 Sanchez Preethi Eugene
Sanchez Preethi Eugene Vadde Sudhakar Reddy
Vadde Sudhakar Reddy Jamma Trinath
Jamma Trinath- 1Department of Biological Sciences, Birla Institute of Technology and Science-Pilani, Hyderabad Campus, Hyderabad, India
- 2Biochemistry Division, National Institute of Nutrition, Hyderabad, India
The intestinal tract encompasses the largest mucosal surface fortified with a fine layer of intestinal epithelial cells along with highly sophisticated network of the lamina propria immune cells that are indispensable to sustain gut homeostasis. However, it can be challenging to uphold homeostasis when these cells in the intestine are perpetually exposed to insults of both endogenous and exogenous origin. The complex networking and dynamic microenvironment in the intestine demand highly functional cells ultimately burdening the endoplasmic reticulum (ER) leading to ER stress. Unresolved ER stress is one of the primary contributors to the pathogenesis of inflammatory bowel diseases (IBD). Studies also suggest that ER stress can be the primary cause of inflammation and/or the consequence of inflammation. Therefore, understanding the patterns of expression of ER stress regulators and deciphering the intricate interplay between ER stress and inflammatory pathways in intestinal epithelial cells in association with lamina propria immune cells contribute toward the development of novel therapies to tackle IBD. This review provides imperative insights into the molecular markers involved in the pathogenesis of IBD by potentiating ER stress and inflammation and briefly describes the potential pharmacological intervention strategies to mitigate ER stress and IBD. In addition, genetic mutations in the biomarkers contributing to abnormalities in the ER stress signaling pathways further emphasizes the relevance of biomarkers in potential treatment for IBD.
Introduction
The intestine houses a plethora of innocuous microbes that establish a symbiotic relationship in the host. Additionally, constant exposure to the external factors makes it susceptible to invasion by exogenous pathogens ensuing persistent immune response in the gut. Therefore, the lamina propria immune cells must be functionally fine-tuned to differentiate and exhibit tolerance toward commensals and immunity to pathogens. At this juncture, the intestinal epithelial cells (IECs) lining the gut play two major roles: segregation and mediation, conserve gut homeostasis (1); avoid unwarranted immune responses to gut microbes utilizing highly specialized cell types (Paneth cells, goblet cells, enteroendocrine cells, and absorptive epithelial cells) (2–7). Perturbations in the functions of these IECs cause microbial dysbiosis, infiltration and hyperactivation of immune cells in the lamina propria contributing to IBD. IBD is multifactorial whose pathophysiology is unclear and disrupts several aspects such as the physiology, microbiology, immunology, and genetics of the host mimicking a chaotic battlefield. Simply put, the impairment of one aspect causes the annihilation of the other. One such widely reported contributing factor for IBD is ER stress as described below.
ER Stress and UPR
The ER is the primary site for facilitating the appropriate folding of proteins and dispatches them to their respective functional destinations in the IEC with secretory function (2). However, the proteins that shuttle through the ER may aggregate, triggering a highly conserved unfolded protein response (UPR) and establish ER homeostasis in three possible ways (8). A. Transcriptional induction:- increases the protein folding capacity by transcribing chaperones that aid in proper folding; B. Translational attenuation:- reduces protein load in the ER by arresting translational machinery, degrading mRNAs; C. ER-associated degradation:- the unfolded proteins are marked for proteasomal degradation. However, if ER stress persists, the effort to establish homeostasis can be futile triggering apoptosis in IEC (9).
UPR Signal Transducers
UPR pathways function with unique mechanisms of signal transduction operating in parallel utilizing IRE1α, PERK, and ATF6 (9). In their inactive state, these stress sensors are bound to BiP toward the intraluminal domain. Under ER stress, the BiP dissociates, activating IRE1α, PERK, and ATF6 signaling cascades to salvage the distressed cell.
IRE1α is the most evolutionarily conserved transmembrane kinase with endoribonuclease activity (10). The active IRE1α cleaves the 26-nucleotide intron from XBP1 forming functional XBP1s (11), which then enters the nucleus, and regulate UPR-related genes. Interestingly, the XBP1u is degraded rapidly after translation; however, during prolonged stress, XBP1u is reported to accumulate and complex with XBP1s, to promote ubiquitin-mediated degradation of XBP1s in HeLa cells (12). Therefore, a balanced level of XBP1u and XBP1s partly dictates the functional role of IRE1α. Another important regulatory mechanism executed by IRE1α is through IRE1-dependent mRNA decay (RIDD). IRE1α cleaves the transcripts that enter ER through the translocon and prevents accumulation of unfolded proteins in the ER (13). Nevertheless, RIDD can also be deleterious if mRNAs that translate for pro-survival proteins are degraded suggesting that a selective degradation of mRNA is favored. Of note, in the recent past, the ability of RIDD pathway to degrade microRNAs responsible for inhibiting the translation of CASP2 in mouse embryonic fibroblasts (MEF) have also been identified signifying that fine-tuning the availability of non-coding RNAs also contribute to the overall outcome of UPR (14) (Figure 1A).
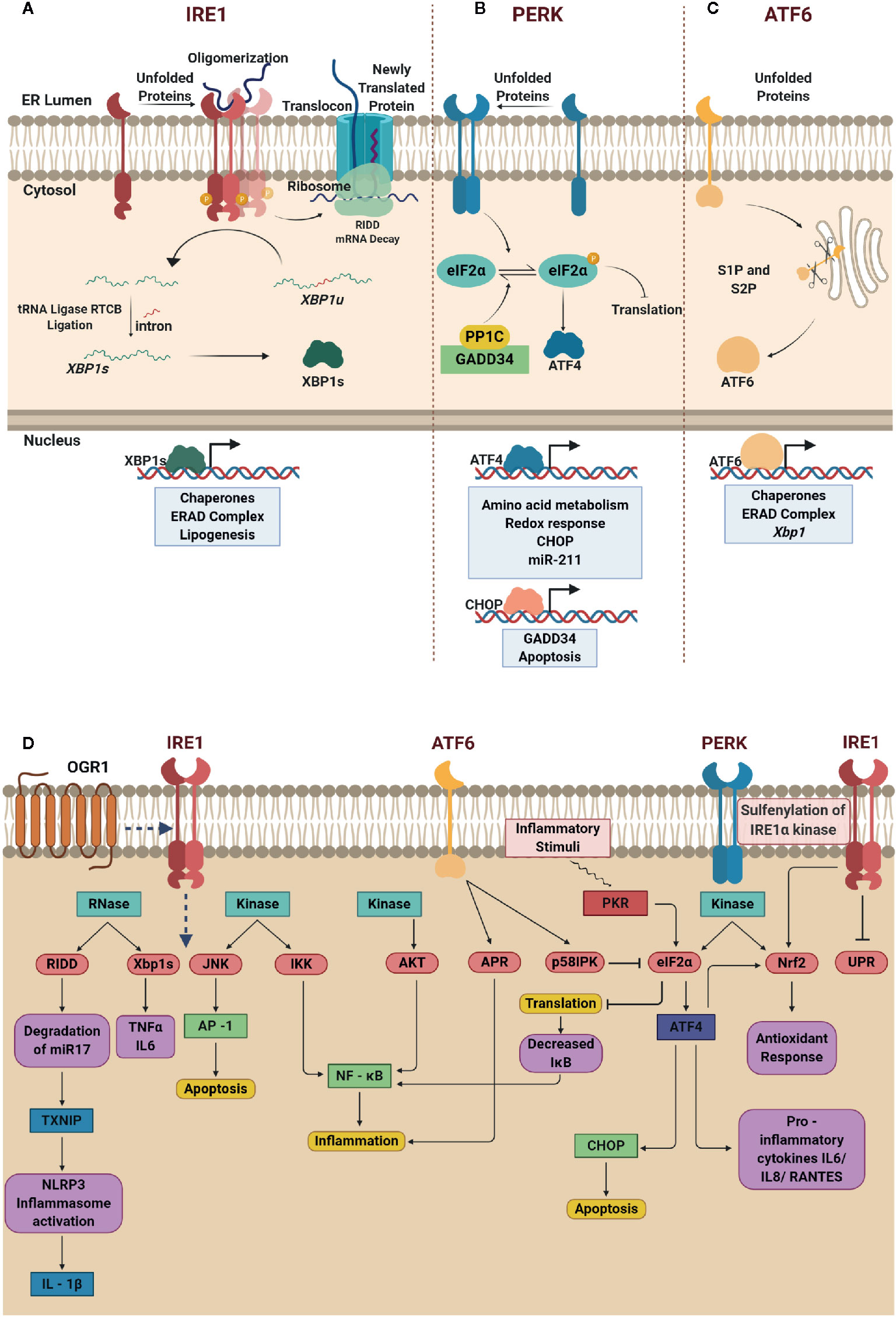
