 Elaine L. Parker
Elaine L. Parker Rachel B. Silverstein
Rachel B. Silverstein Sonam Verma
Sonam Verma Indira U. Mysorekar
Indira U. Mysorekar
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 October 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.522047
This article is part of the Research Topic Perinatal Immunobiology View all 34 articles
The human decidua and placenta form a distinct environment distinguished for its promotion of immunotolerance to infiltrating semiallogeneic trophoblast cells to enable successful pregnancy. The maternal-fetal interface also successfully precludes transmission of most pathogens. This barrier function occurs in conjunction with a diverse influx of decidual immune cells including natural killer cells, macrophages and T cells. However, several viruses, among other microorganisms, manage to escape destruction by the host adaptive and innate immune system, leading to congenital infection and adverse pregnancy outcomes. In this review, we describe mechanisms of pathogenicity of two such viral pathogens, Human cytomegalovirus (HCMV) and Zika virus (ZIKV) at the maternal-fetal interface. Host decidual immune cell responses to these specific pathogens will be considered, along with their interactions with other cell types and the ways in which these immune cells may both facilitate and limit infection at different stages of pregnancy. Neither HCMV nor ZIKV naturally infect commonly used animal models [e.g., mice] which makes it challenging to understand disease pathogenesis. Here, we will highlight new approaches using placenta-on-a-chip and organoids models that are providing functional and physiologically relevant ways to study viral-host interaction at the maternal-fetal interface.
Pregnancy is a unique immunological phenomenon in which the semiallogenic fetus is able to grow in the maternal uterine environment. In order for a successful pregnancy to occur, healthy placentation is necessary to create an environment that is protective for the developing fetus and promotes growth. How immune balance is maintained by maternal and fetal cells to promote the survival of the genetically distinct fetus, while preventing infection by a large number of pathogens, is yet to be fully elucidated (1). This little understood enigma has been the subject of interest and research for decades (2).
Fertilization leads to the creation of single celled embryo which undergoes several successive divisions to form a blastocyst. The blastocyst is made up of two types of cells: the outer trophoblast or trophoectoderm (TE) layer forming the placenta and chorion, and the inner layer or inner cell mass (ICM) forming the embryo proper and amnion (3). The decidua underlying the embryo is called the decidua basalis, which composes the maternal side of the placenta. The maternal-fetal interface is made up of the maternal decidua and fetally-derived placenta. During implantation, the blastocyst attaches to the decidualized endometrium and the outer layer of the blastocyst differentiates into different lineages. The TE gives rise to cytotrophoblast cells (CTBs) which follow villous and extravillous pathways to form the placenta. In the villous pathway, the mononuclear CTBs fuse, creating multinucleated syncytiotrophoblasts (STBs) that establish floating villi (FV). The FV are surrounded by maternal blood, with STBs aiding the provision of nutrients by enabling gas exchange and exchange of secreted pregnancy-related hormones (human chorionic gonadotropin, hCG, human placental lactogen, hPL) at the maternal-fetal interface. Furthermore, CTBs act as anchoring villi for the attachment of the embryo to the uterus. The CTBs present in the cell column of the anchoring villi follow the extravillous pathway and differentiate into interstitial (iCTBs) and endovascular extravillous trophoblast cells (eCTBs). The iCTBs further invade up to the inner third of the myometrium and eCTBs remodel the spiral arteries in low resistance high blood flow to provide nutrients to the developing embryo (3–6). The invasion of trophoblast cells at the maternal-fetal interface occurs in the presence of a large population of maternal immune cells (7). This includes 70% decidual Natural Killer (dNK) cells, 20%–25% macrophages, 3%–10% T cells and 1.7% dendritic cells (8–10). The abundance of decidual cytotoxic T cells and macrophages can vary through the course of pregnancy (11). The abundance of NK cells in the decidua during the first trimester, and through the pregnancy (albeit at lower abundance), implicates them as an essential element in both the promotion of an immunotolerant environment and the control of pathogenic infection during pregnancy (Figure 1). Thus, the paradoxical maternal-fetal interface is admired for both its immunotolerance to semiallogeneic trophoblastic invasion (leading to a successful pregnancy) while remaining remarkably resilient to pathogenic infections. Nevertheless, several pathogen, termed TORCH pathogens (described below), successfully cross the placental barrier and cause devastating infection in the developing fetus (12). In this review, we will look at the interactions between decidual immune cells and specific viral TORCH pathogens and review known mechanisms which may enable viral pathogenesis within the placental environment.
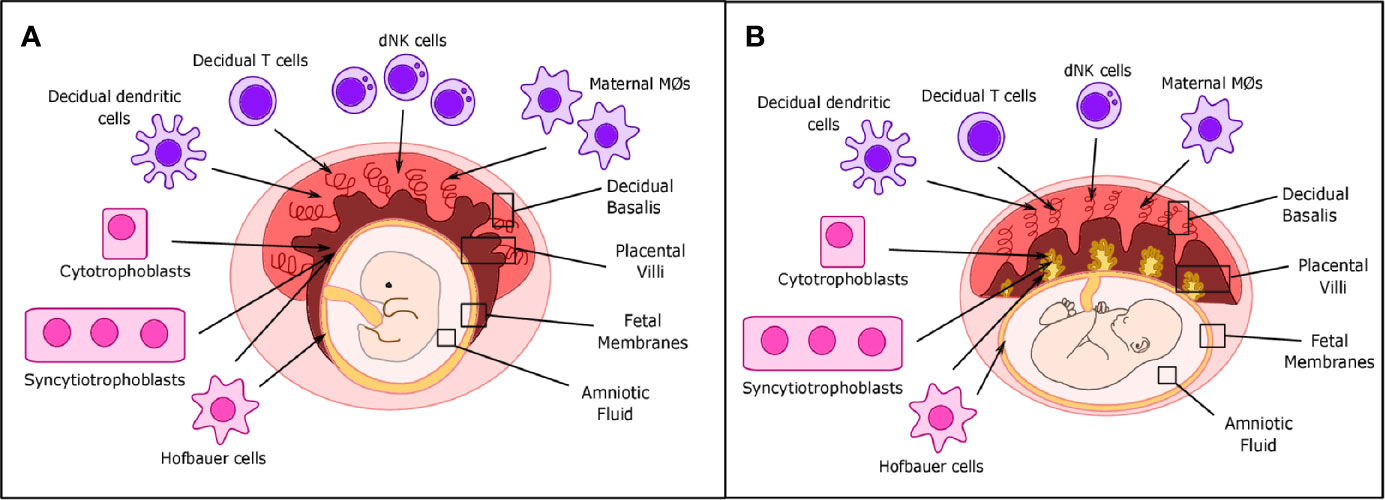
Figure 1 Placental structure and location of decidual immune cells in placental tissues. (A) During the first trimester, decidual NK cells, T cells, and dendritic and maternal macrophages are located primarily in the decidua basalis, while fetal Hofbauer cells (HCs) are primarily located in the placental villi and fetal membranes. Cytotrophoblast (CTBs) can be found in both the decidua basalis and placental villi, while syncytiotrophoblasts (STBs) are only found in the placental villi. (B) In term placentas, location of decidual immune cells is roughly the same, but the number of maternal macrophages and decidual Natural Killer (dNK) cells can be reduced.
TORCH is an acronym defining some of the most common infections associated with vertical transmission. Initially described in 1971, this group contained just 4 pathogens; Toxoplasmosis, Rubella, Cytomegalovirus (CMV) and Herpes simplex type 1 and 2 (13). Since then this group has been broadened to comprise a host of other infections including Listeria monocytogenes, Syphilis, Varicella Zoster virus, Human immunodeficiency virus (HIV), enteroviruses and parvovirus B19 (14). Most recently, following the Zika virus (ZIKV) epidemic in South America resulting in observed congenital anomalies, this group has been further expanded to include ZIKV, with some suggesting renaming this group “TORCHZ” (15). The mechanism by which these “TORCHZ” pathogens are able to circumvent typical clearance by groups of immune cells (e.g. NK cells, macrophages and others) has been studied by many groups over the last few decades in order to elucidate not only routes of pathogenicity but also roles of immune cells within this immune-privileged environment (12). It remains to be proven whether the new emerging viral threat by SARS-COV2 which causes COVID-19 including in pregnant women, will be included in this group of vertically transmitted pathogens (16).
In this review, we will focus on maternal and fetal macrophages, T cells, and NK cells and their relationship with each virus. We will focus on the viruses human Cytomegalovirus (HCMV) and ZIKV, which are known causes of adverse pregnancy outcomes and delve into how they interact with various decidual immune cells to promote their survival and replication. We will examine the timings of pregnancy that appear to be most permissive to pathogenic infection by these viruses and we will look at the role of various immune cells in this context (Figure 2).
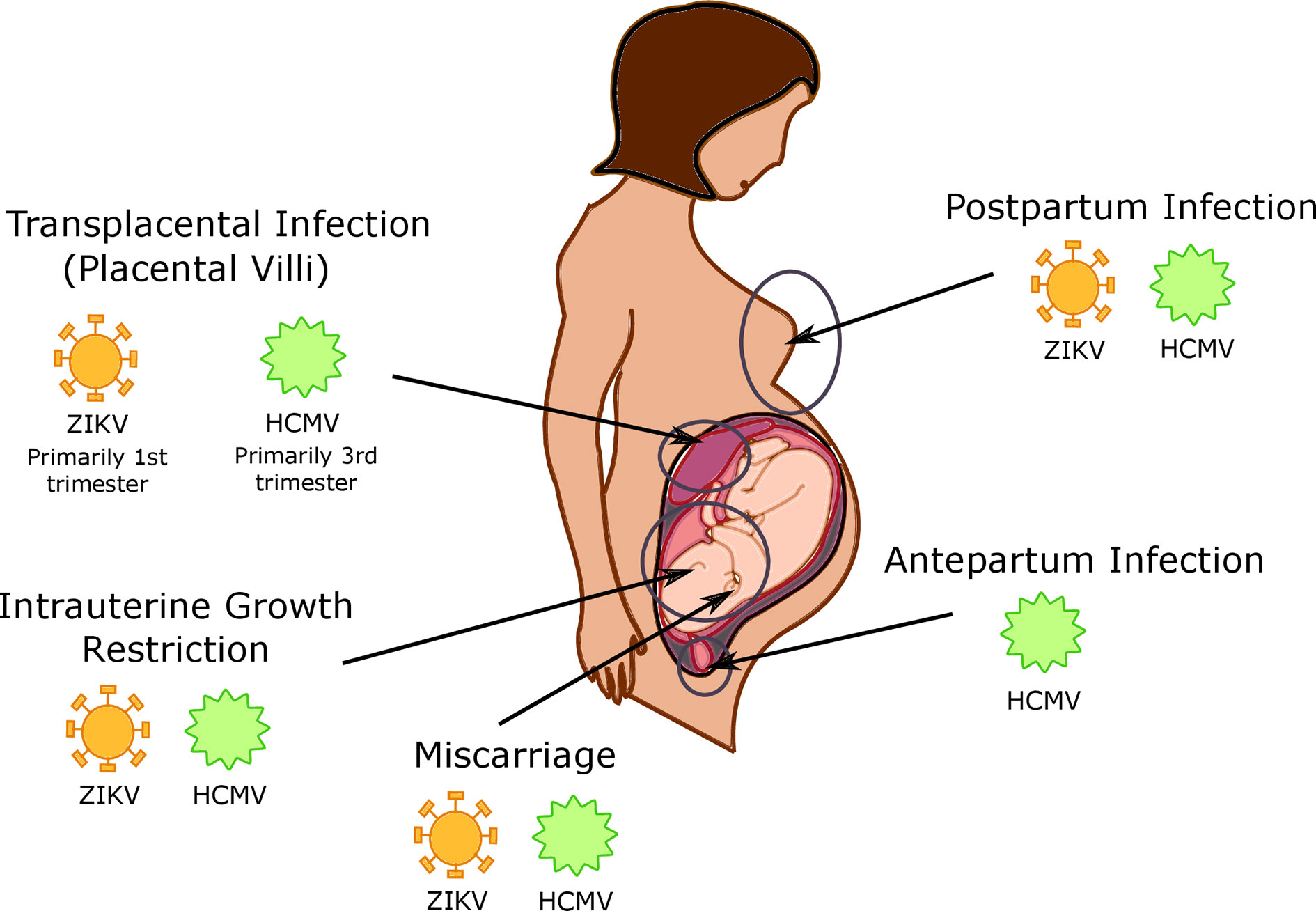
Figure 2 Timing and location of viral infections in the placenta. Human cytomegalovirus (HCMV) is capable of intrapartum infection, as well as postpartum infection during breastfeeding; it is currently unknown whether Zika virus (ZIKV) is capable of this type of transmission as well. HCMV and ZIKV are both capable of transplacental infection, with HCMV infection primarily occurring in the 3rd trimester and ZIKV infection is more common in the 1st trimester. CMV and ZIKV infection additionally cause both intrauterine growth restriction and miscarriage.
