 Padmini Khedoe
Padmini Khedoe Emiel Marges
Emiel Marges Pieter Hiemstra
Pieter Hiemstra Maarten Ninaber
Maarten Ninaber
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 04 September 2020
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01990
This article is part of the Research Topic Interplay Between Immunity and Fibrosis View all 14 articles
Systemic sclerosis (SSc) is an autoimmune connective tissue disease, characterized by immune dysregulation and progressive fibrosis. Interstitial lung disease (ILD) is the most common cause of death among SSc patients and there are currently very limited approved disease-modifying treatment options for systemic sclerosis-related interstitial lung disease (SSc-ILD). The mechanisms underlying pulmonary fibrosis in SSc-ILD are not completely unraveled, and knowledge on fibrotic processes has been acquired mostly from studies in idiopathic pulmonary fibrosis (IPF). The incomplete knowledge of SSc-ILD pathogenesis partly explains the limited options for disease-modifying therapy for SSc-ILD. Fibrosis in IPF appears to be related to aberrant repair following injury, but whether this also holds for SSc-ILD is less evident. Furthermore, immune dysregulation appears to contribute to pro-fibrotic responses in SSc-ILD, perhaps more than in IPF. In addition, SSc-ILD patient heterogeneity complicates the understanding of the underlying mechanisms of disease development, and more importantly, limits correct clinical diagnosis and treatment effectivity. Therefore, there is an unmet need for patient-relevant (in vitro) models to examine patient-specific disease pathogenesis, predict disease progression, screen appropriate treatment regimens and identify new targets for treatment. Technological advances in in vitro patient-relevant disease modeling, including (human induced pluripotent stem cell (hiPSC)-derived) lung epithelial cells, organoids and organ-on-chip technology offer a platform that has the potential to contribute to unravel the underlying mechanisms of SSc-ILD development. Combining these models with state-of-the-art analysis platforms, including (single cell) RNA sequencing and (imaging) mass cytometry, may help to delineate pathogenic mechanisms and define new treatment targets of SSc-ILD.
Systemic sclerosis (SSc) is a devastating disease of unknown etiology, characterized by systemic, immunological, vascular, and fibrotic abnormalities, with a heterogeneous clinical course. Fibrosis, the hallmark of the disease, can affect skin and internal organs, including the lung (1). SSc-related interstitial lung disease (SSc-ILD) is one of the most severe complications and is the main cause of SSc-related deaths (2, 3). Treatment options are limited, although the FDA has recently approved nintedanib for progressive SSc-ILD. The limitation in options for disease-modifying treatments may partly be explained by the incomplete knowledge of the mechanisms underlying lung fibrosis development in SSc-ILD and the heterogeneous course of disease. Furthermore, the optimal type and timing of treatment of SSc-ILD is unknown and it is becoming increasingly clear that the treatment of SSc-ILD patients requires a personalized approach. An unknown subgroup of patients with SSc may benefit from antifibrotic drugs such as nintedanib and related compounds, in addition to their immunosuppressive therapy. Therefore, there is an unmet need for novel biomarkers to predict disease progression and response to treatment (4). As a result, the prognosis for SSc-ILD patients remains extremely poor, and there is an urgent need for better disease-relevant models to identify new treatment targets and for (personalized) drug screening.
Although various experimental animal models of human ILD are currently available, they do not fully represent human disease development, and fibrosis is often reversible (5). Furthermore, whereas pathogenic mechanisms can be studied to some extent using inflammatory cells derived from peripheral blood and bronchoalveolar lavage (BAL) from SSc-ILD patients, insight into the interaction between key players in the pathogenesis of fibrosis is often lacking. Currently, there are only a few available preclinical human in vitro models which recapitulate features of fibrosis, including the complex interplay between (myo)fibroblasts, epithelial cells, inflammatory cells and endothelial cells, and these are reviewed elsewhere (5). The extent to which these models recapitulate fibrosis development in SSc-ILD is unclear.
Here, we discuss how recent advances in human preclinical models may help to unravel pathogenic mechanisms and new treatment targets of SSc-ILD by shortly reviewing the cellular processes that lead to fibrosis in IPF and SSc-ILD (Figure 1).
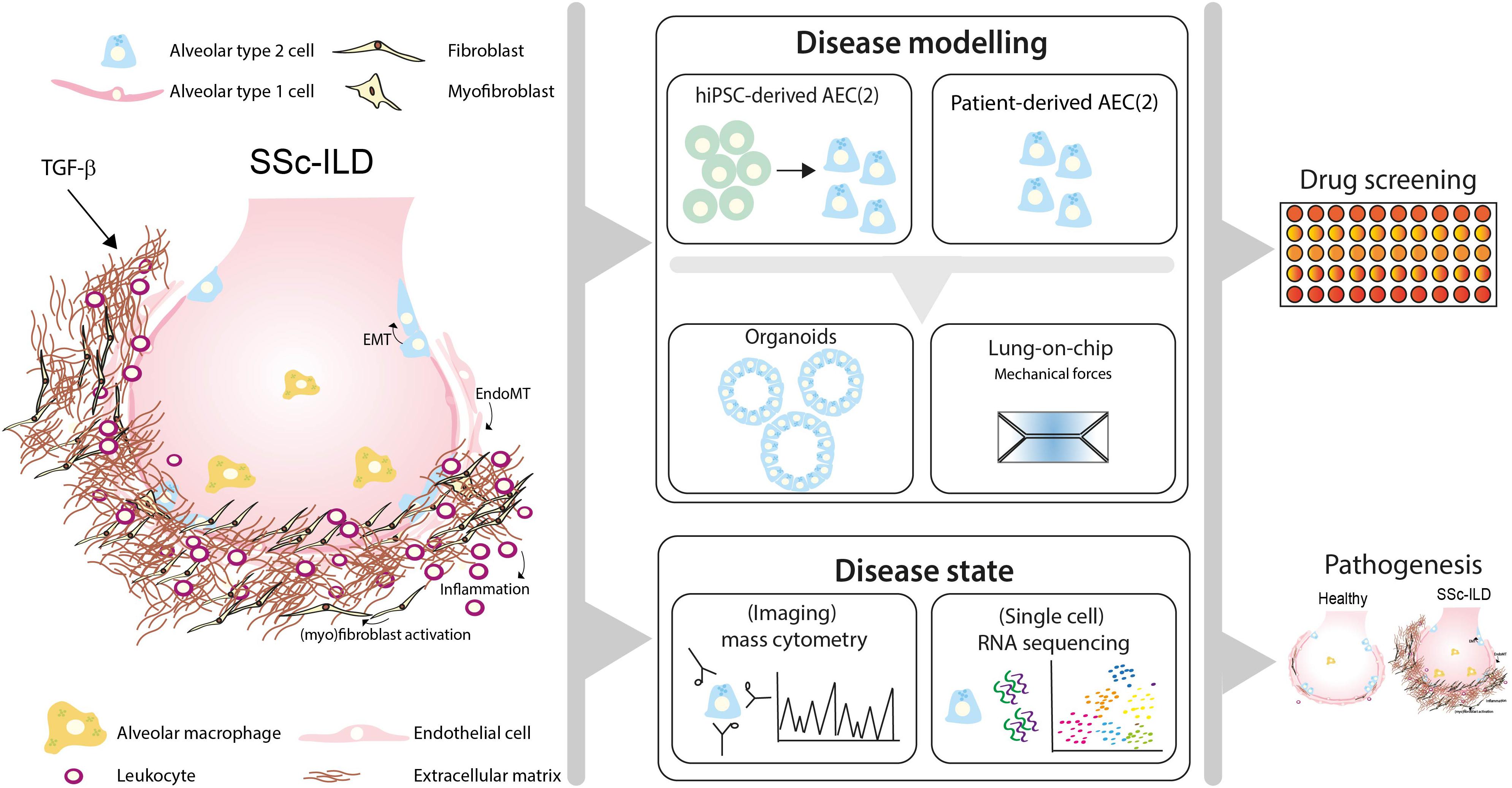
Figure 1. Toward personalized-medicine-based prediction, target identification and drug screening models of SSc-ILD. In SSc-ILD, the interactions and cross-talk between various cell types, including in type 2 alveolar cells (AEC2), AEC1, endothelial cells, fibroblasts and immune cells is disturbed. This may be partly explained by excessive TGF-β signaling activation, leading to AEC1/2 and endothelial cell (EC) transition into a mesenchymal phenotype, (myo)fibroblast activation and proliferation and excessive interstitial extracellular matrix (ECM) deposition. Recruited leukocytes further contribute to the profibrotic environment, leading to lung tissue remodeling and fibrosis. Patient-derived pulmonary cells, either isolated from lung tissue or derived from human induced pluripotent stem cells (hiPSC), can be used to study patient-specific responses in vitro using organoid SSc-ILD disease models. Lung-chip models may add another layer of complexity containing physiological ques, including breathing motions, air-blood flow and stretch, which are often lacking in current available in vitro lung models. Disease-specific features can be measured using cutting-edge techniques such as single-cell-RNA-sequencing and (imaging) mass cytometry. Integrating information derived from SSc-ILD disease models using cells derived from SSc-ILD patients and triggers implicated in disease pathogenesis may lead to novel mechanistic insight and offer a platform for identification of pathogenic processes and drug screening for SSc-ILD.
