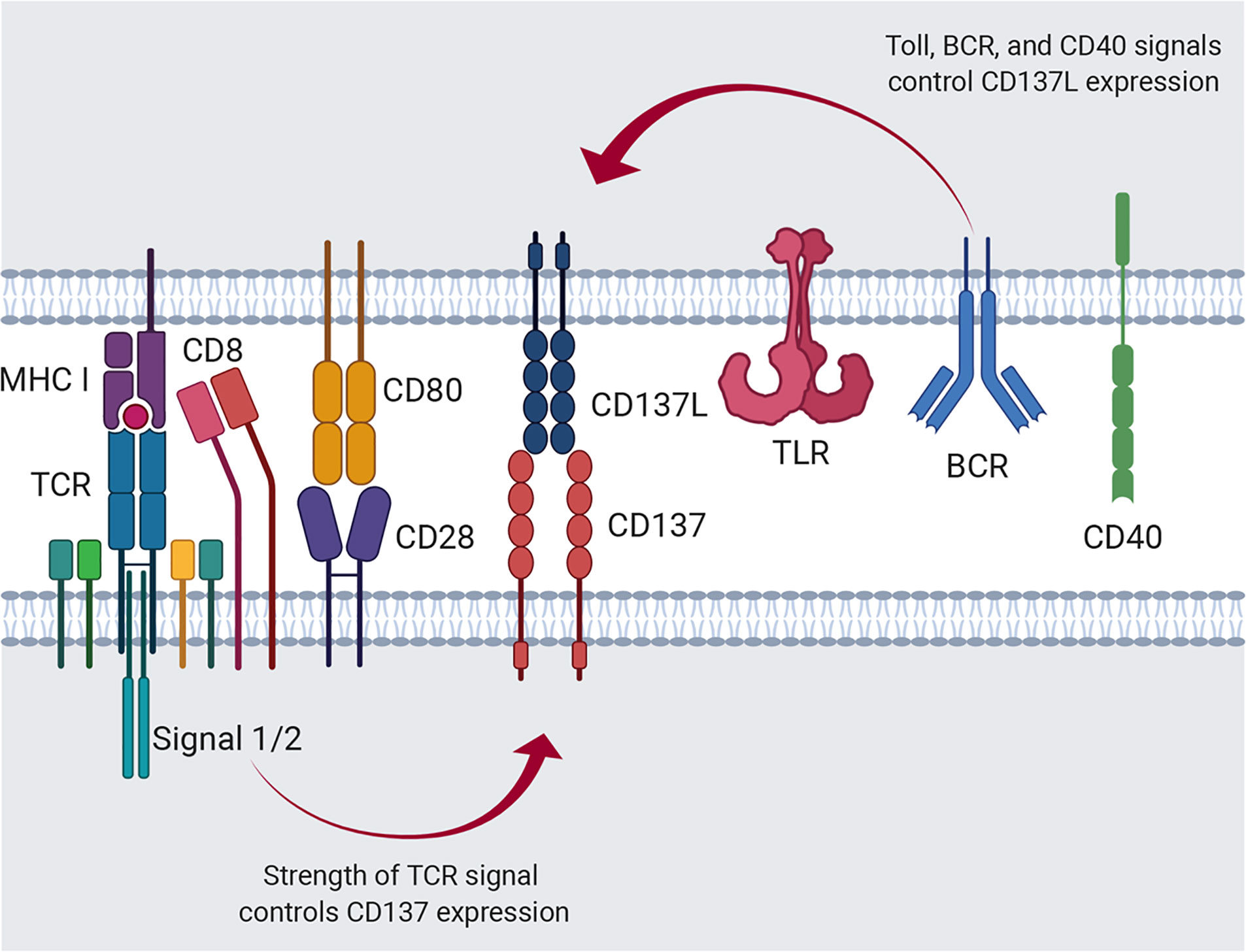
Figure 1 Regulation of CD137 and CD137L expression. Activation of APCs via Toll signals, such as TLR4, B-cell receptor crosslinking, and CD40 ligation induces CD137L expression. Alternatively, antigenic signal provided by the MHC I/peptide complex and costimulation through CD28 induces T-cell activation and CD137 expression.
The strength and/or type of stimulus may also affect CD137L expression on APCs. Consequently, the CD137/CD137L pair can generate diverse cellular responses depending on the spatiotemporal pattern of their expression and the direction of signaling. Numerous parameters contribute to the complexity of the immune responses elicited by the mCD137/CD137L signalosome. However, in this review, we focus on the bidirectional signal transduction pathways evoked by CD137 and CD137L in the context of T-cell/APC interaction.
mCD137 transcript expression is regulated by multiple elements on the promoter (PI, PII, and PIII). There exist multiple mCD137 transcripts. Types I and II are dominant in the mouse. Type I transcript is preferentially induced in stimulated T cells by TCR activation via NF-κB binding on PI. Although type II mRNA is also upregulated in activated T cells via AP-1 binding on PI, it is constitutively expressed in various cells via AP1 binding on the PI, PII, and PIII elements (71). CD28 costimulation strongly upregulates CD137 expression, presumably via binding of both NF-κB and AP-1, as CD28 stimulation sustains TCR signaling pathways such as NF-κB, AP-1, and NFAT (72) (Figure 1). The spliced transcript lacking exon 8 encodes sCD137. It is expressed in activated T cells. It has been suggested that sCD137 may antagonize CD137 signal and/or act as an agonist for CD137L reverse signals, and that sCD137 may antagonize CD137 signaling as a decoy receptor (73, 74). However, Tu et al. reported that sCD137 can deliver CD137L reverse signal in CD137L-positive cells such as APCs and T cells (75). They showed that sCD137 protein levels were elevated in adipose tissues of obese subjects, and sCD137 apparently enhanced obesity-induced adipose inflammation by triggering CD137L reverse inflammatory signals in macrophages and activated T cells.
mCD137L expression is induced not only in activated T cells, but also in activated APCs such as LPS-treated macrophages, LPS- or CD40 mAb-treated splenic DCs, and IgM mAb plus CD40 mAb-treated B cells (24, 67) (Figure 1). However, the molecular mechanism of its regulation at the promoter level remains uncovered. A recent study by Chang et al. suggested that different APC subsets differentially express costimulatory molecules early during viral infection (76). Classical dendritic cells (cDCs) highly express MHC II, CD80, and CD86, whereas inflammatory monocyte-derived APCs (infAPCs), including inflammatory macrophages and DCs, exhibit the highest expression of TNFSF ligands such as GITRL, OX40L, CD137L, and CD70 (76). Moreover, the authors suggested that GITRL on infAPCs provides signal 4 for activated T cells by providing a post-priming survival checkpoint (76). The results of this study elegantly explained the fundamental roles of inducible costimulatory molecules, including GITR/GITRL, OX40/OX40L, and CD137/CD137L, in the survival of activated T cells.
Another report also indicated that infDCs are a distinct subset of DCs that appear during inflammation and are derived from monocytes that differentiate in situ at the site of inflammation and migrate to the lymph nodes to activate T cells, whereas cDCs in lymph nodes generally spend their entire life in secondary lymphoid tissues (77). Given that infAPCs migrate from the site of inflammation while maturing, it is reasonable to expect that infAPCs are mature APCs and cDCs are immature until they start to mature by receiving antigen (Ag) from infAPCs (78). Consequently, inducible costimulatory molecules such as CD137L would be more important for infAPCs than MHC II, CD80, and CD86, which are preferentially expressed on cDCs, because infDCs need to activate T cells and sustain their survival and proliferation.
As mentioned above, CD137 is expressed on activated T cells and CD137L is primarily expressed on myeloid immune cells such as DCs (67). Indeed, when activated CD4+ or CD8+ T cells were analyzed by flow cytometry, CD137 was readily detected on their surface even 24 h after their activation, whereas CD137L was not (67). CD137L expression was induced on mature DCs following their maturation, but was gradually downregulated as CD137 expression increased (43).
Surprisingly, CD137L expression was enhanced on activated T cells and DCs of CD137-deficient mice (26). It is not clear how T cells and DCs downregulate CD137L during their activation or maturation, but it can be speculated that CD137L becomes bound to intracellular CD137, triggering internalization of the CD137-CD137L complexes, and thus, CD137-mediated signals are continuously transmitted into the cells without a requirement for CD137L located on other cells (Figure 2A). Indeed, Martinez-Forero et al. have shown that anti-CD137 mAb bound to CD137 was internalized into an endosomal compartment in vitro and in vivo, and triggered TRAF2-mediated signaling (79). Since CD137-CD137L complexes are internalized into the cells, theoretically, both CD137- and CD137L-mediated signals can be transmitted into the cells; however, which signal is transmitted in vivo remains unclear. Alternatively, CD137:CD137L complexes are exchanged between T:T or T:DC conjugate via trogocytosis—the bidirectional transfer of molecules between interacting cells—and thus accumulate in the cells (80) (Figure 2B). When activated T cells and mature DCs form conjugates, CD137 and CD137L on each cell interact to form the complex. Since TCR triggering induces trogocytosis (80), molecules on the plasma membrane are exchanged between the conjugated cells and thus, the CD137:CD137L complexes move from donor cells to recipient cells and are internalized and accumulated in the recipient cells via receptor-mediated endocytosis. Therefore, it is possible that CD137-mediated signaling in activated T cells is triggered by trans-activation of CD137 by CD137L on DCs in early immune responses, but by cis-activation of CD137 via CD137L in activated T cells in the later phase.
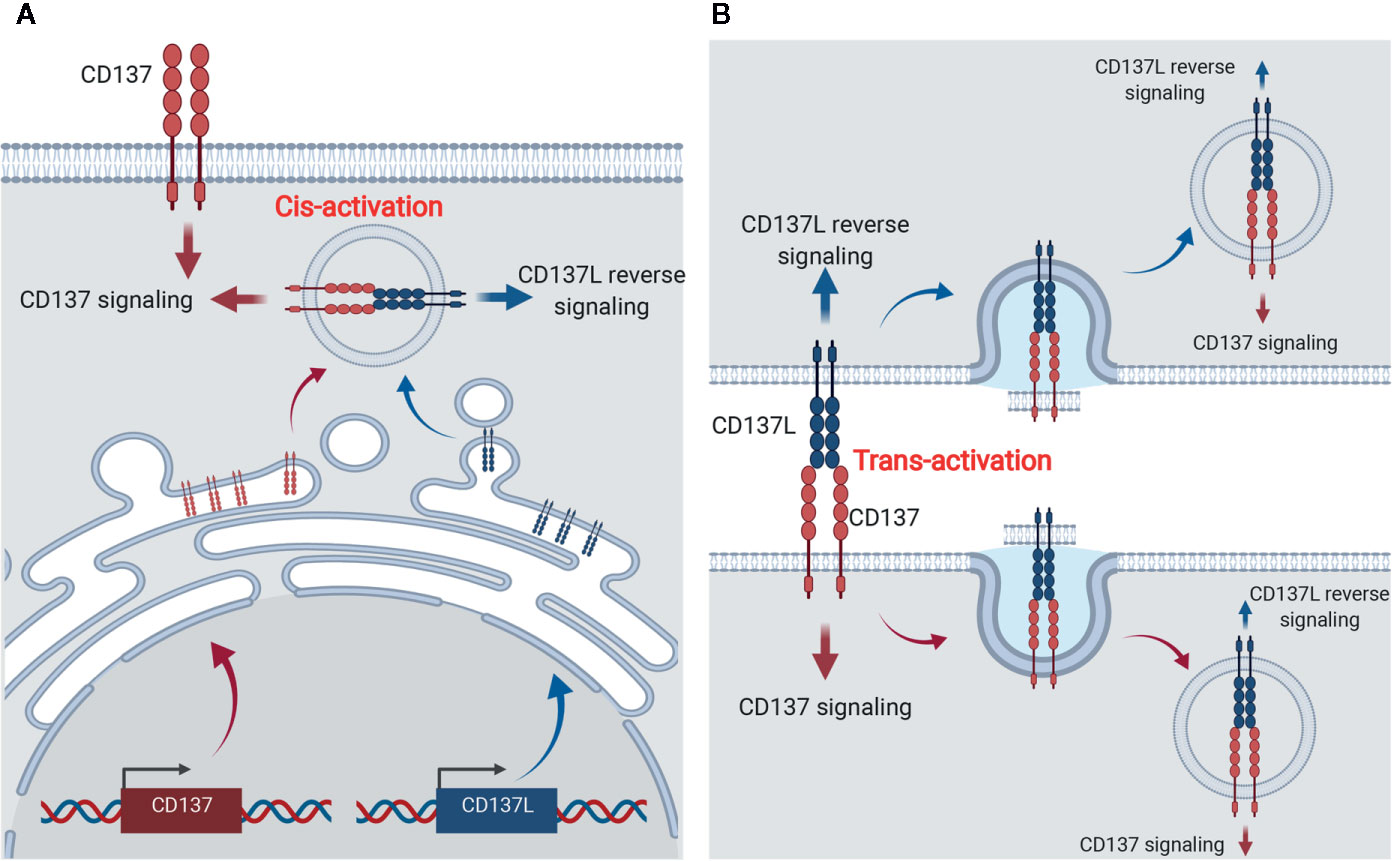
Figure 2 Cis- and trans-activation of CD137 and CD137L. (A) Cis-activation of CD137 and CD137L. Newly synthesized CD137 and CD137L interact within activated cells and thus, bidirectional signals are transmitted into the cells. (B) Trans-activation of CD137 and CD137L. Following the encounter of activated T cells and APCs, CD137, and CD137L are crosslinked and trogocytosis provoked by activation signals induces internalization of the CD137-CD137L complex and thus, bidirectional signals are transmitted into the cells.