In the early first term decidua, 3%–10% of resident leukocytes are T cells with approximately 30%–45% of these T cells being CD4+ (T helper cells) and 45%–75% being CD8+ (cytotoxic) T cells (17–19). Further studies have estimated the decidual CD4+ population to be comprised of about 50% activated memory CD25dim T cells and 5% CD4+ CD25bright FOXP3+ Treg cells. Unlike the peripheral circulation, the decidua has a higher ratio of CD8+ T cells to CD4+ T cells and an overall higher number of CD8+ T cells (20). In addition, approximately 40% of the decidual CD8+ population are effector-memory T cells with reduced perforin and granzyme B in comparison to their peripheral counterparts (21). One study published in 2016 described a small percentage of CD8+ T cells found in uncomplicated term decidua to be viral specific. Though these populations of viral specific CD8+ T cells were 1.3% and 2.2% in the decidual basalis and decidual parietalis respectively, they demonstrated that this was higher than that seen in peripheral blood and postulated a role for their presence in the decidua as one of immunoprotection for the fetus. This study could not conclude upon the origin of these T cells, and whether they were recruited from the periphery or activated in the decidua. In addition, more work remains to be done to establish whether these virus specific CD8+ T cells exist early in pregnancy (22). Another study has described the presence of a small population of CD4+ HLA-G+ T cells which are thought to acquire HLA-G through trogocytosis from decidual dendritic cells. It is thought that these T cells promote immunotolerance at the maternal-fetal interface, and they have been shown to be downregulated in pathologies such as preeclampsia (PE) (23). Therefore, it appears T cells play specific roles in immunity and tolerance. To this end we will look at the role that various populations of T cells may play in either enabling or preventing infection by TORCH pathogens at the maternal-fetal interface.
Macrophages constitute 20-25% of all leukocytes in the first trimester decidua and play an important role in tissue remodeling, angiogenesis, host defense and immunotolerance (24). Macrophages are considered a key link between adaptive and innate immunity, communicating to other immune cells and modulating their activity (25, 26). These cells are therefore vital throughout pregnancy, adapting their phenotype to address the changing requirements of the evolving decidua (27). Tissue resident decidual macrophages are thought to be recruited from monocytes in the peripheral circulation (28). Distinct subtypes of macrophages have been shown to be present in first-trimester decidual tissue exhibiting immunomodulatory, proinflammatory, and tissue remodeling phenotypes and play key roles in protective immunity as well as fetal tolerance (29). Decidual macrophages are known for their highly immunosuppressive phenotype at the maternal-fetal interface, expressing CD206, DC-SIGN and Tim-3 among other receptor markers (30, 31).
In addition to these maternally derived macrophages exist fetal-derived macrophages called Hofbauer cells (HCs), which sit in the stroma of the chorionic villi (32). These HCs are resident in close proximity to fetal vessels and trophoblast cells from the first trimester until birth. HCs could serve as a portal of entry for pathogens from the infected mother (33). Initially during implantation, they appear to have an inflammatory M1 phenotype which has both microbicidal activity and promotes a cell-mediated Th1 cytokine response. Later, they shift to a mixture of both M1 and M2 phenotypes following trophoblastic invasion and remodeling (34, 35). Several studies have implicated HCs in host viral interactions. Here, we look at the reciprocal interactions between HCs, maternal macrophages, and HCMV and ZIKV.
The NK cell population in the peripheral circulation is predominately made up of CD56dim CD16+ cells, which are believed to have a more cytotoxic phenotype (36). Approximately 10% of the peripheral circulation is constituted by CD56bright NK cells, which have a more immunotolerant phenotype (37). In the decidua, these NK cell proportions are reversed; 70-80% of the total lymphocytes are CD56bright CD16- (36). Research has demonstrated a number of dNK subsets within the CD56+CD16- population. It is believed that this distinct immunotolerant population is fundamental to the maintenance of a successful pregnancy, with research postulating both an ability to enable the semiallogenic fetus to thrive while at the same time responding to pathogenic infections. These NK cells reside in the decidua basalis close to invading EVTs and express specific receptors (e.g. KIR receptors, CD94/NKG2A, ILT2) to activate or inhibit EVT function (38). This large population of dNK cells are known to be sustained during the first and second trimester, with their numbers declining toward term (11, 39). Despite the unique immunotolerant phenotype demonstrated by dNK cells, it is evident that this cell population displays a high level of plasticity, gaining cytotoxic function in the presence of specific pathogens (39). One way by which this happens is through activation of dNK cell cytotoxcity via killer cell Ig-like receptor 2DS1 (KIR2DS1). Reduced expression of this receptor has been associated with adverse pregnancy outcomes such as miscarriages and fetal growth restriction and individuals with increased KIR2DS1 expression have shown better outcomes post-viral infections (40). We will explore further the role that NK cells play in specific viral infections in pregnancy
Human cytomegalovirus (HCMV) was first described in 1954 by Margaret Smith, who replicated a virus from two newborn babies who had died from cytomegalic inclusion disease (CID) (41). What we now know as HCMV first came to the attention of Ribbert et al. in 1881, where intranuclear inclusions within large cells were noted in renal and parotid gland cells of stillborn fetuses. These inclusions, often described as ‘owl’s eye inclusions’, were noted to be surrounded by a clear halo (42). HCMV was identified in the 1950s when Smith, Weller and Rowe isolated and cultured HCMV from salivary glands, adenoid tissue and liver biopsies respectively (43, 44). Mechanisms of vertical transmission of HCMV can either be transplacental during gestation or transvaginal during parturition; additionally, there is some evidence for breastmilk transmission (45). HCMV infection is most likely to occur in the third trimester, demonstrating a 30% risk of mother to child transmission in the first trimester compared to a 70% risk in the third trimester (46–48). Congenital HCMV has been estimated to affect 5–20 in every 1,000 live births, with 10% of HCMV positive infants suffering neurological consequences from birth (49). HCMV infection during pregnancy therefore poses a substantial risk to the developing fetus, leading to congenital disease including cerebral abnormalities such as periventricular calcifications, microcephaly, visual impairment, sensorineural hearing loss, neurodevelopmental delay and hepatomegaly (45). Congenital HCMV affects 20,000–40,000 pregnancies annually in the United States and accounts for 25% of all incidents of pediatric sensorineural hearing loss (50–52). It is estimated that the burden of morbidity associated with congenital HCMV infection is greater than that of other common congenital pediatric conditions such as down’s syndrome or fetal alcohol syndrome (53–55). HCMV is also associated with intrauterine growth restriction and miscarriage. There is a great need to understand maternal immunity pathways involved in HCMV infection to develop effective vaccines (56).
HCMV is associated with asymptomatic infection of most of the world’s population and subclinical illness in pregnant mothers. In the US, an estimated 2% of unexposed pregnant women experience primary infection during pregnancy, resulting in congenital infection in 32% of cases from this population (53, 57–61). However, vertical transmission of HCMV is not only seen in mothers with primary infection but also IgG seropositive mothers, who exhibit a 1% rate of congenital HCMV infection. Mechanisms of infection have been studied through analysis of placental tissue from all three trimesters of human gestation. In placental tissues from those suffering from HCMV, necrosis and oedema has been noted associated with severity of congenital disease symptoms. It has also been noted that HCMV infection is often associated with bacterial coinfection with a potentially pathogenic synergism (62). HCMV resides in the chorionic villi, specifically infecting CTBs, STBs and HCs. It is believed that the ability to travel between STBs in the decidua is key to HCMV pathogenesis (63). Many studies have explored the role of the adaptive and innate immune system in HCMV infection. Below we review established interactions between HCMV and immune cells (Figure 3).
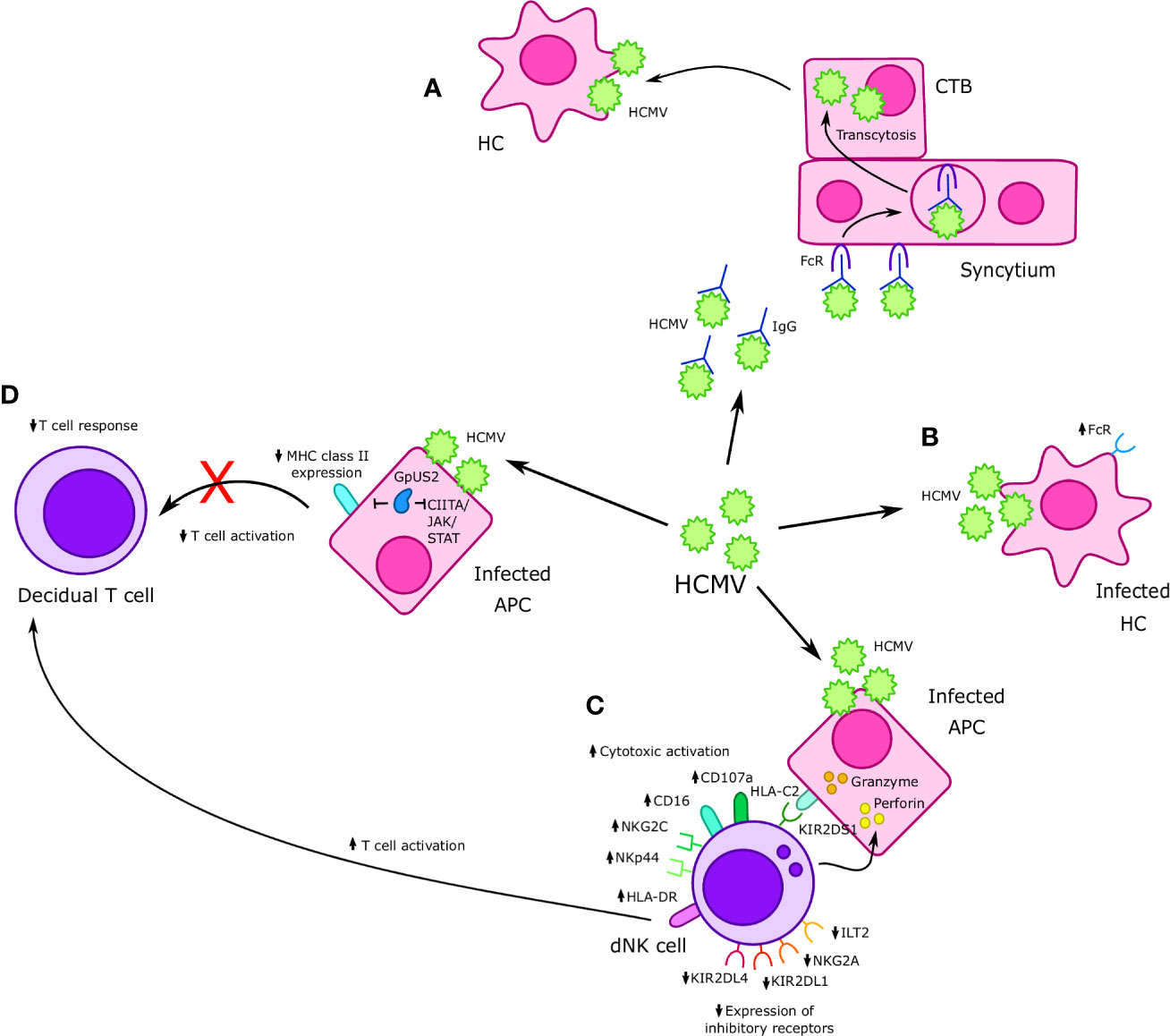
Figure 3 Interactions between Cytomegalovirus (CMV) and immune cells at the maternal-fetal interface. (A) Human cytomegalovirus (HCMV)-IgG complexes undergo FcR mediated transcytosis in the syncytium and infect cytotrophoblast (CTBs); HMCV then goes on to infect Hofbauer cells (HCs) in the placental villi. (B) HCMV infects HCs, resulting in increased FcR expression. (C) Formation of an immunological synapse between decidual Natural Killer (dNK) cells and HCMV infected APCs prompts the release of perforin and granzyme into infected APCs and results in increased expression of cytotoxic receptors NKp44 and NKG2C, as well as other markers of cytotoxicity such as CD16 and CD107a. Furthermore, increased cytotoxicity is accompanied by a reduction in expression of inhibitory receptors such as NKG2A, KIR2DL1, and KIR2DL4. Interaction between KIR2DS1 present on dNK cells and HLA-C on APCs increases dNK cell cytotoxicity in the context of HMCV infection. HMCV infection also results in increased expression of HLA-DR, which may increase T cell activation. (D) APCs infected with HCMV exhibit reduced MHC II expression, which is believed to in turn cause a reduced CD8+ T cell response in cases of vertical transmission. The HCMV protein GpUS2 is also believed to downregulate MHC II expression and CIITA/JAK/STAT signaling.