Figure 1 Schematic depiction of unfolded protein response (UPR) signaling cascade, and the interplay between endoplasmic reticulum (ER) stress response and inflammation. (A) Oligomerization of IRE1α in the presence of unfolded proteins promotes the endoribonuclease activity of IRE1α and unconventional splicing of XBP1 generating functional XBP1 that regulates gene expression. IRE1α promotes RIDD-dependent mRNA decay and reduces protein overload in the ER lumen. (B) Activated PERK drives phosphorylation of eIF2α resulting in translational block. At this juncture, selective IRES dependent translation of ATF4 is promoted to induce chaperones and mitigate oxidative stress as well as apoptosis. (C) Dissociation of BiP from ATF6 leads to translocation of ATF6 from the ER membrane to Golgi promoting its cleavage by S1P and S2P generating functional ATF6 that regulates UPR genes. (D) In IRE1α pathway, the RNase domain is involved in Xbp1 splicing and RIDD mechanism upregulates the expression of pro-inflammatory cytokines TNFα, IL-6 and IL-1β; the kinase domain activates JNK and IKK signaling pathway that results in apoptosis and inflammation respectively. Additionally, activation of proton-sensing OGR1 is responsible for ER-stress mediated response via IRE1α-JNK-XBP1s axis. The kinase activity of ATF6 leads to phosphorylation of AKT ensuing inflammation via NF-κB signaling. The cleaved p50ATF6α acts as a transcription factor and upregulates the expression of APR genes and ER co-chaperone p58IPK that in turn blocks the phosphorylation of eIF2α. PERK is one of the kinases that phosphorylate eIF2α at Ser 51, which enables selective translation of ATF4. ATF4 drives the expression of CHOP and pro-inflammatory cytokines such as IL-6, IL-8, and RANTES. Induced expression of CHOP abrogates pro-survival signaling leading to cell death. Notably, the translational block decreases further translation of IκB ensuing inflammation due to increase in NF-κB. Nrf2, another notable target phosphorylated by PERK that is known to manifest antioxidant response. Created with BioRender.com.
PERK is a type 1 transmembrane serine/threonine kinase, when bound to BiP, remains inactive (10). Dissociation of BiP enables dimerization of PERK and promotes its kinase activity and phosphorylates eIF2α causing a translational block to manage ER stress. Another pivotal step that occurs at this stage is the selective internal ribosomal entry site mediated translation of ATF4 amidst the inhibitory phosphorylation of eIF2α (15). Nuclear translocation of ATF4 promotes GADD34, CHOP and miR-211 expression (16, 17) to mediate UPR in mouse embryonic fibroblasts. Upon resolution of ER stress, GADD34 complexes with PP1C and dephosphorylates eIF2α to restore protein translation. Interestingly, PERK-induced miR-211 abrogates the expression of CHOP/GADD34 suggesting the pro-survival role of miR211 (17). However, if the ER stress remains unresolved, CHOP activates the terminal UPR to induce apoptosis in IEC (18, 19) (Figure 1B).
ATF6 is also a transmembrane kinase, with a basic leucine zipper (bZIP) domain, unlike IRE1α and PERK. Once the BiP dislodges from ATF6, it is trafficked from the ER to Golgi. S1P and S2P cleaves ATF6 releasing the N-terminal cytosolic domain of ATF6 (N) that translocate to the nucleus and promotes the transcription of chaperones, ERAD complex and XBP1 to mitigate ER stress (Figure 1C).
In addition to the three primary signal transducers, a few ER stress transducers belonging to the OASIS family are identified recently (20). These stress sensors share a region of high sequence similarity with ATF6. One such example is CREBH, which is also trafficked from ER to Golgi and proteolyzed by S1P and S2P (21). Studies indicate that ATF6 and CREBH regulate inflammatory gene expression during the early phase of infection or injury.
UPR and Inflammation
Recent studies have extended a clear understanding of the relationship between UPR signaling and inflammation (Figure 1D). Activated IRE1α, in addition to its role as bifunctional enzyme, interacts with TRAF2 to activate JNK and NF-κB (22, 23) regulating inflammatory gene expression. Studies also indicate that XBP1s induces TNFα and IL-6 (24, 25) which in turn activate NF-κB (23, 26); thus amplifying the inflammatory responses. Interestingly, degradation of miR-17, a microRNA that represses the expression of thioredoxin-interacting protein (TXNIP) by IRE1α results in stabilization of TXNIP and expression of IL-1β (27, 28). At the molecular level, identification of IRE1α-TXNIP axis to activate NLRP3 inflammasome, IL-1β expression and programmed cell death (27) through miR-17 degradation hint that ER stress regulates inflammation. Furthermore, activation of IRE1α-GSK3β axis induces the expression of IL-1β and regulates the expression of TNFα (29). It is also a known fact that GSK-3β requires priming kinase to phosphorylate the substrate first for recognition and phosphorylates the phosphorylated substrate at a different site (30). Therefore, it is conjectured that the phosphorylated kinase domain of IRE1α acts as the priming kinase for GSK-3β to phosphorylate the riboendonuclease domain of IRE1α rendering it inactive leaving the hypothesis to be tested as a future prospect. Inhibition of global translation upon activation of PERK lowers the levels of IκBα ensuing massive activation of NF-κB (31). On the other hand, the selectively translated ATF4 binds to IL-6 promoter and regulates its expression (32). Of note, TLR4 signaling is also responsible for the induction of ATF4, independent of ER stressors, resulting in the transactivation of Il6, Il18, and Rantes in macrophages and monocytes (32, 33). Additionally, PERK also directly phosphorylates NRF2, regulating the antioxidant response by nullifying the ROS production during ER stress in fibroblasts (34). Recent investigations, however, direct toward an alternative NRF2 regulation in response to oxidative stress via PERK-eIF2α-ATF4 axis in human cells (NCI-H358) (35). Reversible sulfenylation of cysteine residue (C663) of IRE1α by ROS results in the attenuation of UPR and promotes activation of NRF2-mediated antioxidant response in human cells (36). Overall activation of PERK inhibits eukaryotic translation, prevents the accumulation of unfolded proteins that promote inflammatory gene expression, and regulates apoptosis through ATF4. Interestingly, similar to IRE1α and PERK, ATF6 also contributes to NF-κB signaling through transient phosphorylation of AKT, however, prolonged ER stress resulted in downregulation of AKT phosphorylation (37). The phospho-refractory nature of AKT after ATF6-mediated ER stress is further confirmed by subsequent TLR4 stimulation of ischemic Kupffer cells (38). However, the ability of ATF6 to regulate inflammatory gene expression remains unexplored to a large extent.