SSc-ILD can be regarded as an impaired or otherwise aberrant repair response in the alveolar areas. Proposed mechanisms of fibrosis development and progression include profibrotic responses following lung alveolar epithelial cell (AEC) damage, aberrant alveolar repair (6, 7) and accelerated lung epithelial aging (8). Impaired regenerative capacity of lung progenitor cells, mainly the type 2 alveolar cells (AEC2), are suggested to be key players in the aberrant repair response. In concert with activated endothelial cells (EC) and recruited inflammatory cells, AEC2 orchestrate a profibrotic niche leading to fibroblast activation, myofibroblast formation and excessive extracellular matrix (ECM) deposition. Increased numbers of (myo)fibroblasts and their activation is mainly driven by transforming growth factor-β (TGF-β), whereas in alveolar epithelial type 1 and 2 cells (AEC1/2) and EC, excessive TGF-β may result in epithelial-to-mesenchymal transition (EMT) and endothelial-to-mesenchymal transition (endoMT) (9–12). TGF-β, produced by various cells types, is the central regulator of fibrotic processes and its levels are increased in both IPF and SSc-ILD lungs (13, 14). TGF-β enhances expression of profibrotic and pro-inflammatory cytokines and growth factors, such as tumor necrosis factor-α (TNF-α), platelet derived growth factor (PDGF), Interleukin-1β (IL-1β), and IL-13 (15). Target genes of TGF-β include e.g., α-smooth muscle actin (α-SMA) and connective tissue growth factor (CTGF), which are increased in fibrosis (12). Plasma CTGF levels were reported to be elevated in IPF patients (16) and, together with Vascular Endothelial Growth Factor (VEGF) and TGF-β, act as central mediators in fibrosis (14, 17–19). Altogether, the profibrotic niche leads to aberrant alveolar repair responses, fibroblast foci and excessive extracellular matrix (ECM) production, causing extensive pulmonary remodeling resulting in impairment of lung function.
Since one of the key players in this aberrant repair response is the AEC2, lung organoid assays provide a tool to study the regenerative ability of these lung progenitor cells. Organoids are three-dimensional (3D) self-organizing structures of multiple cell types grown in 3-D matrices, as opposed to a 2D cell culture on culture plates. The 3D matrix contains a mixture of ECM proteins and various growth factors, necessary for expansion, proliferation and differentiation of lung epithelial (progenitor) cells (20). Lung organoids can be cultured for prolonged periods of time, currently up to 2 years, without changes in their karyotype, whilst preserving original (patient) characteristics. An important feature of organoids is that progenitor cells are retained in culture, and have the capacity to proliferate and differentiate into various specialized cell types (21). Furthermore, when generating lung epithelial organoids, the differentiation into bronchial, alveolar or mixed cell types can be achieved by selection of cells, manipulating the cultures with cocktails of cytokines and growth factors, stimulating or inhibiting signaling pathways, and/or by including mesenchymal cells that provide supporting signals for differentiation. Organoid differentiation and behavior may alter in response to a variety of injuries including exposure to SSc-ILD-related cytokines, infectious agents, tobacco smoke, high levels of oxygen and bleomycin (22, 23). This way, lung organoid models are well suited to study early phases of the fibrotic process. TGF-β treatment of airway organoids, for example, resulted in enhanced αSMA expression, and upregulated pro-fibrotic genes including CTGF and fibronectin (24, 25). Importantly, the complexity of the cross-talk between epithelial cells, fibroblasts and inflammatory cells that occur in ILD, can be mimicked in organoid models containing epithelial cells and surrounding cells, including fibroblasts and endothelial cells. This is also illustrated by a study showing that pretreatment of MRC-5 fibroblasts with TGF-β decreases their ability to support mouse epithelial organoid formation (26). Finally, recent advances allowing incorporation of inflammatory cells in lung organoid cultures may contribute to patient-relevant SSc-ILD models.
Fibrosis in SSc-ILD may result from an interplay between autoimmunity, inflammation, alveolar epithelial- and vascular injury (27), which may result in alveolitis. The alveolar and microvascular injurious events drive activation and proliferation of lung-resident immune cells and recruitment of inflammatory cells, including monocytes, neutrophils, mast cells and NK cells (28, 29). Interestingly, monocytes derived from SSc-ILD patients, which were increased in both the circulation and in lungs, exhibit pro-fibrotic properties in vitro upon pro-inflammatory stimulation (30). In addition, fibrotic mediators in SSc-ILD, including fibronectin-EDA and tenascin-C, may trigger activation of Toll Like Receptor 4 (TLR4) (31, 32) and cause uncontrolled ECM deposition in SSc (33), which subsequently potentiates TGF-β signaling, thereby enhancing fibrotic responses (15, 34).
Circulating and pulmonary (auto-reactive) B lymphocytes are important features in SSc-ILD (28, 35, 36) and produce pro-fibrotic mediators including TGF-β and Th2 cytokines (28). Furthermore, auto-antibodies, which are a characteristic feature of SSc, are produced by these autoreactive B cells and may cause local pulmonary damage and are associated with distinct clinical phenotypes of SSc (37). Development of ILD in SSc patients is strongly correlated with the presence of auto-antibodies against topoisomerase I, U11/12, Th/To,PL-7, U1-RNP, whereas anti-RNA polymerase III antibodies are associated with reduced risk of ILD in SSc (38). Also, anti-C1q antibodies were shown in a subgroup of SSc-ILD patients, and were found to be the most important predicting factor for pulmonary fibrosis development in SSc (39). Collectively, especially in SSc-ILD patients these autoreactive B cells may contribute to a profibrotic environment.