HCMV’s ability to infect different populations of macrophages has been demonstrated by several studies. HCMV has been shown to be sequestered by HCs, with placentas from confirmed cases of HCMV infection demonstrating significant hyperplasia of this cell population (64–66). A study investigating vaccine development showed that when neutralizing antibodies are produced against HCMV, rates of HCs infection are decreased (67). A different study utilizing placental explants showed HCMV-IgG immune complexes to undergo Fc Receptor mediated transcytosis as a mechanism to traverse the syncytium to CTBs. HCMV is then taken up by HCs in the placental villi (68). Furthermore, another study by Loenen et al., supports the idea that HCMV genes are able to increase FcR expression on infected cells (69). Another study suggested that HCMV replication in STBs is upregulated in the presence of macrophages (70) by analyzing HCMV replication in STBs alone or when infected STBs were cultured with uninfected placental macrophages. This study also demonstrated elevated levels of HCMV viral titres in co-cultured supernatants when compared to those from STBs cultured alone. This demonstrates that not only do macrophages have the capacity to be infected by HCMV, but also that they may amplify HCMV infection of surrounding cells in the decidua. Some studies have depicted a role for latently infected maternal decidual macrophages in congenital HCMV infection, describing how microbial infections or insults in the placenta may reactivate these macrophages and in turn reactivate HCMV infection (71–73).
The maternal-fetal interface is unique in respect to allogenic interactions with CD8+ T cells. EVTs are known to invade the decidua, evading destruction despite the intrinsic ability of CD8+ T cells to recognize foreign antigen via MHC class I molecules. As discussed previously, one mechanism by which EVTs are believed to evade CD8+ T cell recognition is through a lack of expression of HLA-A and HLA-B, which are key to CD8+ cytotoxic activity. During pregnancy, many viruses have been shown to upregulate maternal CD8+ T cell activity, leading to migration of highly differentiated effector memory T cells to the decidua. Despite many descriptions regarding the role of T cells in HCMV infection in the fetus and the mother, there are few studies identifying their tissue specific role at the maternal-fetal interface (74).
HCMV is thought to limit CD8+ T cell activity through restriction of MHC class II expression on APCs, which in turn may prevent activation of CD8+ and CD4+ T cells (69). This is thought to be mediated through the HCMV protein GpUS2, which may degrade MHC class II glycoproteins or disrupt downstream CIITA/JAK/STAT signaling pathways (69). Crespo et al., in 2016 demonstrated that HCMV did not induce a significant difference in HLA-G expression on either JEG-3 cells or primary EVTs. HLA-G expression has been associated with immunotolerance, and therefore its persistence despite infection may act to protect infected trophoblast cells from cytotoxic destruction (40).
Studies looking at the role of T cells in viral infection at the maternal-fetal interface demonstrated lower T-cell numbers and response in mothers who vertically transmitted HCMV to their offspring when compared to infected mothers who did not transmit HCMV, potentially suggesting an active role for T cells in vertical HCMV transmission (75).
More specifically, a reduction in the number of CD4+CD45RA+IFN-γ+ Treg cells and CD8+CD45RA+IFN-γ+ T cells in mothers who transmitted HCMV to their fetus was noted when contrasted with mothers who were HCMV positive but did not transmit the infection. There was also a measurable blunted T cell response in HCMV infected mothers who vertically transmitted infection, compared to infected mothers who did not transmit the virus (76, 77). In infected mothers, HCMV virus specific T cells have been shown to be elevated in the final trimester when compared to uninfected mothers (78).
Congenital HCMV infection risk is highest for the fetus in the third trimester, with a 72% transmission risk compared to a 30% risk in first trimester (46). This is despite the abundance of immune cells, specifically NK cells, in early pregnancy. dNK cells exposed to HCMV infected decidual fibroblasts are known to alter their phenotype to express higher levels of activating receptors (such as NKG2D and CD94/NKG2C or NKG2E). Uniquely, utilizing in vitro studies, it was noted that decidual NK cells had targeted cytotoxic activity against HCMV infected autologous decidual fibroblasts and heterologous uninfected fibroblast cells, but appeared to spare trophoblast cells (79, 80). This demonstrates a clear cytotoxic effector response by decidual NK cells to HCMV, switching from their typically immunotolerant phenotype with high levels of inhibitory receptor expression (CD94/NKG2A, LIR-1, KIRS), to a cytotoxic phenotype (79, 80). This group also studied the interaction between dNK and HCMV-infected cells using HCMV positive and HCMV negative decidual villous explants. This investigation revealed through fluorescent staining of dNK cells that co-localisation of dNK cells to cells throughout the HCMV positive explant occurred, including synaptic connections which was not seen in HCMV negative explants. This was thought to suggest that the dNK cells were unable to connect with uninfected trophoblasts. This also demonstrates that dNK cells are able to localize and target HCMV infected cells while sparing fetal derived semiallogenic trophoblast cells (80).
dNK cells are unique in their function, both contributing to immunotolerance at the maternal-fetal interface, thereby enabling invasive trophoblastic activity, as well as controlling pathogenic infection (81). This is thought to be mediated by secretion of specific cytokines (79, 82–85). The relatively limited vertical transmission of HCMV during the first trimester of pregnancy, when the population of NK cells is abundant, has led many to speculate about the role NK cells may play in HCMV control (10). Tilburgs and colleagues have recently demonstrated distinct cytotoxic responses in dNK cells to HCMV in first trimester versus at term wherein term pregnancy dNKs harbor reduced efficacy in responding to HCMV–infected cells (86). Siewera et al., suggested that dNK cells undergo a phenotypic transformation to acquire cytotoxic function in the presence of HCMV-infected cells (80). This study proved, through antibody mediated abrogation of the Fas ligand (FasL) and tumor necrosis factor-related apoptosis-induced ligand (TRAIL) on dNK cells, that death of HCMV infected cells is not initiated by dNK cells through these death receptor-ligand pathways. However, this study demonstrated that dNK cells form immunological synapses with HCMV infected fibroblasts, enabling the delivery of perforin/granzyme for cellular destruction. Furthermore, the ability of dNK cells to degranulate in the presence of HCMV infected fibroblasts was demonstrated to be through high levels of CD107a expression, a key cell surface molecule in the mechanism of lytic granule release. dNK cells have also been found to secrete higher quantities of granulysin when compared to peripheral blood NK cells. Upon incubation with infected fibroblast cells, it was noted that CD56bright NK cells decreased from 76.3% to 48%, while there was an elevation in Cd16 expression by NK cells, denoting a transformation to a cytotoxic phenotype. HCMV infected cells have been noted to upregulate expression of natural cytotoxicity receptor (NCRs) NKp44 by almost 2-fold on dNK cells as well as increasing expression of NKG2C. NCRs are associated with activation of the cytotoxic profile of NK cells. Accompanying this was a reduction in NKG2A, KIR2DL1, KIR2DL4, and ILT2 receptor expression, receptors aligned with NK effector inhibitory function (76).
Activating dNK cell receptors such as KIR2DS1, KIR2DS2, KIR2DS5 and KIR3DS1 have been correlated with antiviral activity (40). A study by Crespo et al., demonstrated an increased population of KIR2DS1 + NK cells in the decidua, suggesting an increased activating dNK cell capability in response to HLA-C2, and thereby increased cytotoxic potential. These cells also displayed higher levels of cytolytic molecules when compared to peripheral NK cells. This study demonstrated that KIR2DS1 + dNK cells showed increased cytotoxicity to HCMV infected decidual stromal cells (DSCs) positive for HLA-C2 when compared to KIR2DS1- dNK cells. This was not the case for infected JEG-3 and primary EVT cells, which did not appear to initiate degranulation or cytokine secretion from dNK cells. Despite this, a reduction in the number of infected EVTs in the presence of co-cultured dNK cells was noted, suggesting that dNK may be clearing virus infected EVTs by other means (40). HCMV has been seen to reduce expression of MHC class I, thereby potentially evading CD8+ T cell destruction (87–89). One study reports an initial reduction in HLA-C expression on EVTs in HCMV infection. The possible reason for this is not clear, however this study suggests it could prevent inhibition of NK cells through the HLA-C/KIR2DL1 route, with an additional suggestion of potentially other unknown ligands being upregulated for activation of KIR2DS1, leading to cytotoxic action against infected cells (40).
Another study showed that the potential effect of dNK cell activation on T-cell activation could be mediated via an upregulation in HLA-DR expression upon exposure to HCMV infected fibroblasts (80). Therefore, dNK cells may play a role in congenital HCMV infection by potentially protecting the first trimester fetus from infection via activation of T cell function.
Collectively, these studies indicate varied interactions between dNK cells and HCMV, with many routes by which HCMV may evade clearance as well as a number of ways through which dNK cells may be activated in the presence of HCMV infected cells. Additionally, dNK cells are seen specifically to modulate activity in the context of T cell activation.
Zika Virus (ZIKV) was first isolated in 1947 in Zika Forest, Uganda, from infected Rhesus monkey serum during epidemiological yellow fever research (90, 91). However, the first case of human infection was not reported until 1954, when three patients presented with jaundice and were later confirmed to have rising levels of Zika antibodies (92). Initially, ZIKV was associated with innocuous prodromal illness on the African and Indian subcontinents transmitted by the Aedes aegypti mosquito, leading to an asymptomatic or self-limiting course of infection (93). In 2007, a mild disseminated infection was identified to be ZIKV in over 70% of the population of the Island of Yap (94). Concerns regarding human ZIKV infections were not aroused until 2013 when incidences of neurological deficits associated with ZIKV infection were first described, with almost 30,000 recorded infections noted in French Polynesia (95, 96). Shortly following this in 2015, a ZIKV epidemic began in South America where not only were neurological deficits such as Guillain-Barré syndrome seen, but also spontaneous abortion and congenital malformations such as microcephaly in infants from infected mothers (91). By the end of the 2017 epidemic in Brazil, there were more than 200,000 notifications of ZIKV cases (97). Estimates for infants born with congenital Zika syndrome (CZS) after the 2015-2016 epidemic ranged from 5 to50 in every 10,000 births (98). The threat of a ZIKV epidemic lingers, with WHO reporting 61 countries affected by Aedes aegypti mosquitoes, therefore carrying the potential for ZIKV infection and transmission (99). ZIKV demonstrated continuing global epidemic capacity in India in 2018 (100).
ZIKV belongs to the flavivirus family alongside West Nile virus, Dengue virus, and Yellow fever virus. ZIKV is an enveloped and icosahedral virus with a nonsegmented, 10.7 kb single stranded positive sense RNA genomes (101). This virus is composed of several proteins, categorized as three structural (capsid, pre-membrane and envelope) and seven nonstructural proteins. The seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) are essential for viral replication and assembly, as well as being responsible for the pathogenicity of the virus by binding to transcription and restriction factors (95). The biggest risk of congenital ZIKV infection is for mothers infected during their first trimester (102). ZIKV infection demonstrates wide tissue tropism, with ZIKV successfully infecting the central nervous system, blood, retinal, genital and reproductive tissues including placenta (103–107). ZIKV was thought to be exclusively arthropod transmitted until cases of human-human transmission emerged in nonendemic regions, illustrating a role for sexual transmission (108–110). The presence of ZIKV RNA has also been found in breast milk of ZIKV infected mothers (111–113). However, there are reports which suggest that vertical transmission of ZIKV by breastmilk does not occur in most cases, which suggests the possibility that breastmilk does not have a high enough viral load to infect the newborn (114, 115).
Despite some knowledge regarding ZIKV pathogenesis, its mechanism of infection in placental immune cell types remains limited (116–119). Histopathology of ZIKV infected placentae has shown ZIKV infection in first trimester villous stromal tissue cells, which includes immune cells in the chorionic villi (117, 120, 121). Uniquely, ZIKV was also found to infect CTBs, endothelial cells, fibroblasts and HC in chorionic villi, as well as amniotic epithelial cells and trophoblast progenitor cells (103, 116–118, 122–124) (Figure 4).
