 Hanna Ilchmann-Diounou
Hanna Ilchmann-Diounou Sandrine Menard
Sandrine Menard- Neuro-Gastroenterology and Nutrition Team, Toxalim (Research Centre in Food Toxicology), Université de Toulouse, INRAE, ENVT, INP-Purpan, UPS, Toulouse, France
Autoimmune disorders (ADs) are multifactorial diseases involving, genetic, epigenetic, and environmental factors characterized by an inappropriate immune response toward self-antigens. In the past decades, there has been a continuous rise in the incidence of ADs, which cannot be explained by genetic factors alone. Influence of psychological stress on the development or the course of autoimmune disorders has been discussed for a long time. Indeed, based on epidemiological studies, stress has been suggested to precede AD occurrence and to exacerbate symptoms. Furthermore, compiling data showed that most of ADs are associated with gastrointestinal symptoms, that is, microbiota dysbiosis, intestinal hyperpermeability, and intestinal inflammation. Interestingly, social stress (acute or chronic, in adult or in neonate) is a well-described intestinal disrupting factor. Taken together, those observations question a potential role of stress-induced defect of the intestinal barrier in the onset and/or the course of ADs. In this review, we aim to present evidences supporting the hypothesis for a role of stress-induced intestinal barrier disruption in the onset and/or the course of ADs. We will mainly focus on autoimmune type 1 diabetes, multiple sclerosis and systemic lupus erythematosus, ADs for which we could find sufficient circumstantial data to support this hypothesis. We excluded gastrointestinal (GI) ADs like coeliac disease to privilege ADs not focused on intestinal disorders to avoid confounding factors. Indeed, GIADs are characterized by antibodies directed against intestinal barrier actors.
Introduction
Autoimmune disorders (ADs) are multifactorial diseases involving, genetic, epigenetic, and environmental factors. In the past decades, there has been a continuous rise in the incidence of ADs, which cannot be explained by genetic factors alone. Changes in our lifestyle including diet, hygiene, exposure to social adversity, or pollutants have been suggested to be risk factors for ADs. ADs are associated with defect of the intestinal barrier; and besides nutrition, another environmental factor well described to impair the intestinal barrier is psychological stress. The aim of this review is to compile evidences highlighting a relationship between stress, intestinal barrier disruption, and occurrence of ADs. Even though no causative role of stress-induced intestinal barrier defect on AD onset has been demonstrated so far, the goal of this manuscript is to combine evidences on the basis of a review of the literature and offer a new field of research and perspectives on ADs. We will focus on three of the most studied ADs—autoimmune type 1 diabetes (T1D), systemic lupus erythematosus (SLE), and multiple sclerosis (MS)—for which we could find sufficient evidence supporting our hypothesis, that is, role of psychological stress and defect of intestinal barrier functions.
This review is based on epidemiological and preclinical studies. Numerous excellent and recent reviews treating either ADs and stress, or stress and the intestinal barrier will be quoted to support this hypothesis.
Stress
Stress, firstly described in 1936 by Selye, is defined as a real (physical) or perceived (psychological) threat to homoeostasis, to which the organism has to react by an adaptive response (1).
Life time window, length, and frequency of exposure to stress play pivotal roles in their consequences on the individual pathophysiology. Indeed, acute and chronic stress exposure could occur in early life in a still maturating organism or at adulthood in mature organism. Traumatic experiences can lead to so-called post-traumatic stress disorder (PTSD), a condition in which the patient suffers from anxiety, depression, and flashbacks long after the traumatic experience (2). Persisting stress or inadequate response can lead to harmful maladaptive reactions depending on the kind of stress that will be discussed below. In this review, psychological stress is used as a general term that encompasses several psychological aspects (i.e., anxiety, depression, etc.).
The stress response is orchestrated by hypothalamic–pituitary–adrenal (HPA) axis and sympathetic nervous system (SNS). The neuroendocrine and autonomous responses are mediated by hormones such as epinephrine, norepinephrine, corticotropin-releasing hormone (CRH), adrenocorticotropic hormone (ACTH), glucocorticoids (cortisol in human and corticosterone in rodents) (3, 4). In the past, special attention has been paid to glucocorticoid in stress response and immune regulation. Endogenous glucocorticoids, part of the endocrine stress response, have ubiquitous functions in the development, metabolism, and inflammation. In general, glucocorticoids have been described to dampen immune response all along the inflammation process [for review, see (5)]: they attenuate signaling pathways of many pattern recognition receptors (6, 7), diminish leukocyte transmigration by reducing adhesion molecules (8), decrease the production of chemoattractants (9), program macrophages to anti-inflammatory M2c subtype (high expression of scavenger receptors and secretion of anti-inflammatory cytokines) (10), and decrease T cell response (11, 12), preferentially Th1 and Th17 by promoting Th2 and Treg (13, 14). Owing to their immunosuppressive effects, glucocorticoids have been used to treat various immune-related disorders like ADs.
The literature of the past 60 years has focused on immunosuppressive properties of glucocorticoids, but glucocorticoids can also enhance inflammation and immunity [for review, see (5)]. We will not go into the details of the diverging effects of glucocorticoid on immune response, but part of the explanation might reside in the diversity of glucocorticoid-receptors in different tissues, the presence or absence of 11βHSD, an enzyme inactivating cortisol, the time of glucocorticoid exposure (before or after tissue injury/inflammation) (15), and the dose (16). All those factors might explain that stressful events inducing glucocorticoids release could play a role in AD occurrence that can be treated by exogenous glucocorticoids. As an example, in humans, childhood maltreatment is associated with modified methylation of the glucocorticoid receptor gene NR3C1 in adults in brain and in leucocytes (17–19).
Role of Stress in Autoimmune Disorders (20)
The onset of at least 50% of autoimmune disorders has been attributed to unknown trigger factors. Many retrospective studies observed that most of patients suffering from AD report uncommon emotional stress before disease onset (21). This is obviously a vicious cycle as AD causes stress in patients (22, 23). This review will focus on three of the most studied ADs that will provide sufficient evidence to support the hypothesis of a role of stress-induced intestinal barrier defect on AD onset, that is, T1D, SLE, and MS. T1D is characterized by a defect of insulin production by pancreas owing to an autoimmune response against host pancreatic β-cells (24). SLE is an AD characterized by severe and persistent inflammation that leads to tissue damage in multiple organs (25, 26). MS is a chronic disease affecting the central nervous system and characterized by a defect of the blood–brain barrier and demyelination of the neurons of the central nervous system due to infiltration of auto-reactive T cells (27, 28). The most widely used preclinical MS model is experimental autoimmune encephalomyelitis (EAE).
A potential association between stressful events and T1D has been highlighted already a long time ago when Thomas Willis links, in 17th century, T1D onset to prolonged sorrow (29). Early life stress seems to be of particular risk for T1D development (30, 31). This is in accordance with literature highlighting neonatal maturation of pancreas as critical and vulnerable to stressors (32). Stress in adult has been described to increase incidence of SLE (33) and is able to exacerbate SLE symptoms (physical pain, sleep disturbances, and unemployment) (34). Around 70% of MS patients reported unusual amount of stress before the onset of the disease (35, 36).
Those epidemiological studies suggest that stress could be involved in both triggering and exacerbating ADs. Whether it is dependent on the kind of stress or ADs involved is unknown, and it would be interesting to conduct both retrospective epidemiological studies and preclinical studies to better document the role of stress in ADs. However, some interventional studies suggest that stress management could benefit to AD patients. Indeed, escitalopram (antidepressant) decreases the risk of MS relapsing in women (37). Diazepam (tranquilizer) decreases EAE incidence and histological signs associated with this disease in a mouse model (38). A meta-analysis of 21 trials showed that in the 10 studies of children and adolescents with supportive or counseling therapy, cognitive behavioral therapy and family system therapy reduced glycosylated hemoglobin and as such improved diabetes control (39). Interestingly, in the 11 studies in adults, no beneficial effect of stress management could be observed on T1D (39).
Consequences of Stress on Intestinal Barrier and Systemic Immune Response
Stress can affect various physiological processes. Already Selye observed and others confirmed that the gastrointestinal tract and the immune system are particularly responsive to stress no matter the origin of the stress (1).