In IPF, the contribution of inflammation to IPF pathogenesis is less clear. Whereas the proportion of T- and B- cells was similar in IPF lungs compared to healthy lungs, both CD71– alveolar- and SPP1/osteopontin+ interstitial macrophages were found to be associated with disease severity (40, 41), suggesting that lung-resident macrophages contribute to fibrosis in IPF. In addition, circulating monocytes in patients with IPF appear to be programmed to express fibroblast-stimulating properties prior to entering the lung (42). The contribution of macrophage subtypes in fibrosis development is controversial. Whereas pro-inflammatory M1 macrophages may contribute to dysregulated repair (e.g., in acute exacerbations of IPF) (43, 44), anti-inflammatory M2 macrophages may promote fibroproliferation and progressive fibrosis (42, 44). Furthermore, dysregulated apoptotic cell clearance and imbalanced ECM production and degradation by macrophages may contribute to fibrosis (45, 46).
In addition to enhanced levels of pro-fibrotic mediators, levels of pro-inflammatory cytokines and chemokines (including IL-8, IL-1α, IL-10, macrophage inflammatory protein-1α and MCP-1) and neutrophil-derived alpha-defensins (human neutrophil peptides (HNP), are increased in BALF from SSc-ILD patients (29, 47). In addition, also circulating levels of pro- and anti-inflammatory mediators, including IL-6, IL-10, MCP-1, IL-1RA and TNFα, were found to be increased. Some of these mediators correlate significantly with disease-relevant parameters (47, 48), and especially serum IL-6 is increased in both SSc-ILD and IPF, and may serve as a predictor of fibrosis progression (49). Recent studies intriguingly suggested both in IPF and SSc-ILD patients, an inverse correlation between lung function and myeloid-derived suppressor cell (MDSC) levels, highlighting their potential contribution to lung fibrosis (50). Thus, targeting the MDSC activity may be a novel approach to disease modification, and combined organoid models of epithelial and inflammatory cells may be useful tools to study this. This is illustrated by a study showing that lung organoids, comprised of epithelial and mesenchymal cells, respond to TLR- stimulation by releasing pro−inflammatory cytokines. Addition of primary human monocytes to these stimulated organoids, resulted in their migration toward the organoids and altered their phenotype to an “intermediate−like” phenotype, expressing increased levels of CD14, CD16 and HLA-DR compared to monocytes cultured alone (51).
Collectively these studies show that there are differences in the proposed role of inflammatory and immune cells in the pathogenesis of SSc-ILD and IPF. More complex organoid models, as outlined above, may help to further dissect the specific role of these cells, and the contribution of profibrotic and pro-inflammatory triggers in SSC-ILD. Indeed, whereas incorporation of patient-derived inflammatory and immune cells in organoids is logistically challenging, this may potentially provide preclinical personalized prediction models of disease development, and models for drug screening. In the tumor field, co-culture models of immune cells and tumor cells are used to assess effectivity of immunotherapy (52). Cattaneo et al. showed for example that effector function of autologous tumor-specific T cells could be assessed using peripheral blood-derived T cells and tumor organoids at a personalized level (53). Furthermore, efforts have been made to establish immune-engineered organoid models to study B-cell responses and antibody production (54, 55). Application of such combined organoid models to study (patient-specific) epithelial-inflammatory cell interactions in SSc-ILD, may provide an important step forward in our understanding of this devastating disease.
Accelerated lung aging and senescence of AEC and fibroblasts appear central phenotypes that promote lung fibrosis (8). Polymorphisms in genes involved in the maintenance of telomere length (mutations in TERT, TERC, PARN and RTEL1) are associated with an increased risk of IPF (56). Shortened telomeres, oxidative injury, endoplasmic reticulum (ER) stress, and mitochondrial dysfunction lead to decreased AEC proliferation and secretion of profibrotic mediators (8). In addition, genetic variants in the genes encoding e.g., surfactant protein C (SFPTC) (57, 58) and in the promotor region of mucin 5B (MUC5B), increase the risk of IPF development (59–61), while MUC5B has shown not to contribute to SSc-ILD (62). Whereas genetic susceptibility may be an important driver in IPF, for SSc, this is less clear. Several genetic factors may predispose to development of SSc, of which genetic variants in immune-related genes, including HLA and interferon-related genes, have been found (37). Genetic associations that predispose for development of SSc-ILD specifically, have been found in interferon regulatory factor 5 (IRF5), signal transducer and activator of transcription 4 (STAT4), DNAX accessory molecule 1 (CD226) and interleukin-1 receptor-associated kinase-1 (IRAK1) (63). Genetic variants in monocyte chemoattractant protein (MCP-1) and CTLA-4 were also associated with SSc-ILD (64). Especially, polymorphisms in CTLA-4, which acts as a checkpoint inhibitor, may be of relevance in SSc-ILD patients at risk for developing lung cancer.
Even though adult human cells can be isolated from lungs to study lung epithelial (progenitor) function, (altered) repair and regeneration, in SSc-ILD these are often derived from end-stage disease patients undergoing lung transplantation. To overcome this issue, human induced pluripotent stem cells (hiPSC)-derived lung epithelial cells may provide a personalized platform for assessing disease risk and potential treatment for SSc-ILD. One of the advantages of hiPSC-derived AEC2 is that they can be derived from individual patients from relatively easily accessible sources (urine, blood), enabling a personalized disease and medicine approach. Importantly, hiPSC-AEC2 can be genetically engineered by introducing or correcting mutations using CRISPR-based technology, which may be relevant to study genetic factors contributing to fibrosis in IPF and SSc-ILD (65). These hiPSC-derived AEC2 can be applied in lung organoids which can model pathogenetic processes occurring in ILD (66). Similar to hiPSC, lung organoids can also be derived from embryonic stem cells (ESC). A recent study used this approach with embryonic stem cells in which mutations were introduced in genes associated with Hermansky-Pudlak syndrome, that strongly predisposes to lung fibrosis, and thus demonstrated an essential role for IL-11 in the fibrotic process (67). Whereas generation of hiPSC- or ESC-derived AEC2 can be performed successfully, derivation of long-term primary AEC2 cultures from human lung tissue still is challenging. Recently, Shiraisi et al. successfully cultured AEC2 from human lungs using organoid expansion, which they were able to passage (68). More importantly, they transduced a pulmonary fibrosis-associated mutant SpC (SFPTCΔexon4) protein into AEC2, resulting in the development of similar AEC2 features also observed in IPF patients (68). Combinations of hiPSC-derived AEC2 and lung organoid models may therefore offer a personalized platform to study the contribution of genetic predisposition and environmental cues to fibrosis development in SSc.
Markers of alveolar epithelial injury, including Krebs von den Lungen-6 (KL-6) and surfactant protein-D (SP-D) (27), are increased in serum and may serve as biomarkers in identifying and monitoring SSc-ILD (69, 70). Importantly, exhaled nitric oxide (FeNO) has been proposed as a marker of (alveolar) inflammation in SSc-ILD. FeNO levels were shown to be increased in SSc compared to IPF patients, although it did not discriminate between SSc-patients with or without ILD (71, 72). In addition to the diagnostic potency of the circulating biomarkers and FeNO, microvascular abnormalities are detectable before clinical presentation of fibrosis (28, 73), and may serve as a prognostic marker to monitor disease progression in subgroups of SSc-ILD patients. Furthermore, levels of smooth muscle cells and myofibroblasts are increased in SSc-ILD lung tissue compared to healthy controls, and may also contribute to fibrosis. Finally, pro-fibrotic mediators, including TGF-β, PDGF, CTGF and thrombin, are increased in bronchoalveolar lavage fluid (BALF) or cells derived from BALF (29) and may contribute to altered cellular composition and phenotype in SSc-ILD. Several other potential serum biomarkers are under current investigation, among which serum chemokine [C-C motif] ligand 2 and 18 (74, 75), matrix metalloproteinase-7 and 12 (76, 77) and chemokine [C-X-C motif] ligand 4 (78, 79). Because a complete overview of current serum biomarkers is beyond the scope of this review, we refer the interested reader to a recently published comprehensive review summarizing the clinically used biomarkers, biomarkers under investigation and their link to mechanistic pathways (80). Interestingly, lung organoid models may contribute to identification of novel biomarkers, as was shown by the identification of the essential profibrotic role of IL-11, upon introduction of genes associated with Hermansky-Pudlak syndrome (67).