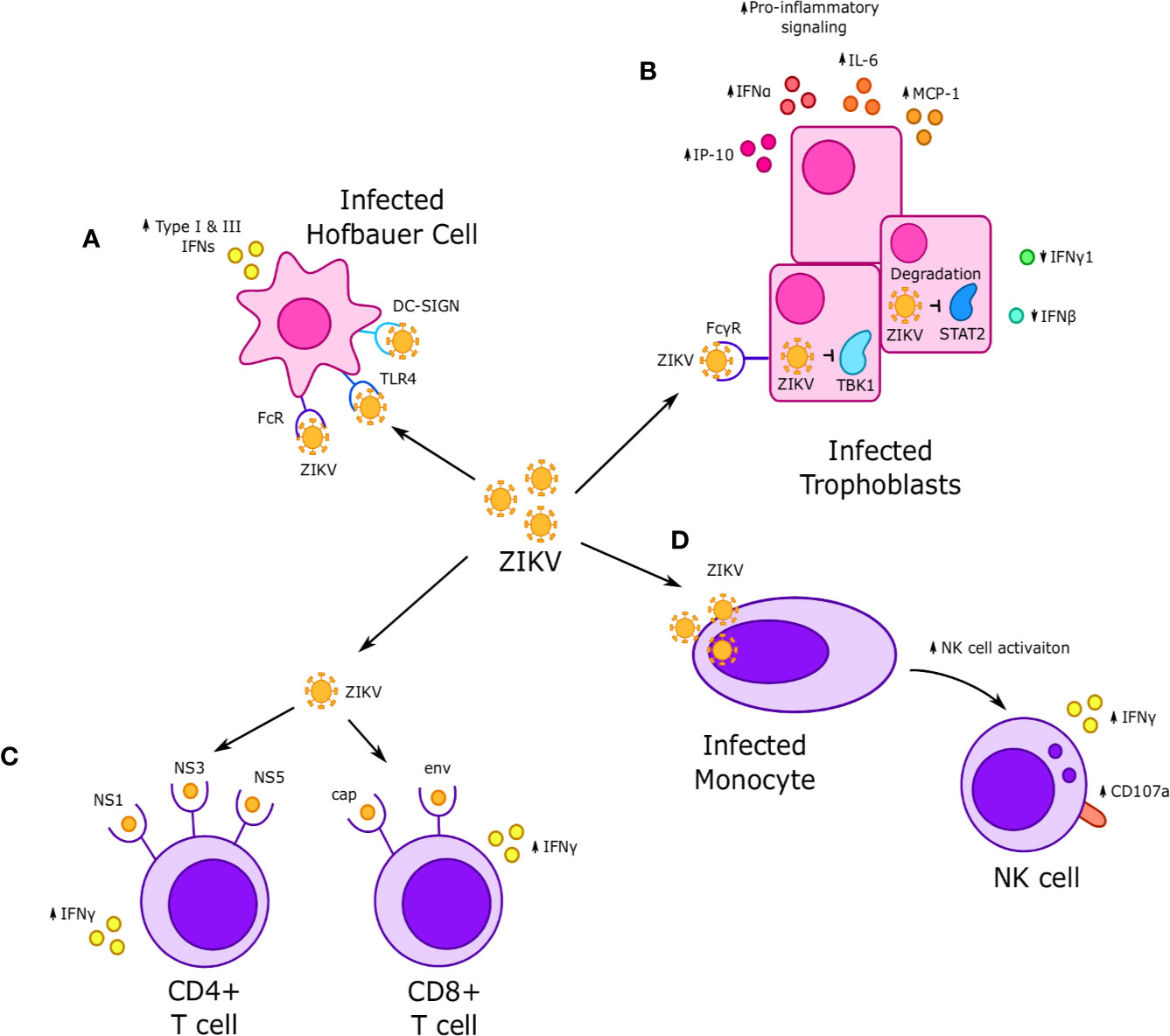
Figure 4 Interactions between Zika virus (ZIKV) and immune cells at the maternal-fetal interface. (A) ZIKV infects Hofbauer cells (HCs) and is thought to achieve replication through Fc, TLR4 and DC-SIGN receptors, which in turn increases secretion of type I and III interferons. (B) ZIKV infection of fetal trophoblasts and fibroblasts occurs through FcγR and results in increased expression of some interferons, such as IFNα, and decreased expression of others, such as IFN-γ and IFN-β. Infection is associated with increased secretion of proinflammatory cytokines such as IP-10, IL-6, and MCP-1. NS5 viral proteins are thought to downregulate interferon-stimulated genes (ISGs) and reduce interferon signaling via STAT2 degradation, while the viral proteins NS1 and NS4B inhibit IFN signaling by downregulating TBK1. (C) CD4+ T cells exhibit a strong response to nonstructural NS1, NS3, and NS5 ZIKV proteins, while CD8+ T cells respond to cap and env ZIKV proteins. In both cases, response to ZIKV proteins was characterized by increased IFN-γ production. (D) Systemic dNK cells exhibit increased activation, including increased IFNγ production and CD107a expression when incubated with ZIKV infected monocytes.
Similar to HCMV, ZIKV has also been shown to infect HCs and CTBs (90, 116, 117). During the first trimester of pregnancy, ZIKV infects HCs, entering the fetal blood stream in order to reside in the placenta. ZIKV uses HCs as a “Trojan Horse”. This strategy is utilized by several viruses in order to cross the blood brain barrier, where the virus infects leukocytes, leading to them being carried across barriers and thereby enabling the propagation and spread of infection (125–127). HCs have been associated with ZIKV spread to the fetus through the “Trojan Horse” route (91). The presence of ZIKV-specific antigen was demonstrated in HCs in confirmed maternal infection. Multiple studies suggest HCs are a crucial step in vertical transmission of ZIKV to fetal cells, demonstrating that HCs are preferentially infected when compared to CTBs (116, 122, 128). Infection of HCs with ZIKV is thought to propagate infection through hyperplasia and proliferation of these cells, leading to persistence of this HC population into later trimesters (116, 122, 128). A study performed on first trimester fetal and maternal tissue showed that ZIKV can replicate in different cell types, such as decidual fibroblasts and macrophages. It can also infect trophoblasts and HCs as well as umbilical cord mesenchymal stem cells, suggesting that the route of ZIKV infection may move from the decidua basalis to the anchoring villi (129). A study performed using blood from 30+ Asian ZIKV infected pregnant women shows that CD14+ monocytes are the primary target of ZIKV infection. These monocytes are resistant to change in M2 phenotype and downregulate type 1 IFN signaling, which induces the expression of different host genes involved in pregnancy complications (130).
In the decidua basalis, ZIKV infects EVTs, macrophages and stromal cells. ZIKV also targets proliferative CTBs in the anchoring villi, however is unlikely to infect STBs due to IFN-λ-mediated antiviral defense mechanisms (131). ZIKV achieves replication within macrophages through FcR, TLR4 and DC-SIGN receptors (116, 132). In vitro studies have demonstrated ZIKV infection to be augmented in HCs by IgG from prior flavivirus exposure through antibody dependent enhancement (ADE) (133). There remain many gaps in knowledge regarding the role for macrophages targeted and infected by ZIKV. A study performed using decidual and chorionic villous tissue from early and mid-gestation human pregnancy shows that ZIKV appears to elevate type I and III IFN expression, which does not occur in HCMV infection (131).
Studies looking at the interaction between ZIKV and T cells in humans are scarce although ZIKV infection has been demonstrated to activate both CD4 and CD8 T cells (134) with specific increases in Vδ2 TCR+ cells which have been implicated in recurrent miscarriages but not associated with ZIKV-induced fetal complications. There have not been notable studies looking specifically at T cell ZIKV communication at the human maternal-fetal interface (135). A recent study examined peripheral T cell responses of 45 confirmed cases of ZIKV infection that had been stimulated with pooled ZIKV peptides from all viral components (136). This study demonstrated responses from both CD4+ and CD8+ T cells to both structural and nonstructural ZIKV components. However, this study particularly showed that CD4+ T cells exhibited a strong response to nonstructural proteins NS1, NS3 and NS5, and CD8+ T cells a strong response to cap and env proteins. This response was demonstrated by marked IFN-γ production from both cell subtypes indicating cell activation (136).
Another case looking at a ZIKV infected individual from the United States demonstrated interactions between the ZIKV NS2 and env proteins with CD4+ and CD8+ T cells, respectively. (137). Furthermore, in a different study, CD4+ T cells of two ZIKV infected individuals showed activity in response to nonstructural proteins (NS1, NS3 and NS5). Consistently, CD8+ T cells were seen to raise activity against the structural protein env (138, 139). These studies demonstrate consistency in the response of CD4+ T cells and CD8+ T cells to ZIKV proteins, revealing CD4+ T cells to specifically respond to particular nonstructural proteins and CD8+ T cells to react to structural proteins, particularly cap and env (136, 138, 139).
Several studies have looked at the ability of DENV-specific CD4+ and CD8+ T cells to be stimulated by the presence of ZIKV peptides in humans (140, 141). These studies showed viral epitopes for specific peptides located in similar regions and structurally conserved across flaviviruses; however, they displayed differences in their sequences (141). Nonetheless, these studies indicated cross-reactivity between the viruses regarding their CD4+ and CD8+ T cell activity. One study demonstrated that CD8+ and CD4+ T cells from DENV positive donors reacted to ZIKV viral peptides, resulting in an upregulation of IFNγ secreting cells. This group also showed that stimulation with ZIKV peptides for those in acute phase of ZIKV infection resulted in recruitment of elevated levels of CD8+ IFN-γ+ T cells (142).
A recent transcriptomics study investigated transcriptional signatures in CD4/CD8 T cells, B, and NK cells and plasmacytoid dendritic cells in patients (nonpregnant) infected with ZIKV (143). Interestingly, they did not note significance transcriptional changes in NK or CD8 T cells in a ZIKV infected background but noted significance alterations in pDCs. Whether pregnancy plus ZIKV infection would affect the immune cell transcriptome in humans remains to be determined.
Studies specifically analyzing interactions between dNK cells and ZIKV in humans once again are lacking. However, studies have looked at ZIKV and its communication with peripheral NK cells. One such study postulated crosstalk between monocytes and NK cells in ZIKV infected patients. The activation of NK cells was associated with the presence of monocytes, which induced expression of IFN-γ and CD107a, key markers of NK cell function. Depletion of monocytes in the peripheral blood reduced the levels of these markers and thus the activation of NK cells (144). There are few studies showing the interaction between ZIKV with NK cells. Glasner et al., showed that ZIKV infection led to activation of MHC class I, which was somehow not sensed by dNK cells and their activating receptors, allowing the virus to escape NK cell-mediated killing. MHC class I expression is triggered through the IFN-β pathway via activation of RIGI-IRF3 (145). However, the mechanism by which NK cells may promote an immunosuppressive environment in the face of ZIKV infection is not clear. Some studies have indicated that interactions between other aspects of the innate immune system and NK cells may be at play in ZIKV pathogenesis.
There are several studies suggesting that pathogenesis of ZIKV is not mediated through decidual immune cells alone but rather conducted, at least in part, through the activation of interferon-stimulated genes (ISGs), which in turn leads to activation of innate host cell immunity (131). ISGs act to specifically target viral replication. Multiple studies have indicated ZIKV stimulation of interferons (IFN) to vary depending on the type of IFN. While type I and III IFNs have been shown to be inhibited by ZIKV, specifically the NS5 component of the pathogen, Type II IFNs have been shown to be upregulated by the virus (146, 147). One study demonstrated that when Type III IFNs were upregulated, specifically IFN-λ1, trophoblast cells were infected with ZIKV at a lower rate. Further, NS1, NS4A and NS4B have been demonstrated to inhibit IFN type I response. This leads to suppression of the TANK binding kinase 1 (TBK1)/IRF3 and JAK-STAT pathway, which in turn results in reduced activation of innate immune responses (148).
Interferon induced transmembrane protein 1 (IFITM1) and IFITM3 specifically are ISGs which act as restriction factors to inhibit ZIKV replication. The mechanism by which the inhibition and activation of innate immunity impacts the recruitment of innate immune cells to the site of infection is not clear. Little is known about the role of NK cells in human ZIKV infection. One study has noted interactions between TLR7, CD81 and IFITM1, postulating that the restriction of ZIKV is associated with inhibitory activity of IFITM1, potentially through activation of NK cells (149, 150). Another group looking at ISGs showed that viperin played a role in ZIKV pathogenesis, with data revealing that when viperin levels were high, ZIKV mRNA levels were low and vice versa (148). NS4 is seen to target directly the Akt-mTOR pathway, leading to reduced signaling from this pathway and subsequent activation of autophagy in host cells (151). ZIKV has been shown to co-opt the autophagy pathway for post-RNA replication capacity and survival (152, 153). Importantly, the NS2B-NS3 protease activity of ZIKV can be blocked by an inhibitor of autophagy, hydroxychloroquine (HCQ) (154). HCQ is an FDA approved drug considered safe to use during pregnancy and could serve as an effective treatment for preventing ZIKV congenital syndrome (124).
The relationship between ZIKV infected cells and attenuated IFN production has been extensively reported, leading to questions regarding the mechanism underlying this association. It has been proposed that ZIKV may infect cells through ADE of infection. Many cells express the Fcγ receptor, and it is thought that viral particles may complex with antibodies and thereby enter into cells via Fcγ receptors (133). Host cells (such as trophoblasts and fibroblasts) infected with ZIKV demonstrate innate immune system activation with a rise in specific IFNs (e.g. IFN-α), but falling levels of others such as IFN-λ1 and IFN-β (155). The elevated levels of proinflammatory cytokines and chemokines, namely IL-6, MCP-1 and IP-10 which are linked to recruitment of immune cells such as monocytes and T cells (155). ZIKV has been shown in multiple studies to downregulate type I IFN signaling and to be active in suppression of antiviral signaling. ZIKV nonstructural proteins NS1 and NS4B inhibit IFN signaling by downregulating levels of TBK1. However, NS2B3 downregulates the JAK-STAT pathway and inhibits apoptosis of ZIKV, and hence inhibits innate antiviral responses (150). One study specifically has implicated the role of the nonstructural ZIKV protein NS5 in promoting ZIKV propagation by targeting STAT2 for degradation, thereby reducing ISG levels (156). This is thought to promote viral replication through a dampened host innate immune cell response. There remains much to be elucidated in terms of ZIKV infection in human pregnancy. New studies are identifying metabolic reprogramming pathways underpinning innate immune responses to ZIKV which opens additional avenues of investigation (157). We refer readers to recent reviews highlighting ZIKV-immune interactions in adverse pregnancy outcomes (119) and ADE (158).
