
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 27 August 2020
Sec. Immunological Memory
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01834
This article is part of the Research Topic Memory T Cells in Chronic Infections and Tumors View all 6 articles
 Wenhui Li1,2
Wenhui Li1,2 Lianjun Zhang1,2*
Lianjun Zhang1,2*Memory T cells persist for long term to mediate robust recall response upon rechallenging with previous encountered pathogens. The memory T cell pool is highly heterogeneous based on distinct phenotypic, functional, and locational properties, and contains discrete subsets, which contribute to diverse immune responses. In this mini-review, we will briefly discuss the distinct subsets of memory T cells and then focus on mitochondria-related metabolic and epigenetic regulations of CD8+ T cell memory formation. In particular, we discuss many aspects of mitochondrial quality control systems (biogenesis, dynamics, etc.) in regulating CD8+ T cell fate decision and antitumor immunity. Importantly, targeting mitochondrial metabolism to boost T cell memory formation and metabolic fitness might represent an attractive strategy to improve cancer immunotherapy including CAR-T therapy.
Memory T cells possess the property of long-term remembrance of priming antigens or pathogens, and evoke a rapid recall response with enhanced magnitude upon antigen reencounter (1). For many years, it is recognized that memory T cell pool is heterogeneous in terms of phenotypic markers, functional traits, epigenetic modifications, and metabolic features (2–4). In particular, human memory T cells were primarily categorized into central memory (Tcm) and effector memory (Tem) subsets, characterized by CD62hiCCR7hi and CD62lowCCR7low phenotype, respectively, and with distinct functional/localization properties (5). Tcm cells have vast proliferative potential and reside in secondary lymphoid organs to invoke robust recall responses, which further differentiate toward effector memory or terminally differentiated effector progenies to protect against infections or undergo self-renew. Tem is commonly found in nonlymphoid tissues and circulate through blood continuously (6). Although Tem cells are less proliferative and could not persist for a long term, they are ready to provide immediate protection at infection sites via producing multiple cytotoxic molecules, including granzyme B (Gzmb), perforin, interferon gamma (IFNγ), tumor necrosis factor (TNF), etc. (5).
Recently, a subset of memory T cells called stem-cell-like memory T cells (Tscm) has been identified both in human and mice (7, 8). Tscm cells exhibit stem-cell-like properties including self-renewal and could further differentiate into memory or effector T cells, which is further validated by a single-cell adoptive transfer that gives rise to diverse progenies upon recall in mice (7–9). On the other hand, tissue-resident memory T cells (Trm) represent the important subset that is permanently embedded into tissues such as skin, lung, gut, brain, and even the tumor sites. Trm cells are characterized by CD103hiCD69hiCD62lowCD27low phenotype and act as the key player for local immune surveillance at the barrier tissues against reinfected pathogens and display accelerated immediate defense via rapid production of Gzmb, among many other effector molecules (10–13).
The origin, formation, and maintenance of distinct subsets of memory T cells are tightly regulated by multiple extrinsic and intrinsic factors. In response to various stimuli, including virus, bacteria, parasites, fungi, and even tumor-derived mutated antigens, naive T cells undergo activation, expansion, and differentiation into distinct progeny of effector/memory T cells to eliminate infections (14). The molecular mechanisms underlying CD8+ T cell memory and effector differentiation have been illustrated at multiple levels including transcriptional regulation, epigenetic modification, and metabolic reprograming (15, 16). In this section, we will particularly focus on mitochondria-dependent metabolic reprograming in CD8+ T cell memory formation.
Naive T cells remain in quiescent state and preserve their survival mainly via oxidative phosphorylation (OXPHOS). Upon TCR activation, naive CD8+ T cells first undergo extensive size increase, followed by proliferative burst and acquisition of cytotoxic functions, which are accompanied by reprogramming of anabolism and catabolism (17, 18). In particular, the prominent aerobic glycolysis takes over within effector CD8+ T cells with relatively low rate of OXPHOS (19) (Figure 1). To this end, a large number of intermediate metabolites produced via glycolysis are engaged to build the macromolecules and support the proliferation burst of effector T cells. Interestingly, memory CD8+ T cells rely heavily on OXPHOS to support their survival and function, in which the fatty acid is predominantly used to fuel fatty acid β-oxidation (FAO) and maintains spare respiratory capacity (SRC). Surprisingly, memory CD8+ T cells prefer to utilize a “futile cycle” of de novo fatty acid synthesis (FAS) and lysosome-based lipid storage in order to maintain long-term survival and supply adequate ATP immediately during antigen rechallenge, rather than direct uptake of fatty acids from the environment (20) (Figure 1). Although both naive and memory T cells rely on OXPHOS, naive T cells harbor less mitochondrial mass and lower SRC as compared to memory T cells (21, 22). Furthermore, CD8+ Trm cells generated from viral-infected skin exhibit increased lipid metabolism dependent on fatty-acid-binding proteins 4 and 5 (FABP4 and FABP5) mediated exogenous lipid uptake and transport in both mouse and human tissues (23).
