 Rong Dong
Rong Dong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 28 July 2020
Sec. Vaccines and Molecular Therapeutics
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01784
This article is part of the Research TopicCoronavirus Disease (COVID-19): Pathophysiology, Epidemiology, Clinical Management and Public Health ResponseView all 400 articles
COVID-19 has recently become the most serious threat to public health, and its prevalence has been increasing at an alarming rate. The incubation period for the virus is ~1–14 days and all age groups may be susceptible to a fatality rate of about 5.9%. COVID-19 is caused by a novel single-stranded, positive (+) sense RNA beta coronavirus. The development of a vaccine for SARS-CoV-2 is an urgent need worldwide. Immunoinformatics approaches are both cost-effective and convenient, as in silico predictions can reduce the number of experiments needed. In this study, with the aid of immunoinformatics tools, we tried to design a multi-epitope vaccine that can be used for the prevention and treatment of COVID-19. The epitopes were computed by using B cells, cytotoxic T lymphocytes (CTL), and helper T lymphocytes (HTL) base on the proteins of SARS-CoV-2. A vaccine was devised by fusing together the B cell, HTL, and CTL epitopes with linkers. To enhance the immunogenicity, the β-defensin (45 mer) amino acid sequence, and pan-HLA DR binding epitopes (13aa) were adjoined to the N-terminal of the vaccine with the help of the EAAAK linker. To enable the intracellular delivery of the modeled vaccine, a TAT sequence (11aa) was appended to C-terminal. Linkers play vital roles in producing an extended conformation (flexibility), protein folding, and separation of functional domains, and therefore, make the protein structure more stable. The secondary and three-dimensional (3D) structure of the final vaccine was then predicted. Furthermore, the complex between the final vaccine and immune receptors (toll-like receptor-3 (TLR-3), major histocompatibility complex (MHC-I), and MHC-II) were evaluated by molecular docking. Lastly, to confirm the expression of the designed vaccine, the mRNA of the vaccine was enhanced with the aid of the Java Codon Adaptation Tool, and the secondary structure was generated from Mfold. Then we performed in silico cloning. The final vaccine requires experimental validation to determine its safety and efficacy in controlling SARS-CoV-2 infections.
In December 2019, COVID-19, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first discovered in China and has rapidly spread across the world. As of 12:00 noon on June 4, a total of 6,392,319 confirmed cases of COVID-19 have been reported globally, including 383,318 deaths. The prevalence of the disease has been increasing at an alarming rate. There were 1,849,852 cases in the United States, 555,383 in Brazil, 431,715 in Russia, 281,270 in the United Kingdom, and 3,275,736 in a number of other countries (1).
The incubation period for the virus is ~1–14 days, and all age groups are susceptible to a fatality rate of about 5.9%. The most common clinical manifestations are low-grade fever, dry cough, fatigue, and gastrointestinal symptoms (2). About half of all patients with COVID-19 develop shortness of breath, and severe cases may rapidly develop SARS, septic shock, difficult-to-correct metabolic acidosis, and coagulation disorders (3). COVID-19 may also affect other organs, most commonly the heart and kidneys (4–6). Some patients may have mild symptoms, without fever, and may recover after 1–4 weeks (7). Other patients may show signs of serious illness and some may die; however, most patients show favorable progress (8). Male individuals with the disease and aged patients have the worst prognosis. In children, the disease is relatively mild (9).
COVID-19 is caused by a novel single-stranded, positive (+) sense RNA beta coronavirus, which is a pathogen of the Coronaviridae family, named SARS-CoV-2 (10). The full-length genome sequences revealed that SARS-CoV-2 has the greatest genetic similarity to bat coronavirus, ~45–90% similarity to severe acute respiratory syndrome-related coronavirus (SARSr-CoV), and a smaller similarity of 20–60% to the Middle East respiratory syndrome-related coronavirus (MERS-CoV) (10). Thus, a bat might be the original host of SARS-CoV-2, but the intermediate host remains undiscovered (10).
The genes of SARS-CoV-2 encode structural proteins and non-structural proteins. Four structural proteins are absolutely vital for viral assembly and invasion of SARS-CoV-2. Spike protein homotrimers constitute the spikes on the viral surface, and these spikes are responsible for attachment to host cells by binding to their receptors (10). The M protein has three transmembrane domains, which determine the shape of the virion, facilitate membrane curvature, and bind to the nucleocapsid. The E protein plays an important role in virion assembly and release, as well as involved in viral pathogenesis. The N protein has two different domains, both of which bind to the viral RNA genome via totally different mechanisms. In addition, some reports have shown that non-structural proteins are essential for the replication of coronaviruses (10).
Vaccination is a vital tool for the control and elimination of the virus, and the development of a vaccine for SARS-CoV-2 remains an urgent need (11). Traditional methods of vaccine development are time-consuming and very labor-intensive (12). The realm of immunoinformatics tools considers the mechanism of the host immune response to yield additional methodologies in the design of vaccine against diseases are cost-effective and convenient, as in silico predictions can reduce the number of experiments needed (13, 14). Dozens of studies have generated epitope-based peptide vaccine of SARS-CoV-2. Baruah and Bose (15) used immunoinformatics tools to discover cytotoxic T lymphocyte (CTL) and B cell epitopes for the spike protein of SARS-CoV-2. Then, Abraham et al. developed a multi-epitope vaccine that was designed using immunoinformatics tools that potentially trigger both CD4+ and CD8+ T-cell immune responses (16).
Although there are many vaccines generated by immunoinformatics tools, most of these are based on spike protein. The spike protein is responsible for attachment to host cells by binding to angiotensin-converting enzyme 2 (ACE2) (17). A vaccine based on the spike protein could induce antibodies to block SARS-COV binding and fusion or neutralize virus infection (18). But there are still many obstacles, spike protein-based SARS vaccine may induce harmful immune responses that cause liver damage of the vaccinated animals (19). Other virus proteins are considered as the candidates for designing vaccine with protective and less harmful immune responses (20). Vaccine-based on structural and non-structural proteins of the virus is revealed potential vaccine inducing protective immune responses (20, 21). Pandey et al. reported the more scientifically rigorous strategy of multi-epitope subunits based on multiple proteins against parasitic and viral diseases, such as malaria, visceral leishmaniasis, and HIV (22–24). In this present, we employed immunoinformatics to predict multiple immunogenic proteins from the SARS-CoV-2 proteome and thereby design a multi-epitope vaccine. These proteins included non-structural and structural sequences of SARS-CoV-2, their reference sequences were retrieved from the National Center for Biotechnology Information (NCBI) database.
The proteins of the SARS-CoV-2 have been reported and reference could get from NCBI (25, 26). The reference sequences of SARS-CoV-2 proteins were retrieved from NCBI Protein Database (https://www.ncbi.nlm.nih.gov/protein) and accession numbers in Table 1, then we stored the reference sequences as a FASTA data type. The proteins with <100 amino acid sequences which are too short to predict epitopes were excluded, the remaining proteins were used for further analysis.

Table 1. Details and antigenicity of SARS-CoV-2 proteins.
VaxiJen is the first server for alignment-independent prediction of protective antigens, which overcome the limitations of alignment-dependent methods (27). To identify the potential antigenicity of SARS-CoV-2 proteins, an online prediction server, VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was used to predict the antigenic values of each protein (28). This identification was applied according to the default parameters of the server. Proteins having antigenicity were sorted according to an antigenic score of ≥ 0.5 (Threshold for this model is 0.5) and were selected for further structural modeling (27).
There are no available experimental structures of SARS-COV-2 proteins, Phyre 2 provide model regions trough a new ab initio folding simulation with no detectable homology (29). The SARS-CoV-2 proteins were modeled by Phyre 2 server (http://www.sbg.bio.ic.ac.uk/phyre2/). Because the SARS-COV-2 proteins with no detectable homology protein to finish the modeling, we chose the intensive search and output the accurate alignment by the alignment of hidden Markov models.
ModRefiner was used by the GalaxyRefine server (http://galaxy.seoklab.org /cgi-bin/submit.cgi?type=REFINE) (30). The structure assessment was performed by the SWISS-MODEL workspace (https://swissmodel.expasy.org/assess) (31). The three dimensional (3D) models were used for the conformational (discontinuous) B-cell epitope predictions while the sequences were utilized in linear B-cell and T-cell epitope predictions.
NetCTL-1.2 is demonstrated to have a high predictive performance (32). The NetCTL 1.2 server (http://www.cbs.dtu.dk/services/NetCTL/) was applied to predict CTL epitopes for the SARS-CoV-2 at the threshold value of 0.75 with high sensitivity and specificity (32). To cover ~90% of the world's population, three supertypes (A2, A3, and B7) were selected based on artificial neural networks, to predict MHC class I binding epitopes (33). The best candidates for the SARS-CoV-2 vaccine construction were sorted for further prediction, based on a half-maximal inhibitory concentration (IC50) < 500 nm and an integrated score. The IC50 < 500 nm represents epitope has a high affinity to receptor. The integrated score indicated the transporter of antigenic peptides (TAP) transport efficiency, class I binding, and proteasomal cleavage prediction (34–36). Then the specific Treg epitopes were screened and excluded by the EpiToolKit (https://epivax.com/).
For MHC class II T cell epitope predictions, The Immune Epitope Database server predicted binders based on the percentile rank or MHC binding affinity (37). The Immune Epitope Database server (IEDB; http://tools.iedb.org/mhcii/) was used to predict helper T lymphocyte (HTL) epitopes (37). We chose the combinatorial approach which recommended by IEDB to predict HTL epitopes. The combinatorial approach combined NN-align, SMM-align, CombLib, Sturniolo, and NetMHCIIpan methods (38–42). The 17 alleles of the human leukocyte antigen (HLA) were selected for the prediction at α and β chains, separately (43). For final construction, epitopes were selected based on their scores (low scores indicated favorable binding), the release of interferon-gamma (IFN-γ), induction of emergent properties, and the IC50 < 500 nm.
The IFN-γ cytokine makes a major contribution to antiviral mechanisms. It excites both native and specific immune responses by activating macrophages and natural killer cells (44). Further, IFN-γ augments the response of MHC to antigens. The IFN-γ epitope server (http://crdd.osdd.net/raghava/ifnepitope/scan.php) was used to recognize IFN-γ epitopes (45). We entered the HTL epitopes with low scores into the IFN-γ epitope server. Positive IFN-γ induction was predicted based on the support vector machine (SVM) hybrid approach. The final HTL epitopes were determined based on IFN-γ induction and MHC Class II binding, both of which facilitate the stimulation of T-helper cells (46).
