 Benoit Pourcet
Benoit Pourcet Hélène Duez
Hélène Duez- University of Lille, Inserm, CHU Lille, Institut Pasteur de Lille, U1011-EGID, Lille, France
The innate immune system senses “non-self” molecules derived from pathogens (PAMPs) as well as endogenous damage-associated molecular patterns (DAMPs) and promotes sterile inflammation that is necessary for injury resolution, tissue repair/regeneration, and homeostasis. The NOD-, LRR- and pyrin domain containing protein 3 (NLRP3) is an innate immune signaling complex whose assembly and activation can be triggered by various signals ranging from microbial molecules to ATP or the abnormal accumulation of crystals, thus leading to IL-1β and IL-18 maturation and secretion. Deregulation of the NLRP3 signaling cascade is associated with numerous inflammatory and metabolic diseases including rheumatoid arthritis, gout, atherosclerosis or type 2 diabetes. Interestingly, the circadian clock controls numerous inflammatory processes while clock disruption leads to or exacerbates inflammation. Recently, the biological clock was demonstrated to control NLRP3 expression and activation, thereby controlling IL-1β and IL-18 secretion in diverse tissues and immune cells, particularly macrophages. Circadian oscillations of NLRP3 signaling is lost in models of clock disruption, contributing to the development of peritonitis, hepatitis, or colitis. Sterile inflammation is also an important driver of atherosclerosis, and targeting the production of IL-1β has proven to be a promising approach for atherosclerosis management in humans. Interestingly, the extent of injury after fulminant hepatitis or myocardial infarction is time-of-day dependent under the control of the clock, and chronotherapy represents a promising approach for the management of pathologies involving deregulation of NLRP3 signaling.
Introduction
Organisms evolved in presence of a recurring daily light–dark cycle generated by the rotation of the Earth. To adapt to this predictable environmental change, they developed an internal clock mechanism that is entrained to and anticipates environmental cues such as light or food availability and optimizes physiological functions by ascribing them to the best time window (1). Many, if not all, physiological pathways and functions are regulated in a daily manner including sleep/active alternance, metabolism, heart rate, brain and muscular activity, to cite a few. More recently, research efforts have been focused on the circadian behavior of the immune system that allows optimization of immune responses throughout the day/night cycle (2), leading to the emerging concept of circadian immunity. As a consequence, alteration of the circadian clock aggravates acute and chronic inflammatory diseases, pointing to new pharmacological approaches (3, 4).
The NLRP3 inflammasome was identified as a critical immune component that orchestrates host immune homeostasis. However, its chronic activation by endogenous danger signals derived from tissue damage and abnormal accumulation of self-components including urea and hydroxyapatite crystals in joints, amyloid fibers in brain or cholesterol crystals in the vascular wall, contributes to the development of a wide variety of diseases (5). Hence, a tight control of its transcription and activation is required to avoid overt deleterious activation.
In this review, we summarize the current knowledge on clock-controlled inflammasome modulation and highlight the underlying mechanisms as well as gaps of knowledge. We discuss several pathological contexts in which clock alteration contributes to NLRP3-driven pathologies and the potential of a (re-) synchronization of the clock to fine-tune NLRP3 activation and restore tissue homeostasis.
Innate Immune System and Pattern Recognition Receptors (PRRs)
The innate immune system is the first line of defense involved in the clearing of invaders like bacteria and viruses and also of abnormal accumulation of self-components including cellular debris or crystals. Immune cells discriminate infectious agents-derived molecules called pathogen-associated molecular patterns (PAMPs) and non-infectious, endogenous “danger molecules” or DAMPS (damage-associated molecular patterns) released by damaged or dying cells following tissue injury. These motifs are specifically recognized by tissue-resident cells such as mast cells, monocytes/macrophages, neutrophils and dendritic cells that express Pattern Recognition Receptors (PRRs). PRRs may be classified depending on their nature, their ligands and their cellular localization [see (6) for review]. Hence, they can be distinguished according to whether they are located at the cytoplasmic membrane (membrane PRRs: Toll-Like Receptors TLRs, C-type lectin receptors CLRs) or in the cytoplasm (cytoplasmic PRRs: NOD-Like Receptors NLRs, RIG-I-like Receptors RLRs and cytosolic DNA sensors CDSs). For instance, TLR-2 and TLR-4 are membrane receptors that are bound by PAMPs such as Gram+ peptidoglycans or Gram- LPS, respectively. Detection of PAMPs by PRRs triggers maturation and activation of immune cells that, in turn, secrete inflammatory factors and stimulate adaptive immunity (7). Non-infectious DAMPs are also recognized by PRRs on innate immune cells and initiate a so-called sterile inflammation. In addition to classical PRRs, numbers of non-PRR transmembrane proteins including Receptor for Advanced glycation endproducts (RAGEs), Triggering Receptors Expressed on Myeloid cells (TREMs), G Protein-Coupled Receptors (GPCRs) and ion channels are able to sense DAMPs and to trigger migration and activation of immune cells (6). PRRs and non-PRRs are involved in sterile inflammation and inflammatory diseases such as ischaemia-reperfusion injury, systemic lupus erythematosus, gout, neurodegenerative diseases, diabetes, colitis, atherosclerosis, hepatitis, rheumatoid arthritis, cancer, lung diseases, and gut diseases (6).
Inflammation is characterized by the production of histamine, cytokines, chemokines, and lipid derivatives (6). Cytokines are immunomodulatory signaling molecules playing a pivotal role in inflammation. The IL-1 cytokine family is composed of several members including IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ (7). Except for IL-1α, IL-1 cytokines are produced as inactive pro-cytokines and require maturation to biologically active forms by enzymatic cleavage. Among those, IL-1β is probably the most studied IL-1 family member because of its central involvement in acute and chronic inflammatory diseases. Pro-IL-1β, the inactive form of IL-1β, is processed by the proteolytic activity of Caspase 1, the predominant IL-1 processing protease. Caspase 1 activity is tightly controlled by cytosolic PRR-constituted inflammasome complex.
NOD-like receptors form the main class of cytosolic PRRs that are activated by diverse exogenous signals including anthrax lethal toxin (NLRP1), bacterial flagellin (NLRC4), double-stranded DNA Absent in Melanoma 2 (AIM2), Toxin-induced modifications of Rho-GTPase (Pyrin). In this regard, NLRP3 is unique because it acts as an intracellular innate immune sensor for a large variety of PAMPS and also DAMPs.
The NLRP3 Inflammasome: A Stress Sensor
The nucleotide-binding domain (NOD)-, Leucine-rich repeat (LRR)- and pyrin domain-containing protein 3 NLRP3 inflammasome was first identified in Cryopyrin-associated periodic syndrome (CAPS) before its implication was recognized in many inflammatory/immune diseases such as gout, atherosclerosis, type 2 diabetes (T2D) and non-alcoholic fatty liver disease (NAFLD) (8), as well as neurodegenerative diseases (Alzheimer and Parkinson diseases) and aging (9–11), and infection by various pathogens (12). The NLRP3 inflammasome is mainly expressed by monocytes/macrophages, neutrophils and dendritic cells, but also by other cell types including hepatocytes (13), neurons (14), cardiomyocytes (15), pancreatic beta cells (16), or endothelial cells (17).
A Two-Step Activation Process
The NLRP3 inflammasome is a macromolecular protein complex whose assembly is hierarchically organized and mostly requires a sensor protein, an adapter protein and an effector protein. The NLRP3 protein is a sensor protein that is composed of a C-terminal leucine rich repeat (LRR) domain, a central oligomerization domain (NOD, nucleotide-binding and oligomerization domain, NACHT) and an N-terminal Pyrin effector domain (PYD). This last PYD interacts with the amino-terminal PYD domain of the apoptosis-associated speck-like protein containing a Caspase recruitment domain (ASC) protein to initiate the inflammasome assembly and the formation of the so-called ASC speck. ASC is playing the role of adapter platform for the Caspase 1 protein thanks to its a carboxy terminal CARD domain that eventually recruits an unprocessed pro-caspase1 (18). Pro-caspase-1 oligomerization on the ASC filament enables proximity-driven autocatalytic caspase-1 maturation.
This complex activation is tightly controlled by a two-step process (Figure 1). A priming step is required to increase gene and protein expression of its components in order to sense stimuli and become activated (19). This priming occurs via ligand binding to PRRs (eg. signals that engage TLRs). These ligands may originate from exogenous sources such as bacterial wall components (Lipopolysaccharides, proteoglycans), or endogenous molecules (oxidized low-density lipoproteins [oxLDL], IL1, TNFα). This priming step is tightly controlled at the transcriptional level by the classical pro-inflammatory NF-κB and AP1 pathways, but also by metabolic sensors such as nuclear receptors including Liver X Receptors (20) and Rev-erb (21). A second step is the activation of the NLRP3 inflammasome in a primed cell/tissue that triggers the NLRP3 multimeric complex assembly that allows caspase 1 maturation and results in caspase1-mediated maturation of the pro-inflammatory interleukin-1β (IL-1β) and IL-18, the release of the mature cytokines, as well as in the so-called pyroptotic cell death (22). This second step may be triggered by a variety of compounds identifying NLRP3 as a wide PAMPs and DAMPs sensor as described below (8, 22, 23).
