 Lan Yang
Lan Yang Jinrong Fu
Jinrong Fu Yufeng Zhou
Yufeng Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 27 August 2020
Sec. Immunological Tolerance and Regulation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01907
This article is part of the Research TopicNovel Advances in Allergy Diagnosis and TreatmentView all 21 articles
The incidence of allergic diseases continues to rise. Cross-sectional and longitudinal studies have indicated that allergic diseases occur in a time-based order: from atopic dermatitis and food allergy in infancy to gradual development into allergic asthma and allergic rhinitis in childhood. This phenomenon is defined as the “atopic march”. Some scholars have suggested that the atopic march does not progress completely in a temporal pattern with genetic and environmental factors. Also, the mechanisms underlying the atopic march are incompletely understood. Nevertheless, the concept of the atopic march provides a new perspective for the mechanistic research, prediction, prevention, and treatment of atopic diseases. Here, we review the epidemiology, related diseases, mechanistic studies, and treatment strategies for the atopic march.
In recent decades, the incidence of allergic diseases has continued to increase, affecting ~20% of the worldwide population, especially children (1). Cross-sectional and longitudinal studies have suggested that allergic diseases occur following a time-based order: from atopic dermatitis (AD) and food allergy in infancy to gradual development into allergic asthma (AA) and allergic rhinitis (AR) in childhood. In terms of anatomic structure, it follows the spatial evolution of skin–gastrointestinal tract–respiratory tract, and this phenomenon is defined as the “atopic march” (2).
Among the allergic diseases mentioned above, some resolve gradually to disappear with age, whereas others continue for many years (3). Some studies have shown that the atopic march does not progress completely in a temporal pattern with genes and the environment (4). Nevertheless, the concept of the atopic march provides a new perspective for the mechanistic research, prediction, prevention, and treatment of allergic diseases.
Here, we review the epidemiology, related diseases, mechanism of action, and treatment strategies of the atopic march.
AD is a chronic recurrent skin disease. Its clinical manifestations are chronic inflammation of the skin, itching, and an impaired skin barrier. AD affects 3% of adults and ~30% of children, and its prevalence tends to increase with age (5). AD occurs in the early years of life. Some epidemiology studies have shown that 45% of affected children had the condition before 6 months of age, 60% before 1 year of age, and up to 85% before 5 years of age (6, 7).
AD etiology is a combination of various factors involving genes and the environment (8). Once external allergens contact a damaged skin barrier, keratinocytes are stimulated to secrete thymic stromal lymphopoietin (TSLP) and other factors in conjunction with Langerhans cells (LCs) to stimulate T-helper type 2 (Th2) immune responses. Then, the body is stimulated to produce non-specific immunoglobulin (Ig)E (if children are exposed to allergens such as mites for a long time, specific IgE may appear). Subsequently, T cells, eosinophils, macrophages, mast cells, and type 2 innate lymphoid cells (ILC2s) infiltrate to secrete cytokines, resulting in local inflammation of the skin (9). AD patients can be classified into two types based on whether the IgE level is increased: intrinsic (normal IgE and non-allergic) and extrinsic (high IgE level associated with increased disease severity). Studies have shown that extrinsic AD increases the risk of developing the atopic march (10, 11).
AA is a common chronic airway disease characterized by the inflammation, hyperresponsiveness, and remodeling of airways (12–15). With modernization and industrialization, AA incidence has increased year by year. This may be because of lifestyle alterations, changes in environmental factors (e.g., increase in indoor dust mites and outdoor pollution), changes in dietary habits, and many other factors. AR involves inflammation of the nasal mucosa (16) and diminishes the quality of life of sufferers (17).
Epidemiologic evidence has revealed a link between AA and AR. A retrospective follow-up study reported the incidence of AR to be higher in AA patients than in non-AA persons (18). In another cohort study, Leynaert et al. demonstrated that 74–81% of AA patients reported AR. Also, they found that AA occurred in 2% of non-AR persons, but in 18.8% of AR patients upon exposure to pollen or animal dander (19).
AR may lead to changes in the function of the lower airways through three main mechanisms. Firstly, stimulation of the nasal mucosa contracts bronchial smooth muscle through the nasal–tracheal reflex. Secondly, various chemical mediators and cytokines released by antigenic stimulation causing nasal mucosa allergy are absorbed into blood, are transported to the lung through circulation, and then act on the trachea and bronchi, causing smooth muscle spasm. Thirdly, nasal inflammatory mediators and secretions are discharged through the nasal passage to the lower airways, resulting in a reduced β-adrenergic receptor functional response (20).
Dharmage et al. found, in infants who have AD within 2 years of age, that the incidence of AA and AR increased significantly during age 6–7 years. In particular, early-onset, persistent, and IgE-positive AD led to a higher risk of developing AA and AR (9). A longitudinal study on a Canadian birth cohort (2,311 children) has shown that AD with sensitization at 1 year of age increased the prevalence of AA and AR at 3 years of age more than 11- and 7-fold, respectively (21). In a recent report from Thailand, 102 children with AD (diagnosed at 1.5 years of age) were reviewed, and subsequently, AR and AA were diagnosed in 61.8 and 29.4%, respectively. Concomitantly, 67% of the AA patients also suffered AR (22). A prospective cohort study (3,124 children aged 1–2 years) reported that, compared with children with no history of AD, those once having AD, particularly moderate-to-severe, early, and persistent AD, were more inclined to develop AA and AR (23).
The discoveries mentioned above strongly support the natural process of the atopic march.
IgE-positive food allergy commonly coexists with AD in early childhood as the earliest manifestation of the atopic march. In 2011, Japanese scholars conducted a retrospective questionnaire survey on freshmen, and they found that AD occurred earlier in those with accompanying food allergy. Also, having food allergy was regarded as the biggest risk factor for the atopic march (24). A family-based cohort study from Chicago revealed that symptomatic food allergy, especially severe or multiple food allergies, was closely related to AA in children aged ≥6 years. Children with food allergy developed AA earlier than those without food allergy (25). A survey of 2,222 infants with AD aged 11.5–25.5 months showed that 64% of children diagnosed with AD within 3 months of birth exhibited an IgE-mediated sensitivity to milk, peanuts, or eggs. Also, in infants <12 months of age, the proportion of infants with sensitivity to eggs, milk, or peanuts increased with AD severity, but this phenomenon was not manifested in children with AD after 1 year of age (26). Among adults with AD, food allergy is relatively rare (27–31). In addition, studies have shown that children sensitive to milk in infancy subsequently exhibited aggravated airway inflammation and increased airway responsiveness to histamine (32, 33). Remarkably, food allergy commonly exists together with AD in infants. Therefore, it is worth exploring whether the link between food allergy and AA or AR is related to AD or is a direct consequence of the food allergy itself.
Eosinophilic esophagitis (EoE) is a chronic esophageal inflammatory disease induced by pollens or food allergens (34). EoE patients are sensitive to allergen avoidance and glucocorticoid therapy. Genome-wide association study (GWAS) data have indicated that EoE shares some susceptible genetic loci with other manifestations of the atopic march, including polymorphisms in the signal transducer and activator of transcription 6 gene (STAT6) and TSLP (35). In addition, epidemiology studies have demonstrated EoE to be associated with other allergic diseases. For example, Mohammad et al. found that, of 449 EoE patients, the prevalence rates of AR, AA, and AD were 61.9, 39, and 46.1%, respectively, and that up to 21.6% of EoE patients developed these three atopic diseases (36). Another study involving 35,528 people reported that those with IgE-positive food allergy were at a higher risk of EoE (37). A birth cohort study involving 130,435 children determined a positive association between EoE and other allergic manifestations (34).
The studies mentioned above suggest the potential of EoE as the fifth “member” of the atopic march, but this hypothesis is controversial. For example, EoE occurs not only in childhood but also after childhood. In addition, EoE can occur in individuals without a history of atopy. Therefore, larger cohorts are needed to study the epidemiologic relationship of EoE with other manifestations of the atopic march and the mechanisms involved (38).
The skin is the foremost barrier for defense against external stimuli, such as pathogens, environmental pollutants, and ultraviolet light. As a component of the innate immune system, the skin has several defensive functions, including microbial, chemical, physical, and immune barrier. These different functions of the skin barrier coordinate with each other to resist external stimuli and maintain skin homeostasis.