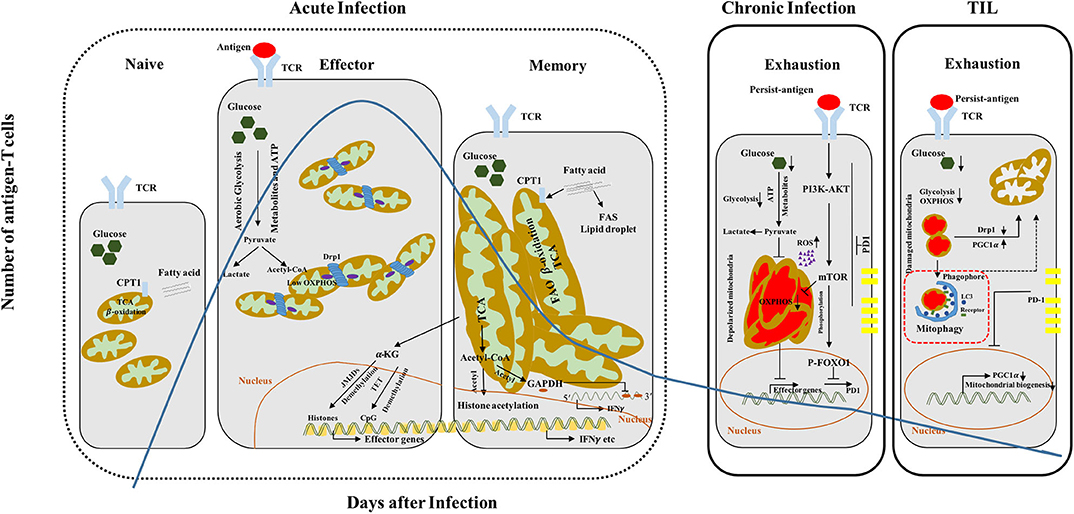
Figure 1. CD8+ T cell differentiation and functionality couples with dynamic metabolic programming. Quiescent naive T cells display small and fragmented mitochondria, which utilize OXPHOS and FAO (CPT1, the rate-limiting enzyme) to maintain their survival. Although effector T cells switch from OXPHOS to aerobic glycolysis to support clonal expansion and effector functions, they also show transiently increased mitochondrial mass accompanied with Drp1-mediated mitochondrial fission. In contrast, memory T cells harbor more fused and elongated mitochondria, which have high spare respiratory capacity (SRC), and predominantly utilize fatty acid to produce energy via β-oxidation, accompanied with fatty acid synthesis (FAS). The effector/memory differentiation process can also be regulated by epigenetic modifications including mitochondrial metabolites (acetyl-CoA and α-KG). Acetyl-CoA provide donor acetyl to histone and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which can be hyperacetylated respectively and promote interferon gamma (IFNγ) expression. α-KG acts as a cofactor of Jumonji C-domain-containing histone demethylases (JMJDs)/ten–eleven translocation (TET), and demethylates histone and DNA with the result of effector genes expression. Exhausted CD8+ T cells show multiple functional or structural alterations in mitochondria. Within CD8+ T cells during chronic infection, larger, structurally defective and depolarized mitochondria are accumulated and accompanied with high ROS production, leading to defective OXPHOS and loss of effector functions. TILs exhibit decreased mitochondrial mass but increased fragmented mitochondria with dysregulated structure and accumulation of ROS, resulting in defective glycolysis and OXPHOS. PD1 can negatively regulate the PGC1α and then inhibit mitochondrial biogenesis. Overexpression of PGC1α or knockdown Drp1 can recover the mitochondrial defects and functions and promote T cell effector functions and antitumor capacity. TCR, T cell receptor; CPT1, carnitine palmitoyltransferase 1; TCA, tricarboxylic acid cycle; acetyl-CoA, acetyl coenzyme A; FAO, fatty acid oxidation; FAS, fatty acid synthesis; OXPHOS, oxidative phosphorylation; PGC1α, peroxisome-proliferator-activated receptor gamma coactivator 1α.
The mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) signaling pathway, the key sensors of intracellular energy status, are also critical regulators of memory CD8+ T cell formation. mTOR complex I (mTORC1) activation is required for protein synthesis and generation of the biomolecules for proliferation. Interestingly, inhibition of mTORC1 by rapamycin promotes FAO and memory CD8+ T cell formation, which was also confirmed by the findings that AMPK (the upstream kinase of mTOR signaling) activator metformin increases the pool of memory CD8+ population via boosting OXPHOS rate (24–26). Of note, complete ablation of mTORC1 activity via genetic deletion of Raptor impairs both effector and memory differentiation. Interestingly, we and others recently demonstrated that functional deficiency of mTORC2 leads to enhanced CD8+ T cell memory formation, which was associated with increased mitochondrial metabolism and FAO (26, 27). Similarly, we found that fine tuning of the mTOR signaling rather than Wnt signaling activation is responsible for the generation of human Tscm (28). Moreover, it has been shown that restrained glycolytic activity or enhanced FAO, which are achieved by inhibition of AKT or overexpression of rate-limiting β-oxidation enzyme CPT1α, favors the formation of memory CD8+ T cells and restricts the effector differentiation (22, 29). Consistently, enhanced glycolytic flux by overexpressing phosphoglycerate mutase-1 (Pgam1) in CD8+ T cells blocks its memory formation (30). Conversely, inhibition of glucose metabolism in the presence of Hk2 inhibitor 2-deoxy-d-glucose favors memory CD8+ T cells' generation and augments antitumor immunity (30). Altogether, those findings strongly suggest that genetic or pharmacological modulation of T cell metabolism may dictate T cell fate decision and reprogram the effector/memory lineage specification.