The limited availability of placental tissues during early pregnancy has always been a challenge for the reproductive biologists, hampering the study of placental physiology and cell to cell interactions. In vitro cell line models can often be biologically distinct and therefore unable to demonstrate enough similarity to replicate the conditions of human pregnancy. In addition, the use of cell line models can fail to reproduce the complexity of the number of cell types and cell interactions present within the decidua. Therefore, functional in vitro 3D models being are developed, for example placenta-on-a chip and organoid cultures, which can mimic in vivo conditions and would be useful to understand the mechanisms of viral host interactions.
The ‘Placenta-on-a-chip’ is a microfluidics model utilizing human trophoblast cells (BeWo) and fetal derived cells (HUVECs and HPVECs) (159, 160). These cell lines are cultured and separated by a semipermeable membrane within flow conditions with the purpose of understanding placental mechanisms and barrier function (159). Recent reports have described the faithfulness of placenta-on-a-chip model to in vivo placental conditions (161). For example, glucose transport using a placenta-on-a-chip model was demonstrated by Lee et al., and Blundell et al., highlighting significant similarity to in vivo glucose transport in the human placenta (159, 160). Placenta-on-a-chip models have also been used to investigate the transport of heparin and anti-hyperglycemic agents such as glyburide using BeWo and human placental villous endothelial cells (162). Recently, the transport of the xenobiotic compound caffeine across the placenta has been studied using this model system, providing new insights into the extent of caffeine transfer from mother to fetus (163).
Bacterial infections have also been studied using this model. Zhu et al., showed that in the presence of Escherichia coli (E. coli), trophoblast cells (BeWo) activated the circulating macrophages on the “maternal” side of the chip to secrete several inflammatory cytokines that mimicked in vivo conditions during pregnancy (164). The impact of common environmental exposures such as titanium dioxide nanoparticles (TiO2-NPs) has also been studied using this 3D placental model showing a series of different placental responses (barrier permeability, oxidative stress, cell apoptosis, and maternal immune cells behavior (165). They showed placental barrier permeability and maternal immune cells to be influenced by even low concentration of NPs (165). Therefore, this simple in vitro model can prove useful in understanding the environmental exposure of NPs during pregnancy and can help in a range of biological studies (165). Recent studies report generation of an organ-on-a-chip model, wherein decidualized human endometrial stromal cells and macrophage cell lines are co-cultured in a microfluidic device and shown to inhibit secretion of TNF-α in response to LPS stimulation (166). These devices have also been used to determine the impact of cytokine secretion by dNK cells on the migration of primary trophoblast cells. These studies illustrate the functionality of microfluidic organ on chip devices to elucidate importance of maternal immune cells in the placenta (167). Thus, the use of fetal membrane on organ-on-a-chip provides a suitable model to explore the impact of pathogenic infections during pregnancy (168, 169).
The use of in vitro trophoblast organoids as a 3D culture model also provides a new tool to understand the mechanism of implantation at the maternal-fetal interface. Recent studies have shown the characterization of these organoids derived from 1st-trimester CTBs (6 to 8 weeks) and suggest their resemblance to primary trophoblast cells (170–172). Due to similarity with the placental architecture, these organoids could be used to study physiological, metabolic and hormonal changes that occur during pregnancy.
The viruses we highlighted in this review, HCMV and ZIKV, do not naturally infect commonly used animal models [e.g., mice] which makes it challenging to understand disease pathogenesis. In particular, there remains a paucity of understanding ZIKV-immune cell interactions during pregnancy. Thus, the employment of placenta-on-a-chip or organ-on-a-chip, and organoid models will be pivotal in providing functional and physiologically relevant ways to study the interaction of immune cells at the maternal-fetal interface with viral pathogens that affect pregnancy.
Both HCMV and ZIKV can be sequestered into fetal macrophages. HCMV implicates HCs in the potential infection of other decidual cells, leading to the promotion of HCMV transcytosis in trophoblasts. ZIKV preferentially infects HCs, persisting in this cell population and potentially mediating infection of other fetal-derived cells. More poignant is the suggestion that decidual macrophages may mediate reactivation of HCMV by acting as a latent reservoir for infection. These studies collectively indicate a central role for macrophages in the pathogenesis of TORCH viruses.
dNK cells have been seen to alter their phenotype to express higher levels of various activating receptors when in the presence of infected decidual fibroblast cells. They also are known for their plasticity in the face of specific pathogens, acquiring more cytotoxic function. KIR2DL1/HLA-C2 has been identified as a mechanism by which dNK cells are activated and display cytoxicity toward HCMV infected cells. It has also been suggested that dNK cell activation may trigger activation of T-cells through upregulating HLA-DR expression on infected fibroblast cells.
ZIKV viral components demonstrate capacity to elicit strong responses from peripheral CD4+ and CD8+ T cells, with NS1, NS3, and NS5 being associated with CD4+ stimulation whereas Cap and Env proteins being associated with CD8+. We also see in this review the importance of Interferon stimulating genes in the restriction of ZIKV replication.
Thus, the implications and outcomes of viral interactions with immune cells at the maternal-fetal interface are varied. We see the importance of the host immune response and recognize the importance of studying mechanisms of pathogenesis in detail to enable targeted therapeutic interventions including vaccines to mitigate the adverse outcomes of viral infections during pregnancy (Figure 5). Finally, we posit that better understanding of the immunological underpinnings of infections at the maternal fetal interface can support the inclusion of pregnant women in trials testing vaccines and therapeutics to compact existing and emerging viral infections.
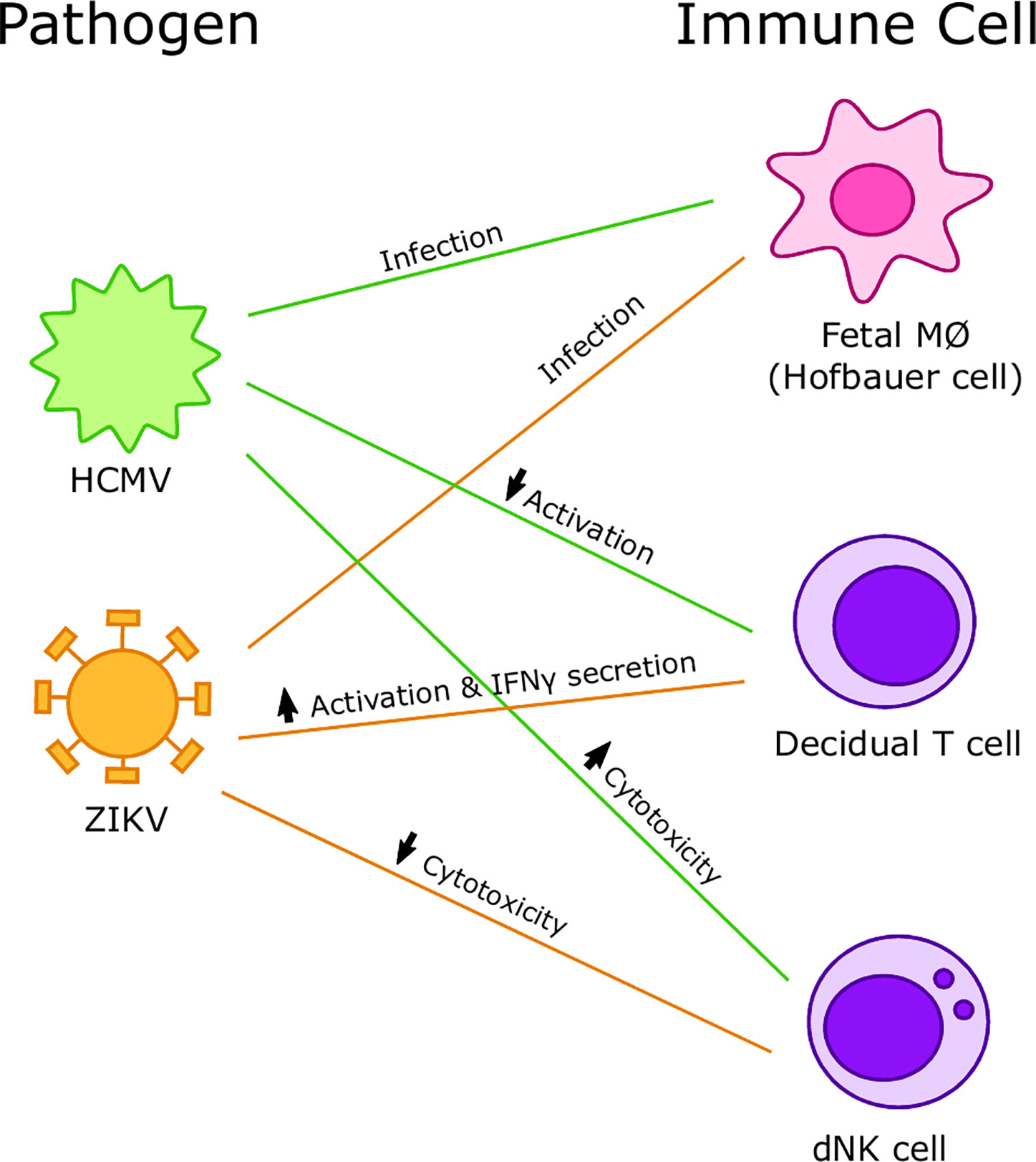
Figure 5 Summary of interactions between viruses and immune cells at the maternal-fetal interface. Human cytomegalovirus (HCMV) preferentially infects Hofbauer cells (HCs) and increases cytotoxicity of decidual Natural Killer (dNK) cells but decreases activation of decidual T cells. Zika virus (ZIKV) preferentially infects HCs and decreases cytotoxicity of dNK cells and increases activation and IFNγ secretion in decidual T cells.
EP, SV, and IM wrote the manuscript. RS generated the figures. All authors contributed to the article and approved the submitted version.
This work was funded in part by a grant from the National Institutes for Health/National Institute for Child Health and Development R01HD091218 to IM. RS was supported by a MARC uSTAR fellowship.
IM serves on the Scientific Advisory Board of Luca Biologics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Rowe JH, Ertelt JM, Xin L, Way SS. Regulatory T cells and the immune pathogenesis of prenatal infection. Reproduction (2013) 146:R191–203. doi: 10.1530/REP-13-0262
2. PrabhuDas M, Bonney E, Caron K, Dey S, Erlebacher A, Fazleabas A, et al. Immune mechanisms at the maternal-fetal interface: perspectives and challenges. Nat Immunol (2015) 16:328–34. doi: 10.1038/ni.3131
3. Burton GJ, Jauniaux E. What is the placenta? Am J Obstet Gynecol (2015) 213:S6 e1, S6–8. doi: 10.1016/j.ajog.2015.07.050
4. Norwitz ER. Defective implantation and placentation: laying the blueprint for pregnancy complications. Reprod BioMed Online (2007) 14 Spec No 1:101–9.