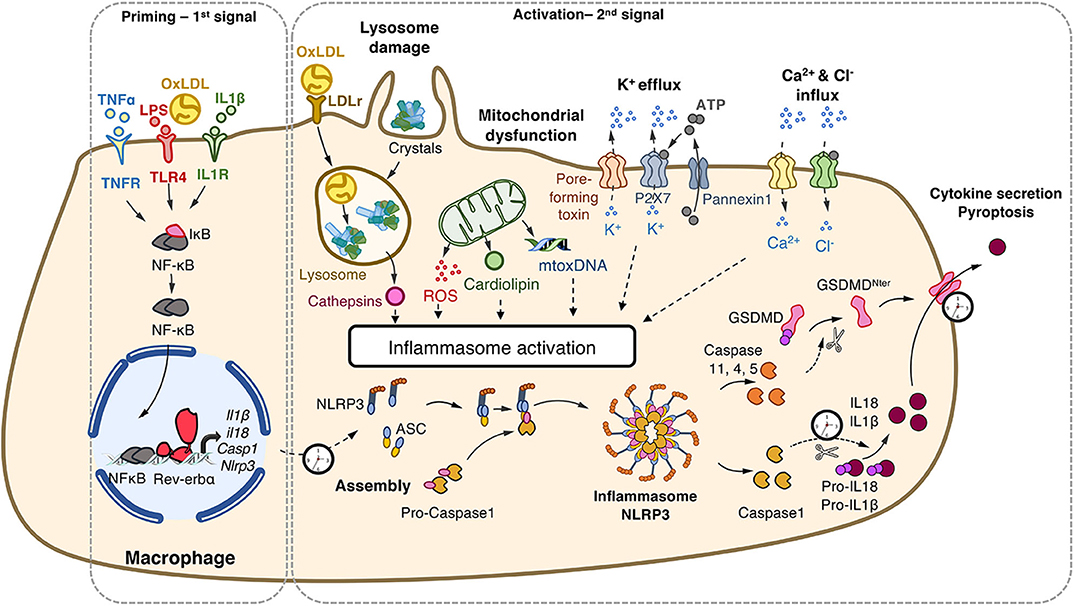
Figure 1. NLRP3 inflammasome priming and activation. The priming (first step) of the NLRP3 inflammasome requires the binding and activation of PRRs (TLRs,.) by PAMPs such as LPS, cytokines or ox-LDL, resulting in the transcription of the NLRP3 inflammasome components. Its activation (second step) is the result of recognition of PAMPs (such as the bacterial pore-forming toxin nigericin) or DAMPs which are released by damaged or dying cells (such as ATP) following injury or metabolic imbalance (such as mtROS), or accumulate in tissues (such as crystals). These lead to lysosomal damage, mitochondrial damages (exposition of cardiolipin, mtDNA) which ultimately modify ion (K+, Ca2+) fluxes. Upon these two-step process, the NLRP3 inflammasome assembles, caspase-1 is activated, Gasdermin-D and pro-IL-1β and pro-IL-18 are cleaved, leading to mature cytokines secretion and cell death by pyroptosis. ASC, apoptosis-associated speck-like protein containing a CARD domain; ATP, adenosine triphosphate; casp, caspase; DAMPs, damage-associated molecular patterns; GSDMD, gasdermin-D; IL, interleukin; IL1R, interleukin-1 receptor; LDL, Low Density Lipoprotein; LDLr; LDL receptor; LPS, lipopolysaccharide; mtoxDNA, mitochondrial oxidized DNA; NFκB, nuclear factor-kappa B; NLRP3, nucleotide-binding, LRR and PYD domains-containing protein 3; Ox-LDL, oxidized low-density lipoproteins; PAMPs, pathogen-associated molecular patterns; PRRs, Pattern Recognition Receptors; ROS, reactive oxygen species; P2X7R, purinergic receptor P2X 7; TLR, Toll-like receptor; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor.
NLRP3: A PAMPs' and DAMPs' Sensor
The NLRP3 inflammasome detects a broad range of DAMPs and PAMPs. Cholesterol crystals that accumulate in the arterial wall during atherosclerosis (24), monosodium urate (MSU) accumulation in joints leading to gout (25) and hydroxyapatite crystals triggering rheumatoid arthritis (5) all activate the NLRP3 inflammasome (Figure 1). The internalization of crystals leads to lysosomal damage and subsequent cathepsins and Ca2+ release that activates NLRP3 in a yet unknown manner. In addition, NLRP3 activation is also triggered by metabolic stresses such as hyperglycemia, some fatty acids and ceramides, and mitochondrial dysfunction, in particular mtROS (26), exposition of cardiolipin (27) or presence of mitochondrial oxidized DNA (28). Bacterial pore-forming toxins such as nigericin act as ionophores promoting K+ efflux which provokes the assembly of the NLRP3 complex, activation of Caspase 1 and the release of mature cytokines (29). Extracellular ATP released by dying cells also results in K+ and Ca2+ fluxes through P2X7 channel opening (23). In the same line, Ca2+ influx into the cytoplasm after mitochondrial reactive oxygen species (mtROS)-mediated cation channel transient receptor potential melastatin 2 (TRPM2) opening has been suggested to trigger the NLRP3 inflammasome assembly and IL-1β production in MSU-stimulated macrophages (28). The NLRP3 inflammasome is also sensing accumulation of aggregates (e.g., β-amyloid, Aβ) as well as metabolic stresses (8). Thus, the NLRP3 inflammasome is considered as a stress sensor that detects loss of homeostasis and abnormal endogenous molecules that signal infection, metabolic abnormalities or tissue damage (23).
Circadian Control of the Immune System
Molecular Organization of the Mammalian Clock
The mammalian clock consists of transcriptional activators and repressors forming interlocked feedback regulatory loops and organized in positive and negative limbs that confer rhythmicity to each other (30) (Figure 2). The positive limb is driven by BMAL1 (Brain and Muscle ARNT-like 1) and CLOCK (Circadian Locomotor Output Cycles Kaput) which heterodimerize and bind to E-boxes in their target gene promoters, amongst which Per and Cry clock genes whose transcription is activated by BMAL1/CLOCK. Period (PER) 1/2/3 and Cryptochrome (CRY) 1/2 form the negative limb. PER and CRY, once they reach sufficient quantity in the cytoplasm, heterodimerize and translocate to the nucleus where they bind BMAL1-CLOCK heterodimers to inhibit BMAL1/CLOCK transcriptional activity in a rhythmic manner (30). This first circuitry is finely tuned by the nuclear receptors Rev-erbs and RORs (31), which compete for binding to the same RevRE/RORE and RevDR2 DNA sequences and regulate gene expression in an opposite manner. While Rev-erbs act as transcriptional repressors, RORs compete with Rev-erbs for DNA binding and activate transcription of common target genes, including Bmal1 (32). Because Rev-erb isotypes display strong circadian rhythmicity in their abundance, this competition for binding to the Bmal1 promoter is rhythmic and contributes to BMAL1 oscillations. It is noteworthy that these transcription factors not only control each other's transcription but also bind to numerous genes containing RORE/RevDR2 or E-boxes, thereby generating rhythmic transcriptional waves in transcriptional programs involved in local tissue functions. For instance, Rev-erb-α controls the expression of E4bp4/Nfil3 in the liver (33, 34) but also in immune cells thereby regulating Th17 immune cell differentiation (35). Rev-erb also represses Cry1 transcription thus controlling both limbs of the clock in a coordinated manner (36).
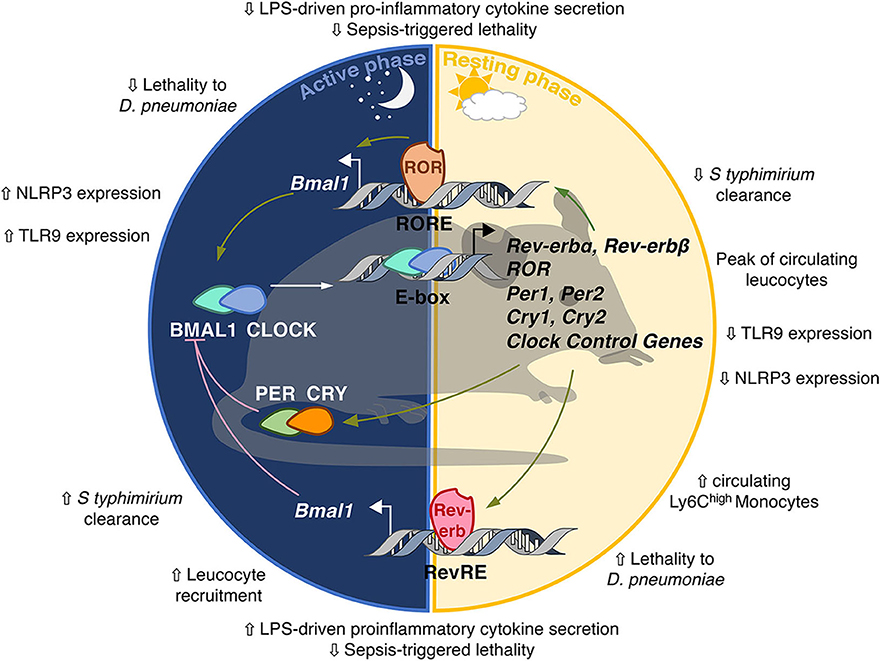
Figure 2. The molecular circadian clock in mammals. The molecular clockwork is formed by transcription–translation feedback loops. The transcription factors BMAL1/CLOCK induce the expression of E-box-containing genes including the negative regulators Period (PER) and Cryptochrome (CRY). In turn, the PER/CRY heterodimer inhibits the transcriptional activity of BMAL1/CLOCK. Once PER and CRY levels are sufficiently low, a new cycle may start. CLOCK/BMAL1 induce the expression of the nuclear receptors Rev-erbα/β and retinoid-related orphan receptor α, β, and γ (RORα/β/γ). Rev-erbs and RORs interact with co-repressors (NCoR) and co-activators (NCoA) and compete for the binding of RevRE/RORE elements in common target genes to repress or activate, respectively, their transcription. Rev-erb and ROR are also able to repress E4BP4/Nfil3 which rhythmically inhibits D-box-dependent transcription. Additional layers of regulation of circadian gene expression include rhythmic histone modifications, circadian chromosomal 3D conformation and post-translational modifications such as acetylation, phosphorylation, sumoylation, O-GlcNacylation.The clock is involved in the control of so-called circadian immunity.
