 Naomi Richardson
Naomi Richardson Sky T. H. Ng
Sky T. H. Ng David C. Wraith
David C. Wraith
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 July 2020
Sec. Alloimmunity and Transplantation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01586
This article is part of the Research Topic New Insights on Liver Transplant and Tolerance View all 18 articles
The liver is a critical organ in controlling immune tolerance. In particular, it is now clear that targeting antigens for presentation by antigen presenting cells in the liver can induce immune tolerance to either autoantigens from the liver itself or tissues outside of the liver. Here we review immune mechanisms active within the liver that contribute both to the control of infectious diseases and tolerance to self-antigens. Despite its extraordinary capacity for tolerance induction, the liver remains a target organ for autoimmune diseases. In this review, we compare and contrast known autoimmune diseases of the liver. Currently patients tend to receive strong immunosuppressive treatments and, in many cases, these treatments are associated with deleterious side effects, including a significantly higher risk of infection and associated health complications. We propose that, in future, antigen-specific immunotherapies are adopted for treatment of liver autoimmune diseases in order to avoid such adverse effects. We describe various therapeutic approaches that either are in or close to the clinic, highlight their mechanism of action and assess their suitability for treatment of autoimmune liver diseases.
The liver is a complex immune-rich organ with a propensity toward tolerance, central to its role in blood filtration and toxin removal. This characteristic is most striking in cases of successful liver transplantation in which patients can be safely weaned off immunosuppression and in multi-organ transplants where transplanting liver alongside other organs including lung and heart prevents multi-organ rejection (1–4).
As the liver receives both arterial blood and blood from the gut via the portal vein, it is regularly exposed to both dietary and microbial antigens, which could establish excessive and prolonged inflammation, tissue damage and fibrosis if unregulated. Therefore, diverse populations of immune cells, stromal cells and hepatocytes work in synergy to resolve localized inflammation and avoid unnecessary immune responses to innocuous stimuli (5, 6). The liver microenvironment is well-adapted to maintain homeostasis due to its unique populations of antigen-presenting cells (APC) with tolerogenic characteristics, feedback mechanisms to control inflammation, high density of innate immune cells and richness of suppressive soluble mediators (summarized in Figure 1 and Table 1).
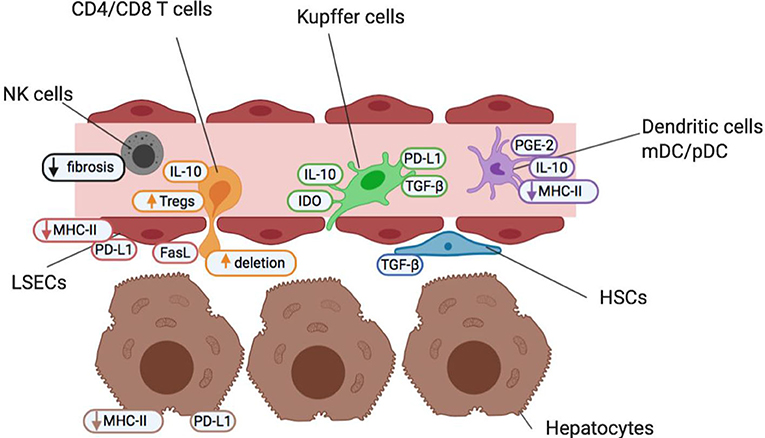
Figure 1. Cells of the liver sinusoid environment and their functions help maintain a state of homeostatic tolerance in the liver. Non-parenchymal resident liver cells including Kupffer cells (green), hepatic stellate cells (HSCs; blue), liver sinusoidal endothelial cells (LSECs; red) and dendritic cells (myeloid mDC and plasmacytoid pDC; purple) are situated within, or in close proximity to, liver sinusoids forming an early detection system to identify pathogens and maintain barrier function. They contribute to the maintenance of a high anti-inflammatory TGF-β and IL-10 cytokine milieu under steady-state conditions and in the face of common bacterial and food antigens to which the liver is continuously exposed. The liver also contains high numbers of innate-like immune cells such as NK cells (gray), and δT cells (not shown). NK cells act as pro-inflammatory agents, and promote the recruitment of effector immune cells, but are also key regulators of fibrosis. Both non-parenchymal antigen-presenting cells and hepatocytes (brown) offer a reduced antigen-presentation capacity and lower levels of costimulation than other antigen-presenting cells elsewhere in the body. This helps promote an environment of low T cell (orange) activation under normal conditions and maintain a state of “active” tolerance, whereby if required, inflammation and T cell activation is readily engaged.
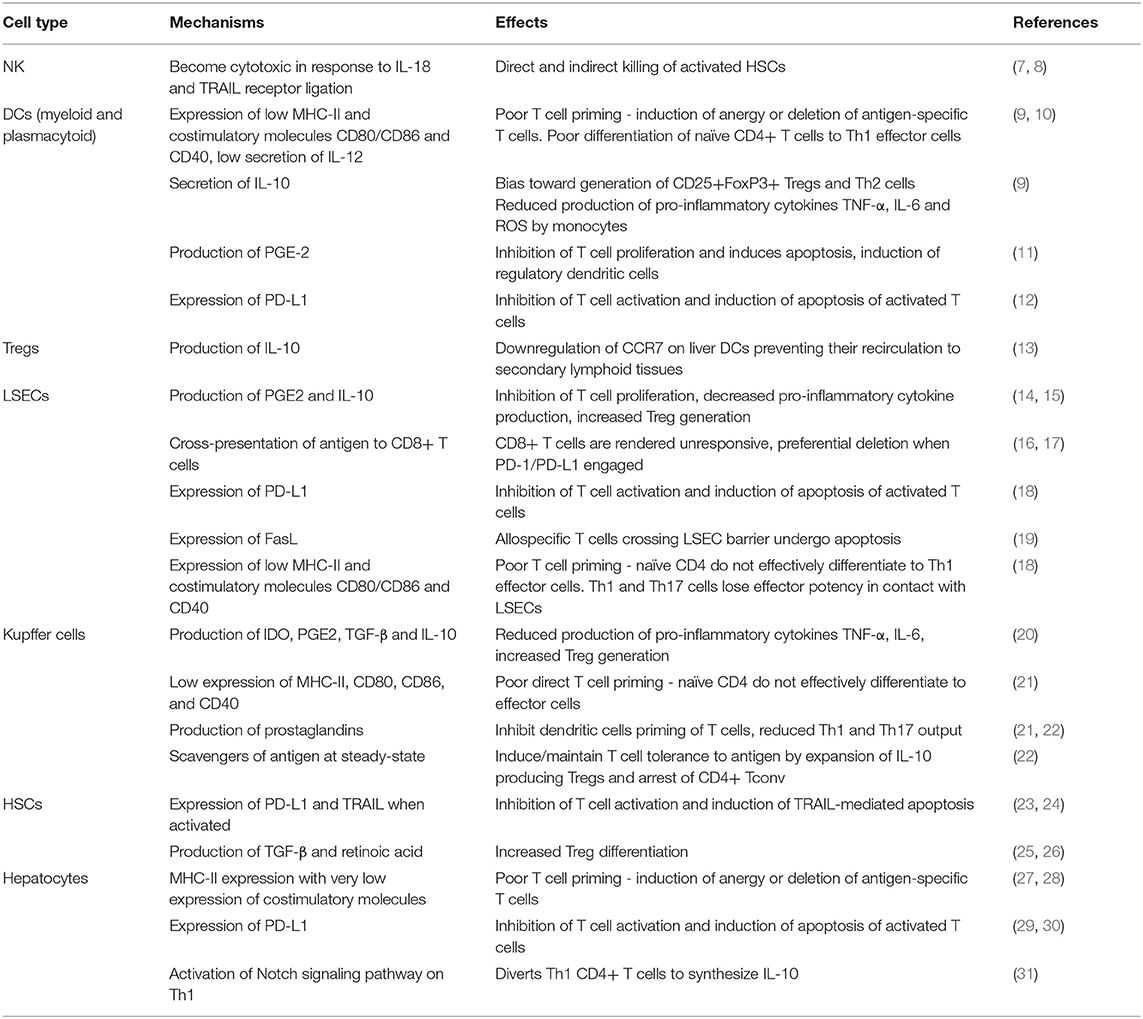
Table 1. Summary of tolerogenic functions exerted by non-parenchymal liver cells and hepatocytes and their physiological effects.
For example, cells of the hepatic sinusoids are continuously exposed to Gram-negative bacterial endotoxin e.g., lipopolysaccharide (LPS), which is detectable in portal vein blood but not systemic circulation (32). These cells when engaging with LPS via Toll-like receptor 4 (TLR4) are adapted to have an increased activating threshold to avoid hyper-active signaling and to better remove LPS from the blood stream (33).
The liver is enriched for innate immune cells which help trigger strong activating signals for inflammation in situations where tolerance is unsuitable, e.g., pathogen infection. Around 50% of liver resident lymphocytes are NK cells (Figure 1, gray), notably higher than in most tissues (34). Similarly, numbers of unconventional T cells, NK-T and γδT cells, are increased in the liver to recognize lipid antigens and bacterial pathogens, respectively (35, 36). Activated NK and NKT cells produce significant amounts of cytokines, including strongly inflammatory TNF-α and GM-CSF in response to viral and bacterial pathogens, to shift the balance from tolerance to inflammation. Activated liver NK cells produce IFN-γ and exert cytotoxicity due to TRAIL receptor binding and in response to IL-18 released by Kupffer cells (7, 8). Intriguingly, cytotoxic NKs also contribute to prevention of fibrosis by IFN-γ dependent arrest and apoptosis of hepatic stellate cells (HSCs) as well as directly killing activated HSC (37, 38). The role of γδT cells in the liver is currently less well-defined, but they are known to accumulate in both human fibrotic liver and experimental liver injury models and are producers of IL-17 (39, 40).
The liver is home to a wide range of APC with a tolerogenic bias, including liver sinusoidal endothelial cells (LSECs; Figure 1, red), resident myeloid and plasmacytoid dendritic cells (mDCs and pDCs; Figure 1, purple), Kupffer cells (KCs; Figure 1, green) and hepatic stellate cells (HSCs; Figure 1, blue). Antigen-presentation and costimulatory capacity of resting APC in the liver is generally low, contributing to the liver's state of active tolerance.
Mouse and human liver resident DCs are tolerogenic under steady-state conditions, as they display a more immature phenotype with significantly lower expression of MHC Class II and CD80/CD86 than DCs found elsewhere (9). When activated by TLR4 ligands, liver DCs produce substantial amounts of anti-inflammatory prostaglandin E2 (PGE2) (11) and IL-10 whereas blood DCs produce almost exclusively inflammatory cytokines. Therefore, liver DCs are less capable to provide sufficiently strong signals required to activate T cells. Instead, DC-T cell interactions generate more CD25+FoxP3+ Tregs and IL-4 producing Th2 cells by an IL-10 dependent mechanism (9). IL-10 also downregulates the expression of CCR7 on circulating DCs preventing their re-circulation to secondary lymphoid tissue (13).
LSECs express both MHC-I and MHC–II, and are as capable at antigen-uptake as DCs (41). They can, therefore, prime CD4+ T cells and cross-present antigen to CD8+ T cells, a function which is modulated by liver IL-10 (14). In both cases, the interaction between LSEC and T cell is biased toward tolerance. Naïve CD4 T cells primed by LSECs do not receive high costimulation, or an IL-12 stimulus from neighboring tolerogenic DCs and, therefore, do not effectively differentiate to Th1 effector cells (42–44). Th1 and Th17 cells when in contact with tolerogenic LSECs are unable to produce high levels of IFN-γ and IL-17, respectively (18). LSECs constitutive expression of PDL-1 when cross-presenting antigen to CD8+ T cells renders these T cells unresponsive and establishes a PDL-1 dependent antigen-specific T cell tolerance in the liver (16, 17). Futhermore, as T cells transmigrate across the LSEC barrier to enter the liver parenchyma, the LSECs are able to detect allospecificity and induce T cell death both directly and indirectly via the Fas/FasL pathway (19, 45).
