 Anna Mondino
Anna Mondino Teresa Manzo
Teresa Manzo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 August 2020
Sec. Immunological Memory
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01915
This article is part of the Research Topic Immune Responses to Persistent or Recurrent Antigens: Implications for Immunological Memory and Immunotherapy View all 8 articles
The generation of immunological memory is a hallmark of adaptive immunity by which the immune system “remembers” a previous encounter with an antigen expressed by pathogens, tumors, or normal tissues; and, upon secondary encounters, mounts faster and more effective recall responses. The establishment of T cell memory is influenced by both cell-intrinsic and cell-extrinsic factors, including genetic, epigenetic and environmental triggers. Our current knowledge of the mechanisms involved in memory T cell differentiation has instructed new opportunities to engineer T cells with enhanced anti-tumor activity. The development of adoptive T cell therapy has emerged as a powerful approach to cure a subset of patients with advanced cancers. Efficacy of this approach often requires long-term persistence of transferred T cell products, which can vary according to their origin and manufacturing conditions. Host preconditioning and post-transfer supporting strategies have shown to promote their engraftment and survival by limiting the competition with a hostile tumor microenvironment and between pre-existing immune cell subsets. Although in the general view pre-existing memory can confer a selective advantage to adoptive T cell therapy, here we propose that also “bad memories”—in the form of antigen-experienced T cell subsets—co-evolve with consequences on newly transferred lymphocytes. In this review, we will first provide an overview of selected features of memory T cell subsets and, then, discuss their putative implications for adoptive T cell therapy.
T cells play a crucial role in immunity against pathogens and cancer. Antigen (Ag) encounter, costimulatory signals, and pro-inflammatory cytokines dictate naïve T cell (TN) activation in secondary lymphoid organs, which is followed by clonal expansion and differentiation of effector T cells (TEFF) (1–3). Although the vast majority of TEFF cells die via apoptosis after antigen clearance, stable populations of memory T cells (TM) can persist over time and ensure a rapid recall response upon further encounter with cognate Ag. The TM pool is composed of several subsets harboring a considerable heterogeneity in trafficking, localization, effector functions, and durability; all features with direct consequences on recall responses.
The growing understanding of the cellular and molecular events underlying their behaviors, functionality, and persistence has instructed the development of defined T cell manufacturing protocols suitable for adoptive T cell therapy (ACT) against cancer and helped predict their behavior and efficacy in vivo. In the context of ACT, tumor-specific TM lymphocytes are generally produced in vitro by the expansion of tumor-infiltrating T cells (TILs) derived from tumor specimens or peripheral blood, or by the genetic engineering of peripheral blood mature T cells with tumor-specific T cell receptor (TCR) or chimeric antigen receptor (CAR). The adoption of ACT envisages several steps: (1) generation of T cell products, (2) conditioning of the host, (3) T-cell transfer, and (4) post-transfer cell support. Each of these steps can have a critical impact on ACT therapeutic efficacy, and vary according to infused T cells’ TM features, and simultaneously shape the immune landscape of the host. Indeed, mounting evidences indicate that the differentiation status of the transferred T cells along with tumor-intrinsic and tumor-extrinsic factors are important determinants of ACT clinical outcome (4). Once (re)infused in patients, tumor-specific T lymphocytes face the challenge to react to tumor lesions, which might vary in anatomical distribution and complexity, in the presence of a plethora of pre-existing TM subsets, which might promote or oppose infused T cell activity. Although the density of CD3+ TILs is generally a favorable prognostic factor for responses to therapy and overall survival of cancer patients, TILs can prove hyporesponsive or exhausted, and as such represent a barrier for ACT. Here, we review some of the seminal characteristics of memory/exhausted T cell subsets [reviewed in details elsewhere (3, 5, 6)] to highlight how pre-existing TM might assist or outcompete newly transferred T cells, and by that represent an advantage or disadvantage for current ACT.
Although TEFF cells mostly disappear upon pathogen/antigen clearance, TM cells survive and patrol against secondary infection or metastatic recurrence in the case of tumors (7, 8). TM cells consist of a collection of distinct subsets of cells with considerable heterogeneity in phenotype, function, location, and trafficking (9, 10). Based on distinctive migratory and effector properties, circulating memory CD4 T cells were initially classified in central memory T cells (TCM cells) and effector memory T cells (TEM cells) (11). CD4 TCM cells, similar to TN cells, express the lymph node and T cell zone homing receptors CD62L and CCR7 and produce substantial amount of IL-2, but lower levels of effector cytokines and cytotoxic molecules (11). A similar phenotype also characterized memory CD8 T cells. CD4 and CD8 TCM cells have good proliferative capacity in response to Ag and ability to self-renew in response to IL-7 and IL-15. Within the long-lived memory subsets, also stem cell memory T cells (TSCM) can be identified for their more naïve-phenotypic qualities and stem cell–like properties including the capacity to reconstitute the entire spectrum of memory and effector T cell subsets (12–15). The long-lived properties of both TCM and TSCM have been considered for effective vaccine design, and exploited in the setting of ACT, where they are associated with improved anti-tumor responses and therapeutic benefit. TEM cells, instead, generally lack CD62L and CCR7, produce effector cytokines, and have higher cytotoxicity when compared with TCM. Although TCM circulate between secondary lymphoid organs and blood, TEM circulate between blood and non-lymphoid tissues, where they persist long after Ag clearance (16, 17). The surface expression of the chemokine receptor CX3CR1 further refines TCM (CX3CR1–) and TEFF (CX3CR1+), (18) and identifies an additional peripheral memory T cell subset (TPM CX3CR1int), which appear to possess the highest steady-state self-renewal capacity of all TM subsets, being able to survey peripheral tissues and return to secondary lymphoid organs, via the lymphatic system. A further distinct subset of TM is constituted by tissue resident memory T cells (TRM cells), which represent the front-line defense in case of reinfection, especially at barrier sites, such as the skin, lung, and gut, having rapid proliferation potential and immediate effector function capacity, (19–21) critically important in cancer immunology (22, 23). These have been described among CD4 and CD8 T cell subsets as being able to remain positioned within non-lymphoid tissues after Ag clearance and lack recirculation capacities (16, 17, 24). CD8 TRM were initially characterized the by expression of CD103 [αE(CD103)β7], CD49a (VLA-1 or α1β1), and the C-type lectin CD69, critical for their retention into tissues, (25, 26) and for recruitment within epithelial tumor regions (27). More recently, data have shown that some CD8 TRM cells lack CD103, and that this integrin is not an absolute marker for residency of CD4 TRM, which also appear more heterogeneous compared with CD8 TRM (28–30). Although the origin of CD4 TRM cells remains debated, recent evidences suggest they might originate from CD4 T follicular helper subsets, which share with CD8 TRM some key features related to their migration, differentiation, and maintenance [reviewed in (31)]. Lastly, it is worth mentioning that also Ag-inexperienced T cells with CD44hiCD122+ memory features have been described both in mice and humans (32, 33). These can arise in the thymus (innate-like memory cells), or in response to lymphopenia (virtual-memory T cell), and can contribute to protective anti-tumor immunity (32–35).
Overall, the heterogeneity of TM subsets, with defined phenotypes, functions, and anatomical distribution, contributes to effective protective immunity. This has to be taken into consideration when developing ACT-based strategies, as adoptively transferred T cell products should closely mimic the behaviors of naturally occurring cells and be endowed with both effector functions, to promote acute tumor debulking, and long-term persistence, to promote surveillance against recurrent disease.
The strength and duration of TCR engagement by cognate peptide–MHC complexes (signal 1), co-stimulation (signal 2), and inflammatory cytokines (signal 3) contribute to naïve T cell priming and TM differentiation (36–38). CD4 TN cells require persistent TCR-peptide/MHCII interactions to achieve maximal clonal expansion (39). This can be regulated by the strength of TCR-peptide/MHCII binding (40) or by repeated contacts with Ag-bearing APCs, (41) and have direct consequences on TEFF cell function (42). In contrast, CD8 TN cells can be “programmed” by short-term access to Ag to allow TEFF differentiation, and prolonged and stable interaction with Ag-bearing APC appear necessary for full T cell activation and memory generation (43). Costimulatory ligand/receptor pairs, able to control the magnitude of the T cell response and the rate of TM development and maintenance, generally provide signal 2. CD28 and members of the tumor necrosis factor receptor (TNFR) family, such as CD27, 4-1BB, and OX-40, in particular impact the formation and/or responsiveness of the memory CD4 and CD8 T cell pool [reviewed in (44)]. The expression of costimulatory receptor can be constitutive, as in the case of CD28 or CD27, or also inducible, as in the case of OX40 or 4-1BB, in response to IL-7 and IL-15, which promote TM survival and homeostatic turnover (44). The importance of costimulation in adopting the appropriate TM feature has also been demonstrated in ACT (45). For instance, exogenous agonistic anti-4-1BB IgG4 significantly promoted the yield of TIL expansion, and programmed them for enhanced survival and effector functions (36). Similar CAR-T cell engineering has evolved to include costimulatory domains in the original CAR construct. Initial studies demonstrated the beneficial effect of including the intracellular domain of CD28 to elicit both TCR and CD28 activation. Later, it became clear that a range of other costimulatory domains, including ICOS, and the TNFR superfamily members 4−1BB, OX40, and CD27, could rather promote long-lived memory cells (46). In addition, the engineering of defined CD28- or 41BB-costimulatory moieties within CAR constructs was proven to favor glycolysis (CD28) or mitochondrial biogenesis and oxidative metabolism (41BB) with direct implication for memory development in vivo (47).
Inflammatory cytokines (signal 3) also contribute to T cell priming. They do so by promoting T cell proliferation, effector functions acquisition, and long-term maintenance of protective immunity. At least three candidate cytokines, IL-12, type-I IFNs, and IFN-γ, have been shown to differentially contribute to CD8 TM cell differentiation (7, 48). In the case of CD4 T cells, according to the type of immunological insult, the host-pathogen interaction, and resulting pro-inflammatory cytokine expression, promote differentiation of various T helper subsets (TH1, TH2, TH17, T follicular helper cells/TFH, T regulatory cells/reg) with pleiotropic effector functions, some degree of plasticity, and various memory-forming potential (31, 49, 50).
Overall, the characterization of the signals dictating T cell differentiation has been instrumental to identify specific manufacturing conditions for ACT. The use of TCR and costimulatory receptor engaging ligands, in combination with common γ chain (γc) cytokines and defined nutrients, can instruct T cells to adopt different TM features, with direct consequences on T cell engraftment and long-term survival.
Any interferences in the three-signal model of T cell activation can result in dysfunctional phenotypes, sometimes endowed with inhibitory functions (51). For instance, defective co-stimulation, negative co-stimulation by inhibitory receptors, or anti-inflammatory cytokines can make T cells anergic and tolerant to cognate Ag, (52, 53) or induce the differentiation of Treg cells. Ag-experienced tolerant T cells, compared with TM, express high levels of co-inhibitory receptors (e.g., PD1, CTLA-4, TIM3, and LAG3) and transcriptional repressor (e.g., EGR1/2, DUSP2), low levels of cytokines and chemokine receptors, and mostly lack effector functions (53). In addition, continuous stimulation through the TCR, typically induced by Ag persistence during chronic infections or cancer, drives CD4 and CD8 TM into a state referred to as T cell exhaustion (TEX cells) (54, 55). TEX cells are characterized by progressive loss of effector functions, metabolic deregulation, poor memory recall, and homeostatic self-renewal (54, 56). They acquire high and sustained expression of different inhibitory receptors, which are not found on TM cells arising after resolution of an acute infection (57). Although CD8 T cell exhaustion was first described in LCMV chronic infection, (58, 59) it is now clear that it occurs in several other chronic infections, (60–62) in autoimmune disorders (63, 64) as well as in cancer (56, 65). TEX cells, along with classical anergic T cells and Treg emerging from the thymus or generated by the conversion of TEFF cells, (66) might induce a dysfunctional state in tumor-infiltrating CTLs (67) and represent barriers to engraftment and function of ACT products. Accordingly, depleting strategies have improved responses to immunotherapy (68).
