 Asmita Pathak†
Asmita Pathak† Sanjay K. Singh
Sanjay K. Singh Douglas P. Thewke
Douglas P. Thewke Alok Agrawal
Alok Agrawal
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 11 August 2020
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01780
This article is part of the Research Topic Diagnostic and Therapeutic Applications of Pentraxin and Pentraxin-Associated Proteins View all 20 articles
The aim of this study was to test the hypothesis that C-reactive protein (CRP) protects against the development of atherosclerosis and that a conformational alteration of wild-type CRP is necessary for CRP to do so. Atherosclerosis is an inflammatory cardiovascular disease and CRP is a plasma protein produced by the liver in inflammatory states. The co-localization of CRP and low-density lipoproteins (LDL) at atherosclerotic lesions suggests a possible role of CRP in atherosclerosis. CRP binds to phosphocholine-containing molecules but does not interact with LDL unless the phosphocholine groups in LDL are exposed. However, CRP can bind to LDL, without the exposure of phosphocholine groups, if the native conformation of CRP is altered. Previously, we reported a CRP mutant, F66A/T76Y/E81A, generated by site-directed mutagenesis, that did not bind to phosphocholine. Unexpectedly, this mutant CRP, without any more conformational alteration, was found to bind to atherogenic LDL. We hypothesized that this CRP mutant, unlike wild-type CRP, could be anti-atherosclerotic and, accordingly, the effects of mutant CRP on atherosclerosis in atherosclerosis-prone LDL receptor-deficient mice were evaluated. Administration of mutant CRP into mice every other day for a few weeks slowed the progression of atherosclerosis. The size of atherosclerotic lesions in the aorta of mice treated with mutant CRP for 9 weeks was ~40% smaller than the lesions in the aorta of untreated mice. Thus, mutant CRP conferred protection against atherosclerosis, providing a proof of concept that a local inflammation-induced structural change in wild-type CRP is a prerequisite for CRP to control the development of atherosclerosis.
Atherosclerosis is a chronic inflammatory disease whose development begins with the dysfunction of the endothelium of the arteries. Endothelial dysfunction leads to infiltration of plasma low-density lipoprotein (LDL) and monocytes in the arterial wall. LDL is subsequently deposited, modified and engulfed by monocyte-derived macrophages. Lipid-laden macrophage foam cells are proinflammatory which enhances the process of the formation of atherosclerotic lesions (1, 2). The inflammatory microenvironment of atherosclerotic lesions is characterized by macrophage activation, hypoxia, and lactate and proton generation, resulting in an acidic extracellular pH (3–7).
C-reactive protein (CRP) is a plasma protein produced by the liver in inflammatory states (8). CRP is also present, co-localized with modified LDL and macrophages, at atherosclerotic lesions in both humans and experimental animals (9, 10). CRP is composed of five identical subunits arranged in a pentameric symmetry. The molecular weight of each subunit is ~23 kDa and there are 206 amino acid residues in each subunit. CRP binds to molecules with exposed phosphocholine (PCh) groups in a Ca2+-dependent manner. There are five PCh-binding sites in the CRP pentamer, one site per subunit (11, 12).
Oxidized LDL (ox-LDL), enzymatically-modified LDL (E-LDL) and acetylated LDL (ac-LDL) are different forms of modified atherogenic LDL that are used in in vitro experiments (1, 2, 13). In the presence of Ca2+, native or recombinant wild-type (WT) CRP interacts with E-LDL due to the exposure of PCh groups on E-LDL, but does not interact with ox-LDL and ac-LDL (14–16). In the absence of Ca2+, WT CRP does not interact with any form of atherogenic LDL (16–19). The native pentameric structure of CRP is altered in response to a variety of experimental conditions (16–23). It has been shown that the pentameric structure of WT CRP is subtly altered by acidic pH and by oxidation, and conformationally altered CRP is capable of binding to atherogenic LDL independent of the PCh-binding site (17–19). Employing E-LDL, it has also been shown that if CRP is bound to atherogenic LDL, it prevents the formation of lipid-laden macrophage foam cells (24). Due to the presence of CRP at atherosclerotic lesions and due to the binding capability of CRP for atherogenic LDL under certain conditions, CRP has been implicated in the development of atherosclerosis (25).
We have previously reported a CRP mutant, F66A/T76Y/E81A, in which Phe66, Thr76, and Glu81 (three critical amino acid residues in the PCh-binding site) were substituted with Ala, Tyr, and Ala, respectively, to abolish the PCh-binding activity of CRP (26). Mutant CRP did not bind to PCh and was used as a tool to investigate the importance of the PCh-binding site in CRP-mediated protection of mice against pneumococcal infection (26). Biochemical characterization of this mutant CRP, in comparison to that of WT CRP, has been published previously (26). The overall structure of mutant CRP was pentameric and the mutation did not affect the stability of the protein in vivo. Mutant CRP circulated freely in the mouse serum and its rate of clearance in vivo was similar to that of WT CRP (26).