Mitochondria are dynamic organelles that undergo fission and fusion to maintain homeostasis and emerge as the hub of innate and adaptive immunity. For instance, mitochondrial DNA (mtDNA) contains plenty of CpG islands, which can be recognized by TLR9 and trigger nuclear factor kappa B (NF-κB) activation and proinflammatory response (31, 32). Mitochondrial antiviral signaling (MAVS), the mitochondrial out-membrane protein, can protect against RNA virus infection via interacting with virus RNA sensor RIG-I and promoting NF-κB and interferon regulatory factor (IRF) activation (33, 34). Herein, we discuss mainly the role of mitochondria in regulating memory CD8+ T cell formation and functionality.
CD8+ T cells with distinct activation or functional states harbor different mitochondrial morphologies to fulfill their metabolic demands and biological functions. Naive CD8+ T cells are characterized by small and fragmented mitochondria, accompanied with relatively lower level of basal energy consumption through mitochondrial OXPHOS (35). Effector T cells have more punctate mitochondria as indicated by increased mitochondrial fission, which lead to lower OXPHOS and higher aerobic glycolytic rate (36) (Figure 1). Moreover, mitochondrial fragmentation also promotes ROS production, which is required for acquisition of effector functions in activated T cells (37). On the other hand, memory CD8+ T cells exhibit increased mitochondrial fusion and mass, which display larger network of elongated mitochondria and may maintain SRC for efficient utilization of fatty acid and rapid proliferation upon recall (36) (Figure 1). Mdivi-1, an inhibitor of mitochondrial fission, enhances respiratory capacity and promotes the formation of memory T cell pool. Consistently, knockdown of Drp1 or overexpression of OPA1, the key mediators of mitochondrial fission and fusion, respectively, promotes CD8+ T cell memory generation (36, 38).
In addition to mitochondrial dynamics, mitochondrial quality can also be regulated by mitochondrial biogenesis, mitophagy, and mitochondrial unfolded protein response (UPRmt) (39). T cells undergo exhaustion during chronic viral infection or within the tumor microenvironment (TME), which was characterized by gradual loss of proliferative capacity and effector function, and accompanied by accumulation of dysfunctional mitochondria (40, 41). Of note, exhausted CD8+ T cells generated upon chronic lymphocytic choriomeningitis virus infection exhibit higher rate of depolarized mitochondria, larger size, and increased ROS level, whereas tumor-infiltrating lymphocytes (TILs), also characterized by exhausted phenotype, show decreased total mitochondrial mass and increased depolarized mitochondria, with fragmented morphology and low level of ROS (42, 43) (Figure 1). Furthermore, dysregulation of mitochondrial homeostasis, indicated by increased ROS level and lower mitochondrial potential, has been observed in exhausted CD8+ T cell from patients with chronic hepatitis B virus (HBV) infections. Scavenger ROS in exhausted T cells by mitochondrion-targeted antioxidants and recovered mitochondrial metabolic capacity by interleukin-12 are confirmed effective approaches to boost antiviral CD8+ T cell functions (44, 45). In contrast to B16 melanoma infiltrated lymphocytes, human CD8+ TILs from clear cell renal cell carcinoma (ccRCC) patients display small, fragmented, and hyperpolarized mitochondria companied with high level of ROS and increased mitochondrial mass with Mitotracker Green (MTG) staining (46). Consequently, the mitochondria demonstrate distinct phenomena in CD8+ TILs from different types of tumor, which may be attributed to tumor heterogeneity and complicated microenvironment. Although TILs showed distinct mitochondrial morphologies, mitochondrial dysfunction and metabolic deficiencies are common features observed in most TILs. Furthermore, a large number of total mitochondria and depolarized mitochondria were observed in early exhausted CD8+ T cells during chronic LCMV clone 13 infection, which can be reversed by PGC1α (the master regulator of mitochondrial biogenesis) overexpression (42). In contrast, transduction of PGC1α into CD8+ TILs promotes mitochondrial biogenesis to maintain the mitochondrial quality and increase the mass. Among these distinct observations, the quality and quantity of mitochondria are both critical parameters in response to diverse stimuli. It is possible that the turnover of the accumulated damaged mitochondria in early exhausted T cells could be stimulated by PGC1α overexpression, which acts to decrease the depolarized mitochondria and in turn maintain the quality and integrity of mitochondria. Intriguingly, it was shown that PD1 could directly suppress PGC1α expression (43). Within the TME, PGC1α expression was repressed in TILs due to activation of AKT signaling and blockade of Foxo1, which is also confirmed by chronic LCMV-infection model. Therefore, neutralization of PD1 boosts PGC1α expression, and overexpression of PGC1α can improve healthy mitochondrial mass and function, and recover antitumor abilities of exhausted T cells (43).