KCs are liver-resident, immobile macrophages located within the sinusoidal lumen. They are hugely abundant, constituting 80% of the body's entire macrophage population and around 35% of non-parenchymal cells within the liver (5). KCs have been found to be essential mediators of homeostatic tolerance in the liver. KCs express significantly lower levels of MHC-II and costimulatory molecules compared to dendritic cells, meaning that they are incapable of sufficiently priming T cells on their own. Notably, they can block dendritic cell priming of antigen-specific T cells in a prostaglandin-dependent manner in vitro (21). Under steady-state conditions, KCs survey the sinusoids for dead cell debris, pathogens and particulates to phagocytose and this surveillance role can both establish tolerance or rapid response to pathogen depending on the physiological context. KCs phagocytose and present non-pathogen derived antigenic particulate matter and generate a skew in liver CD4+ T cells toward non-responsiveness (22). Heymann et al. shed light on the efficacy of KCs to induce tolerance by tracking OVA-loaded liposomes using intra-vital microscopy. KCs were the primary cell type within the liver to internalize labeled particulates and promoted the expansion of CD25+FoxP3+ OVA-specific Tregs in vivo. Both KC depletion and liver inflammation prevented tolerance induction (22).
Their essential sentinel role is further highlighted in mouse models lacking in KCs, where mice are fatally unable to clear a range of bacterial infections (46–48). When encountering pathogen, KCs rapidly release pro-inflammatory cytokines TNF-α, IL-6, and IL-1, promoting the recruitment of granulocytes and neutrophils to clear pathogens (46, 49). Following initial pro-inflammatory function, KCs then express the suppressive mediator IDO and release PGE-2, IL-10, and TGF-β to quench localized inflammation (21, 42, 50).
Targeting KCs to induce antigen-specific tolerance is a promising avenue when considering immunotherapeutic particle delivery for treatment of autoimmune diseases, but would require administration in contexts without liver inflammation. It may therefore not be the most appropriate method for addressing liver autoimmune diseases without prior immune suppressive treatment.
HSCs can also act as APCs and present antigens via MHC-I, MHC-II and CD1d (51). They are powerful producers of TGF-β and retinoic acid within the liver, helping to maintain a generalized immunosuppressive milieu at homeostasis and promoting Treg differentiation and residence within the liver (25, 26). However, when HSCs become activated in the presence of pathogens or strong inflammatory signals, they rapidly metabolize stored Vitamin A and differentiate into myofibroblasts, secreting extra-cellular matrix proteins. Therefore, HSC are key drivers of hepatic fibrosis and associated deterioratio to cirrhosis (52).
Hepatocytes themselves possess tolerogenic properties, as they are MHC-II expressing in the absence or very low expression of costimulatory molecules (27, 28). In mice, hepatocytes in inflammatory conditions can activate a Notch and IFN-γ dependent pathway to divert Th1 CD4+ T cells to synthesize IL-10 (31). PD-L1 is also inducible in hepatocytes by viral infection and by type 1 and type II interferons, mediating apoptosis of activated T cells (29).
At present, it is unclear exactly which of these tolerance-promoting mechanisms fail in the pathogenesis of autoimmune liver diseases, and at which time in disease progression. The consequence of these homeostatic mechanisms failing, however, can be devastating for liver function, impairing tissue regeneration and causing fibrosis. In the case of autoimmune liver diseases, immunological targeting of liver self-antigens catalyzes a system of inflammation and chronic liver disease. It will be important to understand which mechanisms break down in the process of developing autoimmune liver disease, in order to best intervene with tolerance promoting treatments.
Autoimmune liver disease (AILD) can be divided into 3 distinct clinical diseases, autoimmune hepatitis (AIH), primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC). They are distinguished by the molecular and cellular targets of immune pathology alongside the location of observed liver damage (Figure 2). Biliary dominant PBC and PSC affect cholangiocytes lining bile ducts. PBC destroys small, interlobular bile ducts while PSC targets larger bile ducts and is characterized by inflammatory fibrosis in the intrahepatic and extrahepatic biliary tree (53, 54). In AIH, the target is hepatocytes themselves, leading to interface hepatitis and significant lymphocyte infiltration primarily around the portal tracts (55). All 3 diseases will develop to severe liver fibrosis without medical intervention.
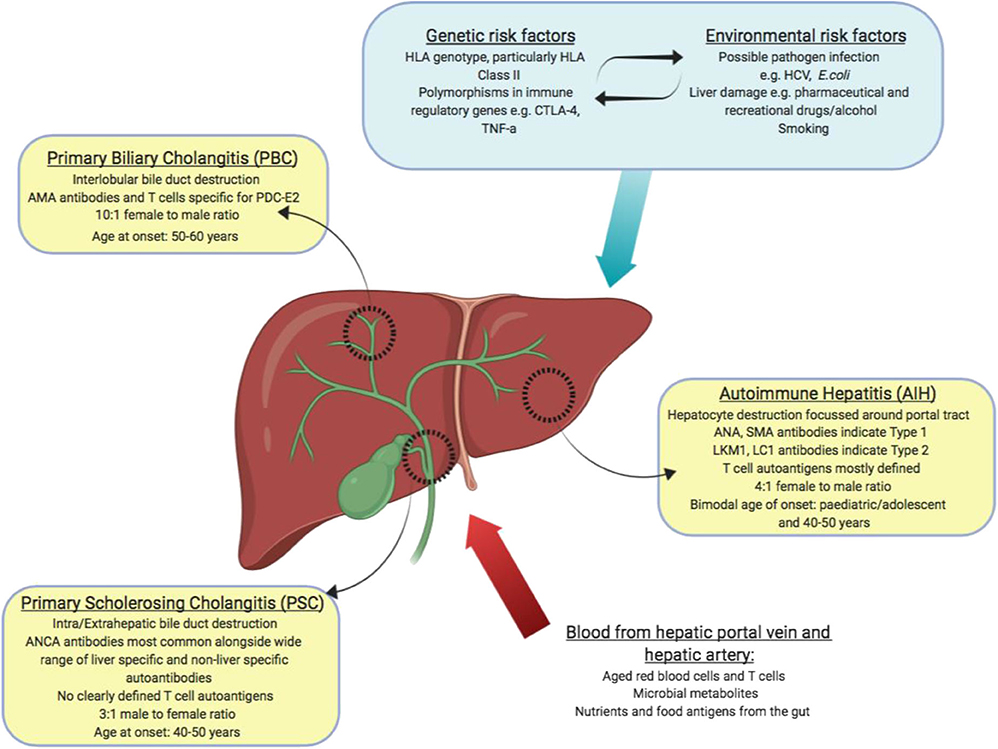
Figure 2. Summary of autoimmune liver diseases: tissues affected, and key features of disease (yellow boxes). Blood borne factors which challenge the maintenance of immune tolerance are listed as inputs (red arrow). Genetic, environmental and lifestyle factors which could affect the maintenance of tolerance are listed as inputs (blue box and arrow).
AIH is a chronic progressive liver-disease that mainly affects women (70–80% cases) and can be diagnosed in adults and children of any age or ethnicity (56). As symptoms and biochemical indicators are widely heterogenous between patients, the International Autoimmune Hepatitis Group (IAIHG) developed a scoring system based on specific criteria to improve early diagnosis (57, 58). Early diagnosis is imperative as cirrhosis is already present at diagnosis of a third of AIH patients and liver cirrhosis is the primary risk factor associated with development of hepatocellular carcinoma (56). AIH is a rare disease affecting between 16 and 20 cases per 100,000 (59–62) but appears to be increasing in prevalence. A long-term Danish study observed an almost 2-fold increase in the annual incidence rate of AIH between 1994 and 2012 (63).
PBC affects around 35/100,000 individuals, and is most common in women (9:1 female: male) and those over 50 years old (64). Reports have also indicated increasing prevalence of PBC over time (65). In around 10% of PBC patients, there will be overlap disease with features of AIH (66).
PSC is lowest in prevalence, and most commonly found in Northern Europe with 5.5–8.5 patients per 100,000 individuals in the UK, which has increased by about 50% since 1991 (67, 68). Unlike AIH and PBC, PSC is more common in men than in women (3:1) and although disease can occur at any age it has a peak incidence around 40 (69).
There is evidence for genetic factors playing a role in pathogenesis of all 3 AILD disease classes, with both major histocompatibility complex HLA genes and non-HLA genes showing disease associations (70). Exactly how HLA confers increased disease risk is unknown, but is presumed to be related to how antigens are presented and recognized by the immune system.
AIH-1 usually presenting in middle age has been linked to HLA-DRB1*0301 and HLA-DRB1*0401 with co-expression of these risk alleles indicating a double-dose effect (71–73). AIH-2 affects around 10% of AIH patients, exhibits a more aggressive phenotype and has been related to the presence of HLA-DRB1*07 and DRB1*03 in cohorts in the UK and Brazil (74). AIH-2 is most commonly diagnosed in childhood and has even been recorded in infants, suggesting a potentially different etiology to AIH1 (75). Around 20% of AIH patients suffer concomitant autoimmune diseases, most commonly thyroiditis (also HLA-DR3), inflammatory bowel disease (IBD), Type 1 diabetes (also HLA-DR4/HLA-DR3) and Addisons disease (HLA-DR4) (76).
PBC susceptibility is highly associated with HLA-DRB1*08 in Europe and North America (77). In contrast HLA-DRB1*11 and HLA-DRB1*13 were found to be protective toward PBC (78).
PSC is generally associated with HLA-DRB1*0301 in Norwegian and British patients (79). In patients with both PSC and inflammatory bowel disease, PSC is also associated with HLA-DRB1*13 but only with individuals with IBD (80).
GWAS studies have also identified association between specific polymorphisms within regulatory genes and AIH, PBC, PSC development. Notably for AIH and PBC but not for PSC, these include CTLA-4 and TNF-α genes (81–86) which are identified in similar studies of wide ranging autoimmune disorders (87, 88). TNF-α is located in the HLA-DR/DP locus; therefore, its appearance in GWAS studies of autoimmune diseases is unsurprising. However, at present it is unclear whether its influence is merely by association via linkage disequilibrium, or whether its function and downstream signaling actively contributes to the strong correlation of certain HLA haplotypes to autoimmunity. A further interesting correlation between TNF-α and CTLA-4 noted that single nucleotide polymorphism (SNP) rs1800629 of the TNF-α gene, leading to increased TNF-α production, amplified the CTLA4 SNP risk associated with rs231725, and that the combination of both SNPs was significantly more common in PBC patients compared to healthy controls (84). Studies in PBC have also identified common variant in IL-12 and IL-12R which indicate a role for aberrant IL-12 signaling in disease pathogenesis (89).
The specific triggers that lead to development of AILD are as yet poorly understood, due to the complex nature of genetic and environmental (drug and foreign pathogen) influences. It is thought that environmental stressors on a background of genetic predisposition in the form of HLA haplotypes and general tolerogenic “fitness” (Tregs and feedback loops) could help establish chronic autoimmune liver injury. AILD patients commonly present other autoimmune diseases, suggesting that immune dysregulation is not isolated to the liver in these cases.