Allergens can enter the body through damaged skin to cause sensitization, which is defined as “transcutaneous sensitization” (39). Transcutaneous sensitization can cause AD and, subsequently, AA and AR (5). Studies have shown that epicutaneous disruption induces sensitization after exposure to peanut and egg allergens (40, 41). Spergel et al. demonstrated that repeated cutaneous exposure to egg allergens induced AD-like skin inflammation and AA-like bronchial hyper-responsiveness in a mouse model (41). Emerging studies now suggest that the skin barrier protein filaggrin and epithelial cell-derived cytokines such as TSLP, IL-25, and IL-33 might be related to the progression of the atopic march.
Filaggrin, a barrier protein, has important roles in the integrity of the stratum corneum in terms of structure and composition. Mutations in the filaggrin gene (FLG) can impair the barrier function of the skin and induce an allergic response (42, 43). Several studies have shown patients with impaired or reduced levels of filaggrin to be more susceptible to food sensitization (44–46). Moreover, FLG mutations increase the risk of early and severe AD and of AA in individuals who have had AD (47–49).
TSLP is an interleukin (IL)-7-like epithelial cell-derived cytokine which regulates the Th2 response (50). Zhang et al. found that TSLP overexpression in keratinocytes aggravated AA-like airway inflammation in mice subjected to ovalbumin (OVA) sensitization intraperitoneally and OVA challenge intranasally (51). Another in vivo study demonstrated that keratinocytic TSLP was essential to induce a Th2 response in the skin and to trigger aeroallergen-challenged AA phenotypes (52). In addition, Noti et al. found that the effect of TSLP was enough to develop experimental EoE-like phenotypes in mice (53). Also, they found that TSLP in skin facilitated food allergy (54).
IL-33 is derived from epithelial cells and acts on macrophages, ILC2s, Th2 cells, mast cells, and basophils through the suppression of tumorigenicity 2/IL-1 receptor accessory protein heterodimer (ST2/IL1RL1) (55–60). Several studies have explored the roles of IL-33 in allergic diseases and found high expressions of IL-33 in the skin or airway epithelial cells in AD or airway inflammation (61–63). Blockade of ST2 expression can alleviate food allergy in peanut- and OVA-challenged models (64, 65).
IL-25 is also an epithelial cell-derived cytokine (66–68). Kim et al. found that IL-25 inhibited filaggrin expression in the skin and aggravated skin inflammation by coordinating with Th2 cytokines (69). Lee et al. reported OVA/alum-sensitized allergic diarrhea to be inhibited in mice lacking IL-17RB, the receptor of IL-25, whereas IL-25 overexpression in the intestine accelerated the development of allergic diarrhea (70). Kang et al. found that the mRNA expression of IL-25 was upregulated in rat lungs in a TiO2-induced model of airway inflammation (71).
In conclusion, allergens (including food and aeroallergens) enter the skin through the damaged skin barrier. Then, they stimulate skin epithelial cells to release TSLP, IL-25, and IL-33. This action activates some immune cells in the dermis [e.g., basophils, mast cells, dendritic cells (DCs), eosinophils, ILC2] to secrete cytokines, and subsequently, Th2 cells are generated and IgE production in local lymph nodes occurs. Th2 cells can secrete more type 2 cytokines (e.g., IL-4) to activate more ILC2 and eosinophils, and IgE can act on mast cells and basophils. This positive feedback causes skin inflammation and AD (72). Furthermore, IgE, Th2, TSLP, IL-25, and IL-33 might enter the digestive and respiratory tracts through blood circulation to facilitate the development of AA, AR, and food allergy if allergens are re-encountered (73, 74) (Figure 1). Therefore, skin barrier dysfunction might be a potential mechanism underlying the atopic march.
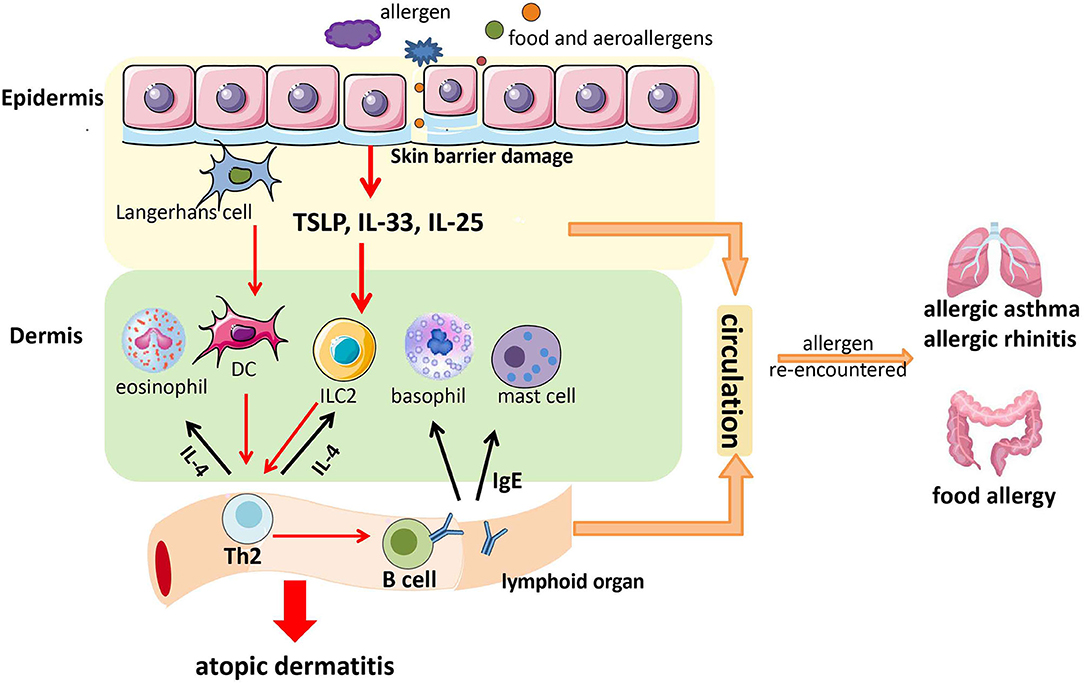
Figure 1. A possible model of the contribution of skin barrier dysfunction to the atopic march. Allergens (including food allergens and aeroallergens) enter the skin through the damaged skin barrier. Then, they stimulate epithelial cells in the skin to release thymic stromal lymphopoietin (TSLP), interleukin (IL)-25, and IL-33. This action activates some immune cells in the dermis (e.g., basophils, mast cells, DCs, eosinophils, and ILC2) to secrete cytokines, followed by the generation of T-helper type 2 (Th2) cells and immunoglobulin E (IgE) production in local lymph nodes. Th2 cells can secrete more type 2 cytokines (e.g., IL-4) to activate more ILC2 and eosinophils, and IgE can act on mast cells and basophils. This positive feedback causes skin inflammation and atopic dermatitis (AD). Furthermore, IgE, Th2 cells, TSLP, IL-25, and IL-33 might enter the digestive tract and respiratory tract through blood circulation to facilitate the development of allergic asthma (AA), allergic rhinitis (AR), and food allergy if allergens are re-encountered.
Many microorganisms are colonized in the intestine, skin, and respiratory tract (75) and influence health and disease. Several studies have suggested that microbiome alteration plays roles in atopic diseases locally or peripherally.
Kennedy et al. observed the skin microbiome dysbiosis in early life of AD patients, and they also found that colonization with commensal Staphylococci at 2 months was related to a lower risk of AD at 1 year of age (76). Forno et al. and Abrahamsson et al. reported that children who had AD at 6 months (77) and 2 years (78) of age had decreased intestinal microbial diversity at 1 month of life. A study of the KOALA birth cohort demonstrated that infants with Clostridium difficile colonization in the gut at 1 month of life were inclined to develop AD and other atopic diseases (79). Azad et al. demonstrated that infants who had a positive skin prick test for food sensitization at 1 year had lower gut microbial richness at 3 months (80). Abrahamsson et al. reported that infants with low diversity in intestinal flora at 1 month of age were inclined to develop AA at school age (81). In addition, Teo et al. determined that microbiome alteration following respiratory infections during infancy might contribute to the development of AA (82).