Autophagy is important for maintaining cell homeostasis via highly selective self-degradation process to clear infected pathogens, aggregated proteins, and damaged or superfluous organelles (mitochondria, ribosome, peroxisome, etc.) (47). Interestingly, T-cell-specific deficiency of Atg5 or Atg7 impairs memory formation without damaging effector differentiation. Along the same line, T cells with Atg7 deletion harbor increased mitochondrial mass and enhanced ROS level, due to failure of removal of the damaged mitochondria (48). These findings highlight the significance of mitochondrial homeostasis through autophagy during CD8+ T cell memory differentiation and maintenance. Mitophagy, an important regulatory mechanism to maintain the mitochondrial quality and integrity, can selectively eliminate dysfunctional or superfluous mitochondria (49). Nevertheless, the role of mitophagy is less studied and remains largely unknown during CD8+ T cell memory formation. The mitophagy receptor BNIP3L (also called NIX) can directly interact with LC3 and recruit autophagic vacuole to mitochondria, followed by fusion with lysosome and degradation of dysfunctional mitochondria (50). A recent study demonstrated that NIX-mediated mitophagy could promote CD8+ T cell effector memory differentiation by preventing HIF1α accumulation and maintaining long-chain fatty acid metabolism (51). Given the accumulation of small fragmented mitochondria in CD8+ T cells within the TME, it remains to be investigated how does mitophagy regulate T cell exhaustion and antitumor immunity.
Multiple pairs of transcription factors have been well demonstrated to regulate CD8+ T cell effector or memory differentiation (3). For instance, the transcriptional factor T-bet is required for the generation of KLRGhi terminated effector T cells, whereas Eomes foster memory T cell generation (52, 53). Moreover, Id2/Id3 and Blimp-1/Bcl-6 are also critical regulators of CD8+ T cell effector or memory progenies (54, 55). Besides the transcriptional regulation, epigenetic modifications including chromatin remodeling, DNA methylation, and histone modifications (methylation, acetylation, phosphorylation, ubiquitination, etc.) represent another important layer to modulate gene expression patterns and dictate CD8+ T cell fate decision (15). DNA methylation predominantly occurs at clusters of CpG dinucleotides, which is also called CpG island and located in gene promoters (56). High frequency of CpG methylation acts as a repressive gene expression marker and affects the chromatin accessibility and transcription factor docking. In memory CD8+ T cells, DNA methylation at the promoter of IFNγ, Gzmb, and IL2 loci is increased and correlates with suppressed gene expression (57). However, Tcf7, a critical transcription factor in memory and naive T cells, and other memory genes (CD62L and CD127), are demethylated and show enhanced expression as compared to activated or effector cells (58). On the other hand, effector T cells show dramatically increased expression of multiple effector genes such as IFNγ, TNF, IL2, and Gzmb, due to reduced level of methylation at their promoter or enhancer region (59). Although the methylation landscape of memory T cells is similar with that of naive T cells, memory T cells are capable of maintaining their epigenetic landscape at effector genes loci, but fewer methylations occur at the same genes in naive T cells, indicating that memory T cells retain an effector-like signature in order to rapidly acquire cytotoxic ability and formation of effector T cells upon recall (60, 61).
Histone modification represents another important layer of chromatin structure modulation, which impacts subsequent transcription factor docking and gene expression. Hyperacetylation, such as H3K9Ac, may improve the chromatin accessibility and enhance the effector gene expression in memory T cells upon recall. Genome-wide analysis of histone methylation shows diverse methylated sites at arginine and lysine; the dimethylated or trimethylated H3K4, H3K9, and H3K27 on the gene loci of effector/memory T cells are involved in the formation and maintenance of T cell memory. Generally, the expression level of effector genes is associated with higher level of H3K4me3 and low level of H3K27me3 in memory T cells upon recall. Thus, these observations support the notion that memory T cells are epigenetically imprinted to mediate an accelerated recall response.