Although initially described in the context of infectious diseases, tumor-associated TRM should also be carefully considered as their role in oncology has now been established (22, 69). In general terms, TRM produce effector molecules such as IFN-γ, TNF-α, and IL-2 more rapidly than other memory T cells, (70) orchestrating the recruitment of auxiliary immune cells to the infected site (71). In tumor immunity, TRM have nevertheless been reported to play controversial functions. For instance, CD8 TRM cells driven by autoimmune vitiligo or primed by active vaccination conferred protection against melanomas, and their intra-tumoral representation was correlated with favorable prognosis in a variety of cancers (72, 73). In other studies, instead, tumor-associated CD103+ CD8 TRM showed regulatory properties (CTLA-4 and IL-10 expression), and were found to adopt a dysfunctional phenotype over time by expressing the highest levels of PD-1, TIM-3, CTLA-4, and LAG-3 (74). It should, however, be noted that tumor-associated TRM can respond to anti-PD-1 treatment and were described to exert potent cytotoxic and effector functions in melanoma patients (75). Likewise, tumor-associated CD103+CD4 TRM have been described to be highly enriched for tumor-specific T cells (76) and suppress tumor growth through the secretion of TNF-α and IFN-γ or direct killing of tumor cells (77, 78). Again, these cells express high levels of co-inhibitory receptors, and yet whenever activated in appropriate conditions (such as by providing agonistic stimulation of CD27 or CD28 co-stimulatory molecules and/or immune checkpoint blockers) (79) might represent valuable allies and support tumor rejection. Otherwise, TRM cells may be an obstacle to ACT products, as they might compete for local resources. For instance, TRM cells are highly sensitive to IL-15, a cytokine which newly transferred T cells depend on (80). Thus, a better understanding of the molecular mechanisms mediating CD4 and CD8 TRM differentiation and interplay will allow harnessing the protective capacity of these memory subsets and modulate their activity in the context of ACT. In this respect, tumor-resident CD4 TRM might reveal useful to provide local help to CD8 ACT products, and by that their function and survival.
Adoptive T cell therapy for tumor therapy is generally provided in the context of allogeneic or autologous settings. In the case of HLA-matched allogeneic donors, mature T cells comprise undefined TN and TM populations transferred at the time of stem cell transplant or shortly after (81–83). According to their nature and TM composition, allogeneic T cells can provide graft-versus-tumor effects and also graft-versus-host disease owing to the presence of T cell reactive to minor histocompatibility antigens (84). In the autologous settings, instead, T cell products can be generated by the expansion of tumor-reactive cells isolated from tumor specimens (TILs) or by the genetic engineering of peripheral blood T cells with TCR or CAR, an antibody-derived single-chain variable fragment fused to T cell signaling domain(s), specific for tumor-specific/associated Ags (46, 85, 86). The phenotype of ACT T cell products can impact on therapy efficacy (87). Given that natural and manufactured TM share similar requirements, they might compete for space, nutrients, cytokines, or TCR-engaging ligands in vivo (Figure 1). According to manufacturing culture conditions, T cells can acquire various TM features. Which TM cell subset represents the most effective in driving durable cures in cancer patients has been debated and remains to be fully elucidated and might vary according to the cancer type/state. Preclinical and clinical studies have shown that less differentiated TSCM and TCM display better expansion, persistence, and antitumor activity in vivo when compared with fully differentiated TEFF (88–90). Accordingly, in retrospective analysis of ACT trials, more favorable objective clinical responses were found with less differentiated T cell products (88, 91). Although initial studies adopted IL-2 to support the in vitro expansion of engineered T cells, it soon became evident that T cell products had limited survival potential when transferred in vivo (92). Rather, shorter expansion times, and the use of IL-7, IL-15, and IL-21 provided T cells with longer persistence in vivo (92). As TSCM and TCM are found in limited number in the peripheral blood and at the tumor site, in vitro methods have been defined to generate them starting from TN precursors. These include polyclonal activation (αCD3/28 antibody−conjugated beads), homeostatic cytokines (IL-7, IL-15, and IL-21), (93–95) inhibitors of specific signaling pathways (such as GSK3β, AKT, or mTOR) (96, 97) or epigenetic regulators, (98) and nutrients/metabolites aimed at arresting terminal differentiation and promoting memory stem cell phenotype, during ex vivo T cell expansion. The same extracellular cues that guide the manufacturing tumor-reactive lymphocyte continue to affect the activity of adoptively transferred T cells, which, as introduced in the previous section, compete with pre-existing TM subsets and the tumor microenvironment (TME), once re-infused in patients. We argue that this competition might have direct consequences on in vivo differentiation and survival of ACT products and their therapeutic effect (Figure 2).
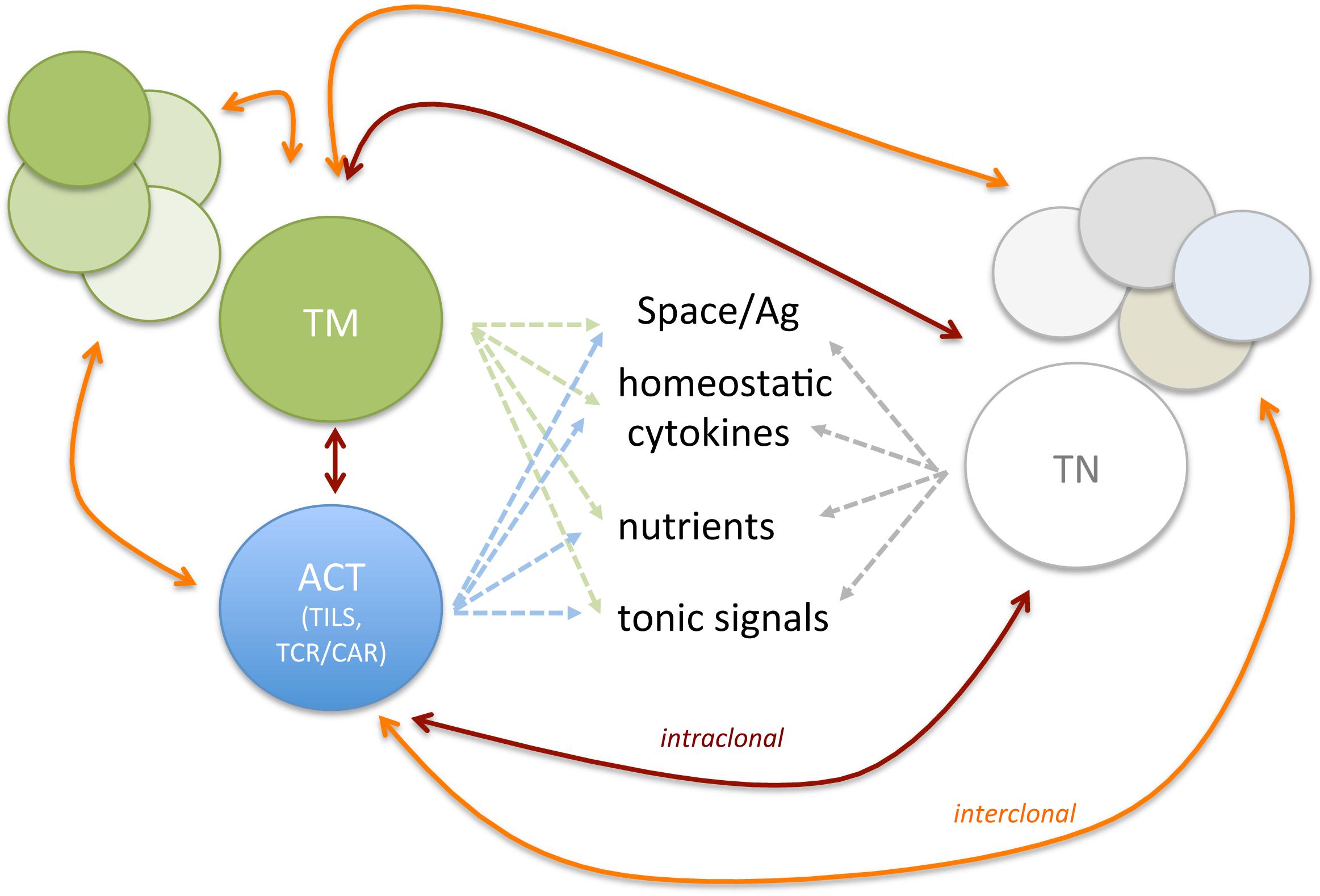
Figure 1. Endogenous and ACT T cell subsets compete for space and resources. Are new comers strong enough to outcompete long-term resident? The figure depicts possible competition among TN, Ag-experienced T cells (TCM, TEM, TEFF, TRM, TEX, and Treg), and ACT. Both intra-clonal competition (among T cell subsets expressing the same TCR, red lines) and inter-clonal competition (among T cell subsets with different TCR, orange lines) might shape persistence of newly infused T cells and the composition of endogenous T cell repertoire.
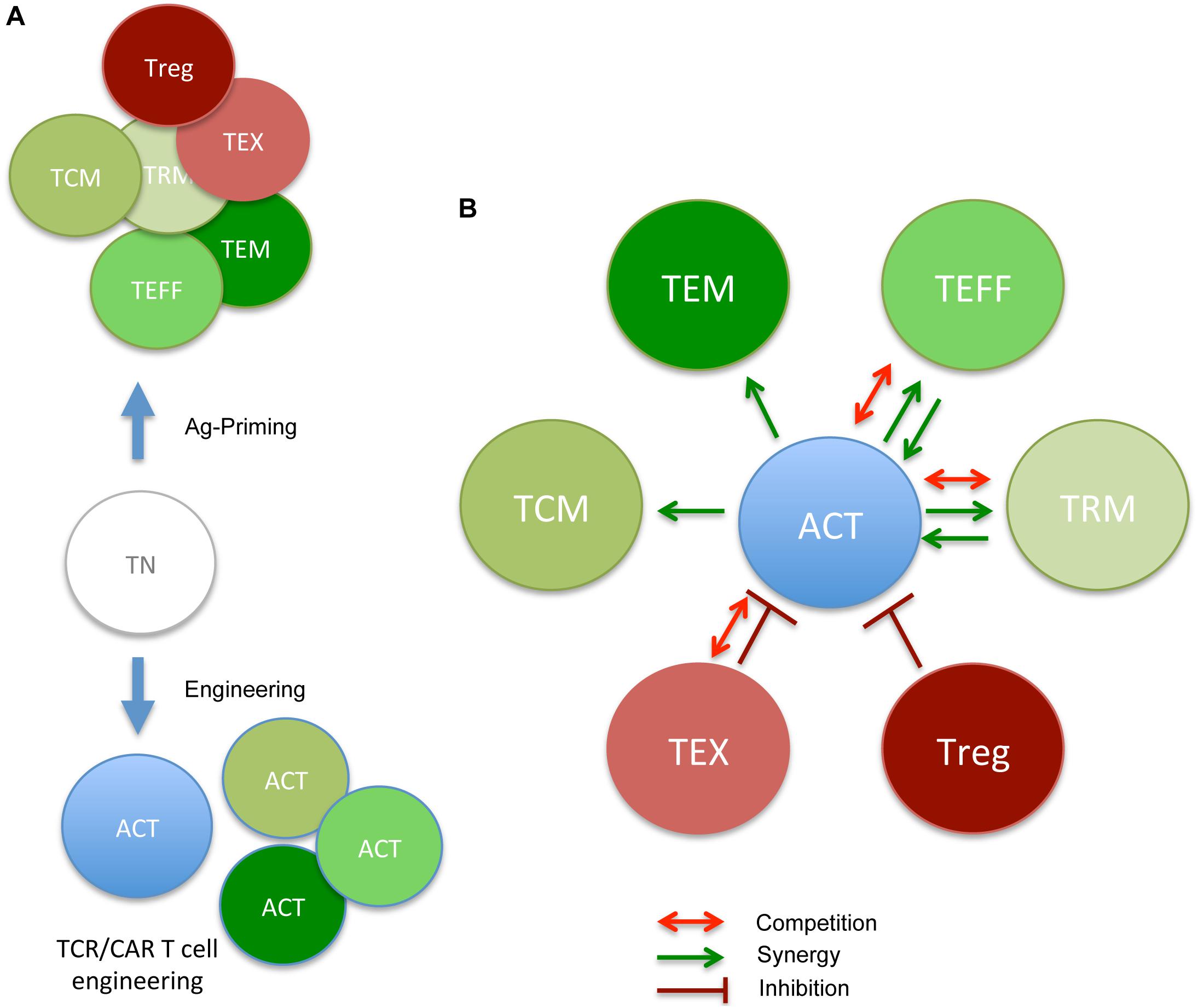
Figure 2. ACT-derived T cell products and cross-talk with pre-existing memory/Ag-experienced subsets (the good, the bad, and the ugly neighbors). (A) During in vivo Ag priming or in vitro engineering/expansion, TN acquire diverse phenotypes, reflecting various degrees of differentiation (TCM, TEM, TEFF, TRM, TEX, and Treg). Each subset reveals defined requirements for proper effector function and survival. (B) In the context of ACT, newly infused T cells will interact with pre-existing subsets, and either benefit from good memory subsets or take advantage of them (TCM, TEM, TEFF, and TRM) in promoting therapeutic anti-tumor responses (green arrows) or be inhibited by nasty ones (TEX and Treg). In some cases, competing for nutrients/survival signal (Figure 1) might also dim ACT efficacy (red arrows). Putative collaborative and antagonistic relationship between pre-existing memory subsets and ACT cell products are depicted.