The rhythmicity observed in gene transcription is not only due to cyclic binding of these transcription factors but also to circadian variations in histone marks and chromatin organization at regulatory regions (30, 37). Beside epigenetic control, dynamic 3D chromatin architecture is another layer of circadian genome function (38). Additionally, post-translational modifications such as phosphorylation, SUMOylation, O-Glc-Nacylation are necessary to ensure the stability of these transcription factors and thus the pace and robustness of the clockwork [(39) for review].
Biological Clocks in the Immune System
Virtually all mammalian cell types harbor a functional circadian clock, leading to circadian oscillations in the transcriptome, proteome, and ultimately cell/tissue function. The central pacemaker is located in the suprachiasmatic nucleus of the hypothalamus. It receives light information and synchronizes clocks throughout the body according to this time cue. The clock is present in immune cells including macrophages, lymphocytes and neutrophils as well as in lymphoid tissues such as the spleen and lymph nodes (40). The number of circulating leukocytes oscillates diurnally, peaking during the rest phase, due to circadian variations in haematopoietic cell egress from bone marrow which preferentially occurs at the onset of this phase (Figure 3) (41, 42). In addition, tissue leucocytes display circadian variations mainly due to oscillations in their rolling and adhesion to the endothelium and infiltration into tissues which predominantly occurs at the onset of the active phase (43, 44). In parallel, immune cell functions such as cytokines production, phagocytosis of exogenous particles or response to pathogens also display daily oscillations resulting in time-of-day-dependent difference in the susceptibility to septic shock or injury (45–48). This temporal organization is meant to ensure an optimization of the immune response in order to maintain or rapidly and efficiently restore homeostasis after infection or injury/tissue damage. Consequently, clock disruption has often been associated with inflammatory diseases (Figure 4).
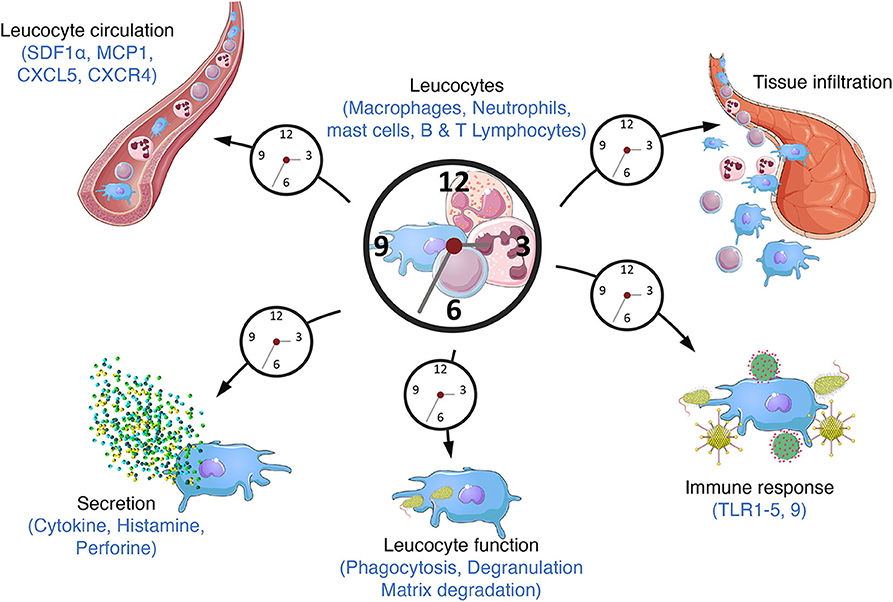
Figure 3. Circadian immunity. The molecular clock regulates a large number of immune functions throughout the day/night cycle such as cytokine secretion, phagocytosis, response to pathogens (bacterium, parasite) through the expression of TLR9 or the TLR4 pathway and danger signals through the regulation of the NLRP3 inflammasome. Circulating leucocyte number also varies along the day altogether with their mobilization from bone marrow and their recruitment into tissues.
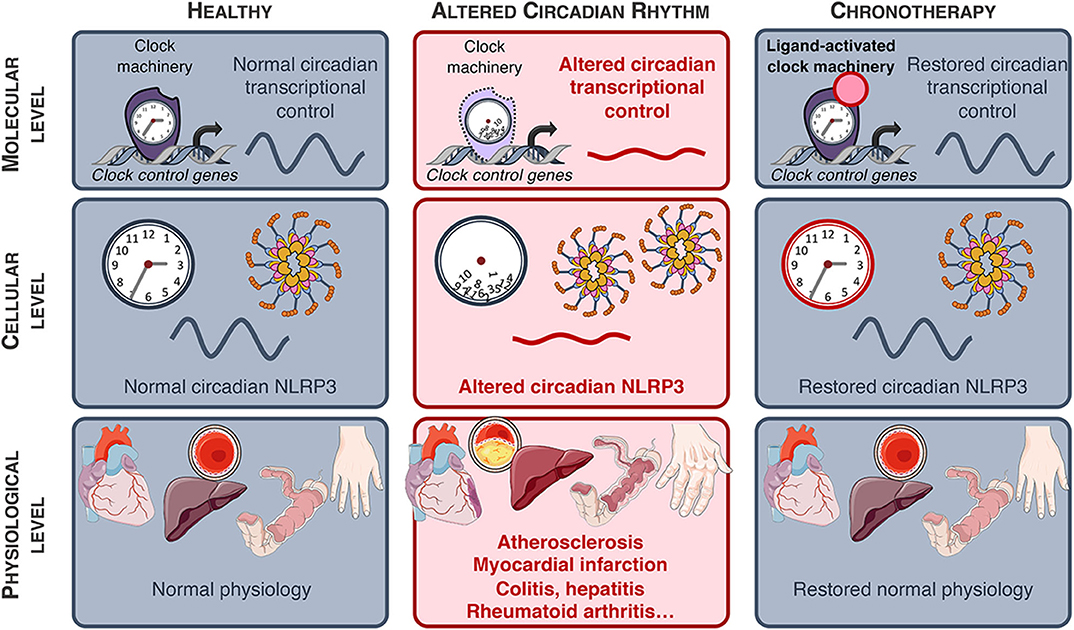
Figure 4. NLRP3-associated diseases. NLRP3-associated diseases and potential for innovative chronotherapies.
Circadian Control of the NLRP3 Inflammasome and Implication in Physiology and Pathologies
Expression of the NLRP3 inflammasome complex components is low and increased transcription is achieved during the priming step. It was recently demonstrated that the mRNA expression of the components of the NLRP3 inflammasome oscillates in a daily manner under the control of Rev-erbα in peritoneal mouse macrophages in vivo, reaching a peak (or zenith) during the active phase, corresponding to the lowest expression (or nadir) of Rev-erbα mRNA (21). Similar oscillations were observed in primary mouse bone marrow-derived and human monocyte-derived macrophages synchronized by a serum shock ex vivo, while circadian oscillations in Nlrp3 mRNA were lost upon Rev-erbα ablation. NLRP3 protein amounts were accordingly regulated upon modulation of Rev-erbα activity (21). Strikingly, alteration in the NLRP3 pathway provoked by impairment of Rev-erbα expression triggers alterations in IL-1β and IL-18 secretion in peritoneum (21). Additionally, Rev-erbα also regulates the NLRP3 activation step. Indeed, Rev-erbα ablation led to increased speck formation, caspase-1 cleavage and NLRP3-induced caspase-1 mediated maturation and secretion of IL-1β and IL-18 in LPS-primed macrophages activated with nigericin or ATP. By contrast, activation of Rev-erb by its natural (heme) or pharmacological ligands reduced the secretion of these pro-inflammatory cytokines. Mechanistically, Rev-erbα binds to specific response elements in Nlrp3 and Il1β gene promoters to silence their expression, and controls the NLRP3 inflammasome assembly and caspase 1 maturation (21) (Figure 1). In an in vivo model of acute sterile peritonitis induced by intraperitoneal administration of LPS and a concomitant injection of alum to specifically activate the NLRP3 pathway, IL-1β and IL-18 plasma levels were higher in Rev-erbα-deficient mice toward the end of the resting phase when Rev-erbα expression is highest in the wild-type controls, whereas the difference was lost during the active phase when Rev-erbα is nearly absent. Accordingly, and although this was not studied at different times of the day, Rev-erbα inhibition with an antagonist exacerbates the severity of LPS-induced acute lung injury by increasing NLRP3-dependent IL-1β secretion (49). These data demonstrate the rhythmic regulation of the NLRP3 inflammasome expression and activation, and suggest that Rev-erb pharmacological modulation may exert beneficial action in acute or chronic inflammatory diseases in which the NLRP3 inflammasome is over-activated, as detailed below.