Further analysis of F66A/T76Y/E81A mutant CRP, presented here, revealed that mutant CRP had inadvertently gained the ability to bind to any protein that was immobilized on microtiter plates, including atherogenic forms of LDL, without the need for any further inflammatory milieu-dependent or acidic pH-induced structural change. We, therefore, hypothesized that mutant CRP might show an atheroprotective effect in murine models of atherosclerosis. We reasoned that even if there was no acidic pH around injected mutant CRP in the available murine models, mutant CRP would be able to recognize and bind atherogenic LDL and exert a protective effect on the development of atherosclerosis. Accordingly, in this study, we evaluated the effects of mutant CRP (F66A/T76Y/E81A) on the development of atherosclerosis employing LDL receptor-deficient (Ldlr−/−) mice, an animal model commonly used to investigate molecules involved in human atherosclerosis.
The construction of F66A/T76Y/E81A mutant CRP cDNA used in this study has been reported previously (26). Mutant CRP was expressed in CHO cells using the ExpiCHO Expression System (Thermo Fisher Scientific), according to manufacturer's instructions. As described previously (26), mutant CRP was purified from cell culture supernatant by Ca2+-dependent affinity chromatography on a phosphoethanolamine-Sepharose column, since this CRP mutant does not bind to PCh. Mutant CRP was further purified by gel filtration on a Superose12 column. Eluted mutant CRP was immediately dialyzed against 10 mM Tris-HCl, 150 mM NaCl, pH 7.2 (TBS), containing 2 mM CaCl2, stored at 4°C, and was used within a week. The purity of CRP was confirmed by using denaturing SDS-PAGE. For in vivo experiments, purified mutant CRP was treated with the Detoxi-Gel Endotoxin Removing Gel (Thermo Fisher Scientific) according to manufacturer's instructions. The removal of endotoxin from mutant CRP preparations was confirmed by using the Limulus Amebocyte Lysate kit QCL-1000 (Lonza) according to manufacturer's instructions.
The solid phase ligand-binding assay was used to determine the binding of mutant CRP to immobilized proteins, as described earlier (17–19). Ox-LDL, E-LDL, ac-LDL, factor H (Complement Technology) and amyloid β peptide 1-42 (Bachem, H-1368) were used as protein ligands. Ox-LDL, E-LDL, and ac-LDL were prepared as described previously (17–19, 24). Briefly, microtiter wells were coated with protein ligands (10 μg/ml) diluted in TBS and incubated overnight at 4°C. The unreacted sites in the wells were blocked with TBS containing 0.5% gelatin. Freshly purified mutant CRP was diluted in TBS-Ca (TBS containing 2 mM CaCl2, 0.1% gelatin and 0.02% Tween 20), added to the wells, and incubated overnight at 4°C. Bound mutant CRP was detected by using a polyclonal rabbit anti-human CRP antibody (Millipore Sigma, 235752). HRP-conjugated donkey anti-rabbit IgG (GE Healthcare) was used as the secondary antibody. Color was developed using ABTS as the substrate and the OD405 was read in a plate reader.
Eight-week-old C57BL/6 male ldlr−/− mice (Jackson Lab, 002207) were used in experiments according to protocols approved by and conducted in accordance with the guidelines administered by the Institutional Animal Care and Usage Committee of East Tennessee State University. Sixty mice were fed on a high-fat western-type diet (21% fat, 0.2% cholesterol), purchased from Envigo (TD.88137), for 10 weeks. After 1 week on high-fat diet, mice were divided into two groups of 30 mice in each group: untreated group and mutant CRP-treated group. Mice were injected with either TBS (untreated group) or mutant CRP (50 μg/injection) on alternate days for 9 weeks, via alternating intravenous and intraperitoneal routes (Figure 1A). Blood, heart and aorta were collected at five different time points (weeks 1, 3, 5, 7, and 9), as described previously (27). Briefly, six mice from each group, at each time point, were sacrificed, blood was collected by cardiac puncture, and the plasma was separated and stored frozen. After collecting blood, the heart was perfused with 10% formalin, followed by removing fat from the entire aorta. Heart was excised from the aorta, was cut into two halves, and the upper half with the aortic root was mounted using OCT medium and stored at −80°C. Following heart excision, aorta was excised and stored in 10% formalin at 4°C. The whole experiment was repeated once more employing another 60 mice and a separate, freshly purified batch of mutant CRP.
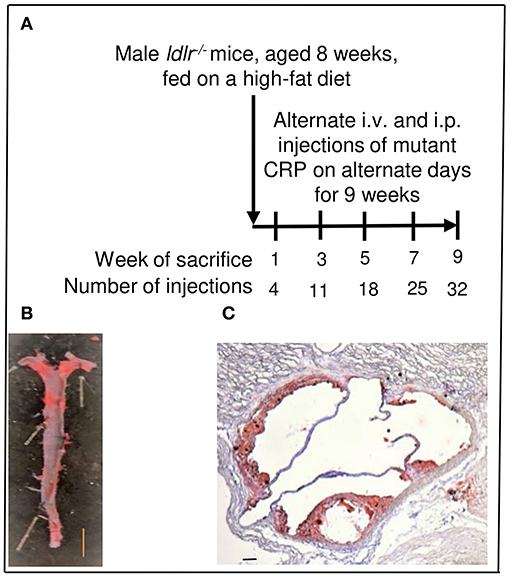
Figure 1. Assessment of atherosclerotic lesions in ldlr−/− mice. (A) Protocol for administration of mutant CRP and for sacrifice of mice after mutant CRP administration. (B) Quantitation of atherosclerotic lesions in the aorta (en face). A representative Sudan IV-stained aorta is shown. The red colored areas are the lesions. Scale bar, 5 mm. (C) Quantitation of atherosclerotic lesions in the aortic root. A representative Oil Red O-stained aortic root section is shown. The red colored areas are the lesions. Scale bar, 100 μm.
