 Seong-Hwan Park1†
Seong-Hwan Park1† Yuseok Moon1,2*
Yuseok Moon1,2*- 1Laboratory of Mucosal Exposome and Biomodulation, Department of Biomedical Sciences, Pusan National University, Yangsan, South Korea
- 2Biomedical Research Institute, Pusan National University, Yangsan, South Korea
Type B 8-keto-trichothecenes are muco-active mycotoxins that exist as inevitable contaminants in cereal-based foodstuffs. Gut-associated inflammation is an early frontline response during human and animal exposure to these mycotoxins. Despite various tools for chemical identification, optimized biomonitoring of sentinel response-associated biomarkers is required to assess the specific proinflammatory actions of 8-keto-trichothecenes in the gut epithelial barrier. In the present study, intoxication with 8-keto-trichothecenes in human intestinal epithelial cells was found to trigger early response gene 1 product (EGR-1) that plays crucial roles in proinflammatory chemokine induction. In contrast, epithelial exposure to 8-keto-trichothecenes resulted in downregulated expression of nuclear factor NF-kappa-B p65 protein, a key transcription factor, during general inflammatory responses in the gut. Based on the early molecular patterns of expression, the inflammation-inducing activity of 8-keto-trichothecenes was quantified using intestinal epithelial cells with dual reporters for EGR-1 and p65 proteins. EGR-1-responsive elements were linked to luciferase reporter while p65 promoter was bound to secretory alkaline phosphatase (SEAP) reporter. In response to conventional inflammagens such as endotoxins and cytokines such as TNF-α, both luciferase and SEAP activity were elevated in a dose-dependent manner. However, as expected from the mechanistic evaluation, 8-keto-trichothecene-exposed dual reporters of luciferase and SEAP displayed contrasting expression patterns. Furthermore, 8-keto-trichothecene-elevated EGR-1-responsive luciferase activity was improved by deficiency of PSMA3, an α-type subunit of the 20S proteasome core complex for ubiquitin-dependent EGR-1 degradation. This molecular event-based dual biomonitoring in epithelial cells is a promising supplementary tool for detecting typical molecular inflammatory pathways in response to 8-keto-trichothecenes in the food matrix.
Introduction
Trichothecenes are a group of over 200 sesquiterpenoid fungal metabolites, some of which are health-threatening mycotoxins leading to eukaryotic ribosome stalling (1–4) or cellular ubiquitin depletion (5, 6). Extensive investigation into toxic trichothecenes have been made due to their widespread contamination in the environment (7, 8) and agricultural commodities such as grains (9–11). Moreover, some of trichothecenes are released from mold-infected field crops into surface soil water and drainage systems (7, 8). Trichothecenes are characterized by a tetracyclic scirpenol ring system and are classified into four groups (types A–D), depending on the presence of the functional moiety (12, 13). Field fungi such as Fusarium species mainly produce type A and B trichothecenes. Type A trichothecenes are characterized by a hydroxyl or acyl moiety at the R1, R2, R3, or R5 position of the tetracyclic scirpenol ring. The Type B group toxins are typically named as 8-keto-trichothecenes due to a ketone group at R5. The ribosome-inactivating activity of trichothecenes has been linked to the presence of an intact 9,10 double bond, C-12,13 epoxide, and substitution of both R1 and R2 (13–16).
Epidemiological investigations have demonstrated positive associations between human exposure to trichothecene and gastrointestinal disorders including gastroenteritis and esophageal cancer (17–19). In particular, the 8-keto-trichothecenes such as deoxynivalenol (DON) and nivalenol (NIV) can produce a variety of mycotoxicosis in humans and animals. Acutely intoxicated animals display severe injuries in gastrointestinal tract and highly-proliferating immune-related cells, leading to diarrhea, vomiting, gastrointestinal hemorrhage ad leukocytopenia (10, 20–22). In addition to the direct tissue injuries, DON-exposed mice show cytokine storm syndromes during mucosal and systemic intoxication (23–26). Epithelial responses are the early gastrointestinal event in response to diet-derived contaminants. The gut epithelial linings sense luminal insults and facilitate molecular reprograming during mucosal and systemic exposure to trichothecenes (27–29). The epithelial barrier responds to luminal insults that activate stress-responsive cellular signaling such as mitogen-activated kinases or other pathologic biochemical pathways. The activated stress kinases then alter profiles of immediately responsive genes such as early growth response gene 1 (EGR-1), which contribute to maintaining tissue integrity during pathologic states by itself or through its regulated gene products (30–32). Moreover, in response to mucosal microbial antigens, pattern recognition receptors are activated and transmit danger signals via proinflammatory transcription factors mainly including nuclear factor-κB (NF-κB) (33, 34). EGR-1 is also involved in diverse inflammatory cellular events including inflammatory cell migration and cytokine production (35–38).
In spite of a variety of detection methods for trichothecene metabolites, conventional analytical technologies are often insufficient to identify their biological activity in the contaminated matrix. We thus developed an enterocyte-based bioassay system measuring epithelial stress responses to 8-keto-trichothecenes. In the present study, we evaluated EGR-1 as a crucial biomarker in the epithelial exposure to 8-keto-trichothecenes. An optimized EGR-1-reporting system was constructed in human intestinal cells by combining with a representative inflammatory biomarker, NF-κB, in response to the unavoidable trichothecenes. This challenge could provide useful biomaterials to measure the early biological activities of proinflammatory trichothecenes in food, feed, and the environment.
Materials and Methods
Cell Culture Conditions and Reagents
The intestinal epithelial cell line, HCT-8 (passage 13), was purchased from ATCC (Rockville, MD, USA). Based on our previous report (39), HCT-8 cells were maintained in RPMI 1640 medium (Welgene, Daegu, South Korea) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS, Welgene), 50 units/ml penicillin and 50 μg/ml streptomycin (Welgene) in a 5% CO2 humidified incubator at 37°C. Cell counting was performed by trypan blue dye exclusion (Sigma-Aldrich chemical company, St. Louis, MO, USA) using a hemocytometer. All other chemicals were obtained from Sigma-Aldrich except for TNF-α (Peprotech, Rocky Hill, NJ, USA).
Mouse Experiments
C57BL/6 mice (6 weeks old with an average weight of 16–18 g) were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Based on our previous report (40), mice were acclimated for 14 days prior to the experiments and maintained at 25°C and 45–55% relative humidity under 12-h light/dark cycles. Mice were housed three per cage and provided sufficient food and water. Animal care and experimental procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee. Mice were maintained in environmentally protected cages consisting of a transparent polypropylene body and a stainless-steel wire top cover. This animal study was approved by the Pusan National University Institutional Animal Care and Use Committee (PNU-IACUC) (PNU-2013-0291).
Immunohistochemistry
Based on our previous report (41), formalin-fixed paraffin-embedded tissues were cut, deparaffinized, and rehydrated. The tissue sections were heated in 10 mM sodium acetate (pH 9.0) for 5 min at 121°C for antigen retrieval. To remove endogenous peroxidase, tissues were bathed in a 1% (v/v) H2O2-PBS solution for 15 min at room temperature in the dark. The samples were then washed with 0.1% TBS-T, and blocked with 3% (w/v) FBS in PBS for 1 h. The blocked samples were incubated overnight at 4°C with the primary antibodies (diluted at 1:200). After washing three times with 0.1% TBS-T, samples were incubated with the horseradish peroxidase-conjugated secondary antibody (diluted at 1:200) for 2 h at room temperature and then washed three times with 0.1% TBS-T. The bound antibodies were identified using freshly prepared substrate buffer (0.05% [w/v] diaminobenzidine (DAB; Sigma-Aldrich) and 0.01% [v/v] H2O2 in PBS) for 5 min. After a final wash in distilled water, the sections were counterstained with hematoxylin (Bioworld, Dublin, Ohio, USA) solution for 1 min and dehydrated. Sections were examined at various magnifications using an Eclipse Ts2R (Nikon Instruments Inc., Melville, NY, USA). The ratio of the target protein-expressing cells was measured using HistoQuest software (TissueGnostics, Vienna, Austria).
Western Immunoblot Analysis
Protein expression levels were compared by western immunoblot analysis using rabbit polyclonal anti-β-actin (actin) antibody, rabbit polyclonal anti-EGR-1, rabbit polyclonal anti-NF-κB p65 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and rabbit polyclonal phosphor-NF-κB p65 (Cell Signaling Technology, Beverly, MA, USA). Based on our previous report (42), the cells were washed with ice-cold phosphate buffer, lysed in boiling lysis buffer [1% (w/v) SDS, 1.0 mM sodium ortho-vanadate, and 10 mM Tris, pH 7.4], and sonicated for 5 s. Lysates containing proteins were quantified using a BCA protein assay kit (Welgene). Next, 15 μg of proteins were separated by Bio-Rad mini gel electrophoresis, and transferred to a PVDF membrane (Pall Corporation, Port Washington, NY, USA). The blots were then blocked for 1 h with 5% skimmed milk in Tris-buffered saline plus Tween 0.1% (TBST). The samples were probed with each antibody (diluted at 1:1,000) for an additional 2 h at room temperature or overnight at 4°C. After washing three times with TBST, blots were incubated with horseradish-conjugated secondary antibody (diluted at 1:5,000) for 2 h and then washed thrice with TBST. Finally, the proteins were detected using a pico-enhanced peroxidase detection system (ELPIS Biotech., Inc., Taejon, South Korea).