Characteristic of all autoimmune diseases, AIH and PBC have autoantibodies present in patient's circulation. In both diseases, there are a some well-defined autoantibodies that are used to diagnose patients; however, the autoantigens that these antibodies are specific for is less well-defined. In contrast, PSC patients do not possess defined liver-specific autoantibodies. The strongest biomarker associated with PSC is elevated serum alkaline phosphatase levels, indicative of cholestasis (69, 90). PSC is usually diagnosed by MRI imaging of the biliary tree to identify cholestasis and/or strictures (69, 91, 92). Up to 80% of PSC patients also present with inflammatory bowel disease (IBD), indicating a general gastrointestinal inflammatory phenotype (93). Taken alongside the fact that PSC is more common in men and has less strong HLA associations, the lack of known autoantibodies calls into question whether the disease is strictly autoimmune, or whether it is autoinflammatory in nature (94).
Suspected AIH patients are scored according to International AIH Group published criteria to determine a diagnosis of AIH (57, 58). For clinical and research purposes, patients are grouped into AIH-1 or AIH-2 by the presence of different autoantibody profiles to liver antigens. The definitive clinical distinction between AIH subtypes is challenging, and age-matched patients usually follow similar trajectories and treatment protocols regardless of patient autoantibody profiles (95).
The vast majority (≈75%) of AIH-1 patients are positive for anti-nuclear antibodies (ANA) and/or anti-smooth muscle antibodies (SMA) (62, 63). However, these autoantibodies are not limited to AIH-1 patients and the autoantigens responsible are not well-defined (96, 97). ANA can react to histones, ribonucleoproteins ds-DNA and chromatin (98). SMA also have a range of specificities, predominantly to F-actin (99, 100). The remainder of patients who lack ANA or SMA antibodies, but present with liver disease pathology in accordance with the IAIHG diagnosis criteria, may possess other defined autoantibodies including anti-perinuclear neutrophil cytoplasmic antibodies (pANCA), anti-liver cytosol (LC-1), anti-soluble liver antigen/liver-pancreas (SLA/LP) and/or asialoglycoprotein receptor (ASGPR). Of note, SLA/LP is present in around 30% of AIH patients and has been identified in both adults and children (101–103). SLA/LP autoantibodies are specific to the autoantigen SLA/LP/tRNP(Ser)Sec (104, 105) and is therefore the only defined autoantigen implicated in AIH-1.
AIH-2 is rarely seen as a newly-diagnosed disease in adult cohorts but is reported to represent around 30% of pediatric AIH patients (106). AIH-2 has a less varied autoantigen profile and is diagnosed predominantly by the presence of anti-liver kidney microsomal antibody (LKM-1) and to a lesser extent anti-liver cytosol antibody (LC-1), specific to the liver proteins cytochrome P450 2D6 (CYP2D6) and formiminotransferase cyclodeaminase (FTCD), respectively (74, 107). Both T cell and B cell epitope mapping studies of CYP2D6 have been published, providing evidence that CYP2D6-reactive lymphocytes circulate in AIH-2 patients but not in healthy people (74, 108). Again, neither LKM-1 or LC-1 autoantibodies are restricted to AIH-2 – notably LKM-1 antibodies are detected in 5–10% of chronic HCV patients (101, 109) with an identified homologous sequence between HCV and CYP2D6 judged to be the cause (102).
The success of antigen-specific immunotherapies in re-establishing tolerance is reliant on having strong knowledge of the autoantigens underpinning immune pathology. Therefore, with our current understanding of AIH disease, it is likely that the most appropriate immediate targets for AIH-2 are CYP2D6, FTCD and for AIH-1 SLA/LP/tRNP(Ser)Sec. To be applicable to the majority of AIH-1 patients, however, detailed antigen profiling of AMA and SMA targets is required but has proved to be extremely challenging thus far.
PBC is diagnosed by the presence of highly-specific anti-mitochondrial antibodies (AMA) against the pyruvate dehydrogenase complex (PDCE2) (110–112). Over 90% of PBC patients are positive for AMA antibodies (113). PDCE2 is expressed at detectable levels on biliary epithelial cells in PBC but not in healthy individuals (114, 115). A minority of PBC patients are AMA negative, however, histological analyses of the bile ducts reveal no difference in pathology and presentation of PDCE2 between AMA positive and AMA negative PBC patients (114). Interestingly, PBC is also associated with prior urinary tract infections which are most frequently caused by E.coli (116–118). It is thought that E.coli induces B and T cell cross reactive responses to human PDCE2 by molecular mimicry (115).
In the case of AIH and PBC the presence of reliable autoantibodies to known autoantigens, and lymphocytes specific to these autoantigens found in patients provides vital evidence that supports targeting autoreactive cells in patients could have therapeutic benefit.
The clinical options to treat AILDs are limited once diagnosis is confirmed. The current front-line treatments center on broad immunosuppressive agents and ursodeoxycholic acid (UDCA) – a biliary protective drug of which the mechanism of action is still poorly understood.
In AIH, randomized controlled trials from the 1970's helped establish the mainstay treatment options of corticosteroids (PRED) and azathioprine (AZA) (119–121). Today, 50 years later, the treatment plan is almost identical to these early trials. This is sufficient to obtain biochemical disease remission and to prevent further liver damage in around 80% of AIH-1 patients (122). However, this level of immunosuppression commonly causes side effects including Cushingoid features, weight gain and gastrointestinal issues. For the vast majority of patients immunosuppressive therapy is lifelong, bringing a range of side effects, including osteoporeosis (especially problematic in middle aged women), diabetes mellitus, an increased risk of infections and risk of both hepatocellular and extra-hepatic cancers (123). Despite treatment, de novo cirrhosis occurs in around 14% of patients increasing the likelihood patients progress to transplant or hepatocellular carcinoma (124, 125). Adolescents often display poor treatment regime compliance, leading to the highest rate of relapse of any age group; therefore, an approach which causes fewer side effects, would be particularly welcome in this cohort (126). A recent trial using the corticosteroid budesonide with AZA indicated improved efficacy to PRED and a much improved adverse effect profile (127). So far, this is yet to be translated to a change in clinical treatment practices for AIH.
The primary course of treatment for PBC is UDCA (128). UDCA slows PBC disease progression by protecting cholangiocytes and hepatocytes from damage (129). UDCA significantly improves transplant free survival (130, 131); however, up to 40% of patients treated with UDCA have an insufficient response to treatment (132, 133), therefore in the long term, a liver transplant is often required. Even with a liver transplant, PBC recurs in around 30% of patients after 10 years (134–136). A recent development in approved PBC treatment is administration of obeticholic acid, particularly in patients refractory to or intolerant of UDCA. Obeticholic acid significantly improved liver function tested by alkaline phosphatase levels in patients with insufficient UDCA responses, with 69% of treated patients achieving a 20% reduction in ALP vs. only 8% of patients treated with UDCA alone (137, 138).
There are no effective treatments for PSC that have been proven to improve transplant free survival. There is no clear evidence that UDCA can treat PSC despite multiple clinical trials (139, 140). Trials applying other immunosuppressants to PSC, including prednisolone, budesonide, azathioprine, cyclosporin, methotrexate, mycophenolate, and tacrolimus have not shown efficacy (141). Drugs that antagonize the effects of anti-TNF-α such as pentoxifylline, etanercept and anti TNF-α monoclonal antibodies are also ineffective (141). Patients may undergo several of these pharmacological interventions in an attempt to quench biliary pathology, yet for most the only long-term option is liver transplantation. The mean time from diagnosis to liver transplantation/death is 9–12 years (90, 142). Unfortunately, PSC is expected to reoccur in 20–25% of patients over a 5–10 year period (136, 143, 144).
There is certainly an unmet need for improved treatment options with increased efficacy in hard to treat groups particularly pediatric AIH patients, refractory PBC patients and PSC patients. With the current understanding of the features of PSC, it is not clear that its pathogenesis is autoimmune, thus without the identification of autoantibodies and autoantigens relevant to PSC it will not be possible to generate antigen-specific immunotherapies for these patients. For AIH and PBC patients, however, there is sufficient evidence that antigen-specific immunotherapies could have real therapeutic value, and in contrast to systemic immunosuppressive drugs these should have a more specific mechanism of action that does not threaten the general health and immune capacity of the patient. The need for antigen-specific immunotherapies becomes ever more important as the world faces highly infectious agents such as the SARS-CoV-2 virus: such pathogens clearly endanger anyone taking immunosuppressive drugs.
Antigen-specific immunotherapy has been practiced in the field of allergy for more than 100 years (145, 146). Recently, there has been increasing interest in the development of antigen-specific approaches for specific immunotherapy of autoimmune conditions (schematic summary in Figure 3). This follows evidence that treatment of experimental animals with antigens can lead to amelioration of disease (146). Currently these approaches target CD4 T cell recognition of self-antigens. This is because CD4 T cells control the generation of all of the tissue damaging mechanisms associated with autoimmunity including pathogenic autoantibodies, antigen-driven inflammation and self-antigen specific CD8 T cells. It is not the focus of this review to discuss the mechanisms of action underpinning each approach aiming to induce antigen-specific tolerance; as these has been described comprehensively recently elsewhere (147, 148). We have briefly summarized within Table 2 the proposed mechanisms of action for each approach in development or in the clinic.
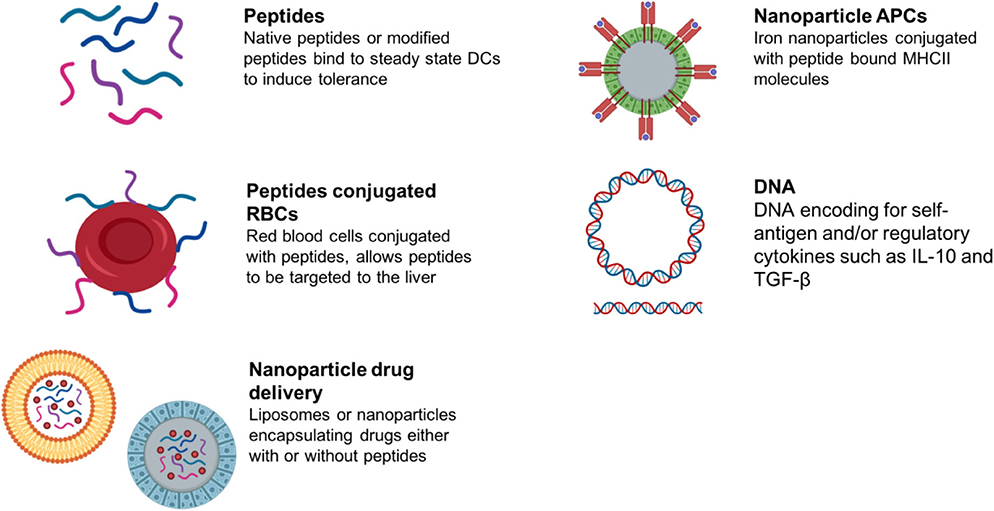
Figure 3. Summary of antigen-specific immunotherapy approaches in preclinical/clinical development.
