 Mireya Becerra-Diaz
Mireya Becerra-Diaz Mason Song
Mason Song Nicola Heller
Nicola Heller
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 August 2020
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01698
Androgens, the predominant male sex hormones, drive the development and maintenance of male characteristics by binding to androgen receptor (AR). As androgens are systemically distributed throughout the whole organism, they affect many tissues and cell types in addition to those in male sexual organs. It is now clear that the immune system is a target of androgen action. In the lungs, many immune cells express ARs and are responsive to androgens. In this review, we describe the effects of androgens and ARs on lung myeloid immune cells—monocytes and macrophages—as they relate to health and disease. In particular, we highlight the effect of androgens on lung diseases, such as asthma, chronic obstructive pulmonary disease and lung fibrosis. We also discuss the therapeutic use of androgens and how circulating androgens correlate with lung disease. In addition to human studies, we also discuss how mouse models have helped to uncover the effect of androgens on monocytes and macrophages in lung disease. Although the role of estrogen and other female hormones has been broadly analyzed in the literature, we focus on the new perspectives of androgens as modulators of the immune system that target myeloid cells during lung inflammation.
The immune system is essential for maintaining homeostasis within tissues and organs and protecting them against threats, such as harmful pathogens or cancerous transformation (1). It comprises both innate and adaptive components. The innate immune system is made up of the innate lymphoid (innate lymphoid cells [ILCs], natural killer cells [NKs], and lymphoid tissue inducers [LTi]) and innate myeloid subsets (2, 3). The innate immune system consists of a network of immune cells and molecules that provide rapid, first-line defense against pathogens. In contrast, the adaptive immune response, made up of B and T lymphocytes (4), takes days or even weeks to become established (5). Innate immune cells express pattern recognition receptors that recognize unique and conserved pathogen-associated molecular patterns such as lipopolysaccharide (LPS), viral ssRNA, and fungal β-glucan (6). B and T cells have evolved to recognize a finer repertoire of self- and nonself-antigens that facilitate pathogen-specific actions, immunologic memory generation, and host immune homeostasis regulation (4). To accomplish this, the adaptive immune response involves a tightly regulated interplay between T and B lymphocytes and antigen-presenting cells of the myeloid lineage, such as dendritic cells (DCs), monocytes, and macrophages (4). Myeloid cells arise from the bone marrow. The type and magnitude of the immune response is influenced by biological sex and age (7), and therefore differs between males and females. Sex differences in the function of the immune system arise from both genetic (chromosomal) sex differences and differences mediated by the action of male and female sex hormones. Because the concentration of sex hormones changes over the lifespan and throughout the course of the menstrual cycle in women, the function of the immune system also changes during different stages of life. Innate myeloid immune cells, like other cell types, express sex hormone receptors and are responsive to sex hormones (8).
Sex hormones are synthesized from cholesterol through a defined enzymatic cascade, predominately in the gonads and the adrenal glands (9). Sex hormones are also produced in other tissues, including the brain, placenta, mammary glands, liver, and adipose tissue (9–11). In addition to driving sexual development of egg and sperm production, sex hormones are responsible for the development of male and female secondary sexual characteristics, like breast development and growth of facial hair, that occur during puberty (12). Androgens include testosterone, dihydrotestosterone (DHT), androstenedione, androstenediol, and dehydroepiandrosterone (DHEA), with DHT being the most potent (13). The concentration of androgens in circulation is about seven-fold higher in adult men than in adult women (14, 15). Estradiol and progesterone are the predominant female sex hormones (16) synthesized by the ovaries and adrenal glands. Both male and female sex hormones are bound to the plasma proteins, albumin and sex hormone binding globulin (SHBG), and only a small percentage exists as free hormone (1–2%). Thus, the bioavailability of sex hormones is regulated by their biosynthesis and also the amount of albumin and SHBG.
Importantly, sex hormones mediate not only anatomic differences between women and men but also direct sex differences in immune responses, leading to different risks for immunologic diseases (17). Overall, women have a greater risk for autoimmune diseases (such as systemic sclerosis and systemic lupus erythematosus) (18), whereas men are more likely to die of infectious and parasitic diseases (19). Moreover, men have a greater risk of non-reproductive cancers (20–22). Both gender and sex are important mediators of these and other health and disease differences observed between men and women. While gender refers to the array of socially constructed roles, attitudes, personality traits, and behaviors, sex represents a biological characteristic of an individual (23), including the hormonal milieu and chromosome complement (22). In general, estrogens are considered to have proinflammatory properties and androgens are thought to have anti-inflammatory properties (24). In the United States (25) and worldwide (26), relevant evidence highlights important epidemiologic sex differences in incidence, susceptibility, and severity of a number of diseases that affect the respiratory tract. In this review, we will focus on how male sex hormones, the androgens, modulate the response of myeloid cells in the lung and how this modulation impacts the outcome of different diseases of the lung.
Biological sex mediates differences in the incidence and pathophysiology of lung diseases. These differences arise from sex differences in the structure and function of the lung itself, and also in the immune cells that populate the lung and are recruited to it during inflammation. Before birth, the female lung has several structural advantages over the male lung. Surfactant is produced earlier, and, although the female lung is smaller, it has more alveoli per unit area. Neonatal females have higher expiratory flow rates than do male neonates when corrected for size. Thus, male sex is a major risk factor for the development of respiratory distress syndrome, bronchopulmonary dysplasia in neonates (27–30), and asthma in childhood (31, 32).
In addition to the contribution of structural differences of the lung between the sexes, sex differences in lung function and lung diseases are also dependent on the action of sex hormones. We have summarized some broad concepts that define how testosterone and estrogen affect lung macrophage function and how this may contribute to the outcome of particular lung diseases in Figure 1. As testosterone rises after puberty, the immunosuppressive effects of this hormone on protective immune responses to infectious diseases in males can worsen pulmonary disease. This would be exemplified by tuberculosis or influenza. Some of these effects are a result of androgen effects on critical inflammatory macrophage functions although the effects on the adaptive immune system also have a significant contribution to the overall outcome. Thus, testosterone appears to play a key immunoregulatory role in lung macrophages. Testosterone's immunoregulatory properties also appear to be dependent on the amount of cellular expression of AR and on the concentration of the hormone. Low concentrations of testosterone have been noted in patients with asthma, COPD, and tuberculosis. Low testosterone may also be linked to insufficient control of tissue-damaging inflammatory responses seen in COPD and pulmonary fibrosis. Estrogen tends to promote wound healing responses in macrophages. Dysregulation of wound healing responses and overactive tissue remodeling macrophages in the lung could be broadly used to describe the Th2 response in allergic asthma, which is worse in women. Cancer could also be considered an aberrant wound healing response driven by M2-like tumor associated macrophages. We have highlighted here how sex hormones contribute to changes in lung macrophage function that contribute to lung disease. However, it should be pointed out that not every sex difference in lung disease is due to direct effects on macrophages but on the broader coordinated immune response as a whole.
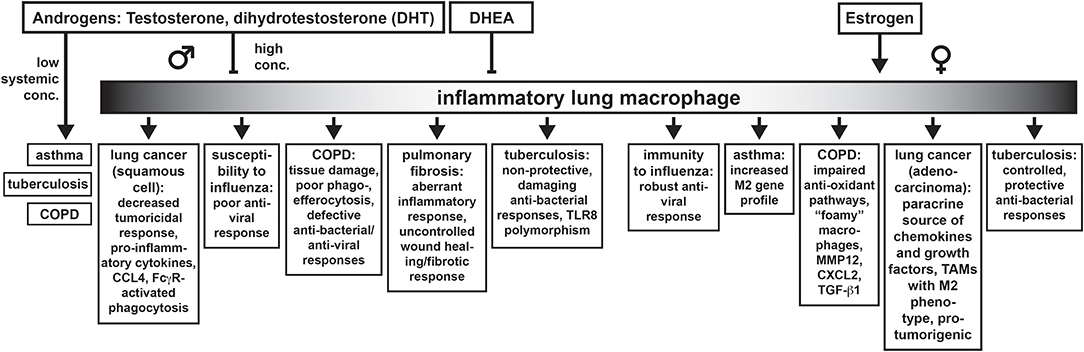
Figure 1. Sex differences in lung diseases discussed in this Review and how they may be connected to the effects of androgens (and estrogens) on inflammatory macrophages in the lung.
Before puberty, the structural differences in the lung, as well as gender differences, likely account for the higher incidence of asthma in boys than in girls. With the onset of puberty, male and female sex hormones and their effects on the structural cells of the lung and on the immune system contribute to the incidence of asthma (33, 34). The incidence and severity of asthma are greater in adult women than in adult men (35, 36) and greater in female than in male mice (37, 38). Female sex hormones, such as estrogen, appear to worsen asthma, although a straightforward correlation between amount of female sex hormone and asthma symptoms has not been concluded. Androgens have multiple immunoregulatory and bronchodilatory functions and may contribute to, or be biomarkers for, better lung function in men (39). Accordingly, serum testosterone is low in men with moderate to severe asthma (39–41). In one study, each 25 ng/dL increase in serum testosterone correlated with a 3% (95% CI, 1%-4%; P = 0.002) decrease in the likelihood of having asthma (42). On the other hand, high concentrations of testosterone and cyclic AMP in sputum of asthmatic women during the luteal phase of the menstrual cycle were thought to play a role in premenstrual exacerbations (43). The idea that sex hormones may be a causal factor in asthma was significantly strengthened by a recent study of 7,615 adults that quantified serum sex hormones and asthma outcomes (44). That study showed that low testosterone in both women and men was associated with an increased incidence of asthma. The other interesting finding was that higher testosterone was protective against asthma in obese women. Obesity is a risk factor for asthma (45–47). Therefore, how high body mass index (BMI) and circulating sex hormones together affect asthma requires further investigation.
Another androgen, dehydroepiandrosterone (DHEA), also known as androstenolone, is an endogenous steroid hormone and one of the most abundant circulating steroids in humans. It is a precursor for the synthesis of both testosterone and estrogen. DHEA is sulfated at the C3β position into DHEA-S by the action of the sulfotransferase enzymes SULT2A1 and SULT1E1 in the adrenal glands. The amount of DHEA-S in the circulation is ~250–300 times those of DHEA. DHEA became of interest to the asthma field because women with severe asthma had very low concentrations of DHEA-S (48) and DHEA-S concentration correlated with lung function (33). Interestingly, DHEA-S is suppressed by oral or inhaled glucocorticoids, the mainstay therapy for asthma (49). Human DHEA peaks at around age 20 and then follows an age-dependent decline until they reach prepubertal concentrations. Reduced secretion of DHEA with age has been related to a number of age-associated conditions. Replacement of DHEA has been considered as a possible therapeutic that could activate protective responses in an aging immune system. DHEA is known to downregulate Th2-inflammatory cytokines while upregulating IL-2 synthesis (50, 51) in concanavalin A-stimulated peripheral blood mononuclear cells from adult males with atopic dermatitis (52, 53). Thus, it was hypothesized that it would be a useful treatment for atopic diseases including asthma and the results of the clinical trials for DHEA in asthma patients show promise. The results are discussed in a later section titled “Effects of androgen exposure on monocytes, macrophages in humans with lung disease”.
Sex differences also have been reported in chronic obstructive pulmonary disease (COPD), a heterogeneous, chronic, and progressive respiratory disorder that includes chronic bronchitis and emphysema (54). Chronic exposure of the airways to insults, such as cigarette smoke, leads to epithelial cell injury, destruction of pulmonary capillary vasculature, acceleration of epithelial cell senescence, and airway remodeling. The loss of lung compliance ultimately leads to COPD (55, 56). COPD was previously thought to affect mostly elderly men, primarily because of the higher prevalence of smoking in men. However, as smoking rates increased in women, the number of COPD cases in women exceeded that of men (57). These differences are not only based on gender, as women develop more severe COPD with early-onset disease (<60 years) and have greater susceptibility to COPD with lower tobacco exposure (58). Moreover, increasing age in female smokers leads to a faster annual decline in forced expiratory volume in the first second when compared to that of male smokers, even when they smoke fewer cigarettes (59). Similarly, pulmonary fibrosis is another lung disease that manifests sex differences (60), with men being more affected than women (61, 62). It is characterized by destruction of the pulmonary parenchyma and deposition of extracellular matrix with alterations in phenotype of both fibroblasts and alveolar epithelial cells (63).
The lungs are also the target of respiratory viruses such as influenza A (“flu”), respiratory syncytial virus, and coronaviruses, such as severe acute respiratory syndrome and the Middle East respiratory syndrome. The viruses infect the airway epithelial cells and cause damage to the epithelial barrier by themselves or as a result of the immune response to the viral infection. Sex differences have been noted in the immune response to influenza A virus and to the influenza vaccine. In general, women have a more robust protective immune response to influenza virus and vaccine than do men. Although this elevated response is helpful in clearing virus, women of reproductive age also experience higher mortality and hospitalizations (64–68), possibly from collateral tissue damage to the lungs. The vigorous immune response in women also means that women experience more adverse events after vaccination (69). Indeed, a systems biology approach identified that high testosterone was correlated with a blunted response to the flu vaccine in men (24). As testosterone wanes in elderly men, mortality increases (70). Since the male immune response to the virus is also less robust, the incidence of seasonal flu is generally higher in men than in women in developed countries, according to the World Health Organization (71). It is not yet known how fluctuations in sex hormones across the menstrual cycle and lifespan affect the immune system's response to the influenza virus in humans. Mouse studies have revealed that estrogen is protective at high, but not low, concentrations (72, 73). On the other hand, testosterone replacement in gonadectomized or aged male mice enhanced survival rates (74). Despite these findings in mouse models, studies examining the effect of sex hormones on cellular and molecular mechanisms in human immune cells during influenza infection are lacking.
Like influenza infection, tuberculosis (TB), a lung disease caused by Mycobacterium tuberculosis, exhibits notable sex differences in the number of cases worldwide, with men being almost twice as frequently affected than women (75, 76). Both sex and gender differences impact the incidence of TB. Although TB affects less women than men in adulthood (75), women in their economically active years (15–59 years old) have a higher TB incidence compared to women in other age groups (77). This indicates that factors associated with gender, such as exposure to the bacteria, are important in this disease. However, because male predominance does not occur in children, this suggests that biological factors such as male sex hormones also play a significant role (75). This is supported by a study of medically castrated men who experienced a significantly smaller proportion of death from TB, 8.1% compared to 20.6% in intact men (78). Understanding how androgens lead to the greater susceptibility of men to TB is critical, as TB is still one of the leading fatal infectious diseases worldwide and may also may favor the development of other diseases, such as lung cancer (79).
Lung cancer is a very complex disease that depends on a number of variants such as sex, gender, race, and socioeconomic status (80). The development of lung cancer is also related to environmental factors, such as pollution due to industrialization and urbanization (81). An additional gender-associated risk factor, significantly linked to developing lung cancer, is cigarette smoking (80). Historically, more men develop lung cancer and suffer lung cancer-associated deaths compared to women (80). However, the incidence of lung cancer has changed notably in both women and men. In men, lung cancer incidence started to increase in the 1920s and started to decrease in the early 1990s, while in women, the mortality rates and incidence began to rise in the 1960s (80). Changes in smoking habits in the last several decades with a rise in the number of women who smoke correlate with an increase in the incidence of lung cancer in this demographic group (80). Smoking is definitely a key factor in the development of lung cancer; however, recent studies show a higher incidence of lung cancer in young women compared to young men (82, 83), even when the prevalence of cigarette smoking among young women has approached but not exceeded that among men (84). This suggests that the higher incidence of lung cancer in women is not explained simply by gender differences in smoking habits: a deeper analysis of differences mediated by sex, such as greater sensitivity to tobacco smoke in women is warranted (85, 86).
Furthermore, men and women develop different specific types of lung cancer. Malignant mesothelioma is more common in men, while women develop more adenocarcinoma (87), particularly non-small cell lung cancer (NSCLC) (88). Women have a superior survival rate for lung cancer compared to men (89). Tumor-associated macrophages are critical in tumor progression yet how androgens influence macrophage behavior in lung cancer and in responses to treatment must be addressed more deeply to develop better therapies and increase survival rates in men.
The lungs are a primary interface with the external environment. The delicate structures needed for gas exchange make them susceptible to damage from invading pathogens and toxic molecules. Some insults to the lung can lead to the development of chronic conditions such as allergic asthma. As a protective mechanism, alveolar macrophages clear the air space of infectious, toxic, or allergenic particles to maintain homeostasis in the alveoli. Thus, alveolar macrophages have a dual function as inflammatory cells, phagocytosing and killing inhaled bacteria or viruses, and also as controllers of the inflammatory immune response, minimizing alveolar damage. Resident alveolar macrophages are seeded embryonically from yolk sac and fetal liver monocytes (90–92). In asthma and other lung diseases, recruited alveolar macrophages derived from blood monocytes can turn into pathogenic cells, worsening the condition (93, 94). Mouse alveolar macrophages are characterized by high surface expression of Siglec F and produce TGFβ. TGFβ both supports AM development (95) and their maintenance of immune homeostasis by induction of Tregs and suppression of B and T cell proliferation. Another important function of AM is the clearance of surfactant. AM from male and female mice respond differently to surfactant protein A (SP-A) (96, 97). SP-A acts as an opsonin and is important in clearance of pathogens. Sex differences in AM responses to surfactant could affect bacterial clearance and regulate the production of proinflammatory mediators. The molecular mechanisms that mediate these differences and how sex hormones change this important AM function is an open question.