Reverse Transcription and Real Time PCR
Based on our previous reports (40, 41), RNA was extracted using RiboEx (GeneAll Biotech, Seoul, South Korea) according to the manufacturer's instructions. RNA (3 μg) from each sample was transcribed to cDNA using Prime RT premix (Genetbio, Nonsan, South Korea). During real time PCR, 6-carboxyl fluorescein was used as the fluorescent reporter dye to detect the amplified cDNA. Real time PCR was conducted using an iCycler Thermal Cycler (Bio-Rad) with the following conditions: denaturation at 95°C for 15 min followed by cycles of denaturation at 95°C for 20 s, annealing at 59°C for 30 s, and elongation at 72°C for 30 s. Each sample was tested in triplicate to ensure statistical significance. Quantification of relative gene expression was performed using the comparative Ct method. The Ct-value is defined as the point at which a statistically significant increase in fluorescence occurs. The number of PCR cycles (Ct) required for the 6-carboxylfluorescein intensities to exceed the threshold of just above the background level was calculated for the test and reference reactions. In all experiments, GAPDH was used as the endogenous control. The 5′ forward and 3′ reverse PCR primers for amplifying each gene were as follows: human p65, 5′-GGC GAG AGG AGC ACA GAT AC-3′ and 5′-ATC TTG AGC TCG GCA GTG TT-3′; human PSMA3, 5′-GCT CAA TCG GCA CTG GGT AT-3′ and 5′-GGC CACA TCT GTC TGC AAG AT-3′; and human GAPDH, 5′-TCA ACG GAT TTG GTC GTA TT-3′ and 5′-CTG TGG TCA TGA GTC CTT CC-3′.
Construction of Plasmids
Empty vector or shRNA of PSMA3 (shPSMA3) were constructed using the pMKO.1 GFP vector purchased from Addgene (a gift from William Hahn). The inserted PSMA3 shRNA targeted the sequence: 5′-TAA AGC TTT TGA ACT AGA A-3′. The luciferase reporter construct, pEBS14-LUC plasmid, which contained four EGR-1 binding sites, was provided by Dr. Seung-Joon Baek (University of Tennessee). Human p65 promoter (−999/+92) was generated by RT-PCR using human genomic DNA from HCT-8 cells with the following primers: 5′-AAG ACG AAT TCA GAG AAA ATA AAA A-3′ and 5′-TGC ACT ACA GAC GAG CCA TT-3′. The resulting 1,091 bp construct was cloned by Kpn1/Xho1 excision, and inserted in the sense orientation into the reporter plasmid, pGL4.14-SEAP-hygro, in which the luciferase gene cassette was replaced with the SEAP gene cassette through HindIII and HpaI excision followed by blunt end formation using DNA polymerase I (New England Biolabs, Ipswich, MA, USA). The flag-tagged SR-IkB expression vector has been described previously (43).
Luciferase Assay
Based on our previous report (40), cells were washed with cold PBS, lysed with passive lysis buffer (Promega), and then centrifuged at 13,800 × g for 10 min. The supernatant was collected and stored at −80°C until measurement. Luciferase activity was measured with a model TD-20/20 dual mode luminometer (Turner Designs, Sunnyvale, CA) after briefly mixing the supernatant (10 μl) with 40 μl of firefly luciferase assay substrate solution, followed by stopping with 50 μl of Renilla luciferase stop solution (Promega). The firefly luciferase activity was divided by the Renilla luciferase activity for normalization.
Secreted Alkaline Phosphatase (SEAP) Assay
Transformed HCT-8 cells were seeded in a 24-well-plate at a density 5 × 104 cells/well and were then treated with various concentrations of mycotoxins and other chemicals for 24 or 72 h at 37°C. Based on our previous report (13), the collected culture medium (400 μl) was heated at 65°C for 5 min and then 100 μl of the heated supernatant was mixed with 100 μl of 2× SEAP assay buffer (2M diethanolamine, 1 mM MgCl2, 20 mM L-homoarginine). The mixture was incubated at 37°C for 10 min and the reaction was terminated by adding 20 μl of 120 mM p-nitrophenylphosphate dissolved in 1× SEAP assay buffer. The final mixture was further incubated at 37°C in the dark. The absorbance of the reaction mixture was measured at 405 nm using an enzyme-linked immunosorbent assay (ELISA) reader (Molecular Devices, Sunnyvale, CA, USA).
Statistics and Reproducibility
Statistical analyses were performed using GraphPad Prism v. 8.01 (La Jolla, CA, USA). For comparative analysis of two groups of data, Student's t-test was performed. For comparative analysis of multiple groups, data were subjected to analysis of variance (ANOVA) with Newman-Keuls method as a post-hoc ANOVA assessment. All in vitro evaluations are representative of two or three independent experiments. Details of the number of biological replicates and the assays are given in each figure legend.
Results
8-keto-trichothecenes Induce EGR-1 and Chemokine Expression in Intestinal Epithelial Cells
The proinflammatory actions of 8-keto-trichothecenes initiate the deleterious progress of acute and chronic diseases in humans and domestic animals. In response to toxic insult, the stress-responsive transcription factor EGR-1 was assessed as an early trigger of inflammatory gene expression. Gastrointestinal exposure to DON, the most prevalent trichothecene in the food chain, caused notable induction of the EGR-1 protein in the gut epithelial layer compared to that in the lamina propria (Figure 1A). Next, we assessed the proinflammatory chemokine expression in trichothecene-exposed HCT-8 cells, which are widely used human intestinal epithelial cells (IEC) to model inflammatory and infectious diseases (44, 45). In particular, the ileocecum of the small intestine, which is the source of the HCT-8 cell line, is one of the regions that is most susceptible to ribosomal inactivation-associated xenobiotic stress (46, 47). EGR-1 is a crucial transcription factor of DON-induced proinflammatory cytokines, as proven by the expression of interleukin-8 (IL-8), chemokine (C-X-C motif) ligand 1 (CXCL1), and monocyte chemoattractant protein-1 (MCP-1) in human IECs (Figure 1B). In addition to DON, other trichothecenes and mycotoxins were also compared for their effects on EGR-1 expression in HCT-8 cells.