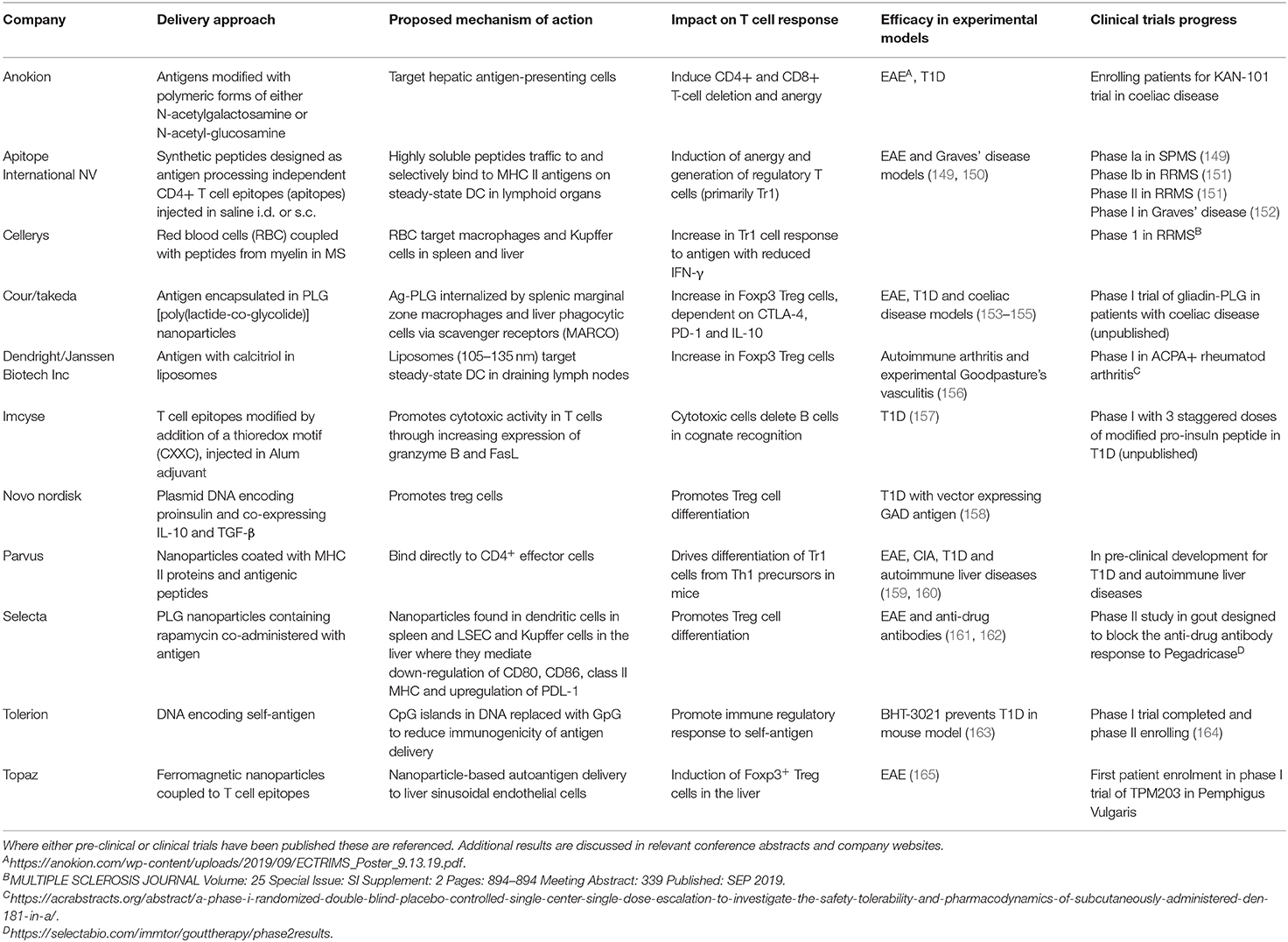
Table 2. This table summarizes the current status of pre-clinical and clinical developments of antigen-specific immunotherapies for autoimmune diseases.
Allergic desensitization involves administration of increasing and repeated doses of allergen, often a crude extract of the allergen material. Early attempts to treat autoimmune diseases in a similar way were not successful with intact antigen inducing pathogenic autoantibodies (166, 167) or driving tissue damaging cytotoxic T cells (168). To ensure safety and efficacy, autoantigens must be modified in such a way as to protect the recipient from exacerbation of the autoimmune response or they must be fragmented so as to avoid engagement with pathogenic autoimmune mechanisms. A preferred approach is to use short fragments of antigens (synthetic peptides) designed to modulate CD4 T cells but lacking either the structural integrity to engage pathogenic B cells or the peptide sequences to engage CD8 T cells.
It is important to appreciate that the mammalian adaptive immune system is poised to respond to foreign antigens but in the steady-state is adapted to limit autoimmune responses to the individual's own antigens. Responsibility for distinguishing between self and foreign antigens falls primarily on dendritic cells (169). In the steady-state, these cells are capable of binding the many fragments of self-antigens that are contained within the lymphoid pool (170). Steady-state dendritic cells presenting self-antigens are tolerogenic. It is only when these cells encounter foreign antigens in the context of microbial pattern-associated molecular patterns (e.g., LPS, bacterial DNA etc.) that they present antigen in an immunogenic rather than a tolerogenic fashion.
There are now a variety of clinical trials in progress that target steady-state/immature dendritic cells either in vivo or in vitro. The in vitro approach involves the generation of myeloid-derived dendritic cells treated with immunosuppressive agents, such as vitamin D3, to maintain a tolerogenic phenotype. The cells are then treated with peptides from self-antigens and reinjected into the patient (171–173). Alternatively, antigens can be coupled to dendritic cell targeting antibodies (e.g., anti—Dec205) for in vivo targeting (174). Our own work has focused on designing peptides that target steady-state dendritic cells directly. Early studies showed that some but not all known CD4 T cell epitopes induce tolerance when injected into experimental mice (175). Peptides must bind directly to MHC Class II and adopt the same conformation as the naturally processed epitope (176). Those peptides that do not mimic the naturally processed epitope fail to induce tolerance in relevant T cells. This implies that tolerogenic peptides bind directly to MHC Class II on or in steady state dendritic cells without further processing. Recent work from our laboratory has shown that such antigen-processing independent epitopes (apitopes) selectively bind to peptide receptive MHC class II molecules on steady-state dendritic cells but not to MHC Class II on the surface of B cells or monocytes. This is explained by the distinct, peptide-receptive nature of MHC Class II molecules on steady-state dendritic cells (177). Furthermore, tolerogenic peptides are detectable on steady state DCs up to 5 days after administration (178). We have shown that apitopes induce tolerance by induction of anergy in self-antigen reactive T cells and the expansion of antigen-specific Tr1 cells (179–182).
Alternative approaches for targeting “tolerogenic” APCs in vivo include combining antigen with liposomes, red blood cells or nanoparticles (Summarized in Table 2). These target different antigen-presenting cells in lymphoid organs or the liver depending on the size of the material or nanoparticle. This determines their modus operandi.
There is increasing evidence that nanoparticles of different sizes transit to and are taken up by different APCs according to their size. Berkland et al. have shown that particles > 200 nm are retained in the liver while those <4 nm are rapidly excreted (183). This evidence would pair well with evidence from Kupffer cell studies that these cells establish tolerance by phagocytosing particulate material and presenting antigenic fragments (21, 22). Such small particles rapidly drain from sites of injection into blood and lymph and particles of 4–10 nm penetrate lymph node cortex where they can interact with steady-state DCs. In contrast, particles > 100 nm are retained in the sub-capsular space where they will be processed by macrophages.
Santamaria et al. have developed artificial APCs (Navacims) based on nanoparticles coated with MHC Class II and antigenic peptide (159). The mechanism of action is in principle the same as apitope immunotherapy, both establish immunological tolerance by inducing IL-10 expressing CD4 T cells through a negative feedback mechanism (159, 160, 181, 184, 185). The resulting Tr1 cells are characterized by the expression of the immunosuppressive genes such as IL10 and co-inhibitory receptors (186, 187). The Tr1 cells induced by Navacims, however, also express inflammatory cytokines such as TNF-α, IL5, and GM-CSF (188). In contrast, Tr1 cells derived from apitope immunotherapy do not express TNF-α, IL5, or GM-CSF (182). Their recent studies serve as a valuable proof of concept, as antigenic peptides identified by in silico binding predictions from PDC-E2 loaded onto IAg7 MHC-nanoparticles are able to ameliorate PBC-like liver damage.
These Navacims, MHCII-based nanomedicines displaying epitopes from mitochondrial, endoplasmic reticulum, or cytoplasmic antigens associated with primary biliary cholangitis or autoimmune hepatitis can suppress disease progression in various murine models in an organ- rather than disease-specific manner (160). The improvement in liver score was shown to be IL-10 and TGF-β dependent. However, none of these liver disease models fully recapitulates the human condition. Furthermore, the T cell epitopes restricted by murine MHC class II molecules are unlikely to resemble those binding HLA-DR and DQ molecules i.e., relevant to human disease
However, Navacims do not work prophylactically to prevent disease onset, this is in contrast to apitope immunotherapy which is effective before as well as after disease onset (160, 179, 182). The bystander suppression demonstrated by loading the artificial APCs with PDC-E2 peptides and supressing the response against the CYP2D6 antigen and vice versa is intriguing and suggests that bystander suppression can influence different autoimmune conditions within the same tissue (189).
At this stage, it is too early to compare the safety and efficacy of the various approaches shown in Table 2. It is likely that different approaches will prove more or less effective for control of different immune pathologies and diseases. It is of paramount importance, however, to apply three tests to these approaches.
1. What is the mechanism of action? It will be critical to fully understand the mechanism by which these approaches induce antigen-specific tolerance both in experimental models and in patients.
2. Which approaches induce bystander suppression? For diseases like Graves' disease, we know precisely what the target antigen is. However, for most autoimmune diseases we do not know which antigen is targeted by the immune system to initiate the disease. For many others, antibodies specific for self-antigens are associated with disease but may or may not have a role in immune pathology. Furthermore, in most autoimmune conditions, epitope spreading leads to the generation of an immune response to a range of antigens within the same tissue (190). In order to account for epitope spreading, we and others have shown that certain immune regulatory mechanisms, such as Tr1 cells, mediate bystander suppression (191). By targeting antigen A within a tissue and eliciting immunosuppressive regulatory T cells, we can control the immune response to antigens B, C, D etc. within the same tissue.
3. Which approach permits repeated antigen administration? Apitope has now conducted clinical trials in multiple sclerosis and Graves' disease. In both cases, protection from immune pathology was observed but the patients treated did not enter a permanent state of tolerance (149, 151). Protection was seen for up to 1 month after the last dose of peptide which correlates well with the duration of tolerance observed in euthymic mice (192). It may well be that humans have evolved to require continued exposure to antigens in order to maintain tolerance. For this reason, it is likely that repeated administration of the different tolerogenic materials described in Table 2 will be required. A successful therapeutic approach must avoid induction of anti-drug antibodies or non-specific immune suppression.
There is already substantial progress in the quest for specific immunotherapies for autoimmune liver diseases. With this in mind, our laboratory is designing putative disease-altering apitopes from the dominant human autoantigens associated with PBC and type 2 AIH.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the University of Birmingham and grants from the Children's Liver Disease Foundation (NR) and by the Wellcome Trust and the Leona M and Harry B Helmsley Charitable Trust (SN).