Both cell-intrinsic and cell-extrinsic events shape TM differentiation, survival, and functions. In this paragraph, we review some of the available evidence that support the impact of clonal abundance, TCR affinity, and availability of cytokines and nutrients on TM behavior and their possible implication in the host–ACT TM cross-talk. As TILs and TCR-engineered T cells recognize MHC restricted antigens, while CAR-T cells bind surface antigens via MHC-independent mechanisms, the impact of such critical determinants might vary and be relative to each given ACT product.
In natural immunity, initial clonal abundance, i.e., the frequency at which any given TCR is represented within the polyclonal repertoire, and the relative TCR affinity for cognate antigens have been shown to give rise to intraclonal and interclonal competition and shape the TM repertoire [recently discussed in (99)]. Data have shown that natural differences in the size of foreign Ag-specific T cell populations have direct consequences on the magnitude of the effector and memory response (100, 101). Intraclonal competition has been shown to affect the expansion and accumulation of both TN and TM naïve cells and to block the proliferation of adoptively transferred CD8 T cells (102). In cases where T cells exceed the relatively narrow physiologic range, intraclonal competition was observed with consequences on overall TM survival and expansion potential (103). Among several factors, competition for Ag and/or access to Ag-bearing APC played a role, (104–106) which caused suboptimal T cell expansion, and defective generation of effector and memory T cells against foreign pathogen and tumors (107–109). In the case of CD8 T cells specific for a tumor-associated self-Ag, increasing naïve T cell frequency at supra-physiological precursor frequencies was reported to ameliorate responses to tumor-specific vaccination and therapeutic effects. Nevertheless, above a certain threshold, intraclonal competition was observed, and this limited protective immunity (110). We also found, in a model of spontaneous prostate cancer development, that increasing the number of TCR-transduced T cells or repeating their administration over time did not significantly increase the relative abundance nor their therapeutic potential (111). Of note, in some models, sequential administration of tumor-specific CTL proved more efficacious than the single initial injection of an equivalent CTL number (112). Thus, data support the notion that intraclonal competition regulates both naturally occurring and ACT-derived antigen-specific CD8 TM.
Intraclonal competition was also reported to regulate relative abundance of TN and TM CD4 T cells (and in the case of tumor/self Ag-specific naïve CD4 T cells). By transferring different numbers of tumor-associated self-Ag-specific TCR transgenic CD4 T cells, Malandro and co-authors showed that initial precursor frequency inversely correlated with in vivo expansion and functional outcomes. Although at low precursor frequency CD4 T cells specific for a tumor-associated self-Ag underwent robust proliferation, they acquired an irreversible exhausted phenotype (113). In contrast, at higher precursor frequencies, T cells showed a poor expansion potential, but preserved an optimal cytokine-secretion profile and antitumor activity. Interestingly, the authors showed that above a certain threshold, intraclonal CD4–CD4 T cell help cooperation became evident (113). It is tempting to speculate that increasing the frequency of tumor-specific CD4 T helper cells might provide a selective advantage to both naturally occurring and ACT-derived TM. Whether increasing the availability of CD4 T cell help might be beneficial to ACT approaches, and whether predefined CD4 and/or CD8 T cell compositions at various degree of differentiation would ameliorate ACT therapeutic potential is currently being investigated in a number of ongoing trials (91, 114).
Availability of the cognate antigen or access to cross-reactive self-Ag appears to be a critical determinant in dictating clonal abundance within a memory subset and/or the emergence of high/low affinity TCRs. In the case of CD4 T cells, weak TCR signals from self-peptide MHCII ligands are important for TM survival, and this becomes limiting in the presence of high frequencies of CD4 T cells specific for the same ligand (103, 115). In the case of CD8 cells, this is critical for shaping the TM repertoire, but less so for TM maintenance, which predominantly depends on cytokines such as IL-15 and IL-7 (116, 117). Nevertheless, TCR affinity/avidity contributes to clonal selection. It regulates access to vital co-stimulatory molecules, cytokines, and nutrients, (99) and it determines that clonotypes with higher affinity (and slower TCR–ligand dissociation rates) acutely increase in frequencies within TM pools, above levels reached during the course of anti-viral immunity starting from naïve precursors (118, 119). During acute murine cytomegalovirus (CMV) infection, for instance, a subset of high-avidity virus-specific CD8 T cells typically increases in size (clonal dominance) and simultaneously establishes a large pool of effector memory T cells able to outcompete lower avidity CD8 T cells (120, 121). This mechanism has now been defined as “memory inflation” and documented over the course of several viral infections (122, 123). In a recent study, Schober and colleagues studied TCR repertoire evolution in the context of latent CMV infection and found that, although high-affinity TCRs dominated T cell responses at early times after infection, low affinity TCRs emerged over time, owing to cellular differentiation and senescence (and not exhaustion) of high affinity ones (124). In a recent publication, Poschke and coauthors exploited TCR deep sequencing to characterize TILs before and after in vitro culture and found that dominant T cell clones were lost during TIL culture because of poor expansion potential, in favor of less represented ones (125). The authors argue that spatial heterogeneity of the tumor T cell repertoire, as well as differences in intrinsic in vitro growth capacity between individual T cell clones, influenced the T cell preparation. In melanoma, the most abundant T cell clones were found to be tumor reactive, and yet, neo-antigen-reactive T cells, of possible highest affinity, were gradually lost during TIL expansion. Whether this occurs also over the course of natural evolution of anti-tumor immunity and/or under the pressure of ACT remains to be determined. Yet, it is tempting to speculate that in some cases a switch in dominance toward low-affinity TCR T cells might indeed take place during the editing process, especially under the pressure of high-affinity ACT T cells and precede and/or account for final tumor escape.
The remodeling of the host T cell repertoire was observed in ACT for both mouse and human CMV. In mice, adoptively transferred T cells were shown to restrict the repertoire of host-derived T cells via competitive mechanisms, supporting clonal dominance of TM ACT cells over endogenous memory cells (126). In the setting of anti-CMV-specific ACT, TM influenced further responses by endogenous CMV-specific T cells in organ transplant recipients (127). This was best observed in patients who had an unbiased TCR repertoire before the transplant, and correlated with therapy efficacy, likely owing to further expansion of viral-specific T cells. Instead, non-responding recipients revealed a pre-transplant biased peripheral T cell repertoire, which was not influenced by ACT. This indicates the ability of ACT to promote a restructuring of the T cell pool, given proper immune cell representation pre-ACT. Because adoptively transferred T cells synergize with endogenous responses (128) (discussed in following paragraph), future studies would be needed to understand how TCR/CAR-T cell impact on the representation of pre-existing and newly generated tumor-reactive TM cells.
Thus, when considering ACT, a detailed characterization of host immune competence might help predict efficacy, and also instruct optimal T cell product composition. Of note, CAR-T cells might be expected to be less sensitive to the competition with endogenous TM subsets than TILs or TCR redirected T cells, at least for TCR engaging ligands and deriving tonic stimulation. In the case of CAR-T cells, antigen-independent tonic signaling has been shown to result from spontaneous clustering of CAR molecules. However, in contrast to tonic TCR signaling, this event was associated with augmented T cell apoptosis, exhaustion, and impaired antitumor effects (129, 130). As preclinical studies suggest that CAR–TCR interactions are a prerequisite for optimal CAR-driven T cell activation, (131) it remains possible that survival of CAR-TM cells is controlled by the same signals that support TCR T cells. If this would be the case, then also CAR-T cell might be sensitive to surrounding endogenous TM subsets.
In the context of natural immunity, competition for γc cytokines, principally IL-7 and IL-15, regulates the balance of CD4 and CD8 TN and TM cells, homeostatic proliferation, and survival (132–134). Only those T cells receiving sufficient signaling escape the apoptotic process and proliferate (135–137). IL-7 mostly supports CD4 and CD8 TM, whereas IL-15 also promotes their homeostatic proliferation. Given the higher expression of IL-2Rβ, CD8 TM cells are more sensitive to IL-15, and outcompete CD4 TM and TN cell subsets during acute infections and homeostatic proliferation (138, 139).
T cells transferred in the setting of ACT are also sensitive to γc-cytokine availability. Accordingly, cytokines like IL-2, IL-7, IL-15, and IL-21 are fundamental both for generation of the ACT cell products and to increase the efficacy and the duration of the anti-tumor response in vivo. Administration of low-dose IL-2 to the patient after ACT therapy has generally been used to enhance the in vivo persistence of the newly adoptively transferred T cells which has been shown to translate in a favorable clinical outcome (140). Although initial studies adopted IL-2 to support the in vitro expansion of engineered T cells, it soon became evident that T cell product had limited survival when transferred in vivo (92). Rather, shorter expansion times and the use of IL-7 and IL-15 mediated the selective expansion of CD4 and CD8 T cells, while limiting the representation of Treg cells, resulting in longer persistence in vivo (141–143). Because of their capacity to support, both in vivo and ex vivo, TM cell generation and homeostatic proliferation, IL-7 and IL-15 are now being evaluated in human clinical trials. IL-21 also plays an important role in ACT manufacturing based on its ability to significantly enhance the ex vivo generation and TCR affinity of TM cells (144, 145).
As in the case of TCR engaging ligands, endogenous and ACT TM might compete for cytokine availability. Thus, once more the T cell repertoire before ACT might influence ACT efficacy, and vice versa recently infused T cells could impact on endogenous subsets. Accordingly, lymphodepletion by various pre-conditioning protocols has been shown to promote both the engraftment and the long-term persistence of TILs, TCR, (142, 146, 147) and also of CAR-T cells (148–150) by lowering the number of immune cell subsets, which compete with transferred tumor-reactive TM for the γc-cytokine binding (142). Clinical trials in melanoma patients first indicated that TIL persistence was improved by preconditioning of the patients with lymphodepleting strategies based on total body irradiation (151, 152). Preclinical models further demonstrated that lymphodepleting regimens improve engraftment and functionality of transferred T cells prolonging their survival, via IL-15-dependent signaling (153). The competition for homeostatic cytokines might also evolve in a “metabolic competition” that might render ACT TM dysfunctional and metabolically exhausted. Indeed, IL-15 also regulates TM metabolic stability (154). In this respect, data indicate that IL-7 administration following cyclophosphamide preconditioning supports the expansion of polyfunctional tumor-specific CD4 T cells (155). Thus, in this scenario, we speculate that lymphodepletion is able to contribute to effective anti-tumor immunity because, in addition to be critical to eliminate immune-suppressive cells (i.e., MDSCs, TAM, TEX, and Tregs) and decrease the metabolic competition in the TME for IL-7, IL-15, and nutrients, it bears the potential to support both ACT TM and possibly also endogenous TN and TM.