Actors of Intestinal Barrier and Function
Intestinal epithelium is the mammalian organism's biggest surface in contact with the environment. Therefore, intestinal barrier functions are highly diverse and well developed. The intestinal barrier has to fulfill conflicting functions. Indeed, the intestinal barrier allows the transport of nutrient but at the same time filters and defends the organism from harmful luminal content (pathogens, toxins, etc.). Among the main actors of the intestinal barrier we can quote, intestinal microbiota, intestinal epithelium, and immune response (innate and adaptive). All those actors are in close relationship and regulate one another [for review, see (40)]. Intestinal microbiota not only participates in the protection against pathogens colonization but also contributes to maturation of intestinal epithelium and immune system and provides various nutritional compounds (41). The intestinal epithelium is formed by distinct cell types distributed along the crypt–villus axis. Although they all derive from a common stem cell progenitor located in the crypts, their morphology and roles differ [for review, see (42)]. The intestinal epithelium is renewed every 5 days, and this constant renewing confers high plasticity and protection to the intestinal barrier because defective cells are removed rapidly (43). Intestinal permeability is the ability of intestinal epithelium to allow the selective entrance of luminal antigens into the organism (44). Another actor of the intestinal barrier is the intestinal immune system. Gut-associated lymphoid tissue (GALT) represents the inductive site for B and T cells of mucosal intestinal barrier and includes the Peyer patches (PPs), the appendix, and isolated lymphoid follicles (ILFs). The humoral response in the intestines can be divided into four stages: predominant IgA induction in mucosal B cells, recirculation of IgA plasma blasts and homing into the intestinal mucosa, terminal B cell differentiation to plasma cells with local IgA production, and export of IgA through the intestinal epithelial layer [for review, see (45)]. Most intestinal T cells mature in peripheral lymphoid organs where they acquire the expression of intestinal homing receptors to migrate to the effector site of the intestines, that is, the mucosal epithelia and the lamina propria. Intestinal lymphocytes are continuously exposed to food and microbial antigens. These lymphocytes help to maintain the integrity of the intestinal barrier and immune homeostasis. Owing to their close location to luminal antigens, they have dual functions: regulatory functions (i.e., maintaining tolerance toward food antigens and commensal microbiota) and effector functions (i.e., prevention of pathogenic invasion) [for review, see (46)]. Innate lymphoid cells (ILCs) are lymphocytes that do not express the type of diversified antigen receptors expressed on T cells and B cells. ILCs are largely tissue-resident cells participating in tissue homeostasis [for review, see (47)]. A defective intestinal barrier will lead to inappropriate intestinal but also systemic immune response leading to gastrointestinal disorders and to extra-intestinal diseases like autoimmune diseases (48–50).
Intestinal barrier homeostasis is highly regulated, and a defect in microbiota composition could lead to intestinal hyperpermeability and intestinal inflammation. Intestinal inflammation not only will contribute to intestinal hyperpermeability (51) but also will favor microbiota colonization by pathobionts (52). Microbiota, intestinal permeability, and immune response mutually regulate one another making it difficult to define their respective role as cause or consequence in complex established pathologies.
Psychological Stress Impairs Intestinal Barrier
Stress plays a role in the course of gastrointestinal disorders like irritable bowel syndrome (IBS) (53–55) and inflammatory bowel disease (IBD) (56). IBS is a very interesting model to study the consequences of stress on the intestinal barrier. Indeed, the occurrence of stressful events is considered as a contributing factor triggering and/or maintaining IBS (57, 58), suggesting that dysfunctional interactions in the brain–gut axis contribute to the pathophysiology of the disease (59) and as such justifying its new classification as a disorder of the brain–gut interaction (60). In this review, we will focus on the consequences of psychological stress on the intestinal barrier and its consequences on systemic immune response (Figure 1).
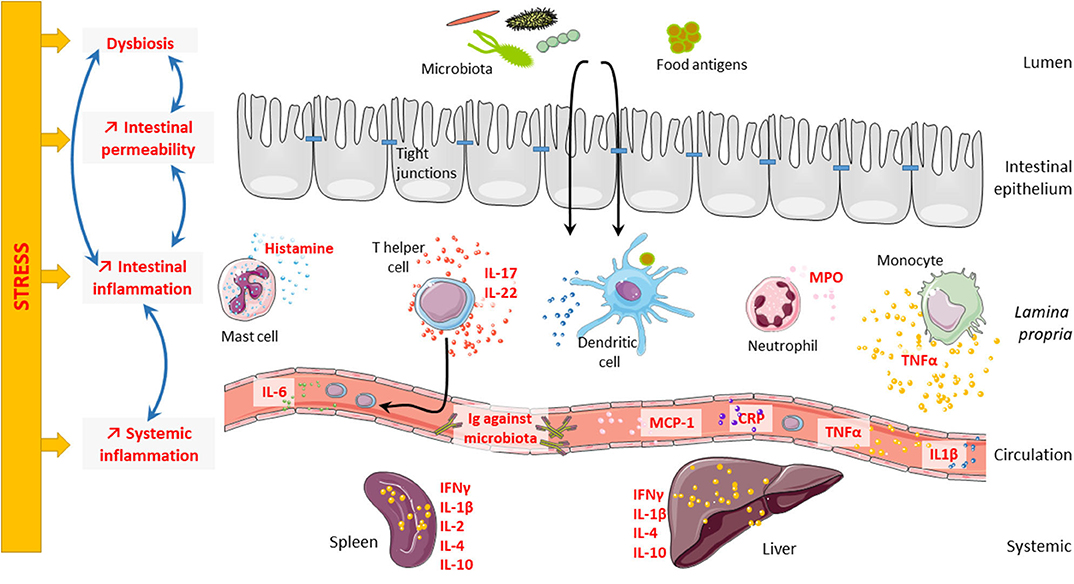
Figure 1. Consequences of stress on intestinal barrier and systemic inflammation. Psychological stress can impair intestinal barrier at different levels. Indeed, stress can lead to microbiota dysbiosis, intestinal hyperpermeability, and intestinal inflammation. Interestingly, all these elements are highly connected and regulate one another. Microbiota dysbiosis can trigger intestinal hyperpermeability and intestinal inflammation; and in contrast, both intestinal hyperpermeability and intestinal inflammation can induce microbiota dysbiosis. Finally, stress can also induce systemic inflammation that might be related to intestinal inflammation.
Microbiota Dysbiosis
Stress is modifying microbiota in animal and human. Neonatal maternal separation induces microbiota dysbiosis in mice at different ages (61, 62). Limited nesting stress alters microbiota in rat pups (63). In adults, chronic water avoidance stress increases susceptibility to indomethacin-induced hyperpermeability in mice, and the effect is transferable via fecal microbiota transfer (64). Germ-free (GF) mice have exaggerated HPA stress response after restraint stress (65), showing the role of microbiota in the regulation of stress response. Mice exposed to social disruption stress have increased circulating IL-6 and MCP-1 levels; these effects were totally abolished by antibiotic treatment showing the importance of microbiota in the induction of stress effects (66).
In humans, decreased total abundance of Actinobacteria, Lentisphaerae, and Verrucomicrobia is associated with PTSD in South African individuals (67). Microbiota dysbiosis has been described in IBS patients [for review, see (68)].
Stress Is Associated With Intestinal Hyperpermeability
In preclinical models and epidemiological studies, stress has been associated with an increase of intestinal permeability. Chronic water avoidance stress increases intestinal permeability and decreases tight junction protein expression in colon of adult rat (69) and overall intestinal permeability in mice (70). Chronic neonatal maternal separation, a model of early life stress, also increases intestinal permeability in rat (63, 71, 72) and mice (73). Maternal separation applied just for one time (acute stress) increases intestinal permeability in rats (74). Combination of different stressors [subacute (isolation, limited movement) and chronic crowding stress] also decreases tight junction mRNA expression in rats (75). In a mouse model of social disruption, a social stressor, bacterial RNA (Lactobacillus spp.), is increased in spleen, which indicates bacterial translocation (76).
In human, acute psychological stress like public speaking has also been shown to induce intestinal hyperpermeability (77). Intestinal hyperpermeability has also been described in IBS patients (78). The stress hormones cortisol (human) and corticosterone (mice) have been shown to mediate stress increased intestinal hyperpermeability as administration of the GR agonist dexamethasone mimics the intestinal hyperpermeability (69, 74).
Stress Exacerbates Intestinal and Systemic Inflammation
Chronic neonatal maternal separation in rats increases cytokine expression, myeloperoxidase activity, and mast cell numbers in colonic tissue and exacerbate TNBS-induced colitis (71). Neonatal maternal separation in mice increases TNFα expression by intestinal tissue in young adult (61) and lipopolysaccharide (LPS)-stimulated TNFα secretion of isolated lamina propria immune cells in aging (62). Acute restraint stress augments histamine release by mast cells (79). Acute acoustic stress increases intestinal IL-17 and IL-22 expression in mice (80).
In human, stress aggravates IBD symptoms including higher release of pro-inflammatory effectors (56). In IBS, an increased state of activation of immune cells has been described even though this observation is under debate (81).
Not only the intestinal immune system is influenced by psychological stress, but there is also evidence for modified systemic immune response without direct proof that inflammatory immune cells were activated in the gastrointestinal tract. Neonatal maternal-deprived rats have increased cytokine expression in liver and spleen (71). Humoral immune response against microbiota is increased in neonatal maternal-deprived mice (62, 73). Social disruption stress in mice increases bacterial translocation and induces circulating IL-6 and MCP-1 (66, 76).
Stress is associated with an increase in pro-inflammatory response as described in PTSD patients (82). A meta-analysis of several studies showed that IL-6, TNFα, and IL-1β secretion are increased in response to acute stress in human (83). Childhood victimization is associated with elevated C-reactive protein (CRP) levels in young adult (84, 85).
Defect of Intestinal Barrier in Autoimmune Disorders (86)
We provided evidence that stress might play a role in onset or course of ADs, and we reviewed the well-documented deleterious role of stress in intestinal barrier functions. We will now summarize the data regarding the defect of the intestinal barrier in ADs. Indeed, the observed defect of the intestinal barrier in ADs is an interesting lead that largely contributes to the rise of the hypothesis, suggesting a contribution of stress-induced intestinal barrier defect in ADs (Figure 2).