The ABCpred (http://crdd.osdd.net/raghava/abcpred/) and BepiPred linear epitope prediction (http://tools.iedb.org/bcell/result/) servers were utilized to predict linear B cell epitopes. The ABCpred server is based on an artificial neural network (ANN) (47, 48). The linear B cell epitopes of the SARS-CoV-2 protein were predicted at a threshold of 0.5. The BepiPred linear epitope prediction server is based on seven methods: (a) Bepipred-1.0 Linear Epitope Prediction; (b) BepiPred-2.0: Sequential B cell Epitope Predictor; (c) Chou and Fasman beta-turn prediction; (d) Emini surface accessibility scale; (e) Karplus and Schulz flexibility scale; and the (f) Kolaskar and Tongaonkar antigenicity scale (49–54). We used these seven methods separately to predict the average threshold. The overlap between ABCpred and BepiPred severs was selected to determine the candidate epitopes for the SARS-CoV-2 vaccine construction (55).
Unlike T-cell epitopes that are linear continuous stretches of residues, B-cell epitopes are generally conformational (discontinuous) (56). In this study, the ElliPro servers (http://tools.iedb.org/ellipro/) were applied to predict the conformational B-cells epitopes (57). The server predicts epitopes based on PI (Protrusion Index) value. The epitope with PI = 0.9 would include 90% of residues with 10% being outside the ellipsoid, discontinues B-cells epitopes with the top PI value was selected for vaccine designing (57).
To develop the final vaccine, epitopes determined by various immunoinformatics software were linked together with the aid of separate linkers. The CTL epitopes were linked by the AAY linker, HTL epitopes by the GPGPG linker, and B cells were linked by the KK linker (48, 58, 59). To increase the vaccine immunogenicity, the β-defensin (45 mer) amino acid sequence was adjoined to the N-terminal of the vaccine with the help of the EAAAK linker (60). The β-defensin peptides provoke innate immunity cells and recruit naive T cells through the chemokine receptor-6 (CCR-6) (61). The pan-HLA DR binding epitopes (13aa) as well as added to the N-terminal of the vaccine with the aid of the same linker (59). The pan-HLA DR binding epitopes in vaccine construct facilitating binding to many different types of mouse and human MHC-II alleles to induce CD4-helper cell responses (59). To enable the intracellular delivery of the modeled vaccine, a TAT sequence (11aa) was appended to C-terminal (62). Linkers (AYY, KK, and GPGPG) play vital roles in producing an extended conformation (flexibility), protein folding, and separation of functional domains, and therefore, make the protein structure more stable (59).
The allergenic proteins induce a harmful immune response, allergenicity of the vaccine should be non-allergic (63). The non-allergic character of the vaccine sequence was evaluated by the AlgPred server (http://www.imtech.res.in/raghava/algpred/) (63). We predicted allergenicity of vaccine sequences choosing a hybrid approach (SVMc+IgE epitope+ARPs BLAST+MAST) with the highest accuracy and sensitivity (63).
The Vaxijen v2.0 server (http://www.ddgpharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was applied to evaluate the antigenicity of the vaccine (27). The antigenicity prediction method was solely based on the physicochemical properties of proteins without recourse to sequence alignment. The precision rate of the server ranged from 70 to 89%.
To determine immune response profile of this multi-epitope vaccine, computational immune simulations were performed by the C-ImmSim online server at (http://kraken.iac.rm.cnr.it/C-IMMSIM/) (64). The C-ImmSim utilizes the Celada-Seiden model for describing both humoral and cellular profiles of a mammalian immune system against designed vaccine. As per the literature, three injections were administrated at different intervals of 1 month. The simulation was performed with default parameters. The vaccine sequence was administered 4 weeks apart. The simulation volume was 1,000, simulation steps was 1,000, random seed was 12,345, and the vaccine injection with no LPS (64).
The ProtParam tool (http://web.expasy.org/protparam/) was used to evaluate the physicochemical properties of the final vaccine protein (65). The physicochemical properties included the number of amino acids, molecular weight, theoretical isoelectric point (pI), amino acid composition, atomic composition, formula, extinction coefficients, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) (66). The molecular weight and theoretical pI were computed by user-entered sequences. The amino acid and atomic compositions were self-explanatory. The extinction coefficient of a protein was based on information about its amino acid composition. The instability index of a protein indirectly indicated the stability of the protein. If the computed instability index of protein was <40, it was regarded as a stable protein, while values >40 were regarded as unstable. In vivo half-life evaluation of proteins was based on the principle of the “N-end rule.” Furthermore, GRAVY is a measurement of the hydrophobic nature of the protein, which is calculated by determining the total hydropathy of all amino acids divided by the number of amino acid residues in the protein.
To avoid inducing pathogenic priming and autoimmunity, the sequence homology of the final vaccine to human protein was screened by BLASTp online server (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (67). An ideal vaccine should have non-sequence to human proteins.
The designed vaccine was a reconstructed protein with no detectable homology (29). Phyre2 incorporates an ab initio folding simulation to model regions of proteins with no detectable homology. The Phyre 2 server (http://www.sbg.bio.ic.ac. uk/phyre2/) was used to predict the three-dimensional structure of the designed vaccine. The server generates a full-length 3D model of a protein sequence by employing both multiple template modeling and simplified ab initio folding simulation (29).
To enhance the overall and partial structural quality of the protein, the output 3D structure of the final vaccine from the Phyre 2 server was further refined by the GalaxyRefine server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) (30). The GalaxyRefine server predicted five refined models of our developed vaccine construct, in which Model 1 was made by the structural perturbation based simply on the clusters of the side chains; whereas, Models 2–5 were generated by deeper perturbations of loops and secondary structural elements (30).
For the assessment of the tertiary structure of the final vaccine protein, a Ramachandran plot was performed by the SWISS-MODEL workspace (https://swissmodel.expasy.org/assess) (31). The Ramachandran plot illuminates favored regions for backbone dihedral angles against amino acid residues in protein structure (31). The Structure Assessment page shows the most relevant scores provided by Molprobity and help we easily identify where residues of low quality lie in their model or structure (31). Then, ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) was employed in the final vaccine protein structure validation. A positive Z-score commonly means an erroneous or erratic section found in the generated 3D protein model (68).
To revealing the binding affinity between the vaccine construct and antigenic recognition receptors of toll-like receptor-3 (TLR3, 2A0Z) and major histocompatibility complex (MHC-I, 4WUU, and MHC-II, 3C5J) present on the surface of immune cells (69). Docking analysis was performed using the ClusPro server (https://cluspro.bu.edu/login.php?redir/queue.php). TLR3 act as receptors for antigenic recognition. The ClusPro server computed the models based on electrostatic interactions and desolvation energy (69). To reconfirm the binding affinity of the designed vaccine construct between these receptors, the PatchDock server (https://bioinfo3d.cs.tau.ac.il/PatchDock/) was used for docking (70). The server predicted the potential complex with the help of three algorithm-molecular shape representations, surface patch matching, filtering, and scoring (70). After the acquisition of the output from the PatchDock server, the complexes were refined by the FireDock algorithm, which predicted the optimal complex with the aid of energy functions (70).
The pdb file of vaccine protein and receptor complex (TLR3, MHC-I, and MHC-II) were used to start the molecular dynamic (MD) simulations. The complexes were placed in a octahedron box of water molecules represented by the three-point charge SPC model, whose boundary is at least 10 Å from any protein atoms. The solvated protein was subsequently neutralized by chloridions. Covalent bonds involving hydrogen atoms were constrained using the LINCS algorithm, and long-range electrostatic interactions were treated with particle-mesh Ewald employing a real-space cutoff of 10 Å. The system was first briefly minimized with backbone atoms restrained to the initial coordinates to remove close contacts, and the restrained system was gradually heated to 300 K under constant volume conditions in 100 ps. Each system was equilibrated for 1 ns using the constant isothermal-isobaric ensemble at 1 atm and 300 K without any restraints. The Parrinello-Rahman barostat and a V-rescale thermostat were used with an integration time step of 2 fs. Production run MD simulations were performed for 10 ns with coordinates recorded every 10 ps. All simulations were performed using GROMACS 2018.2 along with the GROMOS96 54a7 force field (16, 24).
For the purpose of cloning, codon adaptation of the designed vaccine was performed for analyzing the codon usage by the prokaryotic organism (Escherichia coli, E. coli). The Java Codon Adaptation tool (http://www.jcat.de/) was used to optimize codon (71). Then the secondary structure of mRNA was predicted by Mfold (http://unafold.rna.albany.edu/?q=mfold) (72). For raising the expression efficiency of the final vaccine protein, the E. coli K12 strain was chosen. For the valid translation of the vaccine gene, we proofread and avoided rho-independent transcription termination, prokaryote ribosome binding site, and cleavage site of restriction enzymes. Restriction endonuclease sites XhoI and BamHI were appended to N and C terminals of vaccine, respectively. Then, it was inserted into the pET28a (+) vector between the XhoI and BamHI. The flow chart of the designed work is shown in Figure 1.
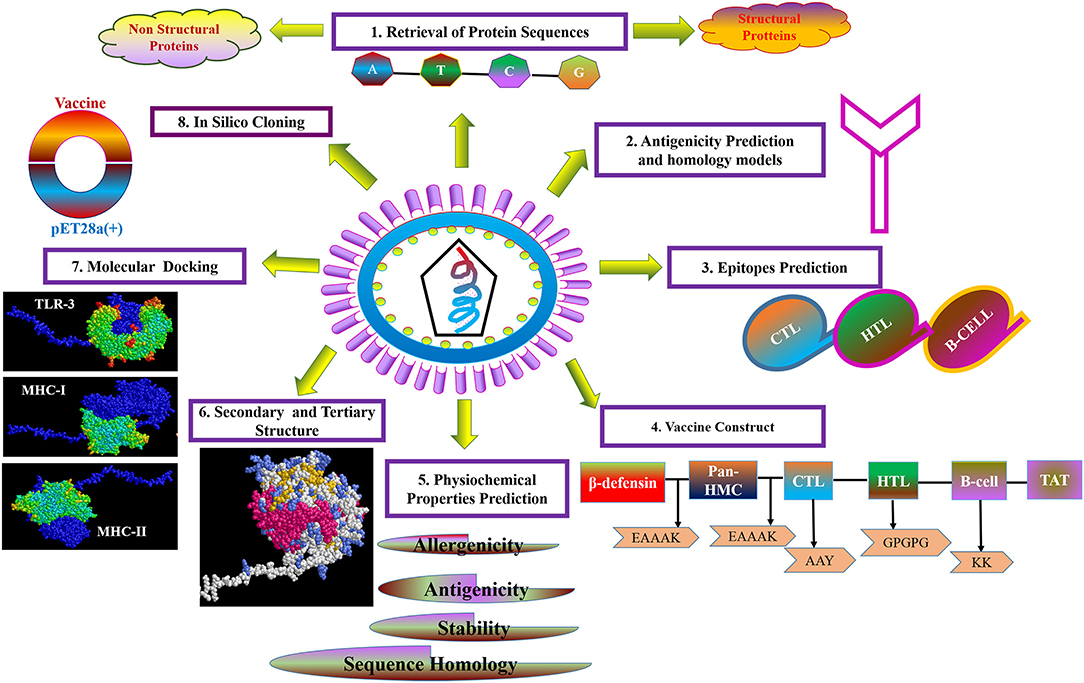
Figure 1. Flow diagram of design strategy, representing the steps of the construct of the multi-epitope subunit vaccine.