Nutrient availability is a requisite for proper TM function and long-term persistence. TM cells own a unique metabolic signature. On Ag encounter, T cells engage OXPHOS, glutaminolysis, and glycolysis to fulfill bioenergetic and biosynthetic demands needed to support proliferation and effector functions. After Ag clearance, TEFF cells reduce their metabolic demands and dependence on glycolysis, and gradually reset back from an anabolic to a catabolic state, typical of long-lived TM cells. At difference with TEFF cells, TCM cells are mostly quiescent and adopt a metabolic profile similar to TN cells, whereby they rely on mitochondrial metabolism and fatty acid oxidation to support their persistence and functions (156, 157). TM cells also possess greater mitochondrial mass and enhanced Spare Respiratory Capacity, (158) two key metabolic features important not only for their development and long-term survival but also for their rapid recall ability.
In the context of ACT, metabolic fitness plays an important role. T cells with high metabolic activity and glycolytic rate are endowed with potent TEFF functions (i.e., capable of cytokine production and rapid proliferation). The identification of the metabolic pathways critical to TM survival and functions supported the concept that in vitro metabolic reprogramming could impact in vivo tumor activity. As a consequence, several approaches targeting T cell metabolism in vitro and in vivo by targeted delivery of metabolism-modulating compounds to the TME have been investigated for effective cancer immunotherapy and recently reviewed (93, 94).
Nevertheless, within the TME, tumor cells and/or other infiltrating immune subsets share metabolic requirements, which are overlapping with those of ACT products. Tumor cells indeed frequently share a similar glycolytic metabolic profile with activated T cells, and compete with them for fundamental nutrients, hindering the ability of effector T cells to meet energetic needs. Two studies showed that highly glycolytic tumors can deplete glucose levels in the TME, dampening the ability of T cells to maintain anabolic growth signaling and produce inflammatory cytokines (159, 160). Moreover, by consuming glucose and producing cAMP, tumors limit TEFF functions and instead promote T cell senescence (161). Additional studies highlight that high rates of amino acid uptake by tumor cells could potentially inhibit the anti-tumor T cell response because amino acids are crucial nutrients that support T cell proliferation and effector functions (162, 163). There is also emerging evidence that some tumors uptake fatty acids with high rates to maintain their proliferation (164). Given that fatty acids have a central role in T cell memory differentiation and function, (165) lipids may be another nutrient that T cells must compete for in the TME.
In the context of this competition, it is worth noticing that both CD8 TRM and TCM cells depend on mitochondrial FAO and OXPHOS. However, to fuel this metabolic pathway, CD8 TRM cells strictly depend on uptake of exogenous fatty acids, (166) whereas CD8 TCM cells rather use extracellular glucose to fuel this process. Thus, CD8 TRM cells might be engaged in a metabolic competition with adoptively transferred CD8 TCM cells to fuel mitochondrial metabolism. Whether such competition for nutrient uptake impacts on ACT product functionality remains to be investigated.
The type of nutrients available to T cells, such as glucose, lipids, and amino acid, influences the differentiation program, functional properties, and shape their ability to control tumor progression (167). Although T cell metabolic reprogramming to a given metabolic fitness might be achieved during manufacturing, it should be considered that programming T cells in vitro might have a detrimental outcome in vivo, as the tumor itself, stromal cells, and/or other infiltrating immune cells compete for critical nutrients. For instance, T cells addicted to glycolysis during in vitro culture might experience nutrient deprivation when transplanted in the host and die because of insufficient glucose availability. Therefore, to mount an effective anti-tumor response, ACT products should retain the metabolic flexibility to adjust to nutrient availability. In this scenario, insights into the metabolic characterization of the TME might help inform in vitro metabolic re-programming of tumor-specific T cells to ameliorate their persistence and long-term survival in a metabolic unfavorable TME.
Pre-clinical and clinical studies have shown that pre-existing memory T cells contribute to the efficacy of immunotherapy and radio-immunotherapy (168–171). Several evidences indicate that also in the case of ACT with TCR/CAR engineered products, host T cells contribute to the therapeutic anti-tumor responses. The heterogeneity within the responding pool of TM, owing to the ability of the cells to integrate several signals (TCR, co-stimulatory molecules, and cytokines), their differentiation status, and their relative fitness in an environment of rapidly expanding cells competing for the same resources, is thus likely to impact on the final therapeutic outcome.
Could then pre-existing memory or on a more general definition, Ag-experienced T cell subsets be stratified according to their impact on ACT efficacy? We speculate that this could be the case, in relation to their relative function in protective responses, as they could improve or hinder ACT products. “Good memories” might be represented by those subsets of cells, endowed with effector capabilities and able to synergize with tumor-specific T cells provided by ACT. For instance, pre-existing TM or TRM might respond to pro-inflammatory signals generated in response to intra-tumoral activation of TCR/CAR engineered T cells, (172) and contribute to anti-tumor immunity by conditioning the immune milieu and/or exerting direct anti-tumor activity. In a recent report, Walsh et al. showed that adoptively transferred T cells synergized with endogenous T cells, which were instrumental to prevent immune escape of Ag-loss variants. Post-transplant tumor-specific vaccination supported better tumor infiltration and prolonged survival of tumor-reactive lymphocytes, likely promoting intra-tumoral responses and Ag cross-presentation (128). Local activation of TRM cells might in turn favor the spreading of circulating CTL responses against tumor-derived neo- and self-antigens (173). We also found that tumor-specific vaccination promoted optimal anti-tumor immunity when applied after allogeneic hematopoietic cell transplantation or ACT with tumor-redirected TCR engineered T cells. Also in these settings, in addition to engineered T cells, non-transduced cell subsets were revealed capable of IFN-γ and Granzyme-B expression (174–177). Similarly, Ma and co-authors found that the implementation of a vaccine boosting via the CAR (using a smart by-specific vaccinable CAR) enhanced CAR T cell expansion and also favored the recruitment of additional specificities (178). It is possible that infiltrating T cells, positively selected in the thymus by virtue of expressing TCRs with a low/intermediate affinity for self-Ags, are indeed able to recognize such antigens on tumor cells, or that TCR/CAR-induced inflammatory environment promotes effector function by bystander cells via non-Ag-specific mechanisms (179–181). In this respect, it is worth mentioning that robust and iterative stimulation of memory self-specific CD8 T cells reverted tolerance to self in the context of acute infection, and promoted anti-tumor immunity, without precipitating autoimmune manifestations (182). This is reminiscent of the synergy between T cells specific for a tumor and a Y chromosome–derived self-antigen in the context of allogeneic hematopoietic cell transplantation and in ACT with TCR-redirected T cells (174, 175). These studies suggest to exploit the use of pre-existing or newly infused TM possibly reactive to self/tumor-associated Ags in ACT of tumors.
Viral-specific memory T cells could also come to help. It is known that both mouse and human tumors are commonly surveyed by memory T cells specific for previously encountered viral infections (183). A recently published manuscript showed that these functional T cells can be specifically reactivated via the local delivery of viral peptides, which caused a local inflammatory environment capable of activating both the innate and adaptive immunity, leading to tumor growth arrest. Immunization with viral peptides sensitized mice to PD-L1 checkpoint blockade promoting the elimination of otherwise resistant tumors (169). Thus, viral-specific memory T cells, if appropriately activated, might synergize with ACT.
Finally, resident TRM, bystander memory subsets, and virtual memory T cells might also play a role, as capable of responding to pro-inflammatory cytokines, known to lower the threshold for T cell activation and/or induce TCR-independent effector functions (180, 184). Supporting this, CAR T cells engineered to express IL-12 and/or IL-18 (TRUCK T cells) have proven more effective than those only expressing the tumor-specific CAR (185, 186). TCR could cross-recognize multiple Ags or respond to unrelated Ags, self-Ag, or environmental Ags (187, 188). Virtual memory T cells can produce IFN-γ and are capable of Ag-independent lytic activity, in response to the inflammatory milieu alone, i.e., when stimulated with IL-12, IL-15, and IL-18 (189).
Although in several instances the cellular and molecular mechanisms at the base of the synergistic effect need further investigation, bystander T memory cells were frequently found within the tumor infiltrates, (180) suggesting that pre-existing memory cells, unrelated to the cognate tumor-associated Ag targeted by TILs or TCR/CAR T cells, might contribute to anti-tumor immunity in ACT settings (Figure 3).
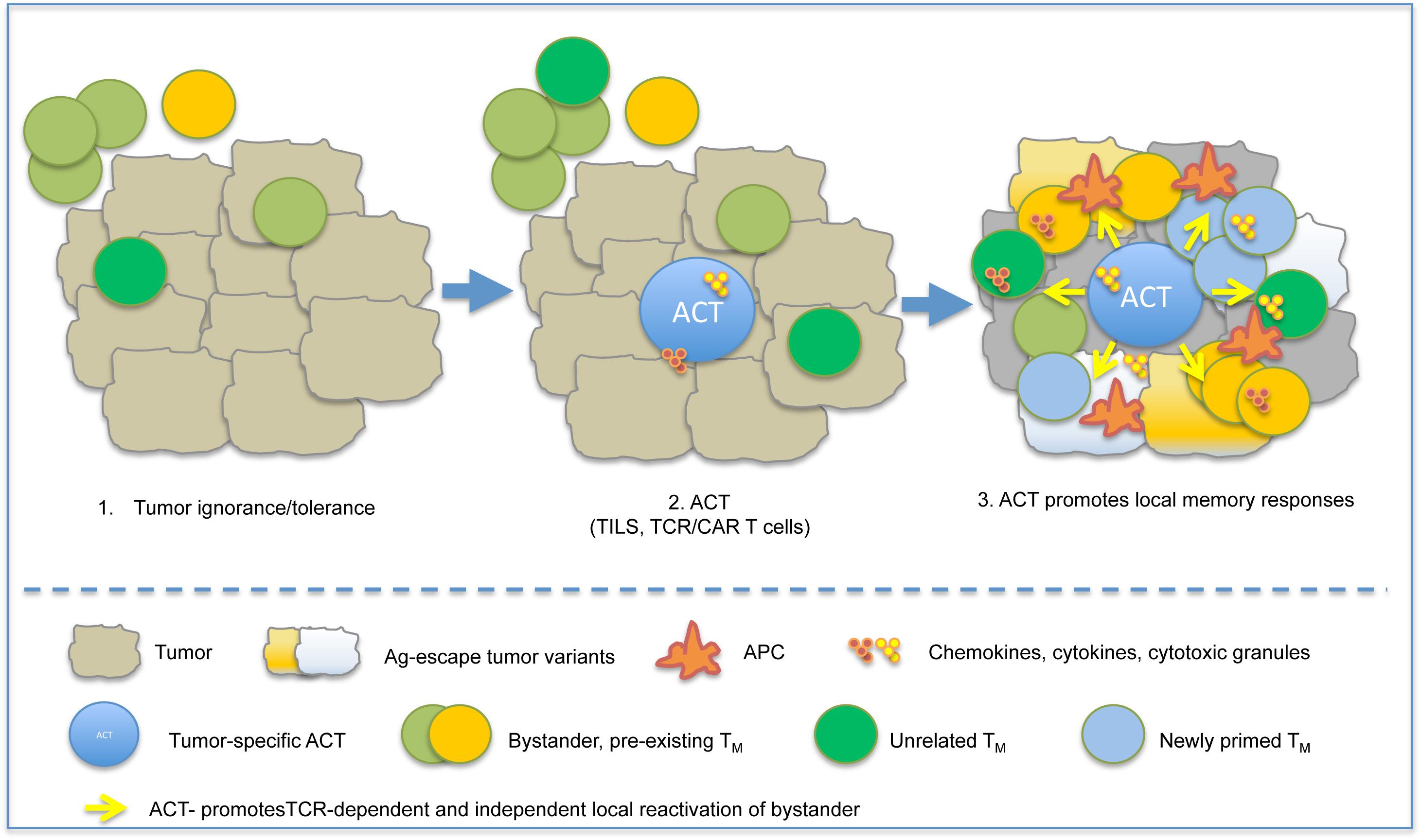
Figure 3. ACT instructs protective immune responses in tumors. A schematic model depicting the possible influence of ACT on pre-existing memory is depicted. Shortly after infusion, ACT-derived TEFF recognize tumor antigen, cause tumor cell death and release of pro-inflammatory cytokines (IFN-γ, TNF-α), and promote local inflammation. This causes recruitment of APC and bystander memory subsets, and their local reactivation by either TCR-dependent or TCR-independent mechanisms.