To measure the size of atherosclerotic lesions in the whole aorta (en face), aorta was cut open longitudinally, stained with Sudan IV, and digitally photographed, as described previously (27). A total of 120 aortae (60 mice for untreated group and 60 mice for mutant CRP-treated group; 12 mice per time point) were processed, stained and photographed. A representative Sudan IV-stained aorta is shown in Figure 1B.
To measure the size of atherosclerotic lesions in the aortic root, 8 μm cross-sections of frozen OCT-embedded heart were collected from the appearance of the aortic valve leaflets to their disappearance (48–72 sections per heart). Every other cross-section of the entire aortic root was stained with Oil Red O for lipids, counterstained with hematoxylin, and digitally photographed, as described previously (27). A total of 120 hearts (60 mice for untreated group and 60 mice for mutant CRP-treated group; 12 mice per time point) were processed. A representative Oil Red O-stained aortic root section is shown in Figure 1C.
Digital photographs were acquired with an Olympus BX41 microscope equipped with a CCD color camera (QImaging). The stained lesion areas were quantified in digital images using the ImageJ software (28). Quantification of the lesion areas in the photographs was performed by two observers blinded to the experimental protocol. Non-parametric test (Mann-Whitney test) using GraphPad Prism software was employed to calculate the p-values.
Sudan IV-stained aorta was first processed to remove the stain, as described earlier (29). Immunostaining of CRP was performed using Vectastain ABC Elite kit (Vector laboratories, PK-6100) according to manufacturer's instructions. CRP was detected by using a polyclonal rabbit anti-human CRP antibody (Millipore Sigma, 235752). Biotinylated goat-anti rabbit IgG was used as the secondary antibody. Color was developed using DAB (Vector laboratories, ImmPACT DAB, SK-4105) as the substrate, according to manufacturer's instructions.
The concentrations of high-density lipoprotein (HDL) and LDL in the plasma were measured using Cholesterol Assay Kit-HDL and LDL/VLDL (Abcam; ab65390) according to manufacturer's instructions. Lipoprotein levels were measured in the pooled plasma samples collected at weeks 1, 3, 5, 7, and 9 (12 mice per time point). Unpaired student t-test was employed to calculate the p-values.
All experiments were performed three times, unless otherwise mentioned, and comparable results were obtained each time. Results of a representative experiment are shown in the figures where the raw data (OD405) were used to plot the curves.
As shown in Figure 2, mutant CRP bound to all three forms of atherogenic LDL immobilized on microtiter wells. The binding occurred at physiological conditions, that is, in the absence of any protein structure-modifying agent, such as acidic pH. Since mutant CRP does not bind to PCh (26), the binding of mutant CRP to either ox-LDL, E-LDL, or ac-LDL was independent of the PCh groups present in atherogenic LDL. In addition to testing the binding of mutant CRP to atherogenic LDL, we also included two other proteins, factor H and amyloid β peptide, in the binding assay. Mutant CRP also bound to factor H and amyloid β peptide in a concentration-dependent manner, similar to its binding to atherogenic LDL. These results suggest that mutant CRP did not recognize immobilized atherogenic LDL per se, but it recognized a pattern, as yet undefined, on immobilized proteins in general.
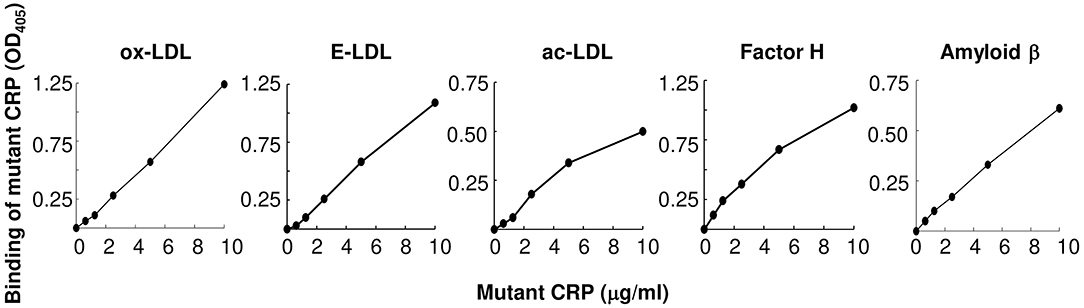
Figure 2. Binding of F66A/T76Y/E81A mutant CRP to immobilized proteins including atherogenic LDL at physiological pH. Microtiter wells were coated with protein ligands as shown. After blocking the unreacted sites in the wells, mutant CRP diluted in TBS-Ca was added to the wells and incubated for 2 h at 37°C. Bound mutant CRP was detected by using a rabbit anti-human CRP antibody and HRP-conjugated donkey anti-rabbit IgG. Color was developed and the OD was read at 405 nm. A representative of three experiments are shown.
