 Rodrigo Abreu
Rodrigo Abreu Pramod Giri
Pramod Giri Fred Quinn
Fred Quinn- Department of Infectious Diseases, University of Georgia, Athens, GA, United States
Tuberculosis (TB) has been a transmittable human disease for many thousands of years, and M. tuberculosis is again the number one cause of death worldwide due to a single infectious agent. The intense 6- to 10-month process of multi-drug treatment, combined with the adverse side effects that can run the spectrum from gastrointestinal disturbances to liver toxicity or peripheral neuropathy are major obstacles to patient compliance and therapy completion. The consequent increase in multidrug resistant TB (MDR-TB) and extensively drug resistant TB (XDR-TB) cases requires that we increase our arsenal of effective drugs, particularly novel therapeutic approaches. Over the millennia, host and pathogen have evolved mechanisms and relationships that greatly influence the outcome of infection. Understanding these evolutionary interactions and their impact on bacterial clearance or host pathology will lead the way toward rational development of new therapeutics that favor enhancing a host protective response. These host-directed therapies have recently demonstrated promising results against M. tuberculosis, adding to the effectiveness of currently available anti-mycobacterial drugs that directly kill the organism or slow mycobacterial replication. Here we review the host-pathogen interactions during M. tuberculosis infection, describe how M. tuberculosis bacilli modulate and evade the host immune system, and discuss the currently available host-directed therapies that target these bacterial factors. Rather than provide an exhaustive description of M. tuberculosis virulence factors, which falls outside the scope of this review, we will instead focus on the host-pathogen interactions that lead to increased bacterial growth or host immune evasion, and that can be modulated by existing host-directed therapies.
Tuberculosis Epidemiology
Despite extensive efforts to control Mycobacterium tuberculosis infections through robust screening and therapeutics programs, the World Health Organization (WHO) reported over 10 million new cases in 2018, with over 1.5 million fatalities, ranking as the leading infectious killer in the world, surpassing HIV in 2017 (1). Worldwide incidence of tuberculosis (TB) has been slowly falling over the last 15 years at an average rate of 1.5% per year and prevalence is estimated to have fallen 42% between 1990 and 2015. Nonetheless, TB incidence remains high in Asia, India and Africa (2). In addition to the high number of active TB cases, approximately one third of the world population is estimated to have latent TB infection with 10% having a lifetime risk of developing active infection (3). With the lack of more sensitive and specific diagnostic tools, latent TB infection is typically identified by a positive immune response to M. tuberculosis antigens (tuberculin skin test or interferon-gamma release assay) in the absence of clinical manifestations. HIV co-infection or immunosuppressive treatment (anti-TNF-α or transplant patients) significantly increases the risk of reactivation to 10% chance every year (2). Out of the 9.6 million TB cases in 2014, more than one million were HIV-positive with about 35% resulting in death. There was a higher incidence rate in Africa where over 30% of all TB cases are in HIV co-infected patients (4).
M. tuberculosis generates systemic infection but is primarily identified in adults as a lung pathogen that interacts to a significant extent with alveolar macrophages and if not cleared, leads to extensive lung inflammation, dissemination and pathology. If active disease develops, symptoms are characterized by persistent cough that can last for several weeks, late day fevers (night sweats), constant fatigue, loss of appetite, and severe weight loss (1, 5, 6). Infection with M. tuberculosis primarily is caused by inhalation of bacilli, transmitted by an actively infected individual. The inhaled bacilli can progress in different stages depending on the host immune system (Figures 1A,B). In 90% of primary infected individuals the host is capable of controlling and resolving the infection (Figure 1D). In latent infection which occurs in ~7–10% of infection cases, mycobacterial replication is minimal and primarily contained in small granulomatous structures until re-activation. Clearance may take up to 3 years, but in some cases it never occurs and the pathogen goes into a life-lasting latent stage that can reactivate in case of immunosuppression (7) (Figure 1E). In primary active TB, M. tuberculosis bacilli migrate to the alveoli where they encounter alveolar macrophages and dendritic cells that actively phagocytize the bacteria and ultimately the bacilli and/or infected phagocytes disseminate to regional lymph nodes (Figure 1C). This first stage can take 3–8 weeks or longer and has no clear manifestation or transmission stage. In a second phase that can last up to 3 months after primary infection, hematogenous dissemination of the bacteria leads to M. tuberculosis spread into the upper and lower lobes of the lung and can cause systemic dissemination including meningitis TB which in many cases is fatal (7) (Figure 1B).
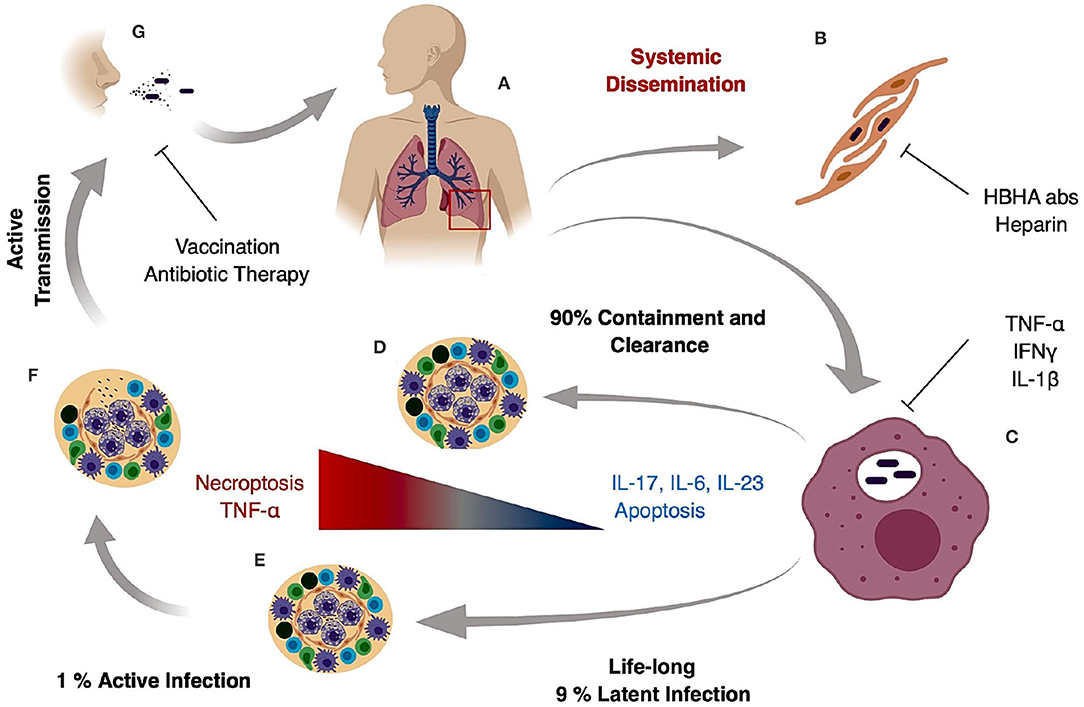
Figure 1. Tuberculosis infection and transmission hallmarks. Inhaled M. tuberculosis bacilli travel to the alveoli where they are phagocytized by alveolar macrophages (A). It is hypothesized that internalization and successful replication within Type II pneumocytes results in systemic dissemination and extrapulmonary TB, which can be decreased by HBHA neutralizing antibodies or heparin treatment (B). In the lung, M. tuberculosis bacilli replicate in alveolar macrophages during early stages of infection (C) and in 90% of the cases the host mounts an appropriate immune response preventing pathogen entry or controlling pathogen growth and replication resulting in bacterial clearance (D). In 9% of the cases, the host develops a life-long latent stage with bacterial containment most prominently maintained inside caseous granulomas (E). In latent cases, a 10% lifetime risk of reactivation due to an improper immune response or immunosuppression can occur. The result is loss of granuloma integrity, M. tuberculosis growth, dissemination, and ultimately infection of the upper lobes (F). Uncontrolled bacterial replication and granuloma caseation (F) augments lung pathology and initiates active aerosol transmission to the next host (G).
Traditional research in M. tuberculosis virulence focused on the comparison of virulent laboratory or clinical strains against the attenuated M. bovis BCG vaccine strain (8). Genetic analysis of M. tuberculosis lab strain H37Rv against BCG revealed 14 regions of differentiation (RD1–RD14) of which three (RD1, RD2, and RD14) are still present in clinical strains of M. bovis (9–12). Within RD3–RD13, multiple genes have been associated with M. tuberculosis virulence both in vivo and in vitro, and have been extensively reviewed elsewhere (12). In this review, we will focus on the host-pathogen interactions that lead to increased bacterial growth in the host that can be modulated by existing host-directed therapies (HDT).
Drug Treatment and Drug Resistance
Drug treatment for TB requires complex drug regimens for long periods of time leading to severe side effects. WHO guidelines recommend the treatment of newly diagnosed TB cases with a six-month regimen of isoniazid, rifampicin, pyrazinamide, and ethambutol during the intensive phase (first 2 months) followed by isoniazid and rifampicin for continuation phase (next 4 months) (13). In cases of TB relapse with a medium- to low-risk of multidrug-resistance the addition of streptomycin to the abovementioned drug regimen during the intensive phase is recommended, followed by a 1-month regimen of isoniazid, rifampicin, pyrazinamide and ethambutol, and a 5-month regimen of isoniazid, rifampicin, and ethambutol (4, 6).
Between 2005 and 2013, only 86% of treatments for newly diagnosed TB cases were successfully completed. This lack of therapy compliance leads to an increase in MDR-TB and XDR-TB cases. In 2014, 3.3% of all new TB cases and 20% of previously treated cases were MDR-TB, accounting for a total of almost 500,000 patients worldwide (2). This increases urgency for the development of new therapeutic strategies through the discovery of new anti-mycobacterial drugs and the identification of HDT that provide a hostile environment for the growth of the organism and promote a protective immune response (4). In fact, novel therapeutic approaches largely centered on host pharmacological targets have been recently reviewed and the focus of grand clinical interest (14–16), but fail to cross-reference with the target bacterial factors. Here we review the known host-pathogen interactions during M. tuberculosis infection, how the bacteria modulate and evade the host immune system, and the currently available HDT that target each mechanism (Table 1).
