 Changhoon Oh
Changhoon Oh Ambika Verma
Ambika Verma Youssef Aachoui
Youssef Aachoui
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 21 August 2020
Sec. Microbial Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01895
This article is part of the Research TopicOutsmarting the Host: How Bacterial Pathogens Modulate Immune Responses in The LungView all 19 articles
The airway epithelium and underlying innate immune cells comprise the first line of host defense in the lung. They recognize pathogen-associated molecular patterns (PAMPs) using membrane-bound receptors, as well as cytosolic receptors such as inflammasomes. Inflammasomes activate inflammatory caspases, which in turn process and release the inflammatory cytokines IL-1β and IL-18. Additionally, inflammasomes trigger a form of lytic cell death termed pyroptosis. One of the most important inflammasomes at the host-pathogen interface is the non-canonical caspase-11 inflammasome that responds to LPS in the cytosol. Caspase-11 is important in defense against Gram-negative pathogens, and can drive inflammatory diseases such as LPS-induced sepsis. However, pathogens can employ evasive strategies to minimize or evade host caspase-11 detection. In this review, we present a comprehensive overview of the function of the non-canonical caspase-11 inflammasome in sensing of cytosolic LPS, and its mechanism of action with particular emphasis in the role of caspase-11 in the lung. We also explore some of the strategies pathogens use to evade caspase-11.
Bacterial infections of the respiratory tract remain a leading cause of global morbidity and mortality (1). As the first line of defense against infection, the airway epithelium and innate immune cells play a critical role in limiting infection. As such, they are especially sensitive to pathogen associated molecular patterns (PAMPs), expressing an array of membrane-bound and cytosolic receptors that recognize specific structural motifs shared by classes of microbes (2). For example, Toll-like receptor 5 (TLR5) and NOD-like receptor 4 (NLRC4) detect and respond to bacterial flagellin. TLR5 detects extracellular flagellin (3), while NLRC4 detects its cytosolic presence (4, 5).
Many pathogens infect cells and exploit intracellular niches to facilitate their replication and spread (6). In turn, host cells have evolved mechanisms to detect and counteract these efforts. For example, intracellular sensors like inflammasomes are expressed in innate immune cells, lung epithelium, and endothelium to enable detection of pathogens and subsequent protective responses (6–8). Inflammasomes lead to the activation of inflammatory caspases, which in turn process and release the inflammatory cytokines IL-1β and IL-18. Additionally, inflammasomes trigger a form of lytic cell death termed pyroptosis that plays important role in clearance of offending pathogens (6).
Virulent intracellular bacteria usually evade intracellular sensors or suppress inflammasome activity by suppressing or modifying the structural features of their PAMPs (9). These strategies diminish the ability of inflammasomes to respond to these pathogens. For instance, Salmonella enterica serovar typhimurium utilizes two pathogenicity islands, SPI-1 and SPI-2, which encode distinct type three secretion systems (T3SSs). The T3SS apparatus encoded by SPI-1 functions primarily during epithelial cell invasion, is co-expressed with flagellin, and is readily detected by the inflammasome NLRC4. In contrast, the SPI-2 T3SS is expressed in the vacuolar compartment of both epithelial cells and macrophages, and is not detected by inflammasomes. S. typhimurium suppresses transcription of flagellin during SPI-2 inducing conditions, thereby avoiding detection by NLRC4 (10, 11). Here, we present a comprehensive overview of the function of the caspase-11 inflammasome in sensing of cytosolic LPS, and its mechanism of action with particular emphasis in the lung. We also explore some of the strategies pathogens use to evade caspase-11.
Caspases are a family of aspartate-specific, cysteine proteases that are activated by homodimerization or cleavage to mediate several cellular events including cell death (12). Cell death is an essential process that maintains tissue homeostasis, and recently, it has emerged as an important defense mechanism against infection (13). Caspases are divided into inflammatory (in mouse these include caspase-1,-11, and -12) or apoptotic caspases (caspase-2,-3,-6,-7,-8, and -9). Apoptotic caspases are further subdivided into initiators (caspase-2,-8, and -9) or executioners (caspase-3,-6, and -7) (12). A discussion of the activation and role of apoptotic caspases during infection is important; however, it is beyond the scope of this review, and we refer the reader to previously published reviews (12).
Inflammatory caspases are able to distinguish virulent from avirulent bacteria, and to alert the immune system to a pathogenic infection. Like apoptotic initiator caspases, inflammatory caspases contain a death fold prodomain termed the caspase activation and recruitment domain (CARD) (12). The CARD domain is essential to the recruitment and activation of inflammatory caspases-1 and-11 within the inflammasomes. Inflammasomes are large, multiprotein complexes typically comprising a NOD-like receptor (NLR) or AIM2-like receptor (ALR) such as NLRP3, NLRC4, or AIM2, sometimes an adaptor protein called ASC, and an executioner caspase, which is typically caspase-1 (6). Several inflammasomes that activate caspase-1 in response to contamination of the cytosol or perturbation of cellular physiology have been characterized, and each one is named after its NLR or ALR protein scaffold. The NLRP3 inflammasome is activated in response to the widest array of stimuli, including particulates, crystals, ATP, K+ efflux, lysosomal disruption, and oxidized mitochondrial DNA. However, the precise signal that NLRP3 detects remains controversial (14). In contrast to the diverse stimuli that activate NLRP3, other inflammasomes respond to a limited set of stimuli. For instance, The AIM2 detects cytosolic dsDNA (15–17), which may be encountered in the cytosol during pathogenic infection. The NLRC4 inflammasome responds to flagellin and the activity of type III secretion system (T3SS) and type IV secretion system (T4SS) (10, 18, 19). Unlike other inflammasomes, activation of NLRC4 involves members of the NAIP subfamily of NLRs, forming mixed NLRC4-NAIP oligomeric inflammasome platforms. The murine NAIP locus is highly polymorphic. C57BL/6 mice encode four NAIPs; NAIP1 binds the T3SS needle, NAIP2 binds the T3SS rod, and NAIP5 and NAIP6 bind flagellins. By contrast, humans have a single NAIP that detects the T3SS needle (18–21). Upon activation, NLRs forming inflammasomes recruit the ASC adaptor, composed of a CARD and a PYD, via homotypic interactions. Additional ASC molecules are incorporated via CARD-CARD and PYD-PYD interactions, until all ASC are collected into a single focus called a “speck.” Attraction of caspase-1 into the ASC speck via CARD-CARD interaction results in caspase-1 dimerization and proximity-induced autoproteolytic processing into p10 and p20 subunits (6). Active caspase-1 processes pro-IL-1β and pro-IL-18 to their mature forms; it additionally triggers a lytic form of programmed cell death called pyroptosis by cleaving and activating the pore-forming protein gasdermin D (GSDMD; discussed in more detail below) (6, 22). Activation of inflammasomes can also promote the release of IL-1α. IL-1α is considered an endogenous danger signal that is widely and constitutively expressed, active in its uncleaved form, and passively released after pyrpotosis (23).