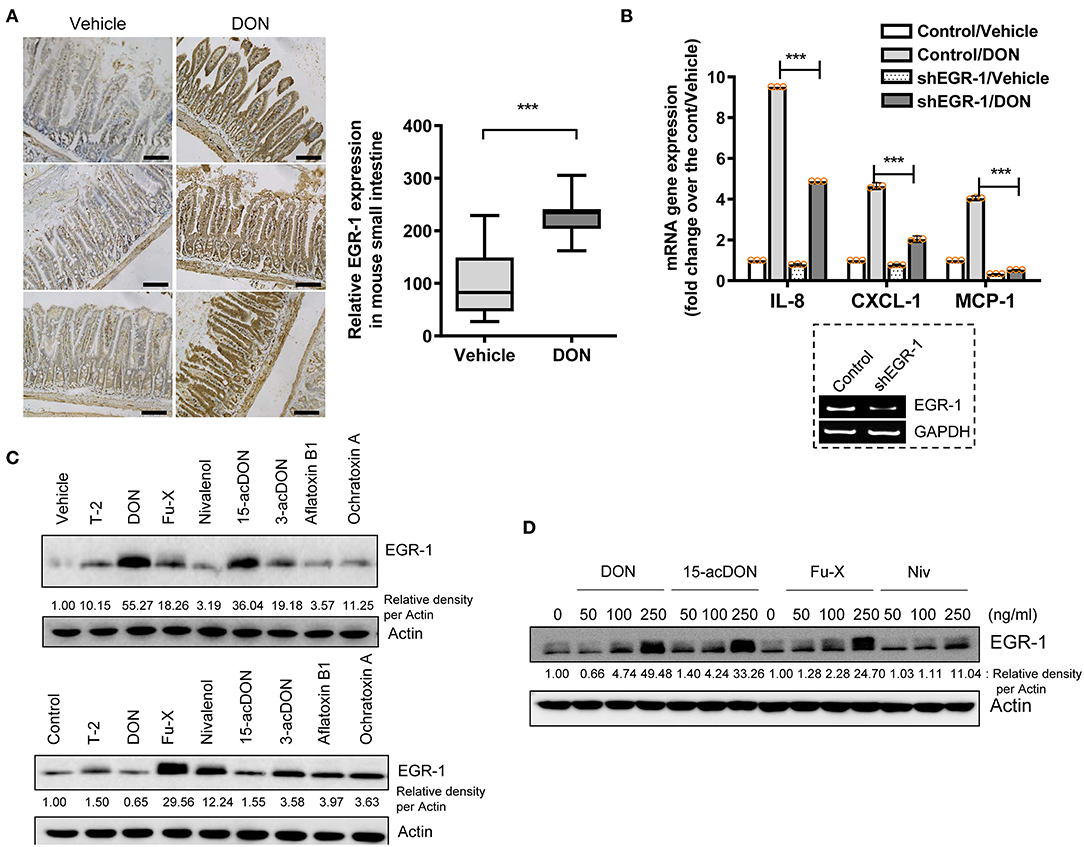
Figure 1. Exposure to 8-keto-trichothecenes induces gut EGR-1 expression. (A) Small intestinal tissues from 6-week-old male mice exposed to vehicle or 25 mg/kg DON for 12 h were subjected to IHC analysis for EGR-1 (n = 4, each group; original magnification, ×200, black scale bar, 0.2 μm). Right panel shows the quantitative analysis of tissue EGR-1 levels in vehicle or DON-treated mice. Results are shown as box-and-whisker plots (min–max) and asterisks (***) indicate significant difference from the control (p < 0.001). (B) Control- or shEGR-1-transfected HCT-8 cells were treated with vehicle or 500 ng/ml DON for 1 h. mRNA expression was measured using RT real-time PCR. Results are shown as mean values ± SD and asterisks (***) indicate a significant difference (p < 0.001, n = 3). The figure in the box represents the relative expression of EGR-1 with vehicle or shEGR-1. (C) HCT-8 cells were treated with vehicle or each mycotoxin at 20% cell growth-inhibitory doses (ID20) (3 ng/ml T-2 toxin, 500 ng/ml DON, 400 ng/ml Fu-X, 300 ng/ml NIV, 300 ng/ml 15-acDON, 2,500 ng/ml 3-acetyl DON, 1 μM aflatoxin B1, or 1 μM Ochratoxin A) for 1.5 h (upper panel) or 5 h (lower panel). (D) HCT-8 cells were exposed to each dose of DON and 15-acDON for 1.5 h and to each dose of Fu-X and NIV for 5 h. Total cell lysates were subjected to western blot analysis.
Other mycotoxins were assessed for their effects on EGR-1 expression in human IECs at 20% cell growth-inhibitory doses (ID20) and optimal induction times (1.5 or 5 h treatment). In particular, 8-keto-trichothecenes such as DON, fusarenon-X or 4-acetyl nivalenol (Fu-X), 15-acetyl DON (15-acDON), and NIV were among the strongest agents to induce EGR-1 expression (Figure 1C). Furthermore, we evaluated the dose responses of EGR-1 induction by these 8-keto-trichothecenes. Treatment with 8-keto-trichothecenes induced EGR-1 expression in human IECs in a dose-dependent manner (Figure 1D). In subsequent experiments, EGR-1 was assumed as a potent early biomarker of proinflammatory insult in response to 8-keto-trichothecenes.
PSMA3 Deficiency Enhanced 8-keto-trichothecene-induced EGR-1 Levels in IECs
To maintain persistent levels of induced EGR-1 as a crucial readout of 8-keto-trichothecene toxicity, its protein stability needs to be improved. Biochemically, proteasome subunit alpha type-3 (PSMA3)/proteasome component C8 (PRC8) is the α-type subunit of the 20S proteasome core complex that interacts with EGR-1, leading to its ubiquitin-dependent proteasome degradation (48). Therefore, we assessed whether PSMA3 deficiency improves 8-keto-trichothecene-induced EGR-1 expression in IECs. PSMA3-suppressed IECs displayed enhanced levels of EGR-1 expression in response to DON exposure (Figure 2A). To evaluate EGR-1-mediated transcriptional regulation, we applied a luciferase reporter construct, pEBS14-LUC plasmid, which contained EGR-1 binding sites. Furthermore, DON-induced EGR-1-mediated transcriptional activity was enhanced in stable PSMA3-suppressed IECs (Figure 2B). Other 8-keto-trichothecenes including 15-acDON, Fu-X, and NIV also increased EGR-1 protein levels (Figures 2C,E,G) and transcription activity (Figures 2D,F,H) in a dose-dependent manner, which was enhanced in PSMA3-deficient IECs compared to the levels in control vector-transfected IECs. In conclusion, regulation of PSMA3 action improved the EGR-1-mediated responses to 8-keto-trichothecenes in human IECs.
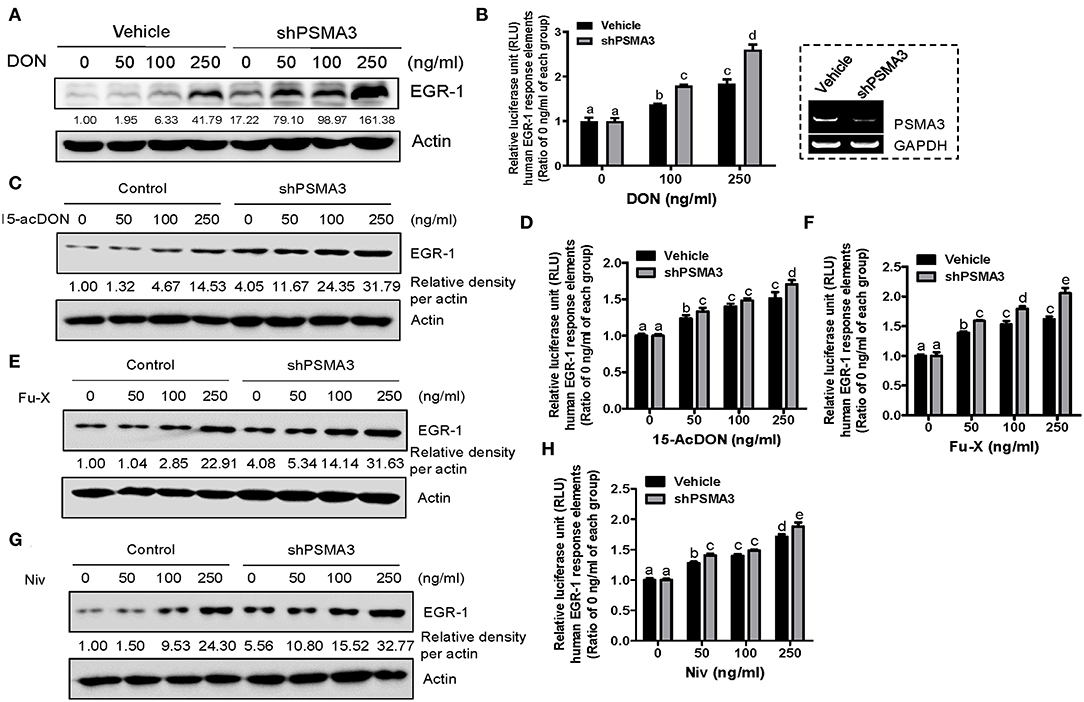
Figure 2. PSMA3 counteracts EGR-1 expression in IECs exposed to 8-keto-trichothecenes. (A,C,E,G) Control or PSMA3-deficient HCT-8 cells were treated with each dose of 8-keto-trichothecene for 1.5 h (A,C) or 5 h (E,G). Total cell lysates were subjected to western blot analysis. (B,D,F,H) Control or PSMA3-deficient HCT-8 cells were transfected with pEBS14-LUC plasmid and treated with each dose of 8-keto-trichothecene for 24 h. The figure in the box represents the efficiency of shPSMA3 expression. Results are shown as mean values ± SD and different letter over each bar represents a significant difference between the groups (p < 0.05, n = 3).
NF-κB Subunit Expression Is Suppressed by 8-keto-trichothecenes in IECs
Although DON is known to trigger inflammatory responses, the levels of p65 as a subunit of NF-κB were significantly reduced by DON treatment in gut epithelia (Figure 3A), consistent with our previous report on p65 suppression by DON at the cellular level (49). DON-treated IECs showed reduced levels of total and phosphorylated p65 (Figure 3B) as well as p65 mRNA (Figure 3C). Since NF-κB is a representative proinflammatory transcription factor in responses to cytokines or microbial components, inhibition of NF-κB signaling significantly attenuated the chemokine induction in response to tumor necrosis factor-α (TNF-α), or other endotoxins such as muramyl dipeptide (MDP) and lipopolysaccharide (LPS) (Figures 3D–F). NF-κB signaling was suppressed using an NF-κB inhibitory construct, super-repressor (SR)-IκB, which expresses a mutated form of IκB, resulting in inhibition of IκB phosphorylation and degradation. In contrast, SR-IκB did not affect the levels of DON-induced proinflammatory chemokines in human IECs (Figures 3D–F). Taken together, DON suppresses p65 mRNA and protein levels even though the toxin induces proinflammatory chemokines in IECs.