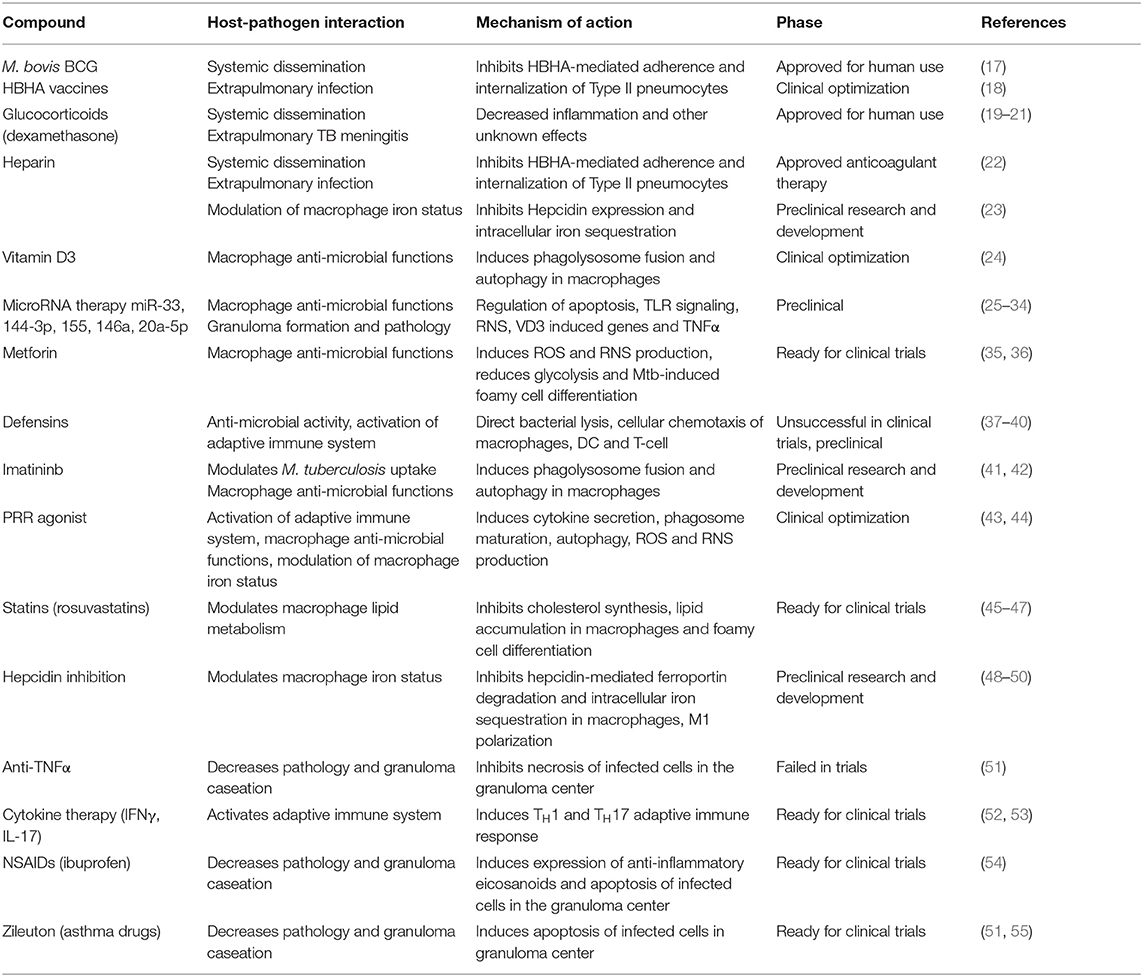
Table 1. Currently available host-directed therapies for tuberculosis.
Systemic Dissemination
M. tuberculosis is primarily known as a pulmonary pathogen; however, it is commonly found to cause disseminated disease with lesions present in many organs and tissues including the spleen, lymph nodes and brain. It can also manifest as a more generalized disseminated form known as miliary TB. The mechanisms involved in dissemination from the lung are not well understood, but M. tuberculosis entry, intracellular replication and necrosis of alveolar epithelial cells is thought to be one mechanism involved in extra-pulmonary dissemination. Attachment to Type II alveolar epithelial cells (pneumocytes) is likely mediated by several bacterial adhesins. One adhesin that has been well studied is the heparin-binding hemagglutinin (HBHA) (18, 22, 56). Inhibiting invasion of Type II pneumocytes with heparin and heparin sulfate, or blocking HBHA function with neutralizing antibodies efficiently prevents M. tuberculosis dissemination (17). Another rare form of extra-pulmonary TB, most prevalent in young children, results in meningitis, the most lethal form of M. tuberculosis infection. M. bovis BCG vaccination remains the main prophylactic approach against TB meningitis with almost 80% protection against this form of disease in young children (57). In fact, meningitis protection in children is one of the main reasons for continuing widespread BCG vaccination programs particularly in endemic areas even in the absence of efficient protection against pulmonary TB. The exact protection mechanisms elicited by M. bovis BCG remain elusive, but elicitation of strong humoral responses against HBHA and other M. tuberculosis surface proteins along with long lasting cellular responses to overlapping internal antigens have been shown to prevent extra-pulmonary dissemination, and consequently meningitis. Nonetheless, in case of dissemination adjunctive glucocorticoid therapy with standard of care anti-mycobacterial drug regiments increases TB meningitis survival rates (19–21).
Macrophage Roles During Infection
Inhibiting entry into target host cells by intracellular pathogens is a frequent therapeutic approach that limits disease pathogenesis. During M. tuberculosis infection this strategy is particularly difficult since a major cell target, alveolar macrophages, are also a crucial player in the host immune response. Upon attachment, alveolar macrophages actively phagocytize M. tuberculosis bacilli through multiple mechanisms, and the internalization pathway greatly influences microbiocidal efficiency (58–60).
During early primary M. tuberculosis infection, direct mycobacterial phagocytosis is mostly mediated by C-type lectin receptors (CLRs) (61, 62). The macrophage mannose receptor (MMR) recognizes M. tuberculosis lipoarabinomannan (63, 64) and is predicted to signal through a putative cytoplasmic tyrosine domain, which phosphorylates and activates CDC42, RHOB, PAK, or ROCK1, involved in actin reorganization, membrane invagination and phagosome formation (62, 65–68). Another CLR involved in M. tuberculosis recognition by macrophages is macrophage-inducible C-type lectin which recognizes trehalose-6,6-dimycolate, an abundant mycobacterial cell wall glycolipid (12, 59, 69). In later stages of infection or in secondary infections, antibody and complement opsonized bacteria are phagocytized through Fc and complement receptors, signaling through a similar mechanism that promotes efficient bacterial killing and controls replication (60, 62). Despite the importance of these receptors in M. tuberculosis cell attachment and phagocytosis, the impact of each mechanism in the outcome of infections is not yet clear. Recently a tyrosine kinase inhibitor used in cancer therapy has been shown to modulate M. tuberculosis uptake and promote bacterial killing in vitro and in vivo (41, 42). Moreover, this drug was particularly effective in combination with anti-mycobacterial drugs, but the exact mechanism remains elusive. It is possible that a decrease in bacterial internalization by macrophages increases antibiotic access to the bacilli, or that inhibition of one specific internalization pathway leads to an alternative uptake mechanism that activates microbiocidal macrophage functions. Currently, Imatinib is the only tyrosine kinase inhibitor tested as a modulator of M. tuberculosis invasion, but other similar drugs presently in trials for cancer therapy (42) might have similar impacts or help clarify the exact mechanism behind bacterial control in vivo.
Granuloma Formation and Pathology
A hallmark of M. tuberculosis infection and pathology is granuloma formation and maintenance. The granuloma is a compact organized immunological structure built of macrophages, monocytes, dendritic cells, neutrophils, epithelioid cells, foamy macrophages, and multi-nucleated giant cells, enclosed by T and B lymphocytes (70). Disease progression results from complex remodeling of the granuloma structure with increased hypoxic necrotic centers rich in lipids and foamy macrophages that fail to control bacterial replication ultimately leading to granuloma caseation (70). The mycobacterial factors leading to granuloma restructuring and rupture are not yet well-described, but the ESX-1 secretion system including ESAT6, and TDM are known to play important roles in the initial steps of granuloma formation (12, 71). Alternatively, TNF-α, IL-6 and complement (C5) are important for cellular recruitment and maintenance of the granuloma structure. In the granuloma center, predominant apoptotic cell death of infected macrophages controls bacterial replication by efferocytosis (72). In contrast, necrosis results in bacterial leakage into the growth permissive extracellular environment, and a characteristic cording phenotype hampers phagocytosis by newly recruited macrophages (70, 71, 73). Efficient M. tuberculosis infection strongly modulates macrophage cell death. In human primary macrophages, M. tuberculosis bacteria significantly modulate expression of microRNAs (miR−145 and miR-20a-5p) regulators of apoptosis, remodeling cell death toward a necroptotic pathway (25, 26, 29).
Efficient bacterial control in the granuloma requires a balanced pro- and anti-inflammatory environment (74). Anti-TNF-α therapy in patients with autoimmune disorders has been shown to increase the risk of TB reactivation (75); however, excessive TNF-α leads to increased macrophage necrosis that results in granuloma caseation (72, 76, 77). Central in the regulation of TNF-α expression during M. tuberculosis infection are pro-inflammatory eicosanoids such as leukotrienes and prostaglandins (51, 55). Excessive leukotrienes promote TNF-α and Type I IFNs that result in increased necrotic cell death, granuloma caseation and cavity formation (55). Alternatively, IL-1 signaling promotes apoptosis and induces prostaglandin expression which counter-regulates the function of Type I IFN (51, 55). Non-steroid anti-inflammatory drugs, such as ibuprofen, induce expression of anti-inflammatory eicosanoids that significantly ameliorates pathology during M. tuberculosis infection in vivo with reduced bacterial load (54). Similarly, leukotriene inhibitors such as Zileuton used for asthma therapy, also reduce bacterial load in M. tuberculosis susceptible animal models (51, 55). Finally, new therapeutic approaches targeting pathologically imbalanced microRNAs are rapidly arising for cancer and inflammatory diseases (78–80). Similar approaches targeting miR-145 and miR-20a-5p might counteract M. tuberculosis anti-apoptotic effects favoring granuloma integrity and bacterial clearance.