DW is Professor of Immunology at the University of Birmingham and CSO and Founder of Apitope International NV.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Benítez C, Londoño MC, Miquel R, Manzia TM, Abraldes JG, Lozano JJ, et al. Prospective multicenter clinical trial of immunosuppressive drug withdrawal in stable adult liver transplant recipients. Hepatology. (2013) 58:1824–35. doi: 10.1002/hep.26426
2. Takatsuki M, Uemoto S, Inomata Y, Egawa H, Kiuchi T, Fujita S, et al. Weaning of immunosuppression in living donor liver transplant recipients. Transplantation. (2001) 72:449–54. doi: 10.1097/00007890-200108150-00016
3. Tisone G, Orlando G, Cardillo A, Palmieri G, Manzia TM, Baiocchi L, et al. Complete weaning off immunosuppression in HCV liver transplant recipients is feasible and favourably impacts on the progression of disease recurrence. J Hepatol. (2006) 44:702–9. doi: 10.1016/j.jhep.2005.11.047
4. Benseler V, McCaughan GW, Schlitt HJ, Bishop GA, Bowen DG, Bertolino P. The liver: a special case in transplantation tolerance. Semin Liver Dis. (2007) 27:194–213. doi: 10.1055/s-2007-979471
5. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. (2013) 14:996–1006. doi: 10.1038/ni.2691
6. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. (2009) 27:147–63. doi: 10.1146/annurev.immunol.021908.132629
7. Taimr P, Higuchi H, Kocova E, Rippe RA, Friedman S, Gores GJ. Activated stellate cells express the TRAIL receptor-2/death receptor-5 and undergo TRAIL-mediated apoptosis. Hepatology. (2003) 37:87–95. doi: 10.1053/jhep.2003.50002
8. Ochi M, Ohdan H, Mitsuta H, Onoe T, Tokita D, Hara H, et al. Liver NK cells expressing TRAIL are toxic against self hepatocytes in mice. Hepatology. (2004) 39:1321–31. doi: 10.1002/hep.20204
9. Bamboat ZM, Stableford JA, Plitas G, Burt BM, Nguyen HM, Welles AP, et al. Human liver dendritic cells promote T cell hyporesponsiveness. J Immunol. (2009) 182:1901–11. doi: 10.4049/jimmunol.0803404
10. Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, et al. Plasmacytoid dendritic cells mediate oral tolerance. Immunity. (2008) 29:464–75. doi: 10.1016/j.immuni.2008.06.017
11. Xia S, Guo Z, Xu X, Yi H, Wang Q, Cao X. Hepatic microenvironment programs hematopoietic progenitor differentiation into regulatory dendritic cells, maintaining liver tolerance. Blood. (2008) 112:3175–85. doi: 10.1182/blood-2008-05-159921
12. Castellaneta A, Sumpter TL, Chen L, Tokita D, Thomson AW. NOD2 ligation subverts IFN-α production by liver plasmacytoid dendritic cells and inhibits their T cell allostimulatory activity via B7-H1 up-regulation. J Immunol. (2009) 183:6922–32. doi: 10.4049/jimmunol.0900582
13. Takayama T, Morelli AE, Onai N, Hirao M, Matsushima K, Tahara H, et al. Mammalian and viral IL-10 enhance C-C chemokine receptor 5 but down-regulate C-C chemokine receptor 7 expression by myeloid dendritic cells: impact on chemotactic responses and in vivo homing ability. J Immunol. (2001) 166:7136–43. doi: 10.4049/jimmunol.166.12.7136
14. Knolle PA, Uhrig A, Hegenbarth S, Löser E, Schmitt E, Gerken G, et al. IL-10 down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells through decreased antigen uptake via the mannose receptor and lowered surface expression of accessory molecules. Clin Exp Immunol. (1998) 114:427–33. doi: 10.1046/j.1365-2249.1998.00713.x
15. Carambia A, Freund B, Schwinge D, Heine M, Laschtowitz A, Huber S, et al. TGF-b-dependent induction of CD4+ CD25+ Foxp3+ Tregs by liver sinusoidal endothelial cells. J Hepatol. (2014) 61:594–9. doi: 10.1016/j.jhep.2014.04.027
16. Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med. (2000) 6:1348–54. doi: 10.1038/82161
17. Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology. (2008) 47:296–305. doi: 10.1002/hep.21965
18. Carambia A, Frenzel C, Bruns OT, Schwinge D, Reimer R, Hohenberg H, et al. Inhibition of inflammatory CD4 T cell activity by murine liver sinusoidal endothelial cells. J Hepatol. (2013) 58:112–8. doi: 10.1016/j.jhep.2012.09.008
19. Tokita D, Shishida M, Ohdan H, Onoe T, Hara H, Tanaka Y, et al. Liver sinusoidal endothelial cells that endocytose allogeneic cells suppress T cells with indirect allospecificity. J Immunol. (2006) 177:3615–24. doi: 10.4049/jimmunol.177.6.3615
20. Kuniyasu Y, Marfani SM, Bin Inayat I, Sheikh SZ, Mehal WZ. Kupffer cells required for high affinity peptide-induced deletion, not retention, of activated CD8+ T cells by mouse liver. Hepatology. (2004) 39:1017–27. doi: 10.1002/hep.20153
21. You Q, Cheng L, Kedl RM, Ju C. Mechanism of T cell tolerance induction by murine hepatic kupffer cells. Hepatology. (2008) 48:978–90. doi: 10.1002/hep.22395
22. Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by kupffer cells. Hepatology. (2015) 62:279–91. doi: 10.1002/hep.27793
23. Yu MC, Chen CH, Liang X, Wang L, Gandhi CR, Fung JJ, et al. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology. (2004) 96:17–24. doi: 10.1002/hep.20488
24. Yang HR, Hsieh CC, Wang L, Fung JJ, Lu L, Qian S. A critical role of trail expressed on cotransplanted hepatic stellate cells in prevention of islet allograft rejection. Microsurgery. (2010) 30:332–7. doi: 10.1002/micr.20697
25. Ichikawa S, Mucida D, Tyznik AJ, Kronenberg M, Cheroutre H. Hepatic stellate cells function as regulatory bystanders. J Immunol. (2011) 186:5549–55. doi: 10.4049/jimmunol.1003917
26. Dunham RM, Thapa M, Velazquez VM, Elrod EJ, Denning TL, Pulendran B, et al. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J Immunol. (2013) 190:2009–16. doi: 10.4049/jimmunol.1201937
27. Herkel J, Jagemann B, Wiegard C, Garcia Lazaro JF, Lueth S, Kanzler S, et al. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocytes. Hepatology. (2003) 37:1079–85. doi: 10.1053/jhep.2003.50191
28. Franco A, Barnaba V, Natali P, Balsano C, Musca A, Balsano F. Expression of class I and class II major histocompatibility complex antigens on human hepatocytes. Hepatology. (1988) 8:449–54. doi: 10.1002/hep.1840080302
29. Mühlbauer M, Fleck M, Schütz C, Weiss T, Froh M, Blank C, et al. PD-L1 is induced in hepatocytes by viral infection and by interferon-α and -γ and mediates T cell apoptosis. J Hepatol. (2006) 45:520–8. doi: 10.1055/s-2006-931751
30. Wahl C, Bochtler P, Chen L, Schirmbeck R, Reimann J. B7-H1 on hepatocytes facilitates priming of specific CD8 T cells but limits the specific recall of primed responses. Gastroenterology. (2008) 135:980–8. doi: 10.1053/j.gastro.2008.05.076
31. Burghardt S, Erhardt A, Claass B, Huber S, Adler G, Jacobs T, et al. Hepatocytes contribute to immune regulation in the liver by activation of the notch signaling pathway in T cells. J Immunol. (2013) 191:5574–82. doi: 10.4049/jimmunol.1300826
32. Lumsden AB, Henderson JM, Kutner MH. Endotoxin levels measured by a chromogenic assay in portal, hepatic and peripheral venous blood in patients with cirrhosis. Hepatology. (1988) 8:232–6. doi: 10.1002/hep.1840080207
33. Catalá M, Antón A, Portolés MT. Characterization of the simultaneous binding of Escherichia coli endotoxin to kupffer and endothelial liver cells by flow cytometry. Cytometry. (1999) 36:123–130. doi: 10.1002/(SICI)1097-032036:2<123::AID–CYTO6>3.0.CO;2-Y
34. Tu Z, Bozorgzadeh A, Crispe IN, Orloff MS. The activation state of human intrahepatic lymphocytes. Clin Exp Immunol. (2007) 149:186–93. doi: 10.1111/j.1365-2249.2007.03415.x
35. Doherty DG, O'Farrelly C. Innate and adaptive lymphoid cells in the human liver. Immunol Rev. (2000) 174:5–20. doi: 10.1034/j.1600-0528.2002.017416.x
36. Gao B, Jeong W II, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. (2008) 47:729–36. doi: 10.1002/hep.22034
37. Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. (2006) 130:435–52. doi: 10.1053/j.gastro.2005.10.055
38. Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, et al. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. (2006) 45:60–71. doi: 10.1016/j.jhep.2005.12.025
39. Martins EB, Graham AK, Chapman RW, Fleming KA. Elevation of γδT lymphocytes in peripheral blood and livers of patients with primary sclerosing cholangitis and other autoimmune liver diseases. Hepatology. (1996) 23:988–93. doi: 10.1053/jhep.1996.v23.pm0008621180
40. Hammerich L, Tacke F. Role of gamma-delta T cells in liver inflammation and fibrosis. World J Gastrointest Pathophysiol. (2014) 5:107–13. doi: 10.4291/wjgp.v5.i2.107
41. Schurich A, Berg M, Stabenow D, Böttcher J, Kern M, Schild H-J, et al. Dynamic regulation of CD8 T cell tolerance induction by liver sinusoidal endothelial cells. J Immunol. (2010) 184:4107–14. doi: 10.4049/jimmunol.0902580
42. Knoll P, Schlaak J, Uhrig A, Kempf P, zum Büschenfelde KHM, Gerken G. Human kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol. (1995) 22:226–9. doi: 10.1016/0168-8278(95)80433-1
43. Kruse N, Neumann K, Schrage A, Derkow K, Schott E, Erben U, et al. Priming of CD4+ T cells by liver sinusoidal endothelial cells induces CD25low forkhead box protein 3- regulatory T cells suppressing autoimmune hepatitis. Hepatology. (2009) 50:1904–13. doi: 10.1002/hep.23191
44. Schildberg FA, Hegenbarth SI, Schumak B, Limmer A, Knolle PA. Liver sinusoidal endothelial cells veto CD8 T cell activation by antigen-presenting dendritic cells. Eur J Immunol. (2008) 38:957–67. doi: 10.1002/eji.200738060
45. Onoe T, Ohdan H, Tokita D, Shishida M, Tanaka Y, Hara H, et al. Liver sinusoidal endothelial cells tolerize T cells across MHC barriers in mice. J Immunol. (2005) 175:139–46. doi: 10.4049/jimmunol.175.1.139
46. Ebe Y, Hasegawa G, Takatsuka H, Umezu H, Mitsuyama M, Arakawa M, et al. The role of kupffer cells and regulation of neutrophil migration into the liver by macrophage inflammatory protein-2 in primary listeriosis in mice. Pathol Int. (1999) 49:519–32. doi: 10.1046/j.1440-1827.1999.00910.x
47. Lee WY, Moriarty TJ, Wong CHY, Zhou H, Strieter RM, Van Rooijen N, et al. An intravascular immune response to borrelia burgdorferi involves kupffer cells and iNKT cells. Nat Immunol. (2010) 11:295–302. doi: 10.1038/ni.1855
48. Helmy KY, Katschke KJ, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. (2006) 124:915–27. doi: 10.1016/j.cell.2005.12.039
49. Chensue SW, Terebuh PD, Remick DG, Scales WE, Kunkel SL. In vivo biologic and immunohistochemical analysis of interleukin-1 alpha, beta and tumor necrosis factor during experimental endotoxemia. Kinetics, kuppfer cell expression, and glucocorticoid effects. Am J Pathol. (1991) 138:395–402.