ER Stress and UPR in Intestinal Inflammation
Continual exposure to a myriad of gut microflora, exogenous antigens, dietary metabolites, and toxins impede the functional ability of IECs. Although nature has bestowed with evolutionarily conserved and sophisticated mechanisms to overcome these impediments, disruption in any of these mechanisms can cause chronic inflammation in the gut. Accordingly, there are two ways to look at the cause for the collapse of these mechanisms: i) IECs are pushed to synthesize copious amounts of proteins, cytokines, and peptides; activating UPR. In this scenario, cells that are competent enough will survive and the rest will succumb to stress. ii) Genetic deficiency of the genes that are involved in UPR, autophagy, secretion, immune response and inflammation can have various impacts and confer a genetic predisposition to IBD owing to decreased protein folding capacity and heightened immune response. Mechanistic studies conducted on murine models deficient in these genes facilitated the understanding of phenotypic outcomes in correlation to IBD (Table 1).
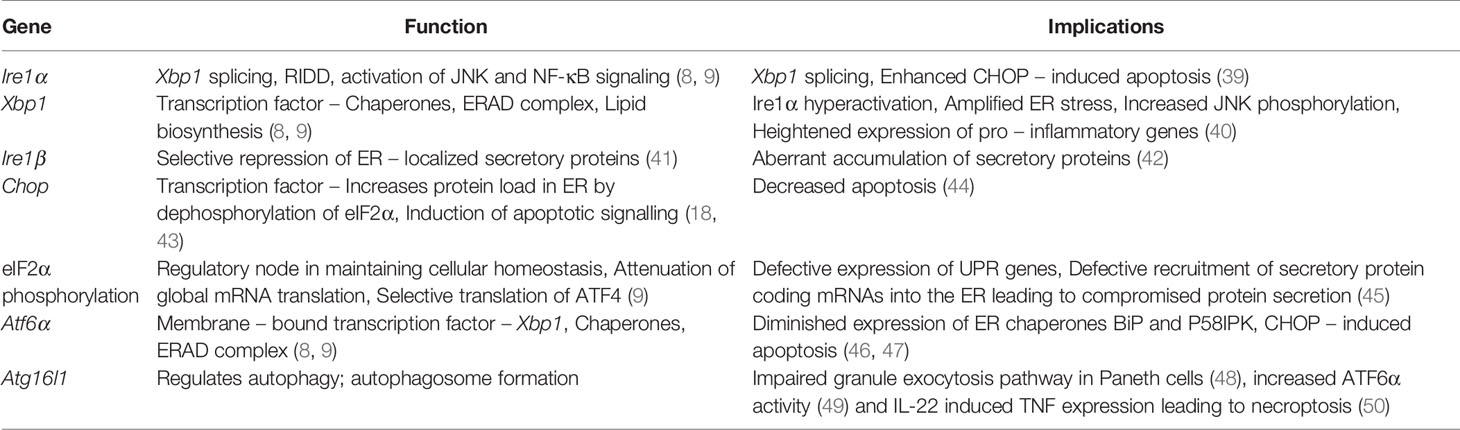
Table 1 List of endoplasmic reticulum (ER) stress-related genes in inflammatory bowel diseases (IBD).
Of the three pathways, IRE1α-XBP1 axis of UPR has been extensively studied and known to play an essential role in regulating immunity and inflammation. Additional evidence from the studies suggests that Xbp1 is linked to IBD. Cell-specific loss of Xbp1 in intestinal epithelial cells (Xbp1ΔIEC) displayed amplified ER stress (40). Additionally, deep-sequencing studies have revealed rare variants/SNPs of the Xbp1 gene that contributes to the susceptibility and severity of the inflammatory disorders in humans (40). Furthermore, Xbp1 deletion resulted in hyperactivation of IRE1α and enhanced the susceptibility to experimentally induced inflammation in mice suggesting a pivotal role of IRE1α in intestinal inflammation. Studies demonstrated that IRE1α recruits TNFR1 during ER stress to activate TNF-independent JNK signaling and apoptosis (51). This was further supported by the fact that the deletion of Tnfr1 in Xbp1ΔIEC mice failed to develop intestinal inflammation and the deletion of Xbp1 in IECs resulted in the apoptotic loss of Paneth cells that maintain homeostasis (40). Of note, ER stress can also activate TNF-independent TNFR1-mediated necroptosis, a programmed RIPK1/RIPK3/MLKL-dependent necrosis, in L929 cells. Inhibition of JNK, however, resulted in the inhibition of both TNFR1-mediated apoptosis and necroptosis (52). On the other hand, the induction of CHOP in Xbp1ΔIEC upregulated the expression of induced NKG2D ligand, which activates natural killer cell-mediated cytotoxicity establishing the involvement of CHOP in innate immune responses (53). Moreover, genetic deletion of IRE1α led to impaired XBP1 splicing and JNK-driven phosphorylation of eIF2α through PERK that promotes apoptotic cell death, suggesting a prominent pro-survival role of IRE1α as well. Furthermore, the study indicated compromised intestinal epithelial barrier integrity, lymphocyte infiltration and induced expression of TNFα, IL-1β and IL-6 leading to the development of spontaneous colitis in the mice (39). Altogether, these studies implicate a homeostatic role of IRE1α in mucosal immunity.
At later stages of ER stress, the PERK-ATF4 axis of UPR is predominantly active and induces CHOP. Whole-body deletion of Chop in mice suppressed the induction of Mac-1, Ero-1α, and caspase-11 with reduced intestinal epithelial cell apoptosis (43, 44). Another important component of the PERK pathway is phosphorylated eIF2α and its role has been studied using IEC-specific non-phosphorylatable S51A mutant AAΔIEC mice (45). The translocation machinery to recruit mRNA into ER has been found to be defective in AAΔIEC mice leading to defective antimicrobial peptides, cryptidin, and lysozyme resulting in the breach of epithelial integrity by commensals and hyperactivation of immune cells. Another notable feature is that a family of protein kinases such as PKR, GCN2, and HRI other than PERK phosphorylate eIF2α and regulate ER stress. Although these protein kinases are activated by different stimuli including infections and inflammatory cytokines, they culminate into phosphorylation of eIF2α at Ser51 emphasizing the importance of eIF2α in maintaining IEC homeostasis (54).
The functional role of Atf6α has been experimented using Atf6-/- mice wherein deletion of Atf6α led to reduced expression of ER chaperones BiP and p58IPK and showed signs of apoptosis (46). p58IPK-/- mice showed amplified ER stress and were more susceptible to DSS-induced colitis (55). Deletion of both Atf6α and p58IPK resulted in embryonic lethality, suggesting either ATF6α or p58IPK is required to oversee protein-folding defects (56). It is important to note that p58IPK is an ER co-chaperone that negatively regulates eIF2α, which in turn down regulates ATF4 and CHOP. As indicated previously, owing to the structural and sequential similarity, Oasis-/- mice developed characteristics of IBD as observed in Atf6-/- mice impacting goblet cell maturation (57, 58). Studies in a similar mouse model elucidated the role of S1P-ATF6 axis in IBD and concluded that missense mutation in Mbtps1 impaired ATF6 arm of UPR (59). Deficiency of Atg16l1 and Xbp1 genes shows increased activity of ATF6α. Interestingly, inhibition of the ATF6α co-activators, CSNK2B and ASCL1 reduced the activity of ATF6α and attenuated CXCL1 and TNFα expression (49). Furthermore, Atg16l1ΔIEC is responsible for IL-22-mediated activation of IFN1-TNF axis and ER stress response (50). Increased levels of TNF potentiate IL-22-induced necroptotic epithelial cell death, contradicting the previously reported protective role of IL-22. In addition, targeting IL-22 in TRUC mice model alleviated ER stress response and colitis (60). Hence, the paradoxical nature of IL-22 challenges its prospect as a treatment for IBD.