Modulation of the Host Adaptive Immune Response
Despite extensive research, it is not yet clear what might be the ideal adaptive immune response leading to efficient control of bacterial replication and clearance with minimal tissue damage (59, 81). Mycobacterium tuberculosis bacteria infect professional antigen-presenting cells with a significant impact on antigen presentation and activation of the adaptive immune response. Dendritic cells infected with M. tuberculosis bacilli have decreased MHC surface expression and impaired antigen processing and presentation to T cells (82, 83). The priming of T helper cells is delayed by M. tuberculosis, and modulation of cytokine secretion by macrophages promotes differentiation of Tregs and secretion of decoy antigens that modulate the humoral response (84, 85). T cell activation and differentiation into TH1, with IL-2 and IL-12, and into TH17 subsets, with IL-6, IL-1β, and IL-23, is essential for M. tuberculosis containment (84). Thus, the effector cytokines produced by these two T helper cell subsets have long been hypothesized as an effective immunomodulatory host-targeted therapy for TB. Despite the long recognized importance of IFNγ producing TH1 cells for an effective adaptive immune response (86, 87), direct IFNγ therapy produced controversial results in TB patients (52). Initial studies with non-tuberculosis mycobacteria-infected patients (atypical pulmonary mycobacteriosis), showed that IFNγ treatment in combination with standard anti-mycobacterial chemotherapy had no impact on sputum cultures, but a pronounced effect in treatment completion rates and decreased lung lesion severity were observed (88). A similar IFNγ treatment study with pulmonary TB patients showed no significant changes in disease morphology as observed by chest radiology results, however the IFNγ treatment did attenuate general disease symptoms such as fever and increased rates of sputum smear conversion (52). Furthermore, other direct cytokine therapies with IL-2 or IFNα also failed to produce conclusive beneficial results during M. tuberculosis infection (53) indicating that single direct cytokine therapy might not be sufficient alone as a HDT approach for anti-TB treatment. Recent studies highlighting the importance of multifunctional TH1 cells capable of producing multiple cytokines (IL-2, TNF-α, and IFNγ) might explain this discrepancy between the importance of some cytokines for an effective host immune response and the inefficacy of these same cytokines in clinical trials (89–91). Instead of direct adaptive immune activation, HDT can also target chemotaxis of innate immune mediators. Defensins are strong chemoattractants of macrophages, dendritic cells and T-cells and promote TH1 responses (37–40). Furthermore, defensin therapy would also directly target extracellular bacteria if administered intranasally (92). Finally, other immunomodulatory therapeutic approaches, focus on Treg downregulation. Infection with M. tuberculosis bacilli promote a tolerogenic immune response and the differentiation of Tregs to facilitate bacterial replication (59). GR1-specific antibodies and denileukin/diftitox efficiently deplete Treg proliferation and other myeloid-derived suppressor cells and significantly enhance anti-mycobacterial drugs effects (93, 94). This is a very active area of research particularly in anti-cancer therapy, but must be approached carefully because breaking host tolerance is frequently associated with severe autoimmune diseases.
Macrophage Activation Signaling
Innate immune cells like macrophages or dendritic cells recognize a myriad of pathogen or danger associated molecular patterns (PAMPS or DAMPS) (62). Efficient microbiocidal functions in macrophages require activation of these stimulatory pathogen recognition receptors (PRR) such as Toll-like (TLR) or Nod-like (NLR) receptors (62). Mycobacterium tuberculosis bacilli evade and modulate PRR signaling to promote recruitment of permissive macrophages and manipulate the host adaptive immune response (58, 59, 95, 96).
Toll-like receptors are abundantly expressed in human macrophages and crucial for early pathogen recognition during infection (97). The relevance of TLR signaling for M. tuberculosis containment is still being assessed (98–100), but it is widely recognized that virulent M. tuberculosis strains modulate and evade TLR signaling (58). Non-pathogenic mycobacterial cell wall glycolipids such as lipoarabinomannan strongly activate TLR2 signaling inducing a strong pro-inflammatory response (101, 102). Contrastingly, similar molecules from M. tuberculosis such as mannose-capped lipoarabinomannan do not activate TLR2 signaling or induce pro-inflammatory cytokines (58, 64). Recent studies focusing on post-transcriptional regulation by microRNA start to shed light on how M. tuberculosis can modulate TLR signaling (27), but the promiscuous activity of microRNAs and the myriad of targets altered during M. tuberculosis infection obstruct the design of a definitive model.
M. tuberculosis bacilli interfere with phagosome maturation, compromise phagosome membrane integrity (103, 104) and some reports describe the bacilli escaping the phagosome and residing in the cytoplasm (105–107). Nod-like receptors are crucial to recognizing cytosolic PAMPS during bacterial infection and play an important role in inducing Type I IFN and inflammasome activation (108). NOD2 recognizes bacterial muramyl dipeptide fragments of the cell wall peptidoglycan in the cytosol and induces autophagy and pro-inflammatory cytokine production (43). However, M. tuberculosis muramyl dipeptides are N-glycolyl modified and modulate NOD2 signaling to an alternative pathway leading to production of Type I IFNs which are not protective during M. tuberculosis infection (109). Furthermore, Type I IFNs antagonize IL-1β and IFNγ host-protective signaling (110).
The use of PRR ligand adjuvants is a particularly active area in vaccine development (111–113), but the use of specific TLR or NLR agonists might also be useful as a HDT. Activation of TLR2 with its specific ligand Pam2Cys rescues TH1 cell exhaustion and significantly ameliorates disease in chronically M. tuberculosis-infected mice (44). Similarly, NOD2 and TLR4 activation significantly enhances the effect of standard anti-mycobacterial drugs isoniazid and rifampicin in M. tuberculosis-infected dendritic cells (43). Alternatively, modulating the expression and activity of miR-146a and miR-155, two microRNAs extensively described as regulators of innate immune cell activation downstream of TLR activation might favor macrophage antimicrobial functions (27). These studies, although preliminary, show the potential of direct PRR activation as an immunomodulatory HDT for TB. Nonetheless, such therapeutic approaches must proceed with care since dysregulated PRR signaling is frequently associated with loss of immune tolerance and as described earlier, development of autoimmune diseases.
Inhibition of Macrophage Microbiocidal Functions
Alveolar macrophages are an important cell target for M. tuberculosis infection. In an ideal immune response, macrophages efficiently phagocytize and control bacterial replication. In this scenario, phagosomes containing live mycobacteria fuse with lysosomes from the Golgi apparatus that lead to an acidified environment, increased reactive oxygen species (ROS) and reactive nitrogen species (RNS) species, and high protease activity (Figure 2). These processes culminate in bacterial killing and clearance (61, 101). However, M. tuberculosis can subsist and replicate inside macrophages by interfering with phagosome maturation and blocking the macrophage microbiocidal mechanisms (65). Generally, M. tuberculosis resorts to three different mechanisms to prevent phagosome killing: phagosome maturation arrest, phagosome evasion, and oxidative and nitrosative stress neutralization.
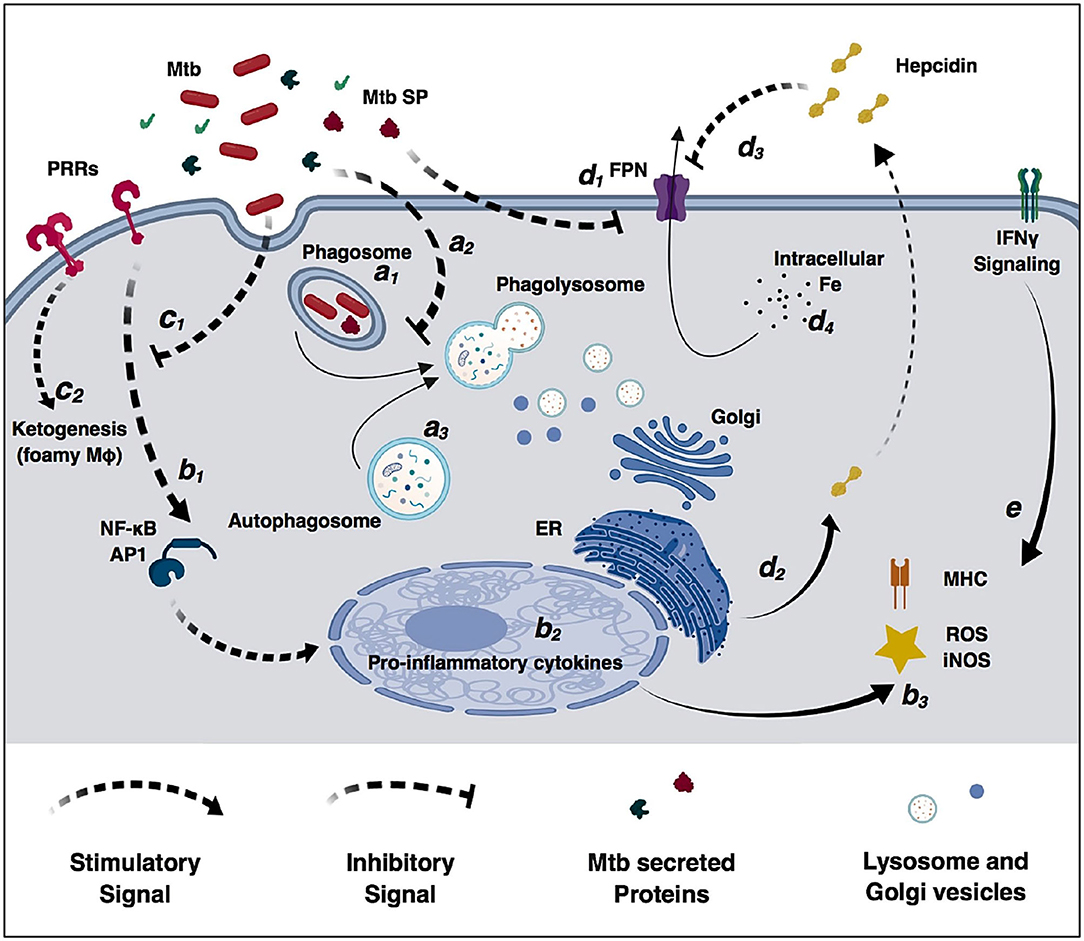
Figure 2. Modulation of macrophage immune functions by M. tuberculosis (Mtb). Bacilli are phagocytized by macrophages through different surface receptors (a1) which greatly influence phagosome maturation and lysosome fusion (a2). M. tuberculosis secreted proteins further inhibit phagosome fusion, but autophagy induction redirects immature phagosomes to the autophagosome (a3) increasing bacterial killing. Macrophages detect pathogen invasion through activation of pathogen-recognition receptors (PRRs) (b1) leading to expression of pro-inflammatory cytokines (b2), increased reactive oxidizing species and activation of the adaptive immune system (b3). However, M. tuberculosis cell-wall glycolipids modulate PRRs signaling (c1), increase lipid accumulation, promote the differentiation in permissive foamy cells (c2) and inhibit cytokine secretion. Infection in macrophages directly decreases ferroportin transcriptional expression (d1), and M. tuberculosis-induced endoplasmic reticulum stress induces hepcidin expression and secretion (d2). Secreted hepcidin binds to ferroportin leading to its internalization and degradation (d3). Decreased surface levels of the iron exported by ferroportin result in increased intracellular iron sequestration in macrophages (d4) that can be redirected to the immature phagosome and used by M. tuberculosis for replication. IFNγ signaling increases macrophages antimicrobial functions and counteracts M. tuberculosis immunomodulatory mechanisms (e).