Additionally, if CD137 is more strongly expressed than CD137L in activated T cells or mature DCs, CD137:CD137L complex formation will gradually become saturated, and newly synthesized CD137 begins to be solely located on the plasma membrane. Indeed, CD137 and CD137L mRNA analysis using data from the ImmGen database (www.immgen.org) showed that CD137 is more strongly expressed than CD137L in activated CD8+ T cells. This speculation not only provides an explanation for the unsolved question why CD137L is not found on the surface of CD137-expressing T cells or DCs, but also adds complexity to the CD137-CD137L interactions.
Although CD137 and CD137L are expressed on various cell types, signaling involving CD137 on T lymphocytes and CD137L on myeloid cells has been studied the most (11, 25). Like other TNFRSF members, CD137 forms a multimer when it binds with CD137L to initiate intracellular signaling. As shown in Figure 3A, human (h)CD137L forms noncovalent trimers, whereas mCD137L forms disulfide bond-linked dimers. The hCD137L homotrimer forms three hydrophobic clefts to which the hCD137 homotrimer binds. However, the mCD137 dimeric quaternary structure allows two mCD137 monomers to bind to each mCD137L protomer (81, 82). Recent studies have shown that galectin-9 (Gal-9), a bivalent lectin expressed together with mCD137 on cells, has a critical role in receptor clustering and mCD137-induced proximal signal transduction (81, 83) (Figure 3B). mCD137 contains four cysteine-rich domains (CDR1–4) (84) and Gal-9 contains N- and C-terminal carbohydrate-binding domains. An N-glycosylated amino acid in CDR4 is involved in interactions between mCD137 and Gal-9 via both the N-and the C-terminal carbohydrate-binding domains (81). This interaction may place the two mCD137 monomers in close vicinity or allow them to cluster, as well as induce a conformational change of the mCD137 cytoplasmic domain that facilitates the initial signaling event, namely binding of TNFR-associated factors (TRAFs) to the cytoplasmic tail of mCD137 (81). Gal-9 also binds to hCD137, which forms a trimeric complex with trimeric hCD137L, and this may induce a conformational change within the cytoplasmic tail of CD137 favoring TRAF binding or clustering (81).
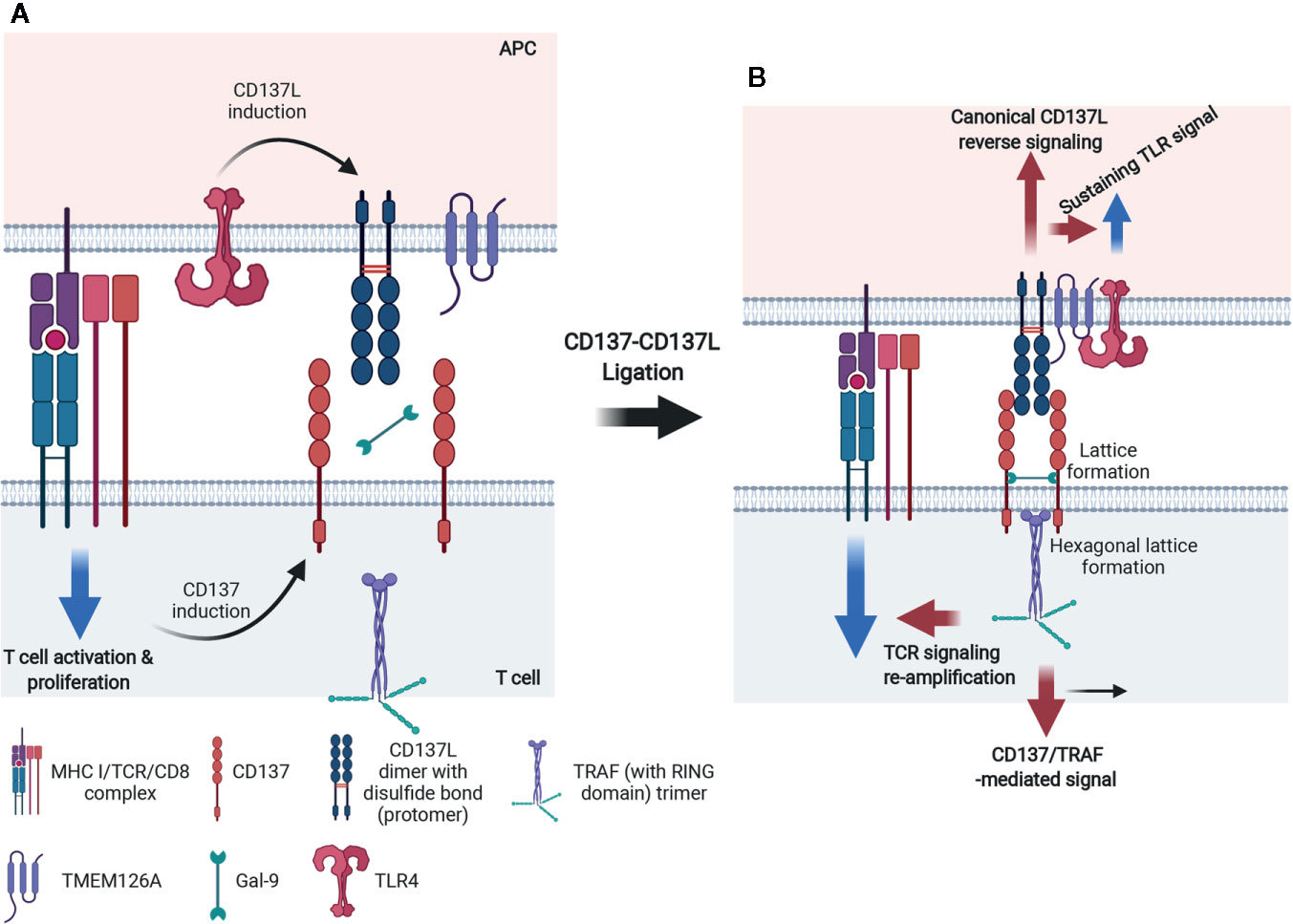
Figure 3 Mechanism of accelerated CD137- and CD137L-mediated signaling in activated T cells and mature APCs. (A) For CD137L on APCs, Toll signals, including TLR4, are essentially required and CD137L is expressed and becomes dimerized (protomer) via a disulfide bond. Simultaneously, APCs provide antigenic information to T cells by triggering TCR signals. Signal 1 via TCR on T cells consecutively induces CD137 expression on T cells. (B) After ligation of CD137 and CD137L, CD137 molecules are multimerized and the CD137 multimer is further stabilized by Gal-9 binding, probably through the formation of a lattice structure. The organized CD137 multimer recruits TRAF1/2 and generates TRAF (with a RING domain) trimer. CD137 signaling through the TRAF trimer has two distinct functions: 1) re-amplification of TCR signaling for mitogenic signals and 2) CD137-TRAF-mediated signals for the survival, proliferation, and differentiation of T cells. For CD137 signals, TLR4 and CD137L seem to form a heteromultimer through TMEM126A binding and thus, TLR4 signaling is sustained and canonical CD137L reverse signaling is transmitted.
A number of TNFRs are not fully activated by soluble ligands or agonistic Abs, but only by being immobilized or crosslinked. Even ligation of dimeric or trimeric CD137 did not generate effective responses (85). Since multimers of a minimum of nine mCD137 monomers bound to mCD137L nonamer generate functional signals in the absence of Gal-9, it seems that clustering of at least nine CD137 monomers is needed to elicit effective signals (81). It has been suggested that small numbers of ligand-free CD137 molecules are pre-assembled in a two-dimensional hexagonal lattice on the cell surface (86), and that Gal-9 may be involved in the formation of this hexagonal structure (83). Gal-9 may provide a structural platform for CD137 to produce initial signaling events via clustering of dimeric or multimeric CD137 (87–90). mCD137L acts together with Gal-9 to aid in the clustering of mCD137 monomers to efficiently initiate mCD137 signaling (81). The Gal-9 lattice may also serve as a docking site for other receptors and signaling molecules (91, 92) (Figure 3B). The modulation of TCR responses induced by exogenous Gal-9 depends on the presence of components of the TCR complex such as the tyrosine kinase, Lck (93). There is no evidence that Gal-9 directly interacts with any of the TCR/CD3 components (93). However, it has been suggested that Gal-9 acts on TCR-Lck signals by linking and assembling specific membrane microdomains of T cells, such as lipid rafts (93).
Homo- or heterotrimers of TRAF1 and TRAF2, which are proximal adaptor molecules for the mCD137 receptor signalosome, associate with the cytoplasmic tails of CD137 upon its ligation with CD137L and relay CD137 activation to distal intracellular signaling events, including NF-κB activation, intracellular Ca2+ mobilization, and ERK1/2 activation (94–96) (Figure 3B). Trimeric TRAFs are made up of combinations of six TRAFs (TRAF1–6); they bind to the cytoplasmic domains of TNFRs and function as scaffold proteins forming complexes with other signaling molecules (97, 98). All TRAFs, except TRAF1, have a RING zinc-finger domain that confers E3 ubiquitin ligase activity, which enables them to regulate the activities of downstream signaling proteins clustered among the TRAFs (99). Homo- or heterotrimers of TRAFs 1, 2, and 3 cluster upon hCD137 activation and stabilize two-dimensional hexagonal lattices of CD137 trimers, forming signalosomes (reviewed in 73). It has been suggested that binding of TRAF2-RING finger dimers as well as cIAP1/2-RING dimers between the TRAF trimers may aid in hexagonal lattice clustering (100–103). In addition, homo- or heterotrimers of TRAFs 1, 2, and 3 recruit various other molecules involved mainly in protein ubiquitination and induce signaling events such as NF-κB, ERK1/2, and p38 MAPK activation [reviewed in (104)].