In the human lung, there appears to be more diversity in the subtypes of lung macrophages compared to mice. The main determinant of the frequency of subtypes of macrophages in humans appears to be their anatomical location within the lung. AM are the predominant immune cells in the lung airways (bronchi and bronchioalveolar space). Flow cytometric panels have employed HLA-DR, CD163, CD169, and CD206 to differentiate between AM, IM and monocytes. Human AM were identified as large, highly autofluorescent CD14- CD16+ cells that also express CD206, CD169, and MARCO (98, 99). There appear to be two populations of AM distinguished by either high or low expression of CD163. More recent approaches to characterize the macrophage populations in the lung involve single-cell transcriptomic analysis (100, 101). Although macrophages show a large variation in the transcriptional phenotype, expression of MARCO, CCL18, APOC1, APOE, PPARG, and MRC1 was found in macrophages from healthy donors (100, 101), while CHI3L1, MARCKS, IL1RN, PLA2G7, MMP9, and SPP1 were highly expressed in macrophages from pulmonary fibrosis patients (101). Thus, a second contributor to diversity is likely the activation state of the cells. There are no data that describe sex differences in human AM responses and the effect of sex hormones on these cells. From our mouse and human MDM studies, we would predict that androgens augment the immune homeostatic functions of these cells in the male lung. Further work is still needed to standardize characterization of the different subpopulations of human lung macrophage populations and their role in maintaining healthy lung function and in disease.
Interstitial macrophages (IMs), are another macrophage population found in the lung. They are a minor population of monocyte-derived macrophages (102), which comprise 30–40% of lung macrophages (103) and are localized in the lung parenchyma (104). IMs contribute to maintaining homeostasis through the spontaneous release of IL-10, a cytokine that dampens inflammation (105). IMs can prevent the development of aberrant type 2 allergic responses triggered by inhaled allergens (104) and have been related to reduction of asthma (106, 107). Different subpopulations of IMs have been found in the lung; however, their characterization has not arrived at a consensus due to difficulties in their identification and isolation. In the mouse lung, different subpopulations of IMs have been described based on the expression of surface markers. One report described three different subpopulations of IMs based on the differential expression of proinflammatory cytokines, chemokine ligands, MHC-II, CD11c, CD206, and Lyve-1 (108); other group identified two subpopulations, based on similar markers but including CX3CR1 (109). Moreover, IMs subpopulations can be also described based on the different anatomic locations these cells populate inside the mouse lung parenchyma (110). Further work is needed to better characterize and define the different IM populations, as the different subtypes may have different functions during the inflammatory process. Smaller in size than their AM counterparts, human IMs express more of the monocytic marker CD14 than AM, perhaps suggesting their monocytic origin, and have lower expression of CD169 than human AM. The responses of IM to androgen will depend on their expression of AR which has not been measured. This will be a challenge due to difficulties in clearly identifying this population (and its subpopulations) from the monocytic, AM and other myeloid populations in the lung.
Monocytes are produced in the bone marrow along with a number of other myeloid cells. Myeloid cells originate from common pluripotent hematopoietic stem cells and represent the major subset of white cells in circulation (111). These cells comprise basophils, neutrophils, eosinophils, DCs, monocytes, and macrophages, among others (112). Monocytes are released into circulation, then blood monocytes are recruited into inflamed tissue and can mature into macrophages or dendritic cells. There are two main subsets of mouse monocytes, “classical” or Ly6Chigh monocytes that originate directly from Ly6C+ precursors, and “non-classical” or Ly6Clow monocytes that derive from Ly6Chigh monocytes (113). The origin of Ly6C low monocytes was demonstrated by Sunderkotter, et al. by tracking the maturation of DiI-labeled Ly-6Chigh monocytes into DiI-labeled Ly6Clow monocytes (114). This process depends on the transcription factor Nr4a1, which regulates the development and survival of Ly6Clow monocytes (113). These two monocyte subsets mirror the human CD14+ classical and CD16+ non-classical monocyte populations, respectively (115). Ly6Chigh monocytes highly express the chemokine receptor CC-chemokine receptor 2 (CCR2), whereas Ly6Clow monocytes highly express CX3CR1 (116). Importantly, CCR2 expression is required for Ly6C+ monocyte egress from the bone marrow into the circulation and entry into non-inflamed and inflamed tissues (117–119) from the blood (120). As monocytes migrate into tissue, they mature into macrophages developing unique, tissue-dependent morphology and functions (121). They lose expression of Ly6C and gain expression of MHC class II, becoming more efficient antigen-presenting cells (122). Some authors have proposed the concept of “tissue monocytes,” which are monocytes that can enter non-lymphoid organs without obligatory differentiation into macrophages. Therefore, monocytes are much more than simply precursors for macrophages.
In human lungs, monocytes, which can be both beneficial and pathogenic in a variety of pulmonary diseases (123), are present at steady state (124). Multiple-color cytometric analysis on cells obtained from different anatomical locations of the lung of healthy subjects (non-smokers with normal lung function and absence of disease or infection) revealed that while intermediate monocytes (CD14+CD16+) are more frequent in the airways, classical monocytes (CD14+CD16−) are more frequent in blood (124). Moreover, the different monocyte subsets produced TNF-α to different degrees upon stimulation with TLR ligands (3,4, and 7/8). Thus, the anatomic location where samples are obtained should be considered and reported when working with human bronchoscopies, as this may alter the type and abundance of monocytes and macrophages found. Accurate identification of monocytes in the lung compartments in humans has been a challenge because monocytic “contamination” from the blood vessels (125, 126). Overcoming this challenge, Desch et al. performed a flow cytometric phenotyping study and identified two additional lung monocyte populations by analyzing lungs obtained from donors who died of non-pulmonary causes (127). CD14+ CD206− CD1c− CD1a− intravascular monocytes were similar to CD14+ blood monocytes and CD14+ CD206+ CD1c− CD1a− monocytes were described as tissue “monocytes.” These studies highlight that we are just at the beginning of understanding the complexity of lung monocyte subtypes and their functions depending on the inflammatory state of the lung.
Other myeloid populations, like DCs, occupy the lung parenchyma at steady state, and their relative numbers change during inflammation. We refer readers to previous excellent reviews in this journal that cover the importance of DCs in immune responses in the lung and how they are affected by sex differences. Therefore, we will not discuss DCs here (2, 128–132).
Polarization is a very important effector characteristic observed in monocytes and macrophages. Polarization refers to the change in phenotype and function of monocytes and macrophages as they are exposed to different inflammatory milieus or factors in the tissue microenvironment. To understand the effects of the differing inflammatory or tissue environments on monocyte-macrophage phenotype and function, researchers have used cytokines and other factors in vitro to mimic different inflammatory and tissue microenvironments. Monocytes and macrophages stimulated with interferon-γ, LPS, TNFα, interleukin (IL)-12, and granulocyte-macrophage colony-stimulating factor promote a pro-inflammatory macrophage phenotype denoted as M1 polarization. The activation state was also known as “classical” activation. M1-polarized macrophages mediate immunity to intracellular infections, such as viruses and bacteria, and they are generally considered tumoricidal (133–136). M1 macrophages accomplish these functions by inducing production of nitric oxide, reactive nitrogen intermediates, reactive oxygen species, and hydrogen peroxide (137–139). In contrast, activation of macrophages with IL-4 or IL-13, as in extracellular parasitic infections and allergic reactions, leads to M2 polarization or “alternative” activation of macrophages (140). M2 macrophages produce inflammatory mediators and chemokines, such as chitinase-like proteins (141), IL-13 (142), CCL17, CCL18, CCL22, and CCL24, which activate Th2 cells and promote eosinophil infiltration into the lungs (143, 144).
In allergic asthma, a Th2-inflammatory response to inhaled allergens drives lung macrophages toward an M2 phenotype. Increased number and percent of M2 macrophages have been correlated with asthma severity and a decline in lung function in humans and mouse models (145–147). Similarly, M2 macrophages are the predominant subset seen in pulmonary fibrosis and are responsible for fibrogenesis (148). During COPD, the number of macrophages in airways, lung parenchyma, bronchoalveolar lavage fluid, and sputum increases (149, 150). This increase may occur as a result of enhanced monocyte recruitment from circulation in response to chemokines such as CCL2 and CXC-chemokine ligand-1, which are increased in the sputum and bronchoalveolar lavage fluid of patients with COPD (151). Unlike in allergic asthma and pulmonary fibrosis, macrophages in COPD are polarized toward an M1 profile (152). In addition to affecting men and women differently, another commonality of COPD is that macrophages both in the alveolar space and in lung tissue present an altered activation phenotype. Different concentrations of cytokines (TNF-α, IL-1β, IL-6, IL-10, IL-12) and chemokines (CCL2, CCL5, CCL7, CCL13, CCL22, IL-8, CXCL9, and CXCL10) are found comparing smokers to healthy subjects (153–161). Thus the external provoking stimulus uniquely shapes macrophage phenotype and function.
While the M1/M2 designations are useful for in vitro studies with stimulation with defined cytokines, the in vivo phenotype of macrophages exists on a spectrum somewhere in between these two well-defined opposing phenotypes or does not fit the paradigm at all. For example, M1 and M2 markers can exist simultaneously within the same cell in some cases (162–164). The key factors dictating the macrophage phenotype or activation state are the stage of the immune response and the soluble factors and interactions in a particular tissue microenvironment. For example, the lung environment is rich in GM-CSF, TGFβ, and PPARγ and is critical for development of mature AMs after birth in both mice (90, 91, 165–169) and humans (170–175). Furthermore, interactions between CD200 on type II alveolar epithelial cells and CD200R on the surface of the AM deliver regulatory signals to the AM to prevent proinflammatory signaling and macrophage activation (176). Thus, macrophage nomenclature has evolved as our understanding of the phenotypes and functions of different types of tissue resident macrophages, recruited monocytes and monocyte-derived macrophages advances. In-depth studies of the effects of androgens and other sex hormones on tissue macrophage plasticity and phenotype have yet to be carried out.
Because androgens are lipophilic steroid hormones, they can easily diffuse across cell membranes without the need for receptor-mediated import (8). Androgens in circulation are found mostly bound to sex hormone-binding globulin and albumin (8). Free (unbound) steroid sex hormones can signal through two different mechanisms: the classical AR, located in the intracellular compartment, and the membrane, or non-classical, AR (8). Androgen binding to classical and non-classical ARs mediates genomic and non-genomic androgen effects, respectively (177). Upon androgen binding, the classical AR undergoes a conformational change and dissociates from heat-shock and other chaperone proteins. An androgen–AR complex is formed that translocates to the nucleus, dimerizes, and binds to androgen responsive elements that modulate the transcription of target genes (178). Importantly, it has been reported that the androgen–AR complex can also mediate non-genomic changes (179) by causing calcium flux and by activating second messenger pathways including ERK, AKT, and MAPK, at least in cell lines (179–181). Whereas, genomic modulation may need hours or days (182), non-genomic modulation can occur within seconds to minutes after androgen exposure, does not involve the complex binding to DNA, and therefore does not affect transcription of target genes (177). DHEA has no known unique receptor and is not a direct AR agonist. It affects immune function but, because it can interact with other sex hormones, it has been difficult to establish its mechanisms of action.
Most studies of androgen–AR complex-mediated gene expression have been carried out in the context of male reproductive tissue in prostate cancer (PCa) (183–185). As previously discussed, immune cells are responsive to sex hormones, and almost all immune cells express sex hormone receptors (8). Mouse monocytes, macrophages (186), and DCs (187) express both classical and non-classical ARs although the vast majority studies do not specifically dissect the role of the two types of AR on the outcomes being measured in the study. Because recent literature has described how sex steroids modulate the functions of DCs (2, 128, 129), we will not discuss it here. We will focus on the importance of androgen–AR regulation of monocyte and macrophage function and how androgen–ARs modulate monocytes and macrophages in lung diseases.
Androgen receptor expression in mouse and human monocytes and macrophages is summarized in Table 1. In general, the expression of the mRNA and protein for classical AR has been assessed, often by non-quantitative means, and non-classical ARs have not been measured.
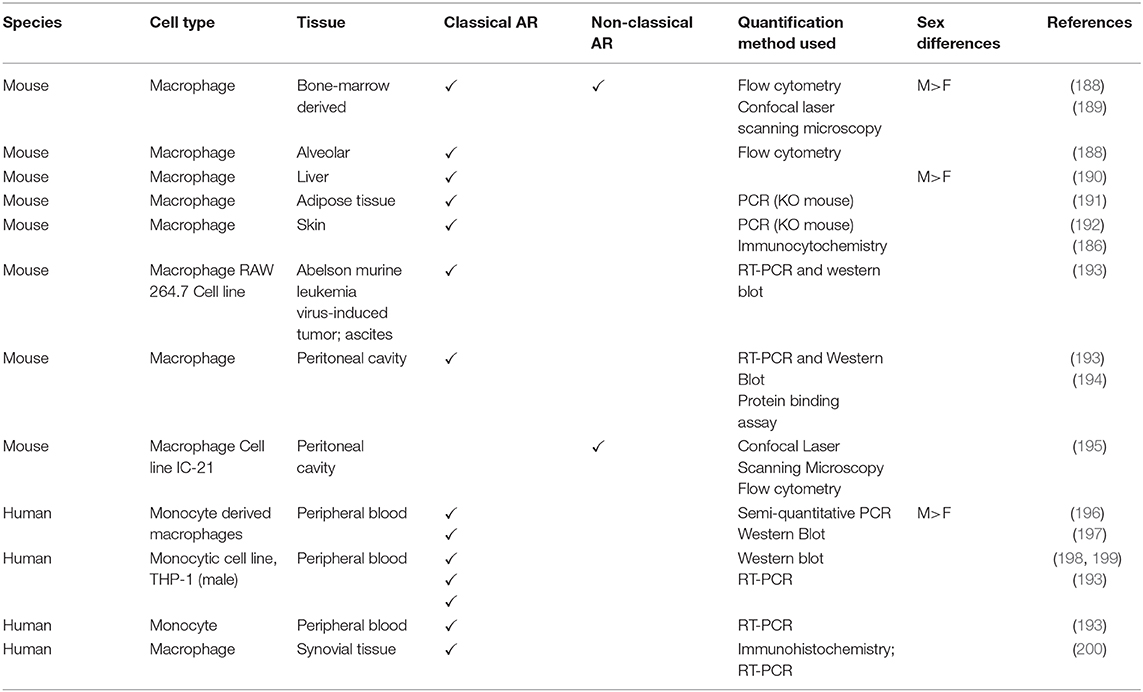
Table 1. Androgen receptor expression in mouse and human monocytes and macrophages.
We have summarized the outcomes of many studies on mouse and human monocyte-macrophages responses in the presence of androgens in Figure 2. In general, monocyte-macrophage exposure to androgen results in a reduction of pro-inflammatory responses (boxed and shaded in green). It is possible that the reduction in inflammation by androgen may be due to AR suppression of estrogen/ERα-driven pro-inflammatory responses. AR was demonstrated to inhibit ERα activity by binding EREs in breast cancer cells (201). Whether this indirect mechanism accounts for the broad immunosuppressive effects of androgens in normal untransformed immune cells is not known. In keeping with reduced pro-inflammatory responses, we found that androgen enhanced IL-4-induced M2 polarization of bone marrow derived and alveolar macrophages in vitro and macrophage-specific deficiency of AR diminished M2 polarization of lung macrophages in vivo (188). In some cases, however, inflammatory responses are increased by androgens (boxed and shaded in red). The different responses may be due to different types of tissue macrophages or experimental system. Monocyte-macrophage responses are dependent on the concentration of the hormone, expression of AR, and upon the inducing stimuli to which the macrophage is exposed. The majority of in vitro studies examining the effects of androgens on monocytes and macrophages have not clearly acknowledged or separated the effect of androgen on membrane ARs and non-classical AR signaling from that of classical ARs. Therefore, we have to assume that the studies described in the section below are a result of classical AR activity unless explicitly investigated or stated. Determining how non-classical AR signaling and androgen-independent activation of AR affects monocyte and macrophage function is a gap in our knowledge that must be addressed in future studies.