Moreover, some studies have shown that microbes regulate atopic diseases by secreting metabolites. Furusawa et al. reported that the short-chain fatty acids produced by several intestinal microorganisms induced the proliferation of colonic regulatory T cells (Tregs) and further ameliorated colitis and allergic responses (83). Dysbiosis of Faecalibacterium prausnitzii, as observed in AD, was found to reduce the production of butyrate and propionate and further destroyed the intestinal mucosa. Then, some toxins permeated into the circulation and induced a Th2 immune response to facilitate skin inflammation and AD development (84). Johnson et al. found that the polysaccharides derived from Bacteroides fragilis induced CD4+Foxp3− T cell activation and further prevented AA onset (85).
The studies mentioned above strongly suggest that microbiome alteration may be involved in the atopic march. However, further studies are needed to determine whether microbiome shifts are a cause or a consequence of the atopic march.
Epigenetic mechanisms can regulate gene expression and constitute the cause of diseases. Several epigenome-wide studies have revealed DNA methylation in blood to be related to food allergy (86, 87) and AA (88). Recently, Peng et al. undertook DNA methylation analyses on the cohorts of the Generation R Study (343 at mid-childhood and 839 newborns) in the Netherlands and Project Viva (396 at mid-childhood and 232 newborns) in the USA. Meta-analyses linked the differential methylation profiles of the peripheral blood of mid-childhood children with food allergens, environmental/inhalant allergens, and atopic sensitization. Multiple methylation site-related genes were enriched to AA pathways, including eosinophil peroxidase (EPX), IL4, interleukin 5 receptor A (IL5RA), and proteoglycan 2 (PRG2). Furthermore, Peng et al. identified several methylation sites of cord blood to be related to allergic phenotypes in mid-childhood and that some methylation sites of cord blood were also present in mid-childhood (89), which suggested a longitudinal time trend.
The findings mentioned above suggest that epigenetics may have roles in allergic diseases. However, these studies show only a correlation between epigenetics and the atopic march. Whether epigenetic change is a cause or a result of the atopic march warrants large and detailed longitudinal studies.
Allergic reactions occur not only in the regions where allergens are in contact directly but also in long-distance, non-contact sites. This may be a systemic reaction of the body, and the mechanisms are incompletely understood. Through literature search, Luo et al. proposed a model of “social events” of cells and molecules to explain the atopic march (90). Epithelial cells, such as epidermal keratinocytes and airway epithelial cells, are the first line of defense against allergen exposure and initiate the inflammatory response by releasing proinflammatory cytokines. Thus, epithelial cells are considered to be key participants in allergic diseases. In this model, Luo and colleagues considered that it is the atopic factors produced by epithelial cells locally, not in the circulation, that drive allergy at different sites and that certain allergens are the irritants that trigger the release of atopic factors at different sites. Zhang et al. reported that TSLP overexpression in keratinocytes induced AD-like symptoms and also aggravated OVA-induced AA manifestations in mice. However, they also found that increased TSLP expression in the skin and, subsequently, peripheral blood was not sufficient to induce lung inflammation (51). The atopic reaction in the lung might be induced by the TSLP derived from the lung epithelia themselves. Therefore, the atopic reaction in the skin and the lung might be the consequence of the “social dysfunction” of homologous epithelia and molecules such as TSLP. Despite its rationality and interpretability, the theory of social events needs sufficient evidence from in vivo and in vitro studies.
Marenholz et al. performed GWAS on 2,428 cases with AD in infancy and AA in childhood and on 17,034 controls. They identified seven susceptible sites associated with the atopic march: FLG [1q21.3], AP5B1/OVOL1 [11q13.1], IL4/KIF3A [5q31.1], IKZF3 [17q21], C11orf30/LRRC329, EFHC1 [6p12.3], and rs99322 [12q21.3] (91).
Bioinformatics analyses by Gupta et al. revealed that the atopic march involved 16 common pathogenic genes: IL4, IL5, TSLP, RNASE3, IL13, IL10, IGHG4, IFNG, CCL11, FCER2, RNASE2, FOXP3, KCNE4, CD4, IL4R, and CCL26 (92). These genes were predicted through large-scale and high-throughput bioinformatics analyses, and their roles in the atopic march need to be determined through further experimentation.
Summarily, Paller et al. have reviewed the multifactorial etiology of the atopic march, including skin barrier damage, microbiome alteration, and epigenetic factors (93), and we consider that “social” dysfunction of cells and molecules, and the interference of other predicted genes, may also contribute to the atopic march (Figure 2). However, further studies are required to detail the relevant mechanisms.
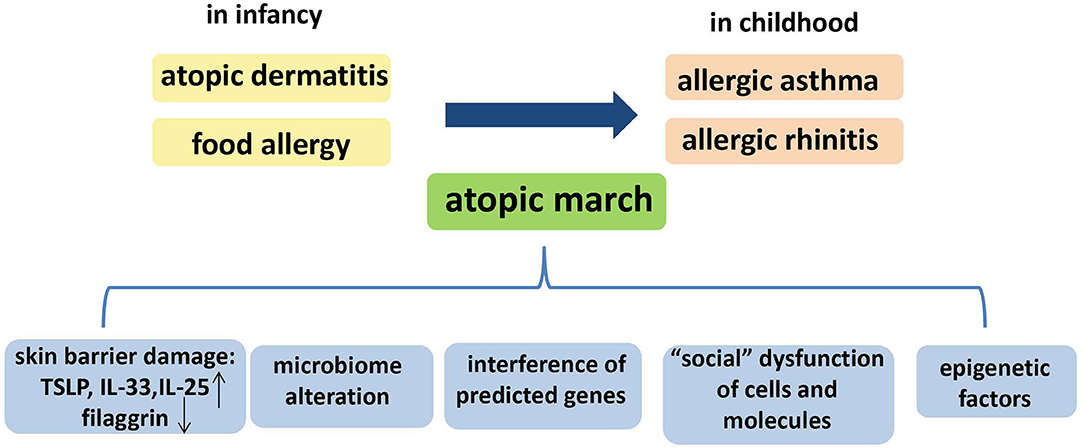
Figure 2. The temporal pattern and the possible mechanisms of the atopic march. The temporal pattern of the atopic march is, in general, from atopic dermatitis (AD) and food allergy in infancy to gradual development into allergic asthma (AA) and allergic rhinitis (AR) in childhood. Several mechanisms could underlie the atopic march: skin barrier damage, microbiome alteration, “social” dysfunction of cells and molecules, epigenetic factors, and interference of other predicted genes.
The modeling process of Leyva-Castillo et al. consisted of two phases. In the first phase, wild-type (WT) BALB/c mice were treated with calcipotriol MC903 plus OVA through epicutaneous sensitization. This led to increased levels of Th2 cytokines, Th17 cytokines, and OVA-specific IgE and IgG1 in serum. In the second phase, MC903-treated (epicutaneous) OVA-sensitized mice underwent intranasal challenge with OVA. These mice exhibited AA-like symptoms with increased mucus secretion, eosinophil infiltration, and expression of Th2 cytokines (52).
In a model established by Han et al., WT BALB/c mice were first treated with OVA plus TSLP via the intradermal route (four times within 2 weeks). After 9 days, the mice were challenged by OVA via the intranasal route for four consecutive days. Consequently, the mice exhibited increased OVA-specific IgE in serum as well as cellular and eosinophil infiltration in the bronchoalveolar lavage fluid. Histopathology showed severe inflammatory infiltrates in mouse lungs. In addition, periodic acid–Schiff staining showed excessive goblet cell metaplasia and mucus secretion (94).
Moreover, one study showed that epicutaneous exposure to Aspergillus fumigates aeroallergens followed by intranasal challenge with A. fumigates induced an allergic nasal response in BALB/c mice (95).
In conclusion, the models mentioned above have one similarity: the skin is used as a sensitization site, consistent with the feature that AD is the initial manifestation during the atopic march. These animal models facilitate the studies of the mechanisms underlying the atopic march.
Despite substantial epidemiologic and experimental evidence, some scholars argued that the prevalence of the atopic march may be overemphasized (96).
First, the methods of the data collection and disease identification initiate one main concern. Considering the cost and time required to make physician diagnoses, allergic disease identification was often based on “yes” or “no” questions. In existing epidemiologic surveys, the diagnosis of AD, AA, and AR simply used “yes” or “no” questionnaires, and some even lacked further physician identification (97–99). In addition, deviations and over-reporting in questionnaire surveys from some individuals led to an overestimation of the disease prevalence (100). Another rebutted criticism of the atopic march is the failure to consider disease heterogeneity or variations. Martinez et al. found that AD patients were at a higher risk of developing transient early AA and persistent AA, not late-onset AA (97). This indicates that the association between AD and AA may be restricted to specific AA subpopulations, not universal. Moreover, some individual-level analyses did not support the typical temporal pattern. At an individual level rather than a large-scale population level, Belgrave et al. demonstrated that only 3.1% of children followed the classical atopic march procession (AD first, followed by AA and then AR) and more than 90% of children with atopic manifestations did not (101).