We have shown that the signaling pathways activated by mCD137 crosslinking are involved in upregulating the expression of anti-apoptotic proteins such as bcl-XL, IL-2, and cyclin D2 in murine T cells (14, 105). These proteins are responsible for CD137-mediated increases in cell survival and expansion, which are relevant to the antiviral and antitumor effects of CD137. The early CD137 signaling pathways also help to relocate lipid rafts to the area of contact between T cells and CD137L-expressing cells. Ligation of CD137 leads to the translocation of TCR pathway proteins such as Lck, pTyr, PKC-θ, PLC-γ1, and SLP-76 to lipid rafts (106). Crosslinking of CD137 recruits CD137 and TRAF2 to lipid rafts and re-activates or sustains TCR signaling. We speculate that Gal-9 binding to CD137 creates a platform to induce redistribution of lipid rafts and subsequent recruitment of TCR and/or TCR signaling proteins, as previously described (106). If that is the case, this could be the molecular mechanism underlying reciprocal and continuous regulation of TCR and CD137 signals: the duration of TCR binding by the antigenic epitope/MHC complex on APC would control the expression of CD137, and its ligation by CD137L on APCs would in turn cluster and assemble sufficient Gal-9/CD137 to affect TCR signaling. Thus, it would be important to examine the involvement of Gal-9 in CD137-induced TCR activation.
Crosslinking of mCD137L with rCD137 or anti-CD137L mAb activates reverse signaling. CD137L reverse signal transduction has been studied in human and murine myeloid cells such as monocytes and macrophages (28, 105, 107). In human cells, the CD137L reverse signaling pathways involve Src tyrosine kinase, PI3K, p38MAPK, ERK1/2, and probably, NF-κB. hCD137L may associate with hTNFR1 to initiate these reverse signals (108). In murine macrophages, crosslinking of CD137L increases tyrosine phosphorylation of PP2-sensitive tyrosine kinases such as Src. Subsequently, it activates mTOR/p70S6K, leading to enhanced cell adhesion, survival, and macrophage colony-stimulating factor expression. It also activates Akt via Src tyrosine kinase/PI3K, as Akt phosphorylation by CD137L ligation is blocked by Src kinase inhibitor, PP2 or PI3K inhibitors, Wortmannin, and LY294002. CD137L reverse signal-mediated Akt activation is responsible for the upregulation of IL-1β transcript expression 107. Using yeast two-hybrid experiments, we found that CD137L binds to the novel transmembrane protein, TMEM126A, a member of the DUF1370 family of proteins, which consists of several hypothetical eukaryotic proteins with approximately 200 residues. Its biological function is unknown (109). TMEM126A exists as multimers in immune cells, but as monomers in liver cells (109), and associates with the C-terminal extracellular domain of CD137L (109). Binding of TMEM126A to CD137L is a prerequisite for CD137L reverse signaling, in which TMEM126A may play the same role as Gal-9 does in CD137 receptor signaling. In unstimulated myeloid cells, endogenous TMEM126A and CD137L colocalize as widespread spots. Upon crosslinking of CD137 with plate-bound rCD137-Fc, TMEM126A and CD137L colocalized predominantly at the points of contact with the culture plate (109). This suggests that TMEM126A aids in the multimerization of CD137L as well as in the linking of CD137L multimers with other signaling molecules in a CD137L signalosome (Figure 3B). CD137L-mediated reverse signals can be as diverse and complex as CD137 receptor signals: a yeast two-hybrid experiment identified proteins and kinases related to ubiquitination, adhesion, and endocytosis as binding candidates (109). In TMEM126A-deficient cells, LPS-induced upregulation of CD54 (ICAM-1), MHC II, CD86, and CD40 expression was diminished, indicating that TMEM126A is involved in TLR4 signaling (110). Flow-cytometric data showed that LPS-induced TMEM126A surface expression increased over time, whereas LPS-induced CD137L expression declined. This implies that TLR4 signaling regulates the cell surface expression of CD137L and TMEM126A. Immunofluorescence data revealed that CD137L/TMEM126A/TLR4 colocalized in spots (109, 110), suggesting that these three proteins may be associated. TMEM126A may also act as a linker between the CD137L signalosome and TLR4 (Figure 3B). LPS treatment induces binding of CD137L to TLR4, and their association triggers MyD88-independent TLR4 signals that prolong TNF production (111, 112). It will be important to examine whether TMEM126A interacts directly with TLR4 as it does with CD137L. This would support the hypothesis that TMEM26A bridges the CD137L signalosome with TLR4. Activation of TLR4 by LPS increases the expression of CD137L and TMEM126A on APCs, producing reverse signals that strengthen innate immune cell function. At the same time, CD137L reinforces TLR4-induced responses, presumably by binding to TMEM126A. In other words, mutual interactions take place between CD137L and TLR4 in APCs, as they do between the CD137 signalosome and TCR in T cells.
Plenty of experimental evidences support the notion that multiple receptor conformations exist, and ligands can select and stabilize the different receptor active states for signal transmission (113–117). In this way, one signal can be selectively produced over others (biased signaling). While signal bias has been extensively studied for G protein-coupled receptors, theoretical and experimental data suggest that it is a general behavior of ligands and receptors (114, 118, 119). Signal bias can be generated by any of the three components of the ligand/receptor/transducer complex, generating ligand bias, receptor bias, and system bias, respectively (Figure 4). Biased signaling through stabilization of select, multiple conformations of the receptor with a wide range of signaling effector molecules can be used in normal physiology to achieve fine control in naturally healthy body systems (116). Ligand bias refers to ligand-induced signal bias through the selection (or induction) of a unique receptor conformation coupled with preferentially adopted transducers. Receptor bias can be produced by altering the receptor to change its ability to bind to specific ligands and/or activate transducers. Receptor mutation and alternative splicing can generate biased signal (117). Receptor homodimerization can produce functional complexity associated with binding and activation cooperativity originating from allosteric interactions between the protomers making up an oligomer and intrinsic ligand efficacy (116). Moreover, in the receptor homodimer context, ligand bias changes with ligand concentration (116, 119). System bias can be established by changing the context of transducers/effectors, such as their expression levels. Since different cell and tissue types have different signaling systems, certain ligands acting on the same receptor can have different signaling properties depending on the cell type, and each receptor can produce unique responses with same ligand because of system bias.
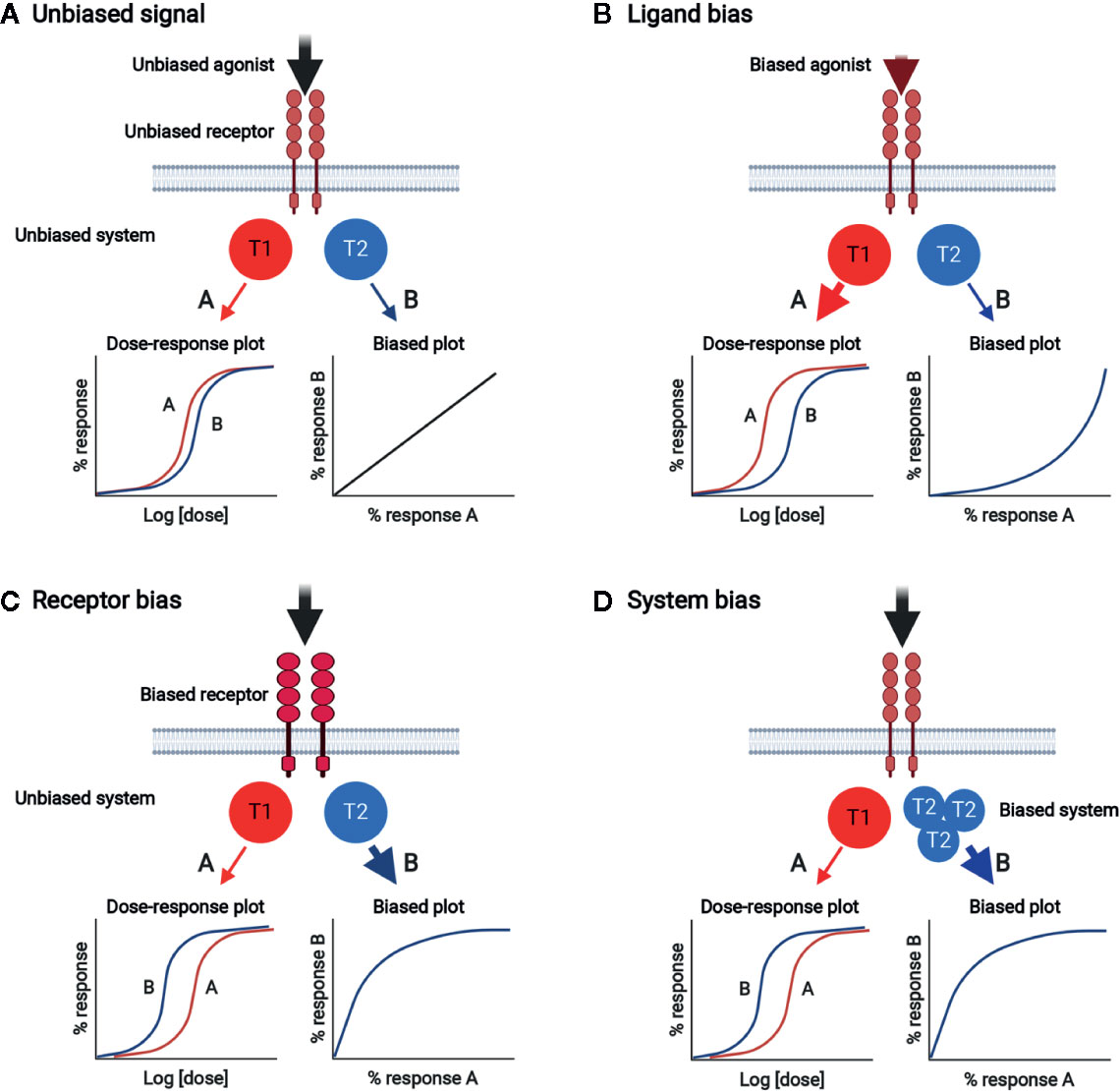
Figure 4 Biased signal generated by ligand bias, receptor bias, or system bias. (A) Unbiased signal. Unbiased agonist binds to unbiased receptor, equally activating signal pathways A and B through unbiased transducers T1 and T2, respectively. The dose-response plot shows the equivalent potencies for pathways A and B. Bias plot displays unbiased signal. (B) Ligand bias. Biased agonist binds and selects a certain unbiased receptor conformation, which relays signal dominantly through T1 rather than T2. The dose-response plot shows that the potency of response via pathway A is stronger than that via pathway B. The bias plot indicates that the signal is biased toward A. (C) Receptor bias. Unbiased agonist binds to biased receptor, which drives signal to pathway A rather than pathway B. The dose-response plot shows that the potency of response via pathway B is stronger than that via pathway A. The bias plot indicates that the signal is biased toward B. (D) System bias. Unbiased agonist binds to unbiased receptor, interacting with and activating more T2 than T1. Signal via pathway B is preferred over that via pathway A. The dose-response plot shows that the potency of response via pathway B is stronger than that via pathway A. The bias plot indicates that the signal is biased toward B.