The total size of all lesion areas in the whole aorta (en face) was measured. Figure 3 shows the combined results of two separate experiments with 12 mice in each group, for each time point. There was no effect of mutant CRP on the en face lesions for the first 5 weeks. The effect of mutant CRP on the lesion size was visible once the disease had progressed further. In comparison to untreated mice, the lesion area in mutant CRP-treated mice was 30.9% less after 7 weeks and 42% less after 9 weeks of CRP administration. As shown, in mutant CRP-treated mice, the lesion area did not increase after 5 weeks unlike in the untreated group where the lesion area kept increasing for another 2 weeks. Similar results were seen when the data from each of the two experiments (6 mice/group/time point/experiment) were analyzed separately (Supplemental Figure 1). We did not determine the specific stages of atherosclerosis at any time point.
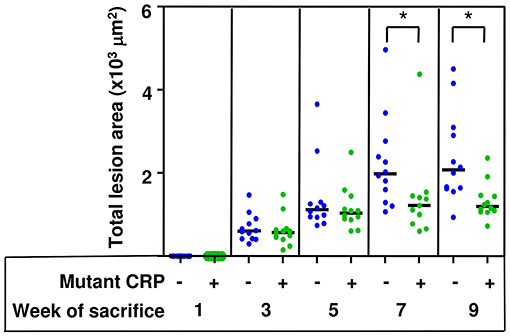
Figure 3. Reduction in the size of atherosclerotic lesions in the aorta of mutant CRP-treated mice. The scatter plot shows the total atherosclerotic lesion area in en face aorta from untreated and mutant CRP-treated mice. Data were collected at five different time points: 1, 3, 5, 7, and 9 weeks after mutant CRP administration (12 mice per group per time point). Each blue dot represents one mouse from the untreated group and each green dot represents one mouse from the mutant CRP-treated group. Horizontal black lines indicate the median total lesion area for each group. Asterisks denote statistically significant difference between untreated and mutant CRP-treated groups (*p ≤ 0.01).
The total size of all lesion areas in the aortic root was measured. Figure 4 shows the combined results of two separate experiments using 12 mice in each group, for each time point. As shown, the disease progressed for the entire duration of 9 weeks in untreated mice. However, the administration of mutant CRP did not affect the lesion size at any time point. There was no statistically significant difference in the lesion size at any time point between untreated and mutant CRP-treated groups of mice. Similar results were seen when the data from each of the two experiments (6 mice/group/time point/experiment) were analyzed separately (Supplemental Figure 2).
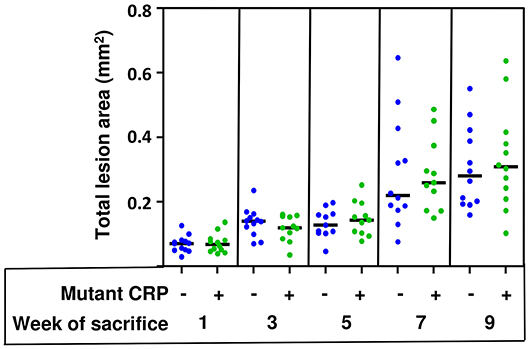
Figure 4. No reduction in the size of atherosclerotic lesions in the aortic root of mutant CRP-treated mice. The scatter plot shows the total atherosclerotic lesion area in aortic root sections from untreated and mutant CRP-treated mice. Data were collected at five different time points: 1, 3, 5, 7, and 9 weeks after mutant CRP administration (12 mice per group per time point). Each blue dot represents one mouse from the untreated group and each green dot represents one mouse from the mutant CRP-treated group. Horizontal black lines indicate the median total aortic root lesion area for each group.
Immunostaining of CRP in the aorta isolated from mice treated with mutant CRP for seven weeks was performed to confirm the presence of administered mutant CRP at the en face atherosclerotic lesions in mutant CRP-treated mice. As shown (Figure 5), the lesions in the aorta of mutant CRP-treated mice were stained while the lesions in the aorta of untreated mice did not stain for CRP. The polyclonal anti-human CRP antibody used in this study does not react with purified murine CRP (data not shown), consistent with the previously published report that anti-human CRP antibodies do not react with murine CRP (30).

Figure 5. Immunostaining of mutant CRP in the aorta. (A) A representative aorta from untreated mice stained for mutant CRP. (B) A representative aorta from mice treated with mutant CRP for 7 weeks, stained for CRP. 1–4 represent an aorta through different stages of staining. (1) Sudan IV-stained aorta. Red colored areas are the lesions. (2) Aorta post-dehydration to remove Sudan IV. White colored areas are the lesion areas. (3) Anti-human CRP antibody-stained aorta. Brown colored areas reflect the presence of mutant CRP in the lesions. (4) Magnified aortic arch area of the anti-human CRP antibody-stained aorta. Red boxes show the lesion areas.
Injecting a total of 1.6 mg of mutant CRP into mouse, in 32 injections over a period of 9 weeks, did not alter the concentration of lipoproteins in the circulation. As shown in Table 1, the concentrations of LDL and HDL in the plasma of untreated and mutant CRP-treated mice were comparable. There was no statistically significant difference in the levels of either LDL or HDL in the plasma at any time point between untreated and mutant CRP-treated groups of mice. Administration of mutant CRP did not affect the body weight. Body weights of untreated and mutant CRP-treated mice were similar at each time point (data not shown).