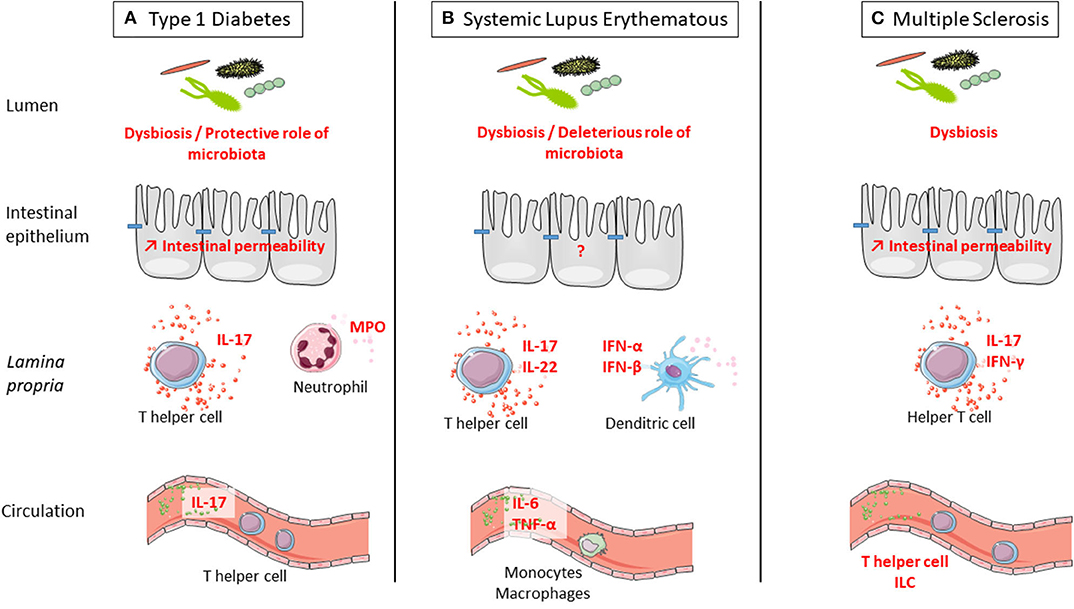
Figure 2. Defect of intestinal barrier and systemic immune response is observed in three examples of autoimmune disorders (ADs): type 1 diabetes (T1D), systemic lupus erythematosus (SLE), and multiple sclerosis (MS). (A) T1D is associated with microbiota dysbiosis, intestinal hyperpermeability, increased IL-17 secretion in the intestine and at systemic level, and increased myeloperoxidase (MPO) in the intestine. Interestingly, colonization by a complex microbiota is protective from type 1 diabetes. (B) SLE is associated with microbiota dysbiosis and increased secretion at the intestinal level of IL-17 and IL-22 by T cells and IFN-α and IFN-β by dendritic cells. At the systemic level, there is a higher secretion of IL-6 and TNF-α by monocytes and macrophages in SLE. Interestingly, colonization by a complex microbiota is deleterious for SLE onset. (C) MS is associated with microbiota dysbiosis, intestinal hyperpermeability, and increased secretion at the intestinal level of IL-17 and IFN-γ by T cells. At the systemic level, the number of innate lymphoid cell (ILC) population was observed in MS. Interestingly, colonization by a complex microbiota is deleterious for MS onset.
Microbiota in Autoimmune Disorders
Microbiota is known to contribute to intestinal mucosal permeability and induction of innate defenses and as such represent a risk factor for ADs (87, 88). A growing body of evidence suggests that intestinal microbiota can affect the incidence and/or severity of immune-mediated extra-intestinal diseases (89). Aberrant microbiota has been described in patients suffering from T1D (90), SLE (88), and MS (91). Knowledge regarding microbiota dysbiosis in AD patients and animal models will be summarized here, and interventional studies, which help to understand the role of microbiota in those diseases, will be discussed at the end of the paragraph.
Increased microbial diversity and low level of butyrate have been observed in feces of pediatric T1D patients (92, 93). In the BABYDIET cohort, early development of islet auto-antibodies is associated with alteration in the composition of mucin-degrading bacteria, that is, increase of Bacteroides and decrease of Akkermansia (94). A reduction of microbial diversity is more pronounced before the time of diabetes onset (95). Fecal transplantation of NOD diabetic microbiota in NOD-resistant mice induced insulitis, suggesting a diabetogenic gut microbial community (96, 97). Antibiotic treatment accelerates disease development (98, 99), suggesting a protective role of microbiota colonization in T1D.
Microbiota dysbiosis has been observed in relapsing–remitting MS patient compared with healthy control (100–103) with no consensus on the involvement of a particular bacterial species. In contrast, another study comparing 16S RNA profiles of feces from MS and healthy patients has not shown any differences (101). Demyelination initiates after colonization with feces of specific pathogen-free mice (104). Microbiota depletion by non-absorbable antibiotics delays the development of EAE by reducing the number of mesenteric Th17 cells (105). GF mice present attenuated symptoms in both spontaneous and induced EAE models (104, 106), suggesting a deleterious role of microbiota colonization in MS. Furthermore, microbiota shapes and predicts the course (chronic-progressive or relapsing–remitting) of EAE in a mouse model (107).
Only a few studies on human SLE microbiome in small cohorts report microbial dysbiosis (108–110), but they are confirmed by preclinical studies in mouse models (110). A study performed in a larger and diversified cohort of SLE patient showed that the severity of disease is associated with more severe microbiota dysbiosis (111).
Interventional Studies: What Do They Tell Us?
Here, we will focus on direct supplementation by living bacteria like probiotic and fecal microbiota transplantation (FMT) treatment but not on indirect interventions like prebiotics or nutritional compounds produced by bacteria, as short chain fatty acids, for example, which may involve indirect effects. Once ADs are diagnosed, the production of antibody against self-antigen will remain and will still damage tissues, but this process could be delayed or reduced. Probiotics are living microorganisms that confer a health benefit to the host (112). Probiotics are known to have beneficial effects on the intestinal barrier (113, 114) and anti-inflammatory properties (115–119) and as such represent an interesting tool to delay, reduce, or even prevent ADs.
Animal studies suggest beneficial effects of probiotics supplementation on EAE via a stimulation of IL-10 production (106, 120–124). Clinical studies showed that a mixture of probiotics improves expanded disability status score and decreased inflammatory markers (125). Regarding T1D, probiotic treatment delays the onset of T1D in an experimental rat model and improves the intestinal barrier (126). Probiotics also protect NOD mice from T1D by reducing intestinal inflammation (127). In humans, it has been demonstrated in the TEDDY (The Environmental Determinants of Diabetes in the Young) cohort that early probiotic supplementation is associated with a decreased risk of islet autoimmunity as compared with late or missing supplementation (128). In animal models for lupus nephritis, probiotic administration lowers inflammatory response in the kidney and intestines in female and castrated males but not in non-castrated males (129).
FMT with microbiota from different diabetes resistant mouse strains delays the onset of T1D in NOD diabetes-prone mice (97). Few studies investigate to role of FMT on AD symptoms, and most of the time the recommendation for FMT treatment was to target associated gastrointestinal troubles. Neurological symptoms are improved and MS progression is paused in three MS patients who underwent FMT treatment for chronic constipation (130). Unfortunately, no data are available on intestinal barrier functions of probiotics and FMT treatments in parallel to beneficial effects on ADs.
Mimicking
Antigens from infectious agents and myelin proteins can share structural similarities called molecular mimicry. This molecular mimicry can be responsible for activation of naïve autoreactive T cells recognizing peptides from infectious agents but also from self-antigens myelin proteins. Cross-reactivity could occur when important motifs are conserved and overall structures of TCR–peptide–MHC interaction are similar, suggesting that cross-reactivity may happen frequently (131). Myelin basic protein, the immunodominant autoantigen of MS, cross react with Epstein–Barr virus (EBV), influenza A virus, herpes simplex virus, human papilloma virus (132), or human herpesvirus-6 (133). Regarding EBV, MS patients seem to have increased antibody titers against certain antigens of the virus than have control subjects even before the onset of MS (134). Despite cross-reactivity, infectious agents can impair self-antigen tolerance by indirect activation (135). It has been showed that an integrase expressed by intestinal Bacteroides encodes a low-avidity mimotope of the pancreatic β-cell autoantigens and as such might participate to T1D onset. Colonization of GF mice with Bacteroides promotes the recruitment of diabetogenic CD8+ T cells to the gut (136).
Intestinal Hyperpermeability in Autoimmune Disorders
In ADs, intestinal hyperpermeability has been described, resulting in an increased entry of luminal antigens derived from food and/or intestinal microbiota or pathogens. The associated inflammation has been suggested to participate in AD onset and/or exacerbation.