Several proteins expressed by M. tuberculosis are capable of inhibiting or blocking phagosome maturation and phagolysosome fusion; e.g., nucleoside diphosphate kinase, a 14 kDa protein isolated from the culture medium, interacts and inactivates Rab7 and Rab5 which are crucial for phagosome-lysosome fusion (12, 60, 114, 115). Similarly, phosphotyrosine protein A (PtpA), a low molecular weight phosphatase, can bind and block the host vacuolar H+-ATPases and dephosphorylate a host vacuolar protein preventing phagosome acidification and maturation (116–118). Aside from these, many other mycobacterial factors have been associated with phagosome maturation or arrest and extensively reviewed elsewhere (12, 60). Until now, IFNγ activation and autophagy induction seem to be the most promising pathways to promote phagosome maturation and phagolysosome fusion (119–121). In vitro macrophage activation with recombinant IFNγ upregulates FcR and CR3 surface expression (122–124), favoring phagocytosis of opsonized bacilli. As mentioned above, this phagocytic pathway promotes phagosome acidification and phagolysosome fusion. The protective effect of vitamin D3 during TB has long been recognized but the mechanisms involved remained elusive (125). Now, we realize that vitamin D3 induces cathelicidin expression in macrophages, an antimicrobial peptide important in phagosome maturation and phagolysosome fusion (24). Furthermore, has-mir-21 has been shown to play a central role in vitamin D3-dependent cathelicidin expression following bacterial infection (126), suggesting that modulation of this specific microRNA might serve as a valuable HDT target. Likewise, imatinib promotes phagosome maturation, lysosome fusion and autophagy (41), a naturally occurring cellular process for recycling and degradation of cytosolic content through vesicular engulfment and lysosome fusion (127). During M. tuberculosis infection, phagosomes containing live bacilli are redirected to the autophagy pathway reactivating lysosome fusion and bacterial killing (119), and here too, microRNAs (miR33, miR-155, and miR144-3p) have been shown to play a crucial role in autophagy regulation (28, 32, 33). Another possible target is the NAD+-dependent histone deacetylase sirtuin 1 (SIRT-1), which was recently shown to be downregulated during M. tuberculosis infection but important for controlling bacterial replication (128). Resveratrol is a phytoalexin present in grapes and berries, frequently commercialized as food supplement and a natural SIRT-1 activator. Resveratrol and a synthetic SIRT-1 activator induce phagolysosome fusion and autophagy, restricting M. tuberculosis growth in vitro and in vivo. Anti-mycobacterial drugs shown to induce autophagy with minimal cell toxicity are a very active HDT research area targeting viral and bacterial infections (119, 129).
For decades intracellular M. tuberculosis bacilli were believed to merely inhibit phagosome maturation, growing and replicating inside this vesicular structure and not escaping into the cytoplasm (63, 84, 130, 131). However, recently M. tuberculosis bacilli have been associated with complete phagosome evasion through permeabilization of the phagosome membrane similar to Shigella or Listeria (105). ESAT6/CF10 proteins, secreted by ESX-1 T7SS, have cell membrane lysis properties (132) and likely are involved in bacterial escape from the phagosome to the cytoplasm in dendritic cells (12, 133, 134). Currently, there are no prospective therapies to target cytosolic bacilli and prevent phagosome evasion, but modulation of the host ubiquitination machinery, inducing autophagy and activating cytosolic PRRs have been shown important for containment of other cytosolic pathogens (135, 136).
Oxidative and nitrosative stress play a crucial role in bacterial clearance in macrophages. In these cells, NOX2 NADPH oxidase releases to the phagosome lumen where through the action of super oxide dismutase (SOD) it is modified into H2O2, generating hydroxyl radicals, singlet oxygen, hypochlorous acid or chloroamines through myeloperoxidase activity (137). In the cytoplasm, increased expression of inducible nitric oxide synthase (NOS2 or iNOS) generates NO− which can diffuse through the membrane to form nitrogen dioxide, peroxynitrite, dinitrogen trioxide, dinitrosyl ion complexes, nitrosothiols, and nitroxyl (138, 139). In the phagosome, ROS and RNS modify lipids, proteins and nucleic acids, culminating in bacterial death (65). In order to survive and replicate in the phagosome M. tuberculosis bacilli upregulate several antioxidant enzymes including superoxide dismutase C (SOD C), catalase-peroxidase-peroxynitritase T (KatG), and thiol peroxidase (TpX). SOD C detoxifies into molecular oxygen or hydrogen peroxide (65, 103), KatG neutralizes the NAPDH-derived peroxides pumped into the phagosome, and TpX generates resistance against macrophage generated RNS (140). As mentioned previously, TNF-α has a putative role during M. tuberculosis infection. In vitro studies with murine macrophages resembling early stages of infection, show that TNF-α-mediated iNOS and ROS induction significantly decreases M. tuberculosis growth (141). Similarly, mycobacterial-induced expression of miR146-a suppresses NO production through negative TNF-α regulation (31). Contrastingly, at later stages of infection, TNF-α induces necrosis of infected cells in the granuloma core leading to bacterial leakage and replication, making direct TNF-α cytokine therapy unsuitable for ROS and iNOS induction (51, 72, 76). Thus, ROS and iNOS inducers with no impact on cell death are a promising HDT approach for TB. Metformin is a FDA approved anti-diabetes drug shown to induce mitochondrial ROS production in M. tuberculosis infected macrophages and to decrease bacterial burden (35, 142). Furthermore, metformin has a positive anti-inflammatory impact decreasing M. tuberculosis-induced lung pathology (143) and positively regulates lipid metabolism (see below). In parallel, future therapeutic approaches focused on mir-146a repression in alveolar macrophages might also promote M. tuberculosis clearance or containment.
Modulation of Lipid Metabolism and Macrophage Phenotype
M. tuberculosis efficiently modulates the macrophage glycolysis pathway and promotes ketogenesis and differentiation into permissive foamy cells (13). Foamy cells are lipid droplet rich macrophages, characteristic of chronic inflammatory diseases and infections (144). In macrophages, M. tuberculosis infection increases glucose uptake and redirects acetyl-CoA from the citric acid cycle to D-3-hydroxybutyrate synthesis, which signals through the anti-lipolytic G protein-coupled receptor GPR109A to induce lipid accumulation and lipid-body formation (13). Furthermore, M. tuberculosis cell wall lipids such as oxygenated ketomycolic and hydroxyl-mycolic acid activate TLR2 and the scavenger receptor MARCO to induce cholesterol uptake with sequestration and lipid droplet accumulation (145, 146) which can serve as a carbon source for M. tuberculosis to persist in nutrient limiting conditions (70, 144). These findings uncovered the cellular similarities of M. tuberculosis infection with other host metabolic diseases such as type II diabetes or hyperlipidemia and open the way to the use of anti-diabetic drugs and statins as possible HDT during TB (51, 73, 143, 147).
As previously described, metformin decreases M. tuberculosis replication in human macrophages through increased ROS production and bacterial killing (35). However, aside from its impact on macrophage oxidative state, metformin also reduces glycolysis efficiency, acetyl-CoA production and possibly ketogenesis in macrophages (36). A parallel therapeutic approach focuses on hypercholesterolemia drugs such as the statins that inhibit cholesterol synthesis and significantly decrease lipid accumulation (45). Despite the initial promising results in animal models treated with statins and antimycobacterial drugs (46, 47), a retrospective analysis with a national medical claim database failed to recognize any beneficial effect of this drug during M. tuberculosis infection (148). More retrospective studies and controlled clinical trials should help clarify the relevance of lipid accumulation and foamy cell differentiation in TB, and help determine if the currently available drugs for diabetes and hyperlipidemia can be effective HDT for M. tuberculosis infection.
Modulation of Macrophage Iron Status
Iron is an essential element in all domains of life as an important cofactor for the synthesis and function of numerous proteins. Upon infection, M. tuberculosis must compete with the host for the same iron pool; M. tuberculosis strains mutated in iron sequestration genes show significantly attenuated growth in vitro and in vivo (149–151). In contrast, increased dietary iron or hemochromatosis is strongly associated with a worse disease prognosis during M. tuberculosis infection (152). Intracellular iron sequestration in macrophages is promoted by M. tuberculosis through two TLR-dependent redundant mechanisms targeting the host iron regulatory proteins hepcidin and ferroportin (23). Ferroportin is the only known iron exporter in mammals, highly expressed in macrophages, enterocytes and hepatocytes (153, 154). During iron overload or inflammation hepcidin secreted from macrophages and hepatocytes binds to ferroportin subsequently leading to its internalization and degradation. This process results in increased intracellular iron sequestration in macrophages, hepatocytes and enterocytes (155–157). Infection by M. tuberculosis in human macrophages directly downregulates ferroportin expression through TLR2 activation, and TLR4-induced endoplasmic reticulum-stress leads to hepcidin secretion which further decreases surface ferroportin. This decrease in ferroportin results in a significant increase in intracellular iron levels (23). Iron chelation therapy is a common strategy to avoid cardiac complications in hemochromatosis and thalassemia patients (158). During M. tuberculosis infection in human macrophages, iron chelation with the FDA approved deferiprone or deferasirox significantly decreases intracellular bacterial replication (159, 160). In vivo, deferasirox intraperitoneal injection during intravenous M. avium infection significantly decreases bacterial burden in the spleen but not in the lung or liver (161). Retrospective studies with hemochromatosis TB patients might unveil the interactions of iron chelation with standard anti-TB drugs regimen. Nonetheless, iron chelation therapy should be approached with care since it will exacerbate anemia due to chronic inflammation. A therapeutic alternative to decreasing iron availability to M. tuberculosis, and simultaneously decreasing the potential anemia, is direct hepcidin inhibition (162–164). Non-anticoagulant heparin significantly decreases hepcidin expression in hepatocytes (165–169), and heparin-mediated hepcidin inhibition decreases intracellular iron levels in human macrophages with pronounced effects in bacterial replication (49). Furthermore, blocking hepcidin function with specific antibodies is currently being tested for treatment of anemia with promising results (170), and could be expanded as a HDT for TB. Similarly, hepcidin blocking with a specific monoclonal antibody might decrease M. tuberculosis and other intracellular siderophilic bacteria replication in macrophages. Additionally, intracellular iron levels have recently been associated with macrophage polarization, with increased intracellular iron sequestration resulting in M2 phenotype (48). Consequently, hepcidin inhibition might not only decrease bacterial replication through nutritional immunity, but also modulate macrophage polarization toward M1 phenotype traditionally correlated with microbiocidal and proinflammatory activity. Further in vitro and in vivo studies will clarify the impact of hepcidin inhibition during M. tuberculosis infection, but the recent studies with other siderophilic bacteria strongly support the hepcidin-ferroportin axis as promising novel HDT for TB.