Caspase-1 was long thought to be the only inflammatory caspase activated within an inflammasome and the sole mediator of pyroptosis because caspase-1-deficient bone marrow macrophages (BMMs) were resistant to inflammasome activation-induced cell death. However, the Dixit lab showed that the process of backcrossing the caspase-1 knockout allele into the C57BL/6 background carried a passenger mutation in caspase-11 from the 129 background, effectively making Casp1-Casp11DKO animals (24). Examination of the single Casp11−/− mice revealed that, caspase-11 drives pyroptosis upon in vitro activation like the closely related caspase-1, but it does not directly mediate IL-1β and IL-18 secretion. Instead, caspase-11 can activate an indirect NLRP3-ASC-caspase-1 pathway leading to IL-1β/IL-18 processing and release (24). The caspase-11 pathway was thus termed the “caspase-11 non-canonical inflammasome” to contrast it with the caspase-1-activating canonical inflammasomes (NLRP3, NLRC4, AIM2, etc.). These revelations ignited interest into the study of caspase-11.
Caspase-11 was identified almost 23 years ago in a mouse cDNA library screen for homologs of caspase-1 (25). Back then, caspase-11 was found to be a key mediator in LPS-induced septic shock model since Casp11−/− mice are more resistant to lethal doses of LPS compared to wild-type mice (25, 26), though its mechanism of activation remained elusive. 15 years later, caspase-11 regained the center stage after Kayagaki et al. demonstrated that exposure to cholera toxin B (CTB) and certain Gram-negative bacteria, such as Escherichia coli, Citrobacter rodentium, and Vibrio cholera, activates caspase-11 to trigger pyroptosis, albeit after long exposure times (~16–24 h) (24). We subsequently showed that caspase-11 specifically differentiates Gram-negative bacteria that invade the cytosol from bacteria that remain extracellular or confined to the vacuole, with the former activating caspase-11 rapidly (27) (Figure 1). However, like the experiments before, vacuolar and extracellular Gram-negative bacteria could still activate caspase-11 after sufficient time (28–31). These observations precipitated interest in the discovery of the identity of the common non-canonical activating agonist(s) and sensor(s). Two independent studies by the Miao and Dixit groups discovered that activation of caspase-11 is induced by LPS that has gained access to the host cytosol (32, 33). Of note, it was later determined that the activation of caspase-11 by CTB in LPS-primed macrophages was triggered by the aberrant delivery of LPS by CTB to the cytosol, rather than the direct action of CTB itself (32). The fact that LPS is the cytosolic ligand that activates caspase-11 also shed light on the role of caspase-11 in septic shock, and why septic shock could occur in the absence of TLR4, the cell-surface LPS receptor (32, 33). Therefore, LPS is detected by two receptors: extracellularly or within the recycling endosomes by TLR4 and in the cytosol via caspase-11. It is also important to note that LPS also co-localizes with intracellular TLR4 in the golgi apparatus or the endoplasmic reticulum (34, 35).
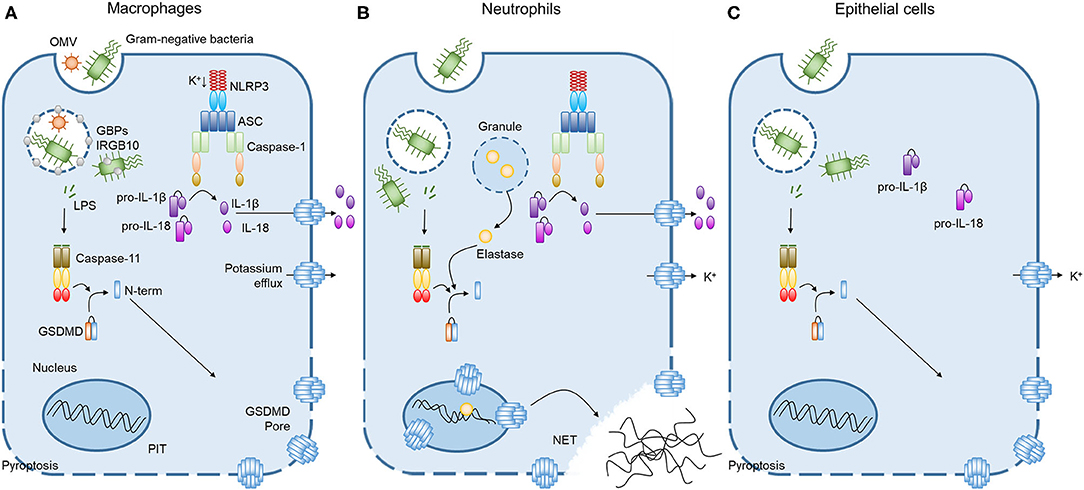
Figure 1. Caspase-11 non-canonical inflammasome activation by intracellular LPS in different cells. (A) Cooperation of GBPchr3 with IRGB10 expose LPS from intracellular bacteria or OMVs in macrophages. Cytoplasmic LPS directly bind to the CARD motif of caspase-11 with their lipid A moiety, leading to oligomerization of caspase-11 to activate the non-canonical inflammasome. Active caspase-11 cleaves GSDMD to release its N-terminal domain, which is subsequently inserted into plasma membrane to form membrane pores. Potassium efflux through GSDMD pores additionally triggers NLRP3 inflammasome, inducing the processing and release of IL-1β and IL-18. Pyroptosis pores in the plasma membrane are large enough to permit soluble proteins to defuse to the extracellular milieu, but small enough to trap organelles and intracellular bacteria. This process is termed the pore-induced intracellular trap (PIT). (B) In neutrophils, both caspase-11 and elastase can cleave GSDMD to trigger neutrophil extracellular traps (NETs). Cleaved GSDMD targets the nucleus in addition to the plasma membrane and drives nuclear permeabilization, chromatin relaxation and plasma membrane rupture in elastase or caspase-11 dependent manner. These processes induce neutrophil to extrude NETs which restrict bacterial replication, dissemination, and enhance bacteria killing. (C) In contrast to the broadly expressed caspase-11 and GSDMD, caspase-1 inflammasome components are not detectable in lung epithelial cells. Therefore, the caspase-11 non-canonical inflammasome activation in lung epithelial cells only triggers pyroptosis without the processing and release of IL-1β and IL-18.