Fulminant Hepatitis
Fulminant hepatitis (FH) is a life-threatening condition characterized by a fast-evolving hepatic dysfunction associated with encephalopathy and coagulopathy (50, 51). Numerous factors such as viral infection, metabolic and genetic diseases as well as absorption of toxic compounds are able to trigger FH, although overdose of acetaminophen still remains the main cause of FH nowadays (52). Acetaminophen accumulation leads to the production of large quantities of toxic metabolites provoking oxidative stress, mitochondrial membrane potential loss and hepatocellular death, the secretion of DAMPs and activation of the NLRP3 inflammasome (52). Increased IL-1β and activation of the NLRP3 inflammasome in macrophages has also been shown in viral hepatitis (53–55). Strikingly, the susceptibility of FH is time-of-day dependent, upon the control of the molecular clock and Rev-erbα was identified as an important regulator of the inflammasome in this context. In mice, ablation of Rev-erbα led to exacerbated fulminant hepatitis, including increased liver damage that was blunted upon administration of the MCC950 specific NLRP3 inhibitor (Table 1) (21, 58). Remarkably, Rev-erbα pharmacological activation dramatically reduced liver injury thereby delaying death and improving the rate of survival from fulminant hepatitis from 10% in the control to 70% in the treated mice (Figure 4) (21).
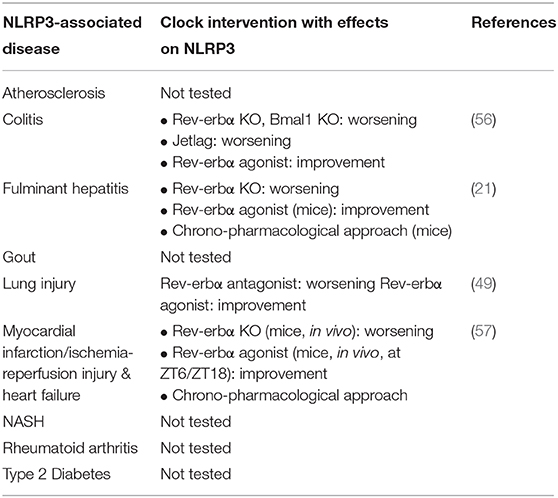
Table 1. NLRP3-associated diseases linked to the clock.
Colitis
Several studies have suggested a role of the NLRP3 inflammasome in inflammatory intestinal diseases and although controversial results were first published, it is now accepted that NLRP3 activation is detrimental in this context. Ablating IL-18 or blocking its signaling reduced the severity of experimental colitis (59, 60). In addition, the NLRP3 inflammasome was identified as a central mediator of intestinal inflammation in dextran sulfate sodium (DSS)-induced colitis (61). Consistent with the previously described role of the clock in the regulation of the NLRP3 activation, DSS-induced colitis was found to be more severe in mouse models of environmental or genetic disruption of the clock (56). Confirming previous results, Rev-erbα-deficient mice were found to display increased activation of the NLRP3 pathway which accounted for the severe phenotype, whereas pharmacological Rev-erb activation attenuated colitis in vivo (Figure 4). Interestingly, when tested in vitro, the Rev-erb agonist seems to be active only on the priming step, and was ineffective at modifying caspase 1 maturation in cultured LPS-primed macrophages activated with ATP. This might be due to the fact that the cells were not synchronized in this study. Still, the effects of Rev-erb activation were abolished by MCC950, a specific inhibitor of the NLRP3 inflammasome activation.
Cardio-Vascular Diseases
Circadian Clock and Blood Vessels Physiology
Circadian clocks reside in the different cell types of blood vessels (62, 63) and participate in vascular function and tone (64). For instance, blood pressure displays circadian oscillations, starting to rise before the rest-to-active transition while being lower during sleep (65), coinciding with the higher frequency of acute cardiovascular events and the exacerbated acute thrombus formation in the early morning hours (66, 67). Circadian oscillations in clock genes expression are attenuated in human atherosclerotic plaque (68), suggesting a mechanistic link between altered clock function and vascular pathologies. Numerous studies revealed that clock disruption (e.g., altered sleep patterns, shift-work) increases cardiovascular risk factors such as dyslipidemia, diabetes, hypertension and lead to cardiovascular diseases including stroke and coronary heart disease (Figure 4) (66, 69). Several studies found a relationship between shift work or acute circadian misalignment and subclinical atherosclerosis, measured by higher intima-media thickness (IMT) and elevated systemic inflammation even after adjustment for age and common risk factors (70–73). Moreover, lower sleep duration and fragmented sleep are independently associated with an increased risk of subclinical coronary and non-coronary atherosclerosis (74).
Clock Control of NLRP3 Inflammasome Activation, IL-1β Production and Atherogenesis
Atherosclerosis is a lipid-driven inflammatory disease of the arterial wall. Infiltration and modification of lipoproteins in the subendothelial space result in their uptake mainly by macrophages, forming foam cells, thus initiating atherosclerotic lesion formation. Then, lipids (fatty acids, ox-LDL, cholesterol crystals...) accumulate as well as inflammatory cells, notably monocyte-derived macrophages, T and B lymphocytes (75–77). Inefficient efferocytotic removal of these foam cells and apoptotic cells promote lesion progression toward advanced lesions with a necrotic core, degradation of the extracellular matrix, migration of smooth muscle cells and in some cases calcification, which may become vulnerable (78). Genetic alteration of the molecular clock contributes to metabolic imbalance and inflammation which promote atherogenesis (79, 80). For instance, BMAL1 modulates lipoprotein production and biliary cholesterol excretion, and its ablation led to hyperlipidemia and atherosclerosis (81). In the same line, Rev-erbα diminishes atherogenic lipoproteins plasma levels (82), modulates the inflammatory profile of macrophages toward an anti-inflammatory phenotype (83) while its activation reduced atherogenesis (84). Accordingly, BMAL1 regulates macrophage polarization as well as the cyclic trafficking of Ly6Chi monocytes and myeloid Bmal1 deletion increased monocyte recruitment and worsened atherosclerosis (Figures 2, 3) (85). Pro-inflammatory recruitment through the CCL2 (MCP-1)-CCR2 axis plays an important role in plaque development (86). In a recent study, McAlpine and colleagues revealed that sleep modulates haematopoiesis while chronic sleep fragmentation in a mouse model prone to atherosclerosis resulted in increased production of Ly6Chigh monocytes and aggravated atherosclerosis development due to increased infiltration to the lesions (Figure 4) (87). In line, disruption of circadian rhythms by chronic jetlag obtained by weekly alternating light-dark cycles with 12 h shifts enhanced atherosclerosis development and increased lesion macrophage content (Figure 4) (88). Interestingly, Winter et al. elegantly showed that myeloid cells are recruited to the lesions in a circadian manner, with a peak during the active-to-rest transition, through the rhythmic deposit of CCL2 on the arterial endothelium by circulating cells. A chrono-pharmacological approach targeting monocyte recruitment via timed inhibition of the CCR2/CCL2 axis during the active phase dampened atherosclerotic lesions development (Figure 4) (89).
In atherosclerotic lesions, oxLDL can prime the macrophage NLRP3 inflammasome by activating TLRs-dependent pathways. In addition, CD36-mediated oxLDL uptake eventually results into intra-lysosomal crystallization. Together with phagocytized extracellular cholesterol, they are thought to trigger macrophage lysosomal damage thus provoking cathepsins release (18). Moreover, defective cholesterol efflux in myeloid cells results in accumulation of unesterified cholesterol which contributes to both priming and activation of the NLRP3 inflammasome, promoting neutrophil recruitment and neutrophil extracellular trap (NET) formation in atherosclerotic plaques (90). The NLRP3 inflammasome activation contributes to the vascular inflammatory response through enhanced production of IL-1α and IL-1β, the latter driving inflammation during early atherogenesis and the evolution of advanced atheroma in mice (91). Canakinumab is an IL-1β-neutralizing antibody approved for the treatment for CAPS-associated symptoms which also reduced the incidence of two other NLRP3-related diseases, arthritis and gout (92). Recently, the CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcome Study) study demonstrated that IL-1β neutralization decreased the incidence of atherosclerotic disease and reduced systemic inflammation in at-risk patients with previous myocardial infarction in the absence of effect on lipids, indicating that suppressing IL-1β contributes to the reduction in cardiovascular risk (93). However, substantial residual inflammatory risk still subsisted after IL-1β neutralization, with on-treatment IL-18 and IL-6 plasma levels associated with future cardiovascular risk (94), advocating for therapies that simultaneously inhibit IL-1β and IL-18. In mice, inhibition of the NLRP3 inflammasome reduces atherogenesis in ApoE−/− or LDLr−/−mice (24, 95). Although plausible, it is currently unclear whether circadian control of NLRP3 inflammasome activation is perturbed within macrophage foam cells from atherosclerotic lesions. In this perspective, a therapy that targets the clock, and particularly Rev-erbα, in a chrono-pharmacological approach would be worth testing as Rev-erbα not only regulates NLRP3 inflammasome expression and activation, reducing both IL-1β and IL-18, but also MCP-1 expression and IL-6 production by macrophages (Figure 4) (47, 96), as well as lipoprotein metabolism, thereby simultaneously impacting both local inflammation and systemic risk factors.