Mitochondrial intermediate metabolites such as citrate, acetyl-CoA, and α-ketoglutarate (α-KG) are derived from Krebs cycle and involved in epigenetic modifications including histone acetylation. Accumulated acetyl-CoA can provide donor acetyl for subsequent histone acetylation, and IFNγ will be transcribed abundantly due to hyperacetylation of its promoter in CD4+ T cells (62). Moreover, acetyl-CoA is also used to acetylate glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which inhibits IFNγ translation via binding the 3′ unfolded protein response (UPR) of its messenger RNA (mRNA), promotes IFNγ production by blocking the interaction (63). During acute bacterial infection, acetate is accumulated in the serum and subsequently uptake by memory CD8+ T cells to accelerate acetyl-CoA production. High concentration of acetyl-CoA provides acetyl group to GAPDH and boost its enzyme activity, which enhances glycolytic flux and cytotoxic capacity (for instance, IFNγ production) (64) (Figure 1). Furthermore, acetate promotes IFNγ production via acetyl-CoA synthesis in glucose-restricted CD8+ T cells and tumor-exposed exhausted T cells, which is attributed to histone acetylation and transcriptional accessibility. Acetyl-CoA synthetase 2 (ACSS2), which can convert acetate to acetyl-CoA, also enhances CD8+ T cell effector functions and IFNγ expression in vivo (65). In addition, α-KG can act as a cofactor for Jumonji C-domain-containing histone demethylases (JMJDs) and ten–eleven translocation (TET) 5-methycytosine hydroxylases, which mediate histone and DNA demethylation, respectively. Levels of α-KG or α-KG–succinate ratios can therefore remodel the epigenomes and gene expression in CD4+ or CD8+ T cells, and accumulation of α-KG drives gene expression associated with effector function and cell differentiation (66, 67). Isocitrate dehydrogenase 1 (IDH1) mutation was also determined to produce α-hydroxyglutarate instead of α-KG, and this results in a block of cell differentiation due to inhibition of histone demethylation (68). Moreover, S-2-hydroxyglutarate, the metabolite enantiomer of 2-hydroxyglutarate, occurs in IDH1/2 mutations or damaged mitochondria and hypoxia treatment, increases the CD8+ T cell recall capacity, and promotes the persistence of adoptively transferred CD8+ T cells and antitumor immunity via modulating DNA/histone methylation and HIF1α stability (69).
Unlike traditional surgical resection, radiation, chemotherapy, and targeted therapies, immunotherapeutic strategies provide novel opportunities for long-term protection against cancer development and recurrence. Immune checkpoint blockade (ICB) and chimeric antigen receptor T cell (CAR-T) therapy represent two successful strategies to treat patients across multiple types of cancer in clinics (70, 71). Given their enhanced expression in TILs, antibodies against either CTLA4, PD1, or in combination can be used to partially recover the impaired T cell functionality and lead to drastic tumor regression both in mouse models and human cancer patients, but only a small fraction of patients benefit from the treatment (72). Although the CAR-T therapy achieved great success in certain hematological cancers, it remains challenging to treat the majority of malignant solid tumors with current CAR-T technology; one key reason is that infiltrated CAR-T undergo metabolic or functional exhaustion in the TME (73). Thus, there is an urgent need to develop novel strategies to enhance CD8+ T cell functionality within the TME. Here, we discuss how can we improve the efficacy of antitumor immunity by modulating mitochondrial metabolism within T cells. In particular, we present the potential mitochondrial targeting strategies that could boost T cell immunity against cancer.
Within the TME, effector T cells are under severe metabolic stress such as nutrient deprivation (low glucose, lower amino acids, etc.), hypoxia, and accumulation of lactate acid and ROS (74, 75). These disadvantages result in dramatic metabolic changes in TILs and may facilitate the evasion of the tumor cells from immune surveillance. To overcome the accumulation of small fragmented mitochondria in CD8+ T cells within the TME, increased mitochondria biogenesis via PGC1α overexpression in T cells boosts the mitochondrial mass and metabolic capacity (glutamine metabolism and OXPHOS) and enhances antitumor immunity (43, 76). Moreover, PGC1α overexpression-mediated antitumor immunity may be combined with anti-PD1 antibody, as suggested by significantly regressed tumor volume with combinatory treatment (76). Bezafibrate, a PGC1α agonist, has been shown to boost antitumor immunity via upregulating mitochondrial OXPHOS and inhibition of apoptosis in MC38-bearing mouse tumor model treated with PD1 blockade (77). Thus, pharmacological induction of mitochondrial biogenesis in T cells may represent a potential therapeutic target for cancer immunotherapy. Of note, it requires further investigation regarding the role of mitophagy in regulating T cell exhaustion and antitumor immunity, in particular to understand the crosstalk between mitophagy and mitochondrial biogenesis in modulating T cell metabolic fitness and functionality. Furthermore, costimulatory or coinhibitory molecules also critically regulate T cell metabolism and mitochondrial phenotype (78). For instance, CD28 signaling during T cell activation stimulates aerobic glycolysis and promotes mitochondrial fusion (79). 4-1BB costimulation can also augment glycolysis via promoting the expression of glucose transporters to support CD8+ T cell proliferation (80). Moreover, 4-1BB costimulation enhanced mitochondrial fusion and biogenesis, which are independent of PGC1α-mediated pathways and p38-MAPK signaling, and resulted in improving metabolic sufficiency and antitumor immunity in CD8+ TILs (81). Furthermore, overexpression of 4-1BB in TILs promotes mitochondrial respiration capacity, associated with increased OPA-1-induced mitochondrial fusion and PGC1α-mediated mitochondrial biogenesis. Indeed, those modified T cells better survive and function under metabolic stress at the TME and trigger enhanced antitumor immunity (81, 82). On the other hand, PD1 enhances FAO via upregulating Cpt1α, and CTLA4 restricts glucose metabolism by reducing AKT phosphorylation in CD4+ T cells (83, 84).