Shortly afterward, the Shao lab found that caspase-11 is the direct sensor of LPS. This was in clear contrast to the caspase-1 canonical inflammasome, where sensor and executioner functions were separated into two proteins, often bridged by the adapter ASC (36). Shi et al. demonstrated that the caspase-11 inflammasome is initiated by the CARD motif of caspase-11 directly binding to the lipid A moiety of LPS. The binding of LPS to the CARD of caspase-11 not only triggers its oligomerization but also its catalytic activation (36, 37).
In humans, caspase-11 is duplicated as caspase-4 and -5, both of which have been shown to detect directly cytosolic LPS (36). The reason for this duplication remains a mystery. One distinction is that caspase-4 expression is high in unstimulated macrophages (38–40), while murine caspase-11 and human caspase-5 are expressed at low and extremely low levels, respectively, without stimulation (39–41). Upon exposure to LPS or other toll agonists, however, macrophages rapidly induce transcription of caspase-11 and caspase- 5. Indeed, caspase-11 is non-responsive in the absence of priming through interferon-β (IFN-β) or interferon-γ (IFN-γ) (24, 27–30). Thus, as one hypothesis, caspase-11 duplication may enable a new priming-independent response through caspase-4.
Inflammatory caspases 11 and its human homologs caspase-4 and 5 cleave the pyroptosis effector protein, GSDMD, separating its N-terminal pore-forming domain from the C-terminal repressor domain (42–44). The N-terminal domain is inserted into the plasma membrane, and then oligomerizes to form large pores in the membrane that drive swelling and membrane rupture (22, 45–47). Additionally, pore formation leads to potassium efflux, which triggers the NLRP3/ASC/caspase-1 inflammasome and subsequent processing of pro-IL-1β and pro-IL-18 to their mature forms in vitro (48–51) (Figure 1); however, this pathway has not yet been confirmed in vivo. Additionally, GSDMD pores allow inflammasomes effectors such as IL-1α, IL-1β, IL-18, and inflammatory mediators such as eicosanoids to be released into the extracellular milieu (22, 52, 53). Although membrane pores release soluble cytosolic contents, they are small enough to retain organelles and entrap bacteria to prevent their dissemination, resulting in a structure called the pore-induced intracellular trap (PIT) (54). The PIT coordinates innate immune responses via complement and scavenger receptors to drive recruitment of and efferocytosis by neutrophils that ultimately kill the invading pathogen (54). GSDMD-dependent pores will ultimately lead to membrane rupture, but before that, cells can potentially repair membrane pores to negatively regulate pyroptosis and cytokines release. Rühl et al. showed that in macrophages, this process is coordinated by the endosomal sorting complexes required for transport (ESCRT). ESCRTs are recruited to damaged membrane in a Ca2+ influx-dependent manner and they remove pores from the plasma membrane (55).
In a conceptual analog to macrophage PITs, the Zychlinsky lab recently found that neutrophil GSDMD can also be cleaved by the serine protease elastase to drive formation of neutrophil extracellular traps (NETs) (56). In this situation, GSDMD cleavage releases the granular proteins including elastase that may enter the nucleus, triggering NETosis, while GSDMD forms pores in the plasma membrane to release these granules and form the NET (56) (Figure 1). Interestingly, the Schroder group showed that neutrophil exposure to cytosolic LPS drives caspase-11 activation and GSDMD cleavage, leading to the extruding of antimicrobial NETs independent of elastase (57). In this case, the combined actions of GSDMD and caspase-11 drive nuclear permeabilization, chromatin relaxation, and plasma membrane rupture prior to NETs release (57). NETs can restrict bacterial replication in the cells, entrap the bacteria to prevent their dissemination and enhance direct bacteria killing through the action of neutrophil granular contents. However, an excess of NETs has been reported for several pulmonary diseases, including asthma, cystic fibrosis (CF), influenza, bacterial pneumonia, and tuberculosis (58).
Caspase-11 and GSDMD are widely expressed in hematopoietic and non-hematopoietic cells, including macrophages, neutrophils, epithelial and endothelial cells. Lung epithelial and endothelial cells express caspase-11 and were found to be sensitive to stimulation with cytosolic LPS (7, 8). Interestingly, lung epithelial cells do not express the caspase-1 inflammasomes components NLRP3 or NLRC4 (7). Therefore, the activation of these cells through the caspase-11 non-canonical inflammasome triggers only pyroptosis without the processing of pro-IL-1β or pro-IL-18 (7) (Figure 1). Activation of caspase-11 can also induces the shedding of intestinal epithelial cells after detection of intracellular S. typhimurium during gut infection (59), and may perform a similar function in lung epithelial cells.
Access of LPS to the cytosol is a key factor during the activation of caspase-11. Cytosol -invasive pathogens such as Burkholderia thailandensis and its more virulent relative Burkholderia pseudomallei use T3SS to escape from the vacuole to the cytosol (60). This exit provides an explanation of how LPS can gain access to the cytosol. However, several Gram-negative bacteria also activate caspase-11 but are not cytosolic. Several studies showed that LPS cytosolic access can also be mediated by the activity of host factors, such as guanylate binding proteins (GBPs) (61, 62). GBPs are cytosolic GTPases that localize to the vacuole or endosome and contribute to cell-autonomous defense against cytosolic and non-cytosolic pathogens via several mechanisms including limiting intracellular bacterial survival and motility (63–66). GBPs are well conserved among vertebrates, and are found in both mice and humans (67). Genes encoding murine GBPs are clustered on chromosome 3 and are known collectively as GBPchr3 (GBP1, GBP2, GBP3, GBP5, and GBP7) or clustered on chromosome 5 (GBP4, GBP6, GBP8, GBP9, GBP10, and GBP11). In contrast, the family is reduced in humans to encode 7 GBPs genes (GBP1-GBP7) that are clustered on chromosome 1 (68–71). Intriguingly, like caspase-11, GBPs are interferon-stimulated genes, and require interferon signaling to be upregulated (62, 68). GBP-dependent effects in the activation of caspase-11 have been established in several intracellular pathogens including, S. typhimurium, Legionella pneumophila, C. rodentium, T3SS-negative Pseudomonas aeruginosa, Vibrio cholerae, Chlamydia trachomatis, and Chlamydia muridarum (61, 62, 72, 73). For instance, macrophages from mice that lack GBPs on Chromosome 3 exhibited less caspase-11-mediated pyroptosis in response to S. typhimurium and L. pneumophila relative to wild-type macrophages (61, 62, 74).
