
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 28 August 2020
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.02109
This article is part of the Research TopicAtypical Functions of Leukocyte Chemoattractant ReceptorsView all 8 articles
 Marco E. Bianchi1,2*
Marco E. Bianchi1,2* Rosanna Mezzapelle1,2*
Rosanna Mezzapelle1,2*The CXCR4 receptor upon binding its ligands triggers multiple signaling pathways that orchestrate cell migration, hematopoiesis and cell homing, and retention in the bone marrow. However, CXCR4 also directly controls cell proliferation of non-hematopoietic cells. This review focuses on recent reports pointing to its pivotal role in tissue regeneration and stem cell activation, and discusses the connection to the known role of CXCR4 in promoting tumor growth. The mechanisms may be similar in all cases, since regeneration often recapitulates developmental processes, and cancer often exploits developmental pathways. Moreover, cell migration and cell proliferation appear to be downstream of the same signaling pathways. A deeper understanding of the complex signaling originating from CXCR4 is needed to exploit the opportunities to repair damaged organs safely and effectively.
The binding of chemokines to G protein-coupled receptors (GPCRs) typically directs cell movement and traffic in and out of specific tissues in developing embryos and adult animals. They are also involved in tumor metastasis and invasion, and in the extension of neurites and axons of neurons (a part of a cell moves, while the cell body stays put). How chemokines and their receptors recruit hematopoietic cells to injured sites and tumors has been intensely investigated, whereas their involvement in the control of cell proliferation is less explored (1). Among chemokine receptors, CXCR4 is the most widely expressed, and is involved in numerous physiological and pathological conditions. CXCR4 is expressed by most cells, including hematopoietic and endothelial cells (ECs), neurons and stem cells (embryonic and adult). Increased levels of CXCR4 are present in cancer cells compared to the normal cells (2, 3). The focus of this mini-review is the emerging role of CXCR4 and its ligands in tissue repair and regeneration, and its relation to cancer cell proliferation. The role of CXCR4 in differentiation, retention, mobilization, migration, and polarization of hematopoietic cells is covered by other excellent reviews (4, 5).
CXCR4 is a 352 amino acid rhodopsin-like GPCR, comprising an extracellular N-terminal domain, 7 transmembrane (TM) helices, 3 extra-cellular loops (ECL), 3 intra-cellular loops (ICL) and an intracellular C-terminal domain (6). CXCR4 can exist in the plasma membrane as a monomer, dimer, higher-order oligomer or nanoclusters (7), although the partitioning and relevance of these different multimerization states has not been addressed in vivo. Several crystal structures of CXCR4 bound to agonists and small molecules are in accordance with the ability of CXCR4 to form homodimers via interactions of the TM5 and TM6 helices (6). TM6 is also implicated in nanoclustering (7). CXCR4 can also form heterodimers with ACKR3 (a related GCPR also known as CXCR7), which have distinctive signaling properties (8).
The canonical ligand of CXCR4 is CXCL12, also known as stromal cell-derived factor 1 (SDF-1) (9, 10). A single gene, CXCL12, codes for six protein isoforms in human (three in mouse), all deriving from alternative splicing of the fourth and final exon. The various forms are differentially expressed and have different affinities to glycosaminoglycans present on the cell surface and in the extracellular matrix (11). CXCL12α, an 89 amino acid protein, is the shorter and most expressed isoform (12, 13). Notably, CXCL12α can exist in monomeric and dimeric forms. CXCL12 only binds to chemokine receptors CXCR4 and ACKR3, itself a CXCR4 interactor; such a restricted receptor selectivity is unusual among chemokines.
The structure of the CXCR4/CXCL12 complex has not yet been determined; a model integrating homology modeling, experimentally derived restraints, and charge swap mutagenesis (14) highlights several contacts between the N-terminal tail of CXCR4 and CXCL12, and the interaction of the N-terminus of CXCL12 with the cavity delimited by the TM helices.
High mobility group box 1 protein (HMGB1) is the archetypal damage-associated molecular pattern (DAMP) molecule; DAMPs are released from dead or severely stressed cells to alert their microenvironment and the innate immune system. HMGB1 can form a heterocomplex with CXCL12 (HMGB1⋅CXL12) that also binds to CXCR4; of note, the conformational rearrangements of CXCR4 differ when triggered by CXCL12 alone or by HMGB1⋅CXCL12, and the complex is over one order of magnitude more potent than CXCL12 alone in inducing cell migration (15). Only the reduced form of HMGB1, where the pair of cysteines in the HMG-box domain A do not form a disulfide bond, binds CXCL12 and interacts with CXCR4 (16). However, a designer form of HMGB1 called 3S-HMGB1, where serines replace all three cysteines, binds to CXCR4 directly and is as effective as HMGB1⋅CXCL12 in promoting cell migration and muscle regeneration (17).
CXCR4 also binds macrophage migration inhibitory factor (MIF), a cytokine involved in the regulation of innate immunity (18). MIF binds to the N-terminal tail of CXCR4 and to the exterior side of TM helices, but not inside the TM pocket (18, 19). MIF also binds to other receptors, including CXCR2, CD74/CD44, and ACKR3 (20), which complicates the dissection of its activities.
Extracellular ubiquitin (eUb), also considered a DAMP, is a CXCL12 antagonist (21). Molecular modeling and mutagenesis suggest that it binds to CXCR4 inside the cavity delimited by TMs (22), but makes contact to CXCR4 residues that are not contributing to CXCL12 binding (23).
Beta-defensin-3 (HBD3) also competes with CXCL12 for CXCR4 binding, and promotes internalization of CXCR4 without inducing calcium flux, ERK phosphorylation, or chemotaxis (24).
Although the above list of actors is long, and multimeric complexes and multiple interactions increase complexity, genetics originated the widespread idea of CXCR4 and CXCL12 as a biunivocal couple: deletion in mice of either the Cxcr4 or Cxcl12 genes causes fetal lethality, defective B-cell lymphopoiesis, impaired bone-marrow myelopoiesis, and abnormal development of the cardiac septum and of the cerebellum (25, 26).
Mice lacking CXCL12 or CXCR4 were first generated in the 1990s; since both die in utero, their ability to regenerate injured tissues was not investigated until later. Depletion of either CXCR4 or CXCL12 with small interfering RNAs injected in injured muscle impairs its regeneration, as does local injection of the CXCR4 antagonist AMD3100 (27), consistent with the expression of both CXCR4 and CXCL12 in skeletal muscle (28), and with impaired myogenesis and depletion of satellite cells in CXCR4 deficient mice (29). Satellite cells are the direct targets of CXCL12 (27).
More recently, CXCR4 and CXCL12 have been shown to control the regeneration of multiple organs and tissues, including lung, heart, liver, and the nervous system.