Even though it is still unclear whether intestinal hyperpermeability is a trigger or a consequence of T1D progression (93, 137, 138), epidemiological and preclinical studies demonstrated that intestinal hyperpermeability occurs before disease onset (139, 140). Reversion of intestinal hyperpermeability by treatment with a zonulin 1 (intestinal homolog of a Vibrio cholerae enterotoxin, which reversibly increases intestinal permeability) inhibitor ameliorates T1D manifestation in rat model (141). Microbial translocation in pancreatic lymph nodes activates NOD2, and IL-17 production in pancreatic lymph nodes and pancreas which contributes to T1D development (142).
Intestinal hyperpermeability precedes EAE onset and increases while disease progresses (143). In this model, increased intestinal permeability is associated with the increase of crypt depth and mucosa thickness in jejunum and ileum, as well as with an overexpression of zonulin 1 (143) as observed for T1D (141, 144).
Intestinal barrier defect and subsequent exposure to microbial products play an important role in the pathology of SLE (145, 146). sCD14, lysozyme, and CXCL16 are markers of antimicrobial response found increased in SLE subject attesting to a defect of the intestinal barrier (147).
Intestinal Inflammation
T1D is associated with increased intestinal myeloperoxidase activity and goblet cell (producing mucus) density, supporting the idea that early intestinal inflammation might lead to intestinal hyperpermeability (148, 149). Many studies suggest that the increased number of Th17 cells is involved in the pathogenesis of autoimmune diabetes. Higher numbers of IL-17 secreting cells are detected in recent-onset T1D-promoting inflammatory response to β-cells (150, 151). Th17 is increased in the peripheral blood of children with T1D (151, 152). In vitro IL-17 potentiates inflammatory and proapoptotic responses on human islets cells (151). Anti-IL-17 treatment reduces islet T cell infiltrates and GAD65 autoantibodies in NOD mice (153). Neutrophil extracellular traps (NETs) might contribute to the generation of ADs by exposing autoantigen (154). The role of NET has been studied particularly in T1D. Indeed, degradation of NETs in the gut prevents immune infiltration of pancreatic islet preserving β-cell mass and systemic inflammation (155).
In a mouse model of SLE developing severe nephritis, α4β7 expressing T cells is increased in PPs and pro-inflammatory cytokines (IL-17, IL-22, IFNα, and β) are much more expressed in distal ileum (156). Furthermore, intestinal monocytes/macrophages of SLE patients have an altered expression of type 1 interferon-stimulated genes, HLA-DR, and Fcγ receptors (157, 158). Monocytes isolated from plasma of SLE patients release higher pro-inflammatory cytokines in response to LPS than do healthy patients (159). More generally, higher production of pro-inflammatory cytokines by monocytes/macrophages has been described in SLE patients [for review, see (160)].
In MS, elevated Th1 and Th17 pro-inflammatory responses are observed in lamina propria, PPs, and mesenteric lymph nodes (143). GF EAE animals produce lower levels of IFNγ and IL-17 in the intestines associated with a higher number of Treg cells (104). Monocolonization of GF animals with segmented filamentous bacteria, IL-17 inducer in gut (161, 162), induces EAE and shows that microbiota can affect neurologic inflammation by recirculation of Th17 to the brain, causing inflammation (106). Autoreactive T cells from gut could migrate in different organs depending on pathologies, to brain in the case of MS, to liver in the case of autoimmune cholestatic liver disease (163, 164), or to the kidney in the case of SLE (165). Interestingly, not only T cells seem to be involved in MS but also circulating ILC. Indeed, a higher number of ILC have been observed in MS patients (166).
Conclusion
As a conclusion, compiling evidences highlight the importance of both intestinal barrier defect and stress in ADs. Stress is well known to have long-lasting deleterious consequences on the intestinal barrier. A transversal research on ADs, stress, and intestinal barrier function would be of great interest and would bring new understanding in the pathophysiology of ADs. Identifying stress-induced intestinal barrier dysfunction as an actor of ADs could bring new possibilities for therapeutic targets and especially preventing strategies toward the spreading epidemic of ADs. Therapeutic strategies suggest that probiotics and FMT treatment might improve AD symptom, but preventive strategies in an at-risk population still need to be explored. In this review, we did not mention autoimmune thyroid diseases (AITDs) that are the most frequent ADs (167). It is difficult to study AITDs by themselves, as they are often observed together with other ADs, which are named polyautoimmunity (168). Then, even though there are sufficient data supporting the role of stress in AITD onset (20), evidences for a defect of intestinal barrier functions in AITD are sparse, and only two studies are available regarding microbiota dysbiosis (169, 170). For those reasons, we did not use AITD to illustrate the hypothesis of this review, supporting a role of stress-induced intestinal barrier disruption in the onset and/or the course of ADs. However, we wanted to mention the case of AITD as data on intestinal barrier function would be of great interest in the future.
Author Contributions
HI-D and SM reviewed the literature, wrote and corrected the manuscript, and drew the figures. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Selye H. Syndrome produced by diverse nocuous agents. Nature. (1936) 138:32. doi: 10.1038/138032a0
2. Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. (2005) 62:593. doi: 10.1001/archpsyc.62.6.593
3. Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci. (2006) 8:383–95. Avialable online at : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3181830/
4. Charmandari E, Tsigos C, Chrousos G. Endocrinology of the stress response. Annu Rev Physiol. (2005) 67:259–84. doi: 10.1146/annurev.physiol.67.040403.120816
5. Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. (2017) 17:233–47. doi: 10.1038/nri.2017.1
6. Miyata M, Lee J-Y, Susuki-Miyata S, Wang WY, Xu H, Kai H, et al. Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK-M. Nat Commun. (2015) 6:6062. doi: 10.1038/ncomms7062
7. Beaulieu E, Morand EF. Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat Rev Rheumatol. (2011) 7:340–8. doi: 10.1038/nrrheum.2011.59
8. Atsuta J, Plitt J, Bochner BS, Schleimer RP. Inhibition of VCAM-1 expression in human bronchial epithelial cells by glucocorticoids. Am J Respir Cell Mol Biol. (1999) 20:643–50. doi: 10.1165/ajrcmb.20.4.3265
9. Mukaida N, Morita M, Ishikawa Y, Rice N, Okamoto SI, Kasahara T, et al. Novel mechanism of glucocorticoid-mediated gene repression. Nuclear factor-κB is target for glucocorticoid-mediated interleukin 8 gene repression. J Biol Chem. (1994) 269:13289–95.
10. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. (2008) 13:453–61. doi: 10.2741/2692
11. Gillis S, Crabtree GR, Smith KA. Glucocorticoid-induced inhibition of T cell growth factor production. II. The effect on the in vitro generation of cytolytic T cells. J Immunol. (1979) 123:1632–8.
12. Gillis S, Crabtree GR, Smith KA. Glucocorticoid-induced inhibition of T cell growth factor production. I. The effect on mitogen-induced lymphocyte proliferation. J Immunol. (1979) 123:1624–31.
13. Elenkov IJ. Glucocorticoids and the Th1/Th2 Balance. Ann N Y Acad Sci. (2004) 1024:138–46. doi: 10.1196/annals.1321.010
14. Liberman AC, Refojo D, Druker J, Toscano M, Rein T, Holsboer F, et al. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein-protein interaction. FASEB J. (2007) 21:1177–88. doi: 10.1096/fj.06-7452com
15. Frank MG, Miguel ZD, Watkins LR, Maier SF. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun. (2010) 24:19–30. doi: 10.1016/j.bbi.2009.07.008
16. Lim H-Y, Müller N, Herold MJ, van den Brandt J, Reichardt HM. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology. (2007) 122:47–53. doi: 10.1111/j.1365-2567.2007.02611.x
17. Perroud N, Paoloni-Giacobino A, Prada P, Olié E, Salzmann A, Nicastro R, et al. Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: a link with the severity and type of trauma. Transl Psychiatry. (2011) 1:e59. doi: 10.1038/tp.2011.60
18. Melas PA, Wei Y, Wong CCY, Sjöholm LK, Åberg E, Mill J, Schalling M, et al. Genetic and epigenetic associations of MAOA and NR3C1 with depression and childhood adversities. Int J Neuropsychopharmacol. (2013) 16:1513–28. doi: 10.1017/S1461145713000102
19. McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonté B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. (2009) 12:342–8. doi: 10.1038/nn.2270
20. Sharif K, Watad A, Coplan L, Lichtbroun B, Krosser A, Lichtbroun M, et al. The role of stress in the mosaic of autoimmunity: an overlooked association. Autoimmun Rev. (2018) 17:967–83. doi: 10.1016/j.autrev.2018.04.005
21. Stojanovich L, Marisavljevich D. Stress as a trigger of autoimmune disease. Autoimmun Rev. (2008) 7:209–13. doi: 10.1016/j.autrev.2007.11.007
22. Shepshelovich D, Shoenfeld Y. Prediction and prevention of autoimmune diseases: additional aspects of the mosaic of autoimmunity. Lupus. (2006) 15:183–90. doi: 10.1191/0961203306lu2274rr
23. Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B. Immunity and behavior: antibodies alter emotion. Proc Natl Acad Sci USA. (2006) 103:678–83. doi: 10.1073/pnas.0510055103
24. Wang Z, Xie Z, Lu Q, Chang C, Zhou Z. Beyond genetics: what causes type 1 diabetes. Clin Rev Allergy Immunol. (2017) 52:273–86. doi: 10.1007/s12016-016-8592-1
25. Teruel M, Alarcón-Riquelme ME. The genetic basis of systemic lupus erythematosus: what are the risk factors and what have we learned. J Autoimmun. (2016) 74:161–75. doi: 10.1016/j.jaut.2016.08.001
26. Giannelou M, Mavragani CP. Cardiovascular disease in systemic lupus erythematosus: a comprehensive update. J Autoimmun. (2017) 82:1–12. doi: 10.1016/j.jaut.2017.05.008
27. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. (2009) 9:393–407. doi: 10.1038/nri2550
28. Selmi C, Barin JG, Rose NR. Current trends in autoimmunity and the nervous system. J Autoimmun. (2016) 75:20–9. doi: 10.1016/j.jaut.2016.08.005
29. Willis T. Pharmaceutice Rationalis: or, an Exercitation of the Operations of Medicines in Humane Bodies. Shewing the Signs, Causes, and Cures of Most Distempers Incident Thereunto: In Two Parts As Also a Treatise of the Scurvy and the Several Sorts Thereof, With Their Symptoms, Causes and Cure…. London: Printed for T. Dring, C. Harper, and J. Leigh, and are to be sold by R. Clavell (1679). p. 1621–75. Available online at: https://quod.lib.umich.edu/e/eebo2/A66509.0001.001?view=toc.