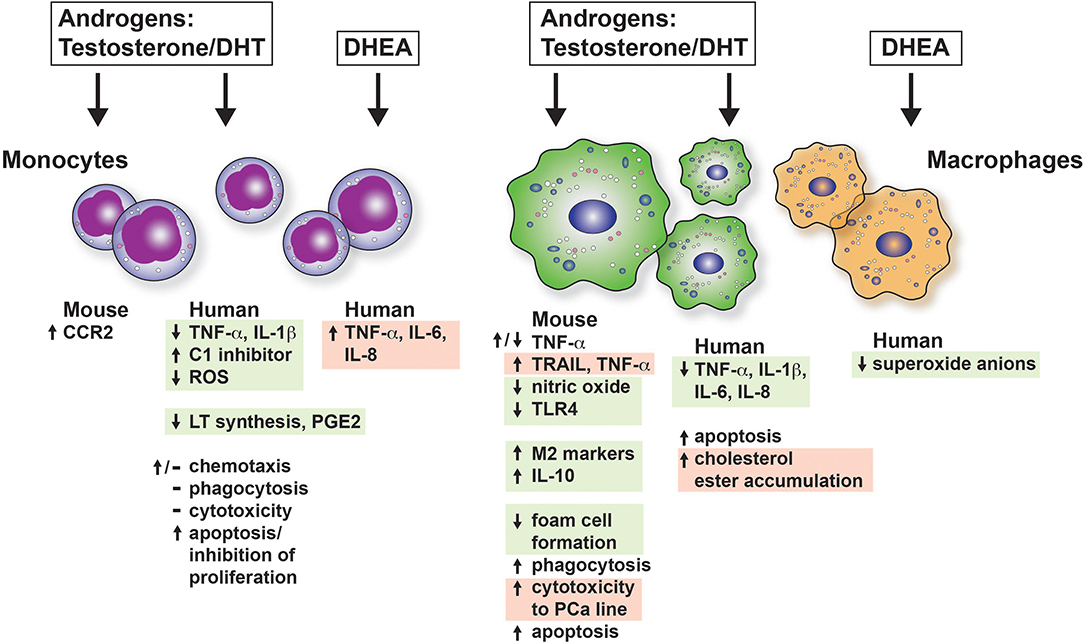
Figure 2. The effects of androgens on mouse and human monocytes and macrophages in vitro. The consequences of androgen exposure of monocytes and macrophages are shown. Green boxes indicate that the effect is to dampen proinflammatory responses or be considered immunosuppressive. The red boxes indicate a response that would increase inflammation. CCR2, C-C chemokine receptor type 2; DHT, dihydroxytestosterone; DHEA, dihydroepiandrosterone; PCa, prostate cancer; PGE2, prostaglandin E2; ROS, reactive oxygen species.
Androgens modulate the expression of proinflammatory molecules such as TNFα in mouse monocytes and macrophages. In 2009, Lai et al. (192) demonstrated that LPS-induced production of TNFα was decreased in BMM lacking classic ARs. Moreover, they found that AR, in the presence of DHT, induced TNF-α promoter activity (192). On the other hand, several reports have suggested the contrary. In one study that used splenic macrophages from midline laparotomy trauma-hemorrhaged mice, DHT suppressed TNF-α production from LPS-stimulated cells (202). This effect was also observed in the mouse macrophage cell line J774 (203), in which testosterone inhibited TNF-α production. In addition, testosterone also decreased expression of the proinflammatory molecule nitric oxide in response to LPS in the mouse macrophage cell lines RAW 264.7 (204) and J774 (203), but it enhanced the expression of IL-10 in the latter.
Other molecules important in monocyte-macrophage functions are also affected by androgens. For example, the expression of CCR2 was enhanced in mouse monocytes by androgens and thereby enhanced chemotaxis (192). However, suppressing AR with siRNA in prostate cells increased macrophage recruitment via CCL2 upregulation, which might promote prostate cancer (205). Phagocytosis was increased by testosterone in rat peritoneal macrophages at 10−12 M but not at concentrations lower or higher than 10−12 M (206). Cytotoxicity of RAW macrophages to the mouse prostate cancer cell line, TRAMP C2, was enhanced by DHT alone (193). This was attributed to enhanced expression of the M1 polarization markers, TRAIL and TNF-α, in the macrophages. Testosterone (100, 200, and 400 nM) induced apoptosis in mouse BMM through Fas-FasL (207) and activation of caspase 3, 8 and poly (ADP-ribose) polymerase (208).
In terms of M2 polarization of macrophages, we showed recently that in vitro exposure of BMM to DHT prior to IL-4 stimulation enhanced Chi3l3 and Arg1 gene expression, as well as production of YM1 (188). Androgen amplified the M2 phenotype by increasing IL-4-mediated M2 polarization. Our results were similar to those found in response to IL-4 in the RAW cell line (209). This enhanced M2 macrophage polarization correlated with decreased TLR4 expression and sensitivity to a TLR4-specific ligand observed in testosterone-treated RAW cells (210).
Taken together, these observations suggest that androgens and ARs can either promote or suppress inflammatory properties of mouse macrophages, depending on the external environmental conditions, AR expression, and concentration of hormone. Overall, androgens are more likely to reduce polarization of M1 macrophages. This could represent an important mechanism by which inflammatory pathways are downregulated in males. The opposite effects seen in different inflammatory contexts highlight the need for a deeper and broader study of the androgen/AR-mediated modulation of monocytes and macrophages, as these cells participate in both the initial and late phases of immune responses in a variety of diseases. Most of the studies analyzing the role of AR have focused on prostate cancer, primarily in transformed cell lines (211–213) but macrophages are vital in cancer development and metastasis (205). Furthermore, it is important to consider that opposing effects could result from differential activation of either classical or non-classical (AR-independent) effects (195, 214) which have been rarely studied to date.
Androgens affect a number of key monocyte and macrophage functions. Studies of androgen receptor function in human monocytes and macrophages have focused primarily on the roles of male sex and sex hormones in promoting atherosclerotic foam cell formation (196) and inhibiting cutaneous wound healing (186, 215). Foam cells are a type of macrophage localized in the blood vessel walls where they engorge cholesterol (216). Foam cells exhibit enhanced inflammatory cytokine secretion and cause atheroma, contributing to cardiovascular disease (216, 217). The effect of androgen on monocytes and macrophages in other immune-mediated human diseases where monocytes and macrophages play a role has been neglected.
The degree of AR expression in monocytes and macrophages is likely the primary determinant of responsiveness, although most studies examining responses to androgens do not quantify AR expression (see Table 1). The expression or action of androgens on non-classical ARs in human monocytes and macrophages has yet to be examined carefully. Most studies assume that the outcomes that are measured are a result of the activity of classical AR. Sex differences in AR content may also play a role in responsiveness. This fact highlights the importance of considering the sex of cells in all in vitro studies to accurately assess how sex hormones affect the responses of monocytes and macrophages.
Apoptosis was significantly greater in human THP-1 cells cultured for 7 days with 10 nM testosterone than in control cells or cells treated with estradiol (E2), owing to a reduction in proliferating cell nuclear antigen, induction of poly-ADP ribose polymerase-cleaved, an increase in IκB-α, and a decrease in phosphorylated IκB-α (218). E2, in contrast, promoted cell survival. Other studies noted concentration- and time-dependent regulation of apoptosis in THP-1 cells, with an increase in the proto-oncogene Bax and Fas (219). Androgen exposure inhibited proliferation of the human monoblastic leukemia cell line U937, depending on the concentration and time of exposure (220). Cell cycle arrest occurred at the G2/M phase, although another study measured no effect of testosterone on PMA-differentiated U937 cells (221). How testosterone regulates apoptosis and survival of untransformed primary human monocytes and MDMs has not been well-studied. Toxicity was observed when monocytes were differentiated into macrophages over 8 days in the presence of 0.1 mg/mL androgen, but not at lower concentrations of the hormone (222). Testosterone reduced the viability of monocytes from a healthy control and a patient with systemic lupus erythematosus in a concentration-dependent fashion (223, 224). These two studies highlight the importance of concentration in studies of sex hormones. An additional example is the finding that E2 enhances TNF-α secretion from antigen-stimulated T-cells at low concentrations and inhibits secretion at high concentrations (225). IL-1β-induced NF-κB activation is also inhibited at high but not at low E2 concentrations (226). Hence, it is important to carry out in vitro studies of sex hormone responses over a wide range of physiologic concentrations of sex hormones.
In general, androgens have a suppressive effect on proinflammatory cytokine expression in monocytes and MDMs. This finding is consistent with the idea that the immune system of females produces cytokines in response to pathogens and insults more robustly than that of males. Monocyte or MDM expression of TNF-α, IL-1β, IL-6, and IL-8 is reduced in the presence of testosterone (227–229). Many studies in this field have relied on human cell lines, such as THP-1 and U937, with or without PMA-induced differentiation into macrophages, and differentiated HL-60 cells, although primary monocytes and MDMs have been used in a few cases (224, 230). Another immunoregulatory function of testosterone is the upregulation and secretion of C1 inhibitor (C1INH) from monocytes (231). C1INH is a 105 kDa plasma protein whose main function is inhibition of the complement system to prevent spontaneous activation. Thus, testosterone keeps complement activation in check. Another mechanism by which testosterone limits inflammation is by decreasing the generation of reactive oxygen species generation from differentiated HL-60 cells. Interestingly, the production of reactive oxygen species in response to zymosan, but not LPS, was inhibited by testosterone (228).
In terms of allergic immune responses, metabolism of arachidonic acid into inflammatory leukotrienes (LTs) via the 5-lipoxygenase (5-LO) pathway is sex-dependent in human monocytes. Pergola et al. (232) reported that primary human peripheral blood monocytes from women synthesize more 5-LO product than do the same cells from men. 5α-DHT (10 nM) suppressed LT synthesis in female cells to the levels observed in males. ERK activation by androgens reduced phospholipase D activity in monocytes and impaired 5-LO product formation by reducing active diacylglycerides. The other branch of arachidonic acid metabolism is the cyclooxygenase (COX) pathway, which generates prostaglandins. Prostaglandin E2 (PGE2), one of the most abundant COX products produced by the airway epithelium and smooth muscle (233, 234), can either stimulate or suppress immune cell function. Testosterone reduced PGE2 production in monocytes obtained from heparinized peripheral blood of healthy adults and incubated for 24 h with LPS (235).
A few studies have examined the effect of DHEA on human monocytes and macrophages. In the presence of LPS, DHEA induced IL-6 and TNF-α production by primary human monocytes and IL-8 and TNF-α production by THP-1 cells (236). In these experiments, DHEA counteracted the effects of cortisol and the glucocorticoid receptor on LPS-induced IL-6 and TNF-α by inducing expression of the scaffolding protein RACK1 (Receptor for Activated C Kinase 1) in THP-1 cells and primary human monocytes (237). RACK1 is involved in multiple signal transduction cascades, including the MAPK, protein kinase C, and Src signaling pathways. RACK1 shuttles proteins around the cell, anchors proteins at particular locations, and is involved in cell migration (238). In contrast, DHEA added to alveolar macrophages lavaged from 11 non-smoking asbestos workers significantly reduced superoxide anion release in vitro (239), consistent with its role in dampening Th2-inflammation (240). Therefore, the effect of DHEA on monocytes and macrophages may be stimulus-dependent and needs more in-depth investigation.
The formation of foam cells (lipid-filled macrophages) is generally associated with the pathogenesis of cardiovascular diseases, such as atherosclerosis. However, foam cells are also found in patients with silicosis (241) and other fibrotic lung diseases (242) and in tuberculosis. Alveolar macrophages take up extracellular and intracellular lipids in response to inhaled silica, vaping products (243), and Mycobacterium tuberculosis (244). Furthermore, the metabolism of fatty acids by macrophages by β-oxidation for sustained energy production is a key feature of the functional phenotype of macrophages with a pro-resolving, tissue reparative (M2) phenotype. Therefore, we have included how androgens modulate foam cell formation and lipid handling in macrophages as part of this discussion.
Macrophages from men and those exposed to testosterone favor the processes of lipid handling and foam cell formation, supporting evidence that atherosclerosis is a male-dominant disease when age is taken into account (245). Atherosclerotic plaques composed of a number of different immune cells form in blood vessel walls. In advanced stages of atherosclerosis, macrophages in plaques take up oxidized low-density lipoprotein (LDL), creating foam cells. Eventually, cholesterol crystals accumulate, trigger inflammation and plaque rupture. The role of sex in the inflammatory events of atherosclerosis has been reviewed elsewhere (246). In vitro studies have sought to ascertain how testosterone promotes these processes by utilizing primary MDMs. In MDMs from healthy men, androgen treatment was shown to upregulate genes involved with lipoprotein processing, transporter proteins, cell-surface adhesion, and other pathways, but none of these genes were upregulated in female macrophages (247). The marked sex specificity of androgen effects on human macrophage gene expression is most likely related to sex differences in MDM AR expression. Similarly, treatment of MDMs with modified and native LDL led to changes in expression of mRNAs involved in homeostatic regulation of lipid metabolism, depending on the sex of macrophage donors (248).
Functionally, androgen-treated MDMs from men but not women accumulate cholesteryl esters (196). Male macrophages exhibit increased rates of lysosomal acetylated LDL degradation and upregulated expression of scavenger receptor class B type I (249), increasing high-density lipoprotein (3)-induced cholesterol efflux. The expression of AR in monocytes/macrophages also upregulates lectin-type oxidized LDL receptor 1 molecules that are involved in foam cell formation (198). However, Corcoran et al. (250) observed no effect of testosterone on cholesterol content or efflux from MDMs of healthy male and postmenopausal female donors (age 50–70 years). Because their study used healthy donors, it is possible that the absence of other health-related factors, such as smoking, poor health, and genetic risk factors for coronary heart disease in the healthy blood donors may have produced these results.
Chemotaxis of THP-1 cells was diminished when androgen receptor was knocked down by siRNA suggesting a role for AR in migration of monocytes (198). The authors identified TNF-α as a key AR-regulated molecule important in monocyte migration. In contrast, a handful of studies have tested the effect of testosterone on primary human monocyte phagocytosis and migration, but no effect was found (222, 251–253). Testosterone did not change the cytotoxic capacity of monocytes from male donors (age range 18–40 years) to lyse red cells sensitized with IgG antibodies (254).
Most studies that have used mouse models to investigate sex differences in lung diseases have focused on the role of estrogen and estrogen receptors (255–257). The importance of androgen and ARs in lung disease has been poorly studied. Earlier studies were directed at modulating monocyte and macrophage functions unconnected to AR function, as 15 years ago it was believed that mouse macrophages did not express classical ARs (189). Nevertheless, recent studies have examined sex differences in mouse models of allergic asthma, COPD, and influenza.
We and others have reported sex differences in mouse models of allergic lung inflammation (37, 38, 188, 255). Some of the observed differences have been clearly attributed to the effect of androgens. We showed that DHT reconstitution of castrated male mice reduced overall lung inflammation (188). A reduction of total serum IgE and total immune cell recruitment to the lungs, specifically eosinophils, revealed the regulatory effect of androgens on several cell types. However, the unexpected enhancement of the production of the canonical M2 macrophage marker involved in eosinophil recruitment (258, 259), YM1, by DHT in alveolar macrophages (188) showed that androgens have a regulatory or an activating effect depending on the cell type. We demonstrated that deletion of classical ARs on monocytes and macrophages (ARfloxLysMCre mice) resulted in reduced inflammation (less eosinophil recruitment to the alveolar space), along with less mucus production and lung cell infiltrate, despite no differences in serum testosterone level between AR-sufficient and ARfloxLysMCre mice (188). This finding indicates the importance of androgens as modulators of M2 macrophage polarization and the critical role of these cells in allergic lung inflammation. Other recent studies have shown that testosterone has an anti-inflammatory role in a mouse model of allergic lung inflammation induced by house dust mite but focused on other cell types in lung, such as Th2 (260) and ILC2 cells (261, 262). Similarly, high concentrations of androgens in circulation have been related to a decrease in the expression of TNFα and other proinflammatory cytokines, such as IL-6 and IL-1β, in rodent macrophage cell models and in human monocytes (203, 223, 224, 230, 263). How androgen and ARs impact functions on IMs still needs to be studied. At the time this review was written, no reports on AR expression in IMs were found. However, we hypothesize that as IMs are derived from blood monocytes (102), but once in the tissue they develop an intermediate size and phenotype between monocytes and AM (103, 264), their expression of AR could be somewhere in between. Therefore, androgen and ARs could regulate the functions and activation of these cells. This requires further study, as IMs are a constitutive macrophage population in the lung, and may play a role mediating sex differences in lung diseases.
Mouse models have also shown that sex differences affect COPD. In 2016, Tam et al. (265) reported that smoke-induced COPD is characterized by small airway remodeling in female but not male mice and that ovariectomy before smoke exposure ameliorates the disease. Another study focusing on α-1 antitrypsin deficiency, the leading genetic cause of emphysema, also uncovered a higher susceptibility of female mice for this condition (266). However, these studies did not determine if androgens mediate resistance to COPD, or if the key to the observed sex differences is ovarian sex hormones. Thus, the role that androgens play in COPD and COPD models remains unclear.