Surgical removal of one lung or part of it (pneumonectomy, PNX) is compensated by alveolar regrowth/regeneration in the remaining lung. After PNX, activated platelets trigger lung regeneration by binding to pulmonary capillary endothelial cells (PCECs) and supplying CXCL12 to activate CXCR4 and ACKR3 on their surface (30). PCECs activate AKT, proliferate and express the membrane metalloproteinase MMP14, which releases ligands that promote the proliferation of progenitor type II alveolar epithelial cells, and eventually alveolar regrowth. Endothelial cells are direct targets of CXCL12 via CXCR4, since genetic silencing of Cxcr4 and Ackr3 in PCECs impairs lung regeneration.
The mammalian heart cannot regenerate in adults, but it can in neonate mice (31). In myocardial infarction (MI), coronary arteries get obstructed, and must regenerate to support continued heart function. A unique CXCR4/CXCL12-dependent process termed “artery reassembly” allows the formation of an alternative (collateral) artery network to bypass obstructed or severed coronary arteries (32). In the mouse, within a few days after ligation of the left coronary artery on day 2 after birth, individual arterial endothelial cells (ECs) migrate out of the existing arteries, proliferate and then coalesce with capillaries, forming collateral arteries that connect branches of the right and left coronary arteries. A similar process reconnects severed arteries after the resection of the apex of the neonatal heart. Artery reassembly does not occur in adult hearts, but injection of a single dose of CXCL12 in the infarcted area promotes collateral formation and functional recovery of the heart. Notably, deletion of Cxcl12 capillary ECs or Cxcr4 in arterial ECs impairs artery reassembly; CXCL12 is not basally expressed in ECs, but hypoxia induces its expression. Thus, during artery reassembly different ECs are both source and target of CXCL12, via CXCR4.
Adult zebrafish hearts do regenerate, and coronary revascularization initiates within hours of injury. After cryoinjury, new coronaries regenerate both superficially around the injured area and intra-ventricularly toward the cardiac lumen, and act as a scaffold for proliferating cardiomyocytes (33). Epicardial cells express Cxcl12b after injury, as a consequence of hypoxia and HIF-1α activation. ECs in both superficial and intra-ventricular coronaries have a common origin and both express CXCR4, but inhibiting CXCR4 pharmacologically or deleting Cxcr4 in the whole heart limits superficial, and not intra-ventricular, regeneration.
The liver is capable of continuous turnover and regeneration, which is overridden by fibrosis, cirrhosis and hepatic failure only after chronic or overwhelming injury. CXCL12 is constitutively expressed in healthy liver, and its expression increases following acute or chronic injury. Liver sinusoidal endothelial cells (LSEC) and hepatic stellate cells (HSC) are important sources of CXCL12 in liver disease. HSC and mesenchymal stem cells mainly respond via CXCR4, while LSEC express both CXCR4 and ACKR3. CXCL12 can activate HSC and recruit bone marrow mesenchymal cells, which promote liver fibrosis; in LSEC, CXCL12 signals via the physical association of CXCR4 and ACKR3 to activate eventually the transcription factor Id1, which orchestrates pro-regenerative responses, such as production of Wnt2 and hepatocyte growth factor (HGF) (34). Liver regeneration is abrogated by genetic silencing of either ACKR3 or CXCR4 in LSEC, or by chronic injuries that lead to excessive CXCR4 and reduced ACKR3 expression. In vitro, CXCL12 induces dose-dependent proliferation of human liver-derived stellate LX-2 cells, mediated by PI3K/Akt and Erk1/2 pathways (35).
The peripheral nervous system has retained throughout evolution the capability to regenerate. Recently, CXCL12 was found to promote the structural and functional recovery of the neuromuscular junction after degeneration of the motor axon terminal (36). CXCL12 is synthetized and released by peri-synaptic Schwann cells, and acts on CXCR4 re-expressed upon injury on the tip of the motor axon. CXCL12 also supports the functional and anatomical recovery of the sciatic nerve after crush injury; of special note, the small molecule NUCC-390, a CXCR4 agonist (37), also promotes nerve regeneration (38).
The central nervous system, in contrast, has a limited ability to regenerate, mostly dependent on neural progenitor cells (NPCs). Astrocytes are the main source of CXCL12 in the brain (39); CXCR4 is expressed on NPCs and CXCL12 appears to stimulate directly their in vitro proliferation and differentiation into neurons (40–42), via PI3K-Erk1/2 (43) and/or AKT/FOXO3α (44) activation. However, Li at al. (45) found no CXCL12-induced proliferation of NPC cells from E12 mouse embryos. CXCR4 activation by CXCL12 promotes the differentiation of human embryonic stem cells into neural stem cells (46) and then helps to maintain their stemness (47).
Overall, these studies implicate CXCR4 and CXCL12 in the regeneration of multiple organs, via CXCL12 release from various sources and CXCR4 activation on endothelial and progenitor cells, which then go on to proliferate; so far, a role of CXCR4 activation on parenchymal cells is not convincingly proven nor excluded. Hematopoietic and mesenchymal cells also contribute to tissue regeneration, but in this case the role played by the CXCL12/CXCR4 system appears limited to directing their chemotaxis to the damaged site.
The existence of the HMGB1⋅CXCL12 complex was first inferred from the ability of HMGB1 to promote the migration of endothelial, hematopoietic and mesenchymal cells (15) via CXCR4; the complex was then biochemically characterized (48). The complex was also found to promote the regeneration of skeletal muscle, since the reduced HMGB1 expression in Hmgb1+/− mice delays muscle regeneration (49), whereas the injection of exogenous reduced HMGB1 accelerates muscle, bone and liver repair in mouse (17, 50). Several cell-specific responses are involved, including the proliferation of satellite cells, skeletal stem cells and hepatocytes. The requirement for HMGB1, as opposed to CXCL12 alone, is supported by several observations: injection of CXCL12 alone promotes abnormal bone regeneration, with a larger fracture callus without a concomitant increase in bone mineral density and mechanical strength (50); local injection of glycyrrhizin, a HMGB1 inhibitor (51), delays bone fracture healing; injection of 3s-HMGB1, a mutant form of HMGB1 that can bind to CXCR4 in the absence of CXCL12, mimics the biological effects of HMGB1⋅CXCL12, including the promotion of in vitro myogenesis (17).
Remarkably, systemic injection of fully reduced HMGB1 (frHMGB1) or 3S-HMGB1 predisposes muscle and bone to regeneration/repair even if injected 2 weeks before injury (50), by inducing the transitioning of resting stem cells to a dynamic state of the cell cycle, intermediate between G0 and G1, termed “GAlert” (52). In contrast to deeply quiescent G0 stem cells, GAlert stem cells are more metabolically active, contain higher levels of ATP and mitochondrial DNA, are larger and poised to enter the cell cycle when exposed to activating signals. Activation mTORC1 is both necessary and sufficient for the transitioning to the GAlert state (53), and rapamycin, an mTORC inhibitor, interferes with HMGB1-induced transitioning to GAlert (54). Multiple stem cell types (SSCs, satellite cells and hematopoietic stem cells) in mice subject to bone fracturing or muscle damage transition to the GAlert state, and this requires HMGB1⋅CXCL12, since stem cells in HMGB1-deficient mice do not transition to the GAlert state after injury unless exogenous HMGB1 is provided.