5. James JL, Stone PR, Chamley LW. The effects of oxygen concentration and gestational age on extravillous trophoblast outgrowth in a human first trimester villous explant model. Hum Reprod (2006) 21:2699–705. doi: 10.1016/S1472-6483(10)61464-2
6. Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature (2018) 563:347–53. doi: 10.1038/s41586-018-0698-6
7. Moffett A, Chazara O, Colucci F. Maternal allo-recognition of the fetus. Fertil Steril (2017) 107:1269–72. doi: 10.1016/j.fertnstert.2017.05.001
8. Mor G, Cardenas I, Abrahams V, Guller S. Inflammation and pregnancy: the role of the immune system at the implantation site. Ann N Y Acad Sci (2011) 1221:80–7. doi: 10.1111/j.1749-6632.2010.05938.x
9. Whitelaw PF, Croy BA. Granulated lymphocytes of pregnancy. Placenta (1996) 17:533–43. doi: 10.1016/S0143-4004(96)80070-1
10. Manaster I, Mandelboim O. The unique properties of uterine NK cells. Am J Reprod Immunol (2010) 63:434–44. doi: 10.1111/j.1600-0897.2009.00794.x
11. Williams PJ, Searle RF, Robson SC, Innes BA, Bulmer JN. Decidual leucocyte populations in early to late gestation normal human pregnancy. J Reprod Immunol (2009) 82:24–31. doi: 10.1016/j.jri.2009.08.001
12. Pereira L. Congenital Viral Infection: Traversing the Uterine-Placental Interface. Annu Rev Virol (2018) 5:273–99. doi: 10.1146/annurev-virology-092917-043236
13. Nahmias AJ WK, Stewart JA, Herrmann KL, Flynt WJ. The ToRCH complex-perinatal infections associated with toxoplasma and rubella, cytomegol- and herpes simplex viruses. Pediatr Res (1971) 5:405–6. doi: 10.1203/00006450-197108000-00144
14. Neu N, Duchon J, Zachariah P. TORCH infections. Clin Perinatol (2015) 42:77–103. viii. doi: 10.1016/j.clp.2014.11.001
15. Coyne CB, Lazear HM. Zika virus - reigniting the TORCH. Nat Rev Microbiol (2016) 14:707–15. doi: 10.1038/nrmicro.2016.125
16. Verma S, Carter EB, Mysorekar IU. SARS-CoV2 and pregnancy: An invisible enemy? Am J Reprod Immunol (2020) e13308. doi: 10.1111/aji.13308
17. Nancy P, Erlebacher A. T cell behavior at the maternal-fetal interface. Int J Dev Biol (2014) 58:189–98. doi: 10.1387/ijdb.140054ae
18. Tilburgs T, Scherjon SA, van der Mast BJ, Haasnoot GW, Voort-Maarschalk MV, Roelen DL, et al. Fetal-maternal HLA-C mismatch is associated with decidual T cell activation and induction of functional T regulatory cells. J Reprod Immunol (2009) 82:148–57. doi: 10.1016/j.jri.2009.05.003
19. Mjosberg J, Berg G, Jenmalm MC, Ernerudh J. FOXP3+ regulatory T cells and T helper 1, T helper 2, and T helper 17 cells in human early pregnancy decidua. Biol Reprod (2010) 82:698–705. doi: 10.1095/biolreprod.109.081208
20. Tilburgs T, van der Mast BJ, Nagtzaam NM, Roelen DL, Scherjon SA, Claas FH. Expression of NK cell receptors on decidual T cells in human pregnancy. J Reprod Immunol (2009) 80:22–32. doi: 10.1016/j.jri.2009.02.004
21. Tilburgs T, Schonkeren D, Eikmans M, Nagtzaam NM, Datema G, Swings GM, et al. Human decidual tissue contains differentiated CD8+ effector-memory T cells with unique properties. J Immunol (2010) 185:4470–7. doi: 10.4049/jimmunol.0903597
22. van Egmond A, van der Keur C, Swings GM, Scherjon SA, Claas FH. The possible role of virus-specific CD8(+) memory T cells in decidual tissue. J Reprod Immunol (2016) 113:1–8. doi: 10.1016/j.jri.2015.09.073
23. Hsu P, Santner-Nanan B, Joung S, Peek MJ, Nanan R. Expansion of CD4(+) HLA-G(+) T Cell in human pregnancy is impaired in pre-eclampsia. Am J Reprod Immunol (2014) 71:217–28. doi: 10.1111/aji.12195
24. Nagamatsu T, Schust DJ. The contribution of macrophages to normal and pathological pregnancies. Am J Reprod Immunol (2010) 63:460–71. doi: 10.1111/j.1600-0897.2010.00813.x
25. Manaster I, Mizrahi S, Goldman-Wohl D, Sela HY, Stern-Ginossar N, Lankry D, et al. Endometrial NK cells are special immature cells that await pregnancy. J Immunol (2008) 181:1869–76. doi: 10.4049/jimmunol.181.3.1869
26. Sayama S, Nagamatsu T, Schust DJ, Itaoka N, Ichikawa M, Kawana K, et al. Human decidual macrophages suppress IFN-gamma production by T cells through costimulatory B7-H1:PD-1 signaling in early pregnancy. J Reprod Immunol (2013) 100:109–17. doi: 10.1016/j.jri.2013.08.001
27. Jena MK, Nayak N, Chen K, Nayak NR. Role of Macrophages in Pregnancy and Related Complications. Arch Immunol Ther Exp (Warsz) (2019) 67:295–309. doi: 10.1007/s00005-019-00552-7
28. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5:953–64. doi: 10.1038/nri1733
29. Houser BL, Tilburgs T, Hill J, Nicotra ML, Strominger JL. Two unique human decidual macrophage populations. J Immunol (2011) 186:2633–42. doi: 10.4049/jimmunol.1003153
30. Svensson J, Jenmalm MC, Matussek A, Geffers R, Berg G, Ernerudh J. Macrophages at the fetal-maternal interface express markers of alternative activation and are induced by M-CSF and IL-10. J Immunol (2011) 187:3671–82. doi: 10.4049/jimmunol.1100130
31. Chabtini L, Mfarrej B, Mounayar M, Zhu B, Batal I, Dakle PJ, et al. TIM-3 regulates innate immune cells to induce fetomaternal tolerance. J Immunol (2013) 190:88–96. doi: 10.4049/jimmunol.1202176
32. Reyes L, Wolfe B, Golos T. Hofbauer Cells: Placental Macrophages of Fetal Origin. Results Probl Cell Differ (2017) 62:45–60. doi: 10.1007/978-3-319-54090-0_3
33. Goldstein J, Braverman M, Salafia C, Buckley P. The phenotype of human placental macrophages and its variation with gestational age. Am J Pathol (1988) 133:648–59.
34. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci (2008) 13:453–61. doi: 10.2741/2692
35. Jaiswal MK, Mallers TM, Larsen B, Kwak-Kim J, Chaouat G, Gilman-Sachs A, et al. V-ATPase upregulation during early pregnancy: a possible link to establishment of an inflammatory response during preimplantation period of pregnancy. Reproduction (2012) 143:713–25. doi: 10.1530/REP-12-0036
36. Moffett-King A. Natural killer cells and pregnancy. Nat Rev Immunol (2002) 2:656–63. doi: 10.1038/nri886
37. Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology (2009) 126:458–65. doi: 10.1111/j.1365-2567.2008.03027.x
38. Liu S, Diao L, Huang C, Li Y, Zeng Y, Kwak-Kim JY. The role of decidual immune cells on human pregnancy. J Reprod Immunol (2017) 124:44–53. doi: 10.1016/j.jri.2017.10.045
39. Jabrane-Ferrat N. Features of Human Decidual NK Cells in Healthy Pregnancy and During Viral Infection. Front Immunol (2019) 10:1397. doi: 10.3389/fimmu.2019.01397
40. Crespo AC, Strominger JL, Tilburgs T. Expression of KIR2DS1 by decidual natural killer cells increases their ability to control placental HCMV infection. Proc Natl Acad Sci U.S.A. (2016) 113:15072–7. doi: 10.1073/pnas.1617927114
41. Smith MG. Propagation in tissue cultures of a cytopathogenic virus from human salivary gland virus (SGV) disease. Proc Soc Exp Biol Med (1956) 92:424–30. doi: 10.3181/00379727-92-22498
42. Ho M. The history of cytomegalovirus and its diseases. Med Microbiol Immunol (2008) 197:65–73. doi: 10.1007/s00430-007-0066-x
43. Wilkinson GW, Davison AJ, Tomasec P, Fielding CA, Aicheler R, Murrell I, et al. Human cytomegalovirus: taking the strain. Med Microbiol Immunol (2015) 204:273–84. doi: 10.1007/s00430-015-0411-4
44. Griffiths P, Baraniak I, Reeves M. The pathogenesis of human cytomegalovirus. J Pathol (2015) 235:288–97. doi: 10.1002/path.4437
45. Nassetta L, Kimberlin D, Whitley R. Treatment of congenital cytomegalovirus infection: implications for future therapeutic strategies. J Antimicrob Chemother (2009) 63:862–7. doi: 10.1093/jac/dkp083
46. Enders G, Daiminger A, Bader U, Exler S, Enders M. Intrauterine transmission and clinical outcome of 248 pregnancies with primary cytomegalovirus infection in relation to gestational age. J Clin Virol (2011) 52:244–6. doi: 10.1016/j.jcv.2011.07.005
47. Bodeus M, Hubinont C, Goubau P. Increased risk of cytomegalovirus transmission in utero during late gestation. Obstet Gynecol (1999) 93:658–60. doi: 10.1016/S0029-7844(98)00538-9
48. Ehrnst A. HIV in pregnant women and their offspring: evidence for late transmission. Lancet (1991) 338:203–7. doi: 10.1016/0140-6736(91)90347-R
49. Bialas KM, Permar SR. The March towards a Vaccine for Congenital CMV: Rationale and Models. PloS Pathog (2016) 12:e1005355. doi: 10.1371/journal.ppat.1005355
50. Dollard SC, Keyserling H, Radford K, Amin MM, Stowell J, Winter J, et al. Cytomegalovirus viral and antibody correlates in young children. BMC Res Notes (2014) 7:776. doi: 10.1186/1756-0500-7-776
51. Rosenthal LS, Fowler KB, Boppana SB, Britt WJ, Pass RF, Schmid DS, et al. Cytomegalovirus shedding and delayed sensorineural hearing loss: results from longitudinal follow-up of children with congenital infection. Pediatr Infect Dis J (2009) 28:515–20. doi: 10.1097/INF.0b013e318198c724
52. Grosse SD, Ross DS, Dollard SC. Congenital cytomegalovirus (CMV) infection as a cause of permanent bilateral hearing loss: a quantitative assessment. J Clin Virol (2008) 41:57–62. doi: 10.1016/j.jcv.2007.09.004
53. Cannon MJ, Davis KF. Washing our hands of the congenital cytomegalovirus disease epidemic. BMC Public Health (2005) 5:70. doi: 10.1186/1471-2458-5-70
54. Fowler KB. Congenital cytomegalovirus infection: audiologic outcome. Clin Infect Dis (2013) 57 Suppl 4:S182–184. doi: 10.1093/cid/cit609
55. Morton CC, Nance WE. Newborn hearing screening–a silent revolution. N Engl J Med (2006) 354:2151–64. doi: 10.1056/NEJMra050700
56. Permar SR, Schleiss MR, Plotkin SA. Advancing Our Understanding of Protective Maternal Immunity as a Guide for Development of Vaccines To Reduce Congenital Cytomegalovirus Infections. J Virol (2018) 92. doi: 10.1128/JVI.00030-18
57. Stagno S, Pass RF, Cloud G, Britt WJ, Henderson RE, Walton PD, et al. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA (1986) 256:1904–8. doi: 10.1001/jama.256.14.1904
58. Hyde TB, Schmid DS, Cannon MJ. Cytomegalovirus seroconversion rates and risk factors: implications for congenital CMV. Rev Med Virol (2010) 20:311–26. doi: 10.1002/rmv.659
59. Cannon MJ, Westbrook K, Levis D, Schleiss MR, Thackeray R, Pass RF. Awareness of and behaviors related to child-to-mother transmission of cytomegalovirus. Prev Med (2012) 54:351–7. doi: 10.1016/j.ypmed.2012.03.009
60. Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol (2007) 17:253–76. doi: 10.1002/rmv.535
61. Staras SA, Flanders WD, Dollard SC, Pass RF, McGowan JE Jr, Cannon MJ. Cytomegalovirus seroprevalence and childhood sources of infection: A population-based study among pre-adolescents in the United States. J Clin Virol (2008) 43:266–71. doi: 10.1016/j.jcv.2008.07.012
62. Weisblum Y, Panet A, Haimov-Kochman R, Wolf DG. Models of vertical cytomegalovirus (CMV) transmission and pathogenesis. Semin Immunopathol (2014) 36:615–25. doi: 10.1007/s00281-014-0449-1
63. Fisher S, Genbacev O, Maidji E, Pereira L. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: implications for transmission and pathogenesis. J Virol (2000) 74:6808–20. doi: 10.1128/JVI.74.15.6808-6820.2000
64. Satosar A, Ramirez NC, Bartholomew D, Davis J, Nuovo GJ. Histologic correlates of viral and bacterial infection of the placenta associated with severe morbidity and mortality in the newborn. Hum Pathol (2004) 35:536–45. doi: 10.1016/j.humpath.2004.01.015
65. Weisblum Y, Panet A, Zakay-Rones Z, Haimov-Kochman R, Goldman-Wohl D, Ariel I, et al. Modeling of human cytomegalovirus maternal-fetal transmission in a novel decidual organ culture. J Virol (2011) 85:13204–13. doi: 10.1128/JVI.05749-11
66. Schwartz DA, Khan R, Stoll B. Characterization of the fetal inflammatory response to cytomegalovirus placentitis. An immunohistochemical study. Arch Pathol Lab Med (1992) 116:21–7.