Along this line, it should be mentioned that unfortunately, also “bad memories” exist, which may impair the development of new memories. This was shown to be the case in the well-recognized phenomenon of “original antigenic sin,” where an existing immune response prevents the initiation of a later, cross-reactive but independent immune response. This phenomenon, which was described for antiviral CD8 T cell immune responses (190, 191) and in vaccinated mice, owing to the ability of pre-existing effector cells to eliminate Ag-bearing dendritic cells, might play a role in limiting propagation of anti-tumor protective memory responses. Accordingly, we found that anti-tumor CD4 T cell responses limited efficacy of active vaccination in tumor-bearing mice, (192) supporting the possibility that in some cases pre-existing memory might counteract a newly born one.
Although in real life it is well conceivable that these “bad memories” might protect individuals from recurrent infection/disease, in the context of tumor immunity, they might represent an obstacle to therapeutic efficacy.
Finally, even if anergic and TEX cells, putative representative of bad memory T cell subsets, might appear to have no apparent function, they might consume useful resources needed for the efficacy of ACT cell products. This has been shown for Treg subsets, which, for instance, can induce TEFF cell senescence by competing for glucose and inducing DNA damage (193). Hence, this subset might prove to be an “ugly neighbor,” capable of providing active suppression and inhibiting tumor recognition by ACT T cell products right from the start.
Given the aforementioned evidence, should pre-existent memory be considered before adoptive T therapy? The authors believe that it should.
The degree of heterogeneity within a T cell pool depends on the integration of signals from the TCR, co-stimulatory molecules, cytokines and nutrients, and also on the relative cell fitness within an environment competing for the same resources. Newly infused T cells would need to face and adapt to such pre-existing conditions, to engraft and exert proper anti-tumor activity. The concept of immunological memory foresees that pre-existing memory T cells would be beneficial for protection against reinfection with the same pathogens, because Ag-specific memory T cells would be numerically increased when compared to endogenous ones, have widened anatomical distribution, and respond more quickly, conferring rapid clearance of the infectious agent. Nevertheless, previous encounters with the Ag has the potential to generate in addition to good memories (TCM, TEFF, and TRM), also bad and ugly memories (TEX, Treg, and in some instances tumor-associated TRM), with opposite effects on recall responses, and on the efficacy of adoptive T cell therapy. The quality and quantity of adoptively transferred cells are also important parameters to consider when optimizing such a treatment. “The more is the better” might not be the right choice, as cells should find sufficient space and support (TCR/CAR engaging ligands, homeostatic cytokines, nutrients) in spite of host–cell competition. Modeling tumor–immune system competition might help predict responses in vivo (112, 194). Would also provoking immunological amnesia promote therapeutic efficacy of T cell products in the context of ACT? We speculate this might not be entirely the case because as in real life, retaining positive memories might help. The finding that endogenous T cells cooperate with TCR/CAR-redirected T cells supports this statement (128, 195). Rather, strategies suitable to evoke selective amnesia from bad memories (i.e., capable of depleting/inhibiting regulatory subsets, and/or overcome competition for space or nutrients) would empower good ones and amplify therapeutic effects of current ACT products. As an alternative, synthetic biology and genetic engineering might help design T cell products insensitive to competition or able to metabolically adapt to hostile TME. Likewise, understanding and exploiting CD4 and CD8 TM representation, both within the ACT product, and the pre-existing endogenous repertoire, might open new avenues of intervention. Thus, interesting challenges ahead will be to understand the cross-talk and homeostatic regulation between adoptively transferred T cells and endogenous ones to define strategies to eliminate any unneeded immunological bad memories, and take advantage of available local resources. This will foster productive synergies and supportive environments to render ACT products highly functional.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
AM acknowledges the support of Associazione Italiana per la Ricerca sul Cancro (AIRC: IG 2014-15883 and IG 2018-21763) and of TRANSCAN (TRS-2016-00000373). TM is the recipient of a Start Up Grant from Associazione Italiana per la Ricerca sul Cancro (AIRC StartUp 2018-21474).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors AM.
The authors wish to acknowledge the work of all those colleagues that we were not able to cite for space constraints.
1. Williams, MA, Bevan, MJ. Effector and memory CTL differentiation. Annu Rev Immunol. (2007) 25:171–92. doi: 10.1146/annurev.immunol.25.022106.141548
2. Pepper, M, Jenkins, MK. Origins of CD4+ effector and central memory T cells. Nat Immunol. (2011) 12:467–71. doi: 10.1038/ni.2038
3. Jameson, SC, Masopust, D. Understanding subset diversity in T cell memory. Immunity. (2018) 48:214–26. doi: 10.1016/j.immuni.2018.02.010
4. Busch, DH, Fräßle, SP, Sommermeyer, D, Buchholz, VR, Riddell, SR. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. (2016) 28:28–34. doi: 10.1016/j.smim.2016.02.001
5. Hope, JL, Stairiker, CJ, Bae, EA, Otero, DC, Bradley, LM. Striking a balance-cellular and molecular drivers of memory T cell development and responses to chronic stimulation. Front Immunol. (2019) 10:1595. doi: 10.3389/fimmu.2019.01595
6. Martin, MD, Badovinac, VP. Defining memory CD8 T cell. Front Immunol. (2018) 9:2692. doi: 10.3389/fimmu.2018.02692
7. Cui, W, Kaech, SM. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev. (2010) 236:151–66. doi: 10.1111/j.1600-065X.2010.00926.x
8. Harty, JT, Badovinac, VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. (2008) 8:107–19. doi: 10.1038/nri2251
9. Chang, JT, Wherry, EJ, Goldrath, AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. (2014) 15:1104–15. doi: 10.1038/ni.3031
10. Omilusik, KD, Goldrath, AW. Remembering to remember: T cell memory maintenance and plasticity. Curr Opin Immunol. (2019) 58:89–97. doi: 10.1016/j.coi.2019.04.009
11. Sallusto, F, Lenig, D, Förster, R, Lipp, M, Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. (1999) 401:708–12. doi: 10.1038/44385
12. Gattinoni, L, Speiser, DE, Lichterfeld, M, Bonini, C. T memory stem cells in health and disease. Nat Med. (2017) 23:18–27. doi: 10.1038/nm.4241
13. Gattinoni, L, Lugli, E, Ji, Y, Pos, Z, Paulos, CM, Quigley, MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. (2011) 17:1290–7. doi: 10.1038/nm.2446
14. Lugli, E, Gattinoni, L, Roberto, A, Mavilio, D, Price, DA, Restifo, NP, et al. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat Protoc. (2013) 8:33–42. doi: 10.1038/nprot.2012.143
15. Cieri, N, Camisa, B, Cocchiarella, F, Forcato, M, Oliveira, G, Provasi, E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. (2013) 121:573–84. doi: 10.1182/blood-2012-05-431718
16. Masopust, D, Vezys, V, Marzo, AL, Lefrançois, L. Preferential localization of effector memory cells in nonlymphoid tissue. J Immunol. (2014) 291:2413–7. doi: 10.1126/science.1058867
17. Reinhardt, RL, Khoruts, A, Merica, R, Zell, T, Jenkins, MK. Visualizing the generation of memory CD4 T cells in the whole body. Nature. (2001) 410:101–5. doi: 10.1038/35065111
18. Gerlach, C, Moseman, EA, Loughhead, SM, Alvarez, D, Zwijnenburg, AJ, Waanders, L, et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity. (2016) 45:1270–84. doi: 10.1016/j.immuni.2016.10.018
19. Gebhardt, T, Wakim, LM, Eidsmo, L, Reading, PC, Heath, WR, Carbone, FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. (2009) 10:524–30. doi: 10.1038/ni.1718
20. Teijaro, JR, Turner, D, Pham, Q, Wherry, EJ, Lefrançois, L, Farber, DL. Cutting Edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. (2011) 187:5510–4. doi: 10.4049/jimmunol.1102243
21. Masopust, D, Choo, D, Vezys, V, Wherry, EJ, Duraiswamy, J, Akondy, R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. (2010) 207:553–64. doi: 10.1084/jem.20090858
22. Mami-Chouaib, F, Blanc, C, Corgnac, S, Hans, S, Malenica, I, Granier, C, et al. Resident memory T cells, critical components in tumor immunology. J Immunother Cancer. (2018) 6:87. doi: 10.1186/s40425-018-0399-6
23. Amsen, D, Van Gisbergen, KPJM, Hombrink, P, Van Lier, RAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol. (2018) 19:538–46. doi: 10.1038/s41590-018-0114-2
24. Schenkel, JM, Masopust, D. Tissue-resident memory T cells. Immunity. (2014) 41:886–97. doi: 10.1016/j.immuni.2014.12.007
25. Casey, KA, Fraser, KA, Schenkel, JM, Moran, A, Abt, MC, Beura, LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. (2012) 188:4866–75. doi: 10.4049/jimmunol.1200402
26. MacKay, LK, Rahimpour, A, Ma, JZ, Collins, N, Stock, AT, Hafon, ML, et al. The developmental pathway for CD103+ CD8+ tissue-resident memory T cells of skin. Nat Immunol. (2013) 14:1294–301. doi: 10.1038/ni.2744
27. Boutet, M, Gauthier, L, Leclerc, M, Gros, G, De Montpreville, V, Theret, N, et al. β signaling intersects with CD103 integrin signaling to promote T-Lymphocyte accumulation and antitumor activity in the lung tumor microenvironment. Cancer Res. (2016) 76:1757–69. doi: 10.1158/0008-5472.CAN-15-1545
28. Bromley, SK, Yan, S, Tomura, M, Kanagawa, O, Luster, AD. Recirculating memory T cells are a unique subset of CD4 + T cells with a distinct phenotype and migratory pattern. J Immunol. (2013) 190:970–6. doi: 10.4049/jimmunol.1202805
29. Bergsbaken, T, Bevan, MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat Immunol. (2015) 16:406–14. doi: 10.1038/ni.3108
30. Topham, DJ, Reilly, EC. Tissue-resident memory CD8+ T cells: From phenotype to function. Front Immunol. (2018) 9:515. doi: 10.3389/fimmu.2018.00515
31. Nguyen, QP, Deng, TZ, Witherden, DA, Goldrath, AW. Origins of CD4+ circulating and tissue-resident memory T-cells. Immunology. (2019) 157:3–12. doi: 10.1111/imm.13059
32. White, JT, Cross, EW, Burchill, MA, Danhorn, T, McCarter, MD, Rosen, HR, et al. Virtual memory T cells develop and mediate bystander protective immunity in an IL-15-dependent manner. Nat Commun. (2016) 7:11291. doi: 10.1038/ncomms11291
33. Marusina, AI, Ono, Y, Merleev, AA, Shimoda, M, Ogawa, H, Wang, EA, et al. CD4+ virtual memory: antigen-inexperienced T cells reside in the naïve, regulatory, and memory T cell compartments at similar frequencies, implications for autoimmunity. J Autoimmun. (2017) 77:76–88. doi: 10.1016/j.jaut.2016.11.001
34. Jameson, SC, Lee, YJ, Hogquist, KA. Innate memory T cells. Adv Immunol. (2015) 126:173–213. doi: 10.1016/bs.ai.2014.12.001
35. Miller, CH, Klawon, DEJ, Zeng, S, Lee, V, Socci, ND, Savage, PA. Eomes identifies thymic precursors of self-specific memory-phenotype CD8+ T cells. Nat Immunol. (2020) 21:567–77. doi: 10.1038/s41590-020-0653-1
36. Chacon, JA, Wu, RC, Sukhumalchandra, P, Molldrem, JJ, Sarnaik, A, Pilon-Thomas, S, et al. Co-stimulation through 4-1BB/cd137 improves the expansion and function of CD8+ melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One. (2013) 8:e60031. doi: 10.1371/journal.pone.0060031
37. Mempel, TR, Henrickson, SE, Von Andrian, UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. (2004) 427:154–9. doi: 10.1038/nature02238
38. Curtsinger, JM, Schmidt, CS, Mondino, A, Lins, DC, Kedl, RM, Jenkins, MK, et al. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. (1999) 162:3256–62.