In clustered dimeric CD137/CD137L signalosome complex, the expression dynamics of CD137 and CD137L may be important to establish biased signals in a cell. For example, antigen species, duration of antigenic challenge, and/or antigen levels determine their expression levels. The levels, degrees, and species of clustering homodimer of CD137 or CD137L and proximal downstream signaling molecules, such as TRAFs for CD137 and their unknown counterparts for CD137L, may generate receptor bias and ligand bias, as well as system bias. That is, the same CD137L (or CD137) can generate different and unique CD137 signals (or CD137L reverse signals) in a cell depending on antigenic differences. As mentioned above, receptor homodimerization confers receptor bias via allostery-mediated cooperativity for receptor binding and/or activation of downstream transducers (116). This notion is supported by a recent finding that the mCD137L dimeric structure undergoes allosteric changes upon mCD137 binding (82). Receptor homodimerization may also change the pattern of ligand bias with ligand concentration. Along with the intrinsic complexity of the homodimeric CD137/CD137L bidirectional signalosome, steric and topological changes in conformation induced by CD137/CD137L binding in the clustered, multimeric state may lead to the diversity and complexity of receptor/ligand-induced cellular responses (Figure 5). In addition, cellular diversity in expression of CD137/CD137L and transducers and their interactions with other key signaling receptors such as TCR and TLR4 further complicate CD137/CD137L bidirectional signals and related responses. These complexities established by signal bias in CD137/CD137L bidirectional signaling pathways may be optimal for finetuning of the immune response. We assume that CD137/CD137L signalosome-mediated in vivo cellular responses in the immune system may not be completely understood because the experimental tools used to elucidate CD137/CD137L functions, such as knockout mice, Abs, and recombinant proteins, can interfere with the complex organization of CD137/CD137L bidirectional signalosomes.
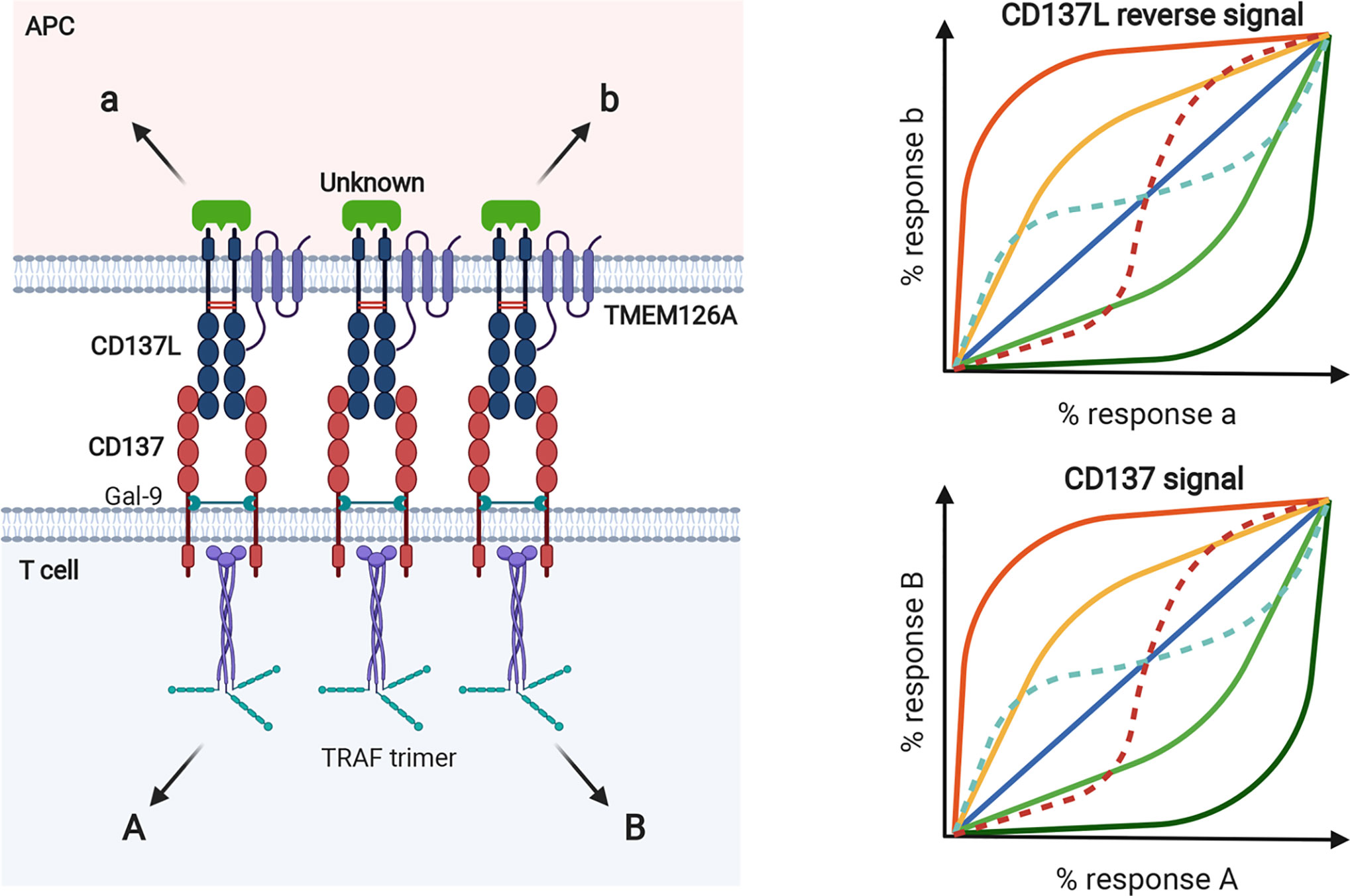
Figure 5 Biased signal in murine CD137/CD137L bidirectional signaling pathways. Expression levels of CD137 and CD137L and their extent of homodimer clustering can establish receptor and ligand bias in T cells and APCs, respectively. In terms of bidirectional signaling, receptors can be ligands for reverse signal and vice versa. Species and levels of transducers, such as TRAF trimer, can generate system bias. Because levels of CD137 and CD137L expression are transcriptionally and posttranslationally regulated by Ag exposure, various cellular responses can be generated by bidirectionally biased signals as shown in the bias plot. In the receptor homodimer context, ligand binding can generate receptor bias via allosteric interactions between protomers, through which ligand binding affinity and/or activation of downstream transducers can be altered. Ligand bias can also change with ligand concentration in the receptor homodimer (dotted lines in the bias plot).
The complexity of CD137/CD137L signaling pathways in T cells and APCs in vivo is even more complex. CD137−/− B6 mice display a reduced humoral response to KLH and reduced cytotoxic T-lymphocyte activity against vesicular stomatitis virus (27). In line herewith, CD137L−/− mice show defects in inducing CD8+ T-cell responses against viral infections, such as influenza (33), MHV-68 (34), and LCMV (35), and further study revealed that secondary rather than primary CD8+ T-cell responses against influenza infection are impaired in these mice (120). Because CD137/CD137L signals augment T-cell responses by preventing activation-induced cell death (AICD) (14), stimulating cell-cycle progression (105) and Th1 responses (121), and accelerating metabolism (122), these data fit the expectation that deficiency of CD137 or CD137L in vivo reduces T-cell immunity.
Although CD137−/− and CD137−/− mice exhibit reduced CD8+ T-cell responses in viral infection models (33–35), their T-cell responses in spontaneous autoimmune disease and tumor models are somehow enhanced (42). CD137 deficiency in MRL/lpr mice accelerated the development of lacrimal gland and skin lesions, enhanced lymphadenopathy, and led to early death, along with increased CD4+ T cells and B-cell activity (38, 39). Tumor growth rates were reduced in CD137−/− mice compared to WT B6 mice in an NK- and CD8+ T cell-dependent manner (42). Moreover, adoptively transferred CD137−/− OVA-specific OT-I and OT-II cells hyperproliferated in WT B6 mice (26, 41), and CD137−/− mice had increased numbers of primary CD8+ T cells and fewer memory CD8+ T cells during chronic/latent infection with mouse CMV (79). These data indicate that the CD137/CD137L axis is capable of both stimulatory and inhibitory cosignaling activities in vivo; however, the mechanisms underlying the enhanced T-cell responses in CD137−/− mice are not completely understood. In humans, CD137 deficiency is highly correlated with autoimmunity, autoimmune lymphoproliferation, common variable immune deficiency, and malignancies (123).
Agonistic anti-CD137 mAb also has dual stimulatory and inhibitory cosignaling activities in vivo. Agonistic anti-CD137 mAb typically enhances CD8+ T-cell responses against immunizing peptides such as HPV E7 (49) and LCMV NP396-404 (35), staphylococcal enterotoxin A (51), and several mouse tumor cells including Ag104A, P815, MC38, and B16-F10 melanoma (51, 53). The oxygen-deprived tumor microenvironment (TME) upregulates CD137 expression via the transcription factor HIF-α in CD4+ and CD8+ tumor-infiltrating T lymphocytes. Hypoxia-induced CD137 expression in tumor-infiltrating T lymphocytes is thought to explain why intratumoral injection of agonistic anti-CD137 mAb can achieve immunotherapeutic antitumor effects with minimum systemic side effects (124). Since triggering the CD137 signal on CD8+ T cells has therapeutic potential in cancer, agonistic anti-CD137 mAbs are under investigation as immunotherapeutic agents in cancer (125, 126). CD137 has been recognized as one of the critical immune checkpoint molecules for tumor Tregs. However, its role in Tregs remains controversial: some studies have suggested that CD137 mAb can block Treg activity (127, 128), whereas one study reported that it enhanced Treg proliferation (129). Recent studies have shown that depletion of Tregs in the TME by depleting anti-CD137 mAb decreased tumor growth, indicating that Tregs in the TME express CD137 to suppress antitumor hyperimmune responses (72, 130). Agonistic anti-CD137 mAb also reduces the incidence and severity of autoimmune diseases including experimental autoimmune encephalitis, collagen-induced arthritis, systemic lupus erythematosus, and experimental autoimmune uveoretinitis (44, 45, 47, 48).
Although CD137 signaling itself seems to be multifaceted in terms of immune response modulation, the contrasting immune responses found in tumor-challenged CD137−/− mice—i.e., enhanced NK- and CD8+ T-cell responses against tumors—need to be fully understood because agonistic anti-CD137 mAbs are being evaluated as antitumor therapeutics based on their positive effects on CD8+ T cells (125, 126). It seems that the enhanced T-cell responses in CD137−/− mice are due to intrinsic effects accumulated during the development of the immune system in the absence of CD137 signals and thus, cannot be simply mimicked when CD137 signals are blocked with antagonistic anti-CD137 mAb in vivo. In addition, since the ligation of CD137-CD137L induces bidirectional signals, agonistic mAb of CD137 or CD137L will enhance the signal via its target, but block the signal through its counterpart (Figure 6), which also increases the complexity of CD137-CD137L signaling.