Whereas, the role of GBPs in general cytosolic sensing is well-established, the precise mechanism of GBPs function in LPS sensing is still the subject of ongoing research. Nonetheless, most studies suggest that GBPs are either recruited to the pathogen-containing vacuoles (PCVs), which might promote lysis of PCVs and consequent exposure of vacuolar LPS to caspase-11, or alternatively, GBPs are recruited to spontaneously ruptured bacteria-containing vacuoles or directly to cytosolic bacteria, where they act to extract or expose lipid A buried in bacterial membranes for recognition by caspase-11 (61, 62, 75, 76). In support of this latter hypothesis, human guanylate binding protein-1 (hGBP1) associates with the surface of intracellular Gram-negative bacterial pathogens after vacuolar escape (77, 78). Then, GBP1 polymerizes to coat the bacteria and initiate the recruitment of GBP2-4 to assemble a platform for caspase-4 activation (77–79). hGBP1 binds directly to LPS (77–79) and exerts a detergent-like effect of clustering LPS, which disrupts bacterial surfaces (79), thereby exposing the membrane-embedded lipid A for caspase-4 detection. In mice, GBPs can serve as a docking platform to recruit another interferon-inducible protein, IRGB10 that can co-localize to the intracellular bacteria to disrupt the bacterial cell membrane and to liberate bacterial ligands, including LPS and DNA, for innate immune recognition (80).
Virulence-associated secretion systems in Gram-negative pathogens can also drive LPS exposure in the cytosol. Gram-negative bacteria produce and release outer membrane vesicles (OMVs) during infection that are internalized by macrophages and function as vehicles that deliver LPS (and several other surface antigens) into the host cytosol (81). LPS on OMVs can trigger caspase-11 activation during E. coli infection. In addition, the OMVs from Gram-negative bacteria S. typhimurium, L. pneumophila, P. aeruginosa, and Shigella flexneri activate caspase-11 in a GBPchr3-dependent manner. In this case, GBPs may disrupt OMVs causing the release of LPS to cytosol where it engages caspase-11 (82).
Since caspase-11 is expressed broadly in the lung tissue, caspase-11-dependent LPS-sensing is poised to play an important role in defense against Gram-negative bacteria. As noted above, pyroptosis accomplishes the dual task of restricting intracellular bacterial growth by lysing infected cells, and releasing the cytokines IL-1β and IL-18 (6), which promote host defense via pleiotropic mechanisms. IL-1α can induce neutrophil recruitment to facilitate bacterial clearance and IL-1β is best known to promote local inflammation and to recruit neutrophils to the site of infection. In turn, IL-18 is best known to induce IFN-γ secretion by natural killer cells and cytotoxic T lymphocytes (83). During systemic infection, caspase-11 is essential for host defense against cytosol-invasive bacteria, including B. thailandensis and B. pseudomallei (27). Mice deficient in caspase-11 are extremely susceptible to even mild infection with B. thailandensis, while wild-type mice are completely resistant to higher doses of infection (27, 39). We showed that the NLRC4 inflammasome detects B. thailandensis, and activates caspase-1 to trigger pyroptosis and IL-18 secretion. Curiously, we found that in vitro, B. thailandensis efficiently triggers both caspase-1 and−11-dependent pyroptosis in macrophages. In vivo, however, caspase-1-dependent pyroptosis plays only a minor role and cannot substitute for caspase-11-dependent pyroptosis. On the other hand, caspase-1-driven IL-18 was needed only to clear early infection. IL-18 promotes a robust and rapid production of IFN-γ, which primes caspase-11. This coordinated response clears even high dose infections in 1 day (39). In the absence of IL-18 signaling, B. thailandensis continues to replicate in vivo for 3 days. This prolonged bacterial burden triggers weak IFN-γ production via another undefined pathway, which is nonetheless sufficient to prime caspase-11 during medium or low dose infection and rescue the mice from lethality. In high dose infections, this pathway comes too little and too late and Il18−/− mice succumb to infection. Intriguingly, while endogenous type I interferons were insufficient to prime caspase-11 in the absence of IFN-γ, injection of poly (I:C), a known inducer of IFN-β, successfully bypassed the IFN-γ requirement and primed caspase-11 in vivo (39). It is important to note that there are no examples in the literature of any pathogen–be it bacterial, fungal, viral, or parasite–in which the magnitude of protection conferred by caspase-11 inflammasomes approaches what is seen with B. thailandensis (84).
Caspase-11 also provides host protection in the lung in response to infection by Gram-negative bacterial pathogens, including B. thailandensis (7, 85), Acinetobacter baumannii (86), Klebsiella pneumoniae (87), L. pneumophila (88), and L. gratiana (88). As mentioned above, we found that B. thailandensis is detected by both caspase-1 and-11 in macrophages. Additionally, Wang et al. found that caspase-1 is the most important in response to B. thailandensis in vitro, and he showed that the absence of caspase-1 completely ablates release of IL-1β and IL-18 as well as pyroptosis of Burkholderia-infected macrophages. In contrast, pyroptosis, IL-1β and IL-18 of macrophages were not affected by deficiency of caspase-11. Interestingly, during pulmonary infection, mice lacking either caspase-1, 11 or GSDMD were significantly more susceptible than wild type mice to intranasal infection with B. thailandensis (7, 85). To account for this phenotype, Wang et al. showed that unlike macrophages, lung epithelial cells do not express caspase-1 canonical inflammasome components and are thus incompetent for caspase-1-mediated pyroptosis (7). Therefore, macrophages are protected by both caspase-1 and−11. However, lung epithelial cells solely depended on caspase-11 to restrict intracellular B. thailandensis replication in these cells (7).