Concluding Remarks
Tuberculosis remains a major public health concern in many areas of the world, and we are still far from achieving eradication. In today's globalized world, MDR- and XDR-TB are every nation's problem and need to be addressed. Novel HDT can help decrease MDR- and XDR-TB either by enhancing the effect of currently available anti-mycobacterial drugs, targeting new mechanisms, circumventing resistance, or by shortening treatment length which would facilitate patient compliance. Over the millennia that M. tuberculosis bacilli have infected humankind, host and pathogen have evolved mechanisms and relationships that greatly influence the outcome of infection. Understanding this evolutionary race and how host-pathogen interactions impact bacterial clearance or host pathology can lead the way to the rational development of new therapeutics that favor a host protective response. The host immune response to M. tuberculosis is a complex network of pro- and anti-inflammatory signals, and it is now clear that targeting a single aspect of the immune response with increased pro-inflammatory signals is not sufficient to treat TB. Most of the promising HDT presented here target many host-pathogen interactions and in some cases seem to induce both pro- and anti-inflammatory responses. As examples: heparin prevents M. tuberculosis invasion of alveolar pneumocytes and systemic dissemination, but also modulates macrophage intracellular iron levels, cytokine secretion and leukocyte recruitment. Hepcidin inhibition decreases intracellular iron levels, but also decreases lipid body formation and modulates cytokine secretion in macrophages. Similarly, metformin and vitamin D3 promote phagolysosome fusion and autophagy, while inducing anti-inflammatory cytokine secretion that prevents excessive lung pathology; and together, these compounds counteract multiple virulence mechanisms used by M. tuberculosis to evade the host immune response and establish infection. Likewise, the emergence of novel RNA delivery technologies will guide the development of RNA-based therapies targeting microRNA pathologically dysregulated during M. tuberculosis infection with broad metabolic targets.
Regardless of the preferred mechanism of action, HDT will most likely always be administered in combination with standard of care anti-mycobacterial drugs. Consequently, it will be important to assess possible drug-drug interactions between HDT and currently approved drug regimens. As example: rifampicin is well known to interact with corticosteroids and oral anticoagulants, that were here presented as possible HDTs.
HDT alone might never be enough to contain and clear M. tuberculosis bacilli in an active TB patient, but incorporating this treatment class will certainly increase the effect of our currently available anti-mycobacterial drugs, and might give our immune system the little push it needs to efficiently contain M. tuberculosis infection.
Author Contributions
RA compiled the literature, conceptualized, wrote the manuscript, and prepared the figures. PG provided guidance, intellectual input, helped write the manuscript, and reviewed the manuscript. FQ provided guidance, intellectual input, helped write the manuscript, and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by an endowment from the University of Georgia Athletic Association (FQ) and Fulbright Ph.D. Scholarship (RA 11/278).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. World Health Organization. Tuberculosis. Available online at: http://www.who.int/immunization/diseases/tuberculosis/en/ (cited September 30, 2017).
2. World Health Organization Others. Global Tuberculosis Report 2016. Geneva: World Health Organization. (2016).
3. Control C for D Prevention others. Reported Tuberculosis in the United States 2014. Atlanta, GA: US Department of Health and Human Services (2015).
4. World Health Organization Others. Global Tuberculosis Report 2017. Geneva: World Health Organization (2017).
5. Daniel TM. The history of tuberculosis. Respir Med. (2006) 100:1862–70. doi: 10.1016/j.rmed.2006.08.006
6. Zumla A, Chakaya J, Centis R, D'Ambrosio L, Mwaba P, Bates M, et al. Tuberculosis treatment and management—an update on treatment regimens, trials, new drugs, and adjunct therapies. Lancet Respir Med. (2015) 3:220–34. doi: 10.1016/S2213-2600(15)00063-6
7. Smith I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin Microbiol Rev. (2003) 16:463–96. doi: 10.1128/CMR.16.3.463-496.2003
8. Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M bovis. J Bacteriol. (1996) 178:1274–82. doi: 10.1128/JB.178.5.1274-1282.1996
9. Lu M, Xia ZY, Bao L. Enhancement of antimycobacterial Th1-cell responses by a Mycobacterium bovis BCG prime-protein boost vaccination strategy. Cell Immunol. (2013) 285:111–7. doi: 10.1016/j.cellimm.2013.10.001
10. Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, et al. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science. (1999) 284:1520–3. doi: 10.1126/science.284.5419.1520
11. Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. (2002) 99:3684–9. doi: 10.1073/pnas.052548299
12. Forrellad MA, Klepp LI, Gioffré A, Sabio y, Garcia J, Morbidoni HR, Santangelo M, de la P, et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. (2013) 4:3–66. doi: 10.4161/viru.22329
13. Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KVS. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe. (2012) 12:669–81. doi: 10.1016/j.chom.2012.09.012
14. Dara Y, Volcani D, Shah K, Shin K, Venketaraman V. Potentials of host-directed therapies in tuberculosis management. J Clin Med. (2019) 8:1166. doi: 10.3390/jcm8081166
15. Palucci I, Delogu G. Host directed therapies for tuberculosis: futures strategies for an ancient disease. Chemotherapy. (2018) 63:172–80. doi: 10.1159/000490478
16. Lee A, Xie YL, Barry CE, Chen RY. Current and future treatments for tuberculosis. BMJ. (2020) 368:m216. doi: 10.1136/bmj.m216
17. Kohama H, Umemura M, Okamoto Y, Yahagi A, Goga H, Harakuni T, et al. Mucosal immunization with recombinant heparin-binding haemagglutinin adhesin suppresses extrapulmonary dissemination of Mycobacterium bovis bacillus Calmette-Guerin (BCG) in infected mice. Vaccine. (2008) 26:924–32. doi: 10.1016/j.vaccine.2007.12.005
18. Pethe K, Alonso S, Biet F, Delogu G, Brennan MJ, Locht C, et al. The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature. (2001) 412:190–4. doi: 10.1038/35084083
19. Thwaites GE, Nguyen DB, Nguyen HD, Hoang TQ, Do TTO, Nguyen TCT, et al. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N Engl J Med. (2004) 351:1741–51. doi: 10.1056/NEJMoa040573
20. Simmons CP, Thwaites GE, Quyen NTH, Chau TTH, Mai PP, Dung NT, et al. The clinical benefit of adjunctive dexamethasone in tuberculous meningitis is not associated with measurable attenuation of peripheral or local immune responses. J Immunol. (2005) 175:579–90. doi: 10.4049/jimmunol.175.1.579
21. Gräb J, Suárez I, van Gumpel E, Winter S, Schreiber F, Esser A, et al. Corticosteroids inhibit Mycobacterium tuberculosis-induced necrotic host cell death by abrogating mitochondrial membrane permeability transition. Nat Commun. (2019) 10:688. doi: 10.1038/s41467-019-08405-9
22. Menozzi FD, Rouse JH, Alavi M, Laude-Sharp M, Muller J, Bischoff R, et al. Identification of a heparin-binding hemagglutinin present in mycobacteria. J Exp Med. (1996) 184:993–1001. doi: 10.1084/jem.184.3.993
23. Abreu R, Quinn F, Giri PK. Role of the hepcidin-ferroportin axis in pathogen-mediated intracellular iron sequestration in human phagocytic cells. Blood Adv. (2018) 2:1089–100. doi: 10.1182/bloodadvances.2017015255
24. Selvaraj P, Anand SP, Harishankar M, Alagarasu K. Plasma 1, 25 dihydroxy vitamin D3 level and expression of vitamin d receptor and cathelicidin in pulmonary tuberculosis. J Clin Immunol. (2009) 29:470–8. doi: 10.1007/s10875-009-9277-9
25. Spizzo R, Nicoloso MS, Lupini L, Lu Y, Fogarty J, Rossi S, et al. miR-145 participates with TP53 in a death-promoting regulatory loop and targets estrogen receptor-alpha in human breast cancer cells. Cell Death Differ. (2010) 17:246–54. doi: 10.1038/cdd.2009.117
26. McGregor RA, Choi MS. microRNAs in the regulation of adipogenesis and obesity. Curr Mol Med. (2011) 11:304–16. doi: 10.2174/156652411795677990
27. Sabir N, Hussain T, Shah SZA, Peramo A, Zhao D, Zhou X. miRNAs in tuberculosis: new avenues for diagnosis and host-directed therapy. Front Microbiol. (2018) 9:602. doi: 10.3389/fmicb.2018.00602
28. Ouimet M, Koster S, Sakowski E, Ramkhelawon B, van Solingen C, Oldebeken S, et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol. (2016) 17:677–86. doi: 10.1038/ni.3434
29. Graff JW, Dickson AM, Clay G, McCaffrey AP, Wilson ME. Identifying functional microRNAs in macrophages with polarized phenotypes. J Biol Chem. (2012) 287:21816–25. doi: 10.1074/jbc.M111.327031
30. Li S, Yue Y, Xu W, Xiong S. MicroRNA-146a Represses Mycobacteria-Induced Inflammatory Response and Facilitates Bacterial Replication via Targeting IRAK-1 and TRAF-6. PLoS ONE. (2013) 8:e81438. doi: 10.1371/journal.pone.0081438
31. Li M, Wang J, Fang Y, Gong S, Li M, Wu M, et al. microRNA-146a promotes mycobacterial survival in macrophages through suppressing nitric oxide production. Sci Rep. (2016) 6:23351. doi: 10.1038/srep24555
32. Guo L, Zhao J, Qu Y, Yin R, Gao Q, Ding S, et al. microRNA-20a inhibits autophagic process by targeting ATG7 and ATG16L1 and favors mycobacterial survival in macrophage cells. Front Cell Infect Microbiol. (2016) 6:134. doi: 10.3389/fcimb.2016.00134
33. Guo L, Zhou L, Gao Q, Zhang A, Wei J, Hong D, et al. MicroRNA-144-3p inhibits autophagy activation and enhances Bacillus Calmette-Guérin infection by targeting ATG4a in RAW264.7 macrophage cells. PLoS ONE. (2017) 12:e0179772. doi: 10.1371/journal.pone.0179772
34. Guo W, Li JT, Pan X, Wei L, Wu JY. Candidate Mycobacterium tuberculosis genes targeted by human microRNAs. Protein Cell. (2010) 1:419–21. doi: 10.1007/s13238-010-0056-4
35. Singhal A, Jie L, Kumar P, Hong GS, Leow MK-S, Paleja B, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med. (2014) 6:263ra159. doi: 10.1126/scitranslmed.3009885
36. Viollet B, Guigas B, Garcia NS, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci. (2012) 122:253–70. doi: 10.1042/CS20110386
37. Lehrer RI, Lichtenstein AK, Ganz T. Defensins: antimicrobial and cytotoxic peptides of mammalian cells. Annu Rev Immunol. (1993) 11:105–28. doi: 10.1146/annurev.iy.11.040193.000541
38. Chaly YV, Paleolog EM, Kolesnikova TS, Tikhonov II, Petratchenko EV, Voitenok NN. Neutrophil alpha-defensin human neutrophil peptide modulates cytokine production in human monocytes and adhesion molecule expression in endothelial cells. Eur Cytokine Netw. (2000) 11:257–66.