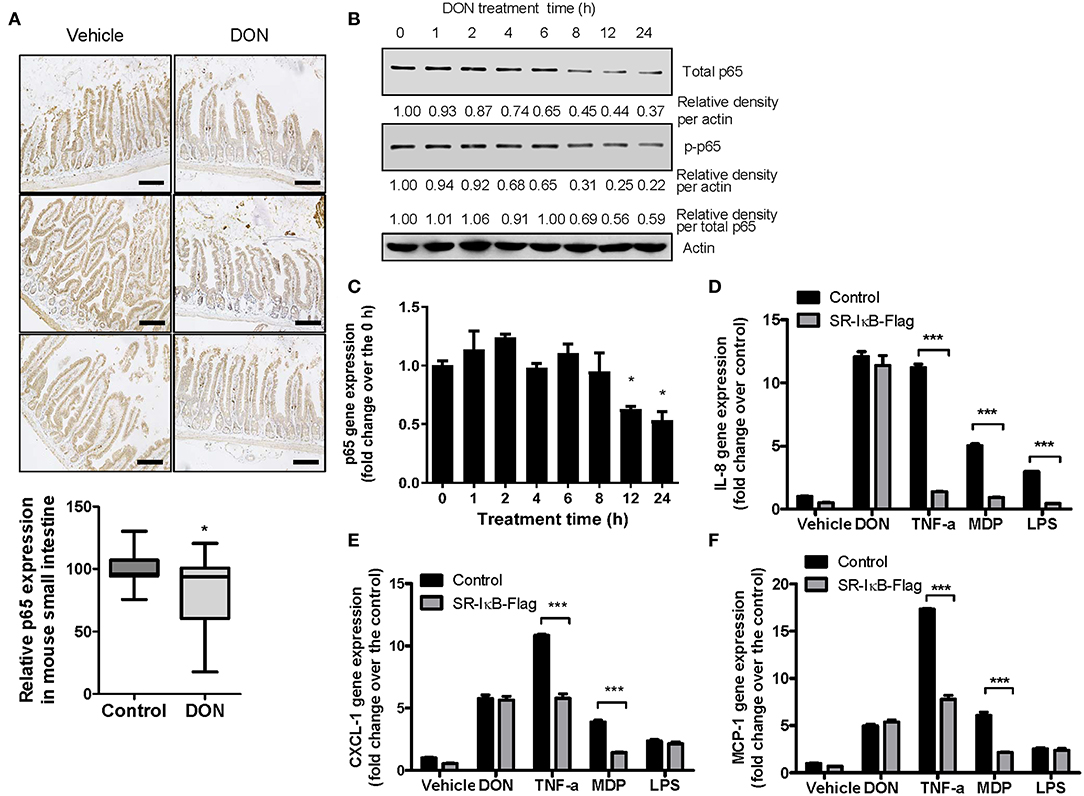
Figure 3. Roles of p65 in the expressions of p65 and chemokines in response to DON or inflammagens in IECs. (A) Small intestinal tissues from 6-week-old male mice exposed to vehicle or 25 mg/kg DON for 12 h were subjected to IHC analysis for p65 (n = 4, each group) (original magnification, × 200, black scale bar, 0.2 μm). Lower panel shows the quantitative analysis of tissue p65 levels in vehicle or DON-treated mice. Results are shown as box-and-whisker plots (min–max) and asterisk (*) indicates a significant difference from the control (p < 0.05, n = 3). (B,C) HCT-8 cells were treated with 500 ng/ml DON for the indicated time. Cell lysates were subjected to western blot analysis (B) and mRNA expression was measured using RT real-time PCR (C). Asterisk (*) indicates a significant difference from the 0 h group (p < 0.05). (D–F) HCT-8 cells transfected with control or SR-IκB-Flag were treated with vehicle, 500 ng/ml DON, 10 ng/ml TNF-α, 10 μg/ml MDP, or 2 μg/ml LPS for 1 h. IL-8 (D), CXCL-1 (E), and MCP-1 (F) mRNA was measured using RT real-time PCR. Results are shown as mean values ± SD and asterisks (***) indicate a significant difference from each control group (p < 0.001, n = 3).
PSMA3 Deficiency Does Not Affect p65 Expression in Response to 8-keto-trichothecenes
As a variety of signaling components for NF-κB activation are regulated by the ubiquitin-linked proteasome system, we assessed the effects of PSMA3 on p65 protein levels in response to 8-keto-trichothecenes in human IECs. PSMA3 deficiency did not significantly influence the total and phosphorylated p65 expression in the absence and presence of DON (Figure 4A). Similar patterns were also observed in response to other 8-keto-trichothecenes (15-acDON, Fu-X, and NIV; Figures 1A–C). Since treatment with 8-keto-trichothecens reduced total levels of p65 protein, the ratio of phospho-p65 to total p65 was not a representative index of NF-κB activation. Instead, we additionally calculated the ratio of phosphor-p65 to stable β-actin. Since DON reduced p65 mRNA levels (Figure 3C), we further measured the p65 transcription activity using cells expressing the human p65 promoter (−999/+92)-containing SEAP reporter. However, PSMA3 was not involved in the transcriptional regulation of p65 expression in response to DON (Figure 4B). Other 8-keto-trichothecenes including 15-acDON, Fu-X, and NIV also displayed suppressive effects on total and phosphorylated p65 protein levels in response to 8-keto-trichothecenes in a dose-dependent manner; however, these were not altered by PSMA3 deficiency in IECs (Figures 1A–C). Similarly, treatment with 15-acDON (Figure 4C), Fu-X (Figure 4D), or NIV (Figure 4E) suppressed the transcriptional activity for p65 expression, which was not affected by PSMA3 deficiency. Taken together, suppression of p65 expression by 8-keto-trichothecenes was independent of PSMA3 levels whereas PSMA3 counteracted the EGR-1 levels in response to 8-keto-trichothecenes in human IECs.
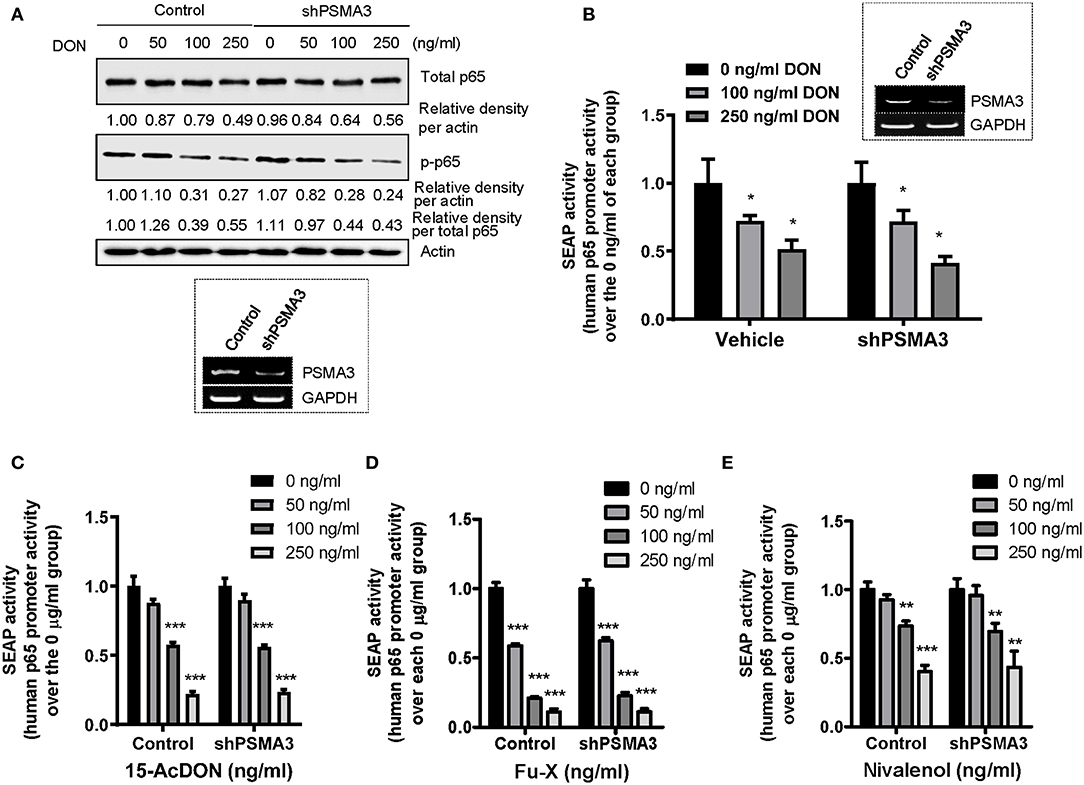
Figure 4. Effects of PSMA3 deficiency on p65 expression in response to 8-keto-trichothecenes. (A) Control- or PSMA3-deficient HCT-8 cells were treated with each dose of DON for 9 h. Cell lysates were subjected to western blot analysis. The figure in the box represents the efficiency of PSMA3 shRNA expression. (B–E) Control- or PSMA3-deficient HCT-8 cells were transfected with human p65 promoter-containing SEAP reporter plasmid and were treated with each dose of 8-keto-trichothecenes for 24 h (DON) (B) or 9 h [15-acDON (C), Fu-X (D), or NIV (E)]. Results are shown as mean values ± SD and asterisks indicate significant differences from each vehicle group (*p < 0.05, **p < 0.01, ***p < 0.001, n = 3–4).