A. baumannii, a pathogenic bacterium that can cause severe pulmonary infection, can activate caspase-1 through the NLRP3 inflammasome, leading to increased IL-1β release by macrophages. However, IL-1β release is partially abrogated in caspase-11-deficient macrophages, indicating that A. baumannii can also activate the caspase-11 non-canonical inflammasomes (86). Furthermore, caspase-11 deficiency completely ablates IL-1α secretion and reduces the cytotoxicity of A. baumannii in macrophages (86). Therefore, these data suggest that caspase-11 deficiency may impair the innate immune response to A. baumannii infection. Indeed, caspase-11-deficient mice register higher bacterial burdens in the BALF and lung tissue during A. baumannii infection. Additionally, caspase-11 deficiency causes mice to exhibit exacerbated pulmonary pathology due to extensive neutrophil Infiltration (86). Similarly, pulmonary infection of Casp11−/− with K. pneumoniae, showed that lack of caspase-11-dependent release of IL-1α impeded neutrophil recruitment in the early stage of K. pneumoniae infection and was accompanied by impaired bacterial clearance as well as exaggerated pulmonary pathological changes (87). Therefore, caspase-11 functions in the lung by promoting pyroptosis to prevent the bacteria from establishing an intracellular niche in macrophages and the lung epithelial. Additionally, caspase-11 pyroptosis can drive IL-1α and IL-1β release to recruit neutrophils that eventually will efferocytose and eliminate the offending bacteria.
Caspase-11-dependent pyroptosis and cytokines are important for combating respiratory infections. However, their excessive activation may contribute to the development of severe diseases in the lung. Acute lung injury (ALI) is a leading cause of death in bacterial sepsis (8). ALI is characterized by massive destruction of the lung endothelial barrier which results in lung edema, influx of pro-inflammatory leukocytes and sever hypoxemia (8). During endotoxemia, high concentrations of LPS may persist and aberrantly localize to the cytoplasm, triggering the hyper-activation of caspase-11 and resulting in massive pyroptosis. The hyper-activation of caspase-11 drives endotoxin shock independently of TLR4 (32, 33). During an ALI model, Cheng et al. show that the activation of caspase-11 with LPS causes severe endothelial pyroptosis that results in ALI (8). Exposure to LPS has also been shown to play a major role in asthma development (89) and recently, caspase-11 response was implicated as a critical contributor to asthma (90). Using the ovalbumin model of allergic airway inflammation, Zaslona et al. found that expression of caspase-11 is elevated in the lung of wild type mice with allergic airway inflammation. Interestingly, they found that caspase-11-deficient mice are strongly resistant to this pathology (90). However, how caspase-11 is activated in this model isn't clear. Zaslona et al. suggest that the lung is a non-sterile environment, and LPS released from naturally occurring lung bacteria might activate caspase-11, leading to airway inflammation and asthma in susceptible individuals.
In patients with cystic fibrosis (CF), P. aeruginosa can cause chronic airway infection that is characterized by an exaggerated pro-inflammatory cytokine response and sustained, neutrophil-dominant inflammation (91). Previous studies have demonstrated a role of NLRP3 and NLRC4 inflammasome activation in response to P. aeruginosa infection in CF patients (73, 92). However, P. aeruginosa strains isolated from chronically infected CF patients have either defective T3SS or are non-motile, enabling them to evade NLRC4-caspase-1 inflammasome detection (93–95). Balakrishan et al. showed that caspase-11 can detect these T3SS-negative P. aeruginosa isolates (73). However, whether this detection takes place in vivo to drive inflammation and disease in CF has yet to be determined.
Caspase-11 protects against lethal infection mediated by cytosol-invasive pathogens (27), it is particularly important for protection against ubiquitous environmental bacteria that have not evolved to escape cytosolic detection, such as the aforementioned B. thailandensis. However, it is not surprising that a number of organisms have evolved specific strategies to avoid the activation of this innate immune signaling pathway. These strategies include limiting LPS access to the cytosol, modifying the structural features of LPS to avoid binding to caspase-11, or suppressing caspase-11 and/or GBPs activity.
As noted above, preventing access of LPS to the cytosol using vacuolar niches or by remaining extracellular are prominent ways to avoid caspase-11 activation. For instance, S. typhimurium and L. pneumophila use T3SS and T4SS, respectively, to translocate effector proteins that work in concert to establish and maintain an intracellular vacuolar growth niche. Loss of the S. typhimurium SifA or L. pneumophila SdhA effectors causes rupture of the vacuole and release of bacteria into the cytosol (96–98). As we might expect, these two bacteria are poorly detected by caspase-11, while their isogenic mutants for these proteins are readily detected by caspase-11, with half the attenuation of the S. typhimurium ΔsifA mutant in mice explained by caspase-11 activation alone (27).
Another strategy is to modify the chemical structure of LPS to avoid detection. Caspase-11 uses its CARD domain to bind penta- or hexa-acylated lipid A moieties on LPS (32, 33, 36). As such, several tetra-acyl lipid A structures from pathogens like Francisella novicida, Yersinia pestis, and S. flexenri have been shown to evade caspase-11/4 detection (33, 99). Indeed, during pulmonary infection, bacteria often appear to exploit these structural requirements in order to evade caspase-11. For example, F. novicida, a Gram-negative cytosolic bacterium, is not detected by caspase-11 (33). F. novicida initially synthesizes a penta-acylated lipid A structure with two phosphates, but then removes the 4' phosphate and 3′ acyl chain resulting in under-acylated LPS that does not activate caspase-11 (100, 101). A similar strategy is used by Y. pestis, which removes two acyl chains from its lipid A upon transition from growth at 25°C to 37°C (102). Caspase-11 detects hexa-acylated lipid A from Y. pestis grown at 25°C, but not tetra-acylated lipid A from bacteria grown at 37°C (33). Surprisingly, Lagrange et al. found that one of the human homologs of caspase-11, caspase-4, is able to respond to tetra-acylated LPS from F. novicida (103), suggesting that the caspase-11 duplication in human may have evolved to accommodate a broader LPS repertoire than that of mice.
Other strategies to evade caspase-11 detection may involve directly inhibiting the caspase-11 signaling pathway, as in the case of S. flexneri, which uses its OspC3 effector to specifically inhibit LPS detection by caspase-4 (104). Additionally, Shigella delivers an array of effector factors such as the ubiquitin ligase effector IpaH9.8 to target GBPs for proteasomal degradation (64, 65). Finally, host factors also can suppress the activity of caspase-11. For instance, L-adrenaline inhibits caspase-11 inflammasome activation through the ADRA2B receptor and the intracellular cAMP metabolism pathway in mouse macrophages or human monocytes (105). It is plausible that pathogens may manipulate this host metabolic pathway to their advantage.