39. Lillard JW, Boyaka PN, Chertov O, Oppenheim JJ, McGhee JR. Mechanisms for induction of acquired host immunity by neutrophil peptide defensins. Proc Natl Acad Sci USA. (1999) 96:651–6. doi: 10.1073/pnas.96.2.651
40. Rivas-Santiago CE, Rivas-Santiago B, León DA, Castañeda-Delgado J, Hernández Pando R. Induction of β-defensins by l-isoleucine as novel immunotherapy in experimental murine tuberculosis. Clin Exp Immunol. (2011) 164:80–9. doi: 10.1111/j.1365-2249.2010.04313.x
41. Napier RJ, Rafi W, Cheruvu M, Powell KR, Zaunbrecher MA, Bornmann W, et al. Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe. (2011) 10:475–85. doi: 10.1016/j.chom.2011.09.010
42. Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharm Exp Ther. (2005) 315:971–9. doi: 10.1124/jpet.105.084145
43. Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN. NOD-2 and TLR-4 signaling reinforces the efficacy of dendritic cells and reduces the dose of TB drugs against Mycobacterium tuberculosis. J Innate Immun. (2016) 8:228–42. doi: 10.1159/000439591
44. Chodisetti SB, Gowthaman U, Rai PK, Vidyarthi A, Khan N, Agrewala JN. Triggering through toll-like receptor 2 limits chronically stimulated T-helper type 1 cells from undergoing exhaustion. J Infect Dis. (2014) 211:486–96. doi: 10.1093/infdis/jiu472
45. Turgeon RD, Barry AR, Pearson GJ. Familial hypercholesterolemia. Can Family Phys. (2016) 62:32–7.
46. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin therapy reduces the Mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. (2013) 209:754–63. doi: 10.1093/infdis/jit550
47. Skerry C, Pinn ML, Bruiners N, Pine R, Gennaro ML, Karakousis PC. Simvastatin increases the in vivo activity of the first-line tuberculosis regimen. J Antimicrob Chemother. (2014) 69:2453–7. doi: 10.1093/jac/dku166
48. Agoro R, Taleb M, Quesniaux VFJ, Mura C. Cell iron status influences macrophage polarization. PLoS ONE. (2018) 13:e0196921. doi: 10.1371/journal.pone.0196921
49. Abreu R, Essler L, Loy A, Quinn F, Giri P. Heparin inhibits intracellular Mycobacterium tuberculosis bacterial replication by reducing iron levels in human macrophages. Sci Rep. (2018) 8:7296. doi: 10.1038/s41598-018-25480-y
50. Handa P, Thomas S, Morgan-Stevenson V, Maliken BD, Gochanour E, Boukhar S, et al. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis. J Leukoc Biol. (2019) 105:1015–26. doi: 10.1002/JLB.3A0318-108R
51. Wallis RS, Hafner R. Advancing host-directed therapy for tuberculosis. Nat Rev Immunol. (2015) 15:255–63. doi: 10.1038/nri3813
52. Dawson R, Condos R, Tse D, Huie ML, Ress S, Tseng C-H, et al. Immunomodulation with recombinant interferon-gamma-1b in pulmonary tuberculosis. PLoS ONE. (2009) 4:e6984. doi: 10.1371/journal.pone.0006984
53. Johnson JL, Ssekasanvu E, Okwera A, Mayanja H, Hirsch CS, Nakibali JG, et al. Randomized trial of adjunctive interleukin-2 in adults with pulmonary tuberculosis. Am J Respir Crit Care Med. (2003) 168:185–91. doi: 10.1164/rccm.200211-1359OC
54. Critchley JA, Young F, Orton L, Garner P. Corticosteroids for prevention of mortality in people with tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis. (2013) 13:223–37. doi: 10.1016/S1473-3099(12)70321-3
55. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. (2014) 511:99–103. doi: 10.1038/nature13489
56. Zimmermann N, Saiga H, Houthuys E, Moura-Alves P, Koehler A, Bandermann S, et al. Syndecans promote mycobacterial internalization by lung epithelial cells. Cell Microbiol. (2016) 18:1846–56. doi: 10.1111/cmi.12627
57. Kumar R, Dwivedi A, Kumar P, Kohli N. Tuberculous meningitis in BCG vaccinated and unvaccinated children. J Neurol Neurosurg Psychiatry. (2005) 76:1550–4. doi: 10.1136/jnnp.2005.065201
58. Kugelberg E. Immune evasion: mycobacteria hide from TLRs. Nat Rev Immunol. (2014) 14:62–3. doi: 10.1038/nri3604
59. Goldberg MF, Saini NK, Porcelli SA. Evasion of innate and adaptive immunity by Mycobacterium tuberculosis. Mol Genet Mycobacteria Second Ed. (2014) 747–72. doi: 10.1128/9781555818845.ch36
60. Hmama Z, Peña-D'iaz S, Joseph S, Av-Gay Y. Immunoevasion and immunosuppression of the macrophage by Mycobacterium tuberculosis. Immunol Rev. (2015) 264:220–32. doi: 10.1111/imr.12268
61. Liu PT, Modlin RL. Human macrophage host defense against Mycobacterium tuberculosis. Curr Opin Immunol. (2008) 20:371–6. doi: 10.1016/j.coi.2008.05.014
62. Ernst JD. Macrophage receptors for Mycobacterium tuberculosis. Infect Immun. (1998) 66:1277–81. doi: 10.1128/IAI.66.4.1277-1281.1998
63. Kang PB, Azad AK, Torrelles JB, Kaufman TM, Beharka A, Tibesar E, et al. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med. (2005) 202:987–99. doi: 10.1084/jem.20051239
64. Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, van Crevel R. Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol. (2011) 2011:405310. doi: 10.1155/2011/405310
65. Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. (2015) 264:182–203. doi: 10.1111/imr.12266
66. Yonekawa A, Saijo S, Hoshino Y, Miyake Y, Ishikawa E, Suzukawa M, et al. Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity. (2014) 41:402–13. doi: 10.1016/j.immuni.2014.08.005
67. Rajaram MV, Arnett E, Azad AK, Guirado E, Ni B, Gerberick AD, et al. M. tuberculosis-initiated human mannose receptor signaling regulates macrophage recognition and vesicle trafficking by FcR-gamma-Chain, Grb2, and SHP-1. Cell Rep. (2017) 21:126–40. doi: 10.1016/j.celrep.2017.09.034
68. Geijtenbeek TBH, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. (2009) 9:465–79. doi: 10.1038/nri2569
69. Yang Y, Kulka K, Montelaro RC, Reinhart TA, Sissons J, Aderem A, et al. A hydrolase of trehalose dimycolate induces nutrient influx and stress sensitivity to balance intracellular growth of Mycobacterium tuberculosis. Cell Host Microbe. (2014) 15:153–63. doi: 10.1016/j.chom.2014.01.008
70. Orme IM, Basaraba RJ. The formation of the granuloma in tuberculosis infection. Semin Immunol. (2014) 26:601–9. doi: 10.1016/j.smim.2014.09.009
71. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol. (2012) 3:411. doi: 10.3389/fimmu.2012.00411
72. Srinivasan L, Ahlbrand S, Briken V. Interaction of Mycobacterium tuberculosis with host cell death pathways. Cold Spring Harb Perspect Med. (2014) 4:a022459. doi: 10.1101/cshperspect.a022459
73. Ndlovu H, Marakalala MJ. Granulomas and inflammation: host-directed therapies for tuberculosis. Front Immunol. (2016) 7:434. doi: 10.3389/fimmu.2016.00434
74. Etna MP, Giacomini E, Severa M, Coccia EM. Pro-and anti-inflammatory cytokines in tuberculosis: a two-edged sword in TB pathogenesis. Semin Immunol. (2014) 26:543–51. doi: 10.1016/j.smim.2014.09.011
75. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor-alpha-neutralizing agent. N Engl J Med. (2001) 345:1098–104. doi: 10.1056/NEJMoa011110
76. Dorhoi A, Kaufmann SH. Tumor necrosis factor alpha in mycobacterial infection. Semin Immunol. (2014) 26:203–9. doi: 10.1016/j.smim.2014.04.003
77. Petruccioli E, Petrone L, Vanini V, Sampaolesi A, Gualano G, Girardi E, et al. IFNγ/TNFα specific-cells and effector memory phenotype associate with active tuberculosis. J Infect. (2013) 66:475–86. doi: 10.1016/j.jinf.2013.02.004
78. Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. (2010) 327:198–201. doi: 10.1126/science.1178178
79. Takeshita F, Patrawala L, Osaki M, Takahashi R, Yamamoto Y, Kosaka N, et al. Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol Ther. (2010) 18:181–7. doi: 10.1038/mt.2009.207
80. Kotsinas A, Sigala F, Garbis SD, Galyfos G, Filis K, Vougas K, et al. MicroRNAs determining inflammation as novel biomarkers and potential therapeutic targets. Curr Med Chem. (2015) 22:2666–79. doi: 10.2174/0929867322666150716113304
81. Jasenosky LD, Scriba TJ, Hanekom WA, Goldfeld AE. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunol Rev. (2015) 264:74–87. doi: 10.1111/imr.12274
82. Harding JS, Rayasam A, Schreiber HA, Fabry Z, Sandor M. Mycobacterium-infected dendritic cells disseminate granulomatous inflammation. Sci Rep. (2015) 5:15248. doi: 10.1038/srep15248
83. Mihret A. The role of dendritic cells in Mycobacterium tuberculosis infection. Virulence. (2012) 3:654–9. doi: 10.4161/viru.22586
84. Orme I. Adaptive immunity to mycobacteria. Curr Opin Microbiol. (2004) 7:58–61. doi: 10.1016/j.mib.2003.11.002