Myocardial Infarction and the Circadian Control of NLRP3 Expression and Activation
The NLRP3 Inflammasome Is Activated Upon Acute Myocardial Ischemia/Reperfusion Injury
Myocardial infarction (MI) is one of the leading causes of death worldwide and is associated with a poor quality of life, acknowledging the increased interest in finding novel therapeutics to reduce reperfusion injury and preserve cardiac function. Despite improvement in reperfusion and treatment strategies that have led to higher survival rates, fibrosis and adverse left ventricular remodeling consecutive to reperfusion injury leads to cardiac contractile dysfunction and eventually heart failure (97).
Acute myocardial infarction (AMI) initiates a sterile inflammatory response that enables necrotic cardiomyocyte debris removal, angiogenesis and wound healing (98); however, this inflammatory response also promotes cell death by pyroptosis, expanding infarct size, and results in fibrosis and adverse ventricular remodeling. Then, refined intervention to rapidly attenuate this inflammatory burst is desirable (99). IL-1β and IL-18 are rapidly increased upon MI. Interestingly, administration of IL-1β- or IL-18-neutralizing antibody inhibits cardiomyocyte apoptosis, reduces infarct size and improves cardiac dysfunction after MI in mice (100, 101). In line, reduction of IL-1β production in caspase1- and in ASC-deficient mice upon ischemia/reperfusion is associated with a marked reduction in the infarct size, left ventricle remodeling and myocardial fibrosis (102). These data suggested that activation of the inflammasome may provoke further tissue damage through caspase-1-mediated production and release of IL-1β. Sandanger et al. confirmed that Nlrp3 deletion in mice leads to reduced infarct size and preservation of cardiac function in isolated perfused hearts subjected to acute I/R ex vivo (103).
Expression of the NLRP3 inflammasome components is very low and priming is induced during ischemic injury by cellular debris. NLRP3 is then activated by extracellular ATP as well as cardiolipin and mtDNA released by dying cells from damaged tissue after acute ischemic injury, or within minutes of reperfusion due to sudden surge of reoxygenation-induced ROS production and mitochondrial damage (Figure 1) (98). The NLRP3 inflammasome also senses extracellular-mediated efflux of K+ in cardiac fibroblasts upon hypoxia (103). In addition, the NLRP3 inflammasome is activated by numerous danger signals stemming from co-morbidities such as high glucose and lipid levels and derivatives (ceramides, advanced glycation products, which may lead to chronic activation of the NLRP3 inflammasome locally or in other organs) (104).
Inhibition of the NLRP3 Activation as a Novel Strategy to Reduce Myocardial I/R Injury?
Strategies to inhibit the activation of NLRP3 in the early reperfusion period after ischemic MI to reduce infarct size, avoid adverse remodeling and fibrosis and ameliorate cardiac function have been tested. Several inhibitors of the NLRP3 inflammasome activation have been developed, enabling pharmacological intervention in animal models undergoing AMI. Administration of the NLRP3 inhibitor MCC950 (58) lowers infarct size and area at risk (105, 106). Remarkably, NLRP3 inhibition by MCC950 treatment was associated with preserved left ventricle (LV) ejection fraction (LVEF), reduced fibrosis and myocardial immune cell infiltration. Interestingly however, the benefit of administering the NLRP3 inhibitor before AMI or within 1 h of reperfusion was lost when the NLRP3 inhibitor was given after 3 h of reperfusion, suggesting that inhibition should be achieved at time of NLRP3 assembly and activation (107). Other NLRP3 inhibitors have been shown to reduce infarct size in mouse models of myocardial ischemia/reperfusion. Among them, OLT1177 reduces infarct size in mice (108) and is currently in Phase 1b in a randomized, double-blinded, placebo-controlled, safety, and pharmacodynamics study in 30 subjects with stable systolic heart failure (HF) with impaired LVEF.
Circadian Rhythms in Cardiac Biology and Diseases
Daily oscillations of blood pressure and heart rate are reduced or lost in cardiomyocyte-specific Clock-mutant (109) acknowledging the important role of cardiomyocyte clock machinery in cardiac function. Consistently, circadian disruption due to either environmental out-of-sync stimuli or genetic manipulation of clock genes results in cardiomyopathies, cardiac dysfunction, arrhythmia, and reduced survival (110–113) and for review (114) (Figure 4). Additionally, circadian variations are seen in the onset and frequency of myocardial infarction, stroke and sudden death (115, 116), as well as in the severity of the diseases (117). Furthermore, environmental circadian disruption adversely impacts cardiac remodeling and function, increases macrophage infiltration and led to cardiac hypertrophy in mice undergoing MI (Figure 4) (118). The circadian influence in the tolerance to I/R injury was corroborated in mice undergoing I/R at the resting-to-active and active-to-rest transitions. The former led to exacerbated infarct size, and subsequent fibrosis and adverse cardiac remodeling. This time-of-day difference in the tolerance to I/R was markedly attenuated in cardiomyocyte-specific circadian clock mutant mice (119). In humans, it was recently assessed whether myocardial tolerance of I/R differed depending on the timing of aortic valve replacement surgery, as measured by the occurrence of major adverse cardiovascular events (cardiovascular death, myocardial infarction, and admission to hospital for acute heart failure). Expectedly, perioperative myocardial injury was better tolerated when patients underwent surgery in the afternoon (120). Interestingly, targeting the circadian clock through pharmacological modulation of Rev-erbα/β in mice was able to reduce myocardial I/R injury ex vivo in an isolated Langendorff-perfused mouse heart model of hypoxia-reperfusion, thus providing new therapeutic ways to dampen the adverse outcome of cardiac I/R injury. Whether Rev-erbα might be an interesting target to reduce cardiac dysfunction was further established in a model of transaortic constriction-induced heart failure (121) as well as in a mouse model of AMI (122). In this later report, the author suggested that blunted inflammation and reduced recruitment of neutrophils and pro-inflammatory macrophages upon pharmacological Rev-erbα modulation may, at least in part, contribute to the benefit of targeting Rev-erbα. Remarkably, Schloss et al. elegantly demonstrated that infarct size is higher at ZT13 vs. ZT5 when the number of cardiac Ly6Chigh monocytes is highest likely because of increased CCR2-mediated recruitment of these cells, and that blocking the CCR2-CCL2 axis blunted the time-of-day variations in infarct size (123). These data, together with the observation that Rev-erbα controls macrophage NLRP3 activation (21), point to a possible role of monocyte/macrophage Rev-erbα in I/R tolerance. In a recent report, Martino and colleagues questioned the cell-specific role of Rev-erbα in the protective effect of Rev-erbα activation. They found that activating Rev-erbα at time of reperfusion in wild-type mice limits infarct expansion, improves cardiac function and outcomes, and reduced recruitment of neutrophils and macrophages as well as cardiac NLRP3 inflammasome activation. However, myeloid cells were unlikely to account for this beneficial effect as shown by bone marrow transfer experiments (57). Instead, Rev-erbα may downregulate the NLRP3 inflammasome in cardiac fibroblasts although further studies using cell-specific mutant mice are necessary to pinpoint the exact contribution of each cell types. More importantly, pharmacological Rev-erbα activation showed the greatest benefit when given at time of reperfusion, whenever it happened during the active (ZT18) or resting (ZT6) phase, although the benefit was greater at ZT6 corresponding to maximal Rev-erbα expression (Figure 4). This suggests that beyond the time of treatment and potentially differential effect on the pace of the clock, Rev-erbα-regulated inhibition of NLRP3 before or at reperfusion may hold promise to reduce myocardial I/R damages, whenever the time at which the patient will undergo cardiac surgery or experience MI.
Conclusion
In this review, we have highlighted the relationship between circadian immunity and the NLRP3 inflammasome pathway. As a central sensor of tissue damages and metabolic imbalance, NLRP3 plays a pivotal role in tissue homeostasis in many tissues including liver, heart and the vasculature. Sustained activation of NLRP3 by exogenous or endogenous triggers thus aggravates chronic inflammatory diseases such as atherosclerosis (24) or worsen acute inflammatory conditions such as fulminant hepatitis (21) or myocardial infarction (121). As such, the NLRP3 inflammasome represents an innovative target, as exemplified by the use of NLRP3 inhibitors in several disease models. However, MCC950 displays hepatotoxic properties, advocating for the development of alternative NLRP3 inhibitory strategy (124). Strikingly, because NLRP3 is controlled by the clock machinery, the time of exposure to intruders and their sensing has a dramatic impact on the inflammatory response amplitude, the disease outcome and its resolution. As such, a chrono-pharmacological approach targeting NLRP3 may have greater benefits for the treatment of NLRP3-driven diseases (Figure 4). Since pathological tissues often display distinct circadian oscillation patterns compared to healthy tissue, such strategies would allow to target NLRP3 specifically in pathological areas and then preserve homeostasis in healthy tissue and thus reduce adverse effects. Several targets should be considered, either the NLRP3 pathway itself or NLRP3-regulating clock components such as Rev-erbα. Finally, alteration of NLRP3 pathway is involved in other diseases including diabetes, Alzheimer disease, gout, rheumatoid arthritis, or asthma (5). Clock-driven NLRP3 resynchronisation may represent an additional approach to help treating these diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Some of our work included in this review manuscript was supported by INSERM, the ANR-Labex-EGID (EGID, ANR-10-LABX-46), the CPER-CTRL Institut Pasteur de Lille, the Fondation Francophone pour la recherche sur le diabète (FFRD) together with the Fédération Française des Diabétiques (AFD) AstraZeneca, Eli Lilly, Merck Sharp & Dohme (MSD), Novo Nordisk & Sanofi; the Région Hauts-de-France/FEDER (Chronoregeneration), Association Française contre les Myopathies (AFM), Société Francophone du Diabète (SFD), Fondation de France and the ANR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the members of the HD's team for helpful discussions.