39. Obst, R, Van Santen, HM, Mathis, D, Benoist, C. Antigen persistence is required throughout the expansion phase of a CD4+ T cell response. J Exp Med. (2005) 201:1555–65. doi: 10.1084/jem.20042521
40. Kim, C, Wilson, T, Fischer, KF, Williams, MA. Sustained interactions between T cell receptors and antigens promote the differentiation of CD4+ memory T cells. Immunity. (2013) 39:508–20. doi: 10.1016/j.immuni.2013.08.033
41. Henrickson, SE, Perro, M, Loughhead, SM, Senman, B, Stutte, S, Quigley, M, et al. Antigen availability determines CD8+ T cell-dendritic cell interaction kinetics and memory fate decisions. Immunity. (2013) 39:496–507. doi: 10.1016/j.immuni.2013.08.034
42. Lanzavecchia, A, Sallusto, F. Progressive differentiation and selection of the fittest in the immune response. Nat Rev Immunol. (2002) 2:982–7. doi: 10.1038/nri959
43. Mehlhop-Williams, ER, Bevan, MJ. Memory CD8+ T cells exhibit increased antigen threshold requirements for recall proliferation. J Exp Med. (2014) 211:345–56. doi: 10.1084/jem.20131271
44. Croft, M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. (2009) 9:271–85. doi: 10.1038/nri2526
45. Croft, M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol. (2003) 3:609–20. doi: 10.1038/nri1148
46. June, CH, O’Connor, RS, Kawalekar, OU, Ghassemi, S, Milone, MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
47. Kawalekar, OU, O’Connor, RS, Fraietta, JA, Guo, L, McGettigan, SE, Posey, AD, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
48. Mescher, MF, Curtsinger, JM, Agarwal, P, Casey, KA, Gerner, M, Hammerbeck, CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. (2006) 211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x
49. Pepper, M, Pagán, AJ, Igyártó, BZ, Taylor, JJ, Jenkins, MK. opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity. (2011) 35:583–95. doi: 10.1016/j.immuni.2011.09.009
50. Choi, YS, Yang, JA, Yusuf, I, Johnston, RJ, Greenbaum, J, Peters, B, et al. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol. (2013) 190:4014–26. doi: 10.4049/jimmunol.1202963
51. Harding, FA, McArthur, JG, Gross, JA, Raulet, DH, Allison, JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. (1992) 356:607–9. doi: 10.1038/356607a0
52. Schietinger, A, Greenberg, PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. (2014) 35:51–60. doi: 10.1016/j.it.2013.10.001
53. Reading, JL, Gálvez-Cancino, F, Swanton, C, Lladser, A, Peggs, KS, Quezada, SA. The function and dysfunction of memory CD8 + T cells in tumor immunity. Immunol Rev. (2018) 283:194–212. doi: 10.1111/imr.12657
54. Wherry, EJ, Blattman, JN, Murali-Krishna, K, van der Most, R, Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. (2003) 77:4911–27. doi: 10.1128/jvi.77.8.4911-4927.2003
55. McLane, LM, Abdel-Hakeem, MS, Wherry, EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
56. van der Leun, AM, Thommen, DS, Schumacher, TN. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer. (2020) 20:218–32. doi: 10.1038/s41568-019-0235-4
57. Barber, DL, Wherry, EJ, Masopust, D, Zhu, B, Allison, JP, Sharpe, AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. (2006) 439:682–7. doi: 10.1038/nature04444
58. Zajac, AJ, Blattman, JN, Murali-Krishna, K, Sourdive, DJD, Suresh, M, Altman, JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. (1998) 188:2205–13. doi: 10.1084/jem.188.12.2205
59. Gallimore, A, Glithero, A, Godkin, A, Tissot, AC, Plückthun, A, Elliott, T, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. (1998) 187:1383–93. doi: 10.1084/jem.187.9.1383
60. Day, CL, Kaufmann, DE, Kiepiela, P, Brown, JA, Moodley, ES, Reddy, S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. (2006) 443:350–4. doi: 10.1038/nature05115
61. Lechner, F, Wong, DKH, Dunbar, PR, Chapman, R, Chung, RT, Dohrenwend, P, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. (2000) 191:1499–512. doi: 10.1084/jem.191.9.1499
62. Ye, B, Liu, X, Li, X, Kong, H, Tian, L, Chen, Y. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis. (2015) 6:e1694. doi: 10.1038/cddis.2015.42
63. Sharpe, AH, Wherry, EJ, Ahmed, R, Freeman, GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. (2007) 8:239–45. doi: 10.1038/ni1443
64. McKinney, EF, Lee, JC, Jayne, DRW, Lyons, PA, Smith, KGC. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. (2015) 523:612–6. doi: 10.1038/nature14468
65. Pauken, KE, Wherry, EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. (2015) 36:265–76. doi: 10.1016/j.it.2015.02.008
66. Josefowicz, SZ, Lu, L-F, Rudensky, AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
67. Bauer, CA, Kim, EY, Marangoni, F, Carrizosa, E, Claudio, NM, Mempel, TR. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest. (2014) 124:2425–40. doi: 10.1172/JCI66375
68. Ohue, Y, Nishikawa, H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. (2019) 110:2080–9. doi: 10.1111/cas.14069
69. Smazynski, J, Webb, JR. Resident memory-like tumor-infiltrating lymphocytes (TILRM): latest players in the immuno-oncology repertoire. Front Immunol. (2018) 9:174. doi: 10.3389/fimmu.2018.01741
70. Oja, AE, Piet, B, Helbig, C, Stark, R, Van Der Zwan, D, Blaauwgeers, H, et al. Trigger-happy resident memory CD4 + T cells inhabit the human lungs. Mucosal Immunol. (2018) 11:654–67. doi: 10.1038/mi.2017.94
71. Ariotti, S, Hogenbirk, MA, Dijkgraaf, FE, Visser, LL, Hoekstra, ME, Song, JY, et al. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science. (2014) 346:101–5. doi: 10.1126/science.1254803
72. Djenidi, F, Adam, J, Goubar, A, Durgeau, A, Meurice, G, de Montpréville, V, et al. CD8 + CD103 + tumor–infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol. (2015) 194:3475–86. doi: 10.4049/jimmunol.1402711
73. Webb, JR, Milne, K, Watson, P, DeLeeuw, RJ, Nelson, BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker cd103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res. (2014) 20:434–44. doi: 10.1158/1078-0432.CCR-13-1877
74. Boddupalli, CS, Bar, N, Kadaveru, K, Krauthammer, M, Pornputtapong, N, Mai, Z, et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight. (2016) 1:e88955. doi: 10.1172/jci.insight.88955
75. Webb, JR, Milne, K, Nelson, BH. PD-1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol Res. (2015) 3:926–35. doi: 10.1158/2326-6066.CIR-14-0239
76. Oja, AE, Piet, B, Van Der Zwan, D, Blaauwgeers, H, Mensink, M, De Kivit, S, et al. Functional heterogeneity of CD4+ tumor-infiltrating lymphocytes with a resident memory phenotype in NSCLC. Front Immunol. (2018) 9:2654. doi: 10.3389/fimmu.2018.02654
77. Quezada, SA, Simpson, TR, Peggs, KS, Merghoub, T, Vider, J, Fan, X, et al. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. (2010) 207:637–50. doi: 10.1084/jem.20091918
78. Friedman, KM, Prieto, PA, Devillier, LE, Gross, CA, Yang, JC, Wunderlich, JR, et al. Tumor-specific CD4+ melanoma tumor-infiltrating lymphocytes. J Immunother. (2012) 35:400–8. doi: 10.1097/CJI.0b013e31825898c5
79. Blanc, C, Hans, S, Tran, T, Granier, C, Saldman, A, Anson, M, et al. Targeting resident memory T cells for cancer immunotherapy. Front Immunol. (2018) 9:1722. doi: 10.3389/fimmu.2018.01722
80. Edwards, J, Wilmott, JS, Madore, J, Gide, TN, Quek, C, Tasker, A, et al. CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy-naïve melanoma patients and expand significantly during anti-PD-1 treatment. Clin Cancer Res. (2018) 24:3036–45. doi: 10.1158/1078-0432.CCR-17-2257
81. Czechowicz, A, Weissman, IL. Purified hematopoietic stem cell transplantation: the next generation of blood and immune replacement. Hematol Oncol Clin North Am. (2011) 30:159–71. doi: 10.1016/j.hoc.2010.11.006
82. Demirer, T, Barkholt, L, Blaise, D, Pedrazzoli, P, Aglietta, M, Carella, AM, et al. Transplantation of allogeneic hematopoietic stem cells: an emerging treatment modality for solid tumors. Nat Clin Pract Oncol. (2008) 5:256–67. doi: 10.1038/ncponc1104
83. D’Souza, A, Lee, S, Zhu, X, Pasquini, M. Current use and trends in hematopoietic cell transplantation in the united states. Biol Blood Marrow Transplant. (2017) 26:e177–82. doi: 10.1016/j.bbmt.2017.05.035
84. Dickinson, AM, Wang, XN, Sviland, L, Vyth-Dreese, FA, Jackson, GH, Schumacher, TNM, et al. In situ dissection of the graft-versus-host activities of cytotoxic T cells specific for minor histocompatibility antigens. Nat Med. (2002) 8:410–4. doi: 10.1038/nm0402-410
85. Yang, JC, Rosenberg, SA. Adoptive T-cell therapy for cancer. Adv Immunol. (2016) 130:279–94. doi: 10.1016/bs.ai.2015.12.006
86. Hammerl, D, Rieder, D, Martens, JWM, Trajanoski, Z, Debets, R. Adoptive T cell therapy: new avenues leading to safe targets and powerful allies. Trends Immunol. (2018) 39:921–36. doi: 10.1016/j.it.2018.09.004
87. Klebanoff, CA, Gattinoni, L, Restifo, NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother. (2012) 35:651–60. doi: 10.1097/CJI.0b013e31827806e6
88. Xu, Y, Zhang, M, Ramos, CA, Durett, A, Liu, E, Dakhova, O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. (2014) 123:3750–9. doi: 10.1182/blood-2014-01-552174
89. Wang, X, Naranjo, A, Brown, CE, Bautista, C, Wong, CW, Chang, WC, et al. Phenotypic and functional attributes of lentivirus-modified CD19-specific Human CD8+ central memory T cells manufactured at clinical scale. J Immunother. (2012) 35:689–701. doi: 10.1097/CJI.0b013e318270dec7
90. Terakura, S, Yamamoto, TN, Gardner, RA, Turtle, CJ, Jensen, MC, Riddell, SR. Generation of CD19-chimeric antigen receptor modified CD8 + T cells derived from virus-specific central memory T cells. Blood. (2012) 119:72–82. doi: 10.1182/blood-2011-07-366419
91. Sommermeyer, D, Hudecek, M, Kosasih, PL, Gogishvili, T, Maloney, DG, Turtle, CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. (2016) 30:492–500. doi: 10.1038/leu.2015.247
92. Crompton, JG, Sukumar, M, Restifo, NP. Uncoupling T-cell expansion from effector differentiation in cell-based immunotherapy. Immunol Rev. (2014) 257:264–76. doi: 10.1111/imr.12135
93. Hinrichs, CS, Spolski, R, Paulos, CM, Gattinoni, L, Kerstann, KW, Palmer, DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. (2008) 111:5326–33. doi: 10.1182/blood-2007-09-113050
94. Zhou, J, Jin, L, Wang, F, Zhang, Y, Liu, B, Zhao, T. Chimeric antigen receptor T (CAR-T) cells expanded with IL-7/IL-15 mediate superior antitumor effects. Protein Cell. (2019) 10:764–9. doi: 10.1007/s13238-019-0643-y
95. Pilipow, K, Roberto, A, Roederer, M, Waldmann, TA, Mavilio, D, Lugli, E. IL15 and T-cell stemness in T-cell-based cancer immunotherapy. Cancer Res. (2015) 75:5187–93. doi: 10.1158/0008-5472.CAN-15-1498