85. Gallegos AM, Pamer EG, Glickman MS. Delayed protection by ESAT-6-specific effector CD4+ T cells after airborne M. tuberculosis infection. J Exp Med. (2008) 205:2359–68. doi: 10.1084/jem.20080353
86. Lee J, Kornfeld H. Interferon-gamma regulates the death of M. tuberculosis infected macrophages. J Cell Death. (2010) 3:1–11. doi: 10.4137/JCD.S2822
87. Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. (2007) 96:41–101. doi: 10.1016/S0065-2776(07)96002-2
88. Milanés-Virelles MT, Garcia-Garcia I, Santos-Herrera Y, Valdés-Quintana M, Valenzuela-Silva CM, Jiménez-Madrigal G, et al. Adjuvant interferon gamma in patients with pulmonary atypical Mycobacteriosis: a randomized, double-blind, placebo-controlled study. BMC Infect Dis. (2008) 8:17. doi: 10.1186/1471-2334-8-17
89. Kaveh DA, Bachy VS, Hewinson RG, Hogarth PJ. Systemic BCG immunization induces persistent lung mucosal multifunctional CD4 TEM cells which expand following virulent mycobacterial challenge. PLoS ONE. (2011) 6:e21566. doi: 10.1371/journal.pone.0021566
90. Derrick SC, Yabe IM, Yang A, Morris SL. Vaccine-induced anti-tuberculosis protective immunity in mice correlates with the magnitude and quality of multifunctional CD4 T cells. Vaccine. (2011) 29:2902–9. doi: 10.1016/j.vaccine.2011.02.010
91. Forbes EK, Sander C, Ronan EO, McShane H, Hill AV, Beverley PC, et al. Multifunctional, high-level cytokine-producing Th1 cells in the lung, but not spleen, correlate with protection against Mycobacterium tuberculosis aerosol challenge in mice. J Immunol. (2008) 181:4955–64. doi: 10.4049/jimmunol.181.7.4955
92. Dong H, Lv Y, Zhao D, Barrow P, Zhou X. Defensins: the case for their use against mycobacterial infections. J Immunol Res. (2016) 2016:7515687. doi: 10.1155/2016/7515687
93. Gupta S, Cheung L, Pokkali S, Winglee K, Guo H, Murphy JR, et al. Suppressor cell-depleting immunotherapy with denileukin diftitox is an effective host-directed therapy for tuberculosis. J Infect Dis. (2017) 215:1883–7. doi: 10.1093/infdis/jix208
94. Knaul JK, Jörg S, Oberbeck-Mueller D, Heinemann E, Scheuermann L, Brinkmann V, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med. (2014) 190:1053–66. doi: 10.1164/rccm.201405-0828OC
95. Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature. (2014) 505:218–22. doi: 10.1038/nature12799
96. Cambier C, O'Leary SM, O'Sullivan MP, Keane J, Ramakrishnan L. Phenolic glycolipid facilitates mycobacterial escape from microbicidal tissue-resident macrophages. Immunity. (2017) 47:552–65. doi: 10.1016/j.immuni.2017.08.003
97. Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. (2002) 168:554–61. doi: 10.4049/jimmunol.168.2.554
98. Quesniaux V, Fremond C, Jacobs M, Parida S, Nicolle D, Yeremeev V, et al. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. (2004) 6:946–59. doi: 10.1016/j.micinf.2004.04.016
99. Reiling N, Hölscher C, Fehrenbach A, Kroger S, Kirschning CJ, Goyert S, et al. Cutting edge: Toll-like receptor (TLR) 2-and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. (2002) 169:3480–4. doi: 10.4049/jimmunol.169.7.3480
100. Hölscher C, Reiling N, Schaible UE, Hölscher A, Bathmann C, Korbel D, et al. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2,-4 and-9. Eur J Immunol. (2008) 38:680–94. doi: 10.1002/eji.200736458
101. Xu G, Wang J, Gao GF, Liu CH. Insights into battles between Mycobacterium tuberculosis and macrophages. Protein. (2014) 5:728–36. doi: 10.1007/s13238-014-0077-5
102. Saraav I, Singh S, Sharma S. Outcome of Mycobacterium tuberculosis and Toll-like receptor interaction: immune response or immune evasion? Immunol Cell Biol. (2014) 92:741–6. doi: 10.1038/icb.2014.52
103. Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. (2009) 7:355–66. doi: 10.1038/nrmicro2128
104. Clemens DL. Characterization of the Mycobacterium tuberculosis phagosome. Trends Microbiol. (1996) 4:113–8. doi: 10.1016/0966-842X(96)81528-9
105. van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, et al. M tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. (2007) 129:1287–98. doi: 10.1016/j.cell.2007.05.059
107. Simeone R, Sayes F, Song O, Gröschel MI, Brodin P, Brosch R, et al. Cytosolic access of Mycobacterium tuberculosis: critical impact of phagosomal acidification control and demonstration of occurrence in vivo. PLoS Pathog. (2015) 11:e1004650. doi: 10.1371/journal.ppat.1004650
108. Saleh M. The machinery of Nod-like receptors: refining the paths to immunity and cell death. Immunol Rev. (2011) 243:235–46. doi: 10.1111/j.1600-065X.2011.01045.x
109. Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA, et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog. (2009) 5:e1000500. doi: 10.1371/journal.ppat.1000500
110. Moreira-Teixeira L, Sousa J, McNab FW, Torrado E, Cardoso F, Machado H, et al. Type I IFN inhibits alternative macrophage activation during Mycobacterium tuberculosis infection and leads to enhanced protection in the absence of IFN-γ signaling. J Immunol. (2016) 197:4714–26. doi: 10.4049/jimmunol.1600584
111. Da Costa C, Walker B, Bonavia A. Tuberculosis vaccines-state of the art, and novel approaches to vaccine development. Int J Infect Dis. (2015) 32:5–12. doi: 10.1016/j.ijid.2014.11.026
112. Orme IM. tuberculosis vaccine types and timings. Clin Vaccine Immunol. (2015) 22:249–57. doi: 10.1128/CVI.00718-14
113. Pahari S, Kaur G, Aqdas M, Negi S, Chatterjee D, Bashir H, et al. Bolstering immunity through pattern recognition receptors: a unique approach to control tuberculosis. Front Immunol. (2017) 8:906. doi: 10.3389/fimmu.2017.00906
114. Chopra P, Singh A, Koul A, Ramachandran S, Drlica K, Tyagi AK, et al. Cytotoxic activity of nucleoside diphosphate kinase secreted from Mycobacterium tuberculosis. Eur J Biochem. (2003) 270:625–34. doi: 10.1046/j.1432-1033.2003.03402.x
115. Sun J, Wang X, Lau A, Liao T-YA, Bucci C, Hmama Z. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages. PLoS ONE. (2010) 5:e8769. doi: 10.1371/journal.pone.0008769
116. Bach H, Wong D, Av-Gay Y. Mycobacterium tuberculosis PtkA is a novel protein tyrosine kinase whose substrate is PtpA. Biochem J. (2009) 420:155–62. doi: 10.1042/BJ20090478
117. Bach H, Papavinasasundaram KG, Wong D, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell host Microbe. (2008) 3:316–22. doi: 10.1016/j.chom.2008.03.008
118. Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci USA. (2011) 108:19371–6. doi: 10.1073/pnas.1109201108
119. Bento CF, Empadinhas N, Mendes V. Autophagy in the fight against tuberculosis. DNA Cell Biol. (2015) 34:228–42. doi: 10.1089/dna.2014.2745
120. Jo E-K. Autophagy as an innate defense against mycobacteria. Pathog Dis. (2013) 67:108–18. doi: 10.1111/2049-632X.12023
121. Chin KL, Anis FZ, Sarmiento ME, Norazmi MN, Acosta A. Role of interferons in the development of diagnostics, vaccines, and therapy for tuberculosis. J Immunol Res. (2017) 2017:5212910. doi: 10.1155/2017/5212910
122. Fairchild KD, Hudson RG, Douglas SD, McKenzie SE, Polin RA. Effect of gamma interferon on expression of Fc gamma receptors in monocytes of newborn infants and adults. Clin Diagn Lab Immunol. (1996) 3:464–9. doi: 10.1128/CDLI.3.4.464-469.1996
123. Keskinen P, Ronni T, Matikainen S, Lehtonen A, Julkunen I. Regulation of HLA class I and II expression by interferons and influenza A virus in human peripheral blood mononuclear cells. Immunology. (1997) 91:421–9. doi: 10.1046/j.1365-2567.1997.00258.x
124. Zhou F. Molecular mechanisms of IFN- γ to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol. (2009) 28:239–60. doi: 10.1080/08830180902978120
125. Lalvani A, Connell DW. Dissecting the immunological, antimicrobial and clinical effects of vitamin D therapy in tuberculosis. Pathog Glob Health. (2012) 106:378–9. doi: 10.1179/2047772412Z.00000000087
126. Liu PT, Wheelwright M, Teles R, et al. MicroRNA-21 targets the vitamin D-dependent antimicrobial pathway in leprosy. Nat Med. (2012) 18:267–73. doi: 10.1038/nm.2584
127. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. (2015) 16:461–72. doi: 10.1038/nrm4024
128. Cheng CY, Gutierrez NM, Marzuki MB, Lu X, Foreman TW, Paleja B, et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci Immunol. (2017) 2:aaj1789. doi: 10.1126/sciimmunol.aaj1789