Dual Reporter System Represents 8-keto-trichothecene-specific Patterns of Inflammatory Regulation
We determined that 8-keto-trichothecenes induced EGR-1 while suppressing p65 promoter activity in IECs. In addition, PSMA3 deficiency enhanced the EGR-1 levels in response to 8-keto-trichothecenes in IECs. Based on these mechanistic evidences, we established a cell-based bioassay system representing the proinflammatory actions of 8-keto-trichothecenes in IECs. Control and PSMA3-deficient cells were stably transfected with dual reporter plasmids including pEBS14-LUC and a human p65 promoter (−999/+92)-containing SEAP reporter plasmid. The pEBS14-LUC plasmid was constructed to detect EGR-1-mediated transcriptional activity whereas SEAP in medium was assessed to quantify p65 promoter activity in response to inflammagens (Figure 5A). We also performed the dual reporter bioassay in response to a cytokine or bacterial endotoxin as positive proinflammatory triggers. We calculated the effective doses for enhancing reporter production by 50% (ED50) or the inhibitory doses for reducing reporter production by 50% (ID50) in the dual reporter system. In response to bacterial cell wall components including LPS or MDP (the minimal bioactive peptidoglycan motif), or tumor necrosis factor-α (TNF-α), both EGR-1-mediated transcription and p65 promoter activities were elevated in a dose-dependent manner (Figures 5B–D). Moreover, PSMA3 deficiency improved the dose responses of EGR-1-mediated transcription to the conventional proinflammatory triggers, particularly MDP, compared to those in the controls. In contrast, PSMA3 deficiency attenuated the p65 promoter activation in response to three inflammagens, indicating that PSMA3 is a positive regulator of NF-κB signaling activation by endotoxins or TNF-α. In response to 8-keto-trichothecenes, EGR-1-mediated transcription was elevated whereas p65 promoter activity was suppressed in a dose-dependent manner (Figure 6). Furthermore, the sensitivity of EGR-1-mediated transcription in response to 8-keto-trichothecenes was enhanced by PSMA3 deficiency. However, the half-inhibitory dose (ID50) for p65 promoter activity was not significantly altered by PSMA3 deficiency in the presence of 8-keto-trichothecenes. In conclusion, the dual reporter system in PSMA3-deficient IECs was promising for the biomonitoring of typical inflammatory regulation by 8-keto-trichothecenes, which was differentiated by the responses to conventional inflammatory triggers including endotoxins and cytokines.
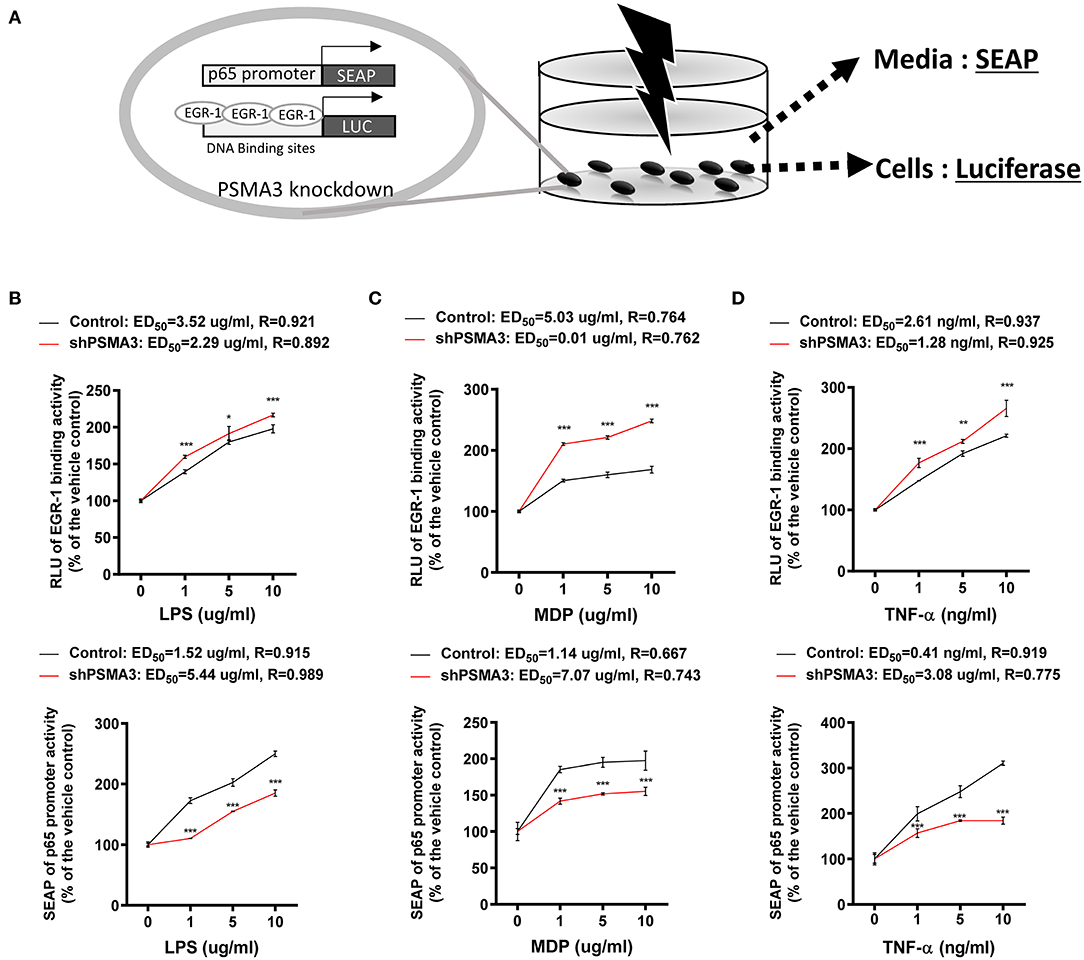
Figure 5. Effects of inflammagens on the dual reporter system in human IECs. (A) A putative scheme for the mechanism of the dual reporter system (SEAP for p65 transcription and luciferase for EGR-1-mediated transcription) to detect 8-keto-trichothecenes in PSMA3-deficient IECs. (B) Control- or PSMA3-deficient HCT-8 with dual reporters were treated with each dose of LPS (B), MDP (C), or TNF-α (D) for 72 h. Levels of SEAP in media and intracellular luciferase were simultaneously measured after exposure. Results are shown as mean values ± SD and asterisks indicate significant differences from the control group at each dose (*p < 0.05, **p < 0.01, ***p < 0.001, n = 3).
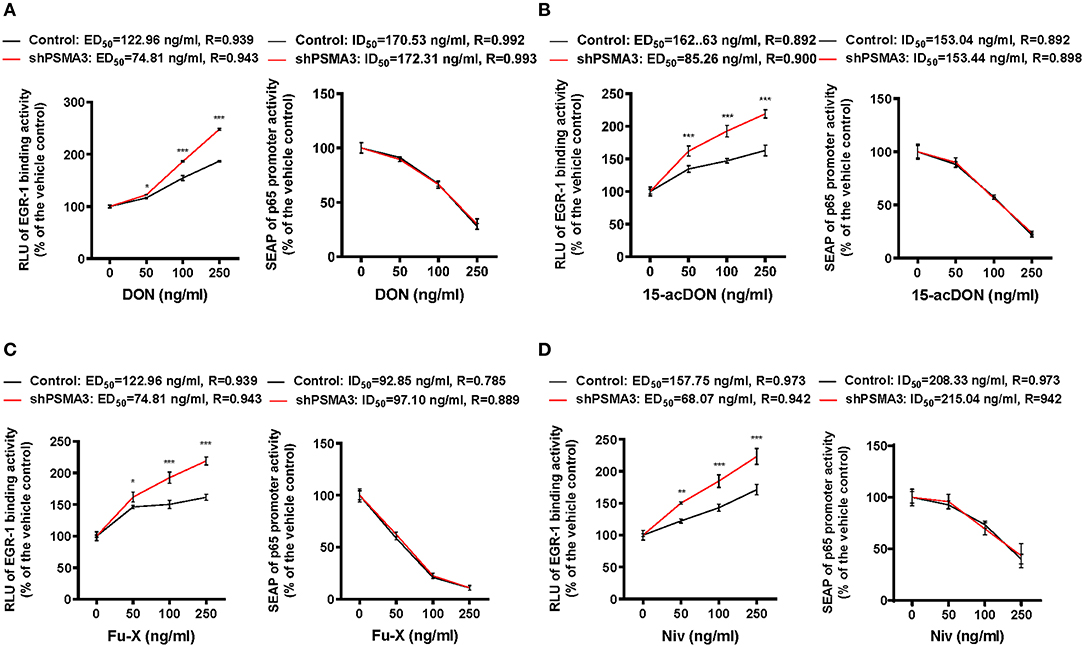
Figure 6. Effects of 8-keto-trichothecenes on the dual reporter system in human IECs. Control- or PSMA3-deficient HCT-8 with dual reporters were treated with each dose of DON (A), 15-acDON (B), Fu-X (C), or NIV (D) for 24 h. Levels of SEAP in media and intracellular luciferase were simultaneously measured after exposure. Results are shown as mean values ± SD and asterisks indicate significant differences from the control group at each dose (*p < 0.05, **p < 0.01, ***p < 0.001, n = 3).