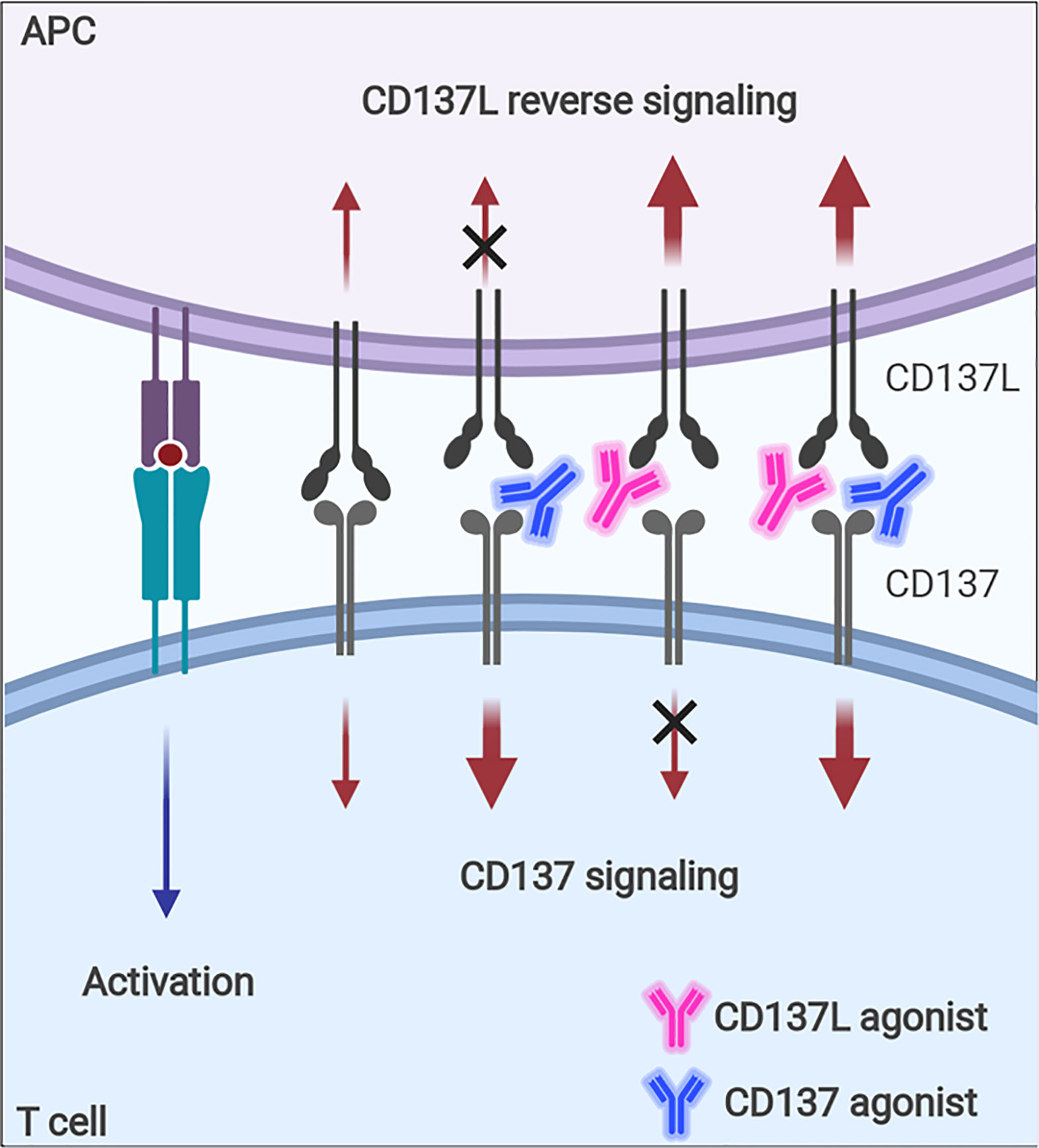
Figure 6 Immune modulation with CD137 or CD137L agonist. Signaling via CD137 or CD137L into activated T cells and APCs can be modulated with agonistic mAb. Since the ligation of CD137 and CD137 transmits bidirectional signals, CD137 agonist will enhance CD137 signal, but block CD137L-mediated signal, whereas CD137L agonist will have the opposite effects.
Since CD137L is an endogenous ligand that binds to CD137, it is conceivable that the CD137/Gal-9/TCR signaling complex may interact with the CD137L/TMEM126A/TLR4 signaling complex at immunological synapses (Figure 7A), which are nanoscale gaps between T lymphocytes and APCs. In signalosome complexes at immunological synapses, these six proteins may influence one another by modulating each other’s expression level, provoking CD137/CD137L bidirectional signaling, and modifying TCR/TLR4 signaling during APC maturation and subsequent antigen presentation to T cells at peripheral infection sites or in lymphoid organs. Encounters with pathogen-associated molecular patterns (and/or damage-associated molecular patterns) activate pattern recognition receptors on APCs such as TLR4 at peripheral sites, and this usually induces pro-inflammatory responses as well as APC maturation involving increases in the expression of MHC proteins, adhesion molecules, and costimulatory molecules. During APC maturation, CD137L expression is upregulated together with TMEM126A expression (Figure 7B), and CD137L may adopt a specific conformation by forming a complex with TMEM126A and TLR4. This conformation may be needed for prolonged activation of TLR4, as well as for initiating CD137L reverse signals upon engagement with CD137 on T cells at a later time when APCs encounter T cells in lymphoid organs. CD137L reverse signals induce further maturation of APCs, which enhances antigen presentation to T cells. When just-matured APCs meet T cells in lymphoid organs, the MHC/epitope on the APCs binds to the TCR on the T cells, and TCR signals are produced with the help of costimulatory molecules such as CD28/B7. TCR activation induces or enhances CD137 expression (Figure 7B). CD137 then interacts with Gal-9 and adopts a state that enables it to bind to a suitable form of CD137L and initiate CD137 signals. In the complex, crosslinking of CD137/Gal-9 re-recruits TCR signaling molecules and maintains TCR signaling, which promotes the differentiation of naïve T cells to effector and/or memory T cells.
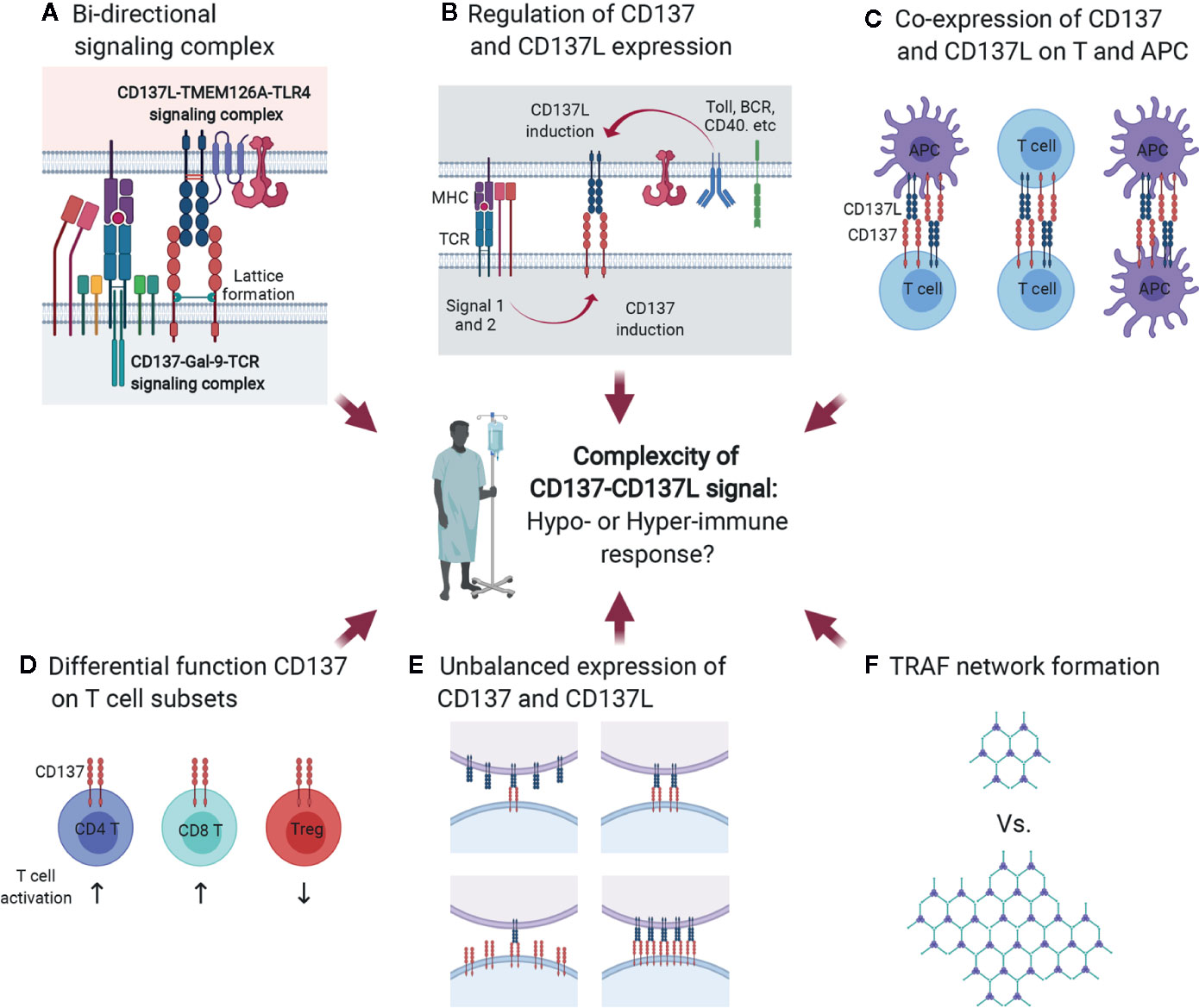
Figure 7 Multiple factors contribute to complexity of CD137-CD137L signal. (A) Bidirectional signaling complex. During direct interaction of activated T cells and APCs, CD137-Gal-9 complex and CD137L-TMEM126A-TLR4 complex simultaneously transmit signals. (B) Multiple factors, including Ag affinity, costimulatory signals, and APC maturation triggers, are involved in CD137 and CD137L expression. (C) Co-expression of CD137 and CD137L. CD137 and CD137L are well known to be expressed on activated T cells and mature APCs, respectively. However, a recent study demonstrated that CD137L is also expressed in activated T cells. Therefore, CD137 and CD137L can interact not only in the T cell-APC conjugate, but also in T cell-T cell or APC-APC conjugates. (D) Differential functions of CD137 in T-cell subsets. CD137 triggering induces proliferation and anti-apoptosis in CD4 and CD8 T cells and Tregs. CD137 triggering enhances CD4 and CD8 T cell-mediated immunity and induces Treg-mediated immune suppression because CD137 signal expands activated Tregs that can suppress the overall immune response. (E) Unbalanced expression of CD137 and CD137L. (F) Limited ligation of CD137 and CD137L leads to inefficient formation of the TRAF network that is required to re-amplify TCR signal and CD137-mediated signal.
The hyperproliferation of CD137-deficient T cells may be due to CD137/CD137L-induced suppression of T-cell proliferation. CD137L reverse signals suppress T-cell activation (29, 131) (Figure 7C). Alternatively, highly expressed CD137 generated by TCR activation with high-affinity Ag may form complexes with TCR/Gal-9. The conformation of TCR/Gal-9/CD137 adopted upon contact with CD137L may tune TCR signals to block their hyperproliferation, but promote their survival and differentiation. Either way, CD137/CD137L may be able to induce optimal T-cell responses during antigen contact. The in vivo situation may be even more complex. CD137 and CD137L bidirectional signals may affect the expression and/or signaling pathways of other costimulatory proteins and/or cytokines. Moreover, TLR4 and TCR signals modified by CD137/CD137L signals may influence other molecules at immunological synapses. Signal bias due to the clustering of dimerized CD137/CD137L, CD137/CD137L expression levels and duration, the degree of binding with cis proteins such as Gal-9, TRAFs, and TMEM126A, and interaction with other key proteins such as TCR and TLR4, gives rise to the complexity of CD137/CD137L bidirectional signalosome-mediated cellular responses (Figures 3 and 7C).