129. Yu X, Li C, Hong W, Pan W, Xie J. Autophagy during Mycobacterium tuberculosis infection and implications for future tuberculosis medications. Cell Signal. (2013) 25:1272–8. doi: 10.1016/j.cellsig.2013.02.011
130. Vergne I, Chua J, Singh SB, Deretic V. Cell biology of Mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol. (2004) 20:367–94. doi: 10.1146/annurev.cellbio.20.010403.114015
131. Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. (2001) 2:569–86. doi: 10.1038/35085034
132. de Jonge MI, Pehau-Arnaudet G, Fretz MM, Romain F, Bottai D, Brodin P, et al. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J Bacteriol. (2007) 189:6028–34. doi: 10.1128/JB.00469-07
133. Abdallah AM, Van Pittius NCG, Champion PAD, Cox J, Luirink J, Vandenbroucke-Grauls CM, et al. Type VII secretion—mycobacteria show the way. Nat Rev Microbiol. (2007) 5:883–91. doi: 10.1038/nrmicro1773
134. Houben ENG, Korotkov KV, Bitter W. Take five - Type VII secretion systems of Mycobacteria. Biochim Biophys Acta. (2014) 1843:1707–16. doi: 10.1016/j.bbamcr.2013.11.003
135. Polajnar M, Dietz MS, Heilemann M, Behrends C. Expanding the host cell ubiquitylation machinery targeting cytosolic Salmonella. EMBO Rep. (2017) 18:1572–85. doi: 10.15252/embr.201643851
136. O'Donnell H, Pham OH, Li L, Atif SM, Lee S-J, Ravesloot MM, et al. Toll-like receptor and inflammasome signals converge to amplify the innate bactericidal capacity of T helper 1 cells. Immunity. (2014) 40:213–24. doi: 10.1016/j.immuni.2013.12.013
137. Minakami R, Sumimotoa H. Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (nox) family. Int J Hematol. (2006) 84:193–8. doi: 10.1532/IJH97.06133
138. Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol. (2004) 2:820–32. doi: 10.1038/nrmicro1004
139. Webb JL, Harvey MW, Holden DW, Evans TJ. Macrophage nitric oxide synthase associates with cortical actin but is not recruited to phagosomes. Infect Immun. (2001) 69:6391–400. doi: 10.1128/IAI.69.10.6391-6400.2001
140. Hu Y, Coates AR. Acute and persistent Mycobacterium tuberculosis infections depend on the thiol peroxidase TpX. PLoS ONE. (2009) 4:e5150. doi: 10.1371/journal.pone.0005150
141. Sato K, Akaki T, Tomioka H. Differential potentiation of anti-mycobacterial activity and reactive nitrogen intermediate-producing ability of murine peritoneal macrophages activated by interferon-gamma (IFN-gamma) and tumour necrosis factor-alpha (TNF-alpha). Clin Exp Immunol. (1998) 112:63. doi: 10.1046/j.1365-2249.1998.00554.x
142. Dutta NK, Pinn ML, Karakousis PC. Metformin adjunctive therapy does not improve the sterilizing activity of the first-line antitubercular regimen in mice. Antimicrob Agents Chemother. (2017) 61:e00652-17. doi: 10.1128/AAC.00652-17
143. Restrepo BI. Metformin: candidate host-directed therapy for tuberculosis in diabetes and non-diabetes patients. Tuberculosis. (2016) 101:S69–72. doi: 10.1016/j.tube.2016.09.008
144. Russell DG, Cardona P-J, Kim M-J, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol. (2009) 10:943–8. doi: 10.1038/ni.1781
145. Geisel RE, Sakamoto K, Russell DG, Rhoades ER. In vivo activity of released cell wall lipids of Mycobacterium bovis bacillus Calmette-Guerin is due principally to trehalose mycolates. J Immunol. (2005) 174:5007–15. doi: 10.4049/jimmunol.174.8.5007
146. Puissegur M-P, Lay G, Gilleron M, Botella L, Nigou J, Marrakchi H, et al. Mycobacterial lipomannan induces granuloma macrophage fusion via a TLR2-dependent, ADAM9-and beta1 integrin-mediated pathway. J Immunol. (2007) 178:3161–9. doi: 10.4049/jimmunol.178.5.3161
147. Yang C-S. Advancing host-directed therapy for tuberculosis. Microbial Cell. (2017) 4:105–7. doi: 10.15698/mic2017.03.565
148. Kang Y, Choi N, Seong J, Heo E, Koo B, Hwang S, et al. The effects of statin use on the development of tuberculosis among patients with diabetes mellitus. Int J Tuberculosis Lung Dis. (2014) 18:717–24. doi: 10.5588/ijtld.13.0854
149. Luo M, Fadeev EA, Groves JT. Mycobactin-mediated iron acquisition within macrophages. Nat Chem Biol. (2005) 1:149–53. doi: 10.1038/nchembio717
150. Sritharan M. Iron homeostasis in Mycobacterium tuberculosis: mechanistic insights into siderophore-mediated iron uptake. J Bacteriol. (2016) 198:2399–409. doi: 10.1128/JB.00359-16
151. Fang Z, Sampson SL, Warren RM, van Pittius NCG, Newton-Foot M. Iron acquisition strategies in mycobacteria. Tuberculosis. (2015) 95:123–30. doi: 10.1016/j.tube.2015.01.004
152. Boelaert JR, Vandecasteele SJ, Appelberg R, Gordeuk VR. The effect of the host's iron status on tuberculosis. J Infect Dis. (2007) 195:1745–53. doi: 10.1086/518040
153. Ganz T, Nemeth E. Regulation of iron acquisition and iron distribution in mammals. Biochim Biophys Acta. (2006) 1763:690–9. doi: 10.1016/j.bbamcr.2006.03.014
154. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. (2004) 306:2090–3. doi: 10.1126/science.1104742
155. Ganz T. Macrophages and iron metabolism. Microbiol Spectr. (2016) 4: 803–12. doi: 10.1128/microbiolspec.MCHD-0037-2016
156. Ganz T. Hepcidin and iron regulation, 10 years later. Blood. (2011) 117:4425–33. doi: 10.1182/blood-2011-01-258467
157. Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol. (2015) 15:500–10. doi: 10.1038/nri3863
158. Poggiali E, Cassinerio E, Zanaboni L, Cappellini MD. An update on iron chelation therapy. Blood Transfus. (2012) 10:411. doi: 10.2450/2012.0008-12
159. Meyer D. Iron chelation as therapy for HIV and Mycobacterium tuberculosis co-infection under conditions of iron overload. Curr Pharm Des. (2006) 12:1943–7. doi: 10.2174/138161206777442164
160. Cronjé L, Edmondson N, Eisenach KD, Bornman L. Iron and iron chelating agents modulate Mycobacterium tuberculosis growth and monocyte-macrophage viability and effector functions. FEMS Immunol. (2005) 45:103–12. doi: 10.1016/j.femsim.2005.02.007
161. Gomes MS, Dom G, Pedrosa J, Boelaert JR, Appelberg R. Effects of iron deprivation on Mycobacterium avium growth. Tuber Lung Dis. (1999) 79:321–8. doi: 10.1054/tuld.1999.0216
162. Steinbicker AU, Muckenthaler MU. Out of balance-systemic iron homeostasis in iron-related disorders. Nutrients. (2013) 5:3034–61. doi: 10.3390/nu5083034
163. Babitt JL, Lin HY. Molecular mechanisms of hepcidin regulation: implications for the anemia of CKD. Am J Kidney Dis. (2010) 55:726–41. doi: 10.1053/j.ajkd.2009.12.030
164. Singh B, Arora S, Agrawal P, Gupta SK. Hepcidin: a novel peptide hormone regulating iron metabolism. Clin Chim Acta. (2011) 412:823–30. doi: 10.1016/j.cca.2011.02.014
165. Poli M, Asperti M, Ruzzenenti P, Regoni M, Arosio P. Hepcidin antagonists for potential treatments of disorders with hepcidin excess. Front Pharmacol. (2014) 5:86. doi: 10.3389/fphar.2014.00086
166. Asperti M, Stuemler T, Poli M, Gryzik M, Lifshitz L, Meyron-Holtz EG, et al. Heparanase overexpression reduces hepcidin expression, affects iron homeostasis and alters the response to inflammation. PLoS ONE. (2016) 11:e0164183. doi: 10.1371/journal.pone.0164183
167. Poli M, Girelli D, Campostrini N, Maccarinelli F, Finazzi D, Luscieti S, et al. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood. (2011) 117:997–1004. doi: 10.1182/blood-2010-06-289082
168. Poli M, Asperti M, Naggi A, Campostrini N, Girelli D, Corbella M, et al. Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo. Blood. (2014) 123:1564–73. doi: 10.1182/blood-2013-07-515221
169. Rochette L, Gudjoncik A, Guenancia C, Zeller M, Cottin Y, Vergely C. The iron-regulatory hormone hepcidin: a possible therapeutic target? Pharmacol Ther. (2015) 146:35–52. doi: 10.1016/j.pharmthera.2014.09.004
Keywords: lipid metabolism, iron metabolism, macrophage, tuberculosis, Mycobacterium
Citation: Abreu R, Giri P and Quinn F (2020) Host-Pathogen Interaction as a Novel Target for Host-Directed Therapies in Tuberculosis. Front. Immunol. 11:1553. doi: 10.3389/fimmu.2020.01553
Received: 27 November 2019; Accepted: 12 June 2020;
Published: 21 July 2020.
Edited by:
Pere-Joan Cardona, Germans Trias i Pujol Health Science Research Institute (IGTP), SpainReviewed by:
Paulo Bettencourt, University of Oxford, United KingdomVarun Jaiswal, Shoolini University of Biotechnology and Management Sciences, India
Copyright © 2020 Abreu, Giri and Quinn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fred Quinn, ZnF1aW5uQHVnYS5lZHU=