References
1. Bass J. Circadian topology of metabolism. Nature. (2012) 491:348–56. 491:348–56. doi: 10.1038/nature11704
2. Scheiermann C, Gibbs J, Ince L, Loudon A. Clocking in to immunity. Nat Rev Immunol. (2018) 18:423–37. doi: 10.1038/s41577-018-0008-4
3. Early JO, Curtis AM. Immunometabolism: is it under the eye of the clock? Semin Immunol. (2016) 28:478–90. doi: 10.1016/j.smim.2016.10.006
4. Man K, Loudon A, Chawla A. Immunity around the clock. Science. (2016) 354:999–1003. 354:999–1003. doi: 10.1126/science.aah4966
5. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
6. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. (2020) 20:95–112. 20:95–112. doi: 10.1038/s41577-019-0215-7
7. Evavold CL, Kagan JC. Inflammasomes: threat-assessment organelles of the innate immune system. Immunity. (2019) 51:609–24. doi: 10.1016/j.immuni.2019.08.005
8. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. (2018) 17:688. 17:688. doi: 10.1038/nrd.2018.149
9. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. (2011) 17:179–88. 17:179–88. doi: 10.1038/nm.2279
10. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. (2013) 493:674–8. 493:674–8. doi: 10.1038/nature11729
11. Youm Y-H, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. (2013) 18:519–32. doi: 10.1016/j.cmet.2013.09.010
12. Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. (2012) 13:325–32. doi: 10.1038/ni.2231
13. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
14. Fann DY-W, Lim Y-A, Cheng Y-L, Lok K-Z, Chunduri P, Baik S-H, et al. Evidence that NF-κB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol Neurobiol. (2018) 55:1082–96. doi: 10.1007/s12035-017-0394-9
15. Yao C, Veleva T, Scott L, Cao S, Li L, Chen G, et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation. (2018) 138:2227–42. 138:2227–42. doi: 10.1161/CIRCULATIONAHA.118.035202
16. Filhoulaud G, Benhamed F, Pagesy P, Bonner C, Fardini Y, Ilias A, et al. O-GlcNacylation links TxNIP to inflammasome activation in pancreatic β cells. Front Endocrinol. (2019) 10:291. 10:291. doi: 10.3389/fendo.2019.00291
17. Xi H, Zhang Y, Xu Y, Yang WY, Jiang X, Sha X, et al. Caspase-1 inflammasome activation mediates Homocysteine-induced Pyrop-apoptosis in endothelial cells. Circ Res. (2016) 118:1525–39. 118:1525–39. doi: 10.1161/CIRCRESAHA.116.308501
18. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in Atherosclerosis. Circu Res. (2018) 122:1722–40. doi: 10.1161/CIRCRESAHA.118.311362
19. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. 183:787–91. doi: 10.4049/jimmunol.0901363
20. Pourcet B, Gage MC, León TE, Waddington KE, Pello OM, Steffensen KR, et al. The nuclear receptor LXR modulates interleukin-18 levels in macrophages through multiple mechanisms. Sci Rep. (2016) 6:25481. doi: 10.1038/srep25481
21. Pourcet B, Zecchin M, Ferri L, Beauchamp J, Sitaula S, Billon C, et al. Nuclear receptor subfamily 1 group D member 1 regulates circadian activity of NLRP3 inflammasome to reduce the severity of fulminant hepatitis in mice. Gastroenterology. (2018) 154:1449–64.e20. 154:1449–64.e20. doi: 10.1053/j.gastro.2017.12.019
22. Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. 19:477–89. doi: 10.1038/s41577-019-0165-0
23. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:3328. doi: 10.3390/ijms20133328
24. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464:1357–61. 464:1357–61. doi: 10.1038/nature08938
25. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. (2006) 440:237–41. 440:237–41. doi: 10.1038/nature04516
26. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. doi: 10.1038/nature09663
27. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. (2013) 39:311–23. 39:311–23. doi: 10.1016/j.immuni.2013.08.001
28. Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun. (2013) 4:1611. 4:1611. doi: 10.1038/ncomms2608
29. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. (2006) 440:228–32. 440:228–32. doi: 10.1038/nature04515
30. Rijo-Ferreira F, Takahashi JS. Genomics of circadian rhythms in health and disease. Genome Med. (2019) 11:82. doi: 10.1186/s13073-019-0704-0
31. Bass J, Lazar MA. Circadian time signatures of fitness and disease. Science. (2016) 354:994–9. doi: 10.1126/science.aah4965
32. Sassone-Corsi P, Christen Y, Lazar MA. Rev-erbs: integrating metabolism around the clock. In: Sassone-Corsi P, Christen Y, editors. A Time for Metabolism and Hormones. Cham: Springer (2016). p. 63–70. doi: 10.1007/978-3-319-27069-2_7
33. Duez H, van der Veen JN, Duhem C, Pourcet B, Touvier T, Fontaine C, et al. Regulation of bile acid synthesis by the nuclear receptor Rev-erbalpha. Gastroenterology. (2008) 135:689–98. 135:689–98. doi: 10.1053/j.gastro.2008.05.035
34. Fang B, Everett LJ, Jager J, Briggs E, Armour SM, Feng D, et al. Circadian enhancers coordinate multiple phases of rhythmic gene transcription in vivo. Cell. (2014) 159:1140–52. 159:1140–52. doi: 10.1016/j.cell.2014.10.022
35. Yu X, Rollins D, Ruhn KA, Stubblefield JJ, Green CB, Kashiwada M, et al. TH17 cell differentiation is regulated by the circadian clock. Science. (2013) 342:727–30. 342:727–30. doi: 10.1126/science.1243884
36. Ukai-Tadenuma M, Yamada RG, Xu H, Ripperger JA, Liu AC, Ueda HR. Delay in feedback repression by cryptochrome 1 is required for circadian clock function. Cell. (2011) 144:268–81. 144:268–81. doi: 10.1016/j.cell.2010.12.019
37. Sassone-Corsi P, Christen Y. The epigenetic and metabolic language of the circadian clock. In: Sassone-Corsi P, Christen Y, editors. A Time for Metabolism and Hormones. (2016). p. 1–11. doi: 10.1007/978-3-319-27069-2_1
38. Kim YH, Marhon SA, Zhang Y, Steger DJ, Won K-J, Lazar MA. Rev-erbα dynamically modulates chromatin looping to control circadian gene transcription. Science. (2018) 359:1274–7. 359:1274–7. doi: 10.1126/science.aao6891
39. Hirano A, Fu Y-H, Ptáček LJ. The intricate dance of post-translational modifications in the rhythm of life. Nat Struct Mol Biol. (2016) 23:1053–60. 23:1053–60. doi: 10.1038/nsmb.3326
40. Curtis AM, Bellet MM, Sassone-Corsi P, O'Neill LA. Circadian clock proteins and immunity. Immunity. (2014) 40:178–86. doi: 10.1016/j.immuni.2014.02.002
41. Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. (2008) 452:442–7. doi: 10.1038/nature06685
42. Lucas D, Battista M, Shi PA, Isola L, Frenette PS. Mobilized hematopoietic stem cell yield depends on species-specific circadian timing. Cell Stem Cell. (2008) 3:364–6. 3:364–6. doi: 10.1016/j.stem.2008.09.004
43. Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang JE, Zhang D, et al. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity. (2012) 37:290–301. 37:290–301. doi: 10.1016/j.immuni.2012.05.021
44. He W, Holtkamp S, Hergenhan SM, Kraus K, de Juan A, Weber J, et al. Circadian expression of migratory factors establishes lineage-specific signatures that guide the homing of leukocyte subsets to tissues. Immunity. (2018) 49:1175–90.e7. 49:1175–90.e7. doi: 10.1016/j.immuni.2018.10.007
45. Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science. (2013) 341:1483–8. 341:1483–8. doi: 10.1126/science.1240636
46. Gibbs J, Ince L, Matthews L, Mei J, Bell T, Yang N, et al. An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat Med. (2014) 20:919–26. 20:919–26. doi: 10.1038/nm.3599
47. Gibbs JE, Blaikley J, Beesley S, Matthews L, Simpson KD, Boyce SH, et al. The nuclear receptor REV-ERBα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Prat Natl Acad Sci USA. (2012) 109:582–7. 109:582–7. doi: 10.1073/pnas.1106750109
48. Kitchen GB, Cunningham PS, Poolman TM, Iqbal M, Maidstone R, Baxter M, et al. The clock gene Bmal1 inhibits macrophage motility, phagocytosis, and impairs defense against pneumonia. Prat Natl Acad Sci USA. (2020) 117:1543–51. doi: 10.1073/pnas.1915932117
49. Yu D, Fang X, Xu Y, Xiao H, Huang T, Zhang Y, et al. Rev-erbα can regulate the NF-κB/NALP3 pathway to modulate lipopolysaccharide-induced acute lung injury and inflammation. Int Immunopharmacol. (2019) 73:312–20. 73:312–20. doi: 10.1016/j.intimp.2019.04.035