Unusually, a study from Italy found an evidence of a “reverse” atopic march. The study included 745 children aged 6–9 years with AA only, without a history of food allergy or AD. After a 9-years follow-up, 20% of the children were found to have developed AD (102). In addition, the prevalence of the atopic march differed in distinct countries. Colombian scholars followed up 326 mother–infant pairs in a birth cohort study, and they found that AA was the most common manifestation by 24 months. The prevalence of recurrent AA was 7.1% at 12 months and reached 14.2% at 24 months. However, allergic symptoms induced by milk, egg, or other food allergens were scarce, only 1.8%, and AD was not observed in any cases (103).
Although these studies refute the concept of atopic march to a certain extent, we cannot deny the contribution of the theory of atopic march to the early prevention, diagnosis, and treatment of allergic diseases. Future research on the atopic march should improve the current data collection and disease identification methods, not only relying on “yes” or “no” questionnaires, take disease subtypes into account, and perform the study in an individual level rather than only in a group level.
Several measures used to prevent and treat allergic diseases are expected to interfere with, delay, and block the natural process of the atopic march.
In most studies, breastfeeding for > 6 months has been recommended because it reduces not only the incidence of AD but also of other allergic diseases (104). A 15-years follow-up study of the German Infant Nutritional Intervention (GINI) cohort has shown that, if breastfeeding is not possible, compared with standard cow's milk formula (CMF), the interventional use of partial whey hydrolyzate (pHF-W) formula and extensive casein hydrolyzate (eHF-C) formula in the first 4 months of life has significant preventive effects on AD, and the eHF-C formula also reduced the prevalence of AA and AR (105). However, the mechanisms underlying the preventative effects of hydrolyzed formulas are unknown. In addition, the assessment of the effects of hydrolyzed formulas was based on parental reports of physicians' diagnosis, not on the clinical examinations. These are the criticisms against the use of hydrolyzed formulas for the prevention of allergic conditions.
Moreover, Wickens et al. found that supplementation with Lactobacillus rhamnosus for the first 2 years of life reduced the prevalence of AD by about half (106). However, further studies are required to handle the uncertainties about whether other probiotics are equally effective and how probiotics exert their effects on allergic diseases.
Furthermore, the Learning Early About Peanut Allergy (LEAP) trial demonstrated that, compared with children who avoided peanut, sustained peanut consumption, beginning in the first 11 months of life, significantly decreased the prevalence of peanut allergy at 60 months of age in infants with high atopic risk (107). In addition, a large-scale population-based prospective study showed that early introduction of cow's milk protein as a supplement to breastfeeding might promote tolerance, reducing the incidence of IgE-mediated cow's milk protein allergy (108).
Exposure to several environmental factors is closely related to having allergic diseases. Studies have shown that smoke in the environment increases children's risk of allergic sensitization and AA (109). Therefore, it is strongly recommended that all parents should stop smoking tobacco. Dust mites, pollen, cockroaches, pet fur, and fungi are common allergens, and avoiding exposure to these allergens can reduce the sensitization of children at high risk. However, it has also been proposed that there was no correlation between house dust mite (HDM) exposure and AA (110), and keeping pets (cats or dogs) in the home in the first year after birth reduced the risk of sensitization to multiple allergens during childhood, but it impaired lung function once cat or dog sensitization has occurred, particularly in children with a family history of AA (111). These controversial views are initiating further research to evaluate their relevance.
Antihistamines are used to relieve itching in AD patients and to prevent skin damage aggravated by scratching. Ketotifen, an H1 antihistamine, significantly lowered AA risk in infants with AD or other pre-asthmatic conditions (112, 113). A double-blind, randomized, placebo-controlled trial showed that compared to placebo, cetirizine significantly reduced the incidence of AA in AD patients sensitized to grass pollen or to HDM (114). However, considering the side effects of antihistamines, a large number of clinical trials are needed to evaluate the security of antihistamines and the effectiveness of interventions in the natural course of allergic diseases.
Glucocorticoid is also an effective anti-inflammatory treatment for allergic diseases, and inhaled glucocorticoids has now become the first-line treatment for AA (115). Although the symptoms of AD and AA can be significantly improved by glucocorticoids, it is prone to relapse after withdrawal. In addition, there are many side effects. Therefore, glucocorticoids should be prescribed with caution by the physicians.
Allergen-specific immunotherapy (ASIT), also known as “desensitization therapy,” can alleviate allergy symptoms for a long time and change the natural course of allergic diseases (116). Several ASIT routes have been documented in preclinical studies, including subcutaneous immunotherapy (SCIT), sublingual immunotherapy (SLIT), epicutaneous immunotherapy (EPIT), and oral immunotherapy (OIT). The recognized mechanism of specific immunotherapy is stimulation of the secretion of IL-10 and transforming growth factor-β from Tregs, promotion of the balance of Th1 cells/Th2 cells, and conversion of IgE to IgG to block the IgE-mediated immune cascade (117, 118). Zhong et al. found that the clinical symptoms and quality of life of AD patients with HDM sensitization could be improved after 2 years of ASIT (119). Besh et al. demonstrated that combining basic therapy with SCIT acquired significantly better results in AA patients compared to basic therapy only (120). Karakoc-Aydiner et al. found that the nasal symptom scores of children with AR were significantly reduced after receiving dust mite allergen vaccine through SCIT or SLIT (121). However, the lack of security greatly limits the development of ASIT. For example, the adverse reactions of SLIT mainly focus on local reactions, such as oromucosal pruritus and gastrointestinal reaction (122). In addition, almost all clinical trials related to OIT are accompanied by one or several serious adverse reactions, such as severe gastrointestinal reactions, systemic allergic reactions, etc. (123). Long-term follow-up of milk OIT patients showed that the complete immune tolerance rate after OIT treatment was only 31% (124, 125). Therefore, further research on ASIT should be directed at the improvement of not only its efficacy but also security.
Omalizumab is a human monoclonal antibody against IgE. In 2003, it was approved for the treatment of severe AA in adolescents and adults. Esquivel et al. demonstrated that omalizumab inhibited rhinovirus infections, illnesses, and exacerbations of AA through specific binding to IgE (126). Dupilumab is a human IgG4 monoclonal antibody against IL-4 receptor subunit alpha (IL-4Rα), and it can inhibit IL-4 and IL-13 signaling pathways by interacting with IL-4Rα (127). Dupilumab has been approved by the US Food and Drug Administration to treat infants with moderate-to-severe AD with poor results from conventional treatment (128). Tezepelumab (AMG 157) is a monoclonal antibody (G2λ) against TSLP. In one clinical trial, tezepelumab treatment for 5–12 weeks blunted inhaled allergen-induced AA attacks (129). Of note is that these targeted therapy medicines are only licensed for use in certain allergic diseases. Although the off-label uses or adjunct to treatment for numerous allergic conditions have acquired encouraging results, their potential efficacy still needs to be evaluated through clinical trials.
Although there are no reliable biomarkers to identify subjects with high risk of atopic march, Davidson et al. have proposed some recommendations recently for future research to explore biomarkers, which would provide some possibilities to examine the atopic march.
The relevant proposals are as follows: (1) to look at the protein, RNA, and lipid signatures in infants before and after AD using multi-omics approaches; (2) to analyze transcriptomics, proteomics, metabolomics, and the cell types of infant blood sequentially; (3) to perform sequential immune profiling of the blood, including serology, cytokine profiles, and the evolution of specific B and T cells; (4) to investigate the microbiomes in the skin and gut from birth; and (5) to consider potential maternal delivery effects for atopy (130).
The global increase of atopic diseases greatly lowers the quality of life. The theory of atopic march facilitates our understandings of the pathophysiology of atopic diseases and further promotes the early detection, prevention, and treatment of children at risk of allergic diseases. Future studies on atopic march would be directed at the following points. Firstly, the methods for data collection should be improved and disease heterogeneity or variations should be considered when performing substantial epidemiologic surveys. Secondly, more detailed and logical mechanisms, including genetic and environmental aspects, should be explored to account for the temporal pattern, which would pave the way for novel approaches for the prevention and timely early treatment of the clinical manifestations, ultimately reducing the allergy burden.