96. Urak, R, Walter, M, Lim, L, Wong, CLW, Budde, LE, Thomas, S, et al. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J Immunother Cancer. (2017) 5:26. doi: 10.1186/s40425-017-0227-4
97. Klebanoff, CA, Crompton, JG, Leonardi, AJ, Yamamoto, TN, Chandran, SS, Eil, RL, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight. (2017) 2:e95103. doi: 10.1172/jci.insight.95103
98. Chou, J, Voong, LN, Mortales, CL, Towlerton, AMH, Pollack, SM, Chen, X, et al. Epigenetic modulation to enable antigen-specific T-cell therapy of colorectal cancer. J Immunother. (2012) 35:131–41. doi: 10.1097/CJI.0b013e31824300c7
99. Kavazoviæ, I, Poliæ, B, Wensveen, FM. Cheating the hunger games: mechanisms controlling clonal diversity of CD8 effector and memory populations. Front Immunol. (2018) 9:2831. doi: 10.3389/fimmu.2018.02831
100. Moon, JJ, Chu, HH, Pepper, M, McSorley, SJ, Jameson, SC, Kedl, RMM, et al. Naive CD4+ T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. (2007) 27:203–13. doi: 10.1016/j.immuni.2007.07.007
101. Obar, JJ, Khanna, KM, Lefrançois, L. Endogenous Naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity. (2008) 28:859–69. doi: 10.1016/j.immuni.2008.04.010
102. Leitão, C, Freitas, AA, Garcia, S. The role of TCR specificity and clonal competition during reconstruction of the peripheral T cell pool. J Immunol. (2009) 182:5232–9. doi: 10.4049/jimmunol.0804071
103. Hataye, J, Moon, JJ, Khoruts, A, Reilly, C, Jenkins, MK. Naïve and memory CD4+ T cell survival controlled by clonal abundance. Science. (2006) 312:114–6. doi: 10.1126/science.1124228
104. Kedl, RM, Rees, WA, Hildeman, DA, Schaefer, B, Mitchell, T, Kappler, J, et al. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med. (2000) 192:1105–14. doi: 10.1084/jem.192.8.1105
105. Quiel, J, Caucheteux, S, Laurence, A, Singh, NJ, Bocharov, G, Ben-Sasson, SZ, et al. Antigen-stimulated CD4 T-cell expansion is inversely and log-linearly related to precursor number. Proc Natl Acad Sci USA. (2011) 108:3312–7. doi: 10.1073/pnas.1018525108
106. Smith, LK, Boukhaled, GM, Condotta, SA, Mazouz, S, Guthmiller, JJ, Vijay, R, et al. Interleukin-10 directly inhibits CD8+ T Cell function by enhancing N-Glycan branching to decrease antigen sensitivity. Immunity. (2018) 48:299–312.e5. doi: 10.1016/j.immuni.2018.01.006
107. Badovinac, VP, Haring, JS, Harty, JT. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8+ T cell response to infection. Immunity. (2007) 26:827–41. doi: 10.1016/j.immuni.2007.04.013
108. Blair, DA, Lefrançois, L. Increased competition for antigen during priming negatively impacts the generation of memory CD4 T cells. Proc Natl Acad Sci USA. (2007) 104:15045–50. doi: 10.1073/pnas.0703767104
109. Marzo, AL, Klonowski, KD, Le Bon, A, Borrow, P, Tough, DF, Lefrançois, L. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat Immunol. (2005) 6:793–9. doi: 10.1038/ni1227
110. Rizzuto, GA, Merghoub, T, Hirschhorn-Cymerman, D, Liu, C, Lesokhin, AM, Sahawneh, D, et al. Self-antigen-specifc CD8 + T cell precursor frequency determines the quality of the antitumor immune response. J Exp Med. (2009) 206:849–66. doi: 10.1084/jem.20081382
111. Manzo, T, Sturmheit, T, Basso, V, Petrozziello, E, Michelini, RH, Riba, M, et al. T cells redirected to a minor histocompatibility antigen instruct intratumoral TNFα expression and empower adoptive cell therapy for solid tumors. Cancer Res. (2017) 77:658–71. doi: 10.1158/0008-5472.CAN-16-0725
112. Khazen, R, Müller, S, Lafouresse, F, Valitutti, S, Cussat-Blanc, S. Sequential adjustment of cytotoxic T lymphocyte densities improves efficacy in controlling tumor growth. Sci Rep. (2019) 9:120308. doi: 10.1038/s41598-019-48711-2
113. Malandro, N, Budhu, S, Kuhn, NF, Liu, C, Murphy, JT, Cortez, C, et al. Clonal abundance of tumor-specific CD4+ T cells potentiates efficacy and alters susceptibility to exhaustion. Immunity. (2016) 44:179–93. doi: 10.1016/j.immuni.2015.12.018
114. Turtle, CJ, Hanafi, LA, Berger, C, Hudecek, M, Pender, B, Robinson, E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. (2016) 8:355ra116. doi: 10.1126/scitranslmed.aaf8621
115. Singh, NJ, Bando, JK, Schwartz, RH. Subsets of nonclonal neighboring CD4+ T cells specifically regulate the frequency of individual antigen-reactive T cells. Immunity. (2012) 37:735–46. doi: 10.1016/j.immuni.2012.08.008
116. Becker, TC, John Wherry, E, Boone, D, Murali-Krishna, K, Antia, R, Ma, A, et al. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med. (2002) 195:1541–8. doi: 10.1084/jem.20020369
117. Carrio, R, Rolle, CE, Malek, TR. Non-redundant role for IL-7R signaling for the survival of CD8+ memory T cells. Eur J Immunol. (2007) 37:3078–88. doi: 10.1002/eji.200737585
118. Malherbe, L, Hausl, C, Teyton, L, McHeyzer-Williams, MG. Clonal selection of helper T cells is determined by an affinity threshold with no further skewing of TCR binding properties. Immunity. (2004) 21:669–79. doi: 10.1016/j.immuni.2004.09.008
119. Savage, PA, Boniface, JJ, Davis, MM. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. (1999) 10:485–92. doi: 10.1016/S1074-7613(00)80048-5
120. Sierro, S, Rothkopf, R, Klenerman, P. Evolution of diverse antiviral CD8+ T cell populations after murine cytomegalovirus infection. Eur J Immunol. (2005) 35:1113–23. doi: 10.1002/eji.200425534
121. Snyder, CM, Cho, KS, Bonnett, EL, van Dommelen, S, Shellam, GR, Hill, AB. Memory inflation during chronic viral infection is maintained by continuous production of short-lived. Funct T Cells Immun. (2008) 29:650–9. doi: 10.1016/j.immuni.2008.07.017
122. Karrer, U, Sierro, S, Wagner, M, Oxenius, A, Hengel, H, Koszinowski, UH, et al. Memory inflation: continous accumulation of antiviral CD8 + T cells over time. J Immunol. (2003) 170:2022–9. doi: 10.4049/jimmunol.171.7.3895-b
123. Morabito, KM, Ruckwardt, TJ, Bar-Haim, E, Nair, D, Moin, SM, Redwood, AJ, et al. Memory inflation drives tissue-resident memory CD8+ T cell maintenance in the lung after intranasal vaccination with murine cytomegalovirus. Front Immunol. (2018) 9:1861. doi: 10.3389/fimmu.2018.01861
124. Schober, K, Voit, F, Grassmann, S, Müller, TR, Eggert, J, Jarosch, S, et al. Reverse TCR repertoire evolution toward dominant low-affinity clones during chronic CMV infection. Nat Immunol. (2020) 21:434–41. doi: 10.1038/s41590-020-0628-2
125. Poschke, IC, Hassel, JC, Rodriguez Ehrenfried, A, Lindner, KAM, Heras-Murillo, I, Appel, LM, et al. The outcome of ex vivo TIL expansion is highly influenced by spatial heterogeneity of the tumor T-cell repertoire and differences in intrinsic in vitro growth capacity between T-cell clones. Clin Cancer Res. (2020). 26:4289–301. doi: 10.1158/1078-0432.ccr-19-3845
126. Turula, H, Smith, CJ, Grey, F, Zurbach, KA, Snyder, CM. Competition between T cells maintains clonal dominance during memory inflation induced by MCMV. Eur J Immunol. (2013) 43:1252–63. doi: 10.1002/eji.201242940
127. Smith, C, Corvino, D, Beagley, L, Rehan, S, Neller, MA, Crooks, P, et al. T cell repertoire remodeling following post-transplant T cell therapy coincides with clinical response. J Clin Invest. (2019) 129:5020–32. doi: 10.1172/JCI128323
128. Walsh, SR, Simovic, B, Chen, L, Bastin, D, Nguyen, A, Stephenson, K, et al. Endogenous T cells prevent tumor immune escape following adoptive T cell therapy. J Clin Invest. (2019) 129:5400–10. doi: 10.1172/JCI126199
129. Long, AH, Haso, WM, Shern, JF, Wanhainen, KM, Murgai, M, Ingaramo, M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
130. Ajina, A, Maher, J. Strategies to address chimeric antigen receptor tonic signaling. Mol Cancer Ther. (2018) 17:1795–815. doi: 10.1158/1535-7163.MCT-17-1097
131. Bridgeman, JS, Ladell, K, Sheard, VE, Miners, K, Hawkins, RE, Price, DA, et al. CD3ζ-based chimeric antigen receptors mediate T cell activation via cis- and trans-signalling mechanisms: Implications for optimization of receptor structure for adoptive cell therapy. Clin Exp Immunol. (2014) 175:258–67. doi: 10.1111/cei.12216
132. Osborne, LC, Abraham, N. Regulation of memory T cells by γc cytokines. Cytokine. (2010) 50:105–13. doi: 10.1016/j.cyto.2009.09.008
133. Surh, CD, Sprent, J. Homeostasis of naive and memory T cells. Immunity. (2008) 29:848–62. doi: 10.1016/j.immuni.2008.11.002
134. Ku, CC, Murakami, M, Sakamoto, A, Kappler, J, Marrack, P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science. (2000) 288:675–8. doi: 10.1126/science.288.5466.675
135. Schluns, KS, Kieper, WC, Jameson, SC, Lefrançois, L. Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat Immunol. (2000) 1:426–32. doi: 10.1038/80868
136. Witherden, D, Van Oers, N, Waltzinger, C, Weiss, A, Benoist, C, Mathis, D. Tetracycline-controllable selection of CD4+ T cells: Half-life and survival signals in the absence of major histocompatibility complex class II molecules. J Exp Med. (2000) 191:355–64. doi: 10.1084/jem.191.2.355
137. Kirberg, J, Berns, A, Von Boehmer, H. Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex-encoded molecules. J Exp Med. (1997) 186:1269–75. doi: 10.1084/jem.186.8.1269
138. van Leeuwen, EM, Sprent, J, Surh, CD. Generation and maintenance of memory CD4+ T Cells. Curr Opin Immunol. (2009) 21:167–72. doi: 10.1016/j.coi.2009.02.005
139. Pepper, M, Linehan, JL, Pagán, AJ, Zell, T, Dileepan, T, Cleary, PP, et al. Different routes of bacterial infection induce long-lived T H 1 memory cells and short-lived T H 17 cells. Nat Immunol. (2010) 11:83–9. doi: 10.1038/ni.1826
140. Yee, C, Thompson, JA, Byrd, D, Riddell, SR, Roche, P, Celis, E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. (2002) 99:16168–73. doi: 10.1073/pnas.242600099
141. Rosenberg, SA, Sportès, C, Ahmadzadeh, M, Fry, TJ, Ngo, LT, Schwarz, SL, et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J Immunother. (2006) 29:313–9. doi: 10.1080/03601270600564088
142. Gattinoni, L, Finkelstein, SE, Klebanoff, CA, Antony, PA, Palmer, DC, Spiess, PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. (2005) 202:907–12. doi: 10.1084/jem.20050732
143. Berger, SC, Berger, M, Hackman, RC, Gough, M, Elliott, C, Jensen, MC, et al. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood. (2009) 114:2417–26. doi: 10.1182/blood-2008-12-189266
144. Li, Y, Bleakley, M, Yee, C. IL-21 Influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol. (2005) 175:2261–9. doi: 10.4049/jimmunol.175.4.2261
145. Dwyer, CJ, Knochelmann, HM, Smith, AS, Wyatt, MM, Rivera, GOR, Arhontoulis, DC, et al. Fueling cancer immunothery with common gamma chain cytokines. Front Immunol. (2019) 10:263. doi: 10.3389/fimmu.2019.00263