In addition to the aforementioned genes, there are other IBD risk genes such as AGR2 and Ormdl3. AGR2 belongs to the PDI family; expressed strongly in tissues that secretes mucus and expressed abundantly in the inflamed mucosa of UC patients (61, 62). Agr2-/- knockout mice developed spontaneous granulomatous ileocolitis (63). In addition to its intracellular role, modifications in the KTEL motif of AGR2 implicate its role in protein secretion (64). To emphasize an interesting hypothesis that inflammation can induce ER stress, the Il10-/- mice model was studied and found that IL-10 mitigates intestinal inflammation during ER stress (65). A recent study, however, demonstrated ER stress in LPS-stimulated macrophages, abrogated the immunosuppressive effects of IL-10 (66). Acidic milieu in IBD activates OGR1 receptor found in abundance lining the mucosal region. Further evidence indicates the role of TNF (67) in the expression of OGR1 that in turn mediates ER stress and exacerbates inflammation via IRE1-JNK-XBP1s axis and blocks autophagy (68). Interestingly, deletion of OGR1 in Il10-/- female mice protected from the development of spontaneous colitis (67).
Microbiota, ER Stress and Inflammation
As mentioned earlier, activation of UPR cascade as a consequence of ER stress potentiates inflammation and IBD (Figure 1D). Of note, evidences report impaired UPR signaling cascade in IBD (69, 70). In the recent past, the impact of diet, nutrients and gut microbiota have been implicated in ER stress and IBD (71). Prevalence of Fusobacterium activates UPR and promotes inflammation in UC patients (72). Probiotic bacteria such as Lactobacillus paracasei ameliorated intestinal inflammation through ER stress-UPR pathway (73). On the contrary, Lactobacillus acidophilus mitigated intestinal inflammation by suppressing NF-κB and thereby inhibiting ER stress (74). Adding to this, methyl deficient diet aggravates DSS-induced colitis by promoting ER stress (75). Interestingly, HFD driven ER stress has been found to be harmful as well as beneficial in ERs stress-mediated inflammation-driven osteoarthritis and liver pathology respectively (76, 77). Likewise, three cancer mice models fed with low protein diet reportedly activated IRE1α/RIG pathway in tumor cells limiting tumor growth (78). DSS treatment disrupts ER homeostasis and membrane integrity (79). Dietary administration of Lachnum Polysaccharide (LEP) to DSS-induced colitis mitigated ER stress-mediated inflammation not only by precluding immune cell infiltration, but also improved epithelial barrier integrity by regulating tight junction (TJ) proteins, mucus layer protecting proteins, and antimicrobial peptides (80). Altogether, these results, suggest a pivotal role for dietary components, microbiota and ER stress in inflammation of the intestine. However, a detailed investigation still remains to understand the molecular association among these to cause IBD.
Potential Therapeutics Targeting ER Stress in IBD
As discussed earlier, dysfunctional ER stress and UPR is one of the contributing factors in the etiology of IBD (40, 81). Therefore, drugs targeting to alleviate ER stress appear as a convincing choice to treat IBD. Chemical chaperones such as TUDCA and 4-PBA augment protein folding and suppress ER stress in IECs in vitro (46, 82). Moreover, oral administration of TUDCA and 4-PBA reduced ER stress in Il10-/- mice and DSS-induced colitis in P58IPK-/- and Atf6-/- mice (46, 83). Recent studies conducted on NEC mouse models showed that TUDCA is capable of reducing the ER stress markers and apoptosis by inhibiting PERK-eIF2α via activation of the PI3K/Akt pathway (84). Salubrinal, a specific eIF2α phosphatase inhibitor, reduces tunicamycin-induced ER stress and TNFR1-independent necroptosis in hepatocytes by selectively preventing eIF2α dephosphorylation (85). The secondary bile acid UDCA protects the intestinal barrier by inducing epithelial cell migration at the site of injury (86) and ameliorates LPS-induced intestinal inflammation (87). In addition, studies conducted on DSS-induced colitis mice model demonstrated the ability of UDCA and LCA to mitigate colonic inflammation by inhibition of epithelial apoptosis (88). Amino acids such as L-glutamine and L-arginine have been reported to regulate proliferation and differentiation of IECs suggesting the role for dietary supplements to regulate ER stress (89, 90). Moreover, it has been proved that L-glutamine and glycine supplementation can salvage IECs from ER stress and apoptosis by improving the intestinal epithelial barrier function (91) upregulating tight junction proteins (92). Plant-based active ingredient berberine (BBR) has long been known to alleviate ER stress response as an alternative to chemical compounds. Accordingly, a recent study asserted the ability of berberine to reduce inflammation and apoptosis in DSS-induced colitis mice model (93). Furthermore, evidences suggest that PERK, and IRE1α inhibitors may be extended to IBD pathogenesis. Accordingly, STF-083010, a small molecule inhibitor that specifically targets IRE1α has been proven to reduce ER stress-driven inflammation in atherosclerosis and diabetes (94, 95). Similarly, pharmacological inhibition of PERK by GSK2656157 and GSK2606414 ameliorate tumor growth and enhance neuroprotection (96–98) and PKR inhibitors such as imoxin and 2-aminopurine reduced ER stress in mouse beta TC-6 cell line (99). Activator of ATF6, Compound 147 has shown to reduce the risk of infarction and preserve cardiac function (100). Nevertheless, the efficacy and implications of these small molecules in ER stress-driven intestinal inflammation remains to be largely unexplored.
Conclusion
The incidence rate of IBD, which has been once considered the disease of the developed nations, is alarmingly at a rise globally. Efforts have been placed to rationalize the root cause by postulating various hypothesis including hygiene (101) and cold chain hypothesis (102) culminating to a single root cause ‘microbial dysbiosis. There are numerous factors at play in disrupting the gut microbiome and integrity of the intestinal barrier (40). As a result, the compromised epithelial barrier allows breaching by microbes and exogenous antigens attracting the attention of the host’s immune system that tries to salvage but ends up damaging the host tissue due to inflammation. Chronic inflammation is one of the hallmark features of IBD, identical to a ‘wildfire’ that if uncontrolled causes collateral damage. There is also growing evidence that deregulated ER stress and UPR signaling pathways can instigate or magnify the inflammatory response in IBD (103–106). Therefore, restoring a robust ER stress and UPR mechanism could be a potential therapeutic target. Nevertheless, the lack of well-demarcated molecular pathways is downright challenging to develop targeted therapies to preclude overlapping adverse effects. However, the development of optimized therapeutics is possible if a profound understanding of the phenotype and pathogenesis of the disease can be established by delineating the cellular and molecular pathways.
Author Contributions
SE and JT wrote the manuscript. VR and JT edited the manuscript into its final format. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by OPERA Award (BITS-Pilani, Hyderabad).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all the authors for their contributions toward better understanding of the ER-stress and implications in IBD, and apologize to those whose work has not been cited due to space constraints.