Thus, HMGB1⋅CXCL12 has similar activities to those reported for CXCL12 in muscle regeneration, but is absolutely required in GAlert transitioning of stem cells. In this context, two questions arise: is HMGB1⋅CXCL12 (as opposed to CXCL12 alone) responsible for the regeneration of most or all tissues? Does HMGB1⋅CXCL12 also promote the proliferation of ECs? Indeed, HMGB1 has been shown to promote the proliferation of ECs of different origin, although the involvement of CXCR4 as the cognate receptor was not investigated (55).
Tumor is an illegitimate tissue that grows out of control because of an altered expression and behavior of pro-proliferative and pro-survival signals. Precisely because tumor tissue is out of balance with the surrounding legitimate tissues, it is also in a state of distress, similar to an injured tissue, and recruits inflammatory cells that support it. Famously, it has been said that a tumor is wound that never heals (56).
Chemokines and their receptors not only drive the trafficking of leukocytes inside the tumor mass but also contribute to most aspects of tumor cell biology (1). High expression of CXCR4 is observed in hematological malignancies (57–59) and in many types of solid tumors, including melanomas and kidney, lung, brain, prostate, breast, pancreas and ovarian tumors (2, 3), where it correlates with poor prognosis (59). Interestingly, the normal tissue adjacent to the CXCR4 overexpressing tumor shows normal or no CXCR4 expression (41), which suggests a differential response of cancer cells to microenvironmental conditions. Expression of CXCR4 and CXCL12 in cancer cells is also controlled by specific microRNAs: CXCL12 by miR-1 (60), miR-9 (61, 62), miR-126 (63), miR-146a (64), and miR-150 (65), whereas miR-200a can increase CXCR4 expression (66).
The expression of CXCR4/CXCL12 in tumors is partially dependent on the hypoxic tumor microenvironment, in a HIF-1α dependent manner (42). As a consequence of CXCL12 release, tumor-associated CXCR4-expressing ECs proliferate (67). CXCR4 is also expressed on putative cancer stem cells populations in various tumors, including renal (68), prostate (69) and non-small lung cancer (70), and affects their clonogenicity and spherogenicity, with adverse effects on prognosis. These CXCL12/CXCR4 effects are similar to the promotion of endothelial and stem cell proliferation in injured tissue.
Moreover, many reports indicate that binding of CXCL12 to CXCR4 on tumor cells of various types enhances their proliferation, both in vitro and in vivo, either via MAPK or PI3K/Akt pathways (54, 71, 72).
Table 1 lists a sample of reports on the role of CXCR4/CXCL12 in tumor cell proliferation (mostly tumor cell lines) (53, 69, 72–80). Targeting of CXCR4 with antibodies or specific inhibitors, most commonly AMD3100, has been intensely investigated; however, AMD3100/Plerixafor/Mozobil has been approved for bone marrow transplantation, but not as anti-cancer treatment.
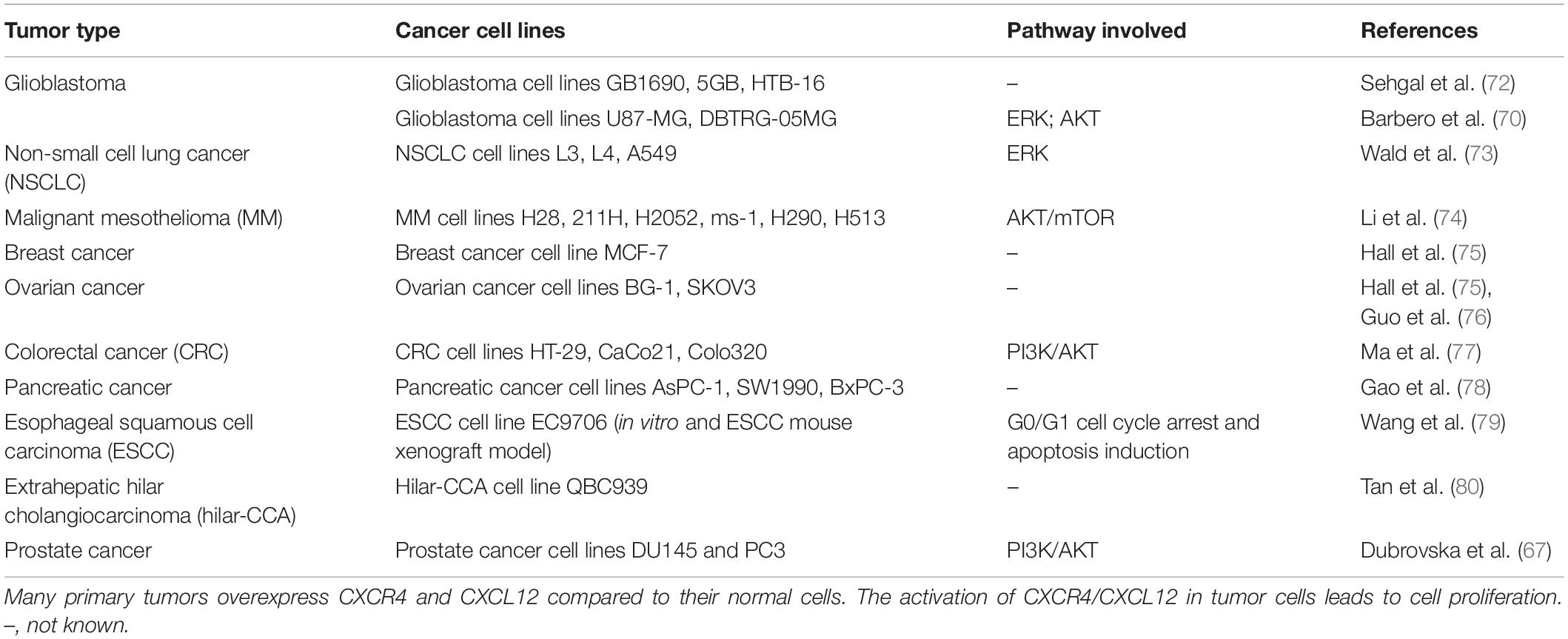
Table 1. CXCR4/CXCL12 axis is involved in cancer cell proliferation.
The preceding sections have highlighted that CXCR4 activation can drive both cell migration and cell proliferation, at least in vascular, progenitor and tumor cells. We will now review current information on the signaling involved.