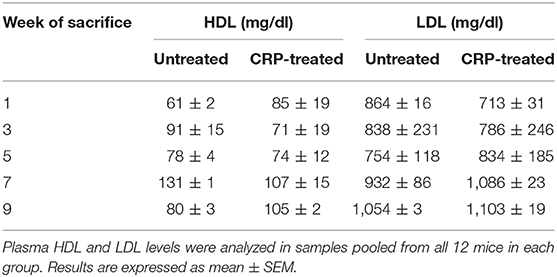
Table 1. Concentration of lipoproteins in the plasma.
In this study, we investigated the effects of mutant CRP F66A/T76Y/E81A, capable of binding to atherogenic LDL but incapable of binding to PCh, on the development of atherosclerosis in male ldlr−/− mice. Our major finding was that the administration of mutant CRP for 7 weeks slowed the progression of atherosclerosis. The total size of atherosclerotic lesions in the whole aorta of mice treated with mutant CRP on alternate days for 7 weeks and beyond was significantly smaller (~40%) than the lesions in the aorta of untreated mice.
Previously, the role of WT CRP in atherosclerosis had been explored, employing a variety of experimental strategies, and 12 papers have been published using CRP from man, mouse and rabbit in both murine and rabbit models of atherosclerosis (31–42). In these studies, both, normal mice and CRP-deficient mice have been employed. Both, normal rabbits and rabbits in which CRP was inhibited by using anti-sense technology have been employed. Both, passively administered CRP and transgenic human CRP have been employed. Three different types of atherosclerosis-prone mice, apoE−/−, ldlr−/−, and apoB100/100 ldlr−/−, have been used in these studies. Two of the 12 papers indicated that both human and murine CRP might be playing an atheroprotective role: human CRP was shown to slow the development of atherosclerosis in the apoB100/100 ldlr−/− mice (36), and the lesion size in CRP-deficient mice on apoE−/− or ldlr−/− background was either equivalent or increased compared to that in normal mice (41). In the other 10 papers, regardless of the experimental strategy used, WT CRP from all species did not show any effect on the development of atherosclerosis in animals, suggesting that WT CRP is neither pro-atherogenic nor anti-atherogenic (25, 43).
The most logical explanation for the observations of zero or nominal effects of WT CRP (31–42) on atherosclerosis in animal models is that the animal models of atherosclerosis do not possess the required inflammatory microenvironment that is needed by CRP to change its structure and be able to interact with atherogenic LDL (10, 18, 25). WT CRP shows no effect because pH near the lesions may not be acidic in animal models and, therefore, the structure of administered or endogenous WT CRP remains unchanged (10, 18, 25). WT CRP treated with acidic pH in vitro was unsuitable for administration into blood circulation of animals since the acidic pH-induced conformational alteration in the pentameric structure of CRP was found to be reversible at physiological pH (17). Therefore, we hypothesized that an in vitro-generated mutant CRP, capable of binding to atherogenic LDL without the requirement of any structural modification in vivo, would be suitable to investigate the mechanism of action of CRP on the development of the disease in animal models (10, 16, 18, 25). Indeed, in one study, monomeric CRP, that is also capable of binding to atherogenic LDL, was employed and the results showed that monomeric CRP was protective against atherosclerosis in apoE−/− mice (34). Thus, our current findings using mutant CRP and previous findings using monomeric CRP, both molecules capable of binding to atherogenic LDL, indicate that structurally altered CRP protects against atherosclerosis; it is just that WT CRP does not exert a protective effect in most animal models (25, 44).
The effect of mutant CRP on the size of atherosclerotic lesions in the en face aorta was obvious since we found that administered mutant CRP had reached the aorta. The staining of aortic roots for the presence of administered mutant CRP provided inconclusive results (data not shown). It is assumed that if mutant CRP reached the aorta, it also reached the aortic root, and if this assumption is correct, then the effect of mutant CRP was site-specific. Site-specific effects of experimental manipulations have been observed in other studies using ldlr−/− mice where the disease developed differently at various lesion-prone sites (45–50). Since the effects of mutant CRP were still observed after week 9 in the en face aorta, it is unlikely that anti-CRP antibodies were produced in response to intravenous administration of mutant CRP that could have inhibited its functions.
The topology of the LDL-binding site and the number of LDL-binding sites on mutant CRP remain undefined. Two possible mechanisms have been proposed for the interaction between conformationally altered CRP and atherogenic LDL. The intrinsically disordered region present in CRP has been shown to participate in the binding of monomeric CRP and atherogenic LDL (51). We proposed that the loosening of the CRP pentamer contributed to the formation of the LDL-binding site (16–18, 25). Recently, it has been suggested that the pentameric assembly of CRP harbors a pronounced plasticity in inter-subunit interactions, which may form the basis for a reversible activation of CRP in inflammation (52). However, it is unknown whether the intrinsically disordered region was exposed or the pentamer was loosened in mutant CRP employed in this study. We speculate that there is only one LDL-binding site per CRP pentamer. However, for the protection of mice against atherosclerosis, it is possible that both sites, the PCh-binding site and the LDL-binding site, participate. The search for a new mutant CRP which can bind to both PCh and to atherogenic LDL is in progress. Any possible similarity between mutant CRP used in this study and previously reported conformationally altered pentameric forms of CRP, pCRP*, and mCRPm (20, 21), is unknown.