Abbreviations
AGR2, Anterior Gradient 2; AKT, Protein Kinase B; APR, Acute Phase Response; ASCL1, Achaete-scute homolog 1; ATF4, Activating Transcription Factor; ATG16L1, Autophagy related 16 like 1; CASP2, Caspase 2; CHOP, C/EBP (CCAAT/Enhancer Binding Protein) Homologous Protein; CREBH, cAMP Responsive Element Binding Protein H; CRP, C – Reactive Protein; CSNK2B, Casein Kinase 2 Beta; DSS, Dextran Sulfate Sodium; eIF2α, Eukaryotic Initiation Factor 2α; ERAD, Endoplasmic Reticulum Associated Protein Degradation; Ero1α, Endoplasmic Reticulum Oxidoreductase 1α; GADD34, Growth Arrest and DNA Damage–Inducible Protein; GCN, General Control Nondepressible 2; HIK, Sensor Histidine Kinase/Response Regulator; IEC, Intestinal Epithelial Cells; IL–1β, Interleukin 1β; IL–6, Interleukin 6; JNK, c-Jun N- terminal Kinase; LCA, Lithocholic Acid; Mac-1, Macrophage-1 antigen; Mbtps1, Membrane Bound Transcription Factor Peptidase, Site 1; MLKL, Mixed Lineage Kinase domain-like Pseudo kinase; MUC2, Mucin 2; NEC, Necrotizing Enterocolitis; NF-κB, Nuclear Factor Kappa – Light Chain Enhancer of Activated B cells; NKG2D, Natural Killer Group 2D; NRF2, Nuclear Factor Erythroid 2–Related Factor; OASIS, Old Astrocyte Specifically Induced Substrate; OGR1, Ovarian cancer G-protein coupled receptor; ORMDL3, ORM1-like protein 3; 4– PBA, 4–Phenyl Butyrate; PERK, Protein Kinase R–Like ER Kinase; PI3K, Phosphatidylinositol 3–kinase; PKR, Protein Kinase R; PP1C, Protein Phosphatase 1C; RIDD, Regulated Ire1–Dependent mRNA Decay; RIPK1/3, Receptor-interacting serine/threonine protein kinase 1/3; S1P, Site 1 Protease; S2P, Site 2 Protease; SAP, Serum Amyloid Protein; SNP, Single Nucleotide Polymorphism; TNFα, Tumor Necrosis Factor α; TNFR1, Tumor Necrosis Factor Receptor 1; TRAF2, TNF Receptor–Associated Factor 2; TRUC, T-bet-/-/RAG2-/-/Ulcerative Colitis; TUDCA, Tauroursodeoxycholic Acid; UDCA, Ursodeoxycholic Acid; ULBP1, UL16 Binding Protein 1; XBP1s, X–box Binding Protein 1 spliced variant; XBP1u, X–box Binding Protein 1 unspliced variant.
References
1. Okumura R, Takeda K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med (2017) 49:e338. doi: 10.1038/emm.2017.20
2. Ouellette AJ. Paneth cells and innate mucosal immunity. Curr Opin Gastroenterol (2010) 26:547–53. doi: 10.1097/MOG.0b013e32833dccde
3. Birchenough GMH, Johansson MEV, Gustafsson JK, Bergström JH, Hansson GC. New developments in goblet cell mucus secretion and function. Mucosal Immunol (2015) 8:712–9. doi: 10.1038/mi.2015.32
4. Aihara E, Engevik KA, Montrose MH. Trefoil Factor Peptides and Gastrointestinal Function. Annu Rev Physiol (2017) 79:357–80. doi: 10.1146/annurev-physiol-021115-105447
5. Graziani F, Pinton P, Olleik H, Pujol A, Nicoletti C, Sicre M, et al. Deoxynivalenol inhibits the expression of trefoil factors (TFF) by intestinal human and porcine goblet cells. Arch Toxicol (2019) 93:1039–49. doi: 10.1007/s00204-019-02425-6
6. Gribble FM, Reimann F. Enteroendocrine Cells: Chemosensors in the Intestinal Epithelium. Annu Rev Physiol (2016) 78:277–99. doi: 10.1146/annurev-physiol-021115-105439
7. Peterson LW, Artis D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat Rev Immunol (2014) 14:141–53. doi: 10.1038/nri3608
8. So JS. Roles of endoplasmic reticulum stress in immune responses. Mol Cells (2018) 41:705–16. doi: 10.14348/molcells.2018.0241
9. Walter P, Ron D. The unfolded protein response: From stress pathway to homeostatic regulation. Sci (80) (2011) 334:1081–6. doi: 10.1126/science.1209038
10. Gardner BM, Pincus D, Gotthardtt K, Gallagher CM, Walter P. Endoplasmic Reticulum Stress Sensing in the Unfolded Protein Response. Cold Spring Harb Perspect Biol (2013) 5:a013169. doi: 10.1101/cshperspect.a013169
11. Tanaka N, Meineke B, Shuman S. RtcB, a novel RNA ligase, can catalyze tRNA splicing and HAC1 mRNA splicing in vivo. J Biol Chem (2011) 286:30253–7. doi: 10.1074/jbc.C111.274597
12. Yoshida H, Oku M, Suzuki M, Mori K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol (2006) 172:565–75. doi: 10.1083/jcb.200508145
13. Maurel M, Chevet E, Tavernier J, Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci (2014) 39:245–54. doi: 10.1016/j.tibs.2014.02.008
14. Upton JP. IRE1a Cleaves Select microRNAs During ER Stress to Derepress Translation of Proapoptotic Caspase-2. Sci (80) (2012) 388:818–22. doi: 10.1126/science.1226191
15. Chan CP, Kok KH, Tang HMV, Wong CM, Jin DY. Internal ribosome entry site-mediated translational regulation of ATF4 splice variant in mammalian unfolded protein response. Biochim Biophys Acta Mol Cell Res (2013) 1833:2165–75. doi: 10.1016/j.bbamcr.2013.05.002
16. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell (2000) 6:1099–108. doi: 10.1016/S1097-2765(00)00108-8
17. Chitnis NS, Pytel D, Bobrovnikova-Marjon E, Pant D, Zheng H, Maas NL, et al. MiR-211 Is a Prosurvival MicroRNA that Regulates chop Expression in a PERK-Dependent Manner. Mol Cell (2012) 48:353–64. doi: 10.1016/j.molcel.2012.08.025
18. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev (2004) 18:3066–77. doi: 10.1101/gad.1250704
19. Palam LR, Baird TD, Wek RC. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J Biol Chem (2011) 286:10939–49. doi: 10.1074/jbc.M110.216093
20. Kondo S, Saito A, Asada R, Kanemoto S, Imaizumi K. Physiological unfolded protein response regulated by OASIS family members, transmembrane bZIP transcription factors. IUBMB Life (2011) 63:233–9. doi: 10.1002/iub.433
21. Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell (2006) 124:587–99. doi: 10.1016/j.cell.2005.11.040
22. Urano F, Wang XZ, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Sci (80) (2000) 287:664–6. doi: 10.1126/science.287.5453.664
23. Wang L, Walia B, Evans J, Gewirtz AT, Merlin D, Sitaraman SV. IL-6 Induces NF-κB Activation in the Intestinal Epithelia. J Immunol (2003) 171:3194–201. doi: 10.4049/jimmunol.171.6.3194
24. Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol (2010) 11:411–8. doi: 10.1038/ni.1857
25. Kim S, Joe Y, Surh YJ, Chung HT. Differential regulation of toll-like receptor-mediated cytokine production by unfolded protein response. Oxid Med Cell Longev (2018) 2018:1–8. doi: 10.1155/2018/9827312
26. Kim JY, Morgan M, Kim DG, Lee JY, Bai L, Lin Y, et al. TNFα-induced noncanonical NF-κB activation is attenuated by RIP1 through stabilization of TRAF2. J Cell Sci (2011) 124:647–56. doi: 10.1242/jcs.075770