In addition to providing mechanistic insight into SSc-ILD pathogenesis, and biomarker identification, preclinical lung organoid models may also be applied to identify new treatment targets and for (personalized) drug screening platforms. So far, treatment of SSc-.ILD is targeted at alveolitis and includes non-selective immunomodulatory drugs like cyclophosphamide, azathioprine and mycophenolate (81, 82), all of which target (T- and/or) B lymphocyte proliferation or trigger cell death (83), as B-lymphocytes have a pivotal role in the pro-inflammatory and profibrotic pathway in SSc-ILD. Standard therapy combined with rituximab, also targeting CD20 on B cells, was shown to be beneficial, with a lower decline of FVC (84), whereas moderate effects were seen on FVC levels and HRCT fibrosis scores (35). The observation that anti-inflammatory and immune suppressive treatment with e.g., prednisone and azathioprine (85), or administration of interferon gamma (86), are clinically not effective, suggests that (late/end-stage) IPF does not result from a primary immunopathogenic process. Recently approved antifibrotic drugs that target selected profibrotic mediators has improved the treatment of IPF (87). The most widely studied drugs in this category are pirfenidone and nintedanib, which were shown to be safe and effective in IPF treatment. Pirfenidone acts through anti-inflammatory and antifibrotic effects, including down-regulation of TGF-β and TNF-α (88), whereas. Nintedanib is a tyrosine kinase inhibitor that inhibits activation of e.g., VEGF, fibroblast growth factor (FGF), and PDGF pathways (89). As nintedanib was recently approved for IPF treatment (90), its effectivity was also examined in SSc-ILD patients (91). Nintedanib demonstrated a clear effect on the annual rate of decline in FVC in SSc-ILD patients (91, 92), suggesting that targeting common pro-fibrotic pathways of lung fibrosis may be of therapeutic relevance (28, 93). Therapeutic targets (under investigation) for IPF may be therefore also be considered for treatment of SSc-ILD.
Regarding treatments under evaluation for IPF, a potential therapeutic target is autotaxin, an enzyme responsible for lysophosphatidic acid (LPA) production (94), both of which are increased in IPF, suggesting that the autotaxin-LPA pathway has a pathogenic role in this disorder (95, 96). Results from a phase-2 trial with GLPG1690, targeting autotaxin, were encouraging, especially regarding decline in FVC (97). Clinical assessment of GLPG1690 as treatment for IPF patients is currently ongoing in a phase-3 trial. A phase 2 trial of pamrevlumab, targeting CTGF (16, 17), also showed a decreased rate in decline of forced vital capacity (FVC) as compared with placebo (98). However, the results of this phase-2 trial must be treated with caution, pending an adequately powered phase 3 study. Furthermore, pentraxin-2, which inhibits differentiation of monocytes toward profibrotic fibrocytes and inhibits TGF-β production (41, 44, 99), may be targeted to stabilize or restore lung function. Results from a phase-2 trial with pentraxin-2 showed a slower decline of FVC compared to placebo (99).
Other treatments that have been evaluated in SSc-ILD include abatacept, tocilizumab, and autologous stem-cell transplantation, and are reviewed elsewhere (6, 7, 84, 91). In daily practice, however, there is no consensus about the right timing of initiating anti-inflammatory and/or antifibrotic treatment and exact patient selection. Furthermore, up to date there are no accepted prognostic models available to identify SSc patients at high risk for the development of lung fibrosis (100). Lung organoid models may therefore be applied to identify new treatment targets and for (personalized) drug screening platforms. Importantly, these models may be used to examine the effect of combined therapies targeting pro-fibrotic and pro-inflammatory pathways simultaneously. Patient-derived lung cancer organoids, for example, were able to predict patient-specific drug responses and drug resistance (101). Such patient-specific models in the future may be used to test drug combinations, drug toxicity and identification of novel drug targets. Furthermore, new pre-clinical models may aid in discriminating SSc patients at high risk for development of ILD.
Current concepts state that impaired or otherwise dysregulated repair of lung epithelial injury is an important driver of (progression of) fibrosis in SSc-ILD, but this is likely not unique to SSc-ILD. Delineating the triggers and/or the dysregulation of repair may reveal features that distinguish SSc-ILD from other ILD. Furthermore, exhaustion and dysfunction of progenitor cells may contribute to the failure in resolving damage. Pharmacological activation of lung progenitor cells may thus be a promising approach for treatment of especially early stage SSc-ILD.
To address these questions, a human lung model that recapitulates essential features of SSc-ILD is required to help understanding mechanisms underlying SSc-ILD and develop new treatments. Availability of such a model, that is also suitable for e.g., screening small-molecule libraries, would be a major step forward. In addition, such a model may provide novel tools for personalized medicine to identify those that may benefit from an anti-inflammatory or an antifibrotic treatment or combined treatment strategies. The need for this is further illustrated by the recent introduction of the antifibrotic drug nintenadib in the treatment of SSc-ILD patients, based on clinical trials showing clear efficacy with a similar slowdown in the rate of lung function decline as found in IPF patients treated with antifibrotic agents (91). SSc-ILD patients with progressive fibrosis may therefore be selected for treatment with these drugs.
The concept of using organoid cultures for selecting individual patients for the optimal treatment is illustrated by the recent publication by Berkers and co-workers (102), in which rectal organoids grown from stem cells provide a tool for personalized medicine in patients suffering from cystic fibrosis. Organoids in this study showed a high correlation between in vitro and in vivo effects of drugs and demonstrated good to excellent predictive value for preclinical identification of response to treatment. Studies such as those by Strikoudis et al., showed that organoids can also be used to study fibrotic lung disease, indicating that organoid technology may help to understand underlying mechanisms of SSc-ILD (Figure 2) (67).