67. Anderholm KM, Bierle CJ, Schleiss MR. Cytomegalovirus Vaccines: Current Status and Future Prospects. Drugs (2016) 76:1625–45. doi: 10.1007/s40265-016-0653-5
68. Maidji E, McDonagh S, Genbacev O, Tabata T, Pereira L. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor-mediated transcytosis. Am J Pathol (2006) 168:1210–26. doi: 10.2353/ajpath.2006.050482
69. Loenen WA, Bruggeman CA, Wiertz EJ. Immune evasion by human cytomegalovirus: lessons in immunology and cell biology. Semin Immunol (2001) 13:41–9. doi: 10.1006/smim.2001.0294
70. Bacsi A, Aranyosi J, Beck Z, Ebbesen P, Andirkó I, Szabó J, et al. Placental macrophage contact potentiates the complete replicative cycle of human cytomegalovirus in syncytiotrophoblast cells: role of interleukin-8 and transforming growth factor-beta1. J Interferon Cytokine Res (1999) 19:1153–60. doi: 10.1089/107999099313091
71. Hahn G, Jores R, Mocarski ES. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc Natl Acad Sci U.S.A. (1998) 95:3937–42. doi: 10.1073/pnas.95.7.3937
72. Soderberg-Naucler C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, Nelson JA. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J Virol (2001) 75:7543–54. doi: 10.1128/JVI.75.16.7543-7554.2001
73. Cook CH, Zhang Y, McGuinness BJ, Lahm MC, Sedmak DD, Ferguson RM. Intra-abdominal bacterial infection reactivates latent pulmonary cytomegalovirus in immunocompetent mice. J Infect Dis (2002) 185:1395–400. doi: 10.1086/340508
74. Tilburgs T, Strominger JL. CD8+ effector T cells at the fetal-maternal interface, balancing fetal tolerance and antiviral immunity. Am J Reprod Immunol (2013) 69:395–407. doi: 10.1111/aji.12094
75. Revello MG, Lilleri D, Zavattoni M, Furione M, Genini E, Comolli G, et al. Lymphoproliferative response in primary human cytomegalovirus (HCMV) infection is delayed in HCMV transmitter mothers. J Infect Dis (2006) 193:269–76. doi: 10.1086/498872
76. Lilleri D, Fornara C, Furione M, Zavattoni M, Revello MG, Gerna G. Development of human cytomegalovirus-specific T cell immunity during primary infection of pregnant women and its correlation with virus transmission to the fetus. J Infect Dis (2007) 195:1062–70. doi: 10.1086/512245
77. Fornara C, Lilleri D, Revello MG, Furione M, Zavattoni M, Lenta E, et al. Kinetics of effector functions and phenotype of virus-specific and gammadelta T lymphocytes in primary human cytomegalovirus infection during pregnancy. J Clin Immunol (2011) 31:1054–64. doi: 10.1007/s10875-011-9577-8
78. Lissauer D, Choudhary M, Pachnio A, Goodyear O, Moss PA, Kilby MD. Cytomegalovirus sero positivity dramatically alters the maternal CD8+ T cell repertoire and leads to the accumulation of highly differentiated memory cells during human pregnancy. Hum Reprod (2011) 26:3355–65. doi: 10.1093/humrep/der327
79. Le Bouteiller P, Siewiera J, Casart Y, Aguerre-Girr M, El Costa H, Berrebi A, et al. The human decidual NK-cell response to virus infection: what can we learn from circulating NK lymphocytes? J Reprod Immunol (2011) 88:170–5. doi: 10.1016/j.jri.2010.12.005
80. Siewiera J, El Costa H, Tabiasco J, Berrebi A, Cartron G, Bouteiller P, et al. Human cytomegalovirus infection elicits new decidual natural killer cell effector functions. PloS Pathog (2013) 9:e1003257. doi: 10.1371/journal.ppat.1003257
81. Crespo AC, van der Zwan A, Ramalho-Santos J, Strominger JL, Tilburgs T. Cytotoxic potential of decidual NK cells and CD8+ T cells awakened by infections. J Reprod Immunol (2017) 119:85–90. doi: 10.1016/j.jri.2016.08.001
82. Koopman LA, Kopcow HD, Rybalov B, Boyson JE, Orange JS, Schatz F, et al. Human decidual natural killer cells are a unique NK cell subset with immunomodulatory potential. J Exp Med (2003) 198:1201–12. doi: 10.1084/jem.20030305
83. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med (2006) 12:1065–74. doi: 10.1038/nm1452
84. Keskin DB, Allan DS, Rybalov B, Andzelm MM, Stern JN, Kopcow HD, et al. TGFbeta promotes conversion of CD16+ peripheral blood NK cells into CD16- NK cells with similarities to decidual NK cells. Proc Natl Acad Sci U.S.A. (2007) 104:3378–83. doi: 10.1073/pnas.0611098104
85. El Costa H, Casemayou A, Aguerre-Girr M, Rabot M, Berrebi A, Parant O, et al. Critical and differential roles of NKp46- and NKp30-activating receptors expressed by uterine NK cells in early pregnancy. J Immunol (2008) 181:3009–17. doi: 10.4049/jimmunol.181.5.3009
86. de Mendonca Vieira R, Meagher A, Crespo ÂC, Kshirsagar SK, Iyer V, et al. Human Term Pregnancy Decidual NK Cells Generate Distinct Cytotoxic Responses. J Immunol (2020) 204:3149–59. doi: 10.4049/jimmunol.1901435
87. Jackson SE, Mason GM, Wills MR. Human cytomegalovirus immunity and immune evasion. Virus Res (2011) 157:151–60. doi: 10.1016/j.virusres.2010.10.031
88. van de Weijer ML, Luteijn RD, Wiertz EJ. Viral immune evasion: Lessons in MHC class I antigen presentation. Semin Immunol (2015) 27:125–37. doi: 10.1016/j.smim.2015.03.010
89. Foley B, De Santis D, Lathbury L, Christiansen F, Witt C. KIR2DS1-mediated activation overrides NKG2A-mediated inhibition in HLA-C C2-negative individuals. Int Immunol (2008) 20:555–63. doi: 10.1093/intimm/dxn013
90. Lazear HM, Diamond MS. Zika Virus: New Clinical Syndromes and Its Emergence in the Western Hemisphere. J Virol (2016) 90:4864–75. doi: 10.1128/JVI.00252-16
91. Zanluca C, de Noronha L, Duarte Dos Santos CN. Maternal-fetal transmission of the zika virus: An intriguing interplay. Tissue Barriers (2018) 6:e1402143. doi: 10.1080/21688370.2017.1402143
92. Macnamara FN. Zika virus: a report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans R Soc Trop Med Hyg (1954) 48:139–45. doi: 10.1016/0035-9203(54)90006-1
93. Ngono AE, Shresta S. Immune Response to Dengue and Zika. Annu Rev Immunol (2018) 36:279–308. doi: 10.1146/annurev-immunol-042617-053142
94. Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med (2009) 360:2536–43. doi: 10.1056/NEJMoa0805715
95. Faria NR, da Silva Azevedo RD, Kraemer MU, Souza R, Cunha MS, Hill SC, et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science (2016) 352:345–9.
96. Cao-Lormeau VM, Roche C, Teissier A, Robin E, Berry AL, Mallet HP, et al. Zika virus, French polynesia, South pacific, 2013. Emerg Infect Dis (2014) 20:1085–6. doi: 10.3201/eid2006.140138
97. Borges ALV, Moreau C, Burke A, Dos Santos OA, Chofakian CB. Women’s reproductive health knowledge, attitudes and practices in relation to the Zika virus outbreak in northeast Brazil. PloS One (2018) 13:e0190024. doi: 10.1371/journal.pone.0190024
98. Lowe R, Barcellos C, Brasil P, Cruz OG, Honório NA, Kuper H, et al. The Zika Virus Epidemic in Brazil: From Discovery to Future Implications. Int J Environ Res Public Health (2018) 15. doi: 10.3390/ijerph15010096
100. Saxena SK, Kumar S, Sharma R, Maurya VK, Dandu HR, Bhatt ML. Zika virus disease in India - Update October 2018. Travel Med Infect Dis (2019) 27:121–2. doi: 10.1016/j.tmaid.2018.10.022
101. Lindenbach BD, Rice CM. Molecular biology of flaviviruses. Adv Virus Res (2003) 59:23–61. doi: 10.1016/S0065-3527(03)59002-9
102. Rather IA, Lone JB, Bajpai VK, Park YH. Zika Virus Infection during Pregnancy and Congenital Abnormalities. Front Microbiol (2017) 8:581. doi: 10.3389/fmicb.2017.00581
103. Miner JJ, Diamond MS. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe (2017) 21:134–42. doi: 10.1016/j.chom.2017.01.004
104. Murray KO, Gorchakov R, Carlson AR, Berry R, Lai L, Natrajan M, et al. Prolonged Detection of Zika Virus in Vaginal Secretions and Whole Blood. Emerg Infect Dis (2017) 23:99–101. doi: 10.3201/eid2301.161394
105. Sun J, Wu D, Zhong H, Guan D, Zhang H, Tan Q, et al. Presence of Zika Virus in Conjunctival Fluid. JAMA Ophthalmol (2016) 134:1330–2. doi: 10.1001/jamaophthalmol.2016.3417
106. Prisant N, Bujan L, Benichou H, Hayot PH, Pavili L, Lurel S, et al. Zika virus in the female genital tract. Lancet Infect Dis (2016) 16:1000–1. doi: 10.1016/S1473-3099(16)30193-1
107. Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell (2016) 18:587–90. doi: 10.1016/j.stem.2016.02.016
108. Foy BD, Kobylinski KC, Foy JL, Blitvich BJ, da Rosa AT, Haddow AD, et al. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg Infect Dis (2011) 17:880–2. doi: 10.3201/eid1705.101939
109. Song BH, Yun S II, Woolley M, Lee YM. Zika virus: History, epidemiology, transmission, and clinical presentation. J Neuroimmunol (2017) 308:50–64. doi: 10.1016/j.jneuroim.2017.03.001
110. Regla-Nava JA, Ngono AE, Viramontes KM, Huynh AT, Wang YT, Nguyen AV, et al. Cross-reactive Dengue virus-specific CD8(+) T cells protect against Zika virus during pregnancy. Nat Commun (2018) 9:3042. doi: 10.1038/s41467-018-05458-0
111. Cavalcanti MG, Cabral-Castro MJ, Gonçalves JL, Santana LS, Pimenta ES, Peralta JM. Zika virus shedding in human milk during lactation: an unlikely source of infection? Int J Infect Dis (2017) 57:70–2. doi: 10.1016/j.ijid.2017.01.042
112. Blohm GM, Lednicky JA, Márquez M, White SK, Loeb JC, Pacheco CA, et al. Evidence for Mother-to-Child Transmission of Zika Virus Through Breast Milk. Clin Infect Dis (2018) 66:1120–1. doi: 10.1093/cid/cix968
113. Dupont-Rouzeyrol M, Biron A, O’Connor O, Huguon E, Descloux E. Infectious Zika viral particles in breastmilk. Lancet (2016) 387:1051. doi: 10.1016/S0140-6736(16)00624-3
114. Mann TZ, Haddad LB, Williams TR, Hills SL, Read JS, Dee DL, et al. Breast milk transmission of flaviviruses in the context of Zika virus: A systematic review. Paediatr Perinat Epidemiol (2018) 32:358–68. doi: 10.1111/ppe.12478
115. Colt S, Garcia-Casal MN, Peña-Rosas JP, Finkelstein JL, Rayco-Solon P, Weise Prinzo ZC, et al. Transmission of Zika virus through breast milk and other breastfeeding-related bodily-fluids: A systematic review. PloS Negl Trop Dis (2017) 11:e0005528. doi: 10.1371/journal.pntd.0005528
116. Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O’Neal JT, et al. Zika Virus Infects Human Placental Macrophages. Cell Host Microbe (2016) 20:83–90. doi: 10.1016/j.chom.2016.05.015
117. Tabata T, Petitt M, Puerta-Guardo H, Michlmayr D, Wang C, Fang-Hoover J, et al. Zika Virus Targets Different Primary Human Placental Cells, Suggesting Two Routes for Vertical Transmission. Cell Host Microbe (2016) 20:155–66. doi: 10.1016/j.chom.2016.07.002
118. Simoni MK, Jurado KA, Abrahams VM, Fikrig E, Guller S. Zika virus infection of Hofbauer cells. Am J Reprod Immunol (2017) 77. doi: 10.1111/aji.12613
119. Teixeira FME, Pietrobon AJ, Oliveira LM, Oliveira L, Sato MN. Maternal-Fetal Interplay in Zika Virus Infection and Adverse Perinatal Outcomes. Front Immunol (2020) 11:175. doi: 10.3389/fimmu.2020.00175
120. Petitt M, Tabata T, Puerta-Guardo H, Harris E, Pereira L. Zika virus infection of first-trimester human placentas: utility of an explant model of replication to evaluate correlates of immune protection ex vivo. Curr Opin Virol (2017) 27:48–56. doi: 10.1016/j.coviro.2017.11.008
121. Miranda J, Martín-Tapia D, Valdespino-Vázquez Y, Alarcón L, Espejel-Nuñez A, Guzmán-Huerta M, et al. Syncytiotrophoblast of Placentae from Women with Zika Virus Infection Has Altered Tight Junction Protein Expression and Increased Paracellular Permeability. Cells (2019) 8. doi: 10.3390/cells8101174
122. Jurado KA, Simoni MK, Tang Z, Uraki R, Hwang J, Householder S, et al. Zika virus productively infects primary human placenta-specific macrophages. JCI Insight (2016) 1. doi: 10.1172/jci.insight.88461