Ligand binding to CXCR4 induces conformational changes that lead to the activation of multiple signaling pathways (Figure 1), originating proximally from the dissociation of heterotrimeric G proteins and from the phosphorylation of the C-terminal cytoplasmic tail of CXCR4. CXCR4 is mainly bound to heterotrimeric Gi proteins, although other G protein classes may transduce CXCR4 binding as well (81). Upon ligand binding, the Gi heterotrimer detaches from the CXCR4 intracellular loops and dissociates into GTP-bound αi and βγ subunits (82, 83). The βγ subunits directly bind and activate phosphatidylinositol-3-OH kinases (PI3K) β or γ, which produce phosphatidylinositol triphosphate (PIP3), and phospholipase C β (PLC-β), which produces inositol-(1,4,5)-trisphosphate (IP3) and diacylglycerol (DAG). The Gαi subunit induces calcium release from intracellular stores and indirectly activates the PI3K-AKT and MEK1/2-Erk1/2 axes (84). Via the production of PIP3, PI3Ks activate the serine-threonine kinase AKT, which can then can phosphorylate many target proteins, most notably glycogen synthase kinase 3 (GSK3), tuberous sclerosis 2 (TSC2), caspase 9 and PRAS40 (AKT1S1), which explains its wide spectrum of downstream effects in promoting cell proliferation, differentiation, apoptosis, angiogenesis, and metabolism (85).
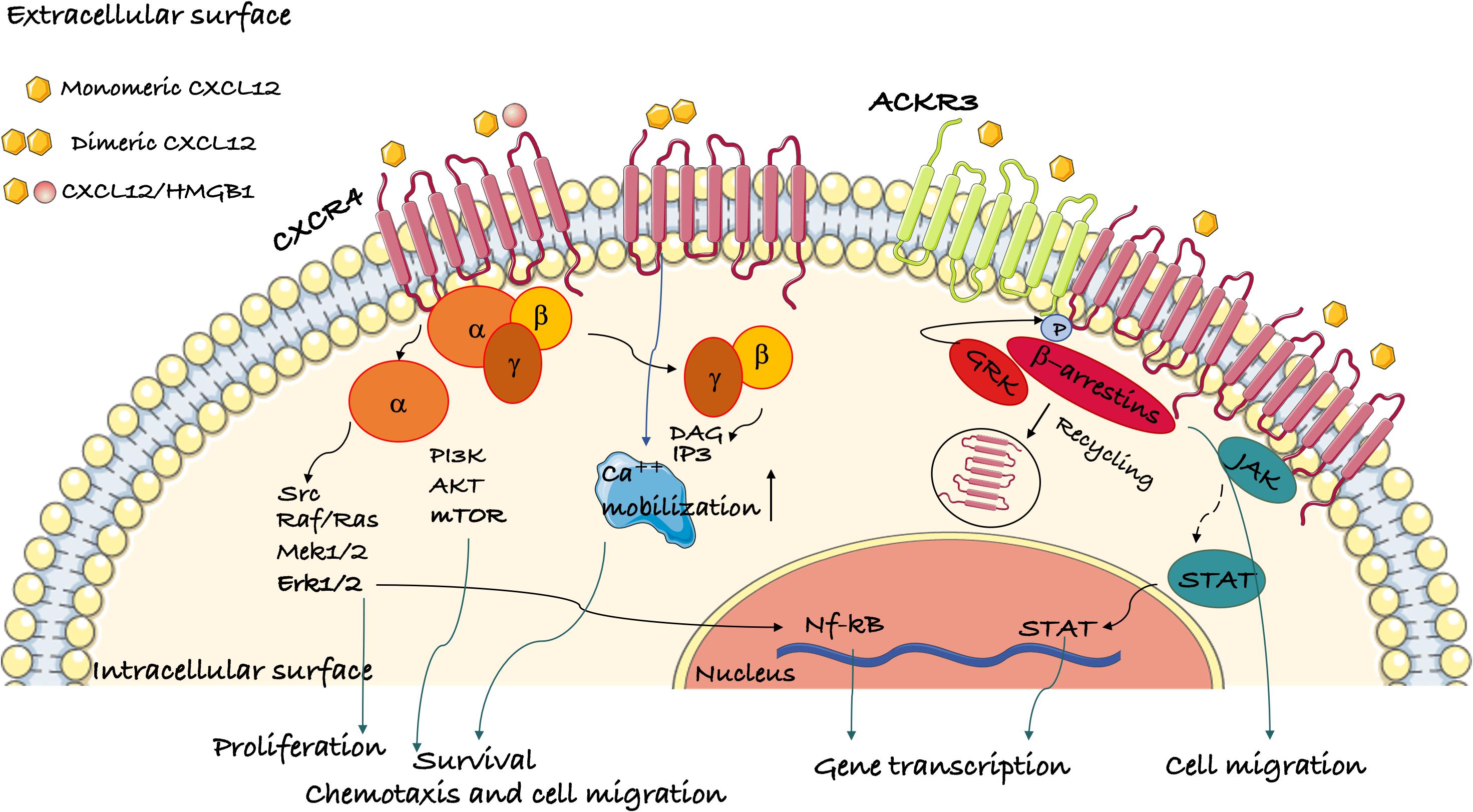
Figure 1. Schematic representation of the signaling pathways activated by CXCR4. Ligand binding to CXCR4 activates G protein subunits and the downstream Ca2+ mobilization from intracellular stores and PI3K/Akt, PLC, and ERK1/2 pathways. This results in gene transcription, cell migration, proliferation and survival. CXCR4 oligomerization can also activate the G-protein independent JAK/STAT pathway. β-arrestins are recruited following GRK phosphorylation of CXR4 and mediate its internalization. ACKR3 is another receptor for CXCL12 that can induce β-arrestin-mediated signaling both by itself or as a heterodimer with CXCR4.
CXCR4 ligand binding induces JAK/STAT activation in a Gα-independent manner (86). GPCR kinases (GRKs) phosphorylate multiple serines/threonines in the cytoplasmic tail of CXCR4. Phosphorylated CXCR4 recruits β-arrestin-1 and -2, which promote CXCR4 internalization (87). Thereafter, CXCR4 can be recycled back to the plasma membrane or sorted to the lysosomes for degradation (88). Of note, the recruitment of β-arrestins to CXCR4 also activates Erk signaling (89).
The binding of CXCL12 to CXCR4-ACKR3 heterodimers activates G protein-independent signaling cascades originating from β-arrestins that potentiate cell migration (8).
Overall, the activation of PI3Ks and Akt supports the proliferation and survival of both normal and cancer cells. mTORC activation underpins the anabolic metabolism that is required for cell growth; indeed, mTORC activation is also necessary for the transitioning of stem cells to the GAlert state.
Notably, the CXCR4-activated pathways that direct cell movement and migration are exactly the same that are involved in cell proliferation, and both processes can be inhibited by the same small molecules. For example, rapamycin is an mTORC inhibitor that blocks cell proliferation, but it inhibits cell migration as well (90, 91). The same is true for PI3K inhibitors (92).