Based on the data obtained from a single regimen for mutant CRP treatment, we conclude that one of the functions of CRP is to confer protection against atherosclerosis. We propose, again, that there is no need to stop the biosynthesis of CRP, and that a drug that can lower cholesterol level but not CRP levels could be superior to statins which reduce both CRP and cholesterol levels (53, 54). These suggestions are supported by the fact that, in rabbits, the inhibition of plasma CRP did not affect the development of atherosclerosis (42). It has also been previously postulated that the deposition of CRP at the atherosclerotic lesions may be independent of the CRP levels in the circulation and that CRP-mediated lipoprotein removal likely underlies the regression of early lesions which occurs continuously throughout life and that CRP should be considered as an anti-atherosclerotic protein (10, 13). A long-term goal should be the discovery and design of small-molecule compounds to aid endogenous native CRP in capturing atherogenic LDL, as proposed earlier (14, 25). Another goal could be to investigate the possible protective effects of mutant CRP used in this study in animal models of other inflammatory diseases.
Our data also provide a proof of concept that a local inflammation-induced structural change in native CRP is a prerequisite for CRP to control the development of atherosclerosis. An appropriate inflammatory microenvironment at the site of LDL deposition seems to be critical for CRP to prevent atherosclerosis. One function of inflammation could be to change the structure of proteins, including CRP. Inflammation is not a silent killer, perhaps, as has been suggested (55).
All datasets generated for this study are included in the article/Supplementary Material.
The animal study was reviewed and approved by University Committee on Animal Care, East Tennessee State University.
AP, SS, and DT performed the experiments. AP and AA analyzed the data. AA conceived and designed the experiments and wrote the paper. All authors contributed to the article and approved the submitted version.
This work was supported by National Institutes of Health Grant AR068787.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01780/full#supplementary-material
CRP, C-reactive protein; LDL, Low-density lipoprotein; Ldlr−/−, LDL receptor-deficient; PCh, Phosphocholine; WT, wild-type.
1. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
2. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. (2009) 27:165–97. doi: 10.1146/annurev.immunol.021908.132620
3. Leake DS. Does an acidic pH explain why low-density lipoprotein is oxidised in atherosclerotic lesions? Atherosclerosis. (1997) 129:149–57. doi: 10.1016/S0021-9150(96)06035-2
4. Björnheden T, Levin M, Evaldsson M, Wiklund O. Evidence of hypoxic areas within the arterial wall in vivo. Arterioscler Thromb Vasc Biol. (1999) 19:870–6. doi: 10.1161/01.ATV.19.4.870
5. Naghavi M, John R, Naguib S, Siadaty MS, Grasu R, Kurian KC, et al. pH heterogeneity of human and rabbit atherosclerotic plaques; a new insight into detection of vulnerable plaque. Atherosclerosis. (2002) 164:27–35. doi: 10.1016/S0021-9150(02)00018-7
6. Sneck M, Kovanen PT, Öörni K. Decrease in pH strongly enhances binding of native, proteolyzed, lipolyzed, and oxidized low density lipoprotein particles to human aortic proteoglycans. J Biol Chem. (2005) 280:37449–54. doi: 10.1074/jbc.M508565200
7. Haka AS, Grosheva I, Chiang E, Buxbaum AR, Baird BA, Pierini LM, et al. Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. Mol Biol Cell. (2009) 20:4932–40. doi: 10.1091/mbc.e09-07-0559
8. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. (1999) 340:448–54. doi: 10.1056/NEJM199902113400607
9. Sun H, Koike T, Ichikawa T, Hatakeyama K, Shiomi M, Zhang B, et al. C-reactive protein in atherosclerotic lesions: Its origin and pathophysiological significance. Am J Pathol. (2005) 167:1139–48. doi: 10.1016/S0002-9440(10)61202-3
10. Agrawal A, Hammond DJ Jr, Singh SK. Atherosclerosis-related functions of C-reactive protein. Cardiovasc Haematol Disord Drug Targets. (2010) 10:235–40. doi: 10.2174/187152910793743841
11. Volanakis JE, Kaplan MH. Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc Soc Exp Biol Med. (1971) 136:612–4. doi: 10.3181/00379727-136-35323
12. Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. (1999) 7:169–77. doi: 10.1016/S0969-2126(99)80023-9
13. Torzewski M. Enzymatically modified LDL, atherosclerosis and beyond: paving the way to acceptance. Front Biosci. (2018) 23:1257–71. doi: 10.2741/4642