LY contributed to collection of references and manuscript preparation. JF and YZ contributed to manuscript modifications. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Key R&D Program of China (2016YFC1305102 to YZ), National Natural Science Foundation of China (81671561, 81974248 to YZ), the International Joint Laboratory Program of National Children's Medical Center (EK1125180109 to YZ), Program for Outstanding Medical Academic Leader (2019LJ19 to YZ), and Shanghai Municipal Planning Commission of Science and Research Fund (201740065 to YZ). Shanghai Pujiang Program (16PJ1401600 to JF). Shanghai Committee of Science and Technology (19ZR1406400 to JF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bantz SK, Zhu Z, Zheng T. The atopic march: progression from atopic dermatitis to allergic rhinitis and asthma. J Clin Cell Immunol. (2014) 5:202. doi: 10.4172/2155-9899.1000202
2. Spergel JM, Paller AS. Atopic dermatitis and the atopic march. J Allergy Clin Immunol. (2003) 112 (6 Suppl.):S118–27. doi: 10.1016/j.jaci.2003.09.033
3. Spergel JM. Epidemiology of atopic dermatitis and atopic march in children. Immunol Allergy Clin North Am. (2010) 30:269–80. doi: 10.1016/j.iac.2010.06.003
4. Zheng T, Yu J, Oh MH, Zhu Z. The atopic march: progression from atopic dermatitis to allergic rhinitis and asthma. Allergy Asthma Immunol Res. (2011) 3:67–73. doi: 10.4168/aair.2011.3.2.67
5. Weidinger S, Novak N. Atopic dermatitis. Lancet. (2016) 387:1109–22. doi: 10.1016/S0140-6736(15)00149-X
6. Spergel JM. Atopic march: link to upper airways. Curr Opin Allergy Clin Immunol. (2005) 5:17–21. doi: 10.1097/00130832-200502000-00005
7. Kay J, Gawkrodger DJ, Mortimer MJ, Jaron AG. The prevalence of childhood atopic eczema in a general population. J Am Acad Dermatol. (1994) 30:35–9. doi: 10.1016/S0190-9622(94)70004-4
9. Dharmage SC, Lowe AJ, Matheson MC, Burgess JA, Allen KJ, Abramson MJ. Atopic dermatitis and the atopic march revisited. Allergy. (2014) 69:17–27. doi: 10.1111/all.12268
10. Novembre E, Cianferoni A, Lombardi E, Bernardini R, Pucci N, Vierucci A. Natural history of “intrinsic” atopic dermatitis. Allergy. (2001) 56:452–3. doi: 10.1034/j.1398-9995.2001.056005452.x
11. Wuthrich B, Schmid-Grendelmeier P. Natural course of AEDS. Allergy. (2002) 57:267–8. doi: 10.1034/j.1398-9995.2002.1n3572.x
12. Redington AE, Howarth PH. Airway wall remodelling in asthma. Thorax. (1997) 52:310–12. doi: 10.1136/thx.52.4.310
13. Kon OM, Kay AB. T cells and chronic asthma. Int Arch Allergy Immunol. (1999) 118:133–5. doi: 10.1159/000024049
14. Carroll N, Elliot J, Morton A, James A. The structure of large and small airways in nonfatal and fatal asthma. Am Rev Respir Dis. (1993) 147:405–10. doi: 10.1164/ajrccm/147.2.405
15. James A. Airway remodeling in asthma. Curr Opin Pulm Med. (2005) 11:1–6. doi: 10.1097/01.mcp.0000146779.26339.d8
16. Hellings PW, Fokkens WJ. Allergic rhinitis and its impact on otorhinolaryngology. Allergy. (2006) 61:656–64. doi: 10.1111/j.1398-9995.2006.01109.x
17. Selnes A, Nystad W, Bolle R, Lund E. Diverging prevalence trends of atopic disorders in Norwegian children. Results from three cross-sectional studies. Allergy. (2005) 60:894–9. doi: 10.1111/j.1398-9995.2005.00797.x
18. Ricci G, Patrizi A, Baldi E, Menna G, Tabanelli M, Masi M. Long-term follow-up of atopic dermatitis: retrospective analysis of related risk factors and association with concomitant allergic diseases. J Am Acad Dermatol. (2006) 55:765–71. doi: 10.1016/j.jaad.2006.04.064
19. Leynaert B, Neukirch C, Kony S, Guenegou A, Bousquet J, Aubier M, et al. Association between asthma and rhinitis according to atopic sensitization in a population-based study. J Allergy Clin Immunol. (2004) 113:86–93. doi: 10.1016/j.jaci.2003.10.010
20. Corren J, Adinoff AD, Irvin CG. Changes in bronchial responsiveness following nasal provocation with allergen. J Allergy Clin Immunol. (1992) 89:611–8. doi: 10.1016/0091-6749(92)90329-Z
21. Tran MM, Lefebvre DL, Dharma C, Dai DW, Lou YWY, Subbarao P, et al. Predicting the atopic march: results from the canadian healthy infant longitudinal development study. J Allergy Clin Immunol. (2018) 141:601–7 e8. doi: 10.1016/j.jaci.2017.08.024
22. Somanunt S, Chinratanapisit S, Pacharn P, Visitsunthorn N, Jirapongsananuruk O. The natural history of atopic dermatitis and its association with Atopic March. Asian Pac J Allergy Immunol. (2017) 35:137–43. doi: 10.1016/j.jaci.2015.12.613
23. von Kobyletzki LB, Bornehag CG, Hasselgren M, Larsson M, Lindstrom CB, Svensson A. Eczema in early childhood is strongly associated with the development of asthma and rhinitis in a prospective cohort. BMC Dermatol. (2012) 12:11. doi: 10.1186/1471-5945-12-11
24. Kijima A, Murota H, Takahashi A, Arase N, Yang L, Nishioka M, et al. Prevalence and impact of past history of food allergy in atopic dermatitis. Allergol Int. (2013) 62:105–12. doi: 10.2332/allergolint.12-OA-0468
25. Schroeder A, Kumar R, Pongracic JA, Sullivan CL, Caruso DM, Costello J, et al. Food allergy is associated with an increased risk of asthma. Clin Exp Allergy. (2009) 39:261–70. doi: 10.1111/j.1365-2222.2008.03160.x
26. Hill DJ, Hosking CS, de Benedictis FM, Oranje AP, Diepgen TL, Bauchau V, et al. Confirmation of the association between high levels of immunoglobulin E food sensitization and eczema in infancy: an international study. Clin Exp Allergy. (2008) 38:161–8. doi: 10.1111/j.1365-2222.2007.02861.x
27. Tse K, Horner AA. Allergen tolerance versus the allergic march: the hygiene hypothesis revisited. Curr Allergy Asthma Rep. (2008) 8:475–83. doi: 10.1007/s11882-008-0088-5
28. Gustafsson D, Sjoberg O, Foucard T. Development of allergies and asthma in infants and young children with atopic dermatitis–a prospective follow-up to 7 years of age. Allergy. (2000) 55:240–5. doi: 10.1034/j.1398-9995.2000.00391.x
29. Lowe AJ, Carlin JB, Bennett CM, Hosking CS, Abramson MJ, Hill DJ, et al. Do boys do the atopic march while girls dawdle? J Allergy Clin Immunol. (2008) 121:1190–5. doi: 10.1016/j.jaci.2008.01.034
30. Burgess JA, Lowe AJ, Matheson MC, Varigos G, Abramson MJ, Dharmage SC. Does eczema lead to asthma? J Asthma. (2009) 46:429–36. doi: 10.1080/02770900902846356
31. Bousquet J, Khaltaev N, Cruz AA, Denburg J, Fokkens WJ, Togias A, et al. Allergic rhinitis and its impact on asthma (ARIA) 2008 update (in collaboration with the World Health Organization, GA(2)LEN and AllerGen). Allergy. (2008) 63 (Suppl. 86):8–160. doi: 10.1111/j.1398-9995.2007.01620.x
32. Malmberg LP, Saarinen KM, Pelkonen AS, Savilahti E, Makela MJ. Cow's milk allergy as a predictor of bronchial hyperresponsiveness and airway inflammation at school age. Clin Exp Allergy. (2010) 40:1491–7. doi: 10.1111/j.1365-2222.2010.03567.x
33. Saarinen KM, Pelkonen AS, Makela MJ, Savilahti E. Clinical course and prognosis of cow's milk allergy are dependent on milk-specific IgE status. J Allergy Clin Immunol. (2005) 116:869–75. doi: 10.1016/j.jaci.2005.06.018
34. Hill DA, Grundmeier RW, Ramos M, Spergel JM. Eosinophilic esophagitis is a late manifestation of the allergic march. J Allergy Clin Immunol Pract. (2018) 6:1528–33. doi: 10.1016/j.jaip.2018.05.010
35. Hirota T, Nakayama T, Sato S, Yanagida N, Matsui T, Sugiura S, et al. Association study of childhood food allergy with genome-wide association studies-discovered loci of atopic dermatitis and eosinophilic esophagitis. J Allergy Clin Immunol. (2017) 140:1713–6. doi: 10.1016/j.jaci.2017.05.034
36. Mohammad AA, Wu SZ, Ibrahim O, Bena J, Rizk M, Piliang M, et al. Prevalence of atopic comorbidities in eosinophilic esophagitis: a case-control study of 449 patients. J Am Acad Dermatol. (2017) 76:559–60. doi: 10.1016/j.jaad.2016.08.068
37. Hill DA, Dudley JW, Spergel JM. The prevalence of eosinophilic esophagitis in pediatric patients with ige-mediated food allergy. J Allergy Clin Immunol Pract. (2017) 5:369–75. doi: 10.1016/j.jaip.2016.11.020
38. Hill DA, Spergel JM. Is eosinophilic esophagitis a member of the atopic march? Ann Allergy Asthma Immunol. (2018) 120:113–4. doi: 10.1016/j.anai.2017.10.003
39. Strugar TL, Kuo A, Seite S, Lin M, Lio P. Connecting the dots: from skin barrier dysfunction to allergic sensitization, and the role of moisturizers in repairing the skin barrier. J Drugs Dermatol. (2019) 18:581.