Mouse studies that have focused on sex differences in influenza showed that at moderate influenza virus A (IAV) loads, morbidity, mortality, and the associated inflammatory response is greater in female than in male mice, but that mortality is similar at higher loads (72, 267, 268). The role of sex hormones was well-addressed in these studies. High levels of estrogen in estrogen-reconstituted female mice protected against lethal IAV doses (72), whereas the lower estrogen levels in intact females were associated with greater inflammatory responses and increased morbidity after infection. Similar observations were made after progesterone replacement (269). In males, a decrease in androgen levels after castration increased morbidity and pathology upon IAV infection, but replacement of testosterone or DHT reduced morbidity, mortality, and inflammation (72, 74). These findings suggest that although estrogen may be protective or detrimental, depending on concentration, androgens may suppress inflammation in a broader way.
Gonadectomy studies in mice have been used to uncover the role of androgens in TB. Similar to observations in castrated men, castrated male mice that displayed greater pro-inflammatory responses in the lung (more TNF-α, IFN γ, IL-12, iNOS, and IL-17) than intact males. IFN-γ-activated macrophages (M1 macrophages) control of TB infection in both human and mouse (270). Ovary removal in females did not impact susceptibility to TB (271), suggesting that testosterone is responsible for male susceptibility to TB. We previously reported that DHT enhances M2 macrophage polarization through AR (188). Therefore, we speculate that the greater male susceptibility to TB could be at least in part mediated by enhanced M2 responses that are poorly protective and decrease protective proinflammatory macrophage responses. Formal studies to address this idea as well as how androgen effects on other key immune cell players in TB are needed.
How androgens affect monocyte and macrophage biology in lung cancer models in mice has not been well-studied. Monocytes and macrophages are important cellular players in tumorigenesis. Tumor-associated macrophages (TAMs) can be classified into two phenotypes that are either pro-inflammatory and tumoricidal (M1-like) or promote tumor growth and suppress anti-tumor immune responses (M2-like) (272–274). As mentioned previously, sex hormones augment M2 macrophage polarization, thus, play an important role in lung carcinogenesis. The greater overall incidence of lung cancer in men could be explained by an enhanced M2 polarization by androgens (188). On the other hand, estrogen has been shown to induce tumor angiogenesis (275). Estrogen signaling though the cAMP, MAPK, and AKT pathways with the consequent phosphorylation of ERK and EGFR signaling, along with the enhanced expression of c-myc and cyclin D, results in NSCLC cell proliferation (276). Mouse models must therefore address the role of androgens on monocytes and macrophage function in the establishment and progression of lung cancer in male and female animals.
Few studies have examined the effect of sex hormones on peripheral blood monocytes and lung macrophages from men and women with asthma or the other lung diseases we have discussed here. In women with asthma, dominance of M2 macrophages in airways and lung tissue has been documented (277) and a connection between female sex and female sex hormones surmised. There is a paucity of literature regarding how introducing or depleting exogenous sex hormones (such as in female-to-male transgender individuals receiving testosterone supplementation or women with estrogen blockade) affects the function of blood monocytes and lung macrophages in men and women with asthma. Most studies correlate concentrations of sex hormones with either inflammatory markers, such as cytokines or chemokines in serum and other fluids, or with lung function measurements. We will summarize below the small number of studies in which androgen concentrations were manipulated in humans and the effects on monocyte or macrophage function.
Hypogonadism in men refers to a deficiency in testosterone production from the testes that results from testicular, hypothalamic, or pituitary abnormalities. Klinefelter's syndrome in men, which is a result of additional X-chromosomes (e.g., XXY), is the most common cause of hypogonadism. Testosterone replacement therapy is the primary treatment option to restore physiologic testosterone levels, typically in the range of 300 to 800 ng/dL. In general, exogenously administered testosterone has a suppressive effect on the proinflammatory immune response from monocytes. For example, spontaneous production of proinflammatory cytokines (IL-1β, IL-6, and TNFα) ex vivo was reduced or completely absent in the monocytes and DCs from men with type-2 diabetes who had partial androgen deficiency and were treated for 12 months with testosterone replacement. This suppression was maintained for 3 more months after testosterone withdrawal (278). Testosterone replacement therapy also is associated with a reduction or complete abrogation of spontaneous ex vivo production of inflammatory cytokines by antigen-presenting cells (279). On the other hand, the circulating monocytes from hypogonadal men treated with testosterone replacement therapy exhibited significantly upregulated expression of CD107b at baseline compared to monocytes from healthy controls. This was also seen after stimulation with CpG oligodeoxynucleotides to mimic bacterial DNA exposure (280). Membrane expression of CD107b, also known as lysosome-associated membrane protein (LAMP)2, is indicative of release of lysosome and/or phagolysosome contents into the extracellular medium, a mechanism that may be involved in killing and/or digesting target cells. These data suggest that testosterone increases the inflammatory function of these cells, an effect that would contrast with its typical role as an immunosuppressant.
The immune system of individuals with Klinefelter's syndrome provides unique insight into the genetic contribution of the X-chromosome and that of diminished testosterone to sex differences in different diseases. Men with Klinefelter's syndrome have an increased risk of developing autoimmune diseases, particularly those that are typically female-dominant, such as rheumatoid arthritis and systemic lupus erythematosus (281). As might be predicted due to the negative effect of lower concentration of testosterone on lung function, men with Klinefelter's syndrome are more likely to be diagnosed with pulmonary diseases, such as COPD and pneumonia (282). Asthma is also reported in these individuals (283–285) and it was successfully controlled with long-acting β-agonists and oral testosterone replacement in one case report (283). At the cellular level, however, cytokine production in stimulated whole blood from Klinefelter's men was similar to that of women (286). These data suggest that the effect of the additional X-chromosome was more dominant than the reduction in circulating androgen in Klinefelter's men. In the same study, however, purified monocytes showed the opposite response: cytokine production from the monocytes of healthy and Klinefelter's men was similar and less robust than that from the monocytes of women. This observation led to the opposite conclusion—that androgen plays a more important role in monocyte cytokine production than does chromosomal complement.
PCOS is a disease characterized by hyperandrogenism, amenorrhea, and polycystic ovaries. The cystic follicles—ovarian theca cells—produce testosterone that causes significant elevations in serum concentrations of testosterone, androstenedione, DHEA, and DHEA-S. In women with PCOS, serum testosterone is in the range of 45–150 ng/dL (2–5 nmol/L) (287), compared with a range of 20–60 ng/dL in healthy, ovulatory women (288). This endocrinopathy is associated with metabolic disorders, such as dyslipidemia, insulin resistance, metabolic syndrome, and cardiovascular complications. Immune function is impaired in women with PCOS, leading to increased secretion of autoantibodies and increased risk of type 1 diabetes, asthma, and thyroid disease (289). Because androgens downregulate the inflammatory responses that contribute to asthma, one might hypothesize that women with PCOS would have less asthma. However, Htet al. (290) found that asthma prevalence was 15.2% in women with PCOS compared to only 10.6% in women without PCOS (P = 0.004). Women both with and without PCOS who had asthma tended to have a higher BMI than those without asthma (290). After multivariable analysis, the authors concluded that both PCOS and high BMI were independently associated with asthma (291). It is therefore possible that testosterone contributes to the chronic inflammatory state that accompanies high BMI and that the metabolic dysfunction overpowers the protective effects of testosterone on asthma development. Few cellular and molecular studies have endeavored to uncover mechanisms that explain the association between asthma and PCOS.
At the cellular level, circulating monocytes from women with PCOS expressed the receptor for advanced glycation end-products (RAGE) more strongly than monocytes from healthy control women (292). AGEs are involved in the pathogenesis of a number of chronic lung diseases, ranging from cystic fibrosis to asthma. RAGE can also bind other alarmins, such as the S100A8/A9 heterodimer (calprotectin) or the high-mobility group box (HMGB)1 protein. Both of these ligands have been implicated in the pathogenesis of allergic asthma (291, 293), as they induce cell proliferation or apoptosis, inflammation, collagen synthesis, and cell migration in many different cell types. The concentration of AGE proteins and testosterone correlated positively, even after controlling for BMI and other metabolic function tests (292). Taken together these two studies suggest that monocytes from women with PCOS would be more responsive to RAGE ligands. This heightened responsiveness could promote cellular inflammatory responses that contribute to asthma pathogenesis. Studies are needed to examine how the increased testosterone in women with PCOS affects circulating monocytes and lung macrophages to increase asthma prevalence in this group.
Testosterone has been administered therapeutically for asthma. In an early study, asthmatic women were given testosterone either daily for 5 days over 2 weeks or daily for 3 days over 2 or more weeks. Although the number of participants in the study was small, 88% saw improved symptoms, with 47% reporting no asthma attacks up to 3 months later (294, 295). No studies have examined the effect of exogeneous testosterone administration on blood monocytes or lung macrophages in men and women with asthma. Testosterone deficiency is also present in patients with COPD (296–304). In a clinical study of exercise and testosterone injection in men with COPD, the interventions did not significantly alter pulmonary function or blood gas variables (305). On the other hand, a retrospective study of two large cohorts of men who commenced testosterone replacement therapy within 12 months of a COPD diagnosis showed a 4.2–9.1% decrease in hospitalizations, dependent on age (306). More work is needed to understand how testosterone and its signaling pathways can be harnessed to alleviate lung disease without affecting reproductive systems or having unwanted metabolic effects.
Asthmatic patients have decreased serum concentration of DHEA and DHEA-S (307–309). Therefore, some clinical trials have tested whether DHEA-S supplementation reduces asthma. Men and women with poorly controlled moderate-to-severe asthma were given nebulized DHEA-S for 6 weeks. This treatment led to a statistically significant improvement in the Asthma Control Questionnaire (ACQ) and trends toward better asthma symptom scores and more symptom-free days and nights (310). Oral DHEA for 2 weeks improved lung function in asthmatic women with low DHEA-S < 200 μg/dL (48). However, neither of these clinical studies examined the cellular component of the disease pre- or post-intervention. DHEA and DHEA-S are also lower in patients with COPD than in healthy controls, and COPD leads to pulmonary hypertension (PH). DHEA supplementation improved the 6-min walk test, pulmonary hemodynamics, and the diffusing capacity of the lungs for carbon monoxide of patients with PH-COPD (311, 312). The therapeutic potential of DHEA is currently being investigated in 24 patients with PH in the EDIPHY (Effects of DHEA in Pulmonary Hypertension) trial. However, outcome measures of this trial do not include examination of the immune cells or the effect of DHEA treatment on those cells. Analysis of immune cell function would add important cellular mechanistic insight to these types of trials and help uncover some of the widespread effects of this hormone on the immune system.
Modulation of monocyte and macrophage function mediated by the interaction of androgen and AR has been examined mostly by correlative studies in humans following lifespan changes in sex hormones or using hormonal manipulation in mouse models of lung disease. Most human-based reports are merely descriptive or correlative and do not consider variables such as age, BMI, and phase of the menstrual cycle as key modulators of circulating sex hormone concentrations. Taking these factors into account should be encouraged if we are to gain a better understanding of the impact of sex hormones in health and disease. Analyses of the function of immune cells from male and female healthy controls and patients with lung diseases are needed to unlock how sex hormones alter the biology of the innate and adaptive immune response.
Studying the role of sex hormones as modulators of the immune system is complex because they interact with other hormonal systems and with one another, and because of the nearly ubiquitous expression of sex hormone receptors in most cells of the body. Males and females have all types of sex steroids, although in different circulating concentrations. In humans, changes in the concentration of sex steroids have implications for lung health and may contribute to disease by affecting the function of the immune system. Female sex hormones have been more widely studied as immune system modulators than have androgens. More focus in the future must be directed to how androgens affect the immune system and the interaction between male and female sex steroids in immune function.
Historically, animal models have used only males as study subjects, leaving females aside out of concern for the variability in results introduced by sexually mature adult females with active estrous cycles. As a result, biomedical and preclinical research has neglected to reflect more than 50% of the world's population. This omission had some notable negative consequences: eight of ten drugs withdrawn by the FDA between 1997 and 2000 had significant health risks to women (313). It was not until 2016 that the NIH addressed this oversight with its requirement to include sex as a biological variable in all research studies (314). The practices of using only male animals, not clearly reporting the sex (and age) of the animals used, and mixing male and female results have obscured a proper understanding of how sex and sex hormones influence normal biology and that of disease states. Moreover, many reports comparing sex as a variable lack strict controls on culture conditions in vitro, which can alter the results. For example, if investigators fail to appreciate that animal serum or pH indicators, such as phenol red, may act as a source of steroids or sex hormone receptor agonists and do not clearly report their use, the interpretation and reproducibility of the experiments can be diminished. We strongly advocate for the use of hormone-free serum or animal serum replacements (for human cell studies) and use of culture medium that does not contain sex steroid receptor agonists. Moreover, rigorous experimentation should include careful and detailed reporting of cell culture conditions, donor sex and age for cell studies, accurate age and sex in animal work (adherence to ARRIVE guidelines), and separate male and female results.
Here, we have highlighted the importance of sex hormones as modulators of monocytes and macrophages and the important role of these innate immune cells in lung diseases where sex differences are apparent. These cells are part of a larger response that includes the adaptive immune system as well as the structural cells of the lung that are all affected by the action of sex steroids. As such, how innate cells like monocytes and macrophages shape the pulmonary immune response and how they resolve lung inflammation differently in the male and female lung and in the presence of different sex steroids needs intensive study. Uncovering the cellular and molecular mechanisms will be crucial for finding new ways to treat different lung diseases depending on the sex of the patient.
MB-D, MS, and NH wrote and revised the manuscript, interpreted the literature, approved and are accountable for all aspects of the final version. All authors contributed to the article and approved the submitted version.
NIH (NHLBI) R01 HL124477 (to NH). Dept of Defense CDMRP W81XWH-16-1-0509 (to NH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. (2010) 125(2 Suppl. 2):S3–23. doi: 10.1016/j.jaci.2009.12.980
2. Gubbels Bupp MR, Jorgensen TN. Androgen-induced immunosuppression. Front Immunol. (2018) 9:794. doi: 10.3389/fimmu.2018.00794
3. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
4. Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. (2010) 125(2 Suppl. 2):S33–40. doi: 10.1016/j.jaci.2009.09.017
5. Farber DL, Netea MG, Radbruch A, Rajewsky K, Zinkernagel RM. Immunological memory: lessons from the past and a look to the future. Nat Rev Immunol. (2016) 16:124–8. doi: 10.1038/nri.2016.13
7. Brodin P, Davis MM. Human immune system variation. Nat Rev Immunol. (2017) 17:21–9. doi: 10.1038/nri.2016.125
8. Sellau J, Groneberg M, Lotter H. Androgen-dependent immune modulation in parasitic infection. Semin Immunopathol. (2019) 41:213–24. doi: 10.1007/s00281-018-0722-9
9. Sanderson JT. The steroid hormone biosynthesis pathway as a target for endocrine-disrupting chemicals. Toxicol Sci. (2006) 94:3–21. doi: 10.1093/toxsci/kfl051
10. Nelson LR, Bulun SE. Estrogen production and action. J Am Acad Dermatol. (2001) 45(3 Suppl.):S116–24. doi: 10.1067/mjd.2001.117432
11. Barakat R, Oakley O, Kim H, Jin J, Ko CJ. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. (2016) 49:488–96. doi: 10.5483/BMBRep.2016.49.9.141
12. Richard E, Jones KHL. Human Reproductive Biology. 4th ed. Waltham, MA: Academic Press (2013). p. 103–18.
13. Marchetti PM, Barth JH. Clinical biochemistry of dihydrotestosterone. Ann Clin Biochem. (2013) 50(Pt 2):95–107. doi: 10.1258/acb.2012.012159
14. Torjesen PA, Sandnes L. Serum testosterone in women as measured by an automated immunoassay and a RIA. Clin Chem. (2004) 50:678 author reply 679–9. doi: 10.1373/clinchem.2003.027565
15. Travison TG, Vesper HW, Orwoll E, Wu F, Kaufman JM, Wang Y, et al. Harmonized reference ranges for circulating testosterone levels in men of four cohort studies in the United States and Europe. J Clin Endocrinol Metab. (2017) 102:1161–73. doi: 10.1210/jc.2016-2935
16. Catenaccio E, Mu W, Lipton ML. Estrogen- and progesterone-mediated structural neuroplasticity in women: evidence from neuroimaging. Brain Struct Funct. (2016) 221:3845–67. doi: 10.1007/s00429-016-1197-x
17. Pennell LM, Galligan CL, Fish EN. Sex affects immunity. J Autoimmun. (2012) 38:J282–91. doi: 10.1016/j.jaut.2011.11.013
18. Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. (2010) 10:594–604. doi: 10.1038/nri2815
19. Owens IP. Ecology and evolution. Sex differences in mortality rate. Science. (2002) 297:2008–9. doi: 10.1126/science.1076813
20. Cook MB, McGlynn KA, Devesa SS, Freedman ND, Anderson WF. Sex disparities in cancer mortality and survival. Cancer Epidemiol Biomarkers Prev. (2011) 20:1629–37. doi: 10.1158/1055-9965.EPI-11-0246
21. Dorak MT, Karpuzoglu E. Gender differences in cancer susceptibility: an inadequately addressed issue. Front Genet. (2012) 3:268. doi: 10.3389/fgene.2012.00268
22. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. (2016) 16:626–38. doi: 10.1038/nri.2016.90
23. World Health Organization. Gender: Definitions. World Health Organization Website. (2018). Available online at: http://www.euro.who.int/en/health-topics/health-determinants/gender/genderdefinitions (accessed 2002).