Lung tissue is a unique epithelial space constantly exposed to a variety of assaults from infectious and non-infectious stimuli. Innate immune detection of, and responses to, intracellular pathogens that invade host cells is paramount in defending against infections. The caspase-11 non-canonical inflammsome plays a key role in this response. It is widely expressed in both hematopoietic and non-hematopoietic cells of the lung, functioning as a general sensor of Gram-negative bacteria whose activation results in protective innate immune effectors including pyroptosis, NETosis, and inflammatory cytokines. Therefore, it is not surprising that bacterial pathogens have evolved several strategies to outsmart caspase-11-mediated sensing of LPS and subsequent activation. Indeed, the importance of this pathway is demonstrated by those species that have not evolved to evade caspase-11, such as B. thailandensis, which strongly triggers caspase-11-dependent effector mechanisms that quickly lead to sterilization of the infection. This remarkable protection highlights an important role of inflammasomes in defense against environmental pathogens. Similarly, pathogens that poorly activate the caspase-11 inflammasome can multiply to uncontrolled levels in the lung, resulting in higher levels of LPS that will eventually access the cell cytosol and hyper-activate caspase-11, leading to LPS induced lung injury and sepsis. Future studies uncovering the structural features of lipid A that activate caspase-11 and the cell-specific roles of the caspase-11 inflammasome in lung tissue could identify new therapeutic avenues to treat infection, sepsis, and immune-mediated pathologies within the lung.
CO, AV, and YA wrote the article. CO and YA contributed to designing the figure. All authors contributed to the article and approved the submitted version.
This work was supported by NIH grant 1K22AI132489-01 (YA), and NIH/NIGMS award P20-GM103625 (YA).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank Alan Tubbs and Stephen Kovacs for their suggestions and editing the manuscript. We also acknowledge the UAMS Center for Microbial Pathogenesis and Host Inflammatory Responses (CMPHIR) for continued support.
1. GBD 2016 Lower Respiratory Infections Collaborators. Estimates of the global, regional, national morbidity. mortality, and aetiologies of lower respiratory infections in 195 countries, 1990-2016: a systematic analysis for the Global burden of disease study 2016. Lancet Infect Dis. (2018) 18:1191–210. doi: 10.1016/S1473-3099(18)30310-4
2. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
3. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. (2001) 410:1099–103. doi: 10.1038/35074106
4. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat Immunol. (2006) 7:569–75. doi: 10.1038/ni1344
5. Franchi L, Amer A, Body-Malapel M, Kanneganti T-D, Özören N, Jagirdar R, Inohara N, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat Immunol. (2006) 7:576–82. doi: 10.1038/ni1346
6. Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol. (2013) 16:319–26. doi: 10.1016/j.mib.2013.04.004
7. Wang J, Sahoo M, Lantier L, Warawa J, Cordero H, Deobald K, et al. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog. (2018) 14:e1007105. doi: 10.1371/journal.ppat.1007105
8. Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. (2017) 127:4124–4135. doi: 10.1172/JCI94495
9. Ulland TK, Ferguson PJ, Sutterwala FS. Evasion of inflammasome activation by microbial pathogens. J Clin Invest. (2015) 125:469–77. doi: 10.1172/JCI75254
10. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA. (2010) 107:3076–80. doi: 10.1073/pnas.0913087107
11. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. (2010) 11:1136–42. doi: 10.1038/ni.1960
12. van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. (2019) 50:1352–64. doi: 10.1016/j.immuni.2019.05.020
13. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. (2017) 17:151–64. doi: 10.1038/nri.2016.147
14. Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
15. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASA. Nature. (2009) 458:514–8. doi: 10.1038/nature07725
16. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. (2009) 458:509–13. doi: 10.1038/nature07710
17. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. (2009) 323:1057–60. doi: 10.1126/science.1169841
18. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. (2011) 477:592–5. doi: 10.1038/nature10394
19. Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. (2011) 477:596–600. doi: 10.1038/nature10510
20. Rayamajhi M, Zak DE, Chavarria-Smith J, Vance RE, Miao EA. Cutting edge: mouse NAIP1 detects the type III secretion system needle protein. J Immunol. (2013) 191:3986–9. doi: 10.4049/jimmunol.1301549
21. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA. (2013) 110:14408–13. doi: 10.1073/pnas.1306376110
22. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. (2017) 27:673–84. doi: 10.1016/j.tcb.2017.05.005
23. Di Paolo NC, Shayakhmetov DM. Interleukin. 1α and the inflammatory process. Nat Immunol. (2016) 17:906–13. doi: 10.1038/ni.3503
24. Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. (2011) 479:117–21. doi: 10.1038/nature10558
25. Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, Shi L, et al. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem. (1996) 271:20580–7. doi: 10.1074/jbc.271.34.20580
26. Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. (1998) 92:501–9. doi: 10.1016/S0092-8674(00)80943-5
27. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. (2013) 339:975–8. doi: 10.1126/science.1230751
28. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. (2012) 490:288–91. doi: 10.1038/nature11419
29. Gurung P, Malireddi RK, Anand PK, Demon D, Vande Walle L, Liu Z, et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem. (2012) 287:34474–83. doi: 10.1074/jbc.M112.401406
30. Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. Fitzgerald. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. (2012) 150:606–19. doi: 10.1016/j.cell.2012.07.007
31. Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci USA. (2013) 110:1851–6. doi: 10.1073/pnas.1211521110
32. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. (2013) 341:1246–9. doi: 10.1126/science.1240248
33. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. (2013) 341:1250–3. doi: 10.1126/science.1240988
34. Hornef MW, Frisan T, vandewalle A, Normark S, Richter-Dahlfors A. Toll-like receptor 4 resides in the golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J Exp Med. (2002) 195:559–70. doi: 10.1084/jem.20011788