40. Strid J, Hourihane J, Kimber I, Callard R, Strobel S. Disruption of the stratum corneum allows potent epicutaneous immunization with protein antigens resulting in a dominant systemic Th2 response. Eur J Immunol. (2004) 34:2100–9. doi: 10.1002/eji.200425196
41. Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J Clin Invest. (1998) 101:1614–22. doi: 10.1172/JCI1647
42. Kezic S, Kemperman PM, Koster ES, de Jongh CM, Thio HB, Campbell LE, et al. Loss-of-function mutations in the filaggrin gene lead to reduced level of natural moisturizing factor in the stratum corneum. J Invest Dermatol. (2008) 128:2117–9. doi: 10.1038/jid.2008.29
43. O'Regan GM, Sandilands A, W.McLean HI, Irvine AD. Filaggrin in atopic dermatitis. J Allergy Clin Immunol. (2008) 122:689–93. doi: 10.1016/j.jaci.2008.08.002
44. Guttman-Yassky E, Zhou L, Krueger JG. The skin as an immune organ: tolerance versus effector responses and applications to food allergy and hypersensitivity reactions. J Allergy Clin Immunol. (2019) 144:362–74. doi: 10.1016/j.jaci.2019.03.021
45. Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med. (2011) 365:1315–27. doi: 10.1056/NEJMra1011040
46. Tan HT, Ellis JA, Koplin JJ, Matheson MC, Gurrin LC, Lowe AJ, et al. Filaggrin loss-of-function mutations do not predict food allergy over and above the risk of food sensitization among infants. J Allergy Clin Immunol. (2012) 130:1211–3 e3. doi: 10.1016/j.jaci.2012.07.022
47. Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. (2006) 38:441–6. doi: 10.1038/ng1767
48. O'Regan GM, Sandilands A, McLean WH, Irvine AD. Filaggrin in atopic dermatitis. J Allergy Clin Immunol. (2009) 124(3 Suppl. 2):R2–6. doi: 10.1016/j.jaci.2009.07.013
49. Hudson TJ. Skin barrier function and allergic risk. Nat Genet. (2006) 38:399–400. doi: 10.1038/ng0406-399
50. Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol. (2010) 11:289–93. doi: 10.1038/ni.1852
51. Zhang Z, Hener P, Frossard N, Kato S, Metzger D, Li M, et al. Thymic stromal lymphopoietin overproduced by keratinocytes in mouse skin aggravates experimental asthma. Proc Natl Acad Sci USA. (2009) 106:1536–41. doi: 10.1073/pnas.0812668106
52. Leyva-Castillo JM, Hener P, Jiang H, Li M. TSLP produced by keratinocytes promotes allergen sensitization through skin and thereby triggers atopic march in mice. J Invest Dermatol. (2013) 133:154–63. doi: 10.1038/jid.2012.239
53. Noti M, Wojno ED, Kim BS, Siracusa MC, Giacomin PR, Nair MG, et al. Thymic stromal lymphopoietin-elicited basophil responses promote eosinophilic esophagitis. Nat Med. (2013) 19:1005–13. doi: 10.1038/nm.3281
54. Noti M, Kim BS, Siracusa MC, Rak GD, Kubo M, Moghaddam AE, et al. Exposure to food allergens through inflamed skin promotes intestinal food allergy through the thymic stromal lymphopoietin-basophil axis. J Allergy Clin Immunol. (2014) 133:1390–9:1399 e1–6. doi: 10.1016/j.jaci.2014.01.021
55. Xu D, Chan WL, Leung BP, Huang F, Wheeler R, Piedrafita D, et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med. (1998) 187:787–94. doi: 10.1084/jem.187.5.787
56. Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: the ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. (2007) 179:2051–4. doi: 10.4049/jimmunol.179.4.2051
57. Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. (2008) 20:791–800. doi: 10.1093/intimm/dxn037
58. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. (2013) 210:2939–50. doi: 10.1084/jem.20130351
59. Siede J, Frohlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 receptor-expressing regulatory T cells are highly activated, Th2 biased and suppress CD4 T cell proliferation through IL-10 and TGFbeta release. PLoS ONE. (2016) 11:e0161507. doi: 10.1371/journal.pone.0161507
60. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. (2010) 463:540–4. doi: 10.1038/nature08636
61. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity. (2015) 42:1005-19. doi: 10.1016/j.immuni.2015.06.006
62. Savinko T, Karisola P, Lehtimaki S, Lappetelainen AM, Haapakoski R, Wolff H, et al. ST2 regulates allergic airway inflammation and T-cell polarization in epicutaneously sensitized mice. J Invest Dermatol. (2013) 133:2522–9. doi: 10.1038/jid.2013.195
63. Savinko T, Matikainen S, Saarialho-Kere U, Lehto M, Wang G, Lehtimaki S, et al. IL-33 and ST2 in atopic dermatitis: expression profiles and modulation by triggering factors. J Invest Dermatol. (2012) 132:1392–400. doi: 10.1038/jid.2011.446
64. Tordesillas L, Goswami R, Benede S, Grishina G, Dunkin D, Jarvinen KM, et al. Skin exposure promotes a Th2-dependent sensitization to peanut allergens. J Clin Invest. (2014) 124:4965–75. doi: 10.1172/JCI75660
65. Galand C, Leyva-Castillo JM, Yoon J, Han A, Lee MS, McKenzie ANJ, et al. IL-33 promotes food anaphylaxis in epicutaneously sensitized mice by targeting mast cells. J Allergy Clin Immunol. (2016) 138:1356–66. doi: 10.1016/j.jaci.2016.03.056
66. Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. (2001) 15:985–95. doi: 10.1016/S1074-7613(01)00243-6
67. Angkasekwinai P, Park H, Wang YH, Wang YH, Chang SH, Corry DB, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. (2007) 204:1509–17. doi: 10.1084/jem.20061675
68. Corrigan CJ, Wang W, Meng Q, Fang C, Eid G, Caballero MR, et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J Allergy Clin Immunol. (2011) 128:116–24. doi: 10.1016/j.jaci.2011.03.043
69. Kim BE, Bin L, Ye YM, Ramamoorthy P, D.Leung YM. IL-25 enhances HSV-1 replication by inhibiting filaggrin expression, and acts synergistically with Th2 cytokines to enhance HSV-1 replication. J Invest Dermatol. (2013) 133:2678–85. doi: 10.1038/jid.2013.223
70. Lee JB, Chen CY, Liu B, Mugge L, Angkasekwinai P, Facchinetti V, et al. IL-25 and CD4(+) TH2 cells enhance type 2 innate lymphoid cell-derived IL-13 production, which promotes IgE-mediated experimental food allergy. J Allergy Clin Immunol. (2016) 137:1216–25 e5. doi: 10.1016/j.jaci.2015.09.019
71. Kang CM, Jang AS, Ahn MH, Shin JA, Kim JH, Choi YS, et al. Interleukin-25 and interleukin-13 production by alveolar macrophages in response to particles. Am J Respir Cell Mol Biol. (2005) 33:290–6. doi: 10.1165/rcmb.2005-0003OC
72. Han H, Roan F, Ziegler SF. The atopic march: current insights into skin barrier dysfunction and epithelial cell-derived cytokines. Immunol Rev. (2017) 278:116–30. doi: 10.1111/imr.12546
73. Hill DA, Spergel JM. The atopic march: critical evidence and clinical relevance. Ann Allergy Asthma Immunol. (2018) 120:131–7. doi: 10.1016/j.anai.2017.10.037