The strategy of vaccine construction is presented in Figure 1.
The proteome of SARS-CoV-2 was retrieved, which comprised 27 proteins. The reference sequences of those proteins were retrieved in the FASTA format and their details are presented in Table 1. Five proteins with <100 amino acid sequences are too short to predict epitopes (ORF6 protein, ORF10 protein, ORF7b protein, nsp11, and envelope protein) were excluded.
In order to develop a subunit vaccine, it is critical to identify candidate proteins that are important for inducing a protective immune response (27). The remaining 22 proteins sequence were relayed to the VaxiJen server to determine their antigenicity based on the antigenic scores (Table 1). Proteins with antigenic scores >0.5 were selected for further analysis (28). Nine proteins, namely ORF7a protein, ORF8 protein, nsp9, nsp6, nsp3, endoRNAse, ORF3a protein, membrane glycoprotein, and nucleocapsid phosphoprotein were finally selected for further epitope prediction.
There is no available experimental structures of these nine proteins, we predicted homology models for the nine proteins applying the normal mode of phyre2 online server. The most suitable templates for the nine proteins were identified to be the PBD entries (Table S1). All of the modeled structures were showed over 90% residues in the Ramachandran favored region Figure S1 and Table S2.
The prediction of CTL epitopes (9 mer) was performed by the NetCTL server. The binder sites were determined based on three supertypes (A2, A3, and B7), with a 95% coverage rate of the world's population. Nine proteins were selected based on antigenicity. One epitope of each supertype was selected based on the highest score and an IC50 value < 500 nm. Then the specific Treg-inducing epitopes were excluded by Epitoolkit. A total of 18 epitopes were selected from nine proteins as the candidates for the construction of the vaccine (Table 2).
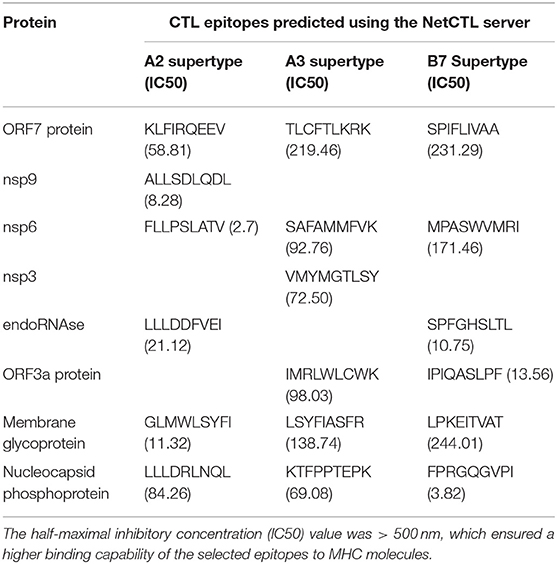
Table 2. Predicted cytotoxic T lymphocyte (CTL) epitopes of SARS-CoV-2 proteins utilized for the construction of a multi-epitope subunit vaccine.
The HTL epitopes (15 mer) were evaluated for three HLA supertypes: HLA-DR (DRB1*01:01, DRB1*07:01, DRB1*09:01, DRB3*01:01, DRB4*01:01); HLA-DQ (DQA1*01:01/DQB1*05:01, DQA1*01:02/DQB1*06:02, DQA1*03:01/DQB1*0:02, DQA1*04:01/DQB1*04:02, DQA1*05:01/DQB1*02:01, DQA1*05:01/DQB1*03:0 1); and HLA-DP (DPA1*01/DPB1*04:01, DPA1*01:03/DPB1*02:01, DPA1*02:01 /DPB1*01:01, DPA1*02:01/DPB1*05:01, DPA1*03:01/DPB1*04:02). We sorted the top epitopes with the lowest scores (low scores indicated the highest binding capability) from three supertypes. The best candidate was then selected based on positive IFN-γ induction and an IC50 < 500 nm. Then the specific Treg-inducing epitopes were excluded by Epitoolkit. Thus, a total of 14 epitopes were selected for vaccine design (Table 3).
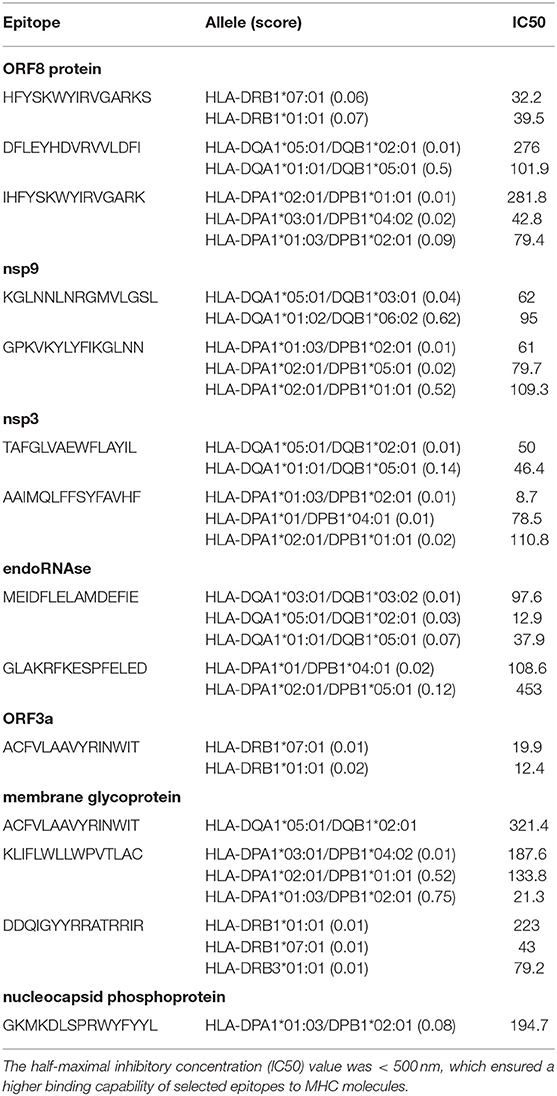
Table 3. Predicted Helper T lymphocyte (HTL) epitopes of SARS-CoV-2 proteins utilized for the construction of a multi-epitope subunit vaccine.
We used the ABCpred and BepiPred servers to identify the line B cell candidate epitopes. All predicted epitopes from both servers were compared, and only the overlapping epitopes were selected for the development of the vaccine. The line epitopes identified by ABCpred had prediction scores ranging from 0.52 to 0.93, and line epitopes identified by BepiPred had prediction scores ranging from 0.5 to 1. Among these line epitopes, only 12 (16 mer) were found to be common or partly common in both servers (Table 4). These 12 line epitopes were selected for vaccine construction (Table 4).
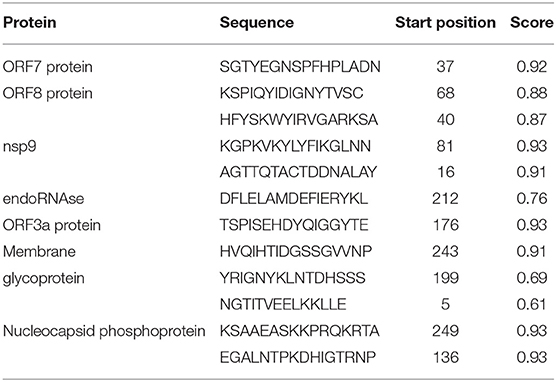
Table 4. Predicted line B cell (BCL) epitopes of SARS-CoV-2 proteins utilized for construction of a multi-epitope subunit vaccine.
The non-continuous B cell epitopes were predicted by the ElliPro severs, a total number of 27 non-continuous B cell epitopes were generated from ElliPro. Amino acid residues, sequence location, the number of residues, and the PI scores of the predicted conformational epitopes are shown in Table 5 and the graphical depiction of these epitopes can be seen in Figure S2. Twenty-four epitopes were excluded because it added the allergenicity of vaccine, three epitopes were marked red and selected for vaccine construction.