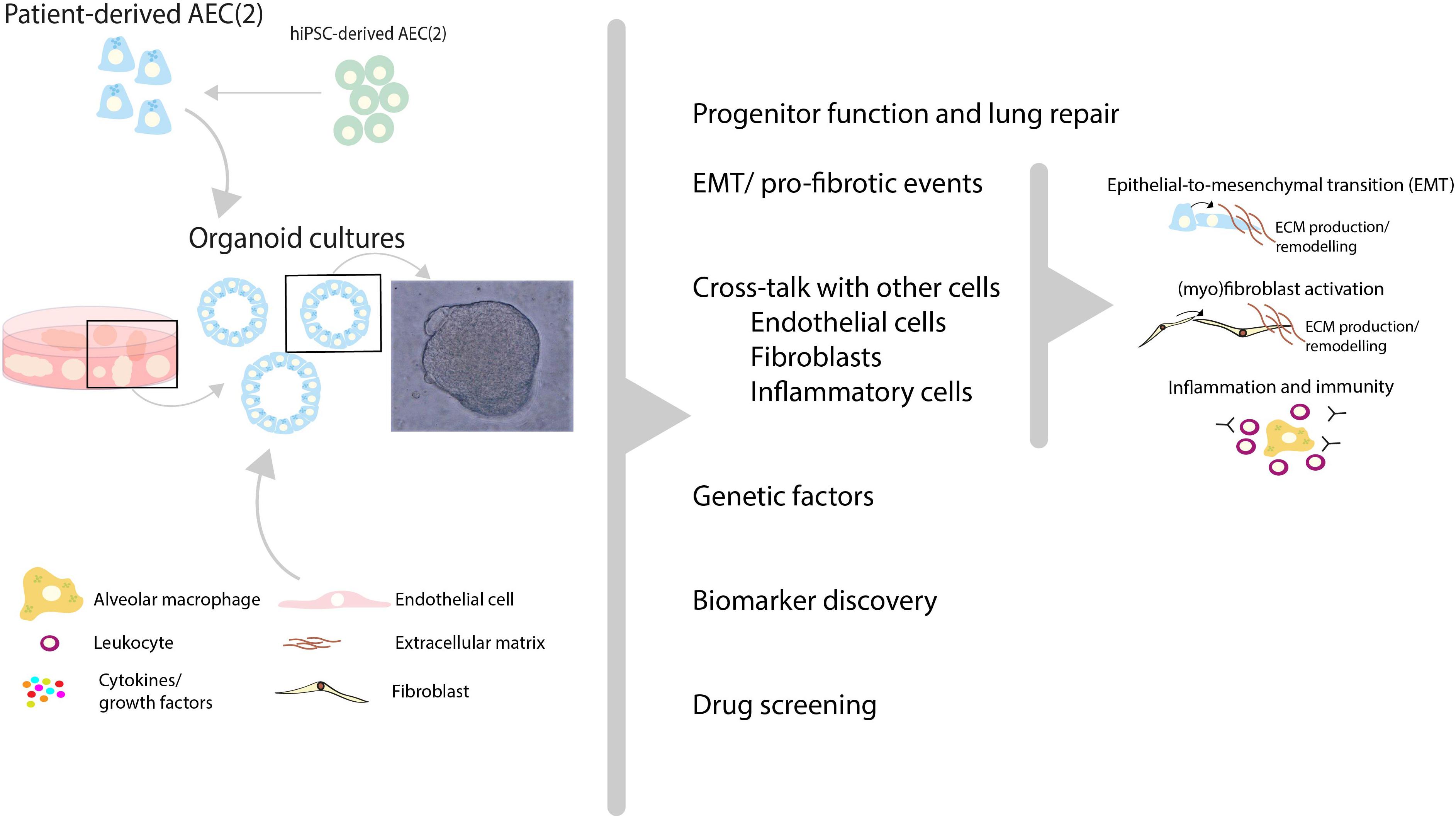
Figure 2. Recent advances in human lung organoid models may help to unravel pathogenic mechanisms and new treatment targets of SSc-ILD. Patient-derived pulmonary cells, either isolated from lung tissue or derived from human induced pluripotent stem cells (hiPSC), can be expanded in vitro using 3D organoid culture models. In these models, a variety of (disease-)relevant characteristics including progenitor cell function, profibrotic features, interactions with other cells (endothelial cells, fibroblasts etc.) and inflammatory cell recruitment can be studied. In addition, in such models the contribution of genetic factors to SSc-ILD can be studied using either patient-derived cells with specific genetic features, or by genetic editing of primary AEC (2) or hiPSC-derived AEC. These 3D lung organoid cultures can be applied to study various aspects of SSc-ILD pathogenic mechanisms and may therefore provide patient-relevant models which may contribute to biomarker and target discovery and aid in personalized drug screening for SSc-ILD.
Important features of fibrotic lung diseases include matrix abnormalities, leading to increased tissue stiffness and consequent altered mechanical forces to which pulmonary cells are exposed, resulting in e.g., increased TGF-β signaling and fibroblast activation (46, 103). These alterations can be mimicked with state-of-the art organs-on-chips (including lung-chips) platforms, which provide us with unprecedented combinations of important features in the lung, including matrix stiffness, mechanical force (breathing pattern), air exposure and co-culture with various relevant cell types (5, 104). Combining organoid and organ-on-chip technology may lead to better patient-relevant culture models, which can be used for drug screening purposes and research into the pathogenesis.
In addition to these advances in in vitro modeling, there are also important developments in analysis of cells in lung tissue that can also be applied in the analysis of the above-mentioned cellular models. Patient-derived cells can for instance be analyzed using single-cell-based RNA sequencing, which has already resulted in the identification of previously undescribed cell populations in SSc-ILD (105). Furthermore, patient-derived cells can now be analyzed at the protein level using (imaging) mass cytometry, enabling multidimensional unbiased analysis of more than 40 markers simultaneously (106) and an unprecedented potential for discovery of molecular culprits of disease. Mass cytometry analyses already revealed distinct immune cell signatures in patients with SSc (107).
In conclusion, the mechanisms underlying pulmonary fibrosis in SSc-ILD are not completely unraveled, and knowledge on fibrotic processes have been acquired mostly from studies in idiopathic pulmonary fibrosis (IPF). Recent advances in human preclinical models and biomedical technology may be used to unravel pathogenic mechanisms and new treatment targets of SSc-ILD (summarized in Figure 1). Developments in in vitro modeling using patient-derived organoid-based culture models and lung-chip platforms, as well as single cell analysis using e.g., single-cell RNA sequencing and (imaging) mass cytometry will be important to further broaden our insight in SSc-ILD pathogenesis and develop better and personalized treatments.
PK and EM drafted the manuscript. PK prepared the figures. PK, EM, PH, MN, and MG edited and revised the manuscript, and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Research conducted by PK is supported by a grant from TAS-ZonMW (40414009816007).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2. Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis. (2007) 66:940–4. doi: 10.1136/ard.2006.066068