27. Lerner AG, Upton JP, Praveen PVK, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab (2012) 16:250–64. doi: 10.1016/j.cmet.2012.07.007
28. Oslowski CM, Hara T, Sullivan-murphy BO, Kanekura K, Lu S, Hara M, et al. Thioredoxin-Interacting Protein Mediates ER Stress-Induced b Cell Death through Initiation of the Inflammasome. Cell Metab (2012) 16:265–73. doi: 10.1016/j.cmet.2012.07.005
29. Kim HJ, Ryter SW, Pae H-O, Kim Y-S, Joe Y, Jeong SO, et al. Endoplasmic Reticulum Stress–Induced IRE1α Activation Mediates Cross-Talk of GSK-3β and XBP-1 To Regulate Inflammatory Cytokine Production. J Immunol (2015) 194:4498–506. doi: 10.4049/jimmunol.1401399
30. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol Ther (2015) 148:114–31. doi: 10.1016/j.pharmthera.2014.11.016
31. Tam AB, Mercado EL, Hoffmann A, Niwa M. ER Stress Activates NF- kB by Integrating Functions of Basal IKK Activity, IRE1 and PERK. PloS One (2012) 7:e45078. doi: 10.1371/journal.pone.0045078
32. Iwasaki Y, Suganami T, Hachiya R, Shirakawa I, Kim-Saijo M, Tanaka M, et al. Activating transcription factor 4 links metabolic stress to interleukin-6 expression in macrophages. Diabetes (2014) 63:152–61. doi: 10.2337/db13-0757
33. Zhang C, Bai N, Chang A, Zhang Z, Yin J, Shen W, et al. ATF4 is directly recruited by TLR4 signaling and positively regulates TLR4-trigged cytokine production in human monocytes. Cell Mol Immunol (2013) 10:84–94. doi: 10.1038/cmi.2012.57
34. Cullinan SB, Diehl JA. PERK-dependent Activation of Nrf2 Contributes to Redox Homeostasis and Cell Survival following Endoplasmic Reticulum Stress. J Biol Chem (2004) 279:20108–17. doi: 10.1074/jbc.M314219200
35. Sarcinelli C, Dragic H, Piecyk M, Barbet V, Duret C, Barthelaix A, et al. ATF4-dependent NRF2 transcriptional regulation promotes antioxidant protection during endoplasmic reticulum stress. Cancers (Basel) (2020) 12:569. doi: 10.3390/cancers12030569
36. Hourihan JM, Moronetti Mazzeo LE, Fernández-Cárdenas LP, Blackwell TK. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol Cell (2016) 63:553–66. doi: 10.1016/j.molcel.2016.07.019
37. Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, et al. Activation of the Akt-NF-κB Pathway by Subtilase Cytotoxin through the ATF6 Branch of the Unfolded Protein Response. J Immunol (2009) 183:1480–7. doi: 10.4049/jimmunol.0900017
38. Rao J, Yue S, Zhu J, Fu Y, Wang X, Busuttil R, et al. Activating Transcription Factor 6 Mediates a Pro-Inflammatory Synergy Between ER Stress and TLR Activation in the Pathogenesis of Liver Ischemia Reperfusion Injury. Am J Transplant (2014) 14:1552–61. doi: 10.1097/00007890-201407151-00135
39. Zhang HS, Chen Y, Fan L, Xi QL, Wu GH, Li XX, et al. The endoplasmic reticulum stress sensor IRE1 α in intestinal epithelial cells is essential for protecting against colitis. J Biol Chem (2015) 290:15237–336. doi: 10.1074/jbc.M114.633560
40. Kaser A, Lee A, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 Links ER Stress to Intestinal Inflammation and Confers Genetic Risk for Human Inflammatory Bowel Disease. Cell (2008) 134:743–56. doi: 10.1016/j.cell.2008.07.021
41. Nakamura D, Tsuru A, Ikegami K, Imagawa Y, Fujimoto N, Kohno K. Mammalian ER stress sensor IRE1 b specifically down-regulates the synthesis of secretory pathway proteins. FEBS Lett (2011) 585:133–8. doi: 10.1016/j.febslet.2010.12.002
42. Tsuru A, Fujimoto N, Takahashi S, Saito M, Nakamura D, Iwano M, et al. Negative feedback by IRE1β optimizes mucin production in goblet cells. Proc Natl Acad Sci U.S.A. (2013) 110:2864–9. doi: 10.1073/pnas.1212484110
43. Li Y, Guo Y, Tang J, Jiang J, Chen Z. New insights into the roles of CHOP-induced apoptosis in ER stress Structure and Properties of C/EBP Homologous Protein Roles of CHOP in ER Stress-Mediated Apoptosis. Acta Biochim Biophys Sin (2014) 46:629–40. doi: 10.1093/abbs/gmu048.Review
44. Namba T, Tanaka KI, Ito Y, Ishihara T, Hoshino T, Gotoh T, et al. Positive role of CCAAT/enhancer-binding protein homologous protein, a transcription factor involved in the endoplasmic reticulum stress response in the development of colitis. Am J Pathol (2009) 174:1786–98. doi: 10.2353/ajpath.2009.080864
45. Cao SS, Wang M, Harrington JC, Chuang BM, Eckmann L, Kaufman RJ. Phosphorylation of eIF2α is dispensable for differentiation but required at a posttranscriptional level for paneth cell function and intestinal homeostasis in mice. Inflammation Bowel Dis (2014) 20:712–22. doi: 10.1097/MIB.0000000000000010
46. Cao SS, Zimmermann EM, Chuang BM, Song B, Nwokoye A, Wilkinson JE, et al. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology (2013) 144:989–1000. doi: 10.1053/j.gastro.2013.01.023
47. Coleman OI, Lobner EM, Bierwirth S, Sorbie A, Waldschmitt N, Rath E, et al. Activated ATF6 Induces Intestinal Dysbiosis and Innate Immune Response to Promote Colorectal Tumorigenesis. Gastroenterology (2018) 155:1539–52. doi: 10.1053/j.gastro.2018.07.028
48. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature (2008) 456:259–63. doi: 10.1038/nature07416
49. Stengel S, Fazio A, Lipinski S, Jahn M, Aden K, Ito G, et al. Activating Transcription Factor 6 Mediates Inflammatory Signals in Intestinal Epithelial Cells Upon Endoplasmic Reticulum Stress. Gastroenterology (2020) 159:1357–74. doi: 10.1053/j.gastro.2020.06.088
50. Aden K, Tran F, Ito G, Sheibani-Tezerji R, Lipinski S, Kuiper JW, et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp Med (2018) 215:2868–86. doi: 10.1084/jem.20171029
51. Yang Q, Kim Y-S, Lin Y, Lewis J, Neckers L, Liu Z-G. Tumour necrosis factor receptor 1 mediates endoplasmic reticulum stress-induced activation of the MAP kinase JNK. EMBO Rep (2006) 7:2–7. doi: 10.1038/sj.embor.7400687
52. Saveljeva S, Mc Laughlin SL, Vandenabeele P, Samali A, Bertrand MJM. Endoplasmic reticulum stress induces ligand-independent TNfR1-mediated necroptosis in L929 cells. Cell Death Dis (2015) 6:e1587. doi: 10.1038/cddis.2014.548
53. Hosomi S, Grootjans J, Tschurtschenthaler M, Krupka N, Matute JD, Flak MB, et al. Intestinal epithelial cell endoplasmic reticulum stress promotes MULT1 up-regulation and NKG2D-mediated inflammation. J Exp Med (2017) 214:2985–97. doi: 10.1084/jem.20162041
54. Cao SS, Song B, Kaufman RJ. PKR protects colonic epithelium against colitis through the unfolded protein response and prosurvival signaling. Inflammation Bowel Dis (2012) 18:1735–42. doi: 10.1002/ibd.22878