Although the various pathways originating from CXCR4 are known, there is ample scope for cell specificity. The human genome encodes 18 different Gα proteins, 5 Gβ proteins and 12 Gγ proteins, and multiple PI3Ks and PLCs, with ample variation of expression in different cell types. Moreover, signaling is enhanced or dampened by dozens of modulators, including scaffold proteins that facilitate the physical interactions of kinases and other enzymes that introduce post-translational modifications. We are unaware of studies that delineate the CXCR4-initiated signaling pathways in cell proliferation down to the specific isoforms and post-translational modifications of the signal transducers involved. Cancer is not the most amenable biological system, since cancer cells have accumulated a number of genetic and epigenetic alterations, often including those of PI3Ks. Cell-specific conditional mutants could be used to investigate CXCR4-controlled proliferation following injury, and this would provide a list of parts in specific cells; even so, we would still miss mechanistic details such as the interaction with modifiers, possible feed-forward and feedback loops and time-dependent signal adaptations like those involving Rac (93) or oscillatory behaviors like those described for NF-κB and p53 (94, 95).
We have discussed several reports showing that CXCR4 can control cell proliferation in addition to directing cell retention and movement, both in physiological processes, such as development and tissue regeneration, and in pathological ones, such as cancer growth. The mechanisms and pathways involved may be broadly similar in all cases, since regeneration often recapitulates developmental processes, and cancer often exploits developmental pathways.
Signal transduction pathways downstream CXCR4 eventually control both cell movement and cell proliferation, which are both dependent on PI3K-Akt and mTORC signaling; the details, however, may vary from cell to cell and in different settings.
So far, the interest has focused on cancer and on drugs that block CXCR4-initiated signaling; we suggest that small molecules that activate CXCR4 signaling or can dissect the effects on cell migration and proliferation may be as useful.
Both authors contributed equally to the writing of the review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Mariagrazia Uguccioni (IRB, Bellinzona), Emilio Hirsch (Università di Torino), Giovanna Musco and Emilie Venéréau (IRCCS San Raffaele Scientific Institute Milan) for discussions and suggestions. This work was supported by grants AIRC IG-18623 and Ministry of Health RF-2016-02363024 to MB; RM is supported by a fellowship from Fondazione Buzzi Unicem.
1. Morein D, Erlichman N, and Baruch, AB. Beyond cell motility: the expanding roles of chemokines and their receptors in malignancy. Front Immunol. (2020) 11:952. doi: 10.3389/fimmu.2020.00952
3. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. (2001) 410:50–6. doi: 10.1038/35065016
4. Pawig L, Klasen C, Weber C, Bernhagen J, and Noels, H. Diversity and inter-connections in the CXCR4 chemokine receptor/ligand family: molecular perspectives. Front Immunol. (2015) 6:429. doi: 10.3389/fimmu.2015.00429
5. Guo F, Wang Y, Liu J, Mok SC, Xue F, and Zhang, W. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene. (2016) 35:816–26. doi: 10.1038/onc.2015.139
6. Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. (2010) 330:1066–71. doi: 10.1126/science.1194396
7. Martínez-Muñoz L, Rodríguez-Frade JM, Barroso R, Sorzano CÓS, Torreño-Pina JA, Santiago CA, et al. Separating actin-dependent chemokine receptor nanoclustering from dimerization indicates a role for clustering in CXCR4 signaling and function. Mol Cell. (2018) 70:106–19.e10. doi: 10.1016/j.molcel.2018.02.034
8. Décaillot FM, Kazmi MA, Lin Y, Ray-Saha S, Sakmar TP, and Sachdev, P. CXCR7/CXCR4 heterodimer constitutively recruits β-arrestin to enhance cell migration. J Biol Chem. (2011) 286:32188–97. doi: 10.1074/jbc.M111.277038
9. Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, and Springer, TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J Exp Med. (1996) 184:1101–9. doi: 10.1084/jem.184.3.1101
10. Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, et al. The CXC chemokine, stromal cell derived factor 1 (SDF-1), is the ligand for LESTR/fusin and prevents infection by lymphocyte-tropic HIV-1 syncytium-inducing strains. Nature. (1996) 382:833–5. doi: 10.1038/382833a0
11. Righetti A, Giulietti M, Sabanovic B, Occhipinti G, Principato G, and Piva, F. CXCL12 and its isoforms: different roles in pancreatic cancer? J Oncol. (2019) 2019:9681698. doi: 10.1155/2019/9681698
12. Yu L, Cecil J, Peng SB, Schrementi J, Kovacevic S, Paul D, et al. Identification and expression of novel isoforms of human stromal cell-derived factor 1. Gene. (2006) 374:174–9. doi: 10.1016/j.gene.2006.02.001
13. Torres R, and Ramirez, JC. A Chemokine targets the nucleus: Cxcl12-gamma isoform localizes to the nucleolus in adult mouse heart. PLoS One. (2009) 4:e7570. doi: 10.1371/journal.pone.0007570
14. Stephens BS, Ngo T, Kufareva I, and Handel, TM. Functional anatomy of the full length CXCR4-CXCL12 complex systematically dissected by quantitative model-guided mutagenesis. Sci Signal. (2020). 13:eaay5024. doi: 10.1126/scisignal.aay5024
15. Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. (2012) 209:551–63. doi: 10.1084/jem.20111739
16. Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. (2012) 209:1519–28. doi: 10.1084/jem.20120189
17. Tirone M, Tran NL, Ceriotti C, Gorzanelli A, Canepari M, Bottinelli R, et al. High mobility group box 1 orchestrates tissue regeneration via CXCR4. J Exp Med. (2018) 215:303–18. doi: 10.1084/jem.20160217
18. Rajasekaran D, Gröning S, Schmitz C, Zierow S, Drucker N, Bakou M, et al. Macrophage migration inhibitory factor-CXCR4 receptor interactions. J Biol Chem. (2016) 291:15881–95. doi: 10.1074/jbc.m116.717751
19. Lacy M, Kontos C, Brandhofer M, Hille K, Gröning S, Sinitski D, et al. Identification of an Arg-Leu-arg tripeptide that contributes to the binding interface between the cytokine MIF and the chemokine receptor CXCR4. Sci Rep. (2018) 8:1–17. doi: 10.1038/s41598-018-23554-5
20. Jankauskas SS, Wong DWL, Bucala R, Djudjaj S, and Boor, P. Evolving complexity of MIF signaling. Cell Signal. (2019) 57:76–88. doi: 10.1016/j.cellsig.2019.01.006
21. Saini V, Marchese A, and Majetschak, M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J Biol Chem. (2010) 285:15566–76. doi: 10.1074/jbc.M110.103408
22. Tripathi A, Saini V, Marchese A, Volkman BF, Tang WJ, and Majetschak, M. Modulation of the CXC chemokine receptor 4 agonist activity of ubiquitin through C-terminal protein modification. Biochemistry. (2013) 52:4184–92. doi: 10.1021/bi400254f
23. Scofield SLC, Daniels CR, Dalal S, Millard JA, Singh M, and Singh, K. Extracellular ubiquitin modulates cardiac fibroblast phenotype and function via its interaction with CXCR4. Life Sci. (2018) 211:8–16. doi: 10.1016/j.lfs.2018.09.012