In addition, the glycolytic capacity and cytokine production of TILs are dampened given the competition for nutrient consumption between tumor cells and effector T cells within the TME (74). Lactate, the product of glycolytic pathway and mostly derived from tumors, directly suppresses T cell proliferation and cytotoxic activity (85). Thus, tumor-imposed metabolic restrictions attenuate the effector functions of TILs and antitumor immunity. TILs suffer from low glucose in the TME and also display fragmented mitochondria, and administration of Mdivi-1 to inhibit mitochondrial fission or knockdown of Drp1 leads to fused mitochondria and increased metabolic fitness in TILs, which thus display superior antitumor effects (36). Thus, reprogramming mitochondrial dynamics by inhibiting mitochondrial fission or promoting fusion may increase the OXPHOS and T cell immunity and represent a potential target for rescuing the effector functions of exhausted CD8+ T cells.
Adoptive cell transfer (ACT) of tumor-antigen-specific T cells could mediate considerable antitumor effects in clinics. Zelig Eshhar's lab first reported CAR-T technology consisting of a single-chain antibody for recognizing the targeted antigen and signaling transduction domain for T cell activation and acquisition of effector functions. To this end, this genetically modified T lymphocytes could directly target and kill the tumor cells that express the cognate antigen of designed antibody (86). Later, the costimulatory CD28 or 4-1BB functional domains were included in the CAR-T engineering and enhanced therapeutic efficacy (87). Thus, the addition of costimulatory domain to CAR is essential for boosting CAR-T activities. Although ICB is an effective approach for treating certain cancers, TILs also exhibit metabolic insufficiency and mitochondrial dysfunction (decreased mitochondrial mass, fragmentation, lower OXPHOS, etc.) (43). In this regard, development of strategies that could recover or boost mitochondrial functions is important to promote T cell or CAR-T antitumor immunity. Consistently, human CAR-T armed with 4-1BB domain show higher rate of expansion, survival, and superior antitumor immunity in vivo due to enhanced mitochondrial mass and fitness. In particular, 4-1BB domain can enhance mitochondrial biogenesis, fatty acid β-oxidation, and SRC (88). Interestingly, a recent study demonstrated that CAR-T with herpes virus entry mediator (HVEM)-derived costimulatory domain exhibits higher mitochondrial respiration and glycolysis compared with CD28 and 4-1BB, which accounts for increased cytotoxic capacity and alleviated exhausted signature (89). Taken together, constructing new generation CAR-T armed with the optimal costimulatory domain to improve mitochondrial quality will be the key to mediate effective antitumor immunity.
Altogether, we believe that mitochondria may act as a key hub in determining CD8 T cell memory differentiation, maintenance, or functional decline (exhaustion) within the TME. Modulating mitochondrial biogenesis, dynamics (fusion/fission), and clearance of dysregulated mitochondria via mitophagy is highly relevant for designing immunotherapeutic strategies against cancer. In addition, rewiring the metabolic communication between distinct cell types within the TME may largely improve the successful rate of CAR-T therapy in solid cancers. Given that certain mitochondrial metabolites are important regulators of epigenetic modifications, fine-tuning mitochondrial quantity/quality/integrity is required for achieving better metabolic fitness or boosting T cell antitumor immunity via epigenetic remodeling the T cell exhaustion signature.
WL and LZ: conception, writing and revision of the manuscript. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
WL was supported by Natural Science Foundation of China (NSFC 31900645) and PUMC Fundamental Research Funds for the Central Universities (3332019113). LZ was supported by Natural Science Foundation of China (NSFC 81971466).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank members of Zhang's lab for helpful discussions.
Tcm, central memory T cell; Tem, effector memory T cell; Tscm, stem cell memory T cell; Trm, resident memory T cell; TCR, T cell receptor; OXPHOS, oxidative phosphorylation; UPRmt, mitochondrial unfolded protein response; ROS, reactive oxygen species; FAO, fatty acid oxidation; FAS, fatty acid synthesis; SRC, spare respiratory capacity; TILs, tumor-infiltrating lymphocytes; ICB, immune checkpoint blockade; ACT, adoptive cell transfer; CAR, chimeric antigen receptor; ECAR, extracellular acidification rate.
1. Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. (1996) 272:54–60. doi: 10.1126/science.272.5258.54
2. Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. (2007) 27:393–405. doi: 10.1016/j.immuni.2007.08.007
3. Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. (2014) 15:1104–15. doi: 10.1038/ni.3031
4. Zhang L, Romero P. Metabolic control of CD8(+) T cell fate decisions and antitumor immunity. Trends Mol Med. (2018) 24:30–48. doi: 10.1016/j.molmed.2017.11.005
5. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. (1999) 401:708–12. doi: 10.1038/44385
6. Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. (2001) 291:2413–7. doi: 10.1126/science.1058867
7. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. (2011) 17:1290–7. doi: 10.1038/nm.2446
8. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. (2017) 23:18–27. doi: 10.1038/nm.4241
9. Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, et al. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity. (2014) 41:116–26. doi: 10.1016/j.immuni.2014.05.018
10. Sheridan BS, Lefrancois L. Regional and mucosal memory T cells. Nat Immunol. (2011) 12:485–91. doi: 10.1038/ni.2029
11. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature. (2012) 483:227–31. doi: 10.1038/nature10851
12. Wilk MM, Misiak A, McManus RM, Allen AC, Lynch MA, Mills KHG. Lung CD4 tissue-resident memory T cells mediate adaptive immunity induced by previous infection of mice with Bordetella pertussis. J Immunol. (2017) 199:233–43. doi: 10.4049/jimmunol.1602051
13. Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. (2010) 207:553–64. doi: 10.1084/jem.20090858
14. Jameson SC, Masopust D. Understanding subset diversity in T cell memory. Immunity. (2018) 48:214–26. doi: 10.1016/j.immuni.2018.02.010
15. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. (2012) 12:749–61. doi: 10.1038/nri3307
16. Gray SM, Kaech SM, Staron MM. The interface between transcriptional and epigenetic control of effector and memory CD8(+) T-cell differentiation. Immunol Rev. (2014) 261:157–68. doi: 10.1111/imr.12205
17. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. (2015) 212:1345–60. doi: 10.1084/jem.20151159
18. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. (2013) 342:1242454. doi: 10.1126/science.1242454
19. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol. (2012) 13:907–15. doi: 10.1038/ni.2386
20. O'Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
21. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. (2009) 460:103–7. doi: 10.1038/nature08097
22. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
23. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. (2017) 543:252–6. doi: 10.1038/nature21379
24. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. (2009) 460:108–12. doi: 10.1038/nature08155
25. Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. AMPKalpha1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol. (2013) 43:889–96. doi: 10.1002/eji.201243008
26. Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, et al. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest. (2015) 125:2090–108. doi: 10.1172/JCI77746
27. Zhang L, Tschumi BO, Lopez-Mejia IC, Oberle SG, Meyer M, Samson G, et al. Mammalian target of rapamycin complex 2 controls CD8 T cell memory differentiation in a foxo1-dependent manner. Cell Rep. (2016) 14:1206–17. doi: 10.1016/j.celrep.2015.12.095
28. Scholz G, Jandus C, Zhang L, Grandclement C, Lopez-Mejia IC, Soneson C, et al. Modulation of mTOR signalling triggers the formation of stem cell-like memory T cells. EBioMedicine. (2016) 4:50–61. doi: 10.1016/j.ebiom.2016.01.019
29. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. (2013) 14:1064–72. doi: 10.1038/ni.2687
30. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. (2013) 123:4479–88. doi: 10.1172/JCI69589
31. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
32. Zhang JZ, Liu Z, Liu J, Ren JX, Sun TS. Mitochondrial DNA induces inflammation and increases TLR9/NF-kappaB expression in lung tissue. Int J Mol Med. (2014) 33:817–24. doi: 10.3892/ijmm.2014.1650
33. Loo YM, Gale M Jr. Immune signaling by RIG-I-like receptors. Immunity. (2011) 34:680–92. doi: 10.1016/j.immuni.2011.05.003
34. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
35. Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. (2016) 24:104–17. doi: 10.1016/j.cmet.2016.06.007
36. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
37. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. (2013) 38:225–36. doi: 10.1016/j.immuni.2012.10.020
38. Baixauli F, Martin-Cofreces NB, Morlino G, Carrasco YR, Calabia-Linares C, Veiga E, et al. The mitochondrial fission factor dynamin-related protein 1 modulates T-cell receptor signalling at the immune synapse. EMBO J. (2011) 30:1238–50. doi: 10.1038/emboj.2011.25
39. Cai Q, Tammineni P. Alterations in mitochondrial quality control in Alzheimer's Disease. Front Cell Neurosci. (2016) 10:24. doi: 10.3389/fncel.2016.00024
41. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. (2015) 6:e1792. doi: 10.1038/cddis.2015.162
42. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity. (2016) 45:358–73. doi: 10.1016/j.immuni.2016.07.008
43. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. (2016) 45:701–3. doi: 10.1016/j.immuni.2016.08.009
44. Fisicaro P, Barili V, Montanini B, Acerbi G, Ferracin M, Guerrieri F, et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat Med. (2017) 23:327–36. doi: 10.1038/nm.4275
45. Schurich A, Pallett LJ, Jajbhay D, Wijngaarden J, Otano I, Gill US, et al. Distinct metabolic requirements of exhausted and functional virus-specific CD8 T cells in the same host. Cell Rep. (2016) 16:1243–52. doi: 10.1016/j.celrep.2016.06.078
46. Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight. (2017) 2:e93411. doi: 10.1172/jci.insight.93411
47. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. (2018) 19:349–64. doi: 10.1038/s41580-018-0003-4
48. Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol. (2014) 15:1152–61. doi: 10.1038/ni.3025
49. Okamoto K. Organellophagy: eliminating cellular building blocks via selective autophagy. J Cell Biol. (2014) 205:435–45. doi: 10.1083/jcb.201402054
50. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. (2007) 104:19500–5. doi: 10.1073/pnas.0708818104
51. Gupta SS, Sharp R, Hofferek C, Kuai L, Dorn GW, 2nd, Wang J, et al. NIX-mediated mitophagy promotes effector memory formation in antigen-specific CD8(+) T cells. Cell Rep. (2019) 29:1862–77 e7. doi: 10.1016/j.celrep.2019.10.032
52. Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. (2003) 302:1041–3. doi: 10.1126/science.1090148
53. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. (2007) 27:281–95. doi: 10.1016/j.immuni.2007.07.010
54. Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. (2011) 12:1221–9. doi: 10.1038/ni.2158
55. Kallies A, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity. (2009) 31:283–95. doi: 10.1016/j.immuni.2009.06.021
56. Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci USA. (2006) 103:1412–7. doi: 10.1073/pnas.0510310103
57. Fitzpatrick DR, Shirley KM, Kelso A. Cutting edge: stable epigenetic inheritance of regional IFN-gamma promoter demethylation in CD44highCD8+ T lymphocytes. J Immunol. (1999) 162:5053–7.