24. Furman D, Hejblum BP, Simon N, Jojic V, Dekker CL, Thiebaut R, et al. Systems analysis of sex differences reveals an immunosuppressive role for testosterone in the response to influenza vaccination. Proc Natl Acad Sci USA. (2014) 111:869–74. doi: 10.1073/pnas.1321060111
25. ALA. Data American Lung Association: Lung Disease Data. (2008) Available online at: http://www.lung.org/assets/documents/publications/lung-disease-data/LDD_2008.pdf (accessed 2008).
26. Pinkerton KE, Harbaugh M, Han MK, Jourdan Le Saux C, van Winkle LS, Martin WJ, et al. Women and lung disease. Sex differences and global health disparities. Am J Respir Crit Care Med. (2015) 192:11–6. doi: 10.1164/rccm.201409-1740PP
27. Farrell PM, Wood RE. Epidemiology of hyaline membrane disease in the United States: analysis of national mortality statistics. Pediatrics. (1976) 58:167–76.
28. Khoury MJ, Marks JS, McCarthy BJ, Zaro SM. Factors affecting the sex differential in neonatal mortality: the role of respiratory distress syndrome. Am J Obstetr Gynecol. (1985) 151:777–82. doi: 10.1016/0002-9378(85)90518-6
29. Stevenson DK, Wright LL, Lemons JA, Oh W, Korones SB, Papile LA, et al. Very low birth weight outcomes of the National institute of child health and human development neonatal research network, january 1993 through December 1994. Am J Obstetr Gynecol. (1998) 179(6 Pt 1):1632–9. doi: 10.1016/S0002-9378(98)70037-7
30. Elsmen E, Hansen Pupp I, Hellstrom-Westas L. Preterm male infants need more initial respiratory and circulatory support than female infants. Acta Paediatr. (2004) 93:529–33. doi: 10.1080/08035250410024998
31. Myers TR. Pediatric asthma epidemiology: incidence, morbidity, and mortality. Respir Care Clin N Am. (2000) 6:1–14. doi: 10.1016/s1078-5337(05)70054-x
32. Bjornson CL, Mitchell I. Gender differences in asthma in childhood and adolescence. J Gend Specif Med. (2000) 3:57–61. doi: 10.1016/S1550-8579(07)80054-4
33. deBoer MD, Phillips BR, Mauger DT, Zein J, Erzurum SC, Fitzpatrick AM, et al. Effects of endogenous sex hormones on lung function and symptom control in adolescents with asthma. BMC Pulm Med. (2018) 18:58. doi: 10.1186/s12890-018-0612-x
34. Kynyk JA, Mastronarde JG, McCallister JW. Asthma, the sex difference. Curr Opin Pulm Med. (2011) 17:6–11. doi: 10.1097/MCP.0b013e3283410038
35. Hansen S, Probst-Hensch N, Keidel D, Dratva J, Bettschart R, Pons M, et al. Gender differences in adult-onset asthma: results from the Swiss SAPALDIA cohort study. Eur Respir J. (2015) 46:1011–20. doi: 10.1183/13993003.02278-2014
36. Dharmage SC, Perret JL, Custovic A. Epidemiology of Asthma in children and adults. Front Pediatr. (2019) 7:246. doi: 10.3389/fped.2019.00246
37. Blacquiere MJ, Hylkema MN, Postma DS, Geerlings M, Timens W, Melgert BN. Airway inflammation and remodeling in two mouse models of asthma: comparison of males and females. Int Arch Allergy Immunol. (2010) 153:173–81. doi: 10.1159/000312635
38. Melgert BN, Postma DS, Kuipers I, Geerlings M, Luinge MA, van der Strate BW, et al. Female mice are more susceptible to the development of allergic airway inflammation than male mice. Clin Exp Allergy. (2005) 35:1496–503. doi: 10.1111/j.1365-2222.2005.02362.x
39. Mohan SS, Knuiman MW, Divitini ML, James AL, Musk AW, Handelsman DJ, et al. Higher serum testosterone and dihydrotestosterone, but not oestradiol, are independently associated with favourable indices of lung function in community-dwelling men. Clin Endocrinol. (2015) 83:268–76. doi: 10.1111/cen.12738
40. Mileva Z, Maleeva A. The serum testosterone level of patients with bronchial asthma treated with corticosteroids and untreated. Vutr Boles. (1988) 27:29–32.
41. Swartz MA, Tschumperlin DJ, Kamm RD, Drazen JM. Mechanical stress is communicated between different cell types to elicit matrix remodeling. Proc Natl Acad Sci USA. (2001) 98:6180–5. doi: 10.1073/pnas.111133298
42. Shepard KV BA, Casale TB, Cardet JC. Elevated testosterone is associated with decreased likelihood of current asthma regardless of gender. In: American Academy of Allergy, Asthma & Immunology Annual Meeting. San Francisco CA. (2019). doi: 10.1016/j.jaci.2018.12.239
43. Matteis M, Polverino F, Spaziano G, Roviezzo F, Santoriello C, Sullo N, et al. Effects of sex hormones on bronchial reactivity during the menstrual cycle. BMC Pulm Med. (2014) 14:108. doi: 10.1186/1471-2466-14-108
44. Han YY, Forno E, Celedon JC. Sex steroid hormones and Asthma in a Nationwide study of US adults. Am J Respir Crit Care Med. (2020) 201:158–66. doi: 10.1164/rccm.201905-0996OC
45. Silva F, Oliveira EE, Ambrosio MGE, Ayupe MC, Souza VP, Gameiro J, et al. High-fat diet-induced obesity worsens TH2 immune response and immunopathologic characteristics in murine model of eosinophilic oesophagitis. Clin Exp Allergy. (2020) 50:244–55. doi: 10.1111/cea.13533
46. Dixon AE, Holguin F. Diet and metabolism in the evolution of Asthma and obesity. Clin Chest Med. (2019) 40:97–106. doi: 10.1016/j.ccm.2018.10.007
47. Tashiro H, Shore SA. Obesity and severe asthma. Allergol Int. (2019) 68:135–42. doi: 10.1016/j.alit.2018.10.004
48. Marozkina N, Zein J, DeBoer MD, Logan L, Veri L, Ross K, et al. Dehydroepiandrosterone supplementation may benefit women with asthma who have low androgen levels: a pilot study. Pulm Ther. (2019) 5:213–20. doi: 10.1007/s41030-019-00101-9
49. Kannisto S, Laatikainen A, Taivainen A, Savolainen K, Tukiainen H, Voutilainen R. Serum dehydroepiandrosterone sulfate concentration as an indicator of adrenocortical suppression during inhaled steroid therapy in adult asthmatic patients. Eur J Endocrinol. (2004) 150:687–90. doi: 10.1530/eje.0.1500687
50. Suzuki T, Suzuki N, Daynes RA, Engleman EG. Dehydroepiandrosterone enhances IL2 production and cytotoxic effector function of human T cells. Clin Immunol Immunopathol. (1991) 61(2 Pt 1):202–11. doi: 10.1016/S0090-1229(05)80024-8
51. Young DG, Skibinski G, Skibinska A, Mason JI, James K. Preliminary studies on the effect of dehydroepiandrosterone (DHEA) on both constitutive and phytohaemagglutinin (PHA)-inducible IL-6 and IL-2 mRNA expression and cytokine production in human spleen mononuclear cell suspensions in vitro. Clin Exp Immunol. (2001) 123:28–35. doi: 10.1046/j.1365-2249.2001.01445.x
52. Chen W, Mempel M, Schober W, Behrendt H, Ring J. Gender difference, sex hormones, and immediate type hypersensitivity reactions. Allergy. (2008) 63:1418–27. doi: 10.1111/j.1398-9995.2008.01880.x
53. McLachlan JA, Serkin CD, Bakouche O. Dehydroepiandrosterone modulation of lipopolysaccharide-stimulated monocyte cytotoxicity. J Immunol. (1996) 156:328–35.
54. Weiss A, Porter S, Rozenberg D, O'Connor E, Lee T, Balter M, et al. Chronic obstructive pulmonary disease: a palliative medicine review of the disease, its therapies and drug interactions. J Pain Symptom Manage. (2020) 60:135–50. doi: 10.1016/j.jpainsymman.2020.01.009
55. MacNee W, Donaldson K. Mechanism of lung injury caused by PM10 and ultrafine particles with special reference to COPD. Eur Respir J Suppl. (2003) 40:47s–51s. doi: 10.1183/09031936.03.00403203
56. Di Stefano A, Caramori G, Ricciardolo FL, Capelli A, Adcock IM, Donner CF. Cellular and molecular mechanisms in chronic obstructive pulmonary disease: an overview. Clin Exp Allergy. (2004) 34:1156–67. doi: 10.1111/j.1365-2222.2004.02030.x
57. Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. (2007) 370:765–73. doi: 10.1016/S0140-6736(07)61380-4
58. Sorheim IC, Johannessen A, Gulsvik A, Bakke PS, Silverman EK, DeMeo DL. Gender differences in COPD: are women more susceptible to smoking effects than men? Thorax. (2010) 65:480–5. doi: 10.1136/thx.2009.122002
59. Gan WQ, Man SF, Postma DS, Camp P, Sin DD. Female smokers beyond the perimenopausal period are at increased risk of chronic obstructive pulmonary disease: a systematic review and meta-analysis. Respir Res. (2006) 7:52. doi: 10.1186/1465-9921-7-52
60. Han MK, Murray S, Fell CD, Flaherty KR, Toews GB, Myers J, et al. Sex differences in physiological progression of idiopathic pulmonary fibrosis. Eur Respir J. (2008) 31:1183–8. doi: 10.1183/09031936.00165207
61. Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. (2006) 61:980–5. doi: 10.1136/thx.2006.062836
62. Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. (2007) 176:277–84. doi: 10.1164/rccm.200701-044OC
63. Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, Cottin V, et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. (2013) 41:1207–18. doi: 10.1183/09031936.00073012
64. Serfling RE, Sherman IL, Houseworth WJ. Excess pneumonia-influenza mortality by age and sex in three major influenza A2 epidemics, United States, 1957-58, 1960 and 1963. Am J Epidemiol. (1967) 86:433–41. doi: 10.1093/oxfordjournals.aje.a120753
65. Eshima N, Tokumaru O, Hara S, Bacal K, Korematsu S, Tabata M, et al. Sex- and age-related differences in morbidity rates of 2009 pandemic influenza A H1N1 virus of swine origin in Japan. PLoS ONE. (2011) 6:e19409. doi: 10.1371/journal.pone.0019409
66. Kumar A, Zarychanski R, Pinto R, Cook DJ, Marshall J, Lacroix J, et al. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA. (2009) 302:1872–9. doi: 10.1001/jama.2009.1496
67. Chitnis AS, Truelove SA, Druckenmiller JK, Heffernan RT, Davis JP. Epidemiologic and clinical features among patients hospitalized in Wisconsin with 2009 H1N1 influenza A virus infections, April to August 2009. WMJ. (2010) 109:201–8.
68. Koegelenberg CF, Irusen EM, Cooper R, Diacon AH, Taljaard JJ, Mowlana A, et al. High mortality from respiratory failure secondary to swine-origin influenza A (H1N1) in South Africa. QJM. (2010) 103:319–25. doi: 10.1093/qjmed/hcq022
69. Tadount F, Doyon-Plourde P, Rafferty E, MacDonald S, Sadarangani M, Quach C. Is there a difference in the immune response, efficacy, effectiveness and safety of seasonal influenza vaccine in males and females? - A systematic review. Vaccine. (2020) 38:444–59. doi: 10.1016/j.vaccine.2019.10.091
70. Roriz-Cruz M, Rosset I, Montero-Odasso M. Lower mortality from H1N1 influenza in older Argentineans: men more affected. J Am Geriatr Soc. (2010) 58:1813–5. doi: 10.1111/j.1532-5415.2010.03045.x
72. Robinson DP, Lorenzo ME, Jian W, Klein SL. Elevated 17beta-estradiol protects females from influenza A virus pathogenesis by suppressing inflammatory responses. PLoS Pathog. (2011) 7:e1002149. doi: 10.1371/journal.ppat.1002149
73. Robinson DP, Hall OJ, Nilles TL, Bream JH, Klein SL. 17β-estradiol protects females against influenza by recruiting neutrophils and increasing virus-specific CD8 T cell responses in the lungs. J Virol. (2014) 88:4711–20. doi: 10.1128/JVI.02081-13
74. Vom Steeg LG, Vermillion MS, Hall OJ, Alam O, McFarland R, Chen H, et al. Age and testosterone mediate influenza pathogenesis in male mice. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L1234–L44. doi: 10.1152/ajplung.00352.2016
75. Neyrolles O, Quintana-Murci L. Sexual inequality in tuberculosis. PLoS Med. (2009) 6:e1000199. doi: 10.1371/journal.pmed.1000199
76. Hertz D, Schneider B. Sex differences in tuberculosis. Semin Immunopathol. (2019) 41:225–37. doi: 10.1007/s00281-018-0725-6
77. Uplekar M, Rangan S, Ogden J. Gender and Tuberculosis Control: Towards a Strategy for Research and Action, WHO/TB/2000.280. Geneva: World Health Organization (1999).