3. Tyndall AJ, Bannert B, Vonk M, Airo P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. (2010) 69:1809–15.
4. Kowal-Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. (2017) 76:1327–39.
5. Sundarakrishnan A, Chen Y, Black LD, Aldridge BB, Kaplan DL. Engineered cell and tissue models of pulmonary fibrosis. Adv Drug Deliv Rev. (2018) 129:78–94. doi: 10.1016/j.addr.2017.12.013
6. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. (2017) 389:1941–52. doi: 10.1016/S0140-6736(17)30866-8
7. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. (2018) 378:1811–23. doi: 10.1056/NEJMra1705751
8. Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med. (2014) 189:1161–72. doi: 10.1164/rccm.201312-2221PP
9. Winters NI, Burman A, Kropski JA, Blackwell TS. Epithelial injury and dysfunction in the pathogenesis of idiopathic pulmonaryfibrosis. Am J Med Sci. (2019) 357:374–8. doi: 10.1016/j.amjms.2019.01.010
10. Salton F, Volpe MC, Confalonieri M. Epithelial(-)mesenchymal transition in the pathogenesis of idiopathic pulmonary fibrosis. Medicina (Kaunas). (2019) 55:83. doi: 10.3390/medicina55040083
11. Hill C, Jones MG, Davies DE, Wang Y. Epithelial-mesenchymal transition contributes to pulmonary fibrosis via aberrant epithelial/fibroblastic cross-talk. J Lung Health Dis. (2019) 3:31–5. doi: 10.29245/2689-999X/2019/2.1149
12. Saito A, Horie M, Nagase T. TGF-beta signaling in lung health and disease. Int J Mol Sci. (2018) 19:2460. doi: 10.3390/ijms19082460
13. Dantas AT, Goncalves SM, de Almeida AR, Goncalves RS, Sampaio MC, Vilar KM, et al. Reassessing the role of the active TGF-beta1 as a biomarker in systemic sclerosis: association of serum levels with clinical manifestations. Dis Markers. (2016) 2016:6064830. doi: 10.1155/2016/6064830
14. Fernandez IE, Eickelberg O. The impact of TGF-beta on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc. (2012) 9:111–6. doi: 10.1513/pats.201203-023AW
15. Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF. Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Discov. (2020) 19:57–75. doi: 10.1038/s41573-019-0040-5
16. Kono M, Nakamura Y, Suda T, Kato M, Kaida Y, Hashimoto D, et al. Plasma CCN2 (connective tissue growth factor; CTGF) is a potential biomarker in idiopathic pulmonary fibrosis (IPF). Clinica Chimica Acta. (2011) 412:2211–5. doi: 10.1016/j.cca.2011.08.008
17. Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. (2012) 5(Suppl. 1):S24. doi: 10.1186/1755-1536-5-S1-S24
18. Barratt SL, Blythe T, Jarrett C, Ourradi K, Shelley-Fraser G, Day MJ, et al. Differential expression of VEGF-Axxx isoforms is critical for development of pulmonary fibrosis. Am J Respir Crit Care Med. (2017) 196:479–93. doi: 10.1164/rccm.201603-0568OC
19. Barratt SL, Flower VA, Pauling JD, Millar AB. VEGF (vascular endothelial growth factor) and fibrotic lung disease. Int J Mol Sci. (2018) 19:1269. doi: 10.3390/ijms19051269
20. Gkatzis K, Taghizadeh S, Huh D, Stainier DYR, Bellusci S. Use of three-dimensional organoids and lung-on-a-chip methods to study lung development, regeneration and disease. Eur Respir J. (2018) 52:1800876. doi: 10.1183/13993003.00876-2018
22. Hogan BL, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CC, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. (2014) 15:123–38. doi: 10.1016/j.stem.2014.07.012
23. Paolicelli G, Luca A, Jose SS, Antonini M, Teloni I, Fric J, et al. Using lung organoids to investigate epithelial barrier complexity and IL-17 signaling during respiratory infection. Front Immunol. (2019) 10:323. doi: 10.3389/fimmu.2019.00323
24. Tan Q, Choi KM, Sicard D, Tschumperlin DJ. Human airway organoid engineering as a step toward lung regeneration and disease modeling. Biomaterials. (2017) 113:118–32. doi: 10.1016/j.biomaterials.2016.10.046
25. Tan Q, Ma XY, Liu W, Meridew JA, Jones DL, Haak AJ, et al. Nascent lung organoids reveal epithelium- and bone morphogenetic protein-mediated suppression of fibroblast activation. Am J Respir Cell Mol Biol. (2019) 61:607–19. doi: 10.1165/rcmb.2018-0390OC
26. Ng-Blichfeldt JP, de Jong T, Kortekaas RK, Wu X, Lindner M, Guryev V, et al. TGF-beta activation impairs fibroblast ability to support adult lung epithelial progenitor cell organoid formation. Am J Physiol Lung Cell Mol Physiol. (2019) 317:L14–28. doi: 10.1152/ajplung.00400.2018
27. Akter T, Silver RM, Bogatkevich GS. Recent advances in understanding the pathogenesis of scleroderma-interstitial lung disease. Curr Rheumatol Rep. (2014) 16:411. doi: 10.1007/s11926-014-0411-1
28. Bagnato G, Harari S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev. (2015) 24:102–14. doi: 10.1183/09059180.00003214
29. Kowal-Bielecka O, Kowal K, Highland KB, Silver RM. Bronchoalveolar lavage fluid in scleroderma interstitial lung disease: technical aspects and clinical correlations: review of the literature. Semin Arthritis Rheum. (2010) 40:73–88. doi: 10.1016/j.semarthrit.2008.10.009
30. Tourkina E, Bonner M, Oates J, Hofbauer A, Richard M, Znoyko S, et al. Altered monocyte and fibrocyte phenotype and function in scleroderma interstitial lung disease: reversal by caveolin-1 scaffolding domain peptide. Fibrogenesis Tissue Repair. (2011) 4:15. doi: 10.1186/1755-1536-4-15
31. Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun. (2016) 7:11703. doi: 10.1038/ncomms11703
32. Kelsh R, You R, Horzempa C, Zheng M, McKeown-Longo PJ. Regulation of the innate immune response by fibronectin: synergism between the III-1 and EDA domains. PLoS One. (2014) 9:e102974. doi: 10.1371/journal.pone.0102974
33. Bhattacharyya S, Varga J. Endogenous ligands of TLR4 promote unresolving tissue fibrosis: Implications for systemic sclerosis and its targeted therapy. Immunol Lett. (2018) 195:9–17. doi: 10.1016/j.imlet.2017.09.011
34. Kang H. Role of MicroRNAs in TGF-beta signaling pathway-mediated pulmonary fibrosis. Int J Mol Sci. (2017) 18:2527. doi: 10.3390/ijms18122527
35. Jordan S, Distler JH, Maurer B, Huscher D, van Laar JM, Allanore Y, et al. Effects and safety of rituximab in systemic sclerosis: an analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis. (2015) 74:1188–94. doi: 10.1136/annrheumdis-2013-204522
36. Lafyatis R, O’Hara C, Feghali-Bostwick CA, Matteson EB. cell infiltration in systemic sclerosis-associated interstitial lung disease. Arthritis Rheum. (2007) 56:3167–8. doi: 10.1002/art.22847
37. Pattanaik D, Brown M, Postlethwaite BC, Postlethwaite AE. Pathogenesis of systemic sclerosis. Front Immunol. (2015) 6:272. doi: 10.3389/fimmu.2015.00272
38. Wells AU, Denton CP. Interstitial lung disease in connective tissue disease–mechanisms and management. Nat Rev Rheumatol. (2014) 10:728–39. doi: 10.1038/nrrheum.2014.149
39. Liaskos C, Rentouli S, Simopoulou T, Gkoutzourelas A, Norman GL, Brotis A. et al. Anti-C1q autoantibodies are frequently detected in patients with systemic sclerosis associated with pulmonary fibrosis. Br J Dermatol. (2019) 181:138–46. doi: 10.1111/bjd.17886
40. Allden SJ, Ogger PP, Ghai P, McErlean P, Hewitt R, Toshner R, et al. The transferrin receptor CD71 delineates functionally distinct airway macrophage subsets during idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2019) 200:209–19. doi: 10.1164/rccm.201809-1775OC
41. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. (2019) 54:1802441. doi: 10.1183/13993003.02441-2018
42. Zhou Y, Peng H, Sun H, Peng X, Tang C, Gan Y, et al. Chitinase 3-like 1 suppresses injury and promotes fibroproliferative responses in Mammalian lung fibrosis. Sci Transl Med. (2014) 6:240ra76. doi: 10.1126/scitranslmed.3007096
43. Pechkovsky DV, Prasse A, Kollert F, Engel KM, Dentler J, Luttmann W, et al. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin Immunol. (2010) 137:89–101. doi: 10.1016/j.clim.2010.06.017
44. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. (2019) 199:1517–36.