Discussion
Based on mechanistic evidences on the regulation of proinflammatory transcription factors by 8-keto-trichothecenes, we developed an IEC-based biomonitoring method to detect the biological activity of trichothecene mycotoxins. Trichothecene-induced chemokine production is regulated by EGR-1 or NF-κB in IECs, which was simulated using cells with dual reporters for these two transcription factors. Moreover, the sensitivity of EGR-1-based biomonitoring was improved by suppressing PSMA3, the α-type subunit of the 20S proteasome core complex. However, PSMA3 was positively involved in p65 promoter response to endotoxins and cytokines. Mechanistically, NF-κB-linked signaling pathways are strongly associated with ubiquitin-proteasome regulation. In particular, ubiquitination reactions of TNF receptor associated factor (TRAF6), IκB kinase γ (IKKγ/NEMO), and Iκα contribute to NF-κB activation via regulation of protein binding or degradation during signaling transduction (50, 51). Despite no known evidences in NF-κB regulation by PSMA3, the present study first identified PSMA3 as a positive regulator of p65-linked signaling in IECs. PSMA3 deficiency decreased the responses of p65-linked promoter activity to conventional inflammagens including endotoxins and TNFα. However, PSMA3 did not contribute to the regulation of p65 expression in response to 8-keto-trichothecenes.
Although treatment with 8-keto-trichothecenes enhanced production of proinflammatory chemokines, it downregulated the key transcription factor NF-κB in gut epithelial cells. Instead, EGR-1 was mainly involved in transcription activation of proinflammatory chemokine genes in the present exposure model. Moreover, our previous investigation demonstrated that DON-induced EGR-1 mediates the induction of peroxisome proliferator-activated receptor γ (PPARγ), which is associated with suppression of pro-inflammatory NF-κB signaling (39). Mechanistically, PPARγ attenuates inflammatory responses by triggering nuclear export of p65 protein in complex with PPARγ (52). Moreover, as another transcriptional regulator of proinflammatory signaling, activating transcription factor 3 (ATF3) is induced as part of the negative feedback loop that modulates pattern recognition receptor-stimulated inflammatory responses (53–55). ATF3 is also induced by DON as part of a transcriptional complex composed of the NF-κB family of transcription factors (29). ATF3 can alter the chromatin structure, thereby restricting access to other transcription factors including NF-κB (53). However, treatment with 8-keto-trichothecenes reduced total levels of p65 protein in human intestinal epithelial cells, which may occur in advance of promoter-binding regulation of NF-κB by either ATF3 or PPARγ. One potent explanation could be a post-transcriptional regulation of p65 since DON treatment downregulates the stability of p65 mRNA (49). In particular, ATF3 leads to the destabilization of p65 mRNA caused by nuclear entrapment of transcript-stabilizing HuR protein via direct interaction with ATF3 (49). In addition to ATF3, PPARγ is a negative regulator of transcript stability since it suppresses the cytoplasmic translocation of HuR (56), which may contribute to reduction in p65 protein in response to 8-keto-trichothecenes in the present study. Suppression of NF-κB could be a beneficial event during inflammation, but which can interfere with the tissue regeneration after injuries since epithelial NF-κB is involved in cellular proliferation and migration (57–60). Thus, it can be speculated that the wound healing process during toxin-induced inflammation can be interfered due to NF-κB suppression by DON. While trichothecene-induced EGR-1 plays pivotal roles in proinflammatory cytokine production, attenuated NF-κB signaling may account for delayed epithelial restitution after gut injury.
The present bioassay system provides high sensitivity to detect 8-keto-trichothecenes, which can be detected at very low doses (<0.1 ppm), far less than the general regulatory limit of 1 ppm. Moreover, the present bioassay system represents the quantitative biological actions of trichothecenes. Until now, most of eukaryotic cell-based bioassays of trichothecenes are based on the detection of the inhibitory effects of the mycotoxins on protein synthesis, secretion, or cell proliferation (13, 61–63). However, the present bioassay indicates the typical biological processes regulated by inflammation-associated transcription factors including EGR-1 and NF-κB in human intestinal epithelial cells. Trichothecene-exposed cells displayed opposite readout patterns of EGR-1 and p65, both of which were elevated by endotoxins and TNF-α. In addition to these two factorials (EGR-1 and p65), regulation of PSMA3 contributed to the enhanced specificity of the dual reporter system. These combinatorial features of responses can indicate the biological identity of 8-keto-trichothecenes in the tested matrix in the present biomonitoring system. Moreover, most (99%) cereal foodstuffs are contaminated with more than 10 mycotoxins. Mixtures of 8-keto-trichothecenes are often found in field crops depending on weather conditions and mold chemotypes. Further, 10–20% of DON and NIV exist in the acetylated form such as 3-acetyl DON, 15-acetyl DON, or Fu-X. For efficient risk evaluation of the 8-keto-trichothecene mixture, the total quantity of the biological actions of toxin mixtures should be calculated based on the mechanism along with the chemical identification. Therefore, the relative risk of chemicals with a common mechanism of action can be assessed by measuring readouts such as the toxic equivalent factor (TEF) through the standard bioassay (13, 64). For example, based on toxin-induced actions such as endoplasmic reticulum stress, a reporter-based TEF can be derived by comparing the ED50 or ID50 of each 8-keto-trichothecene (13). Moreover, TEF-based quantitative and qualitative assessment contributes to the risk characterization such as the total daily intake by humans and animals. The present bioassay using typical patterns of proinflammatory signaling can be standardized with TEF evaluation and further integration for regulatory and industrial uses.
The present bioassay system could be a potential quantitative application for the immunotoxicological determination of 8-keto-trichothecene mixtures in agricultural commodities. However, it needs to be improved for in vitro recapitulation of the realistic pathophysiological gut environment. For instance, the intoxicated tissue contexture is different from two-dimensional culture of intestinal epithelial cells. Local adverse outcomes can be differentiated depending on the accessibility of toxins to epithelial surface in the gut. Therefore, in vivo gut-mimicking culture of three-dimensional monolayer such as intestinal organoids would provide more realistic information on trichothecene stress responses than the simple monolayer model (65, 66). Moreover, dose responses in the static culture model hardly represent the toxicokinetic responses in animals and humans. In particular, local luminal concentration of trichothecenes in the gastrointestinal tract following ingestion is generally much higher than levels in the circulation and other deposition tissues. Moreover, while the proximal parts of the small intestine are the major site of DON absorption (67), DON rarely reaches the distal small intestine and colon at the luminal region. Instead, the distal parts are mainly affected by circulating DON via the basolateral exposure (68). Therefore, additional epithelial readouts from the mucosal signature (69) are warranted to simulate compartment-specific actions of 8-keto-trichothecenesin the gut.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher on reasonable request.
Ethics Statement
The animal study was reviewed and approved by Pusan National University Institutional Animal Care and Use Committee (PNU-IACUC) (PNU-2013-0291).
Author Contributions
Project design and hypotheses were defined by YM and S-HP. S-HP conducted experiments and analyzed the data. YM and S-HP prepared the manuscript. YM supervised the overall project. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A3B05041889).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate Dr. Juil Kim for his kind help for technical assistances in experiments.