50. Perez R V, Swanson C, Morgan M, Erickson K, Hubbard NE, German JB. Portal venous transfusion up-regulates kupffer cell cyclooxygenase activity: a mechanism of immunosuppression in organ transplantation. Transplantation. (1997) 64:135–9. doi: 10.1097/00007890-199707150-00023
51. Winau F, Quack C, Darmoise A, Kaufmann SH. Starring stellate cells in liver immunology. Curr Opin Immunol. (2008) 20:68–74. doi: 10.1016/j.coi.2007.10.006
52. Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: Concept to treatment. J Hepatol. (2015) 62(Suppl. 1):S15–24. doi: 10.1016/j.jhep.2015.02.039
53. Talwalkar JA, Lindor KD. Primary biliary cirrhosis. Lancet. (2015) 386:10003. doi: 10.1016/S0140-6736(15)00154-3
54. Karlsen TH, Boberg KM. Update on primary sclerosing cholangitis. J Hepatol. (2013) 59:571–82. doi: 10.1016/B978-1-4377-0881-3.00042-5
55. Geller SA. Autoimmune hepatitis: histopathology. Clin Liver Dis. (2014) 3:19–23. doi: 10.1002/cld.301
56. European Association for the Study of the Liver. EASL clinical practice guidelines: autoimmune hepatitis. J Hepatol. (2015) 63:971–1004. doi: 10.1016/j.jhep.2015.06.030
57. Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, et al. International autoimmune hepatitis group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. (1999) 31:929–38. doi: 10.1016/S0168-8278(99)80297-9
58. Hennes EM, Zeniya M, Czaja AJ, Parés A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology. (2008) 48:169–76. doi: 10.1002/hep.22322
59. Boberg KM, Aadland E, Jahnsen J, Raknerud N, Stiris M, Bell H. Incidence and prevalence of primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis in a norwegian population. Scand J Gastroenterol. (1998) 33:99–103. doi: 10.1080/00365529850166284
60. Ngu JH, Bechly K, Chapman BA, Burt MJ, Barclay ML, Gearry RB, et al. Population-based epidemiology study of autoimmune hepatitis: A disease of older women? J Gastroenterol Hepatol. (2010) 25:1681–6. doi: 10.1111/j.1440-1746.2010.06384.x
61. Van Gerven NMF, Verwer BJ, Witte BI, Van Erpecum KJ, Van Buuren HR, Maijers I, et al. Epidemiology and clinical characteristics of autoimmune hepatitis in the Netherlands. Scand J Gastroenterol. (2014) 49:1245–54. doi: 10.3109/00365521.2014.946083
62. Werner M, Prytz H, Ohlsson B, Almer S, Björnsson E, Bergquist A, et al. Epidemiology and the initial presentation of autoimmune hepatitis in Sweden: a nationwide study. Scand J Gastroenterol. (2008) 43:1232–40. doi: 10.1080/00365520802130183
63. Grønbæk L, Vilstrup H, Jepsen P. Autoimmune hepatitis in Denmark: incidence, prevalence, prognosis, and causes of death. A nationwide registry-based cohort study. J Hepatol. (2014) 60:612–17. doi: 10.1016/j.jhep.2013.10.020
64. Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol. (2012) 56:1181–8. doi: 10.1016/j.jhep.2011.10.025
65. Boonstra K, Kunst AE, Stadhouders PH, Tuynman HA, Poen AC, van Nieuwkerk KMJ, et al. Rising incidence and prevalence of primary biliary cirrhosis: a large population-based study. Liver Int. (2014) 34:31–38. doi: 10.1111/liv.12434
66. Czaja AJ. The overlap syndromes of autoimmune hepatitis. Dig Dis Sci. (2013) 58:326–43. doi: 10.1007/s10620-012-2367-1
67. Card TR, Solaymani-Dodaran M, West J. Incidence and mortality of primary sclerosing cholangitis in the UK: a POPULATION-BASED COHORT STUDY. J HEPATOL. (2008) 48:939–44. doi: 10.1016/J.JHEP.2008.02.017
68. Liang H, Manne S, Shick J, Lissoos T, Dolin P. Incidence, prevalence, and natural history of primary sclerosing cholangitis in the United Kingdom. Med. (2017) 96:e7116. doi: 10.1097/MD.0000000000007116
69. Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet. (2013) 382:1587–99. doi: 10.1016/S0140-6736(13)60096-3
70. Mells GF, Kaser A, Karlsen TH. Novel insights into autoimmune liver diseases provided by genome-wide association studies. J Autoimmun. (2013) 46:41–54. doi: 10.1016/j.jaut.2013.07.004
71. Donaldson P. Susceptibility to autoimmune chronic active hepatitis: human leukocyte antigens DR4 and A1-B8-DR3 are independent risk factors. Hepatology. (1991) 13:701–6. doi: 10.1002/hep.1840130415
72. Czaja AJ, Carpenter HA, Santrach PJ, Moore BS. Significance of HLA DR4 in type 1 autoimmune. Gastroenterologylogy. (1993) 105:1502–7. doi: 10.1016/0016-5085(93)90157-8
73. Montano-Loza AJ, Carpenter HA, Czaja AJ. Clinical significance of HLA DRB1 *03-DRB1 * 04 in type 1 autoimmune hepatitis. Liver Int. (2006) 26:1201–8. doi: 10.1111/j.1478-3231.2006.01387.x
74. Ma Y, Bogdanos DP, Hussain MJ, Underhill J, Bansal S, Longhi MS, et al. Polyclonal T-cell responses to cytochrome P450IID6 are associated with disease activity in autoimmune hepatitis type 2. Gastroenterology. (2006) 130:868–82. doi: 10.1053/j.gastro.2005.12.020
75. Gregorio G V, Portmann B, Reid F, Donaldson PT, Doherty DG, McCartney M, et al. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology. (1997) 25:541–7. doi: 10.1002/hep.510250308
76. Teufel A, Weinmann A, Kahaly GJ, Centner C, Piendl A, Wörns M, et al. Concurrent autoimmune diseases in patients with autoimmune hepatitis. J Clin Gastroenterol. (2010) 44:208–13. doi: 10.1097/MCG.0b013e3181c74e0d
77. Manns MP, Bremm A, Schneider PM, Notghi A, Gerken G, Prager-Eberle M, et al. HLA DRw8 and complement C4 deficiency as risk factors in primary biliary cirrhosis. Gastroenterology. (1991) 101:1367–73. doi: 10.1016/0016-5085(91)90090-8
78. Invernizzi P, Selmi C, Poli F, Frison S, Floreani A, Alvaro D, et al. Human leukocyte antigen polymorphisms in Italian primary biliary cirrhosis: a multicenter study of 664 patients and 1992 healthy controls. Hepatology. (2008) 48:1906–12. doi: 10.1002/hep.22567
79. Spurkland A, Saarinen S, Boberg KM, Mitchell S, Broome U, Caballeria L, et al. HLA class II haplotypes in primary sclerosing cholangitis patients from five European populations. Tissue Antigens. (1999) 53:459–69. doi: 10.1034/j.1399-0039.1999.530502.x
80. Donaldson PT, Norris S. Immunogenetics in PSC. Best Pract Res Clin Gastroenterol. (2001) 15:611–27. doi: 10.1053/bega.2001.0208
81. Czaja A, Donaldson P. Genetic susceptibilities for immune expression and liver cell injury in autoimmune hepatitis. Immunol Rev. (2000) 174:250–9. doi: 10.1034/j.1600-0528.2002.017401.x
82. Chaouali M, Carvalho A, Tezeghdenti A, Ben Azaiez M, Cunha C, Ghazouani E, et al. Cytotoxic T lymphocyte antigen-4 gene polymorphisms and susceptibility to type 1 autoimmune hepatitis in the tunisian population. Genes Dis. (2018) 5:256–62. doi: 10.1016/j.gendis.2017.12.006
83. Walker EJ, Hirschfield GM, Xu C, Lu Y, Liu X, Lu Y, et al. CTLA4/ICOS gene variants and haplotypes are associated with rheumatoid arthritis and primary biliary cirrhosis in the Canadian population. Arthritis Rheum. (2009) 60:931–7. doi: 10.1002/art.24412
84. Juran BD, Atkinson EJ, Larson JJ, Schlicht EM, Liu X, Heathcote EJ, et al. Carriage of a tumor necrosis factor polymorphism amplifies the cytotoxic T-lymphocyte antigen 4 attributed risk of primary biliary cirrhosis: evidence for a gene-gene interaction. Hepatology. (2010) 52:223–9. doi: 10.1002/hep.23667
85. Ji SG, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet. (2017) 49:269–73. doi: 10.1038/ng.3745
86. Wiencke K, Boberg KM, Donaldson P, Harbo H, Ling V, Schrumpf E, et al. No major effect of the CD28/CTLA4/ICOS gene region on susceptibility to primary sclerosing cholangitis. Scand J Gastroenterol. (2006) 41:586–91. doi: 10.1080/00365520500377870
87. Kristiansen OP, Larsen ZM, Pociot F. CTLA-4 in autoimmune diseases - A general susceptibility gene to autoimmunity? Genes Immun. (2000) 1:170–84. doi: 10.1038/sj.gene.6363655
88. Zhernakova A, Withoff S, Wijmenga C. Clinical implications of shared genetics and pathogenesis in autoimmune diseases. Nat Rev Endocrinol. (2013) 9:646–59. doi: 10.1038/nrendo.2013.161
89. Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med. (2009) 360:2544–55. doi: 10.1056/NEJMoa0810440
90. Broomé U, Olsson R, Lööf L, Bodemar G, Hultcrantz R, Danielsson Å, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. (1996) 38:610–15. doi: 10.1136/gut.38.4.610
91. Lunder AK, Hov JR, Borthne A, Gleditsch J, Johannesen G, Tveit K, et al. Prevalence of sclerosing cholangitis detected by magnetic resonance cholangiography in patients with long-term inflammatory bowel disease. Gastroenterology. (2016) 151:660–9.e4. doi: 10.1053/j.gastro.2016.06.021
92. Schramm C, Eaton J, Ringe KI, Venkatesh S, Yamamura J. Recommendations on the use of magnetic resonance imaging in PSC-A position statement from the international PSC study group. Hepatology. (2017) 66:1675–88. doi: 10.1002/hep.29293
93. Kumagai J, Taida T, Ogasawara S, Nakagawa T, Iino Y, Shingyoji A, et al. Clinical characteristics and outcomes of primary sclerosing cholangitis and ulcerative colitis in Japanese patients. PLoS ONE. (2018) 13:e0209352. doi: 10.1371/journal.pone.0209352
94. Hov JR, Boberg KM, Karlsen TH. Autoantibodies in primary sclerosing cholangitis. World J Gastroenterol. (2008) 14:3781–91. doi: 10.3748/wjg.14.3781
95. Muratori P, Lalanne C, Fabbri A, Cassani F, Lenzi M, Muratori L. Type 1 and type 2 autoimmune hepatitis in adults share the same clinical phenotype. Aliment Pharmacol Ther. (2015) 41:1281–7. doi: 10.1111/apt.13210
96. von Mühlen CA, Tan EM. Autoantibodies in the diagnosis of systemicrheumatic diseases. Semin Arthritis Rheum. (1995) 24:323–58. doi: 10.1016/S0049-0172(95)80004-2
97. McMillan SA, Haire M. The specificity of IgG- and IgM-class smooth muscle antibody in the sera of patients with multiple sclerosis and active chronic hepatitis. Clin Immunol Immunopathol. (1979) 14:256–63. doi: 10.1016/0090-1229(79)90148-X
98. Verstegen G, Duyck MC, Meeus P, Ravelingien I, De Vlam K. Detection and identification of antinuclear antibodies (ANA) in a large community hospital. Acta Clin Belg. (2009) 64:317–23. doi: 10.1179/acb.2009.049
99. Czaja AJ, Cassani F, Cataleta M, Valentini P, Bianchi FB. Frequency and significance of antibodies to actin in type 1 autoimmune hepatitis. Hepatology. (1996) 24:1068–73. doi: 10.1002/hep.510240515
100. Lidman K, Biberfeld G, Fagraeus A, Norberg R, Torstensson R, Utter G, et al. Anti-actin specificity of human smooth muscle antibodies in chronic active hepatitis. Clin Exp Immunol. (1976) 24:266–72.