In general, CD137 signal positively regulates T-cell responses. However, in the case of Tregs, CD137 signal not only increases number of Tregs by enhancing their proliferation, but also temporally neutralizes the suppressive function of activated Tregs, but not naïve Tregs (47). Since CD137 triggering increases the numbers of Tregs and transiently neutralizes their suppressive activity, overall T-cell responses will be strongly inhibited by the suppressive activity of the increased Tregs when CD137L is discontinued (Figure 7D). Therefore, although CD137 signal exerts similar effects such as proliferation enhancement and anti-apoptosis on T-cell subsets, the outcomes of CD137 triggering can differ depending on the function of the T-cell subsets, which increases the complexity of CD137/CD137L signaling.
Another factor contributing to the complexity of CD137/CD137L signaling is unsynchronized CD137 and CD137L expression. Multiple factors, including TCR strength, costimulation, and Toll ligand, are involved in CD137 and CD137 expression on T cells and APCs. If the expression of these molecules is not spatiotemporally synchronized, unbalanced CD137 and CD137L expression may lead to limited ligation of CD137 and CD137L, eventually resulting in inefficient TRAF network formation and delayed T-cell activation (Figures 7E, F).
Immunity against danger signals may be controlled by these web-like interactions. It is difficult to systemically dissect this complexity. However, it is clear the complexity of the molecular interactions gives rise to the immunity required to protect us from the many menaces, and that derangement of the interactions may lead to hypo- or hyperimmune status and to immunopathogenic conditions such as infective inflammation, autoimmune diseases, and possibly, tumors. This may be why contradictory responses have been reported in disease models when anti-CD137 mAb or rCD137L was administered. For example, viral infection or tumors that can escape immune surveillance may create conditions that inhibit the association of CD137 with CD137L and thus suppress T-cell activation. Ligation of T-cell CD137 with agonistic anti-CD137 Ab or rCD137L may reverse the T-cell inhibition induced by viral infection or tumors. In contrast, hyperimmunity in autoimmune diseases may be caused by strong association of CD137 with CD137L. CD137 Abs may restrain this and dampen the CD137/CD137-mediated signals in T cells and APCs.
Sophisticated in vivo mouse experiments using natural antigenic challenge and monitoring changes in mCD137/CD137L expression, localization, and steric structure should provide some answers to the following questions: 1) How are signalosome complexes formed? 2) What is the driving force behind their formation? 3) How do they provide the array of intracellular signals needed for dealing optimally with host immunological cues?
H-WL and BC equally contributed to plan a scheme of this review. H-WL reviewed on bi-directional signaling and proposed potential roles of biased signal in CD137/CD137L complexity. BC reviewed and described about in vivo immune responses mediated by CD137/CD137L and their expression regulation. All authors contributed to the article and approved the submitted version.
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2020R1F1A104819111) to H-WL and a grant of the National Cancer Center of Korea (NCC-191050) to BC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
BioRender (https://biorender.com/) was used to prepare the figures.
2. McCullough KC, Summerfield A. Basic concepts of immune response and defense development. ILAR J (2005) 46:230–40. doi: 10.1093/ilar.46.3.230
3. Janeway CA Jr, Bottomly K, Horowitz J, Kaye J, Jones B, Tite J. Modes of cell:cell communication in the immune system. J Immunol (1985) 135:739s–42s.
4. Oviedo-Orta E, Hoy T, Evans WH. Intercellular communication in the immune system: differential expression of connexin40 and 43, and perturbation of gap junction channel functions in peripheral blood and tonsil human lymphocyte subpopulations. Immunology (2000) 99:578–90. doi: 10.1046/j.1365-2567.2000.00991.x
5. Hodgkin PD, Rush J, Gett AV, Bartell G, Hasbold J. The logic of intercellular communication in the immune system. Immunol Cell Biol (1998) 76:448–53. doi: 10.1046/j.1440-1711.1998.00776.x
6. Kioussis D, Pachnis V. Immune and nervous systems: more than just a superficial similarity? Immunity (2009) 31:705–10. doi: 10.1016/j.immuni.2009.09.009
7. Taams LS, van Eden W, Wauben MH. Antigen presentation by T cells versus professional antigen-presenting cells (APC): differential consequences for T cell activation and subsequent T cell-APC interactions. Eur J Immunol (1999) 29:1543–50. doi: 10.1002/(SICI)1521-4141(199905)29:05<1543::AID-IMMU1543>3.0.CO;2-R
8. Gunzer M, Weishaupt C, Hillmer A, Basoglu Y, Friedl P, Dittmar KE, et al. A spectrum of biophysical interaction modes between T cells and different antigen-presenting cells during priming in 3-D collagen and in vivo. Blood (2004) 104:2801–09. doi: 10.1182/blood-2004-03-1193
9. Wulfing C, Sjaastad MD, Davis MM. Visualizing the dynamics of T cell activation: Intracellular adhesion molecule 1 migrates rapidly to the T cell-B cell interface and acts to sustain calcium levels. Proc Natl Acad Sci USA (1998) 95:6302–07. doi: 10.1073/pnas.95.11.6302
10. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22:240–73. doi: 10.1128/CMR.00046-08
11. Gloria CW, Lin HY, McPherson AJ, Tania H. Watts. Immune regulation by 4-1BB and 4-1BBL: complexities and challenges. Immunol Rev (2009) 229:192–215. doi: 10.1111/j.1600-065X.2009.00765.x
12. Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol (2003) 3:609–20. doi: 10.1038/nri1148
13. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell (2001) 104:487–501. doi: 10.1016/S0092-8674(01)00237-9
14. Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-x(L) and Bfl-1. J Immunol (2002) 169:4882–88. doi: 10.4049/jimmunol.169.9.4882
15. Eissner G, Kirchner S, Lindner H, Kolch W, Janosch P, Grell M, et al. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J Immunol (2000) 164:6193–8. doi: 10.4049/jimmunol.164.12.6193
16. Zhang H, Yan D, Shi X, Liang H, Pang Y, Qin N, et al. Transmembrane TNF-α mediates “forward” and “reverse” signaling, inducing cell death or survival via the NF-κB pathway in Raji Burkitt lymphoma cells. J Leukoc Biol (2008) 84:789–97. doi: 10.1189/jlb.0208078
17. Lückerath K, Kirkin V, Melzer IM, Thalheimer FB, Siele D, Milani W, et al. Immune modulation by Fas ligand reverse signaling: lymphocyte proliferation is attenuated by the intracellular Fas ligand domain. Blood (2011) 117:519–29. doi: 10.1182/blood-2010-07-292722
18. Vinay DS, Kwon BS. Role of 4-1BB in immune responses. Semin Immunol (1998) 10:481–9. doi: 10.1006/smim.1998.0157
19. Pollok KE, Kim YJ, Zhou Z, Hurtado J, Kim KK, Pickard RT, et al. Inducible T cell antigen 4-1BB. Analysis of expression and function. J Immunol (1993) 150:771–81.
20. Melero I, Johnston JV, Shufford WW, Mittler RS, Chen L. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol (1998) 190:167–72. doi: 10.1006/cimm.1998.1396
21. Kienzle G, von Kempis J. CD137 (ILA/4-1BB), expressed by primary human monocytes, induces monocyte activation and apoptosis of B lymphocytes. Int Immunol (2000) 12:73–82. doi: 10.1093/intimm/12.1.73
22. Lindstedt M, Johansson-Lindbom B, Borrebaeck CA. Expression of CD137 (4-1BB) on human follicular dendritic cells. Scand J Immunol (2003) 57:305–10. doi: 10.1046/j.1365-3083.2003.01217.x
23. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4+ CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity (2002) 16:311–23. doi: 10.1016/S1074-7613(02)00280-7
24. Goodwin RG, Din WS, Davis-Smith T, Anderson DM, Gimpel SD, Sato TA, et al. Molecular cloning of a ligand for the inducible T cell gene 4-1BB: a member of an emerging family of cytokines with homology to tumor necrosis factor. Eur J Immunol (1993) 23:2631–41. doi: 10.1002/eji.1830231037
25. Shao Z, Schwarz H. CD137 ligand, a member of the tumor necrosis factor family, regulates immune responses via reverse signal transduction. J Leukoc Biol (2011) 89:21–9. doi: 10.1189/jlb.0510315
26. Eun S-Y, Lee S-W, Xu Y, Croft M. 4-1BB Ligand signaling to T cells limits T Cell activation. J Immunol (2015) 194:134–14. doi: 10.4049/jimmunol.1401383
27. Kwon BS, Hurtado JC, Lee ZH, Kwack KB, Seo SK, Choi BK, et al. Immune responses in 4-1BB (CD137)-deficient mice. J Immunol (2002) 168:5483–90. doi: 10.4049/jimmunol.168.11.5483
28. Kim Y-J, Li G, Broxmeyer HE. 4-1BB ligand stimulation enhances myeloid dendritic cell maturation from human umbilical cord blood CD34+ progenitor cells. J Hematother Stem Cell Res (2002) 11:895–903. doi: 10.1089/152581602321080556
29. Langstein J, Michel J, Fritsche J, Kreutz M, Andreesen R, Schwarz H. CD137 (ILA/4-1BB), a member of the TNF receptor family, induces monocyte activation via bidirectional signaling. J Immunol (1998) 160:2488–94.
30. Langstein J, Schwarz H. Identification of CD137 as a potent monocyte survival factor. J Leukoc Biol (1999) 65:829–33. doi: 10.1002/jlb.65.6.829
31. Kim D-K, Lee SC, Lee H-W. CD137 ligand-mediated reverse signals increase cell viability and cytokine expression in murine myeloid cells: Involvement of mTOR/p70S6 kinase and Akt. Eur J Immunol (2009) 39:2617–28. doi: 10.1002/eji.200939292
32. Martínez Gómez JM, Croxford JL, Yeo KP, Angeli V, Schwarz H, Gasser S. Development of experimental autoimmune encephalomyelitis critically depends on CD137 ligand signaling. J Neurosci (2012) 32:18246–52. doi: 10.1523/JNEUROSCI.2473-12.2012
33. Lin GH, Sedgmen BJ, Moraes TJ, Snell LM, Topham DJ, Watts TH. Endogenous 4-1BB ligand plays a critical role in protection from influenza-induced disease. J Immunol (2009) 182:934–47. doi: 10.4049/jimmunol.182.2.934
34. Fuse S, Bellfy S, Yagita H, Usherwood EJ. CD8+ T cell dysfunction and increase in murine gamma herpes virus latent viral burden in the absence of 4-1BB ligand. J Immunol (2007) 178:5227–36. doi: 10.4049/jimmunol.178.8.5227
35. Tan JT, Whitmire JK, Murali-Krishna K, Ahmed R, Altman JD, Mittler RS, et al. 4-1BB costimulation is required for protective anti-viral immunity after peptide vaccination. J Immunol (2000) 164:2320–5. doi: 10.4049/jimmunol.164.5.2320
36. Tan JT, Whitmire JK, Ahmed R, Pearson TC, Larsen CP. 4-1BB ligand, a member of the TNF family, is important for the generation of antiviral CD8 T cell responses. J Immunol (1999) 163:4859–68.