24. Feng Z, Dubyak GR, Lederman MM, and Weinberg, A. Cutting edge: human β defensin 3-A novel antagonist of the HIV-1 coreceptor CXCR4. J Immunol. (2006) 177:782–6. doi: 10.4049/jimmunol.177.2.782
25. Zou YR, Kottmann AH, Kuroda M, Taniuchi I, and Littman, DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. (1998) 393:595–9. doi: 10.1038/31269
26. Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. (1996) 11:635–8. doi: 10.1038/382635a0
27. Bobadilla M, Sainz N, Abizanda G, Orbe J, Rodriguez JA, Páramo JA, et al. The CXCR4/SDF1 axis improves muscle regeneration through MMP-10 activity. Stem Cells Dev. (2014) 23:1417–27. doi: 10.1089/scd.2013.0491
28. Ratajczak M, Majka M, Kucia M, Drukala J, Pietrzkowski Z, Peiper S, et al. Expression of functional CXCR4 by muscle satellite cells and secretion of SDF-1 by muscle-derived fibroblasts is associated with the presence of both muscle progenitors in bone marrow and hematopoietic stem/progenitor cells in muscles. Stem Cells. (2003) 21:363–71. doi: 10.1017/CBO9781107415324.004
29. Vasyutina E, Stebler J, Brand-Saberi B, Schulz S, Raz E, and Birchmeier, C. CXCR4 and Gab1 cooperate to control the development of migrating muscle progenitor cells. Genes Dev. (2005) 19:2187–98. doi: 10.1101/gad.346205
30. Rafii S, Cao Z, Lis R, Siempos II, Chavez D, Shido K, et al. Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nat Cell Biol. (2015) 17:123–36. doi: 10.1038/ncb3096
31. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. (2011) 331:1078–80. doi: 10.1126/science.1200708
32. Das S, Goldstone AB, Wang H, Farry J, D’Amato G, Paulsen MJ, et al. A unique collateral artery development program promotes neonatal heart regeneration. Cell. (2019) 176:1128–42.e18. doi: 10.1016/j.cell.2018.12.023
33. Marín-Juez R, El-Sammak H, Helker CSM, Kamezaki A, Mullapuli ST, Bibli SI, et al. Coronary revascularization during heart regeneration is regulated by epicardial and endocardial cues and forms a scaffold for cardiomyocyte repopulation. Dev Cell. (2019) 51:503–15.e4. doi: 10.1016/j.devcel.2019.10.019
34. Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. (2014) 505:97–102. doi: 10.1038/nature12681
35. Hong F, Tuyama A, Lee TF, Loke J, Agarwal R, Cheng X, et al. Hepatic stellate cells express functional CXCR4: Role in stromal cell-derived factor-1α-mediated stellate cell activation. Hepatology. (2009) 49:2055–67. doi: 10.1002/hep.22890
36. Negro S, Lessi F, Duregotti E, Aretini P, La Ferla M, Franceschi S, et al. CXCL12α/SDF-1 from perisynaptic Schwann cells promotes regeneration of injured motor axon terminals. EMBO Mol Med. (2017) 9:1000–10. doi: 10.15252/emmm.201607257
37. Mishra RK, Shum AK, Platanias LC, Miller RJ, and Schiltz, GE. Discovery and characterization of novel small-molecule CXCR4 receptor agonists and antagonists. Sci Rep. (2016) 6:1–9. doi: 10.1038/srep30155
38. Zanetti G, Negro S, Megighian A, Mattarei A, Lista F, Fillo S, et al. CXCR4 receptor agonist strongly stimulates axonal regeneration after damage. Ann Clin Transl Neurol. (2019) 6:2395–402. doi: 10.1002/acn3.50926
39. Peng H, Kolb R, Kennedy JE, and Zheng, J. Differential expression of CXCL12 and CXCR4 during human fetal neural progenitor cell differentiation. J Neuroimmune Pharmacol. (2007) 2:251–8. doi: 10.1007/s11481-007-9081-3
40. Shinjyo N, Ståhlberg A, Dragunow M, Pekny M, and Pekna, M. Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem Cells. (2009) 27:2824–32. doi: 10.1002/stem.225
41. Chen Y, Wei Y, Liu J, and Zhang, H. Chemotactic responses of neural stem cells to SDF-1α correlate closely with their differentiation status. J Mol Neurosci. (2014) 54:219–33. doi: 10.1007/s12031-014-0279-6
42. Zhu C, Yao WL, Tan W, and Zhang, CH. SDF-1 and CXCR4 play an important role in adult SVZ lineage cell proliferation and differentiation. Brain Res. (2017) 1657:223–31. doi: 10.1016/j.brainres.2016.06.011
43. Gong X, He X, Qi L, Zuo H, and Xie, Z. Stromal cell derived factor-1 acutely promotes neural progenitor cell proliferation in vitro by a mechanism involving the ERK1/2 and PI-3K signal pathways. Cell Biol Int. (2006) 30:466–71. doi: 10.1016/j.cellbi.2006.01.007
44. Wu Y, Peng H, Cui M, Whitney NP, Huang Y, and Zheng, JC. CXCL12 increases human neural progenitor cell proliferation through Akt-1/FOXO3a signaling pathway. J Neurochem. (2009) 109:1157–67. doi: 10.1111/j.1471-4159.2009.06043.x
45. Li M, Chang CJ, Lathia JD, Wang L, Pacenta HL, Cotleur A, et al. Chemokine receptor CXCR4 signaling modulates the growth factor-induced cell cycle of self-renewing and multipotent neural progenitor cells. Glia. (2011) 59:108–18. doi: 10.1002/glia.21080
46. Zhang L, Hua Q, Tang K, Shi C, Xie X, and Zhang, R. CXCR4 activation promotes differentiation of human embryonic stem cells to neural stem cells. Neuroscience. (2016) 337:88–97. doi: 10.1016/j.neuroscience.2016.09.001
47. Ho SY, Ling TY, Lin HY, Liou JTJ, Liu FC, Chen IC, et al. SDF-1/CXCR4 signaling maintains stemness signature in mouse neural stem/progenitor cells. Stem Cells Int. (2017) 2017:2493752. doi: 10.1155/2017/2493752
48. De Leo F, Quilici G, Tirone M, De Marchis F, Mannella V, Zucchelli C, et al. Diflunisal targets the HMGB1/CXCL12 heterocomplex and blocks immune cell recruitment. EMBO Rep. (2019) 20:e47788. doi: 10.15252/embr.201947788
49. Dormoy-Raclet V, Cammas A, Celona B, Lian XJ, Van Der Giessen K, Zivojnovic M, et al. HuR and miR-1192 regulate myogenesis by modulating the translation of HMGB1 mRNA. Nat Commun. (2013) 4:2388. doi: 10.1038/ncomms3388
50. Lee G, Santo AIE, Zwingenberger S, Cai L, Vogl T, Feldmann M, et al. Fully reduced HMGB1 accelerates the regeneration of multiple tissues by transitioning stem cells to GAlert. Proc Natl Acad Sci USA. (2018) 115:E4463–72. doi: 10.1073/pnas.1802893115
51. Mollica L, De Marchis F, Spitaleri A, Dallacosta C, Pennacchini D, Zamai M, et al. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem Biol. (2007) 14:431–41. doi: 10.1016/j.chembiol.2007.03.007
52. Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, et al. MTORC1 controls the adaptive transition of quiescent stem cells from G0 to GAlert. Nature. (2014) 510:393–6. doi: 10.1038/nature13255
53. Wald O, Izhar U, Amir G, Kirshberg S, Shlomai Z, Zamir G, et al. Interaction between neoplastic cells and cancer-associated fibroblasts through the CXCL12/CXCR4 axis: Role in non-small cell lung cancer tumor proliferation. J Thorac Cardiovasc Surg. (2011) 141:1503–12. doi: 10.1016/j.jtcvs.2010.11.056
54. Wu M, Huang C, Gan K, Huang H, Chen Q, Ouyang J, et al. LRRC4, a putative tumor suppressor gene, requires a functional leucine-rich repeat cassette domain to inhibit proliferation of glioma cells in vitro by modulating the extracellular signal-regulated kinase/protein kinase B/nuclear factor-kB Pathway. Mol Biol Cell. (2006) 17:3534–42.