45. Kolb M, Gauldie J, Bellaye PS. Editorial: extracellular matrix: the common thread of disease progression in fibrosis? Arthritis Rheumatol. (2016) 68:1053–6. doi: 10.1002/art.39569
46. Upagupta C, Shimbori C, Alsilmi R, Kolb M. Matrix abnormalities in pulmonary fibrosis. Eur Respir Rev. (2018) 27:180033. doi: 10.1183/16000617.0033-2018
47. Sakamoto N, Kakugawa T, Hara A, Nakashima S, Yura H, Harada T, et al. Association of elevated alpha-defensin levels with interstitial pneumonia in patients with systemic sclerosis. Respir Res. (2015) 16:148. doi: 10.1186/s12931-015-0308-1
48. Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest. (2010) 90:812–23. doi: 10.1038/labinvest.2010.73
49. De Lauretis A, Sestini P, Pantelidis P, Hoyles R, Hansell DM, Goh NS, et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. J Rheumatol. (2013) 40:435–46. doi: 10.3899/jrheum.120725
50. Fernandez IE, Greiffo FR, Frankenberger M, Bandres J, Heinzelmann K, Neurohr C, et al. Peripheral blood myeloid-derived suppressor cells reflect disease status in idiopathic pulmonary fibrosis. Eur Respir J. (2016) 48:1171–83. doi: 10.1183/13993003.01826-2015
51. Jose SS, De Zuani M, Tidu F, Hortova Kohoutkova M, Pazzagli L, Forte G, et al. Comparison of two human organoid models of lung and intestinal inflammation reveals Toll-like receptor signalling activation and monocyte recruitment. Clin Transl Immunol. (2020) 9:e1131. doi: 10.1002/cti2.1131
52. Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Science. (2019) 364:952–5. doi: 10.1126/science.aaw6985
53. Cattaneo CM, Dijkstra KK, Fanchi LF, Kelderman S, Kaing S, van Rooij N, et al. Tumor organoid-T-cell coculture systems. Nat Protoc. (2020) 15:15–39. doi: 10.1038/s41596-019-0232-9
54. Purwada A, Jaiswal MK, Ahn H, Nojima T, Kitamura D, Gaharwar AK, et al. Ex vivo engineered immune organoids for controlled germinal center reactions. Biomaterials. (2015) 63:24–34. doi: 10.1016/j.biomaterials.2015.06.002
55. Purwada A, Singh A. Immuno-engineered organoids for regulating the kinetics of B-cell development and antibody production. Nat Protoc. (2017) 12:168–82. doi: 10.1038/nprot.2016.157
56. Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. (2007) 356:1317–26. doi: 10.1056/NEJMoa066157
57. Burman A, Tanjore H, Blackwell TS. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. (2018) 68-69:355–65. doi: 10.1016/j.matbio.2018.03.015
58. Dickens JA, Malzer E, Chambers JE, Marciniak SJ. Pulmonary endoplasmic reticulum stress-scars, smoke, and suffocation. FEBS J. (2019) 286:322–41. doi: 10.1111/febs.14381
59. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. (2011) 364:1503–12.
60. Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature. (2014) 505:412–6. doi: 10.1038/nature12807
61. Heukels P, Moor CC, von der Thusen JH, Wijsenbeek MS, Kool M. Inflammation and immunity in IPF pathogenesis and treatment. Respir Med. (2019) 147:79–91. doi: 10.1016/j.rmed.2018.12.015
62. Peljto AL, Steele MP, Fingerlin TE, Hinchcliff ME, Murphy E, Podlusky S, et al. The pulmonary fibrosis-associated MUC5B promoter polymorphism does not influence the development of interstitial pneumonia in systemic sclerosis. Chest. (2012) 142:1584–8. doi: 10.1378/chest.12-0110
63. Stock CJW, De Lauretis A, Visca D, Daccord C, Kokosi M, Kouranos V, et al. Defining genetic risk factors for scleroderma-associated interstitial lung disease : IRF5 and STAT4 gene variants are associated with scleroderma while STAT4 is protective against scleroderma-associated interstitial lung disease. Clin Rheumatol. (2020) 39:1173–9. doi: 10.1007/s10067-019-04922-6
64. Song GG, Lee YH. The CTLA-4 and MCP-1 polymorphisms and susceptibility to systemic sclerosis: a meta-analysis. Immunol Invest. (2013) 42:481–92. doi: 10.3109/08820139.2013.789910
65. Jacob A, Morley M, Hawkins F, McCauley KB, Jean JC, Heins H, et al. Differentiation of human pluripotent stem cells into functional lung alveolar epithelial cells. Cell Stem Cell. (2017) 21:472–88.e10. doi: 10.1016/j.stem.2017.08.014
66. McCauley KB, Hawkins F, Serra M, Thomas DC, Jacob A, Kotton DN. Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of wnt signaling. Cell Stem Cell. (2017) 20:844–57.e6. doi: 10.1016/j.stem.2017.03.001
67. Strikoudis A, Cieslak A, Loffredo L, Chen YW, Patel N, Saqi A, et al. Modeling of fibrotic lung disease using 3D organoids derived from human pluripotent stem cells. Cell Rep. (2019) 27:3709–23.e5. doi: 10.1016/j.celrep.2019.05.077
68. Shiraishi K, Nakajima T, Shichino S, Deshimaru S, Matsushima K, Ueha S. In vitro expansion of endogenous human alveolar epithelial type II cells in fibroblast-free spheroid culture. Biochem Biophys Res Commun. (2019) 515:579–85. doi: 10.1016/j.bbrc.2019.05.187
69. Yanaba K, Hasegawa M, Takehara K, Sato S. Comparative study of serum surfactant protein-D and KL-6 concentrations in patients with systemic sclerosis as markers for monitoring the activity of pulmonary fibrosis. J Rheumatol. (2004) 31:1112–20.
70. Bonella F, Volpe A, Caramaschi P, Nava C, Ferrari P, Schenk K, et al. Surfactant protein D and KL-6 serum levels in systemic sclerosis: correlation with lung and systemic involvement. Sarcoidosis Vasc Diffuse Lung Dis. (2011) 28:27–33.
71. Girgis RE, Gugnani MK, Abrams J, Mayes MD. Partitioning of alveolar and conducting airway nitric oxide in scleroderma lung disease. Am J Respir Crit Care Med. (2002) 165:1587–91. doi: 10.1164/rccm.2104003
72. Paredi P, Kharitonov SA, Loukides S, Pantelidis P, du Bois RM, Barnes PJ. Exhaled nitric oxide is increased in active fibrosing alveolitis. Chest. (1999) 115:1352–6. doi: 10.1378/chest.115.5.1352
73. Trojanowska M. Cellular and molecular aspects of vascular dysfunction in systemic sclerosis. Nat Rev Rheumatol. (2010) 6:453–60. doi: 10.1038/nrrheum.2010.102
74. Elhai M, Hoffmann-Vold AM, Avouac J, Pezet S, Cauvet A, Leblond A, et al. Performance of candidate serum biomarkers for systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol. (2019) 71:972–82. doi: 10.1002/art.40815
75. Wu M, Baron M, Pedroza C, Salazar GA, Ying J, Charles J, et al. CCL2 in the circulation predicts long-term progression of interstitial lung disease in patients with early systemic sclerosis: data from two independent cohorts. Arthritis Rheumatol. (2017) 69:1871–8. doi: 10.1002/art.40171
76. Kennedy B, Branagan P, Moloney F, Haroon M, O’Connell OJ, O’Connor TM, et al. Biomarkers to identify ILD and predict lung function decline in scleroderma lung disease or idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. (2015) 32:228–36.