37. Bertram EM, Dawicki W, Sedgmen B, Bramson JL, Lynch DH, Watts TH. A switch in costimulation from CD28 to 4-1BB during primary versus secondary CD8 T cell response to influenza in vivo. J Immunol (2004) 172:981–8. doi: 10.4049/jimmunol.172.2.981
38. Vinay DS, Kim JD, Asai T, Choi BK, Kwon BS. Absence of 4-1BB gene function exacerbates lacrimal gland inflammation in autoimmune-prone MRL-Faslpr mice. Invest Ophthalmol Vis Sci (2007) 48:4608–15. doi: 10.1167/iovs.07-0153
39. Vinay DS, Choi JH, Kim JD, Choi BK, Kwon BS. Role of endogenous CD137 in the development of systemic lupus erythematosus. Immunology (2007) 122:394–400. doi: 10.1111/j.1365-2567.2007.02653.x
40. Lee SW, Vella AT, Kwon BS, Croft M. Enhanced CD4 T cell responsiveness in the absence of 4-1BB. J Immunol (2005) 174:6803–8. doi: 10.4049/jimmunol.174.11.6803
41. Lee SW, Park Y, Song A, Cheroutre H, Kwon BS, Croft M. Functional dichotomy between OX40 and 4-1BB in modulating effector CD8 T cell responses. J Immunol (2006) 177:4464–72. doi: 10.4049/jimmunol.177.7.4464
42. Choi BK, Kim YH, Kim CH, Kim MS, Kim KH, Oh HS, et al. Peripheral 4-1BB signaling negatively regulates NK cell development through IFN-gamma. J Immunol (2010) 185:1404–11. doi: 10.4049/jimmunol.1000850
43. Humphreys IR, Lee SW, Jones M, Loewendorf A, Gostick E, Price DA, et al. Biphasic role of 4-1BB in the regulation of mouse cytomegalovirus-specific CD8+ T cells. Eur J Immunol (2010) 40:2762–8. doi: 10.1002/eji.200940256
44. Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH, et al. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med (2004) 10:1088–94. doi: 10.1038/nm1107
45. Sun Y, Chen HM, Subudhi SK, Chen J, Koka R, Chen L, et al. Costimulatory molecule-targeted antibody therapy of a spontaneous autoimmune disease. Nat Med (2002) 8:1405–13. doi: 10.1038/nm1202-796
46. Fukushima A, Yamaguchi T, Ishida W, Fukata K, Mittler RS, Yagita H, et al. Engagement of 4-1BB inhibits the development of experimental allergic conjunctivitis in mice. J Immunol (2005) 175:4897–903. doi: 10.4049/jimmunol.175.8.4897
47. Choi BK, Asai T, Vinay DS, Kim YH, Kwon BS. 4-1BB-mediated amelioration of experimental autoimmune uveoretinitis is caused by indoleamine 2,3-dioxygenase-dependent mechanisms. Cytokine (2006) 34:233–42. doi: 10.1016/j.cyto.2006.04.008
48. Sun Y, Lin X, Chen HM, Wu Q, Subudhi SK, Chen L, et al. Administration of agonistic anti-4-1BB monoclonal antibody leads to the amelioration of experimental autoimmune encephalomyelitis. J Immunol (2002) 168):1457–65. doi: 10.4049/jimmunol.168.3.1457
49. Wilcox RA, Flies DB, Zhu G, Johnson AJ, Tamada K, Chapoval AI, et al. Provision of antigen and CD137 signaling breaks immunological ignorance, promoting regression of poorly immunogenic tumors. J Clin Invest (2002) 109:651–9. doi: 10.1172/JCI0214184
50. Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med (1997) 186:47–55. doi: 10.1084/jem.186.1.47
51. Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med (1997) 3:682–5. doi: 10.1038/nm0697-682
52. Myers L, Takahashi C, Mittler RS, Rossi RJ, Vella AT. Effector CD8 T cells possess suppressor function after 4-1BB and Toll-like receptor triggering. Proc Natl Acad Sci U S A (2003) 100:5348–53. doi: 10.1073/pnas.0837611100
53. Kim JA, Averbook BJ, Chambers K, Rothchild K, Kjaergaard J, Papay R, et al. Divergent effects of 4-1BB antibodies on antitumor immunity and on tumor-reactive T-cell generation. Cancer Res (2001) 61:2031–7.
54. Yang Y, Yang S, Ye Z, Jaffar J, Zhou Y, Cutter E, et al. Tumor cells expressing anti-CD137 scFv induce a tumor-destructive environment. Cancer Res (2007) 67:2339–44. doi: 10.1158/0008-5472.CAN-06-3593
55. May KF Jr, Chen L, Zheng P, Liu Y. Anti-4-1BB monoclonal antibody enhances rejection of large tumor burden by promoting survival but not clonal expansion of tumor-specific CD8+ T cells. Cancer Res (2002) 62:3459–65.
56. Sytwu HK, Lin WD, Roffler SR, Hung JT, Sung HS, Wang CH, et al. Anti-4-1BB-based immunotherapy for autoimmune diabetes: lessons from a transgenic non-obese diabetic (NOD) model. J Autoimmun (2003) 21:247–54. doi: 10.1016/S0896-8411(03)00112-4
57. Cooper D, Bansal-Pakala P, Croft M. 4-1BB (CD137) controls the clonal expansion and survival of CD8 T cells in vivo but does not contribute to the development of cytotoxicity. Eur J Immunol (2002) 32:521–9. doi: 10.1002/1521-4141(200202)32:2<521::AID-IMMU521>3.0.CO;2-X
58. Choi BK, Kim YH, Choi JH, Kim CH, Kim KS, Sung YC. Unified immune modulation by 4-1BB triggering leads to diverse effects on disease progression in vivo. Cytokine (2011) 55:420–8. doi: 10.1016/j.cyto.2011.05.015
59. Mittler RS, Bailey TS, Klussman K, Trailsmith MD, Hoffmann MK. Anti-4-1BB monoclonal antibodies abrogate T cell-dependent humoral immune responses in vivo through the induction of helper T cell anergy. J Exp Med (1999) 190:1535–40. doi: 10.1084/jem.190.10.1535
60. Hong HJ, Lee JW, Park SS, Kang YJ, Chang SY, Kim KM, et al. A humanized anti-4-1BB monoclonal antibody suppresses antigen-induced humoral immune response in nonhuman primates. J Immunother (2000) 23:613–21. doi: 10.1097/00002371-200011000-00002
61. Zhu G, Flies DB, Tamada K, Sun Y, Rodriguez M, Fu YX, et al. Progressive depletion of peripheral B lymphocytes in 4-1BB (CD137) ligand/I-Ealpha)-transgenic mice. J Immunol (2001) 167:2671–6. doi: 10.4049/jimmunol.167.5.2671
62. Wu ZQ, Khan AQ, Shen Y, Wolcott KM, Dawicki W, Watts TH, et al. 4-1BB (CD137) differentially regulates murine in vivo protein- and polysaccharide-specific immunoglobulin isotype responses to Streptococcus pneumoniae. Infect Immun (2003) 71:196–204. doi: 10.1128/IAI.71.1.196-204.2003
63. Moens L, Jeurissen A, Mittler RS, Wuyts G, Michiels G, Boon L, et al. Distinct approaches to investigate the importance of the murine 4-1BB-4-1BBL interaction in the antibody response to Streptococcus pneumoniae. J Leukoc Biol (2007) 82:638–44. doi: 10.1189/jlb.1006628
64. Foell J, Strahotin S, O’Neil SP, McCausland MM, Suwyn C, Haber M, et al. CD137 costimulatory T cell receptor engagement reverses acute disease in lupus-prone NZB x NZW F1 mice. J Clin Invest (2003) 111:1505–18. doi: 10.1172/JCI200317662
65. Lee SW, Park Y, So T, Kwon BS, Cheroutre H, Mittler RS, et al. Identification of regulatory functions for 4-1BB and 4-1BBL in myelopoiesis and the development of dendritic cells. Nat Immunol (2008) 9:917–26. doi: 10.1038/ni.1632
66. DeBenedette MA, Chu NR, Pollok KE, Hurtado J, Wade WF, Kwon BS, et al. Role of 4-1BB ligand in costimulation of T lymphocyte growth and its upregulation on M12 B lymphomas by cAMP. J Exp Med (1995) 181:985–92. doi: 10.1084/jem.181.3.985
67. Futagawa T, Akiba H, Kodama T, Takeda K, Hosoda Y, Yagita H, et al. Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int Immunol (2002) 14:275–86. doi: 10.1093/intimm/14.3.275
68. Lee PK, Chang CJ, Lin CM. Lipopolysaccharide preferentially induces 4-1BB ligand expression on human monocyte-derived dendritic cells. Immunol Lett (2003) 9:215–21. doi: 10.1016/j.imlet.2003.08.002
69. Gravestein LA, Borst J. Tumor necrosis factor receptor family members in the immune system. Semin Immunol (1998) 10:423–34. doi: 10.1006/smim.1998.0144
70. Dawicki W, Watts TH. Expression and function of 4-1BB during CD4 versus CD8 T cell responses in vivo. Eur J Immunol (2004) 34:743–51. doi: 10.1002/eji.200324278
71. Kim JD, Kim CH, Kwon BS. Regulation of mouse 4-1BB expression: multiple promoter usages and a splice variant. Mol Cells (2011) 31:141–9. doi: 10.1007/s10059-011-0018-6
72. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood (2018) 131:49–57. doi: 10.1182/blood-2017-06-741041
73. Michel J, Schwarz H. Expression of soluble CD137 correlates with activation induced cell death of lymphocytes. Cytokine (2000) 12:742–6. doi: 10.1006/cyto.1999.0623
74. Furtner M, Straub R, Kruger S, Schwarz H. Levels of soluble CD137 are enhanced in sera of leukemia and lymphoma patients and are strongly associated with chronic lymphocytic leukemia. Leukemia (2005) 19:883–5. doi: 10.1038/sj.leu.2403675
75. Tu TH, Kim CS, Kang JH, Nam-Goong IS, Nam CW, Kim ES, et al. Levels of 4-1BB transcripts and soluble 4-1BB protein are elevated in the adipose tissue of human obese subjects and are associated with inflammatory and metabolic parameters. Int J Obes (Lond) (2014) 38:1075–82. doi: 10.1038/ijo.2013.222
76. Chang YH, Wang KC, Chu KL, Clouthier DL, Tran AT, Torres Perez MS, et al. Dichotomous expression of TNF superfamily ligands on antigen-presenting cells controls post-priming anti-viral CD4+ T cell immunity. Immunity (2017) 47:943–58.e9. doi: 10.1016/j.immuni.2017.10.014
77. Segura E, Amigorena S. Inflammatory dendritic cells in mice and humans. Trends Immunol (2013) 34(9):440–5. doi: 10.1016/j.it.2013.06.001
78. Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity (2006) 25(1):153–62. doi: 10.1016/j.immuni.2006.04.017
79. Martinez-Forero I, Azpilikueta A, Bolaños-Mateo E, Nistal-Villan E, Palazon A, Teijeira A, et al. T cell costimulation with anti-CD137 monoclonal antibodies is mediated by K63-polyubiquitin-dependent signals from endosomes. J Immunol (2013) 190:6694–706. doi: 10.4049/jimmunol.1203010
80. Hudrisier D, Aucher A, Puaux AL, Bordier C, Joly E. Capture of target cell membrane components via trogocytosis is triggered by a selected set of surface molecules on T or B cells. J Immunol (2007) 178:3637–47. doi: 10.4049/jimmunol.178.6.3637
81. Bitra A, Doukov T, Wang J, Picarda G, Benedict CA, Croft M, et al. Crystal structure of murine 4-1BB and its interaction with 4-1BBL support a role for galectin-9 in 4-1BB signaling. J Biol Chem (2018) 293:1317–29. doi: 10.1074/jbc.M117.814905
82. Bitra A, Doukov T, Destito G, Croft M, Zajonc DM. Crystal structure of the 4-1BB/4-1BBL complex reveals an unusual dimeric ligand that undergoes structural changes upon 4-1BB receptor binding. J Biol Chem (2019) 294:1831–45. doi: 10.1074/jbc.RA118.006297
83. Madireddi S, Eun SY, Lee SW, Nemčovičová I, Mehta AK, Zajonc DM, et al. Galectin-9 controls the therapeutic activity of 4-1BB-targeting antibodies. J Exp Med (2014) 211:1433–48. doi: 10.1084/jem.20132687
84. Marsters SA, Frutkin AD, Simpson NJ, Fendly BM, Ashkenazi A. Identification of cysteine-rich domains of the type 1 tumor necrosis factor receptor involved in ligand binding. J Biol Chem (1992) 267:5747–50.