78. Nhamoyebonde S, Leslie A. Biological differences between the sexes and susceptibility to tuberculosis. J Infect Dis. (2014) 209(Suppl. 3):S100–6. doi: 10.1093/infdis/jiu147
79. Molina-Romero C, Arrieta O, Hernandez-Pando R. Tuberculosis and lung cancer. Salud Publica Mex. (2019) 61:286–91. doi: 10.21149/10090
80. Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. (2016) 893:1–19. doi: 10.1007/978-3-319-24223-1_1
81. Mao Y, Yang D, He J, Krasna MJ. Epidemiology of lung cancer. Surg Oncol Clin N Am. (2016) 25:439–45. doi: 10.1016/j.soc.2016.02.001
82. Houston KA, Henley SJ, Li J, White MC, Richards TB. Patterns in lung cancer incidence rates and trends by histologic type in the United States, 2004-2009. Lung Cancer. (2014) 86:22–8. doi: 10.1016/j.lungcan.2014.08.001
83. Lewis DR, Check DP, Caporaso NE, Travis WD, Devesa SS. US lung cancer trends by histologic type. Cancer. (2014) 120:2883–92. doi: 10.1002/cncr.28749
84. Jemal A, Miller KD, Ma J, Siegel RL, Fedewa SA, Islami F, et al. Higher lung cancer incidence in young women than young men in the United States. N Engl J Med. (2018) 378:1999–2009. doi: 10.1056/NEJMoa1715907
85. Kiyohara C, Ohno Y. Sex differences in lung cancer susceptibility: a review. Gend Med. (2010) 7:381–401. doi: 10.1016/j.genm.2010.10.002
86. Zang EA, Wynder EL. Differences in lung cancer risk between men and women: examination of the evidence. J Natl Cancer Inst. (1996) 88:183–92. doi: 10.1093/jnci/88.3.183
87. Hyland RA, Ware S, Johnson AR, Yates DH. Incidence trends and gender differences in malignant mesothelioma in New South Wales, Australia. Scand J Work Environ Health. (2007) 33:286–92. doi: 10.5271/sjweh.1145
88. Rubin JB, Lagas JS, Broestl L, Sponagel J, Rockwell N, Rhee G, et al. Sex differences in cancer mechanisms. Biol Sex Differ. (2020) 11:17. doi: 10.1186/s13293-020-00291-x
89. Velez MA, Burns TF, Stabile LP. The estrogen pathway as a modulator of response to immunotherapy. Immunotherapy. (2019) 11:1161–76. doi: 10.2217/imt-2019-0024
90. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, de Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
91. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. (2014) 15:1026–37. doi: 10.1038/ni.3005
92. van de Laar L, Saelens W, de Prijck S, Martens L, Scott CL, van Isterdael G, et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity. (2016) 44:755–68. doi: 10.1016/j.immuni.2016.02.017
93. Zaslona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, et al. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. (2014) 193:4245–53. doi: 10.4049/jimmunol.1400580
94. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. (2017) 214:2387–404. doi: 10.1084/jem.20162152
95. Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, et al. The cytokine TGF-β promotes the development and homeostasis of alveolar macrophages. Immunity. (2017) 47:903–12.e4. doi: 10.1016/j.immuni.2017.10.007
96. Phelps DS, Umstead TM, Floros J. Sex differences in the response of the alveolar macrophage proteome to treatment with exogenous surfactant protein-A. Proteome Sci. (2012) 10:44. doi: 10.1186/1477-5956-10-44
97. Phelps DS, Umstead TM, Quintero OA, Yengo CM, Floros J. In vivo rescue of alveolar macrophages from SP-A knockout mice with exogenous SP-A nearly restores a wild type intracellular proteome; actin involvement. Proteome Sci. (2011) 9:67. doi: 10.1186/1477-5956-9-67
98. Yu YR, Hotten DF, Malakhau Y, Volker E, Ghio AJ, Noble PW, et al. Flow cytometric analysis of myeloid cells in human blood, bronchoalveolar lavage, and lung tissues. Am J Respir Cell Mol Biol. (2016) 54:13–24. doi: 10.1165/rcmb.2015-0146OC
99. Bharat A, Bhorade SM, Morales-Nebreda L, McQuattie-Pimentel AC, Soberanes S, Ridge K, et al. Flow cytometry reveals similarities between lung macrophages in humans and mice. Am J Respir Cell Mol Biol. (2016) 54:147–9. doi: 10.1165/rcmb.2015-0147LE
100. Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. (2019) 25:1153–63. doi: 10.1038/s41591-019-0468-5
101. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-Cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. (2019) 199:1517–36. doi: 10.1164/rccm.201712-2410OC
102. Sabatel C, Radermecker C, Fievez L, Paulissen G, Chakarov S, Fernandes C, et al. Exposure to bacterial CpG DNA protects from airway allergic inflammation by expanding regulatory lung interstitial macrophages. Immunity. (2017) 46:457–73. doi: 10.1016/j.immuni.2017.02.016
103. Tan SY, Krasnow MA. Developmental origin of lung macrophage diversity. Development. (2016) 143:1318–27. doi: 10.1242/dev.129122
104. Bedoret D, Wallemacq H, Marichal T, Desmet C, Quesada Calvo F, Henry E, et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J Clin Invest. (2009) 119:3723–38. doi: 10.1172/JCI39717
105. Kawano H, Kayama H, Nakama T, Hashimoto T, Umemoto E, Takeda K. IL-10-producing lung interstitial macrophages prevent neutrophilic asthma. Int Immunol. (2016) 28:489–501. doi: 10.1093/intimm/dxw012
106. von Mutius E. The microbial environment and its influence on asthma prevention in early life. J Allergy Clin Immunol. (2016) 137:680–9. doi: 10.1016/j.jaci.2015.12.1301
107. Strachan DP. Hay fever, hygiene, and household size. BMJ. (1989) 299:1259–60. doi: 10.1136/bmj.299.6710.1259
108. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three unique interstitial macrophages in the murine lung at steady state. Am J Respir Cell Mol Biol. (2017) 57:66–76. doi: 10.1165/rcmb.2016-0361OC
109. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. (2019) 363:aau0964. doi: 10.1126/science.aau0964
110. Schyns J, Bai Q, Ruscitti C, Radermecker C, De Schepper S, Chakarov S, et al. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat Commun. (2019) 10:3964. doi: 10.1038/s41467-019-11843-0
111. Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. (1988) 241:58–62. doi: 10.1126/science.2898810
112. Alomari M, Almohazey D, Almofty SA, Khan FA, Al Hamad M, Ababneh D. Role of lipid rafts in hematopoietic stem cells homing, mobilization, hibernation, and differentiation. Cells. (2019) 8:630. doi: 10.3390/cells8060630
113. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
114. Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. (2004) 172:4410–7. doi: 10.4049/jimmunol.172.7.4410
115. Ammari M, Presumey J, Ponsolles C, Roussignol G, Roubert C, Escriou V, et al. Delivery of miR-146a to Ly6C(high) monocytes inhibits pathogenic bone erosion in inflammatory arthritis. Theranostics. (2018) 8:5972–85. doi: 10.7150/thno.29313
116. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19:71–82. doi: 10.1016/S1074-7613(03)00174-2
117. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
118. Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. (2007) 117:902–9. doi: 10.1172/JCI29919
119. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. (2006) 7:311–7. doi: 10.1038/ni1309
120. Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV Jr, Broxmeyer HE, et al. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. (1997) 100:2552–61. doi: 10.1172/JCI119798
121. Jenkins SJ, Hume DA. Homeostasis in the mononuclear phagocyte system. Trends Immunol. (2014) 35:358–67. doi: 10.1016/j.it.2014.06.006
122. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. (2017) 17:349–62. doi: 10.1038/nri.2017.28
123. Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol. (2008) 180:2562–72. doi: 10.4049/jimmunol.180.4.2562
124. Baharom F, Thomas S, Rankin G, Lepzien R, Pourazar J, Behndig AF, et al. Dendritic cells and monocytes with distinct inflammatory responses reside in lung mucosa of healthy humans. J Immunol. (2016) 196:4498–509. doi: 10.4049/jimmunol.1600071
125. Downey GP, Doherty DE, Schwab B 3rd, Elson EL, Henson PM, Worthen GS. Retention of leukocytes in capillaries: role of cell size and deformability. J Appl Physiol. (1990) 69:1767–78. doi: 10.1152/jappl.1990.69.5.1767
126. Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. (2014) 9:209–22. doi: 10.1038/nprot.2014.005
127. Desch AN, Gibbings SL, Goyal R, Kolde R, Bednarek J, Bruno T, et al. Flow cytometric analysis of mononuclear phagocytes in nondiseased human lung and lung-draining lymph nodes. Am J Respir Crit Care Med. (2016) 193:614–26. doi: 10.1164/rccm.201507-1376OC
128. Laffont S, Seillet C, Guery JC. Estrogen receptor-dependent regulation of dendritic cell development and function. Front Immunol. (2017) 8:108. doi: 10.3389/fimmu.2017.00108
129. Mackern-Oberti JP, Jara EL, Riedel CA, Kalergis AM. Hormonal modulation of dendritic cells differentiation, maturation and function: implications for the initiation and progress of systemic autoimmunity. Arch Immunol Ther Exp. (2017) 65:123–36. doi: 10.1007/s00005-016-0418-6
130. Kadel S, Kovats S. Sex hormones regulate innate immune cells and promote sex differences in respiratory virus infection. Front Immunol. (2018) 9:1653. doi: 10.3389/fimmu.2018.01653
131. Kopf M, Schneider C, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol. (2015) 16:36–44. doi: 10.1038/ni.3052
132. Cook PC, MacDonald AS. Dendritic cells in lung immunopathology. Semin Immunopathol. (2016) 38:449–60. doi: 10.1007/s00281-016-0571-3
133. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. (1983) 158:670–89. doi: 10.1084/jem.158.3.670
134. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol. (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
135. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
136. Hamilton TA, Zhao C, Pavicic PG Jr, Datta S. Myeloid colony-stimulating factors as regulators of macrophage polarization. Front Immunol. (2014) 5:554. doi: 10.3389/fimmu.2014.00554
137. Cheenpracha S, Park EJ, Rostama B, Pezzuto JM, Chang LC. Inhibition of nitric oxide (NO) production in lipopolysaccharide (LPS)-activated murine macrophage RAW 264.7 cells by the norsesterterpene peroxide, epimuqubilin A. Mar Drugs. (2010) 8:429–37. doi: 10.3390/md8030429
138. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889–96. doi: 10.1038/ni.1937
139. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
140. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. (1992) 176:287–92. doi: 10.1084/jem.176.1.287
141. Chupp GL, Lee CG, Jarjour N, Shim YM, Holm CT, He S, et al. A chitinase-like protein in the lung and circulation of patients with severe asthma. N Engl J Med. (2007) 357:2016–27. doi: 10.1056/NEJMoa073600
142. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. (2008) 14:633–40. doi: 10.1038/nm1770
143. Byers DE, Holtzman MJ. Alternatively activated macrophages and airway disease. Chest. (2011) 140:768–74. doi: 10.1378/chest.10-2132
144. Siddiqui S, Secor ER Jr, Silbart LK. Broncho-alveolar macrophages express chemokines associated with leukocyte migration in a mouse model of asthma. Cell Immunol. (2013) 281:159–69. doi: 10.1016/j.cellimm.2013.03.001
145. Girodet PO, Nguyen D, Mancini JD, Hundal M, Zhou X, Israel E, et al. Alternative macrophage activation is increased in Asthma. Am J Respir Cell Mol Biol. (2016) 55:467–75. doi: 10.1165/rcmb.2015-0295OC
146. Staples KJ, Hinks TS, Ward JA, Gunn V, Smith C, Djukanovic R. Phenotypic characterization of lung macrophages in asthmatic patients: overexpression of CCL17. J Allergy Clin Immunol. (2012) 130:1404–12.e7. doi: 10.1016/j.jaci.2012.07.023
147. Melgert BN, ten Hacken NH, Rutgers B, Timens W, Postma DS, Hylkema MN. More alternative activation of macrophages in lungs of asthmatic patients. J Allergy Clin Immunol. (2011) 127:831–3. doi: 10.1016/j.jaci.2010.10.045
148. Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang M, et al. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther. (2016) 24:915–25. doi: 10.1038/mt.2016.36
149. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
150. Barnes PJ. Cellular and molecular mechanisms of asthma and COPD. Clin Sci. (2017) 131:1541–58. doi: 10.1042/CS20160487
151. Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROα and MCP-1 in sputum samples from patients with COPD. Thorax. (2002) 57:590–5. doi: 10.1136/thorax.57.7.590
152. Chana KK, Fenwick PS, Nicholson AG, Barnes PJ, Donnelly LE. Identification of a distinct glucocorticosteroid-insensitive pulmonary macrophage phenotype in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2014) 133:207–16.e1–11. doi: 10.1016/j.jaci.2013.08.044
153. Berenson CS, Kruzel RL, Eberhardt E, Dolnick R, Minderman H, Wallace PK, et al. Impaired innate immune alveolar macrophage response and the predilection for COPD exacerbations. Thorax. (2014) 69:811–8. doi: 10.1136/thoraxjnl-2013-203669
154. Chen H, Cowan MJ, Hasday JD, Vogel SN, Medvedev AE. Tobacco smoking inhibits expression of proinflammatory cytokines and activation of IL-1R-associated kinase, p38, and NF-kappaB in alveolar macrophages stimulated with TLR2 and TLR4 agonists. J Immunol. (2007) 179:6097–106. doi: 10.4049/jimmunol.179.9.6097
155. Bozinovski S, Vlahos R, Zhang Y, Lah LC, Seow HJ, Mansell A, et al. Carbonylation caused by cigarette smoke extract is associated with defective macrophage immunity. Am J Respir Cell Mol Biol. (2011) 45:229–36. doi: 10.1165/rcmb.2010-0272OC
156. Metcalfe HJ, Lea S, Hughes D, Khalaf R, Abbott-Banner K, Singh D. Effects of cigarette smoke on Toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages. Clin Exp Immunol. (2014) 176:461–72. doi: 10.1111/cei.12289
157. Frankenberger M, Eder C, Hofer TP, Heimbeck I, Skokann K, Kassner G, et al. Chemokine expression by small sputum macrophages in COPD. Mol Med. (2011) 17:762–70. doi: 10.2119/molmed.2010.00202
158. Hodge S, Matthews G, Mukaro V, Ahern J, Shivam A, Hodge G, et al. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am J Respir Cell Mol Biol. (2011) 44:673–81. doi: 10.1165/rcmb.2009-0459OC
159. Doyle I, Ratcliffe M, Walding A, Vanden Bon E, Dymond M, Tomlinson W, et al. Differential gene expression analysis in human monocyte-derived macrophages: impact of cigarette smoke on host defence. Mol Immunol. (2010) 47:1058–65. doi: 10.1016/j.molimm.2009.11.008
160. Gaschler GJ, Zavitz CC, Bauer CM, Skrtic M, Lindahl M, Robbins CS, et al. Cigarette smoke exposure attenuates cytokine production by mouse alveolar macrophages. Am J Respir Cell Mol Biol. (2008) 38:218–26. doi: 10.1165/rcmb.2007-0053OC
161. Kapellos TS, Bassler K, Aschenbrenner AC, Fujii W, Schultze JL. Dysregulated functions of lung macrophage populations in COPD. J Immunol Res. (2018) 2018:2349045. doi: 10.1155/2018/2349045
162. Trombetta AC, Soldano S, Contini P, Tomatis V, Ruaro B, Paolino S, et al. A circulating cell population showing both M1 and M2 monocyte/macrophage surface markers characterizes systemic sclerosis patients with lung involvement. Respir Res. (2018) 19:186. doi: 10.1186/s12931-018-0891-z
163. Bardi GT, Smith MA, Hood JL. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine. (2018) 105:63–72. doi: 10.1016/j.cyto.2018.02.002
164. Rao Muvva J, Parasa VR, Lerm M, Svensson M, Brighenti S. Polarization of human monocyte-derived cells with vitamin D promotes control of mycobacterium tuberculosis infection. Front Immunol. (2019) 10:3157. doi: 10.3389/fimmu.2019.03157
165. Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. (1994) 264:713–6. doi: 10.1126/science.8171324
166. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. (2001) 15:557–67. doi: 10.1016/S1074-7613(01)00218-7
167. Baker AD, Malur A, Barna BP, Ghosh S, Kavuru MS, Malur AG, et al. Targeted PPAR{gamma} deficiency in alveolar macrophages disrupts surfactant catabolism. J Lipid Res. (2010) 51:1325–31. doi: 10.1194/jlr.M001651
168. Gautier EL, Chow A, Spanbroek R, Marcelin G, Greter M, Jakubzick C, et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J Immunol. (2012) 189:2614–24. doi: 10.4049/jimmunol.1200495
169. Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA. (1994) 91:5592–6. doi: 10.1073/pnas.91.12.5592
170. Bonfield TL, Farver CF, Barna BP, Malur A, Abraham S, Raychaudhuri B, et al. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. Am J Respir Cell Mol Biol. (2003) 29:677–82. doi: 10.1165/rcmb.2003-0148OC
171. Kitamura T, Tanaka N, Watanabe J, Uchida, Kanegasaki S, Yamada Y, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med. (1999) 190:875–80. doi: 10.1084/jem.190.6.875
172. Tanaka N, Watanabe J, Kitamura T, Yamada Y, Kanegasaki S, Nakata K. Lungs of patients with idiopathic pulmonary alveolar proteinosis express a factor which neutralizes granulocyte-macrophage colony stimulating factor. FEBS Lett. (1999) 442:246–50. doi: 10.1016/S0014-5793(98)01668-8
173. Suzuki T, Sakagami T, Rubin BK, Nogee LM, Wood RE, Zimmerman SL, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med. (2008) 205:2703–10. doi: 10.1084/jem.20080990
174. Martinez-Moczygemba M, Doan ML, Elidemir O, Fan LL, Cheung SW, Lei JT, et al. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRα gene in the X chromosome pseudoautosomal region 1. J Exp Med. (2008) 205:2711–6. doi: 10.1084/jem.20080759
175. Sakagami T, Uchida K, Suzuki T, Carey BC, Wood RE, Wert SE, et al. Human GM-CSF autoantibodies and reproduction of pulmonary alveolar proteinosis. N Engl J Med. (2009) 361:2679–81. doi: 10.1056/NEJMc0904077
176. Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol. (2008) 9:1074–83. doi: 10.1038/ni.1637
177. Davey RA, Grossmann M. Androgen receptor structure, function and biology: from bench to bedside. Clin Biochem Rev. (2016) 37:3–15.