101. Dalekos GN, Wedemeyer H, Obermayer-Straub P, Kayser A, Barut A, Frank H, et al. Epitope mapping of cytochrome P4502D6 autoantigen in patients with chronic hepatitis C during α-interferon treatment. J Hepatol. (1999) 30:366–75. doi: 10.1016/S0168-8278(99)80092-0
102. Kerkar N, Choudhuri K, Ma Y, Mahmoud A, Bogdanos DP, Muratori L, et al. Cytochrome P4502D6193-212: a new immunodominant epitope and target of virus/self cross-reactivity in liver kidney microsomal autoantibody type 1-positive liver disease. J Immunol. (2003) 170:1481–9. doi: 10.4049/jimmunol.170.3.1481
103. Manns M, Kyriatsoulis A, Gerken G, Staritz M, Meyer KH, Büschenfelde Z. Characterisation of a new subgroup of autoimmune chronic active hepatitis by autoantibodies against a soluble liver antigen. Lancet. (1987) 329:292–4. doi: 10.1016/S0140-6736(87)92024-1
104. Vitozzi S, Djilali-Saiah I, Lapierre P, Alvarez F. Anti-soluble liver antigen/liver-pancreas (SLA/LP) antibodies in pediatric patients with autoimmune hepatitis. Autoimmunity. (2002) 35:485–92. doi: 10.1080/0891693021000056712
105. Stechemesser E, Klein R, Berg PA. Characterization and clinical relevance of liver-pancreas antibodies in autoimmune hepatitis. Hepatology. (1993) 18:1–9. doi: 10.1002/hep.1840180102
106. Manns MP, Vogel A. Autoimmune hepatitis, from mechanisms to therapy. Hepatology. (2006) 43(2 Suppl. 1):S132–44. doi: 10.1002/hep.21059
107. Longhi MS, Ma Y, Mieli-Vergani G, Vergani D. Aetiopathogenesis of autoimmune hepatitis. J Autoimmun. (2010) 34:7–14. doi: 10.1016/j.jaut.2009.08.010
108. Longhi MS, Hussain MJ, Bogdanos DP, Quaglia A, Mieli-Vergani G, Ma Y, et al. Cytochrome P450IID6-specific CD8 T cell immune responses mirror disease activity in autoimmune hepatitis type 2. Hepatology. (2007) 46:472–84. doi: 10.1002/hep.21658
109. Cassani F, Cataleta M, Valentini P, Muratori P, Giostra F, Francesconi R, et al. Serum autoantibodies in chronic hepatitis C: comparison with autoimmune hepatitis and impact on the disease profile. Hepatology. (1997) 26:561–6. doi: 10.1002/hep.510260305
110. Walker JG, Doniach D, Roitt IM, Sherlock S. Serological tests in diagnosis of primary biliary cirrhosis. Lancet. (1965) 285:827–31. doi: 10.1016/S0140-6736(65)91372-3
111. Gershwin ME, Mackay IR, Sturgess A, Coppel RL. Identification and specificity of a cDNA encoding the 70 kd mitochondrial antigen recognized in primary biliary cirrhosis. J Immunol. (1987) 138:3525–31.
112. Yeaman SJ, Danner DJ, Mutimer DJ, M Fussey SP, W James OF, Bassendine MF. Primary biliary cirrhosis: identification of two major M2 mitochondrial antigens. Lancet. (1988) 331:1067–70. doi: 10.1016/S0140-6736(88)91894-6
113. Vergani D, Alvarez F, Bianchi FB, Cançado ELR, MacKay IR, Manns MP, et al. Liver autoimmune serology: a consensus statement from the committee for autoimmune serology of the international autoimmune hepatitis group. J Hepatol. (2004) 41:677–83. doi: 10.1016/j.jhep.2004.08.002
114. Tsuneyama K, Van De Water J, Van Thiel D, Coppel R, Ruebner B, Nakanuma Y, et al. Abnormal expression of PDC-E2 on the apical surface of biliary epithelial cells in patients with antimitochondrial antibody-negative primary biliary cirrhosis. Hepatology. (1995) 22:1440–6. doi: 10.1002/hep.1840220517
115. Van de Water J, Turchany J, Leung PSC, Lake J, Munoz S, Surh CD, et al. Molecular mimicry in primary biliary cirrhosis. Evidence for biliary epithelial expression of a molecule cross-reactive with pyruvate dehydrogenase complex-E2. J Clin Invest. (1993) 91:2653–64. doi: 10.1172/JCI116504
116. Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. (2005) 42:1194–202. doi: 10.1002/hep.20907
117. Corpechot C, Chrétien Y, Chazouillères O, Poupon R. Demographic, lifestyle, medical and familial factors associated with primary biliary cirrhosis. J Hepatol. (2010) 53:162–9. doi: 10.1016/j.jhep.2010.02.019
118. Morreale M, Tsirigotis M, Hughes MD, Brumfitt W, McIntyre N, Burroughs AK. Significant bacteriuria has prognostic significance in primary biliary cirrhosis. J Hepatol. (1989) 9:149–58. doi: 10.1016/0168-8278(89)90045-7
119. CooK GC, Mulligan R, Sherlock S. Controlled prospective trial of corticosteroid therapy in active chronic hepatitis. QJM. (1971) 40:159–85. doi: 10.1093/oxfordjournals.qjmed.a067264
120. Murray-Lyon IM, Stern RB, Williams R. Controlled trial of prednisone and azathioprine in active chronic hepatitis. Lancet. (1973) 301:735–7. doi: 10.1016/S0140-6736(73)92125-9
121. Soloway RD, Summerskill WH, Baggenstoss AH, Geall MG, Gitnićk GL, Elveback IR, et al. Clinical, biochemical, and histological remission of severe chronic active liver disease: a controlled study of treatments and early prognosis. Gastroenterology. (1972) 63:820–33. doi: 10.1016/S0016-5085(19)33223-8
122. Johnson PJ, Mcfarlane IG, Williams R. Azathioprine for long-term maintenance of remission in autoimmune hepatitis. N Engl J Med. (1995) 333:958–63. doi: 10.1056/NEJM199510123331502
123. Werner M, Almer S, Prytz H, Lindgren S, Wallerstedt S, Björnsson E, et al. Hepatic and extrahepatic malignancies in autoimmune hepatitis. A long-term follow-up in 473 Swedish patients. J Hepatol. (2009) 50:388–93. doi: 10.1016/j.jhep.2008.08.022
124. Hoeroldt B, McFarlane E, Dube A, Basumani P, Karajeh M, Campbell MJ, et al. Long-term outcomes of patients with autoimmune hepatitis managed at a nontransplant center. Gastroenterology. (2011) 140:1980–9. doi: 10.1053/j.gastro.2011.02.065
125. Gleeson D. Long-term outcomes of autoimmune hepatitis. Clin Liver Dis. (2019) 14:24–8. doi: 10.1002/cld.797
126. Floreani A, Liberal R, Vergani D, Mieli-Vergani G. Autoimmune hepatitis: contrasts and comparisons in children and adults - A comprehensive review. J Autoimmun. (2013) 46:7–16. doi: 10.1016/j.jaut.2013.08.004
127. Manns MP, Woynarowski M, Kreisel W, Lurie Y, Rust C, Zuckerman E, et al. Budesonide induces remission more effectively than prednisone in a controlled trial of patients with autoimmune hepatitis. Gastroenterology. (2010) 139:1198–206. doi: 10.1053/j.gastro.2010.06.046
128. Rudic JS, Poropat G, Krstic MN, Bjelakovic G, Gluud C. Ursodeoxycholic acid for primary biliary cirrhosis. Cochrane Database Syst Rev. (2012) 12:CD000551. doi: 10.1002/14651858.CD000551.pub3
129. Poupon R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: an overview of their mechanisms of action. Clin Res Hepatol Gastroenterol. (2012) 36(Suppl. 1):S3–12. doi: 10.1016/S2210-7401(12)70015-3
130. Ter Borg PCJ, Schalm SW, Hansen BE, Van Buuren HR. Prognosis of ursodeoxycholic acid-treated patients with primary biliary cirrhosis. Results of a 10-yr cohort study involving 297 patients. Am J Gastroenterol. (2006) 101:2044–50. doi: 10.1111/j.1572-0241.2006.00699.x
131. Poupon RE, Lindor KD, Cauch-Dudek K, Dickson ER, Poupon R, Heathcote EJ. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology. (1997) 113:884–90. doi: 10.1016/S0016-5085(97)70183-5
132. Corpechot C, Abenavoli L, Rabahi N, Chrétien Y, Andréani T, Johanet C, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. (2008) 48:871–7. doi: 10.1002/hep.22428
133. Kuiper EMM, Hansen BE, de Vries RA, den Ouden-Muller JW, van Ditzhuijsen TJM, Haagsma EB, et al. Improved prognosis of patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology. (2009) 136:1281–7. doi: 10.1053/j.gastro.2009.01.003
134. Carbone M, Neuberger J. Liver transplantation in PBC and PSC: indications and disease recurrence. Clin Res Hepatol Gastroenterol. (2011) 35:446–54. doi: 10.1016/j.clinre.2011.02.007
135. Gilroy RK, Lynch S V, Strong RW, Kerlin P, Balderson GA, Stuart KA, et al. Confirmation of the role of the mayo risk score as a predictor of resource utilization after orthotopic liver transplantation for primary biliary cirrhosis. Liver Transplant. (2000) 6:749–52. doi: 10.1053/jlts.2000.9746
136. Graziadei IW, Wiesner RH, Batts KP, Marotta PJ, Larusso NF, Porayko MK, et al. Recurrence of primary sclerosing cholangitis following liver transplantation. Hepatology. (1999) 8:575–81. doi: 10.1002/hep.510290427
137. Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. (2015) 148:751–61.e8. doi: 10.1053/j.gastro.2014.12.005
138. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. (2016) 375:631–43. doi: 10.1056/NEJMoa1509840
139. Lindor KD, Kowdley K V, Luketic VAC, Harrison ME, McCashland T, Befeler AS, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. (2009) 50:808–14. doi: 10.1002/hep.23082
140. Olsson R, Boberg KM, Schaffalitsky De Muckadell O, Lindgren S, Hultcrantz R, Folvik G, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology. (2005) 129:1464–72. doi: 10.1053/j.gastro.2005.08.017
141. Cullen SN, Chapman RW. Review article: current management of primary sclerosing cholangitis. Aliment Pharmacol Ther. (2005) 21:933–48. doi: 10.1111/j.1365-2036.2005.02407.x
142. Ponsioen CY, Vrouenraets SME, Prawirodirdjo W, Rajaram R, Rauws EAJ, Mulder CJJ, et al. Natural history of primary sclerosing cholangitis and prognostic value of cholangiography in a dutch population. Gut. (2002) 51:562–6. doi: 10.1136/gut.51.4.562
143. Campsen J, Zimmerman MA, Trotter JF, Wachs M, Bak T, Steinberg T, et al. Clinically recurrent primary sclerosing cholangitis following liver transplantation: a time course. Liver Transplant. (2008) 14:181–5. doi: 10.1002/lt.21313
144. Alabraba E, Nightingale P, Gunson B, Hubscher S, Olliff S, Mirza D, et al. A re-evaluation of the risk factors for the recurrence of primary sclerosing cholangitis in liver allografts. Liver Transplant. (2009) 15:330–40. doi: 10.1002/lt.21679
145. Noon L. Prophylactic inoculation against hay fever. Lancet. (1911) 177:1572–3. doi: 10.1016/S0140-6736(00)78276-6