30. Hägglöf B, Blom L, Dahlquist G, Lönnberg G, Sahlin B. The Swedish childhood diabetes study: indications of severe psychological stress as a risk factor for type 1 (insulin-dependent) diabetes mellitus in childhood. Diabetologia. (1991) 34:579–83. doi: 10.1007/BF00400277
31. Thernlund GM, Dahlquist G, Hansson K, Ivarsson SA, Ludvigsson J, Sjöblad S, et al. Psychological stress and the onset of IDDM in children. Diabetes Care. (1995) 18:1323–9. doi: 10.2337/diacare.18.10.1323
32. Portha B, Chavey A, Movassat J. Early-life origins of type 2 diabetes: fetal programming of the beta-cell mass. Exp Diabetes Res. (2011) 2011:1–16. doi: 10.1155/2011/105076
33. Roberts AL, Malspeis S, Kubzansky LD, Feldman CH, Chang S-C, Koenen KC, et al. Association of trauma and posttraumatic stress disorder with incident systemic lupus erythematosus in a longitudinal cohort of women. Arthritis Rheumatol. (2017) 69:2162–69. doi: 10.1002/art.40222
34. Mills SD, Azizoddin D, Racaza GZ, Wallace DJ, Weisman MH, Nicassio PM. The psychometric properties of the perceived stress scale-10 among patients with systemic lupus erythematosus. Lupus. (2017) 26:1218–23. doi: 10.1177/0961203317701844
35. Grant I, Brown GW, Harris T, McDonald WI, Patterson T, Trimble MR. Severely threatening events and marked life difficulties preceding onset or exacerbation of multiple sclerosis. J Neurol Neurosurg Psychiatry. (1989) 52:8–13. doi: 10.1136/jnnp.52.1.8
36. Warren S, Greenhill S, Warren KG. Emotional stress and the development of multiple sclerosis: case-control evidence of a relationship. J Chronic Dis. (1982) 35:821–31. doi: 10.1016/0021-9681(82)90047-9
37. Mitsonis CI, Zervas IM, Mitropoulos PA, Dimopoulos NP, Soldatos CR, Potagas CM, et al. The impact of stressful life events on risk of relapse in women with multiple sclerosis: a prospective study. Eur Psychiatry. (2008) 23:497–504. doi: 10.1016/j.eurpsy.2008.06.003
38. Bibolini MJ, Chanaday NL, Báez NS, Degano AL, Monferran CG, Roth GA. Inhibitory role of diazepam on autoimmune inflammation in rats with experimental autoimmune encephalomyelitis. Neuroscience. (2011) 199:421–8. doi: 10.1016/j.neuroscience.2011.08.076
39. Winkley K, Ismail K, Landau S, Eisler I. Psychological interventions to improve glycaemic control in patients with type 1 diabetes: systematic review and meta-analysis of randomised controlled trials. BMJ. (2006) 333:65. doi: 10.1136/bmj.38874.652569.55
40. Kurashima Y, Kiyono H. Mucosal ecological network of epithelium and immune cells for gut homeostasis and tissue healing. Annu Rev Immunol. (2016) 35:119–47. doi: 10.1146/annurev-immunol-051116-052424
41. Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Reddy DN. Role of the normal gut microbiota. World J Gastroenterol. (2015) 21:8787. doi: 10.3748/wjg.v21.i29.8787
42. Gehart H, Clevers H. Tales from the crypt: new insights into intestinal stem cells. Nat Rev Gastroenterol Hepatol. (2019) 16:19–34. doi: 10.1038/s41575-018-0081-y
43. Gordon JI, Hermiston ML. Differentiation and self-renewal in the mouse gastrointestinal epithelium. Curr Opin Cell Biol. (1994) 6:795–803. doi: 10.1016/0955-0674(94)90047-7
44. Ménard S, Cerf-Bensussan N, Heyman M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol. (2010) 3:247–59. doi: 10.1038/mi.2010.5
45. Mora JR, von Andrian UH. Differentiation and homing of IgA-secreting cells. Mucosal Immunol. (2008) 1:96–109. doi: 10.1038/mi.2007.14
46. Ma H, Tao W, Zhu S. T lymphocytes in the intestinal mucosa: defense and tolerance. Cell Mol Immunol. (2019) 16:216–24. doi: 10.1038/s41423-019-0208-2
47. Rankin L, Joanna Groom, Mielke LA, Seillet C, Belz GT. Diversity, function, and transcriptional regulation of gut innate lymphocytes. Front Immunol. (2013) 4:22. doi: 10.3389/fimmu.2013.00022
48. Annibali V, Policano C, Buscarinu MC, Lionetto L, Mechelli R, Capi M, et al. Intestinal permeability in multiple sclerosis. J Neuroimmunol. (2014) 275:54. doi: 10.1016/j.jneuroim.2014.08.143
49. Secher T, Kassem S, Benamar M, Bernard I, Boury M, Barreau F, et al. Oral administration of the probiotic strain Escherichia coli nissle 1917 reduces susceptibility to neuroinflammation and repairs experimental autoimmune encephalomyelitis-induced intestinal barrier dysfunction. Front Immunol. (2017) 8:1–10. doi: 10.3389/fimmu.2017.01096
50. Li X, Atkinson MA. The role for gut permeability in the pathogenesis of type 1 diabetes - a solid or leaky concept? Pediatr Diabetes. (2015) 16:485–92. doi: 10.1111/pedi.12305
51. Gitter AH, Bendfeldt K, Schulzke J-D, Fromm M. Leaks in the epithelial barrier caused by spontaneous and TNF-α-induced single-cell apoptosis. FASEB J. (2000) 14:1749–53. doi: 10.1096/fj.99-0898com
52. Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, et al. Host-derived nitrate boosts growth of E. coli in the Inflamed Gut. Science (80-). (2013) 339:708–11. doi: 10.1126/science.1232467
53. Lowman BC, Drossman DA, Cramer EM, McKee DC, DC McKee. Recollection of childhood events in adults with irritable bowel syndrome. J Clin Gastroenterol. (1987) 9:324–30. doi: 10.1097/00004836-198706000-00017
54. Videlock EJ, Adeyemo M, Licudine A, Hirano M, Ohning G, Mayer M, et al. Childhood trauma is associated with hypothalamic-pituitary-adrenal axis responsiveness in irritable bowel syndrome. Gastroenterology. (2009) 137:1954–62. doi: 10.1053/j.gastro.2009.08.058
55. Hislop IG. Childhood deprivation: an antecedent of the irritable bowel syndrome. Med J Aust. (1979) 1:372–4. doi: 10.5694/j.1326-5377.1979.tb126963.x
56. Sgambato D, Miranda A, Ranaldo R, Federico A, Romano M. The role of stress in inflammatory bowel diseases. Curr Pharm Des. (2017) 23:3997–4002. doi: 10.2174/1381612823666170228123357
57. Mayer EA, Naliboff BD, Chang L, Coutinho SV. V. Stress and irritable bowel syndrome. Am J Physiol Liver Physiol. (2001) 280:G519–24. doi: 10.1152/ajpgi.2001.280.4.G519
58. Wood JD. Visceral pain: spinal afferents, enteric mast cells, enteric nervous system and stress. Curr Pharm Des. (2011) 17:1573–5. doi: 10.2174/138161211796196918
59. Bonaz BL, Bernstein CN. Brain-gut interactions in inflammatory bowel disease. Gastroenterology. (2013) 144:36–49. doi: 10.1053/j.gastro.2012.10.003
60. Drossman DA. Functional gastrointestinal disorders: history, pathophysiology, clinical features, and Rome IV. Gastroenterology. (2016) 150:1262–79.e2. doi: 10.1053/j.gastro.2016.02.032
61. Riba A, Olier M, Lacroix-Lamandé S, Lencina C, Bacquié V, Harkat C, et al. Paneth cell defects induce microbiota dysbiosis in mice and promote visceral hypersensitivity. Gastroenterology. (2017) 153:1594–606.e2. doi: 10.1053/j.gastro.2017.08.044
62. Ilchmann-Diounou H, Olier M, Lencina C, Riba A, Barretto S, Nankap M, et al. Early life stress induces type 2 diabetes-like features in ageing mice. Brain Behav Immun. (2019) 80:452:63. doi: 10.1016/j.bbi.2019.04.025
63. Moussaoui N, Jacobs JP, Larauche M, Biraud M, Million M, Mayer E, et al. Chronic early-life stress in rat pups alters basal corticosterone, intestinal permeability, and fecal microbiota at weaning: influence of sex. J Neurogastroenterol Motil. (2017) 23:135–43. doi: 10.5056/jnm16105
64. Yoshikawa K, Kurihara C, Furuhashi H, Takajo T, Maruta K, Yasutake Y, et al. Psychological stress exacerbates NSAID-induced small bowel injury by inducing changes in intestinal microbiota and permeability via glucocorticoid receptor signaling. J Gastroenterol. (2017) 52:61–71. doi: 10.1007/s00535-016-1205-1
65. Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu X-N, et al. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol. (2004) 558:263–75. doi: 10.1113/jphysiol.2004.063388
66. Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun. (2011) 25:397–407. doi: 10.1016/j.bbi.2010.10.023
67. Hemmings SMJ, Malan-Müller S, van den Heuvel LL, Demmitt BA, Stanislawski MA, Smith DG, et al. The microbiome in posttraumatic stress disorder and trauma-exposed controls. Psychosom Med. (2017) 79:936–46. doi: 10.1097/PSY.0000000000000512
68. Collins SM. A role for the gut microbiota in IBS. Nat Rev Gastroenterol Hepatol. (2014) 11:497–505. doi: 10.1038/nrgastro.2014.40
69. Zheng G, Wu S-P, Hu Y, Smith DE, Wiley JW, Hong S. Corticosterone mediates stress-related increased intestinal permeability in a region-specific manner. Neurogastroenterol Motil. (2013) 25:e127–39. doi: 10.1111/nmo.12066
70. Cameron HL. Stress impairs murine intestinal barrier function: improvement by glucagon-like peptide-2. J Pharmacol Exp Ther. (2005) 314:214–20. doi: 10.1124/jpet.105.085373
71. Barreau F, Ferrier L, Fioramonti J, Bueno L. Neonatal maternal deprivation triggers long term alterations in colonic epithelial barrier and mucosal immunity in rats. Gut. (2004) 53:501–6. doi: 10.1136/gut.2003.024174
72. Øines E, Murison R, Mrdalj J, Grønli J, Milde AM. Neonatal maternal separation in male rats increases intestinal permeability and affects behavior after chronic social stress. Physiol Behav. (2012) 105:1058–66. doi: 10.1016/j.physbeh.2011.11.024
73. Riba A, Olier M, Lacroix-Lamandé S, Lencina C, Bacquié V, Harkat C, et al. Early life stress in mice is a suitable model for Irritable Bowel Syndrome but does not predispose to colitis nor increase susceptibility to enteric infections. Brain Behav Immun. (2018) 73:403–15. doi: 10.1016/j.bbi.2018.05.024
74. Moussaoui N, Braniste V, Ait-Belgnaoui A, Gabanou M, Sekkal S, Olier M, et al. Changes in intestinal glucocorticoid sensitivity in early life shape the risk of epithelial barrier defect in maternal-deprived rats. PLoS ONE. (2014) 9:1–9. doi: 10.1371/journal.pone.0088382
75. Lauffer A, Vanuytsel T, Vanormelingen C, Vanheel H, Salim Rasoel S, Tóth J, et al. Subacute stress and chronic stress interact to decrease intestinal barrier function in rats. Stress. (2016) 19:225–34. doi: 10.3109/10253890.2016.1154527
76. Lafuse WP, Gearinger R, Fisher S, Nealer C, Mackos AR, Bailey MT. Exposure to a social stressor induces translocation of commensal lactobacilli to the spleen and priming of the innate immune system. J Immunol. (2017) 198:2383–93. doi: 10.4049/jimmunol.1601269
77. Vanuytsel T, van Wanrooy S, Vanheel H, Vanormelingen C, Verschueren S, Houben E, et al. Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut. (2014) 63:1293–9. doi: 10.1136/gutjnl-2013-305690
78. Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke J-D, Serino M, et al. Intestinal permeability – a new target for disease prevention and therapy. BMC Gastroenterol. (2014) 14:189. doi: 10.1186/s12876-014-0189-7
79. Eutamene H, Theodorou V, Fioramonti J, Bueno L. Acute stress modulates the histamine content of mast cells in the gastrointestinal tract through interleukin-1 and corticotropin-releasing factor release in rats. J Physiol. (2003) 553:959–66. doi: 10.1113/jphysiol.2003.052274
80. Miranda S, Roux ME. Acoustic stress induces long term severe intestinal inflammation in the mouse. Toxicol Lett. (2017) 280:1–9. doi: 10.1016/j.toxlet.2017.07.898
81. Barbara G, Cremon C, Carini G, Bellacosa L, Zecchi L, Giorgio R De, et al. The immune system in Irritable Bowel Syndrome. J Neurogastroenterol Motil. (2011) 17:349–59. doi: 10.5056/jnm.2011.17.4.349
82. de Oliveira JF, Wiener CD, Jansen K, Portela LV, Lara DR, Souza LD, de M, et al. Serum levels of interleukins IL-6 and IL-10 in individuals with posttraumatic stress disorder in a population-based sample. Psychiatry Res. (2018) 260:111–5. doi: 10.1016/j.psychres.2017.11.061
83. Marsland AL, Walsh C, Lockwood K, John-Henderson NA. The effects of acute psychological stress on circulating and stimulated inflammatory markers: a systematic review and meta-analysis. Brain Behav Immun. (2017) 64:208–19. doi: 10.1016/j.bbi.2017.01.011
84. Baldwin JR, Arseneault L, Caspi A, Fisher HL, Moffitt TE, Odgers CL, et al. Childhood victimization and inflammation in young adulthood: a genetically sensitive cohort study. Brain Behav Immun. (2017) 67:211–7. doi: 10.1016/j.bbi.2017.08.025
85. Slopen N, Loucks EB, Appleton AA, Kawachi I, Kubzansky LD, Non AL, et al. Early origins of inflammation: an examination of prenatal and childhood social adversity in a prospective cohort study. Psychoneuroendocrinology. (2015) 51:403–13. doi: 10.1016/j.psyneuen.2014.10.016
86. Rizzetto L, Fava F, Tuohy KM, Selmi C. Connecting the immune system, systemic chronic inflammation and the gut microbiome: the role of sex. J Autoimmun. (2018) 92:12–34. doi: 10.1016/j.jaut.2018.05.008
87. Yurkovetskiy LA, Pickard JM, Chervonsky A V. Microbiota and autoimmunity: exploring new avenues. Cell Host Microbe. (2015) 17:548–52. doi: 10.1016/j.chom.2015.04.010
88. Rosser EC, Mauri C. A clinical update on the significance of the gut microbiota in systemic autoimmunity. J Autoimmun. (2016) 74:85–93. doi: 10.1016/j.jaut.2016.06.009
89. Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Heal Dis. (2015) 26:26191. doi: 10.3402/mehd.v26.26191
90. Gianchecchi E, Fierabracci A. On the pathogenesis of insulin-dependent diabetes mellitus: the role of microbiota. Immunol Res. (2017) 65:242–56. doi: 10.1007/s12026-016-8832-8
91. Ochoa-Repáraz J, Magori K, Kasper LH. The chicken or the egg dilemma: intestinal dysbiosis in multiple sclerosis. Ann Transl Med. (2017) 5:145. doi: 10.21037/atm.2017.01.18
92. de Goffau MC, Fuentes S, van den Bogert B, Honkanen H, de Vos WM, Welling GW, et al. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia. (2014) 57:1569–77. doi: 10.1007/s00125-014-3274-0
93. Harbison JE, Roth-Schulze AJ, Giles LC, Tran CD, Ngui KM, Penno MA, et al. Gut microbiome dysbiosis and increased intestinal permeability in children with islet autoimmunity and type 1 diabetes : a prospective cohort study. Pediatr Diabetes. (2019) 20:574–83. doi: 10.1111/pedi.12865
94. Endesfelder D, Engel M, Davis-Richardson AG, Ardissone AN, Achenbach P, Hummel S, et al. Towards a functional hypothesis relating anti-islet cell autoimmunity to the dietary impact on microbial communities and butyrate production. Microbiome. (2016) 4:17. doi: 10.1186/s40168-016-0163-4
95. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyötyläinen T, Hämäläinen A-M, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe. (2015) 17:260–73. doi: 10.1016/j.chom.2015.01.001
96. Brown K, Godovannyi A, Ma C, Zhang Y, Ahmadi-Vand Z, Dai C, et al. Prolonged antibiotic treatment induces a diabetogenic intestinal microbiome that accelerates diabetes in NOD mice. ISME J. (2016) 10:321–32. doi: 10.1038/ismej.2015.114
97. Peng J, Narasimhan S, Marchesi JR, Benson A, Wong FS, Wen L. Long term effect of gut microbiota transfer on diabetes development. J Autoimmun. (2014) 53:85–94. doi: 10.1016/j.jaut.2014.03.005
98. Brugman S, Klatter FA, Visser JTJ, Wildeboer-Veloo ACM, Harmsen HJM, Rozing J, et al. Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia. (2006) 49:2105–8. doi: 10.1007/s00125-006-0334-0
99. Schwartz RF, Neu J, Schatz D, Atkinson MA, Wasserfall C, Comment on: Brugman S, et al. (2006) Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia 49:2105–2108. Diabetologia. (2006) 50:220–1. doi: 10.1007/s00125-006-0526-7
100. Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T, et al. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS ONE. (2015) 10:e0137429. doi: 10.1371/journal.pone.0137429
101. Tremlett H, Fadrosh DW, Faruqi AA, Hart J, Roalstad S, Graves J, et al. Associations between the gut microbiota and host immune markers in pediatric multiple sclerosis and controls. BMC Neurol. (2016) 16:182. doi: 10.1186/s12883-016-0703-3
102. Chen J, Chia N, Kalari KR, Yao JZ, Novotna M, Soldan MMP, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep. (2016) 6:28484. doi: 10.1038/srep28484
103. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun. (2016) 7:12015. doi: 10.1038/ncomms12015
104. Berer K, Mues M, Koutrolos M, Rasbi Z Al, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. (2011) 479:538–41. doi: 10.1038/nature10554
105. Yokote H, Miyake S, Croxford JL, Oki S, Mizusawa H, Yamamura T. NKT cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora. Am J Pathol. (2008) 173:1714–23. doi: 10.2353/ajpath.2008.080622
106. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2011) 108:4615–22. doi: 10.1073/pnas.1000082107
107. Gandy KAO, Zhang J, Nagarkatti P, Nagarkatti M. The role of gut microbiota in shaping the relapse-remitting and chronic-progressive forms of multiple sclerosis in mouse models. Sci Rep. (2019) 9:6923. doi: 10.1038/s41598-019-43356-7
108. He Z, Shao T, Li H, Xie Z, Wen C. Alterations of the gut microbiome in Chinese patients with systemic lupus erythematosus. Gut Pathog. (2016) 8:64. doi: 10.1186/s13099-016-0146-9
109. Hevia A, Milani C, López P, Cuervo A, Arboleya S, Duranti S, et al. Intestinal dysbiosis associated with systemic lupus erythematosus. MBio. (2014) 5:e01548–14. doi: 10.1128/mBio.01548-14
110. Luo XM, Edwards MR, Mu Q, Yu Y, Vieson MD, Reilly CM, et al. Gut microbiota in human systemic lupus erythematosus and a mouse model of lupus. Appl Environ Microbiol. (2018) 84:2288. doi: 10.1128/AEM.02288-17
111. Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis. (2019) 78:947–56. doi: 10.1136/annrheumdis-2018-214856
112. Johnson BM, Gaudreau M-C, Al-Gadban MM, Gudi R, Vasu C. Impact of dietary deviation on disease progression and gut microbiome composition in lupus-prone SNF 1 mice. Clin Exp Immunol. (2015) 181:323–37. doi: 10.1111/cei.12609
113. Abraham BP, Quigley EMM. Probiotics in inflammatory Bowel Disease. Gastroenterol Clin North Am. (2017) 46:769–82. doi: 10.1016/j.gtc.2017.08.003
114. Toumi R, Abdelouhab K, Rafa H, Soufli I, Raissi-Kerboua D, Djeraba Z, et al. Beneficial role of the probiotic mixture Ultrabiotique on maintaining the integrity of intestinal mucosal barrier in DSS-induced experimental colitis. Immunopharmacol Immunotoxicol. (2013) 35:403–9. doi: 10.3109/08923973.2013.790413
115. Riedel C-U, Foata F, Philippe D, Adolfsson O, Eikmanns B-J, Blum S. Anti-inflammatory effects of bifidobacteria by inhibition of LPS-induced NF-kappaB activation. World J Gastroenterol. (2006) 12:3729–35. doi: 10.3748/wjg.v12.i23.3729
116. O'Hara AM, Bhattacharyya A, Mifflin RC, Smith MF, Ryan KA, Scott KG-E, et al. Interleukin-8 induction by Helicobacter pylori in gastric epithelial cells is dependent on apurinic/apyrimidinic endonuclease-1/redox factor-1. J Immunol. (2006) 177:7990–9. doi: 10.4049/jimmunol.177.11.7990
117. Khokhlova EV, Smeianov VV, Efimov BA, Kafarskaia LI, Pavlova SI, Shkoporov AN. Anti-inflammatory properties of intestinal Bifidobacterium strains isolated from healthy infants. Microbiol Immunol. (2012) 56:27–39. doi: 10.1111/j.1348-0421.2011.00398.x
118. Roselli M, Finamore A, Britti MS, Mengheri E. Probiotic bacteria Bifidobacterium animalis MB5 and Lactobacillus rhamnosus GG protect intestinal Caco-2 cells from the inflammation-associated response induced by enterotoxigenic Escherichia coli K88. Br J Nutr. (2006) 95:1177–84. doi: 10.1079/BJN20051681
119. Toumi R, Soufli I, Rafa H, Belkhelfa M, Biad A, Touil-Boukoffa C. Probiotic bacteria lactobacillus and bifidobacterium attenuate inflammation in dextran sulfate sodium-induced experimental colitis in mice. Int J Immunopathol Pharmacol. (2014) 27:615–27. doi: 10.1177/039463201402700418
120. Lavasani S, Dzhambazov B, Nouri M, Fåk F, Buske S, Molin G, et al. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS ONE. (2010) 5:e9009. doi: 10.1371/journal.pone.0009009
121. Ezendam J, de Klerk A, Gremmer ER, van Loveren H. Effects of Bifidobacterium animalis administered during lactation on allergic and autoimmune responses in rodents. Clin Exp Immunol. (2008) 154:424–31. doi: 10.1111/j.1365-2249.2008.03788.x
122. Takata K, Kinoshita M, Okuno T, Moriya M, Kohda T, Honorat JA, et al. The lactic acid bacterium Pediococcus acidilactici suppresses autoimmune encephalomyelitis by inducing IL-10-producing regulatory T cells. PLoS ONE. (2011) 6:e27644. doi: 10.1371/journal.pone.0027644
123. Rezende RM, Oliveira RP, Medeiros SR, Gomes-Santos AC, Alves AC, Loli FG, et al. Hsp65-producing Lactococcus lactis prevents experimental autoimmune encephalomyelitis in mice by inducing CD4+LAP+ regulatory T cells. J Autoimmun. (2013) 40:45–57. doi: 10.1016/j.jaut.2012.07.012
124. Wang Y, Telesford KM, Ochoa-Repáraz J, Haque-Begum S, Christy M, Kasper EJ, et al. An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat Commun. (2014) 5:4432. doi: 10.1038/ncomms5432
125. Kouchaki E, Tamtaji OR, Salami M, Bahmani F, Daneshvar Kakhaki R, Akbari E. Clinical and metabolic response to probiotic supplementation in patients with multiple sclerosis: a randomized, double-blind, placebo-controlled trial. Clin Nutr. (2017) 36:1245–9. doi: 10.1016/j.clnu.2016.08.015
126. Valladares R, Sankar D, Li N, Williams E, Lai K-K, Abdelgeliel AS, et al. Lactobacillus johnsonii N6.2 mitigates the development of Type 1 diabetes in BB-DP rats. PLoS ONE. (2010) 5:e10507. doi: 10.1371/journal.pone.0010507
127. Dolpady J, Sorini C, Di Pietro C, Cosorich I, Ferrarese R, Saita D, et al. Oral probiotic VSL#3 prevents autoimmune diabetes by modulating microbiota and promoting indoleamine 2,3-dioxygenase-enriched tolerogenic intestinal environment. J Diabetes Res. (2016) 2016:7569431. doi: 10.1155/2016/7569431
128. Uusitalo U, Liu X, Yang J, Aronsson CA, Hummel S, Butterworth M, et al. Association of early exposure of probiotics and islet autoimmunity in the TEDDY Study. JAMA Pediatr. (2016) 170:20. doi: 10.1001/jamapediatrics.2015.2757
129. Mu Q, Zhang H, Liao X, Lin K, Liu H, Edwards MR, et al. Control of lupus nephritis by changes of gut microbiota. Microbiome. (2017) 5:73. doi: 10.1186/s40168-017-0300-8
130. Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol. (2012) 9:88–96. doi: 10.1038/nrgastro.2011.244
131. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell. (2014) 157:1073–87. doi: 10.1016/j.cell.2014.03.047
132. Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. (1995) 80:695–705. doi: 10.1016/0092-8674(95)90348-8
133. Tejada-Simon M V, Zang YCQ, Hong J, Rivera VM, Zhang JZ. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol. (2003) 53:189–97. doi: 10.1002/ana.10425
134. Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernán MA, Olek MJ, et al. Epstein-Barr virus antibodies and risk of multiple sclerosis: a prospective study. JAMA. (2001) 286:3083–8. doi: 10.1001/jama.286.24.3083
135. Haring JS, Pewe LL, Perlman S. Bystander CD8 T cell-mediated demyelination after viral infection of the central nervous system. J Immunol. (2002) 169:1550–5. doi: 10.4049/jimmunol.169.3.1550