178. Eder IE, Culig Z, Putz T, Nessler-Menardi C, Bartsch G, Klocker H. Molecular biology of the androgen receptor: from molecular understanding to the clinic. Eur Urol. (2001) 40:241–51. doi: 10.1159/000049782
179. Estrada M, Espinosa A, Muller M, Jaimovich E. Testosterone stimulates intracellular calcium release and mitogen-activated protein kinases via a G protein-coupled receptor in skeletal muscle cells. Endocrinology. (2003) 144:3586–97. doi: 10.1210/en.2002-0164
180. Kang HY, Cho CL, Huang KL, Wang JC, Hu YC, Lin HK, et al. Nongenomic androgen activation of phosphatidylinositol 3-kinase/Akt signaling pathway in MC3T3-E1 osteoblasts. J Bone Miner Res. (2004) 19:1181–90. doi: 10.1359/JBMR.040306
181. Gill A, Jamnongjit M, Hammes SR. Androgens promote maturation and signaling in mouse oocytes independent of transcription: a release of inhibition model for mammalian oocyte meiosis. Mol Endocrinol. (2004) 18:97–104. doi: 10.1210/me.2003-0326
182. Montano LM, Flores-Soto E, Sommer B, Solis-Chagoyan H, Perusquia M. Androgens are effective bronchodilators with anti-inflammatory properties: a potential alternative for asthma therapy. Steroids. (2020) 153:108509. doi: 10.1016/j.steroids.2019.108509
183. Tan MH, Li J, Xu HE, Melcher K, Yong EL. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin. (2015) 36:3–23. doi: 10.1038/aps.2014.18
184. Wadosky KM, Koochekpour S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget. (2017) 8:18550–76. doi: 10.18632/oncotarget.14537
185. Hoang DT, Iczkowski KA, Kilari D, See W, Nevalainen MT. Androgen receptor-dependent and -independent mechanisms driving prostate cancer progression: opportunities for therapeutic targeting from multiple angles. Oncotarget. (2017) 8:3724–45. doi: 10.18632/oncotarget.12554
186. Ashcroft GS, Mills SJ. Androgen receptor-mediated inhibition of cutaneous wound healing. J Clin Invest. (2002) 110:615–24. doi: 10.1172/JCI0215704
187. Thompson MG, Peiffer DS, Larson M, Navarro F, Watkins SK. FOXO3, estrogen receptor alpha, and androgen receptor impact tumor growth rate and infiltration of dendritic cell subsets differentially between male and female mice. Cancer Immunol Immunother. (2017) 66:615–25. doi: 10.1007/s00262-017-1972-4
188. Becerra-Diaz M, Strickland AB, Keselman A, Heller NM. Androgen and androgen receptor as enhancers of M2 macrophage polarization in allergic lung inflammation. J Immunol. (2018) 201:2923–33. doi: 10.4049/jimmunol.1800352
189. Liu L, Benten WP, Wang L, Hao X, Li Q, Zhang H, et al. Modulation of Leishmania donovani infection and cell viability by testosterone in bone marrow-derived macrophages: signaling via surface binding sites. Steroids. (2005) 70:604–14. doi: 10.1016/j.steroids.2005.02.020
190. Bizzaro D, Crescenzi M, Di Liddo R, Arcidiacono D, Cappon A, Bertalot T, et al. Sex-dependent differences in inflammatory responses during liver regeneration in a murine model of acute liver injury. Clin Sci. (2018) 132:255–72. doi: 10.1042/CS20171260
191. Rubinow KB, Houston B, Wang S, Goodspeed L, Ogimoto K, Morton GJ, et al. Androgen receptor deficiency in monocytes/macrophages does not alter adiposity or glucose homeostasis in male mice. Asian J Androl. (2018) 20:276–83. doi: 10.4103/aja.aja_54_17
192. Lai JJ, Lai KP, Chuang KH, Chang P, Yu IC, Lin WJ, et al. Monocyte/macrophage androgen receptor suppresses cutaneous wound healing in mice by enhancing local TNF-alpha expression. J Clin Invest. (2009) 119:3739–51. doi: 10.1172/JCI39335
193. Lee GT, Kim JH, Kwon SJ, Stein MN, Hong JH, Nagaya N, et al. Dihydrotestosterone increases cytotoxic activity of macrophages on prostate cancer cells via TRAIL. Endocrinology. (2019) 160:2049–60. doi: 10.1210/en.2019-00367
194. Ahmadi K, McCruden AB. Macrophage may responses to androgen via its receptor. Med Sci Monit. (2006) 12:BR15–20.
195. Benten WP, Lieberherr M, Stamm O, Wrehlke C, Guo Z, Wunderlich F. Testosterone signaling through internalizable surface receptors in androgen receptor-free macrophages. Mol Biol Cell. (1999) 10:3113–23. doi: 10.1091/mbc.10.10.3113
196. McCrohon JA, Death AK, Nakhla S, Jessup W, Handelsman DJ, Stanley KK, et al. Androgen receptor expression is greater in macrophages from male than from female donors. A sex difference with implications for atherogenesis. Circulation. (2000) 101:224–6. doi: 10.1161/01.CIR.101.3.224
197. Campesi I, Sanna M, Zinellu A, Carru C, Rubattu L, Bulzomi P, et al. Oral contraceptives modify DNA methylation and monocyte-derived macrophage function. Biol Sex Differ. (2012) 3:4. doi: 10.1186/2042-6410-3-4
198. Huang CK, Pang H, Wang L, Niu Y, Luo J, Chang E, et al. New therapy via targeting androgen receptor in monocytes/macrophages to battle atherosclerosis. Hypertension. (2014) 63:1345–53. doi: 10.1161/HYPERTENSIONAHA.113.02804
199. Liu CQ, Wu SZ, Wang ZD, Lai WY, Sun F. Effect of testosterone on expression of androgen receptor in human monocytic cell line THP-1. Di Yi Jun Yi Da Xue Xue Bao. (2004) 24:389–91.
200. Cutolo M, Accardo S, Villaggio B, Barone A, Sulli A, Coviello DA, et al. Androgen and estrogen receptors are present in primary cultures of human synovial macrophages. J Clin Endocrinol Metab. (1996) 81:820–7. doi: 10.1210/jcem.81.2.8636310
201. Peters AA, Buchanan G, Ricciardelli C, Bianco-Miotto T, Centenera MM, Harris JM, et al. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res. (2009) 69:6131–40. doi: 10.1158/0008-5472.CAN-09-0452
202. Schneider CP, Schwacha MG, Samy TS, Bland KI, Chaudry IH. Androgen-mediated modulation of macrophage function after trauma-hemorrhage: central role of 5alpha-dihydrotestosterone. J Appl Physiol. (2003) 95:104–12. doi: 10.1152/japplphysiol.00182.2003
203. D'Agostino P, Milano S, Barbera C, Di Bella G, La Rosa M, Ferlazzo V, et al. Sex hormones modulate inflammatory mediators produced by macrophages. Ann N Y Acad Sci. (1999) 876:426–9. doi: 10.1111/j.1749-6632.1999.tb07667.x
204. Friedl R, Brunner M, Moeslinger T, Spieckermann PG. Testosterone inhibits expression of inducible nitric oxide synthase in murine macrophages. Life Sci. (2000) 68:417–29. doi: 10.1016/S0024-3205(00)00953-X
205. Izumi K, Fang LY, Mizokami A, Namiki M, Li L, Lin WJ, et al. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol Med. (2013) 5:1383–401. doi: 10.1002/emmm.201202367
206. Chao TC, Phuangsab A, Van Alten PJ, Walter RJ. Steroid sex hormones and macrophage function: regulation of chemiluminescence and phagocytosis. Am J Reprod Immunol. (1996) 35:106–13. doi: 10.1111/j.1600-0897.1996.tb00015.x
207. Zhang H, Zhu Z, Liu L, Guo D, Jiang T, Wang L, et al. Upregulation of Fas and FasL expression in testosterone-induced apoptosis of macrophages. Methods Find Exp Clin Pharmacol. (2003) 25:779–84. doi: 10.1358/mf.2003.25.10.793325
208. Jin L, Ai X, Liu L, Wang Z, Cheng Y, Qiao Z. Testosterone induces apoptosis via Fas/FasL-dependent pathway in bone marrow-derived macrophages. Methods Find Exp Clin Pharmacol. (2006) 28:283–93. doi: 10.1358/mf.2006.28.5.990201
209. Ren X, Fu X, Zhang X, Chen S, Huang S, Yao L, et al. Testosterone regulates 3T3-L1 pre-adipocyte differentiation and epididymal fat accumulation in mice through modulating macrophage polarization. Biochem Pharmacol. (2017) 140:73–88. doi: 10.1016/j.bcp.2017.05.022
210. Rettew JA, Huet-Hudson YM, Marriott I. Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol Reprod. (2008) 78:432–7. doi: 10.1095/biolreprod.107.063545
211. Eder IE, Culig Z, Ramoner R, Thurnher M, Putz T, Nessler-Menardi C, et al. Inhibition of LncaP prostate cancer cells by means of androgen receptor antisense oligonucleotides. Cancer Gene Ther. (2000) 7:997–1007. doi: 10.1038/sj.cgt.7700202
212. Li Q, Deng Q, Chao HP, Liu X, Lu Y, Lin K, et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat Commun. (2018) 9:3600. doi: 10.1038/s41467-018-06067-7
213. Murthy S, Wu M, Bai VU, Hou Z, Menon M, Barrack ER, et al. Role of androgen receptor in progression of LNCaP prostate cancer cells from G1 to S phase. PLoS ONE. (2013) 8:e56692. doi: 10.1371/journal.pone.0056692
214. Rahman F, Christian HC. Non-classical actions of testosterone: an update. Trends Endocrinol Metab. (2007) 18:371–8. doi: 10.1016/j.tem.2007.09.004
215. Gilliver SC, Ashworth JJ, Mills SJ, Hardman MJ, Ashcroft GS. Androgens modulate the inflammatory response during acute wound healing. J Cell Sci. (2006) 119(Pt 4):722–32. doi: 10.1242/jcs.02786
216. Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circ Res. (2014) 114:1757–71. doi: 10.1161/CIRCRESAHA.114.301174
217. Linton MRF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, et al. The role of lipids and lipoproteins in atherosclerosis. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, et al. editors. Endotext. NCBI Bookshelf: South Dartmouth, MA (2000).
218. Cutolo M, Capellino S, Montagna P, Ghiorzo P, Sulli A, Villaggio B. Sex hormone modulation of cell growth and apoptosis of the human monocytic/macrophage cell line. Arthritis Res Ther. (2005) 7:R1124–32. doi: 10.1186/ar1791
219. Cutolo M, Sulli A, Craviotto C, Felli L, Pizzorni C, Seriolo B, et al. Modulation of cell growth and apoptosis by sex hormones in cultured monocytic THP-1 cells. Ann N Y Acad Sci. (2002) 966:204–10. doi: 10.1111/j.1749-6632.2002.tb04216.x
220. Mossuz P, Cousin F, Castinel A, Chauvet M, Sotto MF, Polack B, et al. Effects of two sex steroids (17beta estradiol and testosterone) on proliferation and clonal growth of the human monoblastic leukemia cell line, U937. Leuk Res. (1998) 22:1063–72. doi: 10.1016/S0145-2126(98)00101-5
221. Carruba G, D'Agostino P, Miele M, Calabro M, Barbera C, Bella GD, et al. Estrogen regulates cytokine production and apoptosis in PMA-differentiated, macrophage-like U937 cells. J Cell Biochem. (2003) 90:187–96. doi: 10.1002/jcb.10607
222. Viken KE. The effect of steroids on differentiation and function of cultured, mononuclear cells. Acta Pathol Microbiol Scand C. (1976) 84:13–22. doi: 10.1111/j.1699-0463.1976.tb03594.x
223. Kanda N, Tsuchida T, Tamaki K. Testosterone suppresses anti-DNA antibody production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. (1997) 40:1703–11. doi: 10.1002/art.1780400921
224. Kanda N, Tsuchida T, Tamaki K. Testosterone inhibits immunoglobulin production by human peripheral blood mononuclear cells. Clin Exp Immunol. (1996) 106:410–5. doi: 10.1046/j.1365-2249.1996.d01-842.x
225. Gilmore W, Weiner LP, Correale J. Effect of estradiol on cytokine secretion by proteolipid protein-specific T cell clones isolated from multiple sclerosis patients and normal control subjects. J Immunol. (1997) 158:446–51.
226. Harnish DC, Scicchitano MS, Adelman SJ, Lyttle CR, Karathanasis SK. The role of CBP in estrogen receptor cross-talk with nuclear factor-kappaB in HepG2 cells. Endocrinology. (2000) 141:3403–11. doi: 10.1210/endo.141.9.7646
227. Capellino S, Villaggio V, Montagna P, Sulli A, Craviotto C, Cutolo M. [17beta-Estradiol and testosterone influence the mRNA expression and the time course of inflammatory cytokines in activated human monocytic cell line (THP-1)]. Reumatismo. (2005) 57:193–6. doi: 10.4081/reumatismo.2005.193
228. Boje A, Moesby L, Timm M, Hansen EW. Immunomodulatory effects of testosterone evaluated in all-trans retinoic acid differentiated HL-60 cells, granulocytes, and monocytes. Int Immunopharmacol. (2012) 12:573–9. doi: 10.1016/j.intimp.2012.02.008
229. Debelec-Butuner B, Alapinar C, Varisli L, Erbaykent-Tepedelen B, Hamid SM, Gonen-Korkmaz C, et al. Inflammation-mediated abrogation of androgen signaling: an in vitro model of prostate cell inflammation. Mol Carcinog. (2014) 53:85–97. doi: 10.1002/mc.21948
230. Corcoran MP, Meydani M, Lichtenstein AH, Schaefer EJ, Dillard A, Lamon-Fava S. Sex hormone modulation of proinflammatory cytokine and C-reactive protein expression in macrophages from older men and postmenopausal women. J Endocrinol. (2010) 206:217–24. doi: 10.1677/JOE-10-0057
231. Falus A, Feher KG, Walcz E, Brozik M, Fust G, Hidvegi T, et al. Hormonal regulation of complement biosynthesis in human cell lines—I. Androgens and gamma-interferon stimulate the biosynthesis and gene expression of C1 inhibitor in human cell lines U937 and HepG2. Mol Immunol. (1990) 27:191–5. doi: 10.1016/0161-5890(90)90114-F
232. Pergola C, Rogge A, Dodt G, Northoff H, Weinigel C, Barz D, et al. Testosterone suppresses phospholipase D, causing sex differences in leukotriene biosynthesis in human monocytes. FASEB J. (2011) 25:3377–87. doi: 10.1096/fj.11-182758
233. Churchill L, Chilton FH, Resau JH, Bascom R, Hubbard WC, Proud D. Cyclooxygenase metabolism of endogenous arachidonic acid by cultured human tracheal epithelial cells. Am Rev Respir Dis. (1989) 140:449–59. doi: 10.1164/ajrccm/140.2.449
234. Delamere F, Holland E, Patel S, Bennett J, Pavord I, Knox A. Production of PGE2 by bovine cultured airway smooth muscle cells and its inhibition by cyclo-oxygenase inhibitors. Br J Pharmacol. (1994) 111:983–8. doi: 10.1111/j.1476-5381.1994.tb14840.x
235. Miyagi M, Morishita M, Iwamoto Y. Effects of sex hormones on production of prostaglandin E2 by human peripheral monocytes. J Periodontol. (1993) 64:1075–8. doi: 10.1902/jop.1993.64.11.1075
236. Delpedro AD, Barjavel MJ, Mamdouh Z, Bakouche O. Activation of human monocytes by LPS and DHEA. J Interferon Cytokine Res. (1998) 18:125–35. doi: 10.1089/jir.1998.18.125
237. Corsini E, Galbiati V, Papale A, Kummer E, Pinto A, Serafini MM, et al. Role of androgens in dhea-induced rack1 expression and cytokine modulation in monocytes. Immun Ageing. (2016) 13:20. doi: 10.1186/s12979-016-0075-y
238. Adams DR, Ron D, Kiely PA. RACK1, a multifaceted scaffolding protein: structure and function. Cell Commun Signal. (2011) 9:22. doi: 10.1186/1478-811X-9-22
239. Rom WN, Harkin T. Dehydroepiandrosterone inhibits the spontaneous release of superoxide radical by alveolar macrophages in vitro in asbestosis. Environ Res. (1991) 55:145–56. doi: 10.1016/S0013-9351(05)80171-9
240. Choi IS, Cui Y, Koh YA, Lee HC, Cho YB, Won YH. Effects of dehydroepiandrosterone on Th2 cytokine production in peripheral blood mononuclear cells from asthmatics. Korean J Intern Med. (2008) 23:176–81. doi: 10.3904/kjim.2008.23.4.176
241. Hou X, Summer R, Chen Z, Tian Y, Ma J, Cui J, et al. Lipid uptake by alveolar macrophages drives fibrotic responses to silica dust. Sci Rep. (2019) 9:399. doi: 10.1038/s41598-018-36875-2
242. Venosa A, Smith LC, Murray A, Banota T, Gow AJ, Laskin JD, et al. Regulation of macrophage foam cell formation during nitrogen mustard (NM)-induced pulmonary fibrosis by lung lipids. Toxicol Sci. (2019) 172:344–58. doi: 10.1093/toxsci/kfz187
243. Guerrini V, Panettieri RA Jr, Gennaro ML. Lipid-laden macrophages as biomarkers of vaping-associated lung injury. Lancet Respir Med. (2020) 8:e6. doi: 10.1016/S2213-2600(19)30476-X
244. Guerrini V, Prideaux B, Blanc L, Bruiners N, Arrigucci R, Singh S, et al. Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease-specific. PLoS Pathog. (2018) 14:e1007223. doi: 10.1371/journal.ppat.1007223
245. Peters SAE, Muntner P, Woodward M. Sex differences in the prevalence of, and trends in, cardiovascular risk factors, treatment, and control in the United States, 2001 to 2016. Circulation. (2019) 139:1025–35. doi: 10.1161/CIRCULATIONAHA.118.035550
246. Fairweather D. Sex differences in inflammation during atherosclerosis. Clin Med Insights Cardiol. (2014) 8(Suppl. 3):49–59. doi: 10.4137/CMC.S17068
247. Ng MK, Quinn CM, McCrohon JA, Nakhla S, Jessup W, Handelsman DJ, et al. Androgens up-regulate atherosclerosis-related genes in macrophages from males but not females: molecular insights into gender differences in atherosclerosis. J Am Coll Cardiol. (2003) 42:1306–13. doi: 10.1016/j.jacc.2003.07.002
248. Shchelkunova TA, Morozov IA, Rubtsov PM, Samokhodskaya LM, Andrianova IV, Rudimov EG, et al. Effect of sex hormones on levels of mRNAs coding for proteins involved in lipid metabolism in macrophages. Biochemistry (Mosc). (2013) 78:1342–53. doi: 10.1134/S0006297913120043
249. Langer C, Gansz B, Goepfert C, Engel T, Uehara Y, von Dehn G, et al. Testosterone up-regulates scavenger receptor BI and stimulates cholesterol efflux from macrophages. Biochem Biophys Res Commun. (2002) 296:1051–7. doi: 10.1016/S0006-291X(02)02038-7
250. Corcoran MP, Lichtenstein AH, Meydani M, Dillard A, Schaefer EJ, Lamon-Fava S. The effect of 17beta-estradiol on cholesterol content in human macrophages is influenced by the lipoprotein milieu. J Mol Endocrinol. (2011) 47:109–17. doi: 10.1530/JME-10-0158
251. Magri B, Vigano P, Rossi G, Somigliana E, Gaffuri B, Vignali M. Comparative effect of the calcium antagonist verapamil and the synthetic steroids gestrinone and danazol on human monocyte phagocytosis in vitro. Gynecol Obstet Invest. (1997) 43:6–10. doi: 10.1159/000291809
252. Yamada K, Hayashi T, Kuzuya M, Naito M, Asai K, Iguchi A. Physiological concentration of 17 beta-estradiol inhibits chemotaxis of human monocytes in response to monocyte chemotactic protein 1. Artery. (1996) 22:24–35.