146. Sabatos-Peyton CA, Verhagen J, Wraith DC. Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr Opin Immunol. (2010) 22:609–15. doi: 10.1016/j.coi.2010.08.006
147. Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat Biotechnol. (2019) 37:238–51. doi: 10.1038/s41587-019-0015-4
148. Wraith DC. Designing antigens for the prevention and treatment of autoimmune diseases. Curr Opin Chem Eng. (2018) 19:35–42. doi: 10.1016/j.coche.2017.12.004
149. Streeter HB, Rigden R, Martin KF, Scolding NJ, Wraith DC. Preclinical development and first-in-human study of ATX-MS-1467 for immunotherapy of MS. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e93. doi: 10.1212/NXI.0000000000000093
150. Jansson L, Vrolix K, Jahraus A, Martin KF, Wraith DC. Immunotherapy with apitopes blocks the immune response to TSH receptor in HLA-DR transgenic mice. Endocrinology. (2018) 159:3446–57. doi: 10.1210/en.2018-00306
151. Chataway J, Martin K, Barrell K, Sharrack B, Stolt P, Wraith DC. Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology. (2018) 90:e955–62. doi: 10.1212/WNL.0000000000005118
152. Pearce SHS, Dayan C, Wraith DC, Barrell K, Olive N, Jansson L, et al. Antigen-specific immunotherapy with thyrotropin receptor peptides in graves' hyperthyroidism: a phase I study. Thyroid. (2019) 29:1003–11. doi: 10.1089/thy.2019.0036
153. Getts DR, Turley DM, Smith CE, Harp CT, Mccarthy D, Feeney EM, et al. Tolerance induced by apoptotic PD-L1+ and IL-10– producing splenic antigen-coupled leukocytes is induced by macrophages and maintained by T this information is current as regulatory cells. J Immunol. (2011) 187:2405–17. doi: 10.4049/jimmunol.1004175
154. Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD, et al. Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells. J Autoimmun. (2018) 89:112–24. doi: 10.1016/j.jaut.2017.12.010
155. Freitag TL, Podojil JR, Pearson RM, Fokta FJ, Sahl C, Messing M, et al. Gliadin nanoparticles induce immune tolerance to gliadin in mouse models of celiac disease. Gastroenterology. (2020) 158:1667–81.e12. doi: 10.1053/j.gastro.2020.01.045
156. Galea R, Nel HJ, Talekar M, Liu X, Ooi JD, Huynh M, et al. PD-L1– and calcitriol-dependent liposomal antigen-specific regulation of systemic inflammatory autoimmune disease. JCI Insight. (2019) 4:e126025. doi: 10.1172/jci.insight.126025
157. Abrahimians EM, Elst L Vander, Carlier VA, Saint-Remy JM. Thioreductase-containing epitopes inhibit the development of type 1 diabetes in the NOD mouse model. Front Immunol. (2016) 7:67. doi: 10.3389/fimmu.2016.00067
158. Pagni PP, Bresson D, Rodriguez-Calvo T, Hani AB, Manenkova Y, Amirian N, et al. Combination therapy with an anti+IL-1β antibody and GAD65 DNA vaccine can reverse recent-onset diabetes in the RIP-GP mouse model. Diabetes. (2014) 63:2015–25. doi: 10.2337/db13-1257
159. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. (2016) 530:434–40. doi: 10.1038/nature16962
160. Umeshappa CS, Singha S, Blanco J, Shao K, Nanjundappa RH, Yamanouchi J, et al. Suppression of a broad spectrum of liver autoimmune pathologies by single peptide-MHC-based nanomedicines. Nat Commun. (2019) 10:2150. doi: 10.1038/s41467-019-09893-5
161. Maldonado RA, LaMothe RA, Ferrari JD, Zhang A-H, Rossi RJ, Kolte PN, et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci USA. (2015) 112:E156–65. doi: 10.1073/pnas.1408686111
162. Kishimoto TK, Ferrari JD, Lamothe RA, Kolte PN, Griset AP, O'Neil C, et al. Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat Nanotechnol. (2016) 11:890–9. doi: 10.1038/nnano.2016.135
163. Solvason N, Lou Y-P, Peters W, Evans E, Martinez J, Ramirez U, et al. Improved efficacy of a tolerizing DNA vaccine for reversal of hyperglycemia through enhancement of gene expression and localization to intracellular sites. J Immunol. (2008) 181:8298–307. doi: 10.4049/jimmunol.181.12.8298
164. Roep BO, Solvason N, Gottlieb PA, Abreu JR, Harrison LC, Eisenbarth GS, et al. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8 + T cells in type 1 diabetes. Diabetes Technol Ther. (2014) 5:191ra82. doi: 10.1126/scitranslmed.3006103
165. Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, et al. Nanoparticle-based autoantigen delivery to treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J Hepatol. (2015) 62:1349–56. doi: 10.1016/j.jhep.2015.01.006
166. Genain CP, Abel K, Belmar N, Villinger F, Rosenberg DP, Linington C, et al. Late complications of immune deviation therapy in a nonhuman primate. Science. (1996) 274:2054–7. doi: 10.1126/science.274.5295.2054
167. Rapoport B, Banuelos B, Aliesky HA, Hartwig Trier N, McLachlan SM. Critical differences between induced and spontaneous mouse models of graves' disease with implications for antigen-specific immunotherapy in humans. J Immunol. (2016) 197:4560–68. doi: 10.4049/jimmunol.1601393
168. Hanninen A, Braakhuis A, Heath WR, Harrison LC. Mucosal antigen primes diabetogenic cytotoxic T-lymphocytes regardless of dose or delivery route. Diabetes. (2001) 50:771–5. doi: 10.2337/diabetes.50.4.771
169. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. (2003) 21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040
170. Clement CC, Rotzschke O, Santambrogio L. The lymph as a pool of self-antigens. Trends Immunol. (2011) 32:6–11. doi: 10.1016/j.it.2010.10.004
171. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med. (2015) 7:290ra87. doi: 10.1126/scitranslmed.aaa9301
172. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis. (2017) 76:227–34. doi: 10.1136/annrheumdis-2015-208456
173. Willekens B, Presas-Rodríguez S, Mansilla MJ, Derdelinckx J, Lee WP, Nijs G, et al. Tolerogenic dendritic cell-based treatment for multiple sclerosis (MS): a harmonised study protocol for two phase I clinical trials comparing intradermal and intranodal cell administration. BMJ Open. (2019) 9:1–11. doi: 10.1136/bmjopen-2019-030309
174. Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med. (2002) 196:1627–38. doi: 10.1084/jem.20021598
175. Anderton SM, Wraith DC. Hierarchy in the ability of T cell epitopes to induce peripheral tolerance to antigens from myelin. Eur J Immunol. (1998) 28:1251–61. doi: 10.1002/(SICI)1521-4141(199804)28:04<1251::AID-IMMU1251>3.0.CO;2-O
176. Anderton SM, Viner NJ, Matharu P, Lowrey PA, Wraith DC. Influence of a dominant cryptic epitope on autoimmune T cell tolerance. Nat Immunol. (2002) 3:175–81. doi: 10.1038/ni756
177. Santambrogio L, Sato AK, Fischer FR, Dorf ME, Stern LJ. Abundant empty class II MHC molecules on the surface of immature dendritic cells. Proc Natl Acad Sci USA. (1999) 96:15050–5. doi: 10.1073/pnas.96.26.15050
178. Metzler B, Anderton SM, Manickasingham SP, Wraith DC. Kinetics of peptide uptake and tissue distribution following a single intranasal dose of peptide. Immunol Invest. (2000) 29:61–70. doi: 10.3109/08820130009105145
179. Burkhart C, Liu GY, Anderton SM, Metzler B, Wraith DC. Peptide-induced T cell regulation of experimental autoimmune encephalomyelitis: a role for IL-10. Int Immunol. (1999) 11:1625–34. doi: 10.1093/intimm/11.10.1625
180. Sundstedt A, O'Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol. (2003) 170:1240–8. doi: 10.4049/jimmunol.170.3.1240
181. Gabryšová L, Nicolson KS, Streeter HB, Verhagen J, Sabatos-peyton C A, Morgan DJ, et al. Negative feedback control of the autoimmune response through antigen-induced differentiation of IL-10 – secreting Th1 cells. Regulation. (2009) 206:1755–67. doi: 10.1084/jem.20082118
182. Burton BR, Britton GJ, Fang H, Verhagen J, Smithers B, Sabatos-Peyton C A, et al. Sequential transcriptional changes dictate safe and effective antigen-specific immunotherapy. Nat Commun. (2014) 5:4741. doi: 10.1038/ncomms5741
183. Hartwell BL, Antunez L, Sullivan BP, Thati S, Sestak JO, Berkland C. Multivalent nanomaterials: learning from vaccines and progressing to antigen-specific immunotherapies. J Pharm Sci. (2015) 104:346–61. doi: 10.1002/jps.24273
184. Metzler B, Wraith DC. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: influence of MHC binding affinity. Int Immunol. (1993) 5:1159–65. doi: 10.1093/intimm/5.9.1159
185. Gabryšová L, Wraith DC. Antigenic strength controls the generation of antigen-specific IL-10-secreting T regulatory cells. Eur J Immunol. (2010) 40:1386–95. doi: 10.1002/eji.200940151
186. Groux H, O'Garra A, Bigler M, Rouleau M, Antonenko S, De Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. (1997) 389:737–42. doi: 10.1038/39614
187. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med. (2013) 19:739–46. doi: 10.1038/nm.3179
188. Singha S, Shao K, Yang Y, Clemente-Casares X, Solé P, Clemente A, et al. Peptide-MHC-based nanomedicines for autoimmunity function as T-cell receptor microclustering devices. Nat Nanotechnol. (2017) 12:701–10. doi: 10.1038/nnano.2017.56
189. Umeshappa CS, Mbongue J, Singha S, Mohapatra S, Yamanouchi J, Lee JA, et al. Ubiquitous antigen-specific T regulatory type 1 cells variably suppress hepatic and extrahepatic autoimmunity. J Clin Invest. (2020) 130:1823–9. doi: 10.1172/JCI130670
190. Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. (2002) 2:85–95. doi: 10.1038/nri724
Keywords: immunoregulation, liver, autoimmune disease, immunotherapy, T-cell
Citation: Richardson N, Ng STH and Wraith DC (2020) Antigen-Specific Immunotherapy for Treatment of Autoimmune Liver Diseases. Front. Immunol. 11:1586. doi: 10.3389/fimmu.2020.01586
Received: 24 March 2020; Accepted: 15 June 2020;
Published: 21 July 2020.
Edited by:
Gilles Benichou, Harvard Medical School, United StatesReviewed by:
Pere Santamaria, University of Calgary, CanadaCopyright © 2020 Richardson, Ng and Wraith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David C. Wraith, ZC53cmFpdGhAYmhhbS5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.