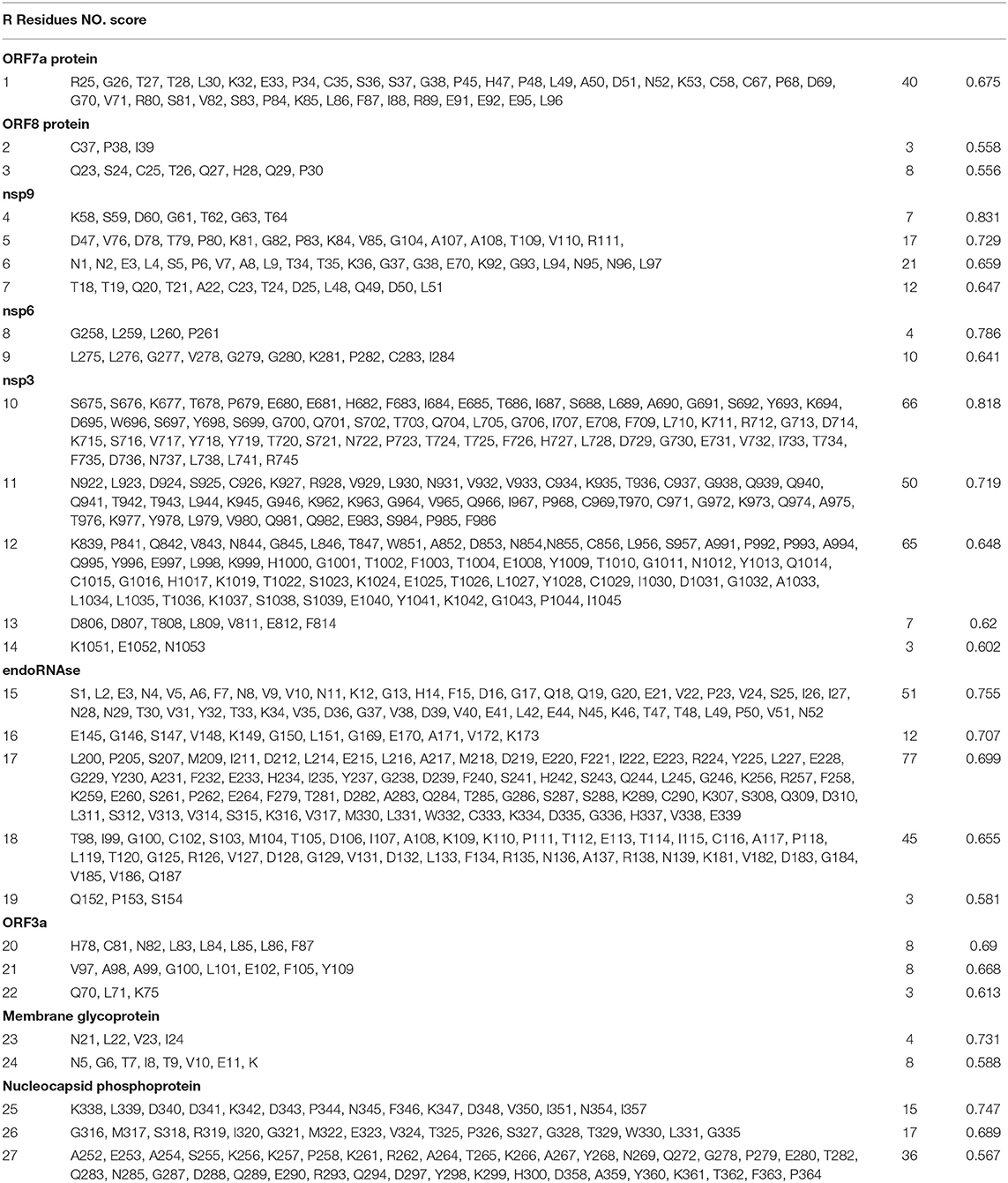
Table 5. Predicted conformational B cell (BCL) epitopes of SARS-CoV-2 proteins.
The best candidate epitopes were used for the construction of the vaccine. A total of 18 CTL epitopes, 14 HTL epitopes, 12 linear, and three non-continuous B cell epitopes were fused together with the aid of linker sequences. The CTL epitopes were linked by AYY (The AAY liner helps the epitopes produce suitable sites for binding to TAP transporter and enhances epitope presentation), the HTL epitopes were combined with the aid of GPGPG (The GPGPG linker stimulate HTL responses and conserve conformational dependent immunogenicity of helpers as well as antibody epitopes), and B cell epitopes were merged with the aid of KK. The final to enhance vaccine immunogenicity, the human β-defensin-3 sequence (45aa) and pan-HLA DR binding epitopes (The pan-HLA DR binding epitopes in vaccine construct facilitating binding to many different types of mouse and human MHC-II alleles to induce CD4-helper cell responses.) was added to the N-terminal of the vaccine with the aid of the EAAK linker. To enable the intracellular delivery of the modeled vaccine, a TAT sequence (11aa) was appended to C-terminal. The vaccine was developed to be 864 amino acids in length (Figure S3). The sequence homology of final vaccine protein to human protein sequence shown that there were no significant alignments (Figure S4).
The allergenic character of the vaccine was determined by the AlgPred server and was based on the hybrid approach (SVMc + IgE epitope + ARPs BLAST + MAST) with a 93.5% coverage. The vaccine was non-allergen with 84% accuracy and 82.78% sensitivity at threshold value was −0.2. Similarly, the antigenic nature of the vaccine construct was evaluated and showed that the protein was a favorable antigen with a global prediction score of a protective antigen of 0.5308 (Probable antigen). The default threshold value for antigenicity was 0.4 in the virus model.
Moreover, the vaccine constructs contained 864 amino acids, and its molecular weight was 95.4 kDa. The theoretical pI was predicted to be 9.71. The vaccine contained 63 negatively charged residues and 125 positively charged residues. The vaccine construct was composed of 13,541 atoms, and its chemical formula was C4395H6791N1153O1174S28. The computed instability index was 32.84, which was <40, classifying the vaccine as a stable protein. The estimated half-life was 1 h in vitro. In vivo, the estimated half-lives in yeast and Escherichia coli are greater 30 min and 10 h, respectively. The aliphatic index of the vaccine construct was 79.29, which suggests a high thermostability. The GRAVY value of the vaccine construct was −0.215, which indicated the hydrophobicity of the protein.
The immune stimulation of the final vaccine was performed using C-ImmSim online server, which gives the immune profiles of the designed vaccine. The proliferation in the secondary and tertian immune response were identified by IgG1 + IgG2 and IgM, as well as, the decreasing of the antigen count IgG + IgM showed the proliferated (Figure 2A). The stimulation result revealed the development of immune response after immunization. B cell population was highly stimulated upon immunization (Figure 2B). Similarly, the cytotoxic and helper T cell levels were proliferated that suggested the development of secondary and tertian immune response (Figures 2C,D). During the exposure time, it was also observed that the production of IFN-γafter immunization (Figure 2E). These results were significant for the immune response against SARS-CoV-2. Hence,
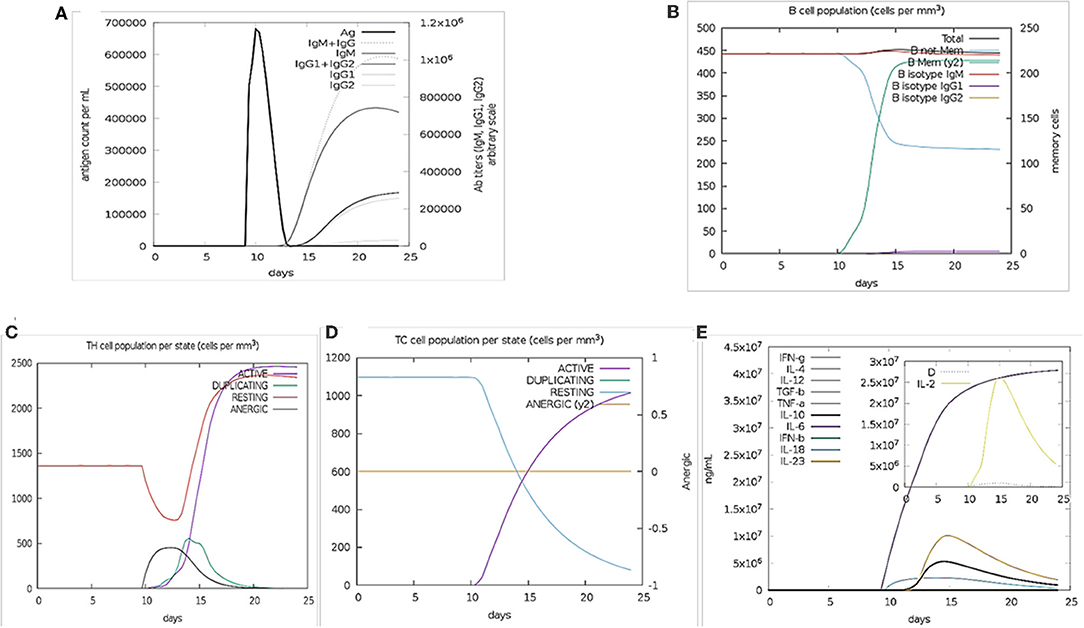
Figure 2. Immune Simulation results by C-ImmSim. (A) The immunoglobulins production represent proliferation of immune response after the vaccine administration. Various subtypes of immunoglobulin are represented as colored peaks. (B) The active B-cell population is observed with the administration of vaccine. (C) The generation of Helper-T cells. (D) The generation of cyototoxic-T cells were found after the vaccine injection. The RESTING indicates to the cells, which were not shown to the antigens while ANERGIC indicates the tolerance level of antigen. (E) The cytokine profile shows that the induced IFN-γlevel upon administration of vaccine. The inset graph indicating the Simpson Index, D of IL- 2. Simpson Index, D was inferred as the measurement of diversity.
The tertiary structure of the full-length vaccine sequence was predicted by Phyre 2, and it was applied for refinement and further analysis. Twenty-five templates were employing modeling as Figure S5 shown. There were three templates from human defensin which were we added in to enhance the immunogenicity, others from virus (Figure S5). The immune epitopes were not structural homology to human proteins that could avoid inducing autoimmune. The secondary structure of the predicted model contained 18% alpha-helix, 21%TM helices 44% beta-sheets, and 27% disordered Figure S6.
To optimize the 3D structure of the modeled protein, the initial model was refined in the GalaxyRefine server. The GalaxyRefine server-generated five models based on the root-mean-square deviation (RMSD) and MolProbity algorithm. The details of the five models are shown in Table S3. Model 1 with the top Ramachandran favored, therefore selected for docking purposes (Figure 3). A model with more residues in the Ramachandran favored region, less in outliers region and rotamer region was considered as a more ideal one. The initial model generated from Phyre 2 server and refine model from GalaxyRefine were evaluated with the aid of the SWISS-MODEL workspace. The initial model was 63.46% of residues in the Ramachandran favored region, 19.49% in the Ramachandran outliers region, and only 10.22% in the rotamer region (Figure 4). The refine model was 89.1% of residues in the Ramachandran favored region, 2.09% in the Ramachandran outliers region, and only 0.15% in the rotamer region (Figure 3). Other favorable parameters of the refined model were as follows: GDT score of 0.9922, RMSD value of 0.260, MolProbability of 2.049, clash score of 8.9, and poor rotamers totaling 0.3 (Table S1).
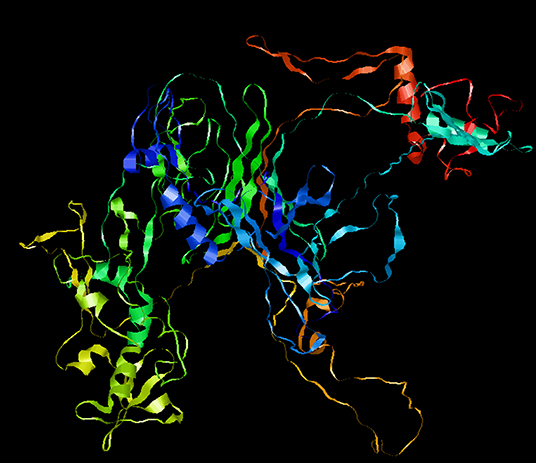
Figure 3. Refinement of the SARS-CoV-2 vaccine construct. Representative 3D image of the tertiary structure of the 2019nCOV vaccine after modeling.