55. Mitola S, Belleri M, Urbinati C, Coltrini D, Sparatore B, Pedrazzi M, et al. Cutting edge: extracellular high mobility group box-1 protein is a proangiogenic cytokine. J Immunol. (2006) 176:12–5. doi: 10.4049/jimmunol.176.1.12
56. Dvorak, HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
57. Burger JA, and Burger, MKT. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath BM stromal cells. Blood. (1999) 94:3658–67.
58. Mohle R, Schittenhelm M, Failenschmid C, Bautz F, Kratz-Albers K, Serve H, et al. Functional response of leukaemic blasts to stromal cell-derived factor-1 correlates with preferential expression of the chemokine receptor CXCR4 in acute myelomonocytic and lymphoblastic leukaemia. Br J Haematol. (2000) 110:563–72. doi: 10.1046/j.1365-2141.2000.02157.x
59. Chatterjee S, Azad BB, and Nimmagadda, S. The intricate role of CXCR4 in cancer (2014). Adv Cancer Res. (2014) 124:31–82. doi: 10.1016/j.immuni.2010.12.017
60. Leone V, D’Angelo D, Rubio I, de Freitas PM, Federico A, Colamaio M, et al. MiR-1 is a tumor suppressor in thyroid carcinogenesis targeting CCND2, CXCR4, and SDF-1α. J Clin Endocrinol Metab. (2011) 96:E1388–98. doi: 10.1210/jc.2011-0345
61. Lu J, Luo H, Liu X, Peng Y, Wang L, Xu X, et al. MIR-9 targets CXCR4 and functions as a potential tumor suppressor in nasopharyngeal carcinoma. Carcinogenesis. (2014) 35:554–63. doi: 10.1093/carcin/bgt354
62. Yu T, Liu K, Wu Y, Fan J, Chen J, Li C, et al. MicroRNA-9 inhibits the proliferation of oral squamous cell carcinoma cells by suppressing expression of CXCR4 via the Wnt/β-catenin signaling pathway. Oncogene. (2014) 33:5017–27. doi: 10.1038/onc.2013.448
63. Liu Y, Zhou Y, Feng X, An P, Quan X, Wang H, et al. MicroRNA-126 functions as a tumor suppressor in colorectal cancer cells by targeting CXCR4 via the AKT and ERK1/2 signaling pathways. Int J Oncol. (2014) 44:203–10. doi: 10.3892/ijo.2013.2168
64. Spinello I, Quaranta MT, Riccioni R, Riti V, Pasquini L, Boe A, et al. MicroRNA-146a and AMD3100, two ways to control CXCR4 expression in acute myeloid leukemias. Blood Cancer J. (2011) 1:e26. doi: 10.1038/bcj.2011.24
65. Liu Z, Ye P, Wang S, Wu J, Sun Y, Zhang A, et al. MicroRNA-150 protects the heart from injury by inhibiting monocyte accumulation in a mouse model of acute myocardial infarction. Circ Cardiovasc Genet. (2015) 8:11–20. doi: 10.1161/CIRCGENETICS.114.000598
66. Toritsuka M, Kimoto S, Muraki K, Landek-Salgado MA, Yoshida A, Yamamoto N, et al. Deficits in microRNA-mediated Cxcr4/Cxcl12 signaling in neurodevelopmental deficits in a 22q11 deletion syndrome mouse model. Proc Natl Acad Sci USA. (2013) 110:17552–7. doi: 10.1073/pnas.1312661110
67. Kryczek I, Lange A, Mottram P, Alvarez X, Cheng P, Hogan M, et al. CXCL12 and vascular endothelial growth factor synergistically induce neonaniogenisis in human ovarian cancers. Cancer Res. (2005) 65:465–72.