74. Li M. Current evidence of epidermal barrier dysfunction and thymic stromal lymphopoietin in the atopic march. Eur Respir Rev. (2014) 23:292–8. doi: 10.1183/09059180.00004314
75. Saad MJ, Santos A, Prada PO. Linking gut microbiota and inflammation to obesity and insulin resistance. Physiology (Bethesda). (2016) 31:283–93. doi: 10.1152/physiol.00041.2015
76. Kennedy EA, Connolly J, Hourihane JO, Fallon PG, McLean WHI, Murray D, et al. Skin microbiome before development of atopic dermatitis: early colonization with commensal staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J Allergy Clin Immunol. (2017) 139:166–72. doi: 10.1016/j.jaci.2016.07.029
77. Forno E, Onderdonk AB, McCracken J, Litonjua AA, Laskey D, Delaney ML, et al. Diversity of the gut microbiota and eczema in early life. Clin Mol Allergy. (2008) 6:11. doi: 10.1186/1476-7961-6-11
78. Abrahamsson TR, Jakobsson HE, Andersson AF, Bjorksten B, Engstrand L, Jenmalm MC. Low diversity of the gut microbiota in infants with atopic eczema. J Allergy Clin Immunol. (2012) 129:434–40, 440 e1–2. doi: 10.1016/j.jaci.2011.10.025
79. Penders J, Thijs C, van den Brandt PA, Kummeling I, Snijders B, Stelma F, et al. Gut microbiota composition and development of atopic manifestations in infancy: the KOALA Birth Cohort Study. Gut. (2007) 56:661–7. doi: 10.1136/gut.2006.100164
80. Azad MB, Konya T, Guttman DS, Field CJ, Sears MR, HayGlass KT, et al. Infant gut microbiota and food sensitization: associations in the first year of life. Clin Exp Allergy. (2015) 45:632–43. doi: 10.1111/cea.12487
81. Abrahamsson TR, Jakobsson HE, Andersson AF, Bjorksten B, Engstrand L, Jenmalm MC. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin Exp Allergy. (2014) 44:842–50. doi: 10.1111/cea.12253
82. Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N, et al. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe. (2015) 17:704–15. doi: 10.1016/j.chom.2015.03.008
83. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
84. Song H, Yoo Y, Hwang J, Na YC, Kim HS. Faecalibacterium prausnitzii subspecies-level dysbiosis in the human gut microbiome underlying atopic dermatitis. J Allergy Clin Immunol. (2016) 137:852–60. doi: 10.1016/j.jaci.2015.08.021
85. Johnson JL, Jones MB, Cobb BA. Bacterial capsular polysaccharide prevents the onset of asthma through T-cell activation. Glycobiology. (2015) 25:368–75. doi: 10.1093/glycob/cwu117
86. Martino D, Joo JE, Sexton-Oates A, Dang T, Allen K, Saffery R, et al. Epigenome-wide association study reveals longitudinally stable DNA methylation differences in CD4+ T cells from children with IgE-mediated food allergy. Epigenetics. (2014) 9:998–1006. doi: 10.4161/epi.28945
87. Hong X, Hao K, Ladd-Acosta C, Hansen KD, Tsai HJ, Liu X, et al. Genome-wide association study identifies peanut allergy-specific loci and evidence of epigenetic mediation in US children. Nat Commun. (2015) 6:6304. doi: 10.1038/ncomms7304
88. Xu CJ, Soderhall C, Bustamante M, Baiz N, Gruzieva O, Gehring U, et al. DNA methylation in childhood asthma: an epigenome-wide meta-analysis. Lancet Respir Med. (2018) 6:379–388. doi: 10.1016/S2213-2600(18)30052-3
89. Peng C, Van Meel ER, Cardenas A, Rifas-Shiman SL, Sonawane AR, Glass KR, et al. Epigenome-wide association study reveals methylation pathways associated with childhood allergic sensitization. Epigenetics. (2019) 14:445–66. doi: 10.1080/15592294.2019.1590085
90. Luo J, Li Y, Gong R. The mechanism of atopic march may be the 'social' event of cells and molecules (Review). Int J Mol Med. (2010) 26:779–85. doi: 10.3892/ijmm_00000525
91. Marenholz I, Esparza-Gordillo J, Ruschendorf F, Bauerfeind A, Strachan DP, Spycher BD, et al. Meta-analysis identifies seven susceptibility loci involved in the atopic march. Nat Commun. (2015) 6:8804. doi: 10.1038/ncomms9804
92. Gupta J, Johansson E, Bernstein JA, Chakraborty R, Khurana Hershey GK, Rothenberg ME, et al. Resolving the etiology of atopic disorders by using genetic analysis of racial ancestry. J Allergy Clin Immunol. (2016) 138:676–99. doi: 10.1016/j.jaci.2016.02.045
93. Paller AS, Spergel JM, Mina-Osorio P, Irvine AD. The atopic march and atopic multimorbidity: many trajectories, many pathways. J Allergy Clin Immunol. (2019) 143:46–55. doi: 10.1016/j.jaci.2018.11.006
94. Han H, Xu W, Headley MB, Jessup HK, Lee KS, Omori M, et al. Thymic stromal lymphopoietin (TSLP)-mediated dermal inflammation aggravates experimental asthma. Mucosal Immunol. (2012) 5:342–51. doi: 10.1038/mi.2012.14
95. Akei HS, Brandt EB, Mishra A, Strait RT, Finkelman FD, Warrier MR, et al. Epicutaneous aeroallergen exposure induces systemic TH2 immunity that predisposes to allergic nasal responses. J Allergy Clin Immunol. (2006) 118:62–9. doi: 10.1016/j.jaci.2006.04.046
96. Busse WW. The atopic march: fact or folklore? Ann Allergy Asthma Immunol. (2018) 120:116–18. doi: 10.1016/j.anai.2017.10.029
97. Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, Morgan WJ. Asthma and wheezing in the first six years of life. The Group Health Medical Associates. N Engl J Med. (1995) 332:133–8. doi: 10.1056/NEJM199501193320301
98. Burgess JA, Dharmage SC, Byrnes GB, Matheson MC, Gurrin LC, Wharton CL, et al. Childhood eczema and asthma incidence and persistence: a cohort study from childhood to middle age. J Allergy Clin Immunol. (2008) 122:280–5. doi: 10.1016/j.jaci.2008.05.018
99. Abo-Zaid G, Sharpe RA, Fleming LE, Depledge M, Osborne NJ. Association of infant eczema with childhood and adult asthma: analysis of data from the 1958 birth cohort study. Int J Environ Res Public Health. (2018) 15:1415. doi: 10.3390/ijerph15071415
100. Soller L, Ben-Shoshan M, Harrington DW, Fragapane J, Joseph L, St Pierre Y, et al. Overall prevalence of self-reported food allergy in Canada. J Allergy Clin Immunol. (2012) 130:986–8. doi: 10.1016/j.jaci.2012.06.029
101. Belgrave DC, Simpson A, Buchan IE, Custovic A. Atopic dermatitis and respiratory allergy: what is the link. Curr Dermatol Rep. (2015) 4:221–7. doi: 10.1007/s13671-015-0121-6
102. Barberio G, Pajno GB, Vita D, Caminiti L, Canonica GW, Passalacqua G. Does a 'reverse' atopic march exist? Allergy. (2008) 63:1630–2. doi: 10.1111/j.1398-9995.2008.01710.x
103. Acevedo N, Sanchez J, Zakzuk J, Bornacelly A, Quiroz C, Alvarez A, et al. Particular characteristics of allergic symptoms in tropical environments: follow up to 24 months in the FRAAT birth cohort study. BMC Pulm Med. (2012) 12:13. doi: 10.1186/1471-2466-12-13
104. Kull I, Bohme M, Wahlgren CF, Nordvall L, Pershagen G, Wickman M. Breast-feeding reduces the risk for childhood eczema. J Allergy Clin Immunol. (2005) 116:657–61. doi: 10.1016/j.jaci.2005.04.028