14. Bhakdi S, Torzewski M, Klouche M, Hemmes M. Complement and atherogenesis: binding of CRP to degraded, nonoxidized LDL enhances complement activation. Arterioscler Thromb Vasc Biol. (1999) 19:2348–54. doi: 10.1161/01.ATV.19.10.2348
15. Chang M-K, Binder CJ, Torzewski M, Witztum JL. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci USA. (2002) 99:13043–8. doi: 10.1073/pnas.192399699
16. Agrawal A, Gang TB, Rusiñol AE. Recognition functions of pentameric C-reactive protein in cardiovascular disease. Mediators Inflamm. (2014) 2014:319215. doi: 10.1155/2014/319215
17. Hammond DJ Jr, Singh SK, Thompson JA, Beeler BW, Rusiñol AE, Pangburn MK, et al. Identification of acidic pH-dependent ligands of pentameric C-reactive protein. J Biol Chem. (2010) 285:36235–44. doi: 10.1074/jbc.M110.142026
18. Singh SK, Thirumalai A, Hammond DJ Jr, Pangburn MK, Mishra VK, Johnson DA, et al. Exposing a hidden functional site of C-reactive protein by site-directed mutagenesis. J Biol Chem. (2012) 287:3550–8. doi: 10.1074/jbc.M111.310011
19. Singh SK, Thirumalai A, Pathak A, Ngwa DN, Agrawal A. Functional transformation of C-reactive protein by hydrogen peroxide. J Biol Chem. (2017) 292:3129–36. doi: 10.1074/jbc.M116.773176
20. Ji S-R, Wu Y, Zhu L, Potempa LA, Sheng F-L, Lu W, et al. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRPm. FASEB J. (2007) 21:284–94. doi: 10.1096/fj.06-6722com
21. Braig D, Nero TL, Koch H-G, Kaiser B, Wang X, Thiele JR, et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat Commun. (2017) 8:14188. doi: 10.1038/ncomms14188
22. Li S-L, Feng J-R, Zhou H-H, Zhang C-M, Lv G-B, Tan Y-B, et al. Acidic pH promotes oxidation-induced dissociation of C-reactive protein. Mol Immunol. (2018) 104:47–53. doi: 10.1016/j.molimm.2018.09.021
23. McFadyen JD, Zeller J, Potempa LA, Pietersz GA, Eisenhardt SU, Peter K. C-reactive protein and its structural isoforms: an evolutionary conserved marker and central player in inflammatory diseases and beyond. Subcell Biochem. (2020) 94:499–520. doi: 10.1007/978-3-030-41769-7_20
24. Singh SK, Suresh MV, Prayther DC, Moorman JP, Rusiñol AE, Agrawal A. C-reactive protein-bound enzymatically modified low-density lipoprotein does not transform macrophages into foam cells. J Immunol. (2008) 180:4316–22. doi: 10.4049/jimmunol.180.6.4316
25. Singh SK, Agrawal A. Functionality of C-reactive protein for atheroprotection. Front Immunol. (2019) 10:1655. doi: 10.3389/fimmu.2019.01655
26. Gang TB, Hammond DJ Jr, Singh SK, Ferguson DA Jr, Mishra VK, Agrawal A. The phosphocholine-binding pocket on C-reactive protein is necessary for initial protection of mice against pneumococcal infection. J Biol Chem. (2012) 287:43116–25. doi: 10.1074/jbc.M112.427310
27. Netherland CD, Pickle TG, Bales A, Thewke DP. Cannabinoid receptor type 2 (CB2) deficiency alters atherosclerotic lesion formation in hyperlipidemic Ldlr-null mice. Atherosclerosis. (2010) 213:102–8. doi: 10.1016/j.atherosclerosis.2010.07.060
28. Schneider CA, Rasband WS, Eliceiri KW. NIH image to imageJ: 25 years of image analysis. Nat Methods. (2012) 9:671–5. doi: 10.1038/nmeth.2089
29. Fu Z, Yan K, Rosenberg A, Jin Z, Crain B, Athas G, et al. Improved protein extraction and protein identification from archival formalin-fixed paraffin-embedded human aortas. Proteomics Clin Appl. (2013) 7:217–24. doi: 10.1002/prca.201200064
30. Maudsley S, Pepys MB. Immunochemical cross-reactions between pentraxins of different species. Immunology. (1987) 62:17–22.
31. Paul A, Ko KWS, Li L, Yechoor V, McCrory MA, Szalai AJ, et al. C-reactive protein accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Circulation. (2004) 109:647–55. doi: 10.1161/01.CIR.0000114526.50618.24
32. Hirschfield GM, Gallimore JR, Kahan MC, Hutchinson WL, Sabin CA, Benson GM, et al. Transgenic human C-reactive protein is not proatherogenic in apolipoprotein E-deficient mice. Proc Natl Acad Sci USA. (2005) 102:8309–14. doi: 10.1073/pnas.0503202102
33. Reifenberg K, Lehr H-A, Baskal D, Wiese E, Schaefer SC, Black S, et al. Role of C-reactive protein in atherogenesis: can the apolipoprotein E knockout mouse provide the answer? Arterioscler Thromb Vasc Biol. (2005) 25:1641–6. doi: 10.1161/01.ATV.0000171983.95612.90
34. Schwedler SB, Amann K, Wernicke K, Krebs A, Nauck M, Wanner C, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation. (2005) 112:1016–23. doi: 10.1161/CIRCULATIONAHA.105.556530
35. Trion A, de Matt MPM, Jukema JW, van der Laarse A, Mass MC, Offerman EH, et al. No effect of C-reactive protein on early atherosclerosis development in apolipoprotein E*3-leiden/human C-reactive protein transgenic mice. Arterioscler Thromb Vasc Biol. (2005) 25:1635–40. doi: 10.1161/01.ATV.0000171992.36710.1e