136. Hebbandi Nanjundappa R, Ronchi F, Wang J, Clemente-Casares X, Yamanouchi J, Sokke Umeshappa C, et al. A gut microbial mimic that hijacks diabetogenic autoreactivity to suppress colitis. Cell. (2017) 171:655–67.e17. doi: 10.1016/j.cell.2017.09.022
137. Odenwald MA, Turner JR. Intestinal permeability defects: is it time to treat? Clin Gastroenterol Hepatol. (2013) 11:1075–83. doi: 10.1016/j.cgh.2013.07.001
138. Buckner JH, Greenbaum CJ. Stacking the deck: studies of patients with multiple autoimmune diseases propelled our understanding of type 1 diabetes as an autoimmune disease. J Immunol. (2017) 199:3011–13. doi: 10.4049/jimmunol.1701299
139. Paun A, Yau C, Danska JS. The influence of the microbiome on type 1 diabetes. J Immunol. (2017) 198:590–5. doi: 10.4049/jimmunol.1601519
140. Kuhn C, Besançon A, Lemoine S, You S, Marquet C, Candon S, et al. Regulatory mechanisms of immune tolerance in type 1 diabetes and their failures. J Autoimmun. (2016) 71:69–77. doi: 10.1016/j.jaut.2016.05.002
141. Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, et al. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci USA. (2005) 102:2916–21. doi: 10.1073/pnas.0500178102
142. de Goffau MC, Luopajarvi K, Knip M, Ilonen J, Ruohtula T, Harkonen T, et al. Fecal microbiota composition differs between children with -cell autoimmunity and those without. Diabetes. (2013) 62:1238–44. doi: 10.2337/db12-0526
143. Nouri M, Bredberg A, Weström B, Lavasani S. Intestinal barrier dysfunction develops at the onset of experimental autoimmune encephalomyelitis, and can be induced by adoptive transfer of auto-reactive T cells. PLoS ONE. (2014) 9:e106335. doi: 10.1371/journal.pone.0106335
144. Visser J, Rozing J, Sapone A, Lammers K, Fasano A. Tight junctions, intestinal permeability, and autoimmunity. Ann N Y Acad Sci. (2009) 1165:195–205. doi: 10.1111/j.1749-6632.2009.04037.x
145. Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science (80-). (2018) 359:1156–61. doi: 10.1126/science.aar7201
146. Mu Q, Tavella VJ, Kirby JL, Cecere TE, Chung M, Lee J, et al. Antibiotics ameliorate lupus-like symptoms in mice. Sci Rep. (2017) 7:13675. doi: 10.1038/s41598-017-14223-0
147. Ayyappan P, Harms RZ, Buckner JH, Sarvetnick NE. Coordinated induction of antimicrobial response factors in systemic lupus erythematosus. Front Immunol. (2019) 10:658. doi: 10.3389/fimmu.2019.00658
148. Neu J, Reverte CM, Mackey AD, Liboni K, Tuhacek-Tenace LM, Hatch M, et al. Changes in intestinal morphology and permeability in the biobreeding rat before the onset of type 1 diabetes. J Pediatr Gastroenterol Nutr. (2005) 40:589–95. doi: 10.1097/01.MPG.0000159636.19346.C1
149. Secondulfo M, Iafusco D, Carratù R, DeMagistris L, Sapone A, Generoso M, et al. Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac, type I diabetic patients. Dig Liver Dis. (2004) 36:35–45. doi: 10.1016/j.dld.2003.09.016
150. Marwaha AK, Crome SQ, Panagiotopoulos C, Berg KB, Qin H, Ouyang Q, et al. Cutting edge: increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol. (2010) 185:3814–8. doi: 10.4049/jimmunol.1001860
151. Honkanen J, Nieminen JK, Gao R, Luopajarvi K, Salo HM, Ilonen J, et al. IL-17 immunity in human type 1 diabetes. J Immunol. (2010) 185:1959–67. doi: 10.4049/jimmunol.1000788
152. Reinert-Hartwall L, Honkanen J, Salo HM, Nieminen JK, Luopajärvi K, Härkönen T, et al. Th1/Th17 plasticity is a marker of advanced β cell autoimmunity and impaired glucose tolerance in humans. J Immunol. (2015) 194:68–75. doi: 10.4049/jimmunol.1401653
153. Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, et al. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes. (2009) 58:1302–11. doi: 10.2337/db08-1113
154. Giaglis S, Hahn S, Hasler P. “The NET Outcome”: are neutrophil extracellular traps of any relevance to the pathophysiology of autoimmune disorders in childhood? Front Pediatr. (2016) 4:97. doi: 10.3389/fped.2016.00097
155. Liang Y, Wang X, He D, You Q, Zhang T, Dong W, et al. Ameliorating gut microenvironment through staphylococcal nuclease-mediated intestinal NETs degradation for prevention of type 1 diabetes in NOD mice. Life Sci. (2019) 221:301–10. doi: 10.1016/j.lfs.2019.02.034
156. Gaudreau M-C, Johnson BM, Gudi R, Al-Gadban MM, Vasu C. Gender bias in lupus: does immune response initiated in the gut mucosa have a role? Clin Exp Immunol. (2015) 180:393–407. doi: 10.1111/cei.12587
157. Hepburn AL, Mason JC, Davies KA. Expression of Fc and complement receptors on peripheral blood monocytes in systemic lupus erythematosus and rheumatoid arthritis. Rheumatology. (2004) 43:547–54. doi: 10.1093/rheumatology/keh112
158. Lee PY, Li Y, Kumagai Y, Xu Y, Weinstein JS, Kellner ES, et al. Type I interferon modulates monocyte recruitment and maturation in chronic inflammation. Am J Pathol. (2009) 175:2023–33. doi: 10.2353/ajpath.2009.090328
159. Jiang W, Zhang L, Lang R, Li Z, Gilkeson G. Sex differences in monocyte activation in systemic lupus erythematosus (SLE). PLoS ONE. (2014) 9:e114589. doi: 10.1371/journal.pone.0114589
160. Li Y, Lee PY, Reeves WH. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Arch Immunol Ther Exp. (2010) 58:355–64. doi: 10.1007/s00005-010-0093-y
161. Gaboriau-Routhiau V, Rakotobe S, Lécuyer E, Mulder I, Lan A, Bridonneau C, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. (2009) 31:677–89. doi: 10.1016/j.immuni.2009.08.020
162. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
163. Wu X, Tian Z. Gut-liver axis: gut microbiota in shaping hepatic innate immunity. Sci China Life Sci. (2017) 60:1191–1196. doi: 10.1007/s11427-017-9128-3
164. Ma H-D, Wang Y-H, Chang C, Gershwin ME, Lian Z-X. The intestinal microbiota and microenvironment in liver. Autoimmun Rev. (2015) 14:183–91. doi: 10.1016/j.autrev.2014.10.013
165. Okamoto A, Fujio K, Tsuno NH, Takahashi K, Yamamoto K. Kidney-infiltrating CD4+ T-cell clones promote nephritis in lupus-prone mice. Kidney Int. (2012) 82:969–79. doi: 10.1038/ki.2012.242
166. Perry JSA, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med. (2012) 4:145ra106. doi: 10.1126/scitranslmed.3004140
167. Weetman A, DeGroot LJ. Autoimmunity to the Thyroid Gland. (2000). Available online at: http://www.ncbi.nlm.nih.gov/pubmed/25905407 (accessed October 17, 2019).
168. Rojas-Villarraga A, Amaya-Amaya J, Rodriguez-Rodriguez A, Mantilla RD, Anaya J-M. Introducing polyautoimmunity: secondary autoimmune diseases no longer exist. Autoimmune Dis. (2012) 2012:1–9. doi: 10.1155/2012/254319
169. Zhou L, Li X, Ahmed A, Wu D, Liu L, Qiu J, et al. Gut microbe analysis between hyperthyroid and healthy individuals. Curr Microbiol. (2014) 69:675–80. doi: 10.1007/s00284-014-0640-6
Keywords: intestinal permeability, psychological stress, type 1 diabetes, multiple sclerosis, systemic lupus erythematosus, microbiota, immune response
Citation: Ilchmann-Diounou H and Menard S (2020) Psychological Stress, Intestinal Barrier Dysfunctions, and Autoimmune Disorders: An Overview. Front. Immunol. 11:1823. doi: 10.3389/fimmu.2020.01823
Received: 17 May 2019; Accepted: 07 July 2020;
Published: 25 August 2020.
Edited by:
Julien Diana, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Gianluigi Giannelli, National Institute of Gastroenterology S. de Bellis Research Hospital (IRCCS), ItalyFederica Facciotti, European Institute of Oncology (IEO), Italy
Copyright © 2020 Ilchmann-Diounou and Menard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandrine Menard, c2FuZHJpbmUubWVuYXJkQGlucmFlLmZy