105. von Berg A, Filipiak-Pittroff B, Schulz H, Hoffmann U, Link E, Sussmann M, et al. Allergic manifestation 15 years after early intervention with hydrolyzed formulas–the GINI Study. Allergy. (2016) 71:210–9. doi: 10.1111/all.12790
106. Wickens K, Black PN, Stanley TV, Mitchell E, Fitzharris P, Tannock GW, et al. A differential effect of 2 probiotics in the prevention of eczema and atopy: a double-blind, randomized, placebo-controlled trial. J Allergy Clin Immunol. (2008) 122:788–94. doi: 10.1016/j.jaci.2008.07.011
107. Du Toit G, Roberts G, Sayre PH, Bahnson HT, Radulovic S, Santos AF, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. (2015) 372:803–13. doi: 10.1056/NEJMoa1414850
108. Katz Y, Rajuan N, Goldberg MR, Eisenberg E, Heyman E, Cohen A, et al. Early exposure to cow's milk protein is protective against IgE-mediated cow's milk protein allergy. J Allergy Clin Immunol. (2010) 126:77–82 e1. doi: 10.1016/j.jaci.2010.04.020
109. Lannero E, Wickman M, van Hage M, Bergstrom A, Pershagen G, Nordvall L. Exposure to environmental tobacco smoke and sensitisation in children. Thorax. (2008) 63:172–6. doi: 10.1136/thx.2007.079053
110. Dick S, Friend A, Dynes K, AlKandari F, Doust E, Cowie H, et al. A systematic review of associations between environmental exposures and development of asthma in children aged up to 9 years. BMJ Open. (2014) 4:e006554. doi: 10.1136/bmjopen-2014-006554
111. Portnoy J, Kennedy K, Sublett J, Phipatanakul W, Matsui E, Barnes C, et al. Environmental assessment and exposure control: a practice parameter–furry animals. Ann Allergy Asthma Immunol. (2012) 108:223 e1–15. doi: 10.1016/j.anai.2012.02.015
112. Iikura Y, Naspitz CK, Mikawa H, Talaricoficho S, Baba M, Sole D, et al. Prevention of asthma by ketotifen in infants with atopic dermatitis. Ann Allergy. (1992) 68:233–6.
113. Bustos GJ, Bustos D, Bustos GJ, Romero O. Prevention of asthma with ketotifen in preasthmatic children: a three-year follow-up study. Clin Exp Allergy. (1995) 25:568–73. doi: 10.1111/j.1365-2222.1995.tb01096.x
114. Warner JO, Businco L, Casimir G, Diepgen TL, Kjellman M, Knol K, et al. Allergic factors associated with the development of asthma the influence of cetirizine in a double-blind, randomised, placebo-controlled trial: first results of ETAC. Early Treatment of the Atopic Child. Pediatr Allergy Immunol. (1998) 9:116–24. doi: 10.1111/j.1399-3038.1998.tb00356.x
116. Jacobsen L, Wahn U, Bilo MB. Allergen-specific immunotherapy provides immediate, long-term and preventive clinical effects in children and adults: the effects of immunotherapy can be categorised by level of benefit -the centenary of allergen specific subcutaneous immunotherapy. Clin Transl Allergy. (2012) 2:8. doi: 10.1186/2045-7022-2-8
117. Kawauchi H, Goda K, Tongu M, Yamada T, Aoi N, Morikura I, et al. Short review on sublingual immunotherapy for patients with allergic rhinitis: from bench to bedside. Adv Otorhinolaryngol. (2011) 72:103–6. doi: 10.1159/000324631
118. Bahceciler NN, Arikan C, Taylor A, Akdis M, Blaser K, Barlan IB, et al. Impact of sublingual immunotherapy on specific antibody levels in asthmatic children allergic to house dust mites. Int Arch Allergy Immunol. (2005) 136:287–94. doi: 10.1159/000083956
119. Zhong H, Deng X, Song Z, Darsow U, Chen W, Chen S, et al. Immunological changes after ASIT in AD allergen-specific immunotherapy and their potential correlation with clinical response in patients with atopic dermatitis patients sensitized to house dust mite. J Eur Acad Dermatol Venereol. (2015) 29:1318–24. doi: 10.1111/jdv.12813
120. Besh O, Besh D, Sorkopud O, Kondratiuk M, Slaba O, Zhakun I, et al. ASIT as the component of bronchial asthma's therapy can improve the adherence to the treatment. Wiad Lek. (2018) 71:849–54.
121. Karakoc-Aydiner E, Eifan AO, Baris S, Gunay E, Akturk E, Akkoc T, et al. Long-term effect of sublingual and subcutaneous immunotherapy in dust mite-allergic children with asthma/rhinitis: a 3-year prospective randomized controlled trial. J Investig Allergol Clin Immunol. (2015) 25:334−42.
122. Canonica GW, Cox L, Pawankar R, Baena-Cagnani CE, Blaiss M, Bonini S, et al. Sublingual immunotherapy: World Allergy Organization position paper 2013 update. World Allergy Organ J. (2014) 7:6. doi: 10.1186/1939-4551-7-6
123. Hofmann AM, Scurlock AM, Jones SM, Palmer KP, Lokhnygina Y, Steele PH, et al. Safety of a peanut oral immunotherapy protocol in children with peanut allergy. J Allergy Clin Immunol. (2009) 124:286–91:291 e1–6. doi: 10.1016/j.jaci.2009.03.045
124. Skripak JM, Nash SD, Rowley H, Brereton NH, Oh S, Hamilton RG, et al. A randomized, double-blind, placebo-controlled study of milk oral immunotherapy for cow's milk allergy. J Allergy Clin Immunol. (2008) 122:1154–60. doi: 10.1016/j.jaci.2008.09.030
125. Keet CA, Frischmeyer-Guerrerio PA, Thyagarajan A, Schroeder JT, Hamilton RG, Boden S, et al. The safety and efficacy of sublingual and oral immunotherapy for milk allergy. J Allergy Clin Immunol. (2012) 129:448–55:455 e1–5. doi: 10.1016/j.jaci.2011.10.023
126. Esquivel A, Busse WW, Calatroni A, Togias AG, Grindle KG, Bochkov YA, et al. Effects of omalizumab on rhinovirus infections, illnesses, and exacerbations of asthma. Am J Respir Crit Care Med. (2017) 196:985–92. doi: 10.1164/rccm.201701-0120OC
127. Gandhi NA, Bennett BL, Graham NM, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov. (2016) 15:35–50. doi: 10.1038/nrd4624
128. Chang HY. Nadeau KC, IL-4Ralpha inhibitor for atopic disease. Cell. (2017) 170:222. doi: 10.1016/j.cell.2017.06.046
129. Gauvreau GM, O'Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med. (2014) 370:2102–10. doi: 10.1056/NEJMoa1402895
130. Davidson WF, Leung DYM, Beck LA, Berin CM, Boguniewicz M, Busse WW, et al. Report from the national institute of allergy and infectious diseases workshop on “Atopic dermatitis and the atopic march: mechanisms and interventions”. J Allergy Clin Immunol. (2019) 143:894–913. doi: 10.1016/j.jaci.2019.01.003
Keywords: atopic dermatitis, asthma, food allergy, allergic rhinitis, atopic march
Citation: Yang L, Fu J and Zhou Y (2020) Research Progress in Atopic March. Front. Immunol. 11:1907. doi: 10.3389/fimmu.2020.01907
Received: 07 May 2020; Accepted: 15 July 2020;
Published: 27 August 2020.
Edited by:
Christiane Hilger, Luxembourg Institute of Health, LuxembourgReviewed by:
Jarmila Celakovska, Charles University, CzechiaCopyright © 2020 Yang, Fu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yufeng Zhou, eWZ6aG91MUBmdWRhbi5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.