146. Wrzesinski, C, Paulos, CM, Gattinoni, L, Palmer, DC, Kaiser, A, Yu, Z, et al. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8+ T cells. J Clin Invest. (2007) 117:492–501. doi: 10.1172/JCI30414
147. Wrzesinski, C, Restifo, NP. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumor immunotherapy. Curr Opin Immunol. (2005) 17:195–201. doi: 10.1016/j.coi.2005.02.002
148. Brentjens, RJ, Rivière, I, Park, JH, Davila, ML, Wang, X, Stefanski, J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. (2011) 118:4817–28. doi: 10.1182/blood-2011-04-348540
149. Turtle, CJ, Hanafi, LA, Berger, C, Gooley, TA, Cherian, S, Hudecek, M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
150. Hirayama, AV, Gauthier, J, Hay, KA, Voutsinas, JM, Wu, Q, Gooley, T, et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood. (2019) 133:1876–87. doi: 10.1182/blood-2018-11-887067
151. Dudley, ME, Wunderlich, JR, Robbins, PF, Yang, JC, Hwu, P, Schwartzentruber, DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. (2002) 298:850–4. doi: 10.1126/science.1076514
152. Restifo, NP, Dudley, ME, Rosenberg, SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. (2012) 12:269–81. doi: 10.1038/nri3191
153. Xu, A, Bhanumathy, KK, Wu, J, Ye, Z, Freywald, A, Leary, SC, et al. IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci. (2016) 6:30. doi: 10.1186/s13578-016-0098-2
154. van der Windt, GJW, Everts, B, Chang, CH, Curtis, JD, Freitas, TC, Amiel, E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8 + T cell memory development. Immunity. (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
155. Ding, ZC, Habtetsion, T, Cao, Y, Li, T, Liu, C, Kuczma, M, et al. Adjuvant IL-7 potentiates adoptive T cell therapy by amplifying and sustaining polyfunctional antitumor CD4+ T cells. Sci Rep. (2017) 7:12168. doi: 10.1038/s41598-017-12488-z
156. Pearce, EL, Walsh, MC, Cejas, PJ, Harms, GM, Shen, H, Wang, LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. (2009) 460:103–7. doi: 10.1038/nature08097
157. Araki, K, Turner, AP, Shaffer, VO, Gangappa, S, Keller, SA, Bachmann, MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. (2009) 460:108–12. doi: 10.1038/nature08155
158. Van Der Windt, GJW, O’Sullivan, D, Everts, B, Huang, SCC, Buck, MD, Curtis, JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA. (2013) 110:14336–41. doi: 10.1073/pnas.1221740110
159. Chang, CH, Qiu, J, O’Sullivan, D, Buck, MD, Noguchi, T, Curtis, JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
160. Ho, PC, Bihuniak, JD, MacIntyre, AN, Staron, M, Liu, X, Amezquita, R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
161. Ye, J, Peng, G. Controlling T cell senescence in the tumor microenvironment for tumor immunotherapy. Oncoimmunology. (2015) 4:e994398. doi: 10.4161/2162402X.2014.994398
162. Sinclair, LV, Rolf, J, Emslie, E, Shi, YB, Taylor, PM, Cantrell, DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. (2013) 14:500–8. doi: 10.1038/ni.2556
163. Hosios, AM, Hecht, VC, Danai, LV, Johnson, MO, Rathmell, JC, Steinhauser, ML, et al. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell. (2016) 36:540–9. doi: 10.1016/j.devcel.2016.02.012
164. Kishton, RJ, Sukumar, M, Restifo, NP. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. (2017) 26:94–109. doi: 10.1016/j.cmet.2017.06.016
165. O’Sullivan, D, vanderWindt, GWJ, Huang, SCC, Curtis, JD, Chang, CH, Buck, MDL, et al. Memory CD8+ T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
166. Pan, Y, Tian, T, Park, CO, Lofftus, SY, Mei, S, Liu, X, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. (2017) 543:252–6. doi: 10.1038/nature21379
167. Manzo, T, Prentice, BM, Anderson, KG, Raman, A, Schalck, A, Codreanu, GS, et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J Exp Med. (2020) 217:e20191920. doi: 10.1084/jem.20191920
168. Crittenden, MR, Zebertavage, L, Kramer, G, Bambina, S, Friedman, D, Troesch, V, et al. Tumor cure by radiation therapy and checkpoint inhibitors depends on pre-existing immunity. Sci Rep. (2018) 8:7012. doi: 10.1038/s41598-018-25482-w
169. Rosato, PC, Wijeyesinghe, S, Stolley, JM, Nelson, CE, Davis, RL, Manlove, LS, et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun. (2019) 10:567. doi: 10.1038/s41467-019-08534-1
170. Chen, PL, Roh, W, Reuben, A, Cooper, ZA, Spencer, CN, Prieto, PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. (2016) 6:827–37. doi: 10.1158/2159-8290.CD-15-1545
171. Taube, JM. Unleashing the immune system: PD-1 and PD-Ls in the pre-treatment tumor microenvironment and correlation with response to PD-1/PD-L1 blockade. Oncoimmunology. (2014) 3:e963413. doi: 10.4161/21624011.2014.963413
172. Jenq, RR, Curran, MA, Goldberg, GL, Liu, C, Allison, JP, van den Brink, MRM. Repertoire enhancement with adoptively transferred female lymphocytes controls the growth of pre-implanted murine prostate cancer. PLoS One. (2012) 7:e35222. doi: 10.1371/journal.pone.0035222
173. Menares, E, Gálvez-Cancino, F, Cáceres-Morgado, P, Ghorani, E, López, E, Díaz, X, et al. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat Commun. (2019) 10:4401. doi: 10.1038/s41467-019-12319-x
174. Manzo, T, Sturmheit, T, Basso, V, Petrozziello, E, Hess Michelini, R, Riba, M, et al. T cells redirected to a minor histocompatibility antigen instruct intratumoral TNFα expression and empower adoptive cell therapy for solid tumors. Cancer Res. (2017) 77:658–71. doi: 10.1158/0008-5472.can-16-0725
175. Hess Michelini, R, Freschi, M, Manzo, T, Jachetti, E, Degl’Innocenti, E, Grioni, M, et al. Concomitant tumor and minor histocompatibility antigen-specific immunity initiate rejection and maintain remission from established spontaneous solid tumors. Cancer Res. (2010) 70:3505–14. doi: 10.1158/0008-5472.can-09-4253
176. Hess Michelini, R, Manzo, T, Sturmheit, T, Basso, V, Rocchi, M, Freschi, M, et al. Vaccine-instructed intratumoral IFN-γ enables regression of autochthonous mouse prostate cancer in allogeneic T-cell transplantation. Cancer Res. (2013) 73:4641–52. doi: 10.1158/0008-5472.can-12-3464
177. Elia, AR, Grioni, M, Basso, V, Curnis, F, Freschi, M, Corti, A, et al. Targeting tumor vasculature with TNF leads effector t cells to the tumor and enhances therapeutic efficacy of immune checkpoint blockers in combination with adoptive cell therapy. Clin Cancer Res. (2018) 24:2171–81. doi: 10.1158/1078-0432.CCR-17-2210
178. Ma, L, Dichwalkar, T, Chang, JYH, Cossette, B, Garafola, D, Zhang, AQ, et al. Enhanced CAR–T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. (2019) 365:162–8. doi: 10.1126/science.aav8692
179. Maurice, NJ, McElrath, MJ, Andersen-Nissen, E, Frahm, N, Prlic, M. CXCR3 enables recruitment and site-specific bystander activation of memory CD8+ T cells. Nat Commun. (2019) 10:4987. doi: 10.1038/s41467-019-12980-2
180. Simoni, Y, Becht, E, Fehlings, M, Loh, CY, Koo, SL, Teng, KWW, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
181. Martin, MD, Jensen, IJ, Ishizuka, AS, Lefebvre, M, Shan, Q, Xue, HH, et al. Bystander responses impact accurate detection of murine and human antigen-specific CD8+ T cells. J Clin Invest. (2019) 129:3894–908. doi: 10.1172/JCI124443
182. Nelson, CE, Thompson, EA, Quarnstrom, CF, Fraser, KA, Seelig, DM, Bhela, S, et al. Robust iterative stimulation with self-antigens overcomes CD8+ T cell tolerance to self- and tumor antigens. Cell Rep. (2019) 28:3092–104.e5. doi: 10.1016/j.celrep.2019.08.038
183. Erkes, DA, Smith, CJ, Wilski, NA, Caldeira-Dantas, S, Mohgbeli, T, Snyder, CM. Virus-specific CD8 + T cells infiltrate melanoma lesions and retain function independently of PD-1 expression. J Immunol. (2017) 198:2979–88. doi: 10.4049/jimmunol.1601064
184. Richer, MJ, Nolz, JC, Harty, JT. Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8+ T cells by enhancing T cell receptor signaling. Immunity. (2013) 38:140–52. doi: 10.1016/j.immuni.2012.09.017
185. Chmielewski, M, Kopecky, C, Hombach, AA, Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. (2011) 71:5697–706. doi: 10.1158/0008-5472.CAN-11-0103
186. Kunert, A, Chmielewski, M, Wijers, R, Berrevoets, C, Abken, H, Debets, R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology. (2017) 7:e1378842. doi: 10.1080/2162402X.2017.1378842
187. Wucherpfennig, KW, Strominger, JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. (1995) 80:695–705. doi: 10.1016/0092-8674(95)90348-8
188. Haluszczak, C, Akue, AD, Hamilton, SE, Johnson, LDS, Pujanauski, L, Teodorovic, L, et al. The antigen-specific CD8 + T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med. (2009) 206:435–48. doi: 10.1084/jem.20081829
189. White, JT, Cross, EW, Kedl, RM. Antigen-inexperienced memory CD8+ T cells: where they come from and why we need them. Nat Rev Immunol. (2017) 17:391–400. doi: 10.1038/nri.2017.34
190. Yang, J, Huck, SP, McHugh, RS, Hermans, IF, Ronchese, F. Perforin-dependent elimination of dendritic cells regulates the expansion of antigen-specific CD8+ T cells in vivo. Proc Natl Acad Sci USA. (2006) 103:147–52. doi: 10.1073/pnas.0509054103
191. Klenerman, P, Hill, A. T cells and viral persistence: lessons from diverse infections. Nat Immunol. (2005) 6:873–9. doi: 10.1038/ni1241
192. Zimmermann, VS, Casati, A, Schiering, C, Caserta, S, Hess Michelini, R, Basso, V, et al. Tumors hamper the immunogenic competence of CD4 + T cell-directed dendritic cell vaccination. J Immunol. (2007) 179:2899–909. doi: 10.4049/jimmunol.179.5.2899
193. Liu, X, Mo, W, Ye, J, Li, L, Zhang, Y, Hsueh, EC, et al. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat Commun. (2018) 9:249. doi: 10.1038/s41467-017-02689-5
194. D’Onofrio, A. A general framework for modeling tumor-immune system competition and immunotherapy: Mathematical analysis and biomedical inferences. Phys D Nonlinear Phenom. (2005) 208:220–35. doi: 10.1016/j.physd.2005.06.032
Keywords: T cell, memory, adoptive T cell immunotherapy of cancer, competition, tumor immunity
Citation: Mondino A and Manzo T (2020) To Remember or to Forget: The Role of Good and Bad Memories in Adoptive T Cell Therapy for Tumors. Front. Immunol. 11:1915. doi: 10.3389/fimmu.2020.01915
Received: 20 December 2019; Accepted: 16 July 2020;
Published: 27 August 2020.
Edited by:
Stefano Caserta, University of Hull, United KingdomReviewed by:
Karl Kai McKinstry, University of Central Florida, United StatesCopyright © 2020 Mondino and Manzo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Mondino, bW9uZGluby5hbm5hQGhzci5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.