References
1. Desjardins AE, Hohn TM, McCormick SP. Trichothecene biosynthesis in Fusarium species: chemistry, genetics, and significance. Microbiol Rev. (1993) 57:595–604. doi: 10.1128/MMBR.57.3.595-604.1993
2. Grove JF. The trichothecenes and their biosynthesis. Fortschr Chem Org Naturst. (2007) 88:63–130.
4. Shifrin VI, Anderson P. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J Biol Chem. (1999) 274:13985–92. doi: 10.1074/jbc.274.20.13985
5. Hanna J, Leggett DS, Finley D. Ubiquitin depletion as a key mediator of toxicity by translational inhibitors. Mol Cell Biol. (2003) 23:9251–61. doi: 10.1128/MCB.23.24.9251-9261.2003
6. Kugler KG, Jandric Z, Beyer R, Klopf E, Glaser W, Lemmens M, et al. Ribosome quality control is a central protection mechanism for yeast exposed to deoxynivalenol and trichothecin. BMC Genomics. (2016) 17:417. doi: 10.1186/s12864-016-2718-y
7. Schenzel J, Schwarzenbach RP, Bucheli TD. Multi-residue screening method to quantify mycotoxins in aqueous environmental samples. J Agric Food Chem. (2010) 58:11207–17. doi: 10.1021/jf102737q
8. Schenzel J, Hungerbuhler K, Bucheli TD. Mycotoxins in the environment: II. Occurrence and origin in Swiss river waters. Environ Sci Technol. (2012) 46:13076–84. doi: 10.1021/es301558v
9. Mirocha CJ, Pathre SV, Schauerhamer B, Christensen CM. Natural occurrence of Fusarium toxins in feedstuff. Appl Environ Microbiol. (1976) 32:553–6. doi: 10.1128/AEM.32.4.553-556.1976
10. Peraica M, Domijan AM. Contamination of food with mycotoxins and human health. Arh Hig Rada Toksikol. (2001) 52:23–35.
11. Sudakin DL. Trichothecenes in the environment: relevance to human health. Toxicol Lett. (2003) 143:97–107. doi: 10.1016/S0378-4274(03)00116-4
12. Ueno Y. Toxicological features of T-2 toxin and related trichothecenes. Fundam Appl Toxicol. (1984) 4(2 Pt 2):S124–32. doi: 10.1093/toxsci/4.2part2.124
13. Yang H, Park SH, Choi HJ, Do KH, Kim J, An TJ, et al. Mechanism-based alternative monitoring of endoplasmic reticulum stress by 8-keto-trichothecene mycotoxins using human intestinal epithelial cell line. Toxicol Lett. (2010) 198:317–23. doi: 10.1016/j.toxlet.2010.07.008
14. Betina V. Structure-activity relationships among mycotoxins. Chem Biol Interact. (1989) 71:105–46. doi: 10.1016/0009-2797(89)90030-6
15. Ehrlich KC, Daigle KW. Protein synthesis by mammalian cells treated with C-3-modified analogs of the 12,13-epoxytrichothecenes T-2 and T-2 tetraol. Appl Environ Microbiol. (1985) 50:914–8. doi: 10.1128/AEM.50.4.914-918.1985
16. Ehrlich KC, Daigle KW. Protein synthesis inhibition by 8-oxo-12,13-epoxytrichothecenes. Biochim Biophys Acta. (1987) 923:206–13. doi: 10.1016/0304-4165(87)90005-5
17. Bhat RV, Beedu SR, Ramakrishna Y, Munshi KL. Outbreak of trichothecene mycotoxicosis associated with consumption of mould-damaged wheat production in Kashmir Valley, India. Lancet. (1989) 1:35–7. doi: 10.1016/S0140-6736(89)91684-X
18. Li FQ, Li YW, Luo XY, Yoshizawa T. Fusarium toxins in wheat from an area in Henan Province, PR China, with a previous human red mould intoxication episode. Food Addit Contam. (2002) 19:163–7. doi: 10.1080/02652030110070058
19. Luo Y, Yoshizawa T, Katayama T. Comparative study on the natural occurrence of Fusarium mycotoxins (trichothecenes and zearalenone) in corn and wheat from high- and low-risk areas for human esophageal cancer in China. Appl Environ Microbiol. (1990) 56:3723–6. doi: 10.1128/AEM.56.12.3723-3726.1990
20. Danicke S, Goyarts T, Doll S, Grove N, Spolders M, Flachowsky G. Effects of the Fusarium toxin deoxynivalenol on tissue protein synthesis in pigs. Toxicol Lett. (2006) 165:297–311. doi: 10.1016/j.toxlet.2006.05.006
21. Pestka JJ, Smolinski AT. Deoxynivalenol: toxicology and potential effects on humans. J Toxicol Environ Health B Crit Rev. (2005) 8:39–69. doi: 10.1080/10937400590889458
22. Rotter BA, Prelusky DB, Pestka JJ. Toxicology of deoxynivalenol (vomitoxin). J Toxicol Environ Health. (1996) 48:1–34. doi: 10.1080/009841096161447
23. Azcona-Olivera JI, Ouyang Y, Murtha J, Chu FS, Pestka JJ. Induction of cytokine mRNAs in mice after oral exposure to the trichothecene vomitoxin (deoxynivalenol): relationship to toxin distribution and protein synthesis inhibition. Toxicol Appl Pharmacol. (1995) 133:109–20. doi: 10.1006/taap.1995.1132
24. Chung YJ, Yang GH, Islam Z, Pestka JJ. Up-regulation of macrophage inflammatory protein-2 and complement 3A receptor by the trichothecenes deoxynivalenol and satratoxin G. Toxicology. (2003) 186:51–65. doi: 10.1016/S0300-483X(02)00605-4
25. Moon Y, Pestka JJ. Vomitoxin-induced cyclooxygenase-2 gene expression in macrophages mediated by activation of ERK and p38 but not JNK mitogen-activated protein kinases. Toxicol Sci. (2002) 69:373–82. doi: 10.1093/toxsci/69.2.373
26. Zhou HR, Harkema JR, Yan D, Pestka JJ. Amplified proinflammatory cytokine expression and toxicity in mice coexposed to lipopolysaccharide and the trichothecene vomitoxin (deoxynivalenol). J Toxicol Environ Health A. (1999) 57:115–36. doi: 10.1080/009841099157818
27. Moon Y, Yang H, Lee SH. Modulation of early growth response gene 1 and interleukin-8 expression by ribotoxin deoxynivalenol (vomitoxin) via ERK1/2 in human epithelial intestine 407 cells. Biochem Biophys Res Commun. (2007) 362:256–62. doi: 10.1016/j.bbrc.2007.07.168
28. Moon Y, Yang H, Park SH. Hypo-responsiveness of interleukin-8 production in human embryonic epithelial intestine 407 cells independent of NF-κB pathway: new lessons from endotoxin and ribotoxic deoxynivalenol. Toxicol Appl Pharmacol. (2008) 231:94–102. doi: 10.1016/j.taap.2008.03.016
29. Yang H, Park SH, Choi HJ, Moon Y. Epithelial cell survival by activating transcription factor 3 (ATF3) in response to chemical ribosome-inactivating stress. Biochem Pharmacol. (2009) 77:1105–15. doi: 10.1016/j.bcp.2008.11.028
30. Dieckgraefe BK, Weems DM. Epithelial injury induces egr-1 and fos expression by a pathway involving protein kinase C and ERK. Am J Physiol. (1999) 276(2 Pt 1):G322–30. doi: 10.1152/ajpgi.1999.276.2.G322
31. Khachigian LM, Collins T. Inducible expression of Egr-1-dependent genes. A paradigm of transcriptional activation in vascular endothelium. Circ Res. (1997) 81:457–61. doi: 10.1161/01.RES.81.4.457
32. Murphy LO, MacKeigan JP, Blenis J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol Cell Biol. (2004) 24:144–53. doi: 10.1128/MCB.24.1.144-153.2004
33. MacDonald TT, Pettersson S. Bacterial regulation of intestinal immune responses. Inflamm Bowel Dis. (2000) 6:116–22. doi: 10.1097/00054725-200005000-00008
34. Ma N, Guo P, Zhang J, He T, Kim SW, Zhang G, et al. Nutrients mediate intestinal bacteria-mucosal immune crosstalk. Front Immunol. (2018) 9:5. doi: 10.3389/fimmu.2018.00005
35. Fitzgerald KA, O'Neill LA. Characterization of CD44 induction by IL-1: a critical role for Egr-1. J Immunol. (1999) 162:4920–7.