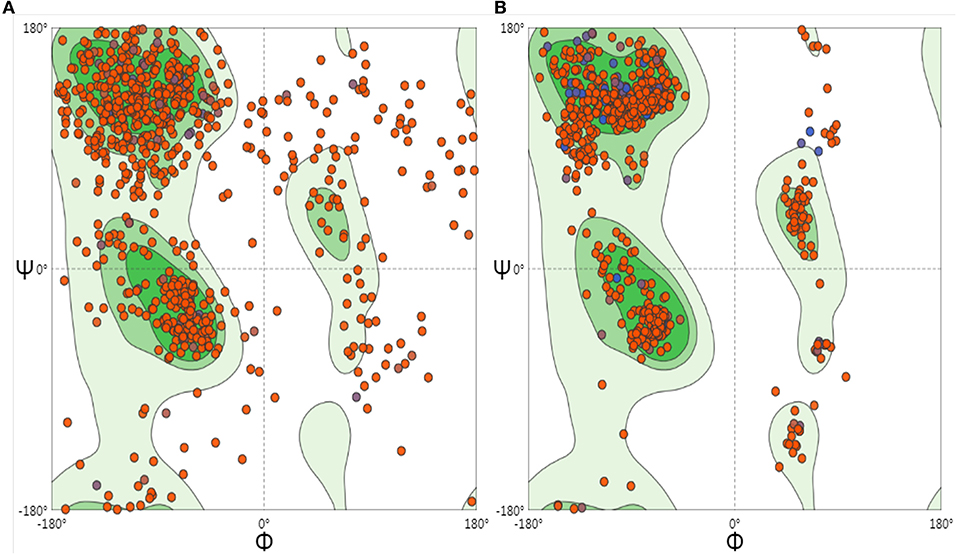
Figure 4. Ramachandran plots to initiate and refine the 3D structure of the vaccine construct illustrated using the SWISS-MODEL/Structure Assessment. (A) Shows the Ramachandran plot of initiate model, (B) shows the Ramachandran plot of refining the model.
The quality and potential errors in the final vaccine 3D model were verified by ProSA-web. The Z-score indicates overall model quality, the model with a lower Z-score was considered as the higher quality one. The z-score of the initial model was −2.81, refine model is −3.64 (Figure 5).
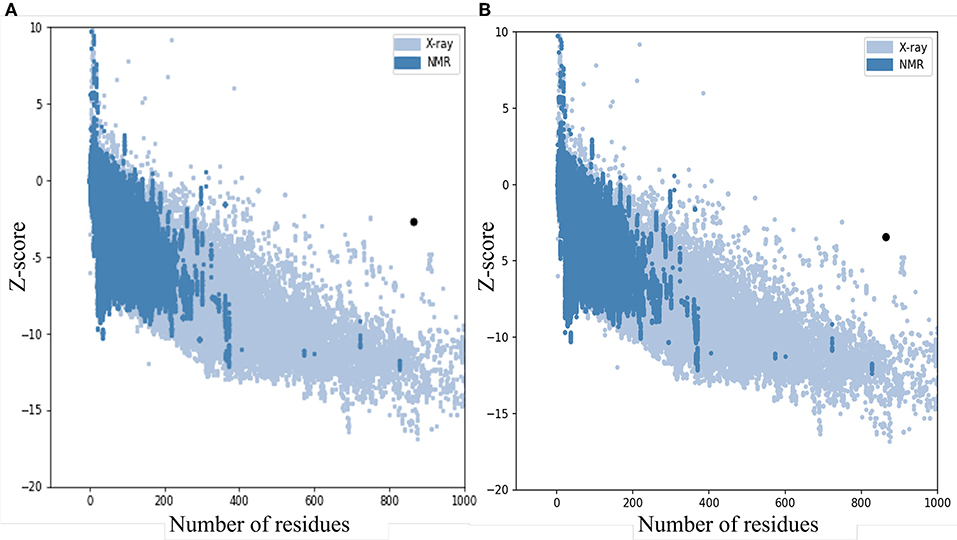
Figure 5. Z-Score plot for the 3D structure of the final vaccine. The Z-score of (A) the initial model is 2.81 and (B) The z-score of the refined model is 3.64, both of two models not in the range of native protein conformation. Z-Score plot contains z-scores of all experimental protein chains in PDB determined by NMR spectroscopy (dark blue) and X-ray crystallography (light blue).
To further evaluate the binding affinity between the developed vaccine construct and the relative antigenic receptors (TLR3, MHC-I, and MHC-II), molecular docking was performed. The server yielded 44 candidate models with different binding energies. Twenty-nine model complexes of TLR3 and COVID-19 vaccine were determined, from which just one complex with the lower binding energy score of −1156.2 was selected to show (Table 6 and Figure 6). A total of 29 model complexes of MHC-I and the COVID-19 vaccine were discovered, and the lowest binding energy score was −1346.8 (Table 6 and Figure 6). A total of 29 complex models of MHC-II and the COVID-19 vaccine were predicted, among which, one model complex with the lowest binding energy score of −1309.1 was chosen to show (Table 6 and Figure 6). Further, the vaccine construct was evaluated using the PatchDock server, which identified different models and produced a score table. The top 10 complexes identified were refined by the FireDock algorithm. Among those top 10 models, the model with the lowest binding energy was further selected to show in this paper. The refinement outcomes of TLR3 and the vaccine complex was solution number 1 with global energy of −38.40, attractive van der Waals energy (VdW) of −26.02, repulsive (VdW) of 8.62, and atomic contact energy of −11.06 (Table 6 and Figure 6). The complex of MHC-I and the vaccine was ranked number nine, with global energy of −22.97, attractive VdW of −26.84, repulsive VdW of 12.82, and atomic contact energy of −1.79 (Table 6 and Figure 6). The complex of MHC-II and the vaccine was ranked number three, with global energy of −27.52, attractive VdW of −26.86, repulsive VdW of 10.93, and atomic contact energy of 0.77 (Table 6 and Figure 6).
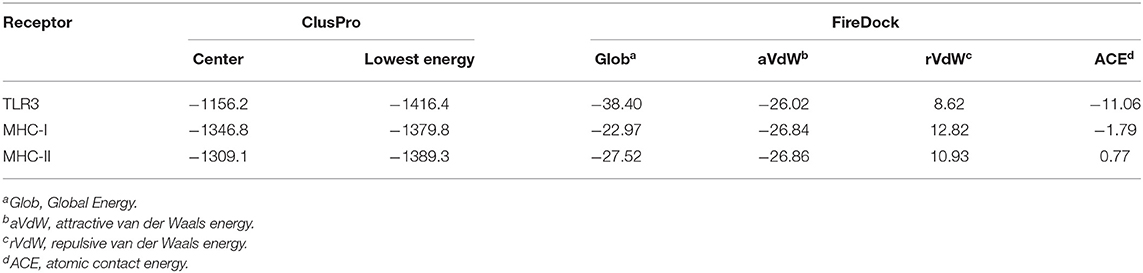
Table 6. Molecular docking of final vaccine constructs with TLR3, Mda5, and MHC-II.
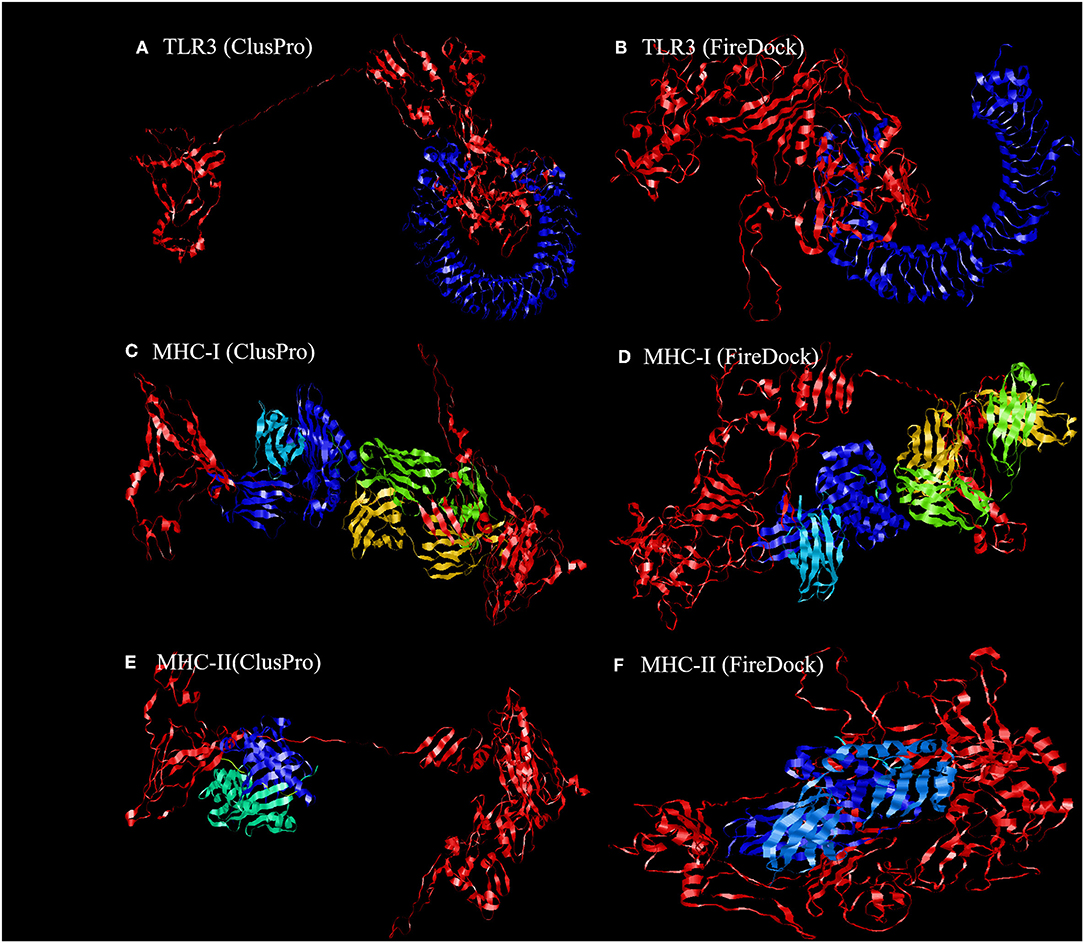
Figure 6. Representation of the ligand-receptor docked complex. (A,C,E) show the molecular docking of the vaccine construct (red color) and TLR-3, MHC-I, and MHC-II receptors (other colors) illustrated using the ClusPro software. (B,D,F) show the molecular docking of the vaccine construct (red color) and TLR-3, MHC-I, and MHC-II receptors (other colors) illustrated using PatchDock to verify the stability of the docked complex.