68. Gassenmaier M, Chen D, Buchner A, Henkel L, Schiemann M, Mack B, et al. CXC chemokine receptor 4 is essential for maintenance of renal cell carcinoma-initiating cells and predicts metastasis. Stem Cells. (2013) 31:1467–76. doi: 10.1002/stem.1407
69. Dubrovska A, Elliott J, Salamone RJ, Telegeev GD, Stakhovsky AE, Schepotin IB, et al. CXCR4 expression in prostate cancer progenitor cells. PLoS One. (2012) 7:e31226. doi: 10.1371/journal.pone.0031226
70. Jung MJ, Rho JK, Kim YM, Jung JE, Jin YB, Ko YG, et al. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene. (2013) 32:209–21. doi: 10.1038/onc.2012.37
71. Thomas RM, Kim J, Revelo-Penafiel MP, Angel R, and Dawson, DWLA. The chemokine receptor CXCR4 is expressed in pancreatic intraepithelial neoplasia. Gut. (2008) 57:1555–60. doi: 10.1136/gut.2007.143941
72. Barbero S, Bonavia R, Bajetto A, Porcile C, Pirani P, Ravetti JL, et al. Stromal cell-derived factor 1α stimulates human glioblastoma cell growth through the activation of both extracellular signal-regulated kinases 1/2 and Aktx1. Cancer Res. (2003) 63:1969–74. doi: 10.1016/j.ygyno.2006.05.045
73. Sehgal A, Keener C, Boynton AL, Warrick J, and Murphy, GP. CXCR-4, a chemokine receptor, is overexpressed in and required for proliferation of glioblastoma tumor cells. J Surg Oncol. (1998) 69:99–104. doi: 10.1002/(SICI)1096-9098(199810)69:23.0.co;2-m
74. Li T, Li H, Wang Y, Harvard C, Tan JL, Au A, et al. The expression of CXCR4, CXCL12 and CXCR7 in malignant pleural mesothelioma. J Pathol. (2011) 223:519–30. doi: 10.1002/path.2829
75. Hall JM, and Korach, KS. Stromal cell-derived factor 1, a novel target of estrogen receptor action, mediates the mitogenic effects of estradiol in ovarian and breast cancer cells. Mol Endocrinol. (2003) 17:792–803. doi: 10.1210/me.2002-0438
76. Guo Q, Gao BL, Zhang XJ, Liu GC, Xu F, Fan QY, et al. CXCL12-CXCR4 axis promotes proliferation migration, invasion, and metastasis of ovarian cancer. Oncol Res. (2015) 22:247–58. doi: 10.3727/096504015X14343704124430
77. Ma J, Sun X, Wang Y, Chen B, Qian L, and Wang, Y. Fibroblast-derived CXCL12 regulates PTEN expression and is associated with the proliferation and invasion of colon cancer cells via PI3k/Akt signaling. Cell Commun Signal. (2019) 17:1–12. doi: 10.1186/s12964-019-0432-5
78. Gao Z, Wang X, Wu K, Zhao Y, and Hu, G. Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/CXCR4 axis. Pancreatology. (2010) 10:186–93. doi: 10.1159/000236012
79. Wang DF, Lou N, Qiu MZ, Lin YB, and Liang, Y. Effects of CXCR4 gene silencing by lentivirus shRNA on proliferation of the EC9706 human esophageal carcinoma cell line. Tumor Biol. (2013) 34:2951–9. doi: 10.1007/s13277-013-0858-0
80. Tan XY, Chang S, Liu W, and Tang, HH. Silencing of CXCR4 inhibits tumor cell proliferation and neural invasion in human hilar cholangiocarcinoma. Gut Liver. (2014) 8:196–204. doi: 10.5009/gnl.2014.8.2.196
81. Rubin, JB. Chemokine signaling in cancer: One hump or two? Semin Cancer Biol. (2009) 19:116–22. doi: 10.1016/j.semcancer.2008.10.001
82. Goldsmith ZG, and Dhanasekaran, DNG. Protein regulation of MAPK networks. Oncogene. (2007) 26:3122–42. doi: 10.1038/sj.onc.1210407
83. Mellado M, Rodríguez-Frade JM, Mañes S, and Martínez-A, C. Chemokine signaling and functional responses: the role of receptor dimerization and TK pathway activation. Annu Rev Immunol. (2001) 19:397–421. doi: 10.1146/annurev.immunol.19.1.397
84. New DC, Wu K, Kwok AWS, and Wong, YH. G protein-coupled receptor-induced Akt activity in cellular proliferation and apoptosis. FEBS J. (2007) 274:6025–36. doi: 10.1111/j.1742-4658.2007.06116.x
85. Mo W, Chen J, Patel A, Zhang L, Chau V, Li Y, et al. CXCR4/CXCL12 mediate autocrine cell- cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell. (2013) 152:1077–90. doi: 10.1016/j.cell.2013.01.053
86. Vila-Coro AJ, Rodríguez-Frade JM, De Ana AM, Moreno-Ortíz MC, Martínez AC, and Mellado, M. The chemokine SDF-lα triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. (1999) 13:1699–710. doi: 10.1096/fasebj.13.13.1699
87. Haribabu B, Richardson RM, Fisher I, Sozzani S, Peiper SC, Horuk R, et al. Regulation of human chemokine receptors CXCR4: role of phosphorylation in desensitization and internalization. J Biol Chem. (1997) 272:28726–31. doi: 10.1074/jbc.272.45.28726
88. Marchese A, Chen C, Kim YM, and Benovic, JL. The ins and outs of G protein-coupled receptor trafficking. Trends Biochem Sci. (2003) 28:369–76. doi: 10.1016/S0968-0004(03)00134-8
89. Lee SH, Hollingsworth R, Kwon HY, Lee N, and Chung, CY. β-arrestin 2-dependent activation of ERK1/2 is required for ADP-induced paxillin phosphorylation at Ser83 and microglia chemotaxis. Glia. (2012) 60:1366–77. doi: 10.1002/glia.22355
90. Delgado-Martín C, Escribano C, Pablos JL, Riol-Blanco L, and Rodríguez-Fernández, JL. Chemokine CXCL12 uses CXCR4 and a signaling core formed by bifunctional akt, extracellular signal-regulated kinase (ERK)1/2, and mammalian target of rapamycin complex 1 (mTORC1) proteins to control chemotaxis and survival simultaneously in mature dendriti. J Biol Chem. (2011) 286:37222–36. doi: 10.1074/jbc.M111.294116
91. Chen G, Chen SM, Wang X, Ding XF, Ding J, and Meng, LH. Inhibition of chemokine (CXC motif) ligand 12/chemokine (CXC motif) receptor 4 axis (CXCL12/CXCR4)-mediated cell migration by targeting mammalian target of rapamycin (mTOR) pathway in human gastric carcinoma cells. J Biol Chem. (2012) 287:12132–41. doi: 10.1074/jbc.M111.302299
92. Fernandis AZ, Prasad A, Band H, Klösel R, and Ganju, RK. Regulation of CXCR4-mediated chemotaxis and chemoinvasion of breast cancer cells. Oncogene. (2004) 23:157–67. doi: 10.1038/sj.onc.1206910
93. Campa CC, Germena G, Ciraolo E, Copperi F, Sapienza A, Franco I, et al. Rac signal adaptation controls neutrophil mobilization from the bone marrow. Sci Signal. (2016) 9:ra124. doi: 10.1126/scisignal.aah5882
94. Zambrano S, de Toma I, Piffer A, Bianchi ME, and Agresti, A. NF-κB oscillations translate into functionally related patterns of gene expression. Elife. (2016) 5:e09100. doi: 10.7554/eLife.09100
Keywords: chemokine, cancer, tissue regeneration, CXCL12, HMGB1, CXCR4
Citation: Bianchi ME and Mezzapelle R (2020) The Chemokine Receptor CXCR4 in Cell Proliferation and Tissue Regeneration. Front. Immunol. 11:2109. doi: 10.3389/fimmu.2020.02109
Received: 11 May 2020; Accepted: 04 August 2020;
Published: 28 August 2020.
Edited by:
José Luis Rodríguez-Fernández, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Vadim Gaponenko, University of Illinois at Chicago, United StatesCopyright © 2020 Bianchi and Mezzapelle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco E. Bianchi, YmlhbmNoaS5tYXJjb0Boc3IuaXQ=; Rosanna Mezzapelle, bWV6emFwZWxsZS5yb3Nhbm5hQGhzci5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.