35. Hornef MW, Normark BH, Vandewalle A, Normark S. Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J Exp Med. (2003) 198:1225–35. doi: 10.1084/jem.20022194
36. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. (2014) 514:187–92. doi: 10.1038/nature13683
37. Lee BL, Stowe IB, Gupta A, Kornfeld OS, Roose-Girma M, Anderson K, et al. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J Exp Med. (2018) 215:2279–88. doi: 10.1084/jem.20180589
38. Kajiwara Y, Schiff T, Voloudakis G, Gama Sosa MA, Elder G, Bozdagi O, et al. A critical role for human caspase-4 in endotoxin sensitivity. J Immunol. (2014) 193:335–43. doi: 10.4049/jimmunol.1303424
39. Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting J-PY, Coers J, et al. Canonical inflammasomes drive IFN-γ to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe. (2015) 18:320–32 doi: 10.1016/j.chom.2015.07.016
40. Lin XY, Choi MS, Porter AG. Expression analysis of the human caspase-1 subfamily reveals specific regulation of the CASP5 gene by lipopolysaccharide and interferon-gamma. J Biol Chem. (2000) 275:39920–6. doi: 10.1074/jbc.M007255200
41. Schauvliege R, vanrobaeys J, Schotte P, Beyaert R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem. (2002) 277:41624–30. doi: 10.1074/jbc.M207852200
42. He W-t, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Research. (2015) 25:1285–98. doi: 10.1038/cr.2015.139
43. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
44. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
45. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. (2016) 35:1766–78. doi: 10.15252/embj.201694696
46. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
47. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. (2016) 535:111–6. doi: 10.1038/nature18590
48. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
49. Kanneganti T-D, Lamkanfi M. K+ drops tilt the NLRP3 inflammasome. Immunity. (2013) 38:1085–8. doi: 10.1016/j.immuni.2013.06.001
50. Ruhl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol. (2015) 45:2927–36. doi: 10.1002/eji.201545772
51. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D'Silva DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol. (2015) 45:2918–26. doi: 10.1002/eji.201545655
52. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin d regulates interleukin-1 secretion from living macrophages. Immunity. (2018) 48:35–44.e6. doi: 10.1016/j.immuni.2017.11.013
53. Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur J Immunol. (2018) 48:584–92. doi: 10.1002/eji.201747404
54. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. (2016) 213:2113–28. doi: 10.1084/jem.20151613
55. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. (2018) 362:956–60. doi: 10.1126/science.aar7607
56. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. (2018) 3:eaar6689. doi: 10.1126/sciimmunol.aar6689
57. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D–dependent neutrophil extracellular traps. Sci Immunol. (2018) 3:eaar6676. doi: 10.1126/sciimmunol.aar6676
58. Porto BN, Stein RT. Neutrophil extracellular traps in pulmonary diseases: too much of a good thing? Front Immunol. (2016) 7:311. doi: 10.3389/fimmu.2016.00311
59. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe. (2014) 16:249–56. doi: 10.1016/j.chom.2014.07.002
60. Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Microbiol. (2006) 4:272–82. doi: 10.1038/nrmicro1385
61. Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, et al. Guanylate binding proteins promote caspase-11–dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci. (2014) 111:6046–51. doi: 10.1073/pnas.1321700111
62. Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature. (2014) 509:366–70. doi: 10.1038/nature13157
63. Kravets E, Degrandi D, Ma Q, Peulen TO, Klumpers V, Felekyan S, et al. Guanylate binding proteins directly attack Toxoplasma gondii via supramolecular complexes. Elife. (2016) 5:e11479. doi: 10.7554/eLife.11479
64. Li P, Jiang W, Yu Q, Liu W, Zhou P, Li J, et al. Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence. Nature. (2017) 551:378–83. doi: 10.1038/nature24467
65. Wandel MP, Pathe C, Werner EI, Ellison CJ, Boyle KB, von der Malsburg A, et al. GBPs inhibit motility of shigella flexneri but are targeted for degradation by the bacterial ubiquitin ligase IpaH9.8. Cell Host Microbe. (2017) 22:507–18.e5. doi: 10.1016/j.chom.2017.09.007
66. Thurston TL, Matthews SA, Jennings E, Alix E, Shao F, Shenoy AR, et al. Growth inhibition of cytosolic Salmonella by caspase-1 and caspase-11 precedes host cell death. Nat Commun. (2016) 7:13292. doi: 10.1038/ncomms13292
67. Man SM, Place DE, Kuriakose T, Kanneganti TD. Interferon-inducible guanylate-binding proteins at the interface of cell-autonomous immunity and inflammasome activation. J Leukoc Biol. (2017) 101:143–50. doi: 10.1189/jlb.4MR0516-223R
68. Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science. (2011) 332:717–21. doi: 10.1126/science.1201711
69. Olszewski MA, Gray J, Vestal DJ. In silico genomic analysis of the human and murine guanylate-binding protein (GBP) gene clusters. J Interferon Cytokine Res. (2006) 26:328–52. doi: 10.1089/jir.2006.26.328
70. Degrandi D, Konermann C, Beuter-Gunia C, Kresse A, Wurthner J, Kurig S, et al. Extensive characterization of IFN-induced GTPases mGBP1 to mGBP10 involved in host defense. J Immunol. (2007) 179:7729–40. doi: 10.4049/jimmunol.179.11.7729
71. Shenoy AR, Kim BH, Choi HP, Matsuzawa T, Tiwari S, MacMicking JD. Emerging themes in IFN-gamma-induced macrophage immunity by the p47 and p65 GTPase families. Immunobiology. (2007) 212:771–84. doi: 10.1016/j.imbio.2007.09.018
72. Finethy R, Jorgensen I, Haldar AK, de Zoete MR, Strowig T, Flavell RA, et al. Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in Chlamydia-infected macrophages. Infect Immun. (2015) 83:4740–9. doi: 10.1128/IAI.00856-15
73. Balakrishnan A, Karki R, Berwin B, Yamamoto M, Kanneganti T-D. Guanylate binding proteins facilitate caspase-11-dependent pyroptosis in response to type 3 secretion system-negative Pseudomonas aeruginosa. Cell Death Discov. (2018) 4:66. doi: 10.1038/s41420-018-0068-z
74. Feeley EM, Pilla-Moffett DM, Zwack EE, Piro AS, Finethy R, Kolb JP, et al. Galectin-3 directs antimicrobial guanylate binding proteins to vacuoles furnished with bacterial secretion systems. Proc Natl Acad Sci USA. (2017) 114:E1698–706. doi: 10.1073/pnas.1615771114