To accomplish the estimate of the stability of the vaccine-receptor complex, we performed the simulation of the docked complexes (vaccine and TLR-3, MHC-I, and MHC-II) with the help of GROMACS. Then, various analysis like energy minimization, pressure assessment, temperature, and potential energy calculations were performed. The temperature and pressure of the simulation system during the production run was around 300 K and 1 atmosphere, respectively, indicating a stable system and successful md run. The temperature and pressure of the three simulation systems (vaccine and TLR-3, MHC-I, and MHC-II complexes) during the production run were around 300 K and 1 atmosphere, respectively, indicating the stable systems and successful MD run (Figures 7A–F). The complex root mean square deviation (RMSD) plot represents the structural fluctuation of the overall structure of the complex of vaccine and immune receptor. The RMSD of vaccine-TLR3 complex has large fluctuation during 0–6 ns simulation. After 6 ns, the RMSD value was kept around 1.25 nm, indicating that the conformation of this complex was stable (Figure 7G). Otherwise, the RMSD of vaccine-MHC-I and -MHC-II complexes has large fluctuation during 0–4 ns simulation. After 4 ns, the RMSD value were kept around 1 nm, indicating that the conformation of the two complexes were stable (Figures 7H,I). Next, the root medium square fluctuation (RMSF) indicates the flexibility of the residue in the docking complex. From the results of vaccine-TLR3, MHC-I, and MHC-II complexes, residue 200–600 has low RMSF value, indicating these residues has low structural flexibility. By contrast, residue 0–200 and 600–800 has relatively higher RMSF value, indicating the larger flexibility during those regions (Figures 7J–L).
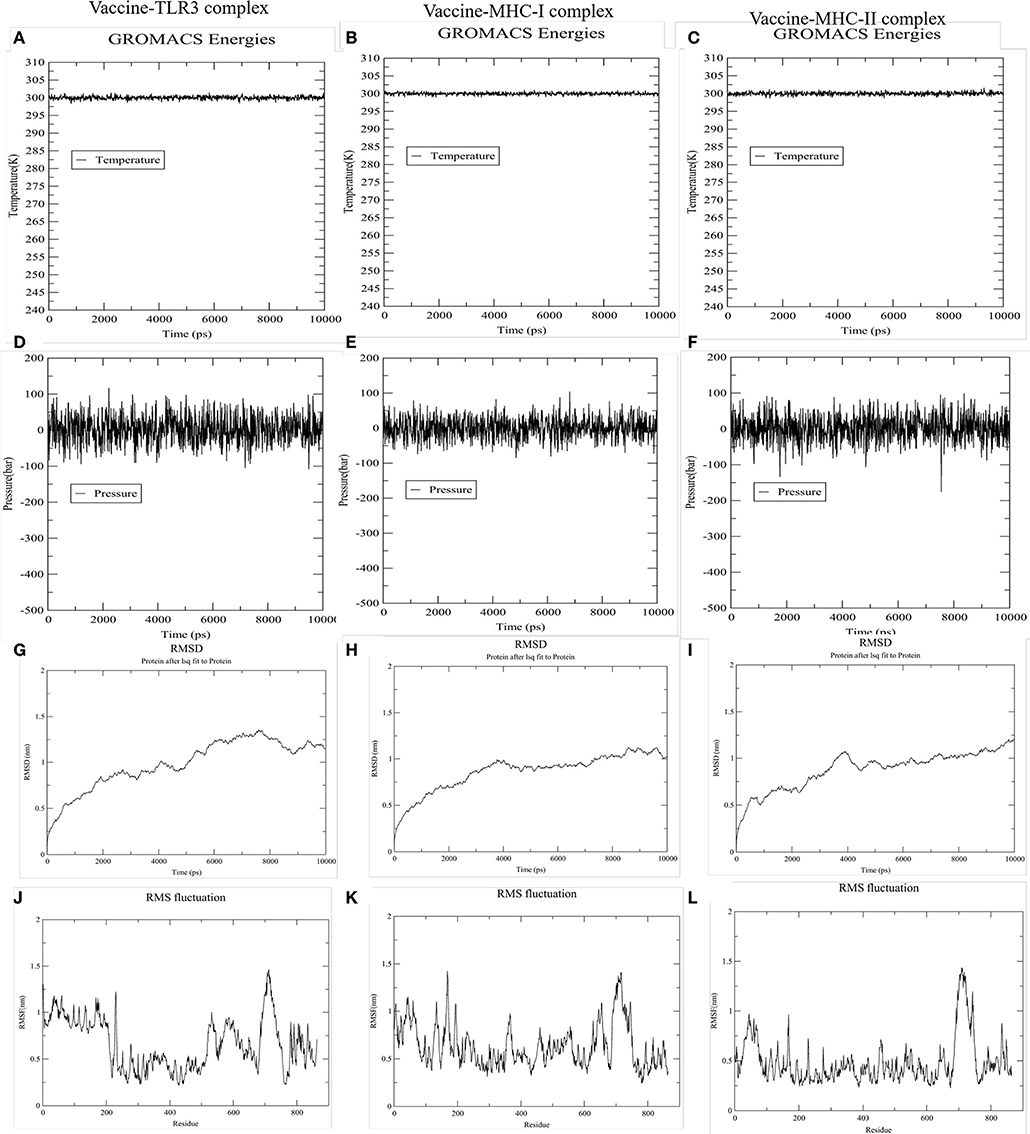
Figure 7. The results of molecular dynamics simulation of vaccine and immune receptors. (A–C) show the equilibration phase ensembles-temperature (constant at 300 k for 100 ps) of the complex of vaccine-TLR3, MHC-I, and MHC-II, respectively. (D–F) represent the pressure (displaying fluctuations at 1 bar value for 100 ps) of the complex of vaccine-TLR3, MHC-I, and MHC-II, respectively. (G–I) suggest the RMSD (root mean square deviation) plots reflect the stability between the vaccine and TLR-3, MHC-I, and MHC-II receptor, separately. Whereas, (J–L) RMSF (root mean square fluctuation) reflect the flexibility and fluctuation of the amino-acids residues in the side chain of docked complexes (the complex of vaccine-TLR3, MHC-I, and MHC-II), separately.
To fuse the final vaccine to an expression vector, codon conversion of the vaccine protein was performed by the Java Codon Adaptation tool. Restriction site XhoI and Bam HI were added to N and C terminals of the codon sequence, then was inserted into the pET28a (+) vector between the XhoI and BamHI (Figure 8). The RNA secondary structure using the Mfold program was generated foldings contain 4,381 base pairs out of 2.3% in the energy dot plot. Mfold predicted an identical secondary structure of 4,381 bp formed by nucleotide fragments (Figure S7).
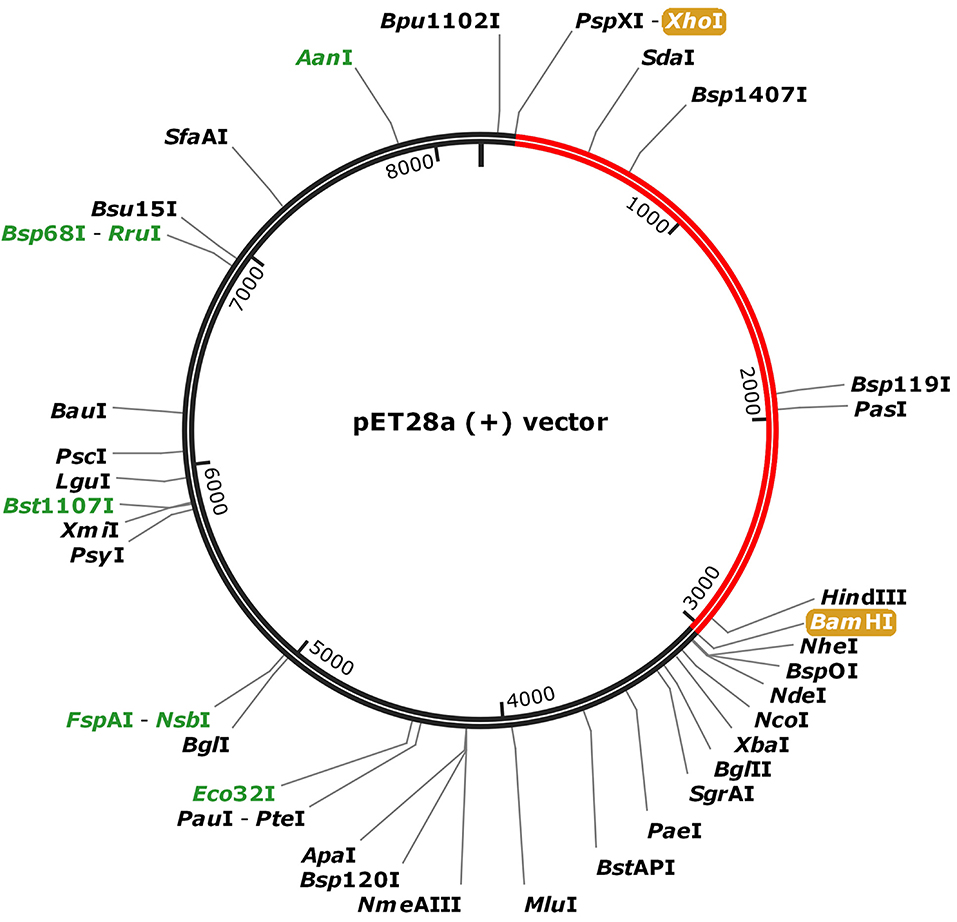
Figure 8. In silico cloning of the SARS-CoV-2 vaccine in the vector, pET28a (+). Red areas represent the COVID-19 vaccine, while the black areas represent the expression vector, pET28a (+).
SARS-CoV-2 is characterized by high infectivity and high transmission speed; thus, a prophylactic vaccine is needed (11). The availability and advantages of the multi-peptide vaccine developed by immunoinformatics methods have been confirmed by previous studies (73, 74). Ojha et al. used the immunoinformatics methods to develop a multiepitope subunit vaccine to Epstein-Barr virus-associated malignancy (73). In recent studies, genomics and proteomics information of SARS-CoV-2 have been retrieved, stored, and utilized (75, 76). In the present research, we tried to develop a multi-epitope subunit prophylactic vaccine of SARS-CoV-2, with the help of immunoinformatics tools.
A line of research have tried to develop the vaccine of SARS-CoV-2 by immunoinformatics tools. Baruah and Bose (15) used immunoinformatics tools to discover cytotoxic T lymphocyte (CTL) and B cell epitopes for the spike protein of SARS-CoV-2. Then, Abraham et al. developed a multi-epitope vaccine that was designed using immunoinformatics tools that potentially trigger both CD4+ and CD8+ T-cell immune responses (16). Most of those research just focus on the spike protein-based vaccine. A vaccine based on the spike protein could induce antibodies to block SARS-COV-2 binding and fusion or neutralize virus infection (18), as well as induce harmful immune responses that cause liver damage (19). Other proteins should be ideal candidates for designing vaccines.