36. Giri RK, Selvaraj SK, Kalra VK. Amyloid peptide-induced cytokine and chemokine expression in THP-1 monocytes is blocked by small inhibitory RNA duplexes for early growth response-1 messenger RNA. J Immunol. (2003) 170:5281–94. doi: 10.4049/jimmunol.170.10.5281
37. Grotegut S, von Schweinitz D, Christofori G, Lehembre F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. Embo J. (2006) 25:3534–45. doi: 10.1038/sj.emboj.7601213
38. Mishra JP, Mishra S, Gee K, Kumar A. Differential involvement of calmodulin-dependent protein kinase II-activated AP-1 and c-Jun N-terminal kinase-activated EGR-1 signaling pathways in tumor necrosis factor-alpha and lipopolysaccharide-induced CD44 expression in human monocytic cells. J Biol Chem. (2005) 280:26825–37. doi: 10.1074/jbc.M500244200
39. Do KH, Choi HJ, Kim J, Park SH, Kim HH, Oh CG, et al. Ambivalent roles of early growth response 1 in inflammatory signaling following ribosomal insult in human enterocytes. Biochem Pharmacol. (2012) 84:513–21. doi: 10.1016/j.bcp.2012.05.015
40. Park SH, Kim J, Do KH, Park J, Oh CG, Choi HJ, et al. Activating transcription factor 3-mediated chemo-intervention with cancer chemokines in a noncanonical pathway under endoplasmic reticulum stress. J Biol Chem. (2014) 289:27118–33. doi: 10.1074/jbc.M114.568717
41. Kim KH, Park SH, Do KH, Kim J, Choi KU, Moon Y. NSAID-activated gene 1 mediates pro-inflammatory signaling activation and paclitaxel chemoresistance in type I human epithelial ovarian cancer stem-like cells. Oncotarget. (2016) 7:72148–66. doi: 10.18632/oncotarget.12355
42. Park SH, Kim J, Kim D, Moon Y. Mycotoxin detoxifiers attenuate deoxynivalenol-induced pro-inflammatory barrier insult in porcine enterocytes as an in vitro evaluation model of feed mycotoxin reduction. Toxicol In Vitro. (2017) 38:108–16. doi: 10.1016/j.tiv.2016.10.003
43. Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, et al. Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol Cell Biol. (1995) 15:2809–18. doi: 10.1128/MCB.15.5.2809
44. Alcantara Warren C, Destura RV, Sevilleja JE, Barroso LF, Carvalho H, Barrett LJ, et al. Detection of epithelial-cell injury, and quantification of infection, in the HCT-8 organoid model of cryptosporidiosis. J Infect Dis. (2008) 198:143–9. doi: 10.1086/588819
45. Thebault S, Deniel N, Marion R, Charlionet R, Tron F, Cosquer D, et al. Proteomic analysis of glutamine-treated human intestinal epithelial HCT-8 cells under basal and inflammatory conditions. Proteomics. (2006) 6:3926–37. doi: 10.1002/pmic.200500714
46. Avantaggiato G, Havenaar R, Visconti A. Evaluation of the intestinal absorption of deoxynivalenol and nivalenol by an in vitro gastrointestinal model, and the binding efficacy of activated carbon and other adsorbent materials. Food Chem Toxicol. (2004) 42:817–24. doi: 10.1016/j.fct.2004.01.004
47. Obremski K, Zielonka L, Gajecka M, Jakimiuk E, Bakula T, Baranowski M, et al. Histological estimation of the small intestine wall after administration of feed containing deoxynivalenol, T-2 toxin and zearalenone in the pig. Pol J Vet Sci. (2008) 11:339–45.
48. Bae MH, Jeong CH, Kim SH, Bae MK, Jeong JW, Ahn MY, et al. Regulation of Egr-1 by association with the proteasome component C8. Biochim Biophys Acta. (2002) 1592:163–7. doi: 10.1016/S0167-4889(02)00310-5
49. Park SH, Do KH, Choi HJ, Kim J, Kim KH, Park J, et al. Novel regulatory action of ribosomal inactivation on epithelial Nod2-linked proinflammatory signals in two convergent ATF3-associated pathways. J Immunol. (2013) 191:5170–81. doi: 10.4049/jimmunol.1301145
50. Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y. TRAF6 autoubiquitination-independent activation of the NFkappaB and MAPK pathways in response to IL-1 and RANKL. PLoS One. (2008) 3:e4064. doi: 10.1371/journal.pone.0004064
51. Chen J, Chen ZJ. Regulation of NF-kappaB by ubiquitination. Curr Opin Immunol. (2013) 25:4–12. doi: 10.1016/j.coi.2012.12.005
52. Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol. (2004) 5:104–12. doi: 10.1038/ni1018
53. Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Roach JC, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. (2006) 441:173–8. doi: 10.1038/nature04768
54. Suganami T, Yuan X, Shimoda Y, Uchio-Yamada K, Nakagawa N, Shirakawa I, et al. Activating transcription factor 3 constitutes a negative feedback mechanism that attenuates saturated Fatty acid/toll-like receptor 4 signaling and macrophage activation in obese adipose tissue. Circ Res. (2009) 105:25–32. doi: 10.1161/CIRCRESAHA.109.196261
55. Whitmore MM, Iparraguirre A, Kubelka L, Weninger W, Hai T, Williams BR. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J Immunol. (2007) 179:3622–30. doi: 10.4049/jimmunol.179.6.3622
56. Park SH, Choi HJ, Yang H, Do KH, Kim J, Moon Y. Repression of peroxisome proliferator-activated receptor gamma by mucosal ribotoxic insult-activated CCAAT/enhancer-binding protein homologous protein. J Immunol. (2010) 185:5522–30. doi: 10.4049/jimmunol.1001315
57. Ishida Y, Kondo T, Kimura A, Matsushima K, Mukaida N. Absence of IL-1 receptor antagonist impaired wound healing along with aberrant NF-kappaB activation and a reciprocal suppression of TGF-beta signal pathway. J Immunol. (2006) 176:5598–606. doi: 10.4049/jimmunol.176.9.5598
58. Egan LJ, de Lecea A, Lehrman ED, Myhre GM, Eckmann L, Kagnoff MF. Nuclear factor-kappa B activation promotes restitution of wounded intestinal epithelial monolayers. Am J Physiol Cell Physiol. (2003) 285:C1028–35. doi: 10.1152/ajpcell.00167.2003
59. Noiri E, Peresleni T, Srivastava N, Weber P, Bahou WF, Peunova N, et al. Nitric oxide is necessary for a switch from stationary to locomoting phenotype in epithelial cells. Am J Physiol. (1996) 270(3 Pt 1):C794–802. doi: 10.1152/ajpcell.1996.270.3.C794
60. Cowan MJ, Coll T, Shelhamer JH. Polyamine-mediated reduction in human airway epithelial migration in response to wounding is PGE2 dependent through decreases in COX-2 and cPLA2 protein levels. J Appl Physiol. (2006) 101:1127–35. doi: 10.1152/japplphysiol.01287.2005
61. Thompson WL, Wannemacher RW Jr. Detection and quantitation of T-2 mycotoxin with a simplified protein synthesis inhibition assay. Appl Environ Microbiol. (1984) 48:1176–80. doi: 10.1128/AEM.48.6.1176-1180.1984
62. Widestrand J, Lundh T, Pettersson H, Lindberg JE. Cytotoxicity of four trichothecenes evaluated by three colorimetric bioassays. Mycopathologia. (1999) 147:149–55. doi: 10.1023/A:1007127919901
63. Yike I, Allan T, Sorenson WG, Dearborn DG. Highly sensitive protein translation assay for trichothecene toxicity in airborne particulates: comparison with cytotoxicity assays. Appl Environ Microbiol. (1999) 65:88–94. doi: 10.1128/AEM.65.1.88-94.1999
64. Tritscher AM, Page SW. The risk assessment paradigm and its application for trichothecenes. Toxicol Lett. (2004) 153:155–63. doi: 10.1016/j.toxlet.2004.04.030
65. Kuratnik A, Giardina C. Intestinal organoids as tissue surrogates for toxicological and pharmacological studies. Biochem Pharmacol. (2013) 85:1721–6. doi: 10.1016/j.bcp.2013.04.016
66. Truskey GA. Human microphysiological systems and organoids as in vitro models for toxicological studies. Front Public Health. (2018) 6:185. doi: 10.3389/fpubh.2018.00185
67. Danicke S, Valenta H, Doll S. On the toxicokinetics and the metabolism of deoxynivalenol (DON) in the pig. Arch Anim Nutr. (2004) 58:169–80. doi: 10.1080/00039420410001667548
68. Diesing AK, Nossol C, Danicke S, Walk N, Post A, Kahlert S, et al. Vulnerability of polarised intestinal porcine epithelial cells to mycotoxin deoxynivalenol depends on the route of application. PLoS One. (2011) 6:e17472. doi: 10.1371/journal.pone.0017472
Keywords: 8-keto-trichothecene, ribotoxic stress, inflammation, epithelium, reporter biomonitoring
Citation: Park S-H and Moon Y (2020) Enterocyte-Based Bioassay via Quantitative Combination of Proinflammatory Sentinels Specific to 8-keto-trichothecenes. Front. Immunol. 11:1530. doi: 10.3389/fimmu.2020.01530
Received: 12 February 2020; Accepted: 10 June 2020;
Published: 16 July 2020.
Edited by:
James J. Pestka, Michigan State University, United StatesReviewed by:
Erwin L. Roggen, Independent Researcher, Lyngby, DenmarkXi Ma, China Agricultural University, China
Copyright © 2020 Park and Moon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuseok Moon, bW9vbkBwbnUuZWR1
†Present address: Seong-Hwan Park, Personalized Genomic Medicine Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Daejeon, South Korea