55. Rutkowski DT, Kang S-W, Goodman AG, Garrison JL, Taunton J, Katze MG, et al. The Role of p58IPK in Protecting the Stressed Endoplasmic Reticulum. Mol Biol Cell (2007) 18:3250–63. doi: 10.1091/mbc.E07
56. Gomez JA, Tyra HM, DeZwaan-McCabe D, Olivier AK, Rutkowski DT. Synthetic embryonic lethality upon deletion of the ER cochaperone p58IPK and the ER stress sensor ATF6α. Biochem Biophys Res Commun (2014) 443:115–9. doi: 10.1016/j.bbrc.2013.11.060
57. Hino K, Saito A, Asada R, Kanemoto S, Imaizumi K. Increased susceptibility to dextran sulfate sodium-induced colitis in the endoplasmic reticulum stress transducer OASIS deficient mice. PloS One (2014) 9:1–2. doi: 10.1371/journal.pone.0088048
58. Asada R, Saito A, Kawasaki N, Kanemoto S, Iwamoto H, Oki M, et al. The endoplasmic reticulum stress transducer OASIS is involved in the terminal differentiation of goblet cells in the large intestine. J Biol Chem (2012) 287:8144–53. doi: 10.1074/jbc.M111.332593
59. Brandl K, Rutschmann S, Li X, Du X, Xiao N, Schnabl B, et al. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc Natl Acad Sci U.S.A. (2009) 106:3300–5. doi: 10.1073/pnas.0813036106
60. Powell N, Pantazi E, Pavlidis P, Tsakmaki A, Li K, Yang F, et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut (2020) 69:578–90. doi: 10.1136/gutjnl-2019-318483
61. Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, et al. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci U.S.A. (2009) 106:6950–5. doi: 10.1073/pnas.0808722106
62. Camarillo GF, Goyon EI, Zuñiga RB, Salas LAS, Escárcega AEP, Yamamoto-Furusho JK. Gene expression profiling of mediators associated with the inflammatory pathways in the intestinal tissue from patients with ulcerative colitis. Mediators Inflammation (2020) 2020:1–11. doi: 10.1155/2020/9238970
63. Zhao F, Edwards R, Dizon D, Afrasiabi K, Mastroianni JR, Geyfman M, et al. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2-/- mice. Dev Biol (2010) 338:270–9. doi: 10.1016/j.ydbio.2009.12.008
64. Bergström JH, Berg KA, Rodríguez-Piñeiro AM, Stecher B, Johansson MEV, Hansson GC. AGR2, an endoplasmic reticulum protein, is secreted into the gastrointestinal mucus. PloS One (2014) 9:e104186. doi: 10.1371/journal.pone.0104186
65. Moschen AR, Gerner RR, Wang J, Klepsch V, Adolph TE, Reider SJ, et al. Lipocalin 2 Protects from Inflammation and Tumorigenesis Associated with Gut Microbiota Alterations. Cell Host Microbe (2016) 19:455–69. doi: 10.1016/j.chom.2016.03.007
66. Hansen IS, Schoonejans JM, Sritharan L, van Burgsteden JA, Ambarus CA, Baeten DLP, et al. ER stress abrogates the immunosuppressive effect of IL-10 on human macrophages through inhibition of STAT3 activation. Inflammation Res (2019) 68:775–85. doi: 10.1007/s00011-019-01261-9
67. De Vallière C, Wang Y, Eloranta JJ, Vidal S, Clay I, Spalinger MR, et al. G protein-coupled pH-sensing receptor OGR1 is a regulator of intestinal inflammation. Inflammation Bowel Dis (2015) 21:1269–81. doi: 10.1097/MIB.0000000000000375
68. Maeyashiki C, Melhem H, Hering L, Baebler K, Cosin-Roger J, Schefer F, et al. Activation of pH-Sensing Receptor OGR1 (GPR68) Induces ER Stress Via the IRE1α/JNK Pathway in an Intestinal Epithelial Cell Model. Sci Rep (2020) 10:1438. doi: 10.1038/s41598-020-57657-9
69. Coope A, Pascoal BL, Botezelli DJ, Ramos da Silva FA, Ayrizono M de LS, Milanski M, et al. ER stress activation in the intestinal mucosa but not in mesenteric adipose tissue is associated with inflammation in Crohn ‘ s disease patients. PloS One (2019) 14:1–19. doi: 10.1371/journal.pone.0223105
70. Tréton X, Pédruzzi E, Hatem DC, Grodet A, Panis Y, Groyer A, et al. Altered Endoplasmic Reticulum Stress Affects Translation in InactiveColon Tissue From Patients With Ulcerative Colitis. Gastroenterology (2011) 141:1024–35. doi: 10.1053/j.gastro.2011.05.033
71. Cao SS. Cellular Stress Responses and Gut Microbiota in Inflammatory Bowel Disease. Gastroenterol Res Pract (2018) 2018:1–13. doi: 10.1155/2018/7192646
72. Cao P, Chen Y, Chen Y, Su W, Zhan N, Dong W. Fusobacterium nucleatum Activates Endoplasmic Reticulum Stress to Promote Crohn’s Disease Development via the Upregulation of CARD3 Expression. Front Pharmacol (2020) 11:106. doi: 10.3389/fphar.2020.00106
73. Choi JH, Moon CM, Shin T-S, Kim EK, McDowell A, Jo M-K, et al. Lactobacillus paracasei-derived extracellular vesicles attenuate the intestinal inflammatory response by augmenting the endoplasmic reticulum stress pathway. Exp Mol Med (2020) 52:423–37. doi: 10.1038/s12276-019-0359-3
74. Kim DH, Kim S, Lee JH, Kim JH, Che X, Ma HW, et al. Lactobacillus acidophilus suppresses intestinal inflammation by inhibiting endoplasmic reticulum stress. J Gastroenterol Hepatol (2019) 34:178–85. doi: 10.1111/jgh.14362
75. Melhem H, Hansmannel F, Bressenot A, Battaglia-Hsu SF, Billioud V, Alberto JM, et al. Methyl-deficient diet promotes colitis and SIRT1-mediated endoplasmic reticulum stress. Gut (2016) 65:595–606. doi: 10.1136/gutjnl-2014-307030
76. Lee MR, Bae SJ, Kim JE, Song BR, Choi JY, Park JJ, et al. Inhibition of endoplasmic reticulum stress in high-fat-diet-induced obese C57BL/6 mice: Efficacy of a novel extract from mulberry (Morus alba) leaves fermented with Cordyceps militaris. Lab Anim Res (2018) 34:288–94. doi: 10.5625/lar.2018.34.4.288
77. Tan L, Harper L, McNulty MA, Carlson CS, Yammani RR. High-fat diet induces endoplasmic reticulum stress to promote chondrocyte apoptosis in mouse knee joints. FASEB J (2020) 34:5818–26. doi: 10.1096/fj.201902746R
78. Rubio-Patiño C, Bossowski JP, De Donatis GM, Chevet E, Villa E, Aira LE, et al. Low-Protein Diet Induces IRE1 a -Dependent Article Low-Protein Diet Induces IRE1 a -Dependent Anticancer Immunosurveillance. Cell Metab (2018) 27:828–42. doi: 10.1016/j.cmet.2018.02.009
79. Toutounji M, Wanes D, El-Harakeh M, El-Sabban M, Rizk S, Naim HY. Dextran sodium sulfate-induced impairment of protein trafficking and alterations in membrane composition in intestinal Caco-2 cell line. Int J Mol Sci (2020) 21:2726. doi: 10.3390/ijms21082726
80. Zong S, Ye Z, Zhang X, Chen H, Ye M. Protective effect of Lachnum polysaccharide on dextran sulfate sodium-induced colitis in mice. Food Funct (2020) 11:846–59. doi: 10.1039/C9FO02719J