In the present report, we selected nine proteins with positive antigenicity for further epitope prediction. All proteins from SARS-CoV-2 with <100 amino acid sequences were excluded, and the antigenic nature of the remaining proteins was evaluated. This method can facilitate the discovery of potential antigens of SARS-CoV-2 when the precise immunity mechanisms are unknown. To design an effective vaccine, we selected the SARS-CoV-2 protein through the above-mentioned methods for epitope prediction. In recently, Asaf et al. reported that identify multiple epitopes for CD4 + 12 and CD8 + T cells based on muti-protein (77). Their protein list was the same as this in our research. In Asaf's report, they just predicted the T cell epitopes, non-B cell, B cell peptide was not predicted (77).
The B cell epitopes are antigenic determinants from the antigen that are recognized by the B cell surface membrane receptor and evoke the production of specific antibodies. The persistent challenge in immunological prediction tools is the prediction of epitopes to a higher level of accuracy (78). To determine accurate linear B cell epitopes from the antigenic proteins, we used two bioinformatics tools based on different algorithms of prediction. We identified nine overlapping linear B cell epitope candidates from two different bioinformatics tools. This method was superior to the prediction of epitopes from a single tool (78). Moreover, we also have predicted the non-continue B-cell epitopes.
The B cell immune response is preferred in the design of a vaccine. However, T cells may also elicit a strong immunoreaction. The vaccine that activates both CTLs and HTLs should be more effective than a vaccine that only targets CTL responses (79). To generate a more effective vaccine, we predicted both CTL epitopes and HTL epitopes. The T cell epitopes were decomposed fragments from the antigen presented by the MHC molecules of T cells and stimulated the production of effector T cells, immunological memory T cells, and IFN-γ. The cell-mediated immune response induced by CTLs plays a vital role in the defense against viral infections through the recognition of intracellular viral pathogens by MHC class I molecules.
In the present report, MHC-I binding epitopes were predicted by choosing A2, A3, and B7 alleles, which cover ~95% of world's population. We selected 18 CTL epitopes. The HTLs play a vital role in the antiviral immune response by producing IFN-γ. Moreover, HTLs are able to induce and maintain CTL responses. Furthermore, 14 HTLs epitopes were chosen based on both the binding capability and IFN-γ induction. Bhattacharya et al. also used the spike protein sequence predicted for MHC-I and MHC-II epitopes of SARS-CoV-2, but not predicted capability of producing IFN-γ (80). The T cell epitopes enhanced IFN-γ inducing capability, which evokes both the native and specific immune responses by activating macrophages and natural killer cells, and augmenting the response of the MHC to the antigen (81, 82).
In this study, the immunogenic epitopes from B cells, CTLs, and HTLs were chosen to develop a more valid, reliable, and effective vaccine against SARS-CoV-2. A multiepitope approach was used by splicing together epitopes with the aid of their respective linkers. To improve the immunogenicity of this multiepitope vaccine, an adjuvant β-defensin and pan-HLA DR binding epitopes (13aa) were fused to the N-terminal with the aid of an EAAAK linker, then A TAT sequence (11aa) was appended to C-terminal with the added of KK. The final vaccine constituted 864 amino acids. The allergenicity, antigenicity, and stability of the designed vaccine constructs were then evaluated. The tertiary structure of the generated vaccine was predicted by using the Phyre 2 server and then refined by the GalaxyRefine server. The binding affinity of complexes of the developed vaccine and receptors, in which TLR-3, MHC-I, and MHC-II (present on the surface of the immune cell) were confirmed by the ClusPro server was based on molecular docking.
Furthermore, to ensure the translation efficiency of the designed vaccine in a specific expression system, the mRNA of the vaccine was enhanced with the aid of the Java Codon Adaptation Tool. The restriction enzyme cutting sites of Xho? and BamH? were then appended to the N and C terminals, respectively. The vaccine sequence was subsequently cloned in pET28a (+), the expression vector. Further experimental validation of the safety and efficacy of the designed vaccine for SARS-CoV-2 is warranted.
All datasets presented in this study are included in the article.
RD and ZC performed the experiments. RD and YZ wrote the paper. YZ and FY edited the final version. All authors participated in the experimental design, data analysis, and agreed with the final version of the paper.
This work was partly supported by grants from the Special Fund for Basic Scientific Research Operating of Central Public Welfare Research Institutes, the Chinese Academy of Medical Sciences (2019PT320003), and Guizhou High-Level Innovative Talents Program [QKHPTRC(2018)5636].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We extend the sincerest appreciation to NHC Key Laboratory of Pulmonary Immunological Diseases, and Guizhou Provincial People's Hospital, for their technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01784/full#supplementary-material
1. World Health Organization. COVID-19. (2020). Available online at: https://www.gisaid.org/epiflu-applications/global-cases-covid-19/ (accessed March 15, 2020).
2. Zhang JJ, Dong X, Cao YY, Yuan YD, Yang YB, Yan YQ, et al. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy. (2020) 2:19. doi: 10.1111/all.14238
3. Harcourt J, Taemin A, Lu X, Kamili S, Sakthivel SK, Murray J, et al. Severe acute respiratory syndrome coronavirus 2 from patient with 2019 novel coronavirus disease, United States. Emerg Infect Dis. (2020) 11:26. doi: 10.3201/eid2606.200516
4. Liu C, Jiang ZC, Shao CX, Zhang HG, Yue HM, Chen ZH, et al. A preliminary study of the relationship between novel coronavirus pneumonia and liver function damage: a multicenter study. Zhonghua Gan Zang Bing Za Zhi. (2020) 28:148–152. doi: 10.3760/cma.j.issn.1007-3418.2020.02.003
5. Wei ZY, Qian HY. Myocardial injury in patients with COVID-19 pneumonia. Zhonghua Xin Xue Guan Bing Za Zhi. (2020) 48:E006. doi: 10.1016/j.lfs.2020.117723
6. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. (2020) 8:475–81. doi: 10.1016/S2213-2600(20)30079-5
7. Liu R, Han H, Liu F, Lv Z, Wu K, Liu Y, et al. Positive rate of RT-PCR detection of SARS-CoV-2 infection in 4,880 cases from one hospital in Wuhan, China, from Jan to Feb 2020. Clin Chim Acta. (2020) 505:172–175. doi: 10.1016/j.cca.2020.03.009
8. Li LQ, Huang T, Wang YQ, Wang ZP, Liang Y, Huang TB, et al. 2019 novel coronavirus patients' clinical characteristics, discharge rate, and fatality rate of meta-analysis. J Med Virol. (2020) 3:12. doi: 10.1002/jmv.25757
9. Dong Y, Mo X, Hu Y, Qi X, Jiang F, Jiang Z, et al. Epidemiology of COVID-19 among children in China. Pediatrics. (2020) 145:e20200702. doi: 10.1542/peds.2020-0702
10. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. (2020) 20:727–33. doi: 10.1056/NEJMoa2001017
11. Lu S. Timely development of vaccines against SARS-CoV-2. Emerg Microbes Infect. (2020) 9:542–4. doi: 10.1080/22221751.2020.1737580
13. Tomar N, De RK. Immunoinformatics: a brief review. Methods Mol Biol. (2014) 1184:23–55. doi: 10.1007/978-1-4939-1115-8_3
14. Oli AN, Obialor WO, Ifeanyichukwu MO, Odimegwu DC, Okoyeh JN, Emechebe GO, et al. Immunoinformatics and vaccine development: an overview. Immunotargets Ther. (2020) 26:13–30. doi: 10.2147/ITT.S241064
15. Baruah V, Bose S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-now. J Med Virol. (2020) 2:5. doi: 10.1002/jmv.25698
16. Abraham PK, Srihansa T, Krupanidhi S, Vijaya SA, Venkateswarulu TC. Design of multi-epitope vaccine candidate against SARS-CoV-2: an in-silico study. J Biomol Struct Dyn. (2020) 1:1–9. doi: 10.1080/07391102.2020.1770127
17. Du L, He Y, Zhou Y, Liu S, Zheng BJ, Jiang S. The spike protein of sars-cov a target for vaccine and therapeutic development. Nat Rev Microbiol. (2009) 3:226–36. doi: 10.1038/nrmicro2090
18. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 3:6. doi: 10.1101/2020.02.19.956581
19. Czub M, Weingart H, Czub S, He R, Cao J. Evaluation of modified vaccinia virus Ankara based recombinant SARS vaccine in ferrets. Vaccine. (2005) 23:2273–9. doi: 10.1016/j.vaccine.2005.01.033
20. Liu WL, Michael LY, Chen C. Bioinformatics analysis of sars-cov m protein provides information for vaccine development. Progr Nat Sci. (2003) 11:1–7. doi: 10.1080/10020070312331344530
21. Sedeyn K, Schepens B, Saelens X. Respiratory syncytial virus non-structural proteins 1 and 2: exceptional disrupters of innate immune responses. PLoS Pathog. (2019) 10:e1007984. doi: 10.1371/journal.ppat.1007984
22. Pandey RK, Ojha R, Mishra A, Kumar Prajapati V. Designing B-cell and T-cell multi-epitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. J Cell Biochem. (2018) 119:7631–42. doi: 10.1002/jcb.27110
23. Pandey RK, Ali M, Ojha R, Bhatt TK, Prajapati VK. Development of multi-epitope driven subunit vaccine in secretory and membrane protein of Plasmodium falciparum to convey protection against malaria infection. Vaccine. (2018) 36:4555–65. doi: 10.1016/j.vaccine.2018.05.082
24. Pandey RK, Bhatt TK, Prajapati VK. Novel immunoinformatics approaches to design a multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci Rep. (2018) 3:4555–65. doi: 10.1038/s41598-018-19456-1
25. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 579:265–9. doi: 10.1038/s41586-020-2008-3
26. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 7798:270–3. doi: 10.1038/s41586-020-2012-7
27. Doytchinova IA, Flower DR. Identifying candidate subunit vaccines using an alignment-independent method based on principal amino acid properties. Vaccine. (2007) 25:866. doi: 10.1016/j.vaccine.2006.09.032