58. Kersh EN, Fitzpatrick DR, Murali-Krishna K, Shires J, Speck SH, Boss JM, et al. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J Immunol. (2006) 176:4083–93. doi: 10.4049/jimmunol.176.7.4083
59. Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol. (2013) 191:3419–29. doi: 10.4049/jimmunol.1301395
60. Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature. (2017) 552:362–7. doi: 10.1038/nature24633
61. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature. (2017) 552:404–9. doi: 10.1038/nature25144
62. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. (2016) 354:481–4. doi: 10.1126/science.aaf6284
63. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
64. Balmer ML, Ma EH, Bantug GR, Grahlert J, Pfister S, Glatter T, et al. Memory CD8(+) T cells require increased concentrations of acetate induced by stress for optimal function. Immunity. (2016) 44:1312–24. doi: 10.1016/j.immuni.2016.03.016
65. Qiu J, Villa M, Sanin DE, Buck MD, O'Sullivan D, Ching R, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep. (2019) 27:2063–74 e5. doi: 10.1016/j.celrep.2019.04.022
66. Kaelin WG Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. (2013) 153:56–69. doi: 10.1016/j.cell.2013.03.004
67. Chisolm DA, Savic D, Moore AJ, Ballesteros-Tato A, Leon B, Crossman DK, et al. CCCTC-binding factor translates interleukin 2- and alpha-ketoglutarate-sensitive metabolic changes in T cells into context-dependent gene programs. Immunity. (2017) 47:251–67 e7. doi: 10.1016/j.immuni.2017.07.015
68. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. (2012) 483:474–8. doi: 10.1038/nature10860
69. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature. (2016) 540:236–41. doi: 10.1038/nature20165
70. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. (2015) 14:561–84. doi: 10.1038/nrd4591
71. Filley AC, Henriquez M, Dey M. CART immunotherapy: development, success, and translation to malignant gliomas and other solid tumors. Front Oncol. (2018) 8:453. doi: 10.3389/fonc.2018.00453
72. Achkar T, Tarhini AA. The use of immunotherapy in the treatment of melanoma. J Hematol Oncol. (2017) 10:88. doi: 10.1186/s13045-017-0458-3
73. Li X, Chen W. Mechanisms of failure of chimeric antigen receptor T-cell therapy. Curr Opin Hematol. (2019) 26:427–33. doi: 10.1097/MOH.0000000000000548
74. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
75. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
76. Dumauthioz N, Tschumi B, Wenes M, Marti B, Wang H, Franco F, et al. Enforced PGC-1alpha expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cell Mol Immunol. (2020). doi: 10.1038/s41423-020-0365-3
77. Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8(+) T cells and facilitates anti-PD-1 therapy. Cancer Immunol Res. (2018) 6:1375–87. doi: 10.1158/2326-6066.CIR-18-0095
78. Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol. (2019) 16:425–41. doi: 10.1038/s41571-019-0203-7
79. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
80. Choi BK, Lee DY, Lee DG, Kim YH, Kim SH, Oh HS, et al. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8(+) T cell proliferation. Cell Mol Immunol. (2017) 14:748–57. doi: 10.1038/cmi.2016.02
81. Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, et al. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. (2018) 215:1091–100. doi: 10.1084/jem.20171068
82. Teijeira A, Labiano S, Garasa S, Etxeberria I, Santamaria E, Rouzaut A, et al. Mitochondrial morphological and functional reprogramming following CD137 (4-1BB) costimulation. Cancer Immunol Res. (2018) 6:798–811. doi: 10.1158/2326-6066.CIR-17-0767
83. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. (2015) 6:6692. doi: 10.1038/ncomms7692
84. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
85. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
86. Gross G, Gorochov G, Waks T, Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc. (1989) 21:127–30.
87. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
88. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:712. doi: 10.1016/j.immuni.2016.02.023
Keywords: CD8 T cell, mitochondria, memory, cancer, immunotherapy
Citation: Li W and Zhang L (2020) Rewiring Mitochondrial Metabolism for CD8+ T Cell Memory Formation and Effective Cancer Immunotherapy. Front. Immunol. 11:1834. doi: 10.3389/fimmu.2020.01834
Received: 15 January 2020; Accepted: 08 July 2020;
Published: 27 August 2020.
Edited by:
Daniel T. Utzschneider, Peter Doherty Institute for Infection and Immunity, AustraliaReviewed by:
Bertram Bengsch, University of Freiburg, GermanyCopyright © 2020 Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lianjun Zhang, emxqQGlzbS5jYW1zLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.