81. Li A, Song N, Riesenberg BP, Li Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front Immunol (2020) 10:3154. doi: 10.3389/fimmu.2019.03154
82. You Y, Wen D, Zhao L, Mei F, Hong Y, Zhou Y. 4 − Phenylbutyric Acid Attenuates Endoplasmic Reticulum Stress − Mediated Intestinal Epithelial Cell Apoptosis in Rats with Severe Acute Pancreatitis. Dig Dis Sci (2019) 64:1535–47. doi: 10.1007/s10620-018-5437-1
83. Shkoda A, Ruiz PA, Daniel H, Kim SC, Rogler G, Sartor RB, et al. Interleukin-10 Blocked Endoplasmic Reticulum Stress in Intestinal Epithelial Cells: Impact on Chronic Inflammation. Gastroenterology (2007) 132:190–207. doi: 10.1053/j.gastro.2006.10.030
84. Li P, Fu D, Sheng Q, Yu S, Bao X, Lv Z. TUDCA attenuates intestinal injury and inhibits endoplasmic reticulum stress - mediated cell apoptosisin necrotizing enterocolitis. Int Immunopharmacol (2019) 74:1056665. doi: 10.1016/j.intimp.2019.05.050
85. Tian RD, Chen YQ, He YH, Tang YJ, Chen GM, Yang FW, et al. Phosphorylation of eIF2α mitigates endoplasmic reticulum stress and hepatocyte necroptosis in acute liver injury. Ann Hepatol (2020) 19:79–87. doi: 10.1016/j.aohep.2019.05.008
86. Golden JM, Escobar OH, Nguyen MVL, Mallicote MU, Kavarian P, Frey MR, et al. Ursodeoxycholic acid protects against intestinal barrier breakdown by promoting enterocyte migration via EGFR- and COX-2-dependent mechanisms. Am J Physiol Gastrointest Liver Physiol (2018) 315:G259–71. doi: 10.1152/ajpgi.00354.2017
87. Ko WK, Lee SH, Kim SJ, Jo MJ, Kumar H, Han IB, et al. Anti-inflammatory effects of ursodeoxycholic acid by lipopolysaccharide-stimulated inflammatory responses in RAW 264.7 macrophages. PloS One (2017) 12:1–12. doi: 10.1371/journal.pone.0180673
88. Lajczak-McGinley NK, Porru E, Fallon CM, Smyth J, Curley C, McCarron PA, et al. The secondary bile acids, ursodeoxycholic acid and lithocholic acid, protect against intestinal inflammation by inhibition of epithelial apoptosis. Physiol Rep (2020) 8:1–11. doi: 10.14814/phy2.14456
89. Crespo I, San-Miguel B, Prause C, Marroni N, Cuevas MJ, González-Gallego J, et al. Glutamine Treatment Attenuates Endoplasmic Reticulum Stress and Apoptosis in TNBS-Induced Colitis. PloS One (2012) 7:e50407. doi: 10.1371/journal.pone.0050407
90. Kong S, Zhang YH, Zhang W. Regulation of intestinal epithelial cells properties and functions by amino acids. BioMed Res Int (2018) 2018:1–10. doi: 10.1155/2018/2819154
91. He Y, Fan X, Liu N, Song Q, Kou J, Shi Y, et al. L-Glutamine Represses the Unfolded Protein Response in the Small Intestine of Weanling Piglets. J Nutr (2019) 149:1904–10. doi: 10.1093/jn/nxz155
92. Fan X, Li S, Wu Z, Dai Z, Li J, Wang X, et al. Glycine supplementation to breast-fed piglets attenuates post-weaning jejunal epithelial apoptosis: a functional role of CHOP signaling. Amino Acids (2019) 51:463–73. doi: 10.1007/s00726-018-2681-9
93. Yan S, Yingchao L, Zhangliu W, Xianli R, Si L, Siyi N, et al. Effect of Berberine from Coptis chinensis on Apoptosis of Intestinal Epithelial Cells in a Mouse Model of Ulcerative Colitis: Role of Endoplasmic Reticulum Stress. Evidence-Based Complement Altern Med (2020) 2020:7 pages. doi: 10.1155/2020/3784671
94. Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, et al. Targeting ABL-IRE1α Signaling Spares ER-Stressed Pancreatic β Cells to Reverse Autoimmune Diabetes. Cell Metab (2017) 25:883–897.e8. doi: 10.1016/j.cmet.2017.03.018
95. Tufanli O, Telkoparan P, Acosta-alvear D, Kocaturk B, Inci U. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc Natl Acad Sci U.S.A. (2017) 114:E1395–404. doi: 10.1073/pnas.1621188114
96. Atkins C, Liu Q, Minthorn E, Zhang S, Figueroa DJ, Moss K, et al. Characterization of a Novel PERK Kinase Inhibitor with Antitumor and Antiangiogenic Activity. Cancer Res (2013) 73:1993–2003. doi: 10.1158/0008-5472.CAN-12-3109
97. Axten M, Medina R, Feng Y, Shu A, Romeril SP, Grant SW, et al. Discovery of 7-Methyl-5-(1-{[3(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a Potent and Selective First-in-Class Inhibitor of Protein Kinase R (PKR)-like Endoplasmic Reticulum Kinase (PERK). J Med Chem (2012) 55:7193–207. doi: 10.1021/jm300713s
98. Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral Treatment Targeting the Unfolded Protein Response Prevents Neurodegeneration and Clinical Disease in Prion-Infected Mice. Sci Transl Med (2013) 5:206ra138. doi: 10.1126/scitranslmed.3006767
99. Yalçin A, Şarkici G, Kolaç UK. PKR inhibitors suppress endoplasmic reticulum stress and subdue glucolipotoxicity-mediated impairment of insulin secretion in pancreatic beta cells. Turkish J Biol (2020) 44:93–102. doi: 10.3906/biy-1909-20
100. Blackwood EA, Azizi K, Thuerauf DJ, Paxman RJ, Plate L, Kelly JW, et al. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat Commun (2019) 10:1–16. doi: 10.1038/s41467-018-08129-2
101. Koloski NA, Bret L, Radford-Smith G. Hygiene hypothesis in inflammatory bowel disease: A critical review of the literature. World J Gastroenterol (2008) 14:165–73. doi: 10.3748/wjg.14.165
102. Hugot JP, Alberti C, Berrebi D, Bingen E, Cézard JP. Crohn’s disease: The cold chain hypothesis. Lancet (2003) 362:2012–5. doi: 10.1016/S0140-6736(03)15024-6
103. Ma A. Unresolved ER Stress Inflames the Intestine. Cell (2008) 1:724–5. doi: 10.1016/j.cell.2008.08.023
104. Mcguckin MA, Eri RD, Das I, Lourie R, Florin TH. ER stress and the unfolded protein response in intestinal inflammation. Am J Physiol Gastrointest Liver Physiol (2010) 298:820–32. doi: 10.1152/ajpgi.00063.2010
105. Coleman OI, Haller D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front Immunol (2019) 10:2825. doi: 10.3389/fimmu.2019.02825
Keywords: endoplasmic reticulum stress, unfolded protein response, apoptosis, inflammation, intestinal epithelial cells
Citation: Eugene SP, Reddy VS and Trinath J (2020) Endoplasmic Reticulum Stress and Intestinal Inflammation: A Perilous Union. Front. Immunol. 11:543022. doi: 10.3389/fimmu.2020.543022
Received: 15 March 2020; Accepted: 28 October 2020;
Published: 25 November 2020.
Edited by:
Deirdre R. Coombe, Curtin University, AustraliaReviewed by:
Luz Pamela Blanco, National Institutes of Health (NIH), United StatesBeate E. Kehrel, University Hospital Münster, Germany
Copyright © 2020 Eugene, Reddy and Trinath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jamma Trinath, dHJpbmF0aEBoeWRlcmFiYWQuYml0cy1waWxhbmkuYWMuaW4=; a2FydW55YS5mcmllbmRAZ21haWwuY29t