77. Manetti M, Guiducci S, Romano E, Bellando-Randone S, Conforti ML, Ibba-Manneschi L, et al. Increased serum levels and tissue expression of matrix metalloproteinase-12 in patients with systemic sclerosis: correlation with severity of skin and pulmonary fibrosis and vascular damage. Ann Rheum Dis. (2012) 71:1064–72. doi: 10.1136/annrheumdis-2011-200837
78. Kafaja S, Clements PJ, Wilhalme H, Tseng CH, Furst DE, Kim GH, et al. Reliability and minimal clinically important differences of forced vital capacity: Results from the Scleroderma Lung Studies (SLS-I and SLS-II). Am J Respir Crit Care Med. (2018) 197:644–52.
79. van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. (2014) 370:433–43. doi: 10.1056/NEJMc1402401
80. Khanna D, Tashkin DP, Denton CP, Renzoni EA, Desai SR, Varga J. Etiology, risk factors, and biomarkers in systemic sclerosis with interstitial lung disease. Am J Respir Crit Care Med. (2020) 201:650–60. doi: 10.1164/rccm.201903-0563CI
81. Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. (2016) 4:708–19.
82. Khanna D, Tashkin DP, Denton CP, Lubell MW, Vazquez-Mateo C, Wax S. Ongoing clinical trials and treatment options for patients with systemic sclerosis-associated interstitial lung disease. Rheumatology (Oxford). (2019) 58:567–79. doi: 10.1093/rheumatology/key151
83. Hall AG, Tilby MJ. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. (1992) 6:163–73. doi: 10.1016/0268-960X(92)90028-O
84. Misra DP, Ahmed S, Agarwal V. Is biological therapy in systemic sclerosis the answer? Rheumatol Int. (2020) 40:679–94. doi: 10.1007/s00296-020-04515-6
85. Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE Jr., Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. (2012) 366:1968–77. doi: 10.1056/NEJMoa1113354
86. King TE Jr., Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L. et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet. (2009) 374:222–8. doi: 10.1016/S0140-6736(09)60551-1
87. Wuyts WA, Antoniou KM, Borensztajn K, Costabel U, Cottin V, Crestani B, et al. Combination therapy: the future of management for idiopathic pulmonary fibrosis? Lancet Respir Med. (2014) 2:933–42. doi: 10.1016/S2213-2600(14)70232-2
88. Kolb M, Bonella F, Wollin L. Therapeutic targets in idiopathic pulmonary fibrosis. Respir Med. (2017) 131:49–57. doi: 10.1016/j.rmed.2017.07.062
89. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2071–82. doi: 10.1056/NEJMoa1402584
90. Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. (2015) 45:1434–45. doi: 10.1183/09031936.00174914
91. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med. (2019) 380:2518–28. doi: 10.1056/NEJMoa1903076
92. Distler O, Brown KK, Distler JHW, Assassi S, Maher TM, Cottin V, et al. Design of a randomised, placebo-controlled clinical trial of nintedanib in patients with systemic sclerosis-associated interstitial lung disease (SENSCIS). Clin Exp Rheumatol. (2017) 35(Suppl. 106):75–81.
93. Renaud L, da Silveira WA, Takamura N, Hardiman G, Feghali-Bostwick C. Prominence of IL6, IGF TLR, and bioenergetics pathway perturbation in lung tissues of scleroderma patients with pulmonary fibrosis. Front Immunol. (2020) 11:383. doi: 10.3389/fimmu.2020.00383
94. Nakanaga K, Hama K, Aoki J. Autotaxin–an LPA producing enzyme with diverse functions. J Biochem. (2010) 148:13–24. doi: 10.1093/jb/mvq052
95. Montesi SB, Mathai SK, Brenner LN, Gorshkova IA, Berdyshev EV, Tager AM, et al. Docosatetraenoyl LPA is elevated in exhaled breath condensate in idiopathic pulmonary fibrosis. BMC Pulm Med. (2014) 14:5. doi: 10.1186/1471-2466-14-5
96. Oikonomou N, Mouratis MA, Tzouvelekis A, Kaffe E, Valavanis C, Vilaras G, et al. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol. (2012) 47:566–74. doi: 10.1165/rcmb.2012-0004OC
97. Maher TM, van der Aar EM, Van de Steen O, Allamassey L, Desrivot J, Dupont S, et al. Safety tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2a randomised placebo-controlled trial. Lancet Respir Med. (2018) 6:627–35. doi: 10.1016/S2213-2600(18)30181-4
98. Richeldi L, Fernandez Perez ER, Costabel U, Albera C, Lederer DJ, Flaherty KR, et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir Med. (2020) 8:25–33. doi: 10.1016/S2213-2600(19)30262-0
99. Raghu G, van den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA, et al. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: a randomized clinical trial. JAMA. (2018) 319:2299–307. doi: 10.1001/jama.2018.6129
100. Cottin V, Brown KK. Interstitial lung disease associated with systemic sclerosis (SSc-ILD). Respir Res. (2019) 20:13. doi: 10.1186/s12931-019-0980-7
101. Kim M, Mun H, Sung CO, Cho EJ, Jeon HJ, Chun SM, et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat Commun. (2019) 10:3991. doi: 10.1038/s41467-019-11867-6
102. Berkers G, Van Mourik P, Vonk AM, Kruisselbrink E, Dekkers JF, De Winter-de Groot KM, et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep (2019) 26:1701–8.e3. doi: 10.1016/j.celrep.2019.01.068
103. Froese AR, Shimbori C, Bellaye PS, Inman M, Obex S, Fatima S, et al. Stretch-induced activation of transforming growth factor-beta1 in pulmonary fibrosis. Am J Respir Crit Care Med. (2016) 194:84–96. doi: 10.1164/rccm.201508-1638OC
104. Nawroth JC, Barrile R, Conegliano D, van Riet S, Hiemstra PS, Villenave R. Stem cell-based lung-on-chips: the best of both worlds? Adv Drug Deliv Rev. (2019) 140:12–32. doi: 10.1016/j.addr.2018.07.005
105. Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Trejo Bittar H, et al. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann Rheum Dis. (2019) 78:1379–87. doi: 10.1136/annrheumdis-2018-214865
106. Ijsselsteijn ME, van der Breggen R, Farina Sarasqueta A, Koning F, De Miranda NA. 40-marker panel for high dimensional characterization of cancer immune microenvironments by imaging mass cytometry. Front Immunol. (2019) 10:2534. doi: 10.3389/fimmu.2019.02534
Keywords: systemic sclerosis, interstitial lung disease, idiopathic pulmonary fibrosis, pathogenesis, organoids, human disease models
Citation: Khedoe P, Marges E, Hiemstra P, Ninaber M and Geelhoed M (2020) Interstitial Lung Disease in Patients With Systemic Sclerosis: Toward Personalized-Medicine-Based Prediction and Drug Screening Models of Systemic Sclerosis-Related Interstitial Lung Disease (SSc-ILD). Front. Immunol. 11:1990. doi: 10.3389/fimmu.2020.01990
Received: 13 March 2020; Accepted: 23 July 2020;
Published: 04 September 2020.
Edited by:
Oliver Distler, University of Zurich, SwitzerlandReviewed by:
Elizabeth Volkmann, University of California, Los Angeles, United StatesCopyright © 2020 Khedoe, Marges, Hiemstra, Ninaber and Geelhoed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Padmini Khedoe, cC5wLnMuai5raGVkb2VAbHVtYy5ubA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.