36. Kovacs A, Tornvall P, Nilsson R, Tegnér J, Hamsten A, Björkegren J. Human C-reactive protein slows atherosclerosis development in a mouse model with human-like hypercholesterolemia. Proc Natl Acad Sci USA. (2007) 104:13768–73. doi: 10.1073/pnas.0706027104
37. Tennent GA, Hutchinson WL, Kahan MC, Hirschfield GM, Gallimore JR, Lewin J, et al. Transgenic human CRP is not pro-atherogenic, pro-atherothrombotic or pro-inflammatory in apoE-/- mice. Atherosclerosis. (2008) 196:248–55. doi: 10.1016/j.atherosclerosis.2007.05.010
38. Torzewski M, Reifenberg K, Cheng F, Wiese E, Küpper I, Crain J, et al. No effect of C-reactive protein on early atherosclerosis in LDLR-/-/human C-reactive protein transgenic mice. Thromb Haemost. (2008) 99:196–201. doi: 10.1160/TH07-10-0595
39. Koike T, Kitajima S, Yu Y, Nishijima K, Zhang J, Ozaki Y, et al. Human C-reactive protein does not promote atherosclerosis in transgenic rabbits. Circulation. (2009) 120:2088–94. doi: 10.1161/CIRCULATIONAHA.109.872796
40. Ortiz MA, Campana GL, Woods JR, Boguslawski G, Sosa MJ, Walker CL, et al. Continously-infused human C-reactive protein is neither proatherosclerotic nor proinflammatory in apolipoprotein E-deficient mice. Exp Biol Med. (2009) 234:624–31. doi: 10.3181/0812-RM-347
41. Teupser D, Weber O, Rao TN, Sass K, Thiery J, Fehling HJ. No reduction of atherosclerosis in C-reactive protein (CRP)-deficient mice. J Biol Chem. (2011) 286:6272–9. doi: 10.1074/jbc.M110.161414
42. Yu Q, Liu Z, Waqar AB, Ning B, Yang X, Shiomi M, et al. Effects of antisense oligonucleotides against C-reactive protein on the development of atherosclerosis in WHHL rabbits. Mediators Inflamm. (2014) 2014:979132. doi: 10.1155/2014/979132
43. Badimon L, Peña E, Arderiu G, Padró T, Slevin M, Vilahur G, et al. C-reactive protein in atherothrombosis and angiogenesis. Front Immunol. (2018) 9:430. doi: 10.3389/fimmu.2018.00430
44. Boncler M, Wu Y, Watala C. The multiple faces of C-reactive protein - physiological and pathophysiological implications in cardiovascular disease. Molecules. (2019) 24:2062. doi: 10.3390/molecules24112062
45. Babaev VR, Patel MB, Semenkovich CF, Fazio S, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in low density lipoprotein receptor-deficient mice. J Biol Chem. (2000) 275:26293–9. doi: 10.1074/jbc.M002423200
46. Schiller NK, Kubo N, Boisvert WA, Curtiss LK. Effect of γ-irradiation and bone marrow transplantation on atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2001) 21:1674–80. doi: 10.1161/hq1001.096724
47. King VL, Szilvassy SJ, Daugherty A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor-/- mice. Arterioscler Thromb Vasc Biol. (2002) 22:456–61. doi: 10.1161/hq0302.104905
48. VanderLaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: Site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. (2004) 24:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0
49. Goel R, Schrank BR, Arora S, Boylan B, Fleming B, Miura H, et al. Site-specific effects of PECAM-1 on atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2008) 28:1996–2002. doi: 10.1161/ATVBAHA.108.172270
50. Melgar-Lesmes P, Sánchez-Herrero A, Lozano-Juan F, de la Torre Hernández JM, Montell E, Jiménez W, et al. Chondroitin sulphate attenuates atherosclerosis in apoE knockout mice involving cellular regulation of the inflammatory response. Thromb Haemost. (2018) 118:1329–39. doi: 10.1055/s-0038-1657753
51. Li H-Y, Wang J, Meng F, Jia Z-K, Su Y, Bai Q-F, et al. An intrinsically disordered motif mediates diverse actions of monomeric C-reactive protein. J Biol Chem. (2016) 291:8795–804. doi: 10.1074/jbc.M115.695023
52. Lv J-M, Chen J-Y, Liu Z-P, Yao Z-Y, Wu Y-X, Tong C-S, et al. Cellular folding determinants and conformational plasticity of native C-reactive protein. Front Immunol. (2020) 11:583. doi: 10.3389/fimmu.2020.00583
53. Voleti B, Agrawal A. Statins and nitric oxide reduce C-reactive protein production while inflammatory conditions persist. Mol Immunol. (2006) 43:891–6. doi: 10.1016/j.molimm.2005.06.045
Keywords: atherosclerosis, C-reactive protein, inflammation, low-density lipoprotein, phosphocholine
Citation: Pathak A, Singh SK, Thewke DP and Agrawal A (2020) Conformationally Altered C-Reactive Protein Capable of Binding to Atherogenic Lipoproteins Reduces Atherosclerosis. Front. Immunol. 11:1780. doi: 10.3389/fimmu.2020.01780
Received: 21 May 2020; Accepted: 03 July 2020;
Published: 11 August 2020.
Edited by:
Kenji Daigo, Nippon Medical School, JapanReviewed by:
Tomohide Takaya, Shinshu University, JapanCopyright © 2020 Pathak, Singh, Thewke and Agrawal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alok Agrawal, YWdyYXdhbEBldHN1LmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.