123. Schwartz DA. Viral infection, proliferation, and hyperplasia of Hofbauer cells and absence of inflammation characterize the placental pathology of fetuses with congenital Zika virus infection. Arch Gynecol Obstet (2017) 295:1361–8. doi: 10.1007/s00404-017-4361-5
124. Liang B, Guida JP, Costa Do Nascimento ML, Mysorekar IU. Host and viral mechanisms of congenital Zika syndrome. Virulence (2019) 10:768–75. doi: 10.1080/21505594.2019.1656503
125. Laval K, Favoreel HW, Poelaert KC, Van Cleemput J, Nauwynck HJ. Equine Herpesvirus Type 1 Enhances Viral Replication in CD172a+ Monocytic Cells upon Adhesion to Endothelial Cells. J Virol (2015) 89:10912–23. doi: 10.1128/JVI.01589-15
126. Paul AM, Acharya D, Duty L, Thompson EA, Le L, Stokic DS, et al. Osteopontin facilitates West Nile virus neuroinvasion via neutrophil “Trojan horse” transport. Sci Rep (2017) 7:4722. doi: 10.1038/s41598-017-04839-7
127. Peluso R, Haase A, Stowring L, Edwards M, Ventura P. A Trojan Horse mechanism for the spread of visna virus in monocytes. Virology (1985) 147:231–6. doi: 10.1016/0042-6822(85)90246-6
128. Rosenberg AZ, Yu W, Hill DA, Reyes CA, Schwartz DA. Placental Pathology of Zika Virus: Viral Infection of the Placenta Induces Villous Stromal Macrophage (Hofbauer Cell) Proliferation and Hyperplasia. Arch Pathol Lab Med (2017) 141:43–8. doi: 10.5858/arpa.2016-0401-OA
129. El Costa H, Gouilly J, Mansuy JM, Chen Q, Levy C, Cartron G, et al. ZIKA virus reveals broad tissue and cell tropism during the first trimester of pregnancy. Sci Rep (2016) 6:35296. doi: 10.1038/srep35296
130. Foo SS, Chen W, Chan Y, Bowman JW, Chang LC, Choi Y, et al. Asian Zika virus strains target CD14(+) blood monocytes and induce M2-skewed immunosuppression during pregnancy. Nat Microbiol (2017) 2:1558–70. doi: 10.1038/s41564-017-0016-3
131. Weisblum Y, Oiknine-Djian E, Vorontsov OM, Haimov-Kochman R, Zakay-Rones Z, Meir K, et al. Zika Virus Infects Early- and Midgestation Human Maternal Decidual Tissues, Inducing Distinct Innate Tissue Responses in the Maternal-Fetal Interface. J Virol (2017) 91. doi: 10.1128/JVI.01905-16
132. Routhu NK, Byrareddy SN. Host-Virus Interaction of ZIKA Virus in Modulating Disease Pathogenesis. J Neuroimmune Pharmacol (2017) 12:219–32. doi: 10.1007/s11481-017-9736-7
133. Bardina SV, Bunduc P, Tripathi S, Duehr J, Frere JJ, Brown JA, et al. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science (2017) 356:175–80. doi: 10.1126/science.aal4365
134. Cimini E, Castilletti C, Sacchi A, Casetti R, Bordoni V, Romanelli A, et al. Human Zika infection induces a reduction of IFN-gamma producing CD4 T-cells and a parallel expansion of effector Vdelta2 T-cells. Sci Rep (2017) 7:6313. doi: 10.1038/s41598-017-06536-x
135. Pardy RD, Richer MJ. Protective to a T: The Role of T Cells during Zika Virus Infection. Cells (2019) 8. doi: 10.3390/cells8080820
136. El Sahly HM, Gorchakov R, Lai L, Natrajan MS, Patel SM, Atmar RL, et al. Clinical, Virologic, and Immunologic Characteristics of Zika Virus Infection in a Cohort of US Patients: Prolonged RNA Detection in Whole Blood. Open Forum Infect Dis (2019) 6:ofy352. doi: 10.1093/ofid/ofy352
137. Ricciardi MJ, Magnani DM, Grifoni A, Kwon YC, Gutman MJ, Grubaugh ND, et al. Ontogeny of the B- and T-cell response in a primary Zika virus infection of a dengue-naive individual during the 2016 outbreak in Miami, FL. PloS Negl Trop Dis (2017) 11:e0006000. doi: 10.1371/journal.pntd.0006000
138. Edupuganti S, Natrajan MS, Rouphael N, Lai L, Xu Y, Feldhammer M, et al. Biphasic Zika Illness With Rash and Joint Pain. Open Forum Infect Dis (2017) 4:ofx133. doi: 10.1093/ofid/ofx133
139. Waggoner JJ, Rouphael N, Xu Y, Natrajan M, Lai L, Patel SM, et al. Pericarditis Associated With Acute Zika Virus Infection in a Returning Traveler. Open Forum Infect Dis (2017) 4:ofx103. doi: 10.1093/ofid/ofx103
140. Lim MQ, Kumaran EA, Tan HC, Lye DC, Leo YS, Ooi EE, et al. Cross-Reactivity and Anti-viral Function of Dengue Capsid and NS3-Specific Memory T Cells Toward Zika Virus. Front Immunol (2018) 9:2225. doi: 10.3389/fimmu.2018.02225
141. Koblischke M, Stiasny K, Aberle SW, Malafa S, Tsouchnikas G, Schwaiger J, et al. Structural Influence on the Dominance of Virus-Specific CD4 T Cell Epitopes in Zika Virus Infection. Front Immunol (2018) 9:1196. doi: 10.3389/fimmu.2018.02083
142. Grifoni A, Pham J, Sidney J, O'rourke PH, Paul S, Peters B, et al. Prior Dengue Virus Exposure Shapes T Cell Immunity to Zika Virus in Humans. J Virol (2017) 91.
143. Sun X, Hua S, Gao C, Blackmer JE, Ouyang Z, Ard K, et al. Immune-profiling of ZIKV-infected patients identifies a distinct function of plasmacytoid dendritic cells for immune cross-regulation. Nat Commun (2020) 11:2421. doi: 10.1038/s41467-020-16217-5
144. Lum FM, Lee D, Chua TK, Tan JJ, Lee CY, Liu X, et al. Zika Virus Infection Preferentially Counterbalances Human Peripheral Monocyte and/or NK Cell Activity. mSphere (2018) 3. doi: 10.1128/mSphereDirect.00120-18
145. Glasner A, Oiknine-Djian E, Weisblum Y, Diab M, Panet A, Wolf DG, et al. Zika Virus Escapes NK Cell Detection by Upregulating Major Histocompatibility Complex Class I Molecules. J Virol (2017) 91. doi: 10.1128/JVI.00785-17
146. Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr., et al. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe (2016) 19:705–12. doi: 10.1016/j.chom.2016.03.008
147. Chaudhary V, Yuen KS, Chan JF, Chan CP, Wang PH, Cai JP, et al. Selective Activation of Type II Interferon Signaling by Zika Virus NS5 Protein. J Virol (2017) 91. doi: 10.1128/JVI.00163-17
148. Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, et al. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep (2016) 17:1766–75. doi: 10.15252/embr.201642627
149. Ganapathiraju MK, Karunakaran KB, Correa-Menendez J. Predicted protein interactions of IFITMs may shed light on mechanisms of Zika virus-induced microcephaly and host invasion. F1000Res (2016) 5:1919. doi: 10.12688/f1000research.9364.1
150. Wu Y, Liu Q, Zhou J, Xie W, Chen C, Wang Z, et al. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discovery (2017) 3:17006. doi: 10.1038/celldisc.2017.14
151. Liang Q, Luo Z, Zeng J, Chen W, Foo SS, Lee SA, et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell (2016) 19:663–71. doi: 10.1016/j.stem.2016.07.019
152. Cao B, Parnell LA, Diamond MS, Mysorekar IU. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J Exp Med (2017) 214:2303–13. doi: 10.1084/jem.20170957
153. Abernathy E, Mateo R, Majzoub K, van Buuren N, Bird SW, Carette JE, et al. Differential and convergent utilization of autophagy components by positive-strand RNA viruses. PloS Biol (2019) 17:e2006926. doi: 10.1371/journal.pbio.2006926
154. Kumar A, Liang B, Aarthy M, Singh SK, Garg N, Mysorekar IU, et al. Hydroxychloroquine Inhibits Zika Virus NS2B-NS3 Protease. ACS Omega (2018) 3:18132–41. doi: 10.1021/acsomega.8b01002
155. Johnson EL, Chakraborty R. HIV-1 at the placenta: immune correlates of protection and infection. Curr Opin Infect Dis (2016) 29:248–55. doi: 10.1097/QCO.0000000000000267
156. Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe (2016) 19:882–90. doi: 10.1016/j.chom.2016.05.009
157. Chen Q, Gouilly J, Ferrat YJ, Espino A, Glaziou Q, Cartron G, et al. Metabolic reprogramming by Zika virus provokes inflammation in human placenta. Nat Commun (2020) 11:2967. doi: 10.1038/s41467-020-16754-z
158. Zimmerman MG, Wrammert J, Suthar MS. Cross-Reactive Antibodies during Zika Virus Infection: Protection, Pathogenesis, and Placental Seeding. Cell Host Microbe (2020) 27:14–24. doi: 10.1016/j.chom.2019.12.003
159. Lee JS, Romero R, Han YM, Kim HC, Kim CJ, Hong JS, et al. Placenta-on-a-chip: a novel platform to study the biology of the human placenta. J Matern Fetal Neonatal Med (2016) 29:1046–54. doi: 10.3109/14767058.2015.1038518
160. Blundell C, Tess ER, Schanzer AS, Coutifaris C, Su EJ, Parry S, et al. A microphysiological model of the human placental barrier. Lab Chip (2016) 16:3065–73. doi: 10.1039/C6LC00259E
161. Pemathilaka RL, Reynolds DE, Hashemi NN. Drug transport across the human placenta: review of placenta-on-a-chip and previous approaches. Interface Focus (2019) 9:20190031. doi: 10.1098/rsfs.2019.0031
162. Blundell C, Yi YS, Ma L, Tess ER, Farrell MJ, Georgescu A, et al. Placental Drug Transport-on-a-Chip: A Microengineered In Vitro Model of Transporter-Mediated Drug Efflux in the Human Placental Barrier. Adv Healthc Mater (2018) 7. doi: 10.1002/adhm.201700786
163. Pemathilaka RL, Caplin JD, Aykar SS, Montazami R, Hashemi NN. Placenta-on-a-Chip: In Vitro Study of Caffeine Transport across Placental Barrier Using Liquid Chromatography Mass Spectrometry. Glob Chall (2019) 3:1800112. doi: 10.1002/gch2.201800112
164. Yin F, Zhu Y, Wang H, Wang L, Yuan J, Qin J. Placental Barrier-on-a-Chip: Modeling Placental Inflammatory Responses to Bacterial Infection. (2018) 9.
165. Yin F, Zhu Y, Zhang M, Yu H, Chen W, Qin J. A 3D human placenta-on-a-chip model to probe nanoparticle exposure at the placental barrier. Toxicol Vitro (2019) 54:105–13. doi: 10.1016/j.tiv.2018.08.014
166. Rogers LM, Anders AP, Doster RS, Gill EA, Gnecco JS, Holley JM, et al. Decidual stromal cell-derived PGE2 regulates macrophage responses to microbial threat. Am J Reprod Immunol (2018) 80:e13032. doi: 10.1111/aji.13032
167. Abbas Y, Oefner CM, Polacheck WJ, Gardner L, Farrell L, Sharkey A, et al. A microfluidics assay to study invasion of human placental trophoblast cells. J R Soc Interface (2017) 14. doi: 10.1098/rsif.2017.0131
168. Gnecco JS, Anders AP, Cliffel D, Pensabene V, Rogers LM, Osteen K, et al. Instrumenting a Fetal Membrane on a Chip as Emerging Technology for Preterm Birth Research. Curr Pharm Des (2017) 23:6115–24. doi: 10.2174/1381612823666170825142649
169. Richardson L, Jeong S, Kim S, Han A, Menon R. Amnion membrane organ-on-chip: an innovative approach to study cellular interactions. FASEB J (2019) 33:8945–60. doi: 10.1096/fj.201900020RR
170. Haider S, Meinhardt G, Saleh L, Kunihs V, Gamperl M, Kaindl U, et al. Self-Renewing Trophoblast Organoids Recapitulate the Developmental Program of the Early Human Placenta. Stem Cell Rep (2018) 11:537–51. doi: 10.1016/j.stemcr.2018.07.004
171. Turco MY, Gardner L, Kay RG, Hamilton RS, Prater M, Hollinshead MS, et al. Trophoblast organoids as a model for maternal-fetal interactions during human placentation. Nature (2018) 564:263–7. doi: 10.1038/s41586-018-0753-3
Keywords: NK cells, T cells, macrophages, Hofbauer cell, human cytomegalovirus, decidua, pregnancy, placenta
Citation: Parker EL, Silverstein RB, Verma S and Mysorekar IU (2020) Viral-Immune Cell Interactions at the Maternal-Fetal Interface in Human Pregnancy. Front. Immunol. 11:522047. doi: 10.3389/fimmu.2020.522047
Received: 20 December 2019; Accepted: 14 September 2020;
Published: 07 October 2020.
Edited by:
David Aronoff, Vanderbilt University, United StatesReviewed by:
Tamara Tilburgs, Cincinnati Children’s Hospital Medical Center, United StatesCopyright © 2020 Parker, Silverstein, Verma and Mysorekar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Indira U. Mysorekar, aW15c29yZWthckB3dXN0bC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.