253. Miyagi M, Aoyama H, Morishita M, Iwamoto Y. Effects of sex hormones on chemotaxis of human peripheral polymorphonuclear leukocytes and monocytes. J Periodontol. (1992) 63:28–32. doi: 10.1902/jop.1992.63.1.28
254. Koopman MG, Fleer A, vd Schaaf DB, van der Meulen FW, von dem Borne AE, Engelfriet CP. Male-female differences in the cytotoxic activity of human monocytes in vitro. Clin Lab Haematol. (1981) 3:45–50. doi: 10.1111/j.1365-2257.1981.tb01308.x
255. Keselman A, Fang X, White PB, Heller NM. Estrogen signaling contributes to sex differences in macrophage polarization during Asthma. J Immunol. (2017) 199:1573–83. doi: 10.4049/jimmunol.1601975
256. Yanagisawa R, Koike E, Win-Shwe TT, Takano H. Oral exposure to low dose bisphenol A aggravates allergic airway inflammation in mice. Toxicol Rep. (2019) 6:1253–62. doi: 10.1016/j.toxrep.2019.11.012
257. Townsend EA, Miller VM, Prakash YS. Sex differences and sex steroids in lung health and disease. Endocr Rev. (2012) 33:1–47. doi: 10.1210/er.2010-0031
258. Raes G, van den Bergh R, de Baetselier P, Ghassabeh GH, Scotton C, Locati M, et al. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J Immunol. (2005) 174:6561. author reply 6561–2. doi: 10.4049/jimmunol.174.11.6561
259. Hoenerhoff MJ, Starost MF, Ward JM. Eosinophilic crystalline pneumonia as a major cause of death in 129S4/SvJae mice. Vet Pathol. (2006) 43:682–8. doi: 10.1354/vp.43-5-682
260. Fuseini H, Yung JA, Cephus JY, Zhang J, Goleniewska K, Polosukhin VV, et al. Testosterone decreases house dust mite-induced Type 2 and IL-17A-mediated airway inflammation. J Immunol. (2018) 201:1843–54. doi: 10.4049/jimmunol.1800293
261. Cephus JY, Stier MT, Fuseini H, Yung JA, Toki S, Bloodworth MH, et al. Testosterone attenuates group 2 innate lymphoid cell-mediated airway inflammation. Cell Rep. (2017) 21:2487–99. doi: 10.1016/j.celrep.2017.10.110
262. Laffont S, Blanquart E, Savignac M, Cenac C, Laverny G, Metzger D, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med. (2017) 214:1581–92. doi: 10.1084/jem.20161807
263. Chao TC, Van Alten PJ, Greager JA, Walter RJ. Steroid sex hormones regulate the release of tumor necrosis factor by macrophages. Cell Immunol. (1995) 160:43–9. doi: 10.1016/0008-8749(95)80007-6
264. Sebring RJ, Lehnert BE. Morphometric comparisons of rat alveolar macrophages, pulmonary interstitial macrophages, and blood monocytes. Exp Lung Res. (1992) 18:479–96. doi: 10.3109/01902149209064341
265. Tam A, Churg A, Wright JL, Zhou S, Kirby M, Coxson HO, et al. Sex differences in airway remodeling in a mouse model of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2016) 193:825–34. doi: 10.1164/rccm.201503-0487OC
266. Joshi R, Ojha M, Lewis J, Fan Q, Monia B, Guo S, et al. Sex-specific differences in emphysema using a murine antisense oligonucleotide model of α-1 antitrypsin deficiency. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L1165–73. doi: 10.1152/ajplung.00502.2018
267. Lorenzo ME, Hodgson A, Robinson DP, Kaplan JB, Pekosz A, Klein SL. Antibody responses and cross protection against lethal influenza A viruses differ between the sexes in C57BL/6 mice. Vaccine. (2011) 29:9246–55. doi: 10.1016/j.vaccine.2011.09.110
268. Hoffmann J, Otte A, Thiele S, Lotter H, Shu Y, Gabriel G. Sex differences in H7N9 influenza A virus pathogenesis. Vaccine. (2015) 33:6949–54. doi: 10.1016/j.vaccine.2015.08.044
269. Hall OJ, Limjunyawong N, Vermillion MS, Robinson DP, Wohlgemuth N, Pekosz A, et al. Progesterone-based therapy protects against influenza by promoting lung repair and recovery in females. PLoS Pathog. (2016) 12:e1005840. doi: 10.1371/journal.ppat.1005840
270. Silver RF, Li Q, Boom WH, Ellner JJ. Lymphocyte-dependent inhibition of growth of virulent Mycobacterium tuberculosis H37Rv within human monocytes: requirement for CD4+ T cells in purified protein derivative-positive, but not in purified protein derivative-negative subjects. J Immunol. (1998) 160:2408–17.
271. Bini EI, Mata Espinosa D, Marquina Castillo B, Barrios Payan J, Colucci D, Cruz AF, et al. The influence of sex steroid hormones in the immunopathology of experimental pulmonary tuberculosis. PLoS ONE. (2014) 9:e93831. doi: 10.1371/journal.pone.0093831
272. Yamaguchi T, Fushida S, Yamamoto Y, Tsukada T, Kinoshita J, Oyama K, et al. Tumor-associated macrophages of the M2 phenotype contribute to progression in gastric cancer with peritoneal dissemination. Gastric Cancer. (2016) 19:1052–65. doi: 10.1007/s10120-015-0579-8
273. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. (2018) 17:887–904. doi: 10.1038/nrd.2018.169
274. Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. (2015) 75:3479–91. doi: 10.1158/0008-5472.CAN-14-3587
275. Siegfried JM, Gubish CT, Rothstein ME, Henry C, Stabile LP. Combining the multitargeted tyrosine kinase inhibitor vandetanib with the antiestrogen fulvestrant enhances its antitumor effect in non-small cell lung cancer. J Thorac Oncol. (2012) 7:485–95. doi: 10.1097/JTO.0b013e31824177ea
276. Hershberger PA, Vasquez AC, Kanterewicz B, Land S, Siegfried JM, Nichols M. Regulation of endogenous gene expression in human non-small cell lung cancer cells by estrogen receptor ligands. Cancer Res. (2005) 65:1598–605. doi: 10.1158/0008-5472.CAN-04-2694
277. Draijer C, Boorsma CE, Robbe P, Timens W, Hylkema MN, Ten Hacken NH, et al. Human asthma is characterized by more IRF5+ M1 and CD206+ M2 macrophages and less IL-10+ M2-like macrophages around airways compared with healthy airways. J Allergy Clin Immunol. (2017) 140:280–3.e3. doi: 10.1016/j.jaci.2016.11.020
278. Corrales JJ, Almeida M, Miralles JM, Orfao A. Persistence of androgenic effects on the production of proinflammatory cytokines by circulating antigen-presenting cells after withdrawal of testosterone treatment in aging type 2 diabetic men with partial androgen deficiency. Fertil Steril. (2009) 92:311–9. doi: 10.1016/j.fertnstert.2008.05.040
279. Corrales JJ, Almeida M, Burgo R, Mories MT, Miralles JM, Orfao A. Androgen-replacement therapy depresses the ex vivo production of inflammatory cytokines by circulating antigen-presenting cells in aging type-2 diabetic men with partial androgen deficiency. J Endocrinol. (2006) 189:595–604. doi: 10.1677/joe.1.06779
280. Corrales JJ, Almeida M, Martin-Martin L, Miralles JM, Orfao A. Testosterone replacement therapy in hypogonadal men is associated with increased expression of LAMP-2 (CD107b) by circulating monocytes and dendritic cells. Clin Endocrinol. (2014) 80:577–84. doi: 10.1111/cen.12338
281. Gravholt CH, Chang S, Wallentin M, Fedder J, Moore P, Skakkebaek A. Klinefelter syndrome: integrating genetics, neuropsychology, and endocrinology. Endocr Rev. (2018) 39:389–423. doi: 10.1210/er.2017-00212
282. Bojesen A, Juul S, Birkebaek NH, Gravholt CH. Morbidity in Klinefelter syndrome: a Danish register study based on hospital discharge diagnoses. J Clin Endocrinol Metab. (2006) 91:1254–60. doi: 10.1210/jc.2005-0697
283. Ladias S, Katsenos S. Klinefelter syndrome and bronchial asthma: Is there any relationship between the low testosterone levels and asthma exacerbations? Lung India. (2018) 35:368–9. doi: 10.4103/lungindia.lungindia_40_18
284. Nan HL, Zhang Y, Chen Z. A case of Klinefelter's syndrome with refractory asthma, diabetes mellitus and rib fracture. Chin Med J. (2013) 126:196.
285. Daly JJ, Rickards DF. Klinefelter's syndrome with Asthma. Lancet J. (1964) 283:P1451–2. doi: 10.1016/S0140-6736(64)92027-6
286. Lefevre N, Corazza F, Valsamis J, Delbaere A, de Maertelaer V, Duchateau J, et al. The number of X chromosomes influences inflammatory cytokine production following toll-like receptor stimulation. Front Immunol. (2019) 10:1052. doi: 10.3389/fimmu.2019.01052
287. Penttila TA, Anttila L, Torma A, Koskinen P, Erkkola R, Irjala K. Serum free testosterone in polycystic ovary syndrome measured with a new reference method. Fertil Steril. (1996) 65:55–60. doi: 10.1016/S0015-0282(16)58027-3
288. Legro RS, Schlaff WD, Diamond MP, Coutifaris C, Casson PR, Brzyski RG, et al. Total testosterone assays in women with polycystic ovary syndrome: precision and correlation with hirsutism. J Clin Endocrinol Metab. (2010) 95:5305–13. doi: 10.1210/jc.2010-1123
289. Glintborg D, Andersen M. Medical comorbidity in polycystic ovary syndrome with special focus on cardiometabolic, autoimmune, hepatic and cancer diseases: an updated review. Curr Opin Obstet Gynecol. (2017) 29:390–6. doi: 10.1097/GCO.0000000000000410
290. Htet TD, Teede HJ, de Courten B, Loxton D, Real FG, Moran LJ, et al. Asthma in reproductive-aged women with polycystic ovary syndrome and association with obesity. Eur Respir J. (2017) 49:1601334. doi: 10.1183/13993003.01334-2016
291. Ding J, Cui X, Liu Q. Emerging role of HMGB1 in lung diseases: friend or foe. J Cell Mol Med. (2017) 21:1046–57. doi: 10.1111/jcmm.13048
292. Diamanti-Kandarakis E, Piperi C, Kalofoutis A, Creatsas G. Increased levels of serum advanced glycation end-products in women with polycystic ovary syndrome. Clin Endocrinol (Oxf). (2005) 62:37–43. doi: 10.1111/j.1365-2265.2004.02170.x
293. Halayko AJ, Ghavami S. S100A8/A9: a mediator of severe asthma pathogenesis and morbidity? Can J Physiol Pharmacol. (2009) 87:743–55. doi: 10.1139/Y09-054
294. Wulfsohn NL, Politzer WM, Henrico JS. Testosterone therapy in bronchial Asthma. S Afr Med J. (1964) 38:170–2.
296. Karadag F, Ozcan H, Karul AB, Yilmaz M, Cildag O. Sex hormone alterations and systemic inflammation in chronic obstructive pulmonary disease. Int J Clin Pract. (2009) 63:275–81. doi: 10.1111/j.1742-1241.2007.01501.x
297. Akbas T, Karakurt S, Unluguzel G, Celikel T, Akalin S. The endocrinologic changes in critically ill chronic obstructive pulmonary disease patients. COPD. (2010) 7:240–7. doi: 10.3109/15412555.2010.496815
298. Svartberg J, Schirmer H, Medbo A, Melbye H, Aasebo U. Reduced pulmonary function is associated with lower levels of endogenous total and free testosterone. The Tromso study. Eur J Epidemiol. (2007) 22:107–12. doi: 10.1007/s10654-006-9095-9
299. Van Vliet M, Spruit MA, Verleden G, Kasran A, Van Herck E, Pitta F, et al. Hypogonadism, quadriceps weakness, and exercise intolerance in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2005) 172:1105–11. doi: 10.1164/rccm.200501-114OC
300. Kamischke A, Kemper DE, Castel MA, Luthke M, Rolf C, Behre HM, et al. Testosterone levels in men with chronic obstructive pulmonary disease with or without glucocorticoid therapy. Eur Respir J. (1998) 11:41–5. doi: 10.1183/09031936.98.11010041
301. Laghi F, Antonescu-Turcu A, Collins E, Segal J, Tobin DE, Jubran A, et al. Hypogonadism in men with chronic obstructive pulmonary disease: prevalence and quality of life. Am J Respir Crit Care Med. (2005) 171:728–33. doi: 10.1164/rccm.200501-037OC
302. Svartberg J. Androgens and chronic obstructive pulmonary disease. Curr Opin Endocrinol Diabetes Obes. (2010) 17:257–61. doi: 10.1097/MED.0b013e3283369da6
303. Snyder PJ. Hypogonadism in elderly men–what to do until the evidence comes. N Engl J Med. (2004) 350:440–2. doi: 10.1056/NEJMp038207
305. Casaburi R, Bhasin S, Cosentino L, Porszasz J, Somfay A, Lewis MI, et al. Effects of testosterone and resistance training in men with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2004) 170:870–8. doi: 10.1164/rccm.200305-617OC
306. Baillargeon J, Urban RJ, Zhang W, Zaiden MF, Javed Z, Sheffield-Moore M, et al. Testosterone replacement therapy and hospitalization rates in men with COPD. Chron Respir Dis. (2019) 16:1479972318793004. doi: 10.1177/1479972318793004
307. Weinstein RE, Lobocki CA, Gravett S, Hum H, Negrich R, Herbst J, et al. Decreased adrenal sex steroid levels in the absence of glucocorticoid suppression in postmenopausal asthmatic women. J Allergy Clin Immunol. (1996) 97(1 Pt 1):1–8. doi: 10.1016/S0091-6749(96)70276-2
308. Feher KG, Koo E, Feher T. Adrenocortical function in bronchial asthma. Acta Med Hung. (1983) 40:125–31.
309. Dunn PJ, Mahood CB, Speed JF, Jury DR. Dehydroepiandrosterone sulphate concentrations in asthmatic patients: pilot study. N Z Med J. (1984) 97:805–8.
310. Wenzel SE, Robinson CB, Leonard JM, Panettieri RA Jr. Nebulized dehydroepiandrosterone-3-sulfate improves asthma control in the moderate-to-severe asthma results of a 6-week, randomized, double-blind, placebo-controlled study. Allergy Asthma Proc. (2010) 31:461–71. doi: 10.2500/aap.2010.31.3384
311. de La Roque ED, Savineau JP, Metivier AC, Billes MA, Kraemer JP, Doutreleau S, et al. Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): a pilot study. Ann Endocrinol. (2012) 73:20–5. doi: 10.1016/j.ando.2011.12.005
312. Zurfluh S, Nickler M, Ottiger M, Steuer C, Kutz A, Christ-Crain M, et al. Association of adrenal hormone metabolites and mortality over a 6-year follow-up in COPD patients with acute exacerbation. Clin Chem Lab Med. (2018) 56:669–80. doi: 10.1515/cclm-2017-0873
313. Lee SK. Sex as an important biological variable in biomedical research. BMB Rep. (2018) 51:167–73. doi: 10.5483/BMBRep.2018.51.4.034
Keywords: androgen, androgen receptor, monocyte, macrophage, asthma, lung, sex difference, sex hormone
Citation: Becerra-Diaz M, Song M and Heller N (2020) Androgen and Androgen Receptors as Regulators of Monocyte and Macrophage Biology in the Healthy and Diseased Lung. Front. Immunol. 11:1698. doi: 10.3389/fimmu.2020.01698
Received: 02 March 2020; Accepted: 25 June 2020;
Published: 07 August 2020.
Edited by:
Flavia Bazzoni, University of Verona School of Medicine and Surgery, ItalyReviewed by:
Paola Parronchi, University of Florence, ItalyCopyright © 2020 Becerra-Diaz, Song and Heller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Heller, bmhlbGxlckBqaG1pLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.