50. Cardoso FS, Marcelino P, Bagulho L, Karvellas CJ. Acute liver failure: an up-to-date approach. J Crit Care. (2017) 39:25–30. doi: 10.1016/j.jcrc.2017.01.003
51. Sarin SK, Choudhury A. Acute-on-chronic liver failure: terminology, mechanisms and management. Nat Rev Gastroenterol Hepatol. (2016) 13:131–49. 13:131–49. doi: 10.1038/nrgastro.2015.219
52. Ichai P, Samuel D. Epidemiology of liver failure. Clin Res Hepatol Gastroenterol. (2011) 35:610–7. doi: 10.1016/j.clinre.2011.03.010
53. Guo S, Yang C, Diao B, Huang X, Jin M, Chen L, et al. The NLRP3 inflammasome and IL-1β accelerate immunologically mediated pathology in experimental viral fulminant hepatitis. PLoS pathogens. (2015) 11:e1005155. doi: 10.1371/journal.ppat.1005155
54. Negash AA, Ramos HJ, Crochet N, Lau DTY, Doehle B, Papic N, et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. (2013) 9:e1003330. 9:e1003330. doi: 10.1371/journal.ppat.1003330
55. Serti E, Werner JM, Chattergoon M, Cox AL, Lohmann V, Rehermann B. Monocytes activate natural killer cells via inflammasome-induced interleukin 18 in response to hepatitis C virus replication. Gastroenterology. (2014) 147:209–20.e3. 147:209–20.e3. doi: 10.1053/j.gastro.2014.03.046
56. Wang S, Lin Y, Yuan X, Li F, Guo L, Wu B. REV-ERBα integrates colon clock with experimental colitis through regulation of NF-κB/NLRP3 axis. Nat Commun. (2018) 9:4246. 9:4246. doi: 10.1038/s41467-018-06568-5
57. Reitz CJ, Alibhai FJ, Khatua TN, Rasouli M, Bridle BW, Burris TP, et al. SR9009 administered for one day after myocardial ischemia-reperfusion prevents heart failure in mice by targeting the cardiac inflammasome. Commun Biol. (2019) 2:353. 2:353. doi: 10.1038/s42003-019-0595-z
58. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. (2015) 21:248–55. doi: 10.1038/nm.3806
59. Hove Ten T, Corbaz A, Amitai H, Aloni S, Belzer I, Graber P, et al. Blockade of endogenous IL-18 ameliorates TNBS-induced colitis by decreasing local TNF-α production in mice. Gastroenterology. (2001) 121:1372–9. doi: 10.1053/gast.2001.29579
60. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell. (2015) 163:1444–56. 163:1444–56. doi: 10.1016/j.cell.2015.10.072
61. Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. (2010) 59:1192–9. 59:1192–9. doi: 10.1136/gut.2009.197822
62. Rudic RD, Curtis AM, Cheng Y, FitzGerald G. Peripheral clocks and the regulation of cardiovascular and metabolic function. Meth Enzymol. (2005) 393:524–39. 393:524–39. doi: 10.1016/S0076-6879(05)93027-9
63. McNamara P, Seo SB, Rudic RD, Sehgal A, Chakravarti D, FitzGerald GA. Regulation of CLOCK and MOP4 by nuclear hormone receptors in the vasculature: a humoral mechanism to reset a peripheral clock. Cell. (2001) 105:877–89. doi: 10.1016/S0092-8674(01)00401-9
64. Panza JA, Epstein SE, Quyyumi AA. Circadian variation in vascular tone and its relation to alpha-sympathetic vasoconstrictor activity. N Engl J Med. (1991) 325:986–90. 325:986–90. doi: 10.1056/NEJM199110033251402
65. Millar-Craig MW, Bishop CN, Raftery EB. Circadian variation of blood-pressure. Lancet. (1978) 1:795–7. doi: 10.1016/S0140-6736(78)92998-7
66. Chellappa SL, Vujovic N, Williams JS, Scheer FAJL. Impact of circadian disruption on cardiovascular function and disease. Trends Endocrinol Metab. (2019) 30:767–79. 30:767–79. doi: 10.1016/j.tem.2019.07.008
67. Tofler GH, Brezinski D, Schafer AI, Czeisler CA, Rutherford JD, Willich SN, et al. Concurrent morning increase in platelet aggregability and the risk of myocardial infarction and sudden cardiac death. N Engl J Med. (1987) 316:1514–8. doi: 10.1056/NEJM198706113162405
68. Lin C, Tang X, Zhu Z, Liao X, Zhao R, Fu W, et al. The rhythmic expression of clock genes attenuated in human plaque-derived vascular smooth muscle cells. Lipids Health Dis. (2014) 13:14–9. 13:14–9. doi: 10.1186/1476-511X-13-14
69. Boggild H, Knutsson A. Shift work, risk factors and cardiovascular disease. Scand J Work Environ Health. (1999) 25:85–99. doi: 10.5271/sjweh.410
70. Puttonen S, Kivimaki M, Elovainio M, Pulkki-Raback L, Hintsanen M, Vahtera J, et al. Shift work in young adults and carotid artery intima-media thickness: the cardiovascular risk in young finns study. Atherosclerosis. (2009) 205:608–13. 205:608–13. doi: 10.1016/j.atherosclerosis.2009.01.016
71. Skogstad M, Mamen A, Lunde L-K, Ulvestad B, Matre D, Aass HCD, et al. Shift work including night work and long working hours in industrial plants increases the risk of atherosclerosis. Int J Environ Res Public Health. (2019) 16:521. doi: 10.3390/ijerph16030521
72. Jankowiak S, Backé E, Liebers F, Schulz A, Hegewald J, Garthus-Niegel S, et al. Current and cumulative night shift work and subclinical atherosclerosis: results of the Gutenberg health study. Int Arch Occup Environ Health. (2016) 89:1169–82. doi: 10.1007/s00420-016-1150-6
73. Morris CJ, Purvis TE, Hu K, Scheer FA. Circadian misalignment increases cardiovascular disease risk factors in humans. Proc Natl Acad Sci USA. (2016) 113:E1402–11. 113:E1402–11. doi: 10.1073/pnas.1516953113
74. Domínguez F, Fuster V, Fernández-Alvira JM, Fernández-Friera L, López-Melgar B, Blanco-Rojo R, et al. Association of sleep duration and quality with subclinical atherosclerosis. J Am Coll Cardiol. (2019) 73:134–44. doi: 10.1016/j.jacc.2018.10.060
75. Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. (2006) 86:515–81. doi: 10.1152/physrev.00024.2005
76. Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. (2010) 74:213–20. doi: 10.1253/circj.CJ-09-0706
77. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. (2010) 10:36–46. doi: 10.1038/nri2675
78. Bäck M, Yurdagul A, Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. (2019) 16:389–406. 16:389–406. doi: 10.1038/s41569-019-0169-2
79. Steffens S, Winter C, Schloss MJ, Hidalgo A, Weber C, Soehnlein O. Circadian control of inflammatory processes in atherosclerosis and its complications. Arterioscler Thromb Vasc Biol. (2017) 37:1022–8. doi: 10.1161/ATVBAHA.117.309374
80. McAlpine CS, Swirski FK. Circadian influence on metabolism and inflammation in atherosclerosis. Circ Res. (2016) 119:131–41. doi: 10.1161/CIRCRESAHA.116.308034
81. Pan X, Bradfield CA, Hussain MM. Global and hepatocyte-specific ablation of Bmal1 induces hyperlipidaemia and enhances atherosclerosis. Nat Commun. (2016) 7:13011. 7:13011. doi: 10.1038/ncomms13011
82. Raspe E, Duez H, Mansen A, Fontaine C, Fievet C, Fruchart JC, et al. Identification of Rev-erbalpha as a physiological repressor of apoC-III gene transcription. J Lipid Res. (2002) 43:2172–9. 43:2172–9. doi: 10.1194/jlr.M200386-JLR200
83. Ma H, Zhong W, Jiang Y, Fontaine C, Li S, Fu J, et al. Increased atherosclerotic lesions in LDL receptor deficient mice with hematopoietic nuclear receptor Rev-erbalpha knock- down. J Am Heart Assoc. (2013) 2:e000235. doi: 10.1161/JAHA.113.000235
84. Sitaula S, Billon C, Kamenecka TM, Solt LA, Burris TP. Suppression of atherosclerosis by synthetic REV-ERB agonist. Biochem Biophys Res Commun. (2015) 460:566–71. 460:566–71. doi: 10.1016/j.bbrc.2015.03.070
85. Huo M, Huang Y, Qu D, Zhang H, Wong WT, Chawla A, et al. Myeloid Bmal1 deletion increases monocyte recruitment and worsens atherosclerosis. FASEB J. (2017) 31:1097–106. 31:1097–106. doi: 10.1096/fj.201601030R
87. McAlpine CS, Kiss MG, Rattik S, He S, Vassalli A, Valet C, et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature. (2019) 566:383–7. 566:383–7. doi: 10.1038/s41586-019-0948-2
88. Schilperoort M, van den Berg R, Bosmans LA, van Os BW, Dollé MET, Smits NAM, et al. Disruption of circadian rhythm by alternating light-dark cycles aggravates atherosclerosis development in APOE*3-Leiden.CETP mice. J Pineal Res. (2020) 68:e12614. 68:e12614. doi: 10.1111/jpi.12614
89. Winter C, Silvestre-Roig C, Ortega-Gomez A, Lemnitzer P, Poelman H, Schumski A, et al. Chrono-pharmacological targeting of the CCL2-CCR2 axis ameliorates atherosclerosis. Cell Metab. (2018) 28:175–82.e5. doi: 10.1016/j.cmet.2018.05.002
90. Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and Atherogenesis. Circulation. (2018) 138:898–912. doi: 10.1161/CIRCULATIONAHA.117.032636