28. Doytchinova IA, Flower DR. Vaxijen: a server for prediction of protective antigens, tumor antigens, and subunit vaccines. BMC Bioinf . (2007) 8:4–10. doi: 10.1186/1471-2105-8-4
29. Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg ZAMJE. The phyre2 web portal for protein modeling, prediction, and analysis. Nat Protocol. (2015) 10:845–58. doi: 10.1038/nprot.2015.053
30. Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. (2012) 40:294–7. doi: 10.1093/nar/gks493
31. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modeling of protein structures and complexes. Nucleic Acids Res. (2018) 46:W296–303. doi: 10.1093/nar/gky427
32. Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic t-lymphocyte epitope prediction. BMC Bioinf . (2007) 8:424. doi: 10.1186/1471-2105-8-424
33. Larsen M, Lundegaard C, Lamberth K, Buus S, Brunak S, Lund O. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol. (2005) 35:2295–303. doi: 10.1002/eji.200425811
34. Lund O, Nielsen M, Kesmir C, Petersen AG, Lundegaard C, Worning P, et al. Definition of supertypes for HLA molecules using clustering of specificity matrices. Immunogenetics. (2004) 55:797–810. doi: 10.1007/s00251-004-0647-4
35. Rana A, Akhter Y. A multi-subunit based, thermodynamically stable model vaccine using combined immunoinformatics and protein structure-based approach. Immunobiology. (2015) 221:544–57. doi: 10.1016/j.imbio.2015.12.004
36. Nielsen M, Lundegaard C, Lund O, Can K. The role of the proteasome in generating cytotoxic t-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. (2005) 57:33–41. doi: 10.1007/s00251-005-0781-7
37. Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, et al. Peptide binding predictions for HLA DR, DP, and DQ molecules. BMC Bioinf . (2010) 11:568–670. doi: 10.1186/1471-2105-11-568
38. Nielsen M, Lund O. Nn-align. An artificial neural network-based alignment algorithm for MHC class ii peptide binding prediction. BMC Bioinf . (2009) 1:296–300. doi: 10.1186/1471-2105-10-296
39. Nielsen M, Lundegaard C, Lund O. Prediction of MHC class ii binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinf . (2007) 8:238. doi: 10.1186/1471-2105-8-238
40. Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, Sette A, et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. (2008) 4:2. doi: 10.1186/1745-7580-4-2
41. Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. (1999) 17:555–61. doi: 10.1038/9858
42. Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, Nielsen M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics. (2015) 67:641–50. doi: 10.1007/s00251-015-0873-y
43. Paul S, Lindestam Arlehamn CS, Scriba TJ, Dillon MB, Oseroff C, Hinz D, et al. Development and validation of a broad scheme for prediction of HLA class II-restricted T cell epitopes. J Immunol Methods. (2015) 422:28–34. doi: 10.1016/j.jim.2015.03.022
44. Russell CD, Unger SA, Walton M, Schwarze J. The human immune response to respiratory syncytial virus infection. Clin Microbiol Rev. (2017) 30:481–502. doi: 10.1128/CMR.00090-16
45. Nina C, Rupal O, Nazia K, Kumar PV. Scrutinizing, Mycobacterium tuberculosis membrane and secretory proteins to formulate multiepitope subunit vaccine against pulmonary tuberculosis by utilizing immunoinformatic approaches. Int J Biol Macromol. (2018) 118:180–8. doi: 10.1016/j.ijbiomac.2018.06.080
46. Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. (2013) 8:30. doi: 10.1186/1745-6150-8-30
47. Saha S, Raghava GPS. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins. (2006) 65:40–8. doi: 10.1002/prot.21078
48. Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. (2017) 45:W24–9. doi: 10.1093/nar/gkx346
49. Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. (2006) 2:2. doi: 10.1186/1745-7580-2-2
50. Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. (1978) 47:45–148. doi: 10.1002/9780470122921.ch2
51. Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. (1985) 55:836–9. doi: 10.1128/JVI.55.3.836-839.1985
52. Karplus PA, Schulz GE. Prediction of chain flexibility in proteins. Naturwissenschaften. (1985) 72:212–3. doi: 10.1007/BF01195768
53. Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. (1990) 276:172–4. doi: 10.1016/0014-5793(90)80535-Q
54. Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from residues with antigenicity and X-ray-derived accessible sites. Biochemistry. (1986) 25:5425–32. doi: 10.1021/bi00367a013
55. Tahir U, Qamar M, Saleem S, Ashfaq UA, Bari A, Anwar F, et al. Epitope-based peptide vaccine design and target site depiction against the Middle East Respiratory Syndrome Coronavirus: an immune-informatics study. J Transl Med. (2019) 17:362–232. doi: 10.1186/s12967-019-2116-8
56. Ferdous S, Kelm S, Baker TS, Shi J, Martin ACR. B-cell epitopes: discontinuity and conformational analysis. Mol Immunol. (2019) 114:643–50. doi: 10.1016/j.molimm.2019.09.014
57. Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics. (2008) 9:514. doi: 10.1186/1471-2105-9-514
58. Pandey RK, Ojha R, Aathmanathan VS, Krishnan M, Prajapati VK. Immunoinformatics approaches to design a novel multiepitope subunit vaccine against HIV infection. Vaccine. (2018) 36:2262–72. doi: 10.1016/j.vaccine.2018.03.042
59. Nezafat N, Ghasemi Y, Javadi G, Khoshnoud MJ, Omidinia E. A novel multi-epitope peptide vaccine against cancer: an in silico approach. Theor Biol. (2014) 349:121–34. doi: 10.1016/j.jtbi.2014.01.018
60. Barh D, Misra AN, Kumar A, Azevedo V. A novel strategy of epitope design in Neisseria gonorrhoeae. Bioinformation. (2010) 5:77–82. doi: 10.6026/97320630005077
61. Mohan T, Sharma C, Bhat AA, Rao DN. Modulation of HIV peptide antigen-specific cellular immune response by synthetic α-and β-defensin peptides. Vaccine. (2013) 31:1707–16. doi: 10.1016/j.vaccine.2013.01.041
62. Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. (1988) 6:1189–93. doi: 10.1016/0092-8674(88)90263-2
63. Saha S, Raghava GP. Nucleic Acids Res. Alfred: prediction of allergenic proteins and mapping of IgE epitopes. Mol Immunol. (2006) 34:202–9. doi: 10.1093/nar/gkl343
64. Nicolas R, Ole L, Massimo B, Filippo C. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE. (2010) 4:e9862. doi: 10.1371/journal.pone.0009862
65. Gasteiger E, Hoogland C, Gattiker A, Duval S, Wilkins MR, Appel RD, et al. Protein identification and analysis tools on the ExPASy server; the proteomics protocols handbook. Humana Press. (2005) 112:531–52. doi: 10.1385/1-59259-890-0:571
66. Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H. Template-based protein structure modeling using the Phyre 2 web server. Nat Protoc. (2012) 7:1511–22. doi: 10.1038/nprot.2012.085
67. González-Pech RA, Stephens TG, Chan CX. Commonly misunderstood parameters of NCBI BLAST and important considerations for users. Bioinformatics. (2019) 15:2697–8. doi: 10.1093/bioinformatics/bty1018
68. Markus W, Manfred J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res Sippl. (2007) 35:W407–10. doi: 10.1093/nar/gkm290
69. Vajda S, Yueh C, Beglov D, Bohnuud T, Mottarella SE, Xia B, et al. New additions to the ClusPro server motivated by CAPRI. Proteins. (2017) 85:435–44. doi: 10.1002/prot.25219
70. Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. (2005) 33:W363–7. doi: 10.1093/nar/gki481
71. Grote A, Hiller K, Scheer M, Munch R, Nortemann B, Hempel DC, et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. (2005) 33:W526–31. doi: 10.1093/nar/gki376
72. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. (2003) 13:3406–15. doi: 10.1093/nar/gkg595
73. Ojha R, Nandani R, Prajapati VK. Contriving multiepitope subunit vaccine by exploiting structural and non-structural viral proteins to prevent Epstein-Barr virus-associated malignancy. J Cell Physiol. (2019) 234:6437–48. doi: 10.1002/jcp.27380
74. Ikram A, Zaheer T, Awan FM, Obaid A, Naz A, Hanif R, et al. Exploring NS3/4A, NS5A, and NS5B proteins to design conserved subunit multi-epitope vaccine against HCV utilizing immunoinformatics approaches. Sci Rep. (2018) 8:16107. doi: 10.1038/s41598-018-34254-5
75. Sah R, Rodriguez-Morales AJ, Jha R, Chu DKW, Gu H, Peiris M, et al. Complete genome sequence of a 2019 novel coronavirus (SARS-CoV-2) strain isolated in Nepal. Microbiol Resour Announc. (2020) 9:e00169–20. doi: 10.1128/MRA.00169-20
76. Wang C, Liu Z, Chen Z, Huang X, Xu M, He T, et al. The establishment of a reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. (2020) 3:13. doi: 10.1002/jmv.25762
77. Asaf P, Dewi H, Matthew M, Michael SR, Lakshmi S, Richard BG. Sequence-based prediction of vaccine targets for inducing T cell responses to SARS-CoV-2 utilizing the bioinformatics predictor RECON. bioRxiv. (2020). doi: 10.1101/2020.04.06.027805
78. Jian H, Bienfang H, Peng Z. Mimotope-based prediction of B-cell epitopes. Methods Mol Biol. (2014) 1184:237–43. doi: 10.1007/978-1-4939-1115-8_13
79. Karpenko O, Huang L, Dai Y. A probabilistic meta-predictor for the MHC class II binding peptides. Immunogenetics. (2008) 1:25–36. doi: 10.1007/s00251-007-0266-y
80. Bhattacharya M, Sharma AR, Patra P, Ghosh P, Sharma G, Patra BC, et al. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J Med Virol. (2020) 2:28. doi: 10.1002/jmv.25736
81. Nagendra R, Hegde S, Gauthami HM, Sampath K, Jagadeesh B. The use of databases, data mining, and immunoinformatics in vaccinology: where are we? Exp Opin Drug Disc. (2017) 17:117–30. doi: 10.1080/17460441.2018.1413088
Keywords: immunoinformatics, epitope prediction, COVID-19, SARS-CoV-2, vaccine
Citation: Dong R, Chu Z, Yu F and Zha Y (2020) Contriving Multi-Epitope Subunit of Vaccine for COVID-19: Immunoinformatics Approaches. Front. Immunol. 11:1784. doi: 10.3389/fimmu.2020.01784
Received: 27 March 2020; Accepted: 03 July 2020;
Published: 28 July 2020.
Edited by:
Rashika El Ridi, Cairo University, EgyptReviewed by:
Ahmad Karkhah, Babol University of Medical Sciences, IranCopyright © 2020 Dong, Chu, Yu and Zha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Zha, emhheWFuNzJAMTI2LmNvbQ==
†ORCID: Rong Dong orcid.org/0000-0003-2372-9833
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.