85. Lang I, Füllsack S, Wyzgol A, Fick A, Trebing J, Arana JA, et al. Binding studies of TNF receptor superfamily (TNFRSF) receptors on intact cells. J Biol Chem (2016) 291:5022–37. doi: 10.1074/jbc.M115.683946
86. Vanamee ÉS, Faustman DL. Structural principles of tumor necrosis factor superfamily signaling. Sci Signal (2018) 11:eaao4910. doi: 10.1126/scisignal.aao4910
87. Wada J, Kanwar YS. Identification and characterization of galectin-9, a novel betagalactoside-binding mammalian lectin. J Biol Chem (1997) 272:6078–86. doi: 10.1074/jbc.272.9.6078
88. Wada J, Ota K, Kumar A, Wallner EI Kanwar YS. Developmental regulation, expression, and apoptotic potential of galectin-9, a beta-galactoside binding lectin. J Clin Invest (1997) 99:2452–61. doi: 10.1172/JCI119429
89. Liu FT, Rabinovic GA. Galectins as modulators of tumour progression. Nat Rev (2005) 5:29–41. doi: 10.1038/nrc1527
90. Rabinovich GA, Liu FT, Hirashima M, Anderson A. An emerging role for galectins in tuning the immune response: lessons from experimental models of inflammatory disease, autoimmunity and cancer. Scand J Immunol (2007) 66:143–58. doi: 10.1111/j.1365-3083.2007.01986.x
91. Bi S, Hong PW, Lee B, Baum LG. Galectin-9 binding to cell surface protein disulfide isomerase regulates the redox environment to enhance T-cell migration and HIV entry. Proc Natl Acad Sci USA (2011) 108:10650–5. doi: 10.1073/pnas.1017954108
92. Tanikawa R, Tanikawa T, Hirashima M, Yamauchi A, Tanaka Y. Galectin-9 induces osteoblast differentiation through the CD44/ Smad signaling pathway. Biochem Biophys Res Commun (2010) 394:317–22. doi: 10.1016/j.bbrc.2010.02.175
93. Lhuillier C, Barjon C, Niki T, Gelin A, Praz F, Morales O, et al. Impact of exogenous galectin-9 on human T cells: CONTRIBUTION OF THE T CELL RECEPTOR COMPLEX TO ANTIGEN-INDEPENDENT ACTIVATION BUT NOT TO APOPTOSIS INDUCTION. J Biol Chem (2015) 290:16797–811. doi: 10.1074/jbc.M115.661272
94. Arch RH, Thompson CB. 4-1BB and OX40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol Cell Biol (1998) 18:558–65. doi: 10.1128/MCB.18.1.558
95. Jang IK, Lee ZH, Kim YJ, Kim SH, Kwon BS. Human 4-1BB (CD137) signals are mediated by TRAF2 and activate nuclear factor-kappa B. Biochem Biophys Res Commun (1998) 242:613–20. doi: 10.1006/bbrc.1997.8016
96. Saoulli K, Lee SY, Cannons JL, Yeh WC, Santana A, Goldstein MD, et al. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4-1BB ligand. J Exp Med (1998) 187:1849–62. doi: 10.1084/jem.187.11.1849
97. Chung JY, Park YC, Ye H, Wu H. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci (2002) 115:679–88.
98. Zapata JM. TNF-receptor-associated factors as targets for drug development. Expert Opin Ther Targets (2003) 7:411–25. doi: 10.1517/14728222.7.3.411
99. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal (2013) 8:7. doi: 10.1186/1750-2187-8-7
100. Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta (2014) 843:47–60. doi: 10.1016/j.bbamcr.2013.05.026
101. Yin Q, Lamothe B, Darnay BG, Wu H. Structural basis for the lack of E2 interaction in the RING domain of TRAF2. Biochemistry (2009) 48:10558–67. doi: 10.1021/bi901462e
102. Zheng C, Kabaleeswaran V, Wang Y, Cheng G, Wu H. Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Mol Cell (2010) 38:101–13. doi: 10.1016/j.molcel.2010.03.009
103. Mace PD, Smits C, Vaux DL, Silke J, Day CL. Asymmetric recruitment of cIAPs by TRAF2. J Mol Biol (2010) 400:8–15. doi: 10.1016/j.jmb.2010.04.055
104. Zapata JM, Perez-Chacon G, Carr-Baena P, Martinez-Forero I, Azpilikueta A, Otano I, et al. CD137 (4-1BB) signalosome: complexity is a matter of TRAFs. Front Immunol (2018) 9:2618. doi: 10.3389/fimmu.2018.02618
105. Lee HW, Nam KO, Park SJ, Kwon BS. 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur J Immunol (2003) 33:2133–41. doi: 10.1002/eji.200323996
106. Nam KO, Kang H, Shin SM, Cho KH, Kwon B, Kwon BS, et al. Cross-linking of 4-1BB activates TCR-signaling pathways in CD8+ T lymphocytes. J Immunol (2005) 174:1898–905. doi: 10.4049/jimmunol.174.4.1898
107. Kang YJ, Kim SO, Shimada S, Otsuka M, Seit-Nebi A, Kwon BS, et al. Cell surface 4-1BBL mediates sequential signaling pathways “downstream” of TLR and is required for sustained TNF production in macrophages. Nat Immunol (2007) 8:601–9. doi: 10.1038/ni1471
108. Moh MC, Lorenzini PA, Gullo C, Schwarz H. Tumor necrosis factor receptor 1 associates with CD137 ligand and mediates its reverse signaling. FASEB J (2013) 27:2957–66. doi: 10.1096/fj.12-225250
109. Bae JS, Choi JK, Moon JH, Kim EC, Croft M, Lee HW. Novel transmembrane protein 126A (TMEM126A) couples with CD137L reverse signals in myeloid cells. Cell Signal (2012) 24:2227–36. doi: 10.1016/j.cellsig.2012.07.021
110. Kim EC, Moon JH, Kang SW, Kwon B, Lee HW. TMEM126A, a CD137 ligand binding protein, couples with the TLR4 signal transduction pathway in macrophages. Mol Immunol (2015) 64:244–51. doi: 10.1016/j.molimm.2014.12.001
111. Ma J, Bang BR, Lu J, Eun SY, Otsuka M, Croft M, et al. The TNF family member 4-1BBL sustains inflammation by interacting with TLR signaling components during late-phase activation. Sci Signal (2013) 6:ra87. doi: 10.1126/scisignal.2004431
112. Bang BR, Kim SJ, Yagita H, Croft M, Kang YJ. Inhibition of 4-1BBL-regulated TLR response in macrophages ameliorates endotoxin-induced sepsis in mice. Eur J Immunol (2015) 45:886–92. doi: 10.1002/eji.201445174
113. Hall DA, Giraldo J. A method for the quantification of biased signalling at constitutively active receptors. Br J Pharmacol (2018) 175:2046–62. doi: 10.1111/bph.14190
114. Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov (2018) 17:243–60. doi: 10.1038/nrd.2017.229
115. Wisler JW, Rockman HA, Lefkowitz RJ. Biased G protein-coupled receptor signaling: changing the paradigm of drug discovery. Circulation (2018) 137:2315–17. doi: 10.1161/CIRCULATIONAHA.117.028194
116. Zhou B. Mathematical modeling of oligomerization and biased signaling of G-protein-coupled receptors. [dissertation/doctoral thesis]. Bellaterra (Spain): Universitat Autonoma de Barcelona (2018).
117. Kenakin T. Biased Receptor Signaling in Drug Discovery. Pharmacol Rev (2019) 71:267–315. doi: 10.1124/pr.118.016790
118. Suomivuori CM, Latorraca NR, Wingler LM, Eismann S, King MC, Kleinhenz ALW, et al. Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science (2020) 367:881–7. doi: 10.1126/science.aaz0326
119. Sun Y, Huang J, Xiang Y, Bastepe M, Jüppner H, Kobilka BK, et al. Dosage-dependent switch from G protein-coupled to G protein-independent signaling by a GPCR. EMBO J (2007) 26:53–64. doi: 10.1038/sj.emboj.7601502
120. Bertram EM, Lau P, Watts TH. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J Immunol (2002) 168:3777–85. doi: 10.4049/jimmunol.168.8.3777
121. Li Q, Carr A, Ito F, Teitz-Tennenbaum S, Chang AE. Polarization effects of 4-1BB during CD28 costimulation in generating tumor-reactive T cells for cancer immunotherapy. Cancer Res (2003) 63:2546–52.
122. Choi BK, Lee DY, Lee DG, Kim YH, Kim SH, Oh HS, et al. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8+ T cell proliferation. Cell Mol Immunol (2017) 14:748–57. doi: 10.1038/cmi.2016.02
123. Somekh I, Thian M, Medgyesi D, Gülez N, Magg T, Gallón Duque A, et al. CD137 deficiency causes immune dysregulation with predisposition to lymphomagenesis. Blood (2019) 134:1510–6. doi: 10.1182/blood.2019000644
124. Palazón A, Martínez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, et al. The HIF-1α hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov (2012) 2:608–23. doi: 10.1158/2159-8290.CD-11-0314
125. Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res (2017) 23:1929–36. doi: 10.1158/1078-0432.CCR-16-1272
126. Segal NH, He AR, Doi T, Levy R, Bhatia S, Pishvaian MJ, et al. Phase I study of single-agent utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in patients with advanced cancer. Clin Cancer Res (2018) 24:1816–23. doi: 10.1158/1078-0432.CCR-17-1922
127. Smith SE, Hoelzinger DB, Dominguez AL, Van Snick J, Lustgarten J. Signals through 4-1BB inhibit T regulatory cells by blocking IL-9 production enhancing antitumor responses. Cancer Immunol Immunother (2011) 60:1775–87. doi: 10.1007/s00262-011-1075-6
128. Choi BK, Bae JS, Choi EM, Kang WJ, Sakaguchi S, Vinay DS, et al. 4-1BB-dependent inhibition of immunosuppression by activated CD4+CD25+ T cells. J Leukoc Biol (2004) 75:785–91. doi: 10.1189/jlb.1003491
129. Zhang P, Gao F, Wang Q. Agonistic anti-4-1BB antibody promotes the expansion of natural regulatory T cells while maintaining Foxp3 expression. Scand J Immunol (2007) 66:435–40. doi: 10.1111/j.1365-3083.2007.01994.x
130. Freeman ZT, Nirschl TR, Hovelson DH, Johnston RJ, Engelhardt JJ, Selby MJ, et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J Clin Invest (2020) 130:1405–16. doi: 10.1172/JCI128672
Keywords: CD137, CD137 ligand, signalosome, bi-directional signal transduction pathways, CD137/CD137L-mediated immune modulation
Citation: Choi BK and Lee H-W (2020) The Murine CD137/CD137 Ligand Signalosome: A Signal Platform Generating Signal Complexity. Front. Immunol. 11:553715. doi: 10.3389/fimmu.2020.553715
Received: 20 April 2020; Accepted: 06 November 2020;
Published: 10 December 2020.
Edited by:
Janos G. Filep, Université de Montréal, CanadaReviewed by:
Alvaro Teijeira, University of Navarra, SpainCopyright © 2020 Choi and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyeon-Woo Lee, aHllb253b29Aa2h1LmFjLmty
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.