91. Vromman A, Ruvkun V, Shvartz E, Wojtkiewicz G, Santos Masson G, Tesmenitsky Y, et al. Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur Heart J. (2019) 40:2482–91. doi: 10.1093/eurheartj/ehz008
92. Alten R, Gram H, Joosten LA, van den Berg WB, Sieper J, Wassenberg S, et al. The human anti-IL-1 beta monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof-of-concept study in patients with rheumatoid arthritis. Arthritis Res Ther. (2008) 10:R67–9. 10:R67–9. doi: 10.1186/ar2438
93. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with Canakinumab for Atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. 377:1119–31. doi: 10.1056/NEJMoa1707914
94. Ridker PM, MacFadyen JG, Thuren T, Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1β inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J. (2019) 41:2153–63. doi: 10.1093/eurheartj/ehz542
95. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slütter B, et al. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol. (2017) 37:1457–61. 37:1457–61. doi: 10.1161/ATVBAHA.117.309575
96. Sato S, Sakurai T, Ogasawara J, Takahashi M, Izawa T, Imaizumi K, et al. A circadian clock gene, Rev-erbα, modulates the inflammatory function of macrophages through the negative regulation of Ccl2 expression. J Immunol. (2014) 192:407–17. 192:407–17. doi: 10.4049/jimmunol.1301982
97. Ibañez B, Heusch G, Ovize M, van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. (2015) 65:1454–71. 65:1454–71. doi: 10.1016/j.jacc.2015.02.032
98. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. (2018) 15:203–14. doi: 10.1038/nrcardio.2017.161
99. Zuurbier CJ, Abbate A, Cabrera-Fuentes HA, Cohen MV, Collino M, De Kleijn DPV, et al. Innate immunity as a target for acute cardioprotection. Cardiovasc Res. (2019) 115:1131–42. 115:1131–42. doi: 10.1093/cvr/cvy304
100. Toldo S, Mezzaroma E, Van Tassell BW, Farkas D, Marchetti C, Voelkel NF, et al. Interleukin-1β blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp Physiol. (2013) 98:734–45. 98:734–45. doi: 10.1113/expphysiol.2012.069831
101. Venkatachalam K, Prabhu SD, Reddy VS, Boylston WH, Valente AJ, Chandrasekar B. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J Biol Chem. (2009) 284:7853–65. doi: 10.1074/jbc.M808824200
102. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. (2011) 123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777
103. Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. (2013) 99:164–74. doi: 10.1093/cvr/cvt091
104. Mastrocola R, Collino M, Penna C, Nigro D, Chiazza F, Fracasso V, et al. Maladaptive modulations of NLRP3 inflammasome and cardioprotective pathways are involved in diet-induced exacerbation of myocardial ischemia/reperfusion injury in mice. Oxid Med Cell Longev. (2016) 2016:3480637–12. 2016:3480637–12. doi: 10.1155/2016/3480637
105. van Hout GPJ, Bosch L, Ellenbroek GHJM, de Haan JJ, van Solinge WW, Cooper MA, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J. (2017) 38:828–36. 38:828–36. doi: 10.1093/eurheartj/ehw247
106. Gao R, Shi H, Chang S, Gao Y, Li X, Lv C, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int Immunopharmacol. (2019) 74:105575. 74:105575. doi: 10.1016/j.intimp.2019.04.022
107. Toldo S, Marchetti C, Mauro AG, Chojnacki J, Mezzaroma E, Carbone S, et al. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int J Cardiol. (2016) 209:215–20. 209:215–20. doi: 10.1016/j.ijcard.2016.02.043
108. Toldo S, Mauro AG, Cutter Z, Van Tassell BW, Mezzaroma E, Del Buono MG, et al. The NLRP3 inflammasome inhibitor, OLT1177 (Dapansutrile), reduces infarct size and preserves contractile function after ischemia reperfusion injury in the mouse. J Cardiovasc Pharmacol. (2019) 73:215–22. 73:215–22. doi: 10.1097/FJC.0000000000000658
109. Bray MS, Shaw CA, Moore MWS, Garcia RAP, Zanquetta MM, Durgan DJ, et al. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol Heart Circ Physiol. (2008) 294:H1036–47. 294:H1036–47. doi: 10.1152/ajpheart.01291.2007
110. Penev PD, Kolker DE, Zee PC, Turek FW. Chronic circadian desynchronization decreases the survival of animals with cardiomyopathic heart disease. Am J Physiol. (1998) 275:H2334–7. 275:H2334–7. doi: 10.1152/ajpheart.1998.275.6.H2334
111. Martino TA, Young ME. Influence of the cardiomyocyte circadian clock on cardiac physiology and pathophysiology. J Biol Rhythms. (2015) 30:183–205. 30:183–205. doi: 10.1177/0748730415575246
112. Martino TA, Oudit GY, Herzenberg AM, Tata N, Koletar MM, Kabir GM, et al. Circadian rhythm disorganization produces profound cardiovascular and renal disease in hamsters. Am J Physiol Regul Integr Comp Physiol. (2008) 294:R1675–83. doi: 10.1152/ajpregu.00829.2007
113. Martino TA, Tata N, Belsham DD, Chalmers J, Straume M, Lee P, et al. Disturbed diurnal rhythm alters gene expression and exacerbates cardiovascular disease with rescue by resynchronization. Hypertension. (2007) 49:1104–13. 49:1104–13. doi: 10.1161/HYPERTENSIONAHA.106.083568
114. Crnko S, Pré Du BC, Sluijter JPG, van Laake LW. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat Rev Cardiol. (2019) 16:437–47. 16:437–47. doi: 10.1038/s41569-019-0167-4
115. Muller JE, Stone PH, Turi ZG, Rutherford JD, Czeisler CA, Parker C, et al. Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med. (1985) 313:1315–22. 313:1315–22. doi: 10.1056/NEJM198511213132103
116. Muller JE, Ludmer PL, Willich SN, Tofler GH, Aylmer G, Klangos I, et al. Circadian variation in the frequency of sudden cardiac death. Circulation. (1987) 75:131–8. 75:131–8. doi: 10.1161/01.CIR.75.1.131
117. Suárez-Barrientos A, López-Romero P, Vivas D, Castro-Ferreira F, Núñez-Gil I, Franco E, et al. Circadian variations of infarct size in acute myocardial infarction. Heart. (2011) 97:970–6. doi: 10.1136/hrt.2010.212621
118. Alibhai FJ, Tsimakouridze EV, Chinnappareddy N, Wright DC, Billia F, O'Sullivan ML, et al. Short-term disruption of diurnal rhythms after murine myocardial infarction adversely affects long-term myocardial structure and function. Circ Res. (2014) 114:1713–22. 114:1713–22. doi: 10.1161/CIRCRESAHA.114.302995
119. Durgan DJ, Pulinilkunnil T, Villegas-Montoya C, Garvey ME, Frangogiannis NG, Michael LH, et al. Short communication: ischemia/reperfusion tolerance is time-of-day-dependent: mediation by the cardiomyocyte circadian clock. Circ Res. (2010) 106:546–50. 106:546–50. doi: 10.1161/CIRCRESAHA.109.209346
120. Montaigne D, Marechal X, Modine T, Coisne A, Mouton S, Fayad G, et al. Daytime variation of perioperative myocardial injury in cardiac surgery and its prevention by Rev-Erbα antagonism: a single-centre propensity-matched cohort study and a randomised study. Lancet. (2018) 391:59–69. 391:59–69. doi: 10.1016/S0140-6736(17)32132-3
121. Zhang L, Zhang R, Tien C-L, Chan RE, Sugi K, Fu C, et al. REV-ERBα ameliorates heart failure through transcription repression. JCI Insight. (2017) 2:e95177. 2:e95177. doi: 10.1172/jci.insight.95177
122. Stujanna EN, Murakoshi N, Tajiri K, Xu D, Kimura T, Qin R, et al. Rev-erb agonist improves adverse cardiac remodeling and survival in myocardial infarction through an anti-inflammatory mechanism. PLoS ONE. (2017) 12:e0189330. doi: 10.1371/journal.pone.0189330
123. Schloss MJ, Hilby M, Nitz K, Guillamat Prats R, Ferraro B, Leoni G, et al. Ly6Chigh monocytes oscillate in the heart during homeostasis and after myocardial infarction-brief report. Arterioscler Thromb Vasc Biol. (2017) 37:1640–5. doi: 10.1161/ATVBAHA.117.309259
Keywords: clock, inflammasome, NLRP3, Rev-erb, RORalpha, circadian immunity
Citation: Pourcet B and Duez H (2020) Circadian Control of Inflammasome Pathways: Implications for Circadian Medicine. Front. Immunol. 11:1630. doi: 10.3389/fimmu.2020.01630
Received: 14 April 2020; Accepted: 18 June 2020;
Published: 31 July 2020.
Edited by:
Christoph Scheiermann, Ludwig Maximilian University of Munich, GermanyReviewed by:
Je-Wook Yu, Yonsei University, South KoreaChiara Agostinis, IRCCS Materno Infantile Burlo Garofolo, Italy
Copyright © 2020 Pourcet and Duez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hélène Duez, aGVsZW5lLmR1ZXpAcGFzdGV1ci1saWxsZS5mcg==