75. Kim BH, Chee JD, Bradfield CJ, Park ES, Kumar P, MacMicking JD. Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nat Immunol. (2016) 17:481–9. doi: 10.1038/ni.3440
76. Pilla-Moffett D, Barber MF, Taylor GA, Coers J. Interferon-inducible GTPases in host resistance, inflammation and disease. J Mol Biol. (2016) 428:3495–513. doi: 10.1016/j.jmb.2016.04.032
77. Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, Shkarina K, et al. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun. (2020) 11:3276. doi: 10.1038/s41467-020-16889-z
78. Wandel MP, Kim BH, Park ES, Boyle KB, Nayak K, Lagrange B, et al. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol. (2020) 21:880–91. doi: 10.1038/s41590-020-0697-2
79. Kutsch M, Sistemich L, Lesser CF, Goldberg MB, Herrmann C, Coers J. Direct binding of polymeric GBP1 to LPS disrupts bacterial cell envelope functions. EMBO J. (2020) 39:e104926. doi: 10.15252/embj.2020104926
80. Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, et al. IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase-11-NLRP3 inflammasomes. Cell. (2016) 167:382–96.e17. doi: 10.1016/j.cell.2016.09.012
81. Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, et al. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell. (2016) 165:1106–19. doi: 10.1016/j.cell.2016.04.015
82. Santos JC, Dick MS, Lagrange B, Degrandi D, Pfeffer K, Yamamoto M, et al. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J. (2018) 37:e98089. doi: 10.15252/embj.201798089
83. Dinarello CA. IL-18: A TH1-inducing, proinflammatory cytokine and new member of the IL-1 family. J Allergy Clin Immunol. (1999) 103(1 Pt 1):11–24. doi: 10.1016/S0091-6749(99)70518-X
84. Maltez VI, Miao EA. Reassessing the evolutionary importance of inflammasomes. J Immunol. (2016) 196:956–62. doi: 10.4049/jimmunol.1502060
85. Wang J, Deobald K, Re F. Gasdermin D protects from melioidosis through pyroptosis and direct killing of bacteria. J Immunol. (2019) 202:3468–73. doi: 10.4049/jimmunol.1900045
86. Wang W, Shao Y, Li S, Xin N, Ma T, Zhao C, et al. Caspase-11 plays a protective role in pulmonary acinetobacter baumannii infection. Infect Immun. (2017) 85:e00350–17. doi: 10.1128/IAI.00350-17
87. Wang J, Shao Y, Wang W, Li S, Xin N, Xie F, et al. Caspase-11 deficiency impairs neutrophil recruitment and bacterial clearance in the early stage of pulmonary Klebsiella pneumoniae infection. Int J Med Microbiol. (2017) 307:490–6. doi: 10.1016/j.ijmm.2017.09.012
88. Cerqueira DM, Pereira MS, Silva AL, Cunha LD, Zamboni DS. Caspase-1 but not caspase-11 Is required for NLRC4-mediated pyroptosis and restriction of infection by flagellated legionella species in mouse macrophages and in vivo. J Immunol. (2015) 195:2303–11. doi: 10.4049/jimmunol.1501223
89. Simpson A, Martinez FD. The role of lipopolysaccharide in the development of atopy in humans. Clin Exp Allergy. (2010) 40:209–23. doi: 10.1111/j.1365-2222.2009.03391.x
90. Zasłona Z, Flis E, Wilk MM, Carroll RG, Palsson-McDermott EM, Hughes MM, et al. Caspase-11 promotes allergic airway inflammation. Nat Commun. (2020) 11:1055. doi: 10.1038/s41467-020-14945-2
91. Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. (1995) 152(6 Pt 1):2111–8. doi: 10.1164/ajrccm.152.6.8520783
92. Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat Commun. (2015) 6:6201. doi: 10.1038/ncomms7201
93. Mahenthiralingam E, Campbell ME, Speert D. Nonmotility P, and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect Immun. (1994) 62:596–605. doi: 10.1128/IAI.62.2.596-605.1994
94. Jain M, Ramirez D, Seshadri R, Cullina JF, Powers CA, Schulert GS, et al. Type III secretion phenotypes of Pseudomonas aeruginosa strains change during infection of individuals with cystic fibrosis. J Clin Microbiol. (2004) 42:5229–37. doi: 10.1128/JCM.42.11.5229-5237.2004
95. Huus KE, Joseph J, Zhang L, Wong A, Aaron SD, Mah TF, et al. Clinical isolates of pseudomonas aeruginosa from chronically infected cystic fibrosis patients fail to activate the inflammasome during both stable infection and pulmonary exacerbation. J Immunol. (2016) 196:3097–108. doi: 10.4049/jimmunol.1501642
96. Beuzón CR, Méresse S, Unsworth KE, Ruíz-Albert J, Garvis S, Waterman SR, et al. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. (2000) 19:3235–49. doi: 10.1093/emboj/19.13.3235
97. Creasey EA, Isberg RR. The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc Natl Acad Sci USA. (2012) 109:3481–6. doi: 10.1073/pnas.1121286109
98. Ge J, Gong YN, Xu Y, Shao F. Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc Natl Acad Sci USA. (2012) 109:6193–8. doi: 10.1073/pnas.1117490109
99. Paciello I, Silipo A, Lembo-Fazio L, Curcurù L, Zumsteg A, Noël G, et al. Intracellular Shigella remodels its LPS to dampen the innate immune recognition and evade inflammasome activation. Proc Natl Acad Sci USA. (2013) 110:E4345–54. doi: 10.1073/pnas.1303641110
100. Wang X, Ribeiro AA, Guan Z, Abraham SN, Raetz CR. Attenuated virulence of a Francisella mutant lacking the lipid A 4'-phosphatase. Proc Natl Acad Sci USA. (2007) 104:4136–41. doi: 10.1073/pnas.0611606104
101. Kanistanon D, Powell DA, Hajjar AM, Pelletier MR, Cohen IE, Way SS, et al. Role of Francisella lipid A phosphate modification in virulence and long-term protective immune responses. Infect Immun. (2012) 80:943–51. doi: 10.1128/IAI.06109-11
102. Rebeil R, Ernst RK, Gowen BB, Miller SI, Hinnebusch BJ. Variation in lipid A structure in the pathogenic yersiniae. Mol Microbiol. (2004) 52:1363–73. doi: 10.1111/j.1365-2958.2004.04059.x
103. Lagrange B, Benaoudia S, Wallet P, Magnotti F, Provost A, Michal F, et al. Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat Commun. (2018) 9:242. doi: 10.1038/s41467-017-02682-y
104. Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, et al. The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe. (2013) 13:570–83. doi: 10.1016/j.chom.2013.04.012
Keywords: caspase-11, gasdermin D, inflammasomes, pore intracellular traps (PIT), Burkholderia thailandensis lung defense, Gbps, LPS
Citation: Oh C, Verma A and Aachoui Y (2020) Caspase-11 Non-canonical Inflammasomes in the Lung. Front. Immunol. 11:1895. doi: 10.3389/fimmu.2020.01895
Received: 06 November 2019; Accepted: 14 July 2020;
Published: 21 August 2020.
Edited by:
Charles S. Dela Cruz, School of Medicine, Yale University, United StatesReviewed by:
Karlhans Fru Che, Karolinska Institutet (KI), SwedenCopyright © 2020 Oh, Verma and Aachoui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youssef Aachoui, eWFhY2hvdWlAdWFtcy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.