 Izabela Sieminska
Izabela Sieminska Jarek Baran
Jarek Baran
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 07 August 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01526
This article is part of the Research Topic Roles of Tumor-Recruited Myeloid Cells in Immune Evasion in Cancer View all 13 articles
Colorectal cancer (CRC) remains one of the most common malignancies diagnosed worldwide. The pathogenesis of CRC is complex and involves, among others, accumulation of genetic predispositions and epigenetic factors, dietary habits, alterations in gut microbiota, and lack of physical activity. A growing body of evidence suggests that immune cells play different roles in CRC, comprising both pro- and anti-tumorigenic functions. Immunosuppression observed during cancer development and progression is a result of the orchestration of many cell types, including myeloid-derived suppressor cells (MDSCs). MDSCs, along with other cells, stimulate tumor growth, angiogenesis, and formation of metastases. This article focuses on MDSCs in relation to their role in the initiation and progression of CRC. Possible forms of immunotherapies targeting MDSCs in CRC are also discussed.
According to the World Cancer Research Foundation, colorectal cancer (CRC) (referring to malignancy of colon, rectum, or anus) is the third most common malignancy worldwide. In 2018, more than 1.8 million new cases of CRC were diagnosed (1). About 20–25% of CRC cases are caused by genetic predispositions, including monogenic mutations in mismatched repairing genes associated with, e.g., DNA repair, the cell cycle, and apoptosis (2). Alongside inherited genetic mutations, epigenetic changes also play a significant role in CRC development (3). The remaining 75–80% of cases develop spontaneously and are related to environmental factors such as lack of physical activity, dietary habits, and smoking or alcohol abuse (4). Currently, alterations in the composition of the gut microbiome and its metabolites (playing a role in damaging local tolerance) are also considered as risk factors for CRC (5). An increased risk of CRC is often associated with chronic inflammation of the mucous membrane, which may lead to cell dysplasia, as was proven for patients with inflammatory bowel disease (IBD) (6).
The role of inflammation in CRC development was further supported by data showing that non-steroidal anti-inflammatory drugs (NSAIDs) may decrease the risk of both CRC and colon polyps, which are considered as a premalignant stage (7, 8). The tumor-infiltrating leukocytes (TILs), especially lymphocytes, contribute to the immunoscore classification, where the density of CD3+ and CD8+ T-cell infiltrate is used as a predictor of anti-tumor response and the prognostic marker in CRC (9, 10). However, further studies have shown that most of the immune cells may actually have a dual activity—anti- and pro-tumor, depending on the signals received from the tumor microenvironment. Interestingly, the so-called myeloid-derived suppressor cells (MDSCs) can switch the polarization of other cells to the status with pro-tumorigenic activity (11).
Already in the early 1900s, it was shown that cancer development is often accompanied by extra-medullary hematopoiesis (EMH) and neutrophilia (12). These “fresh” leukocytes were further characterized by suppressive activity and were called immature myeloid cells (ImC) or myeloid suppressor cells (MSC) (13). Eventually, in 2007, their name was changed to MDSCs (13). These cells represent a heterogeneous population of granulocytes and monocytes that rapidly expand during infection, inflammation, and cancer (14, 15). MDSCs, together with the tumor-associated neutrophils (TANs), tumor-associated macrophages (TAMs), and regulatory dendritic cells, compose the population of myeloid regulatory cells (MRC), strongly cooperating with each other during cancer development, and progression (16). Based on mouse data, the MDSC population has been divided into two subgroups: of monocyte (Mo-MDSCs), defined as CD11b+Ly6G−Ly6Chigh, and polymorphonuclear (PMN-MDSCs), CD11b+Ly6G+Ly6Clow, origin (11, 17, 18). Reflecting MDSC populations already defined in mice, human MDSCs have been described as Lin− HLA-DR−/low CD11b+ CD14− CD15+ CD33+ for PMN-MDSCs and Lin− HLA-DR−/low CD11b+ CD14+ CD15− CD33+ for Mo-MDSCs. Very recently, a population of early-stage MDSCs (e-MDSCs) was detected and defined as Lin− HLA-DR−/low CD11b+ CD14− CD15− CD33+ (17, 19). As their name suggests, these cells possess immunosuppressive function and help cancer to escape the surveillance of the immune system and support further tumor development (17). Most studies point out that the suppressive role of MDSCs in cancer is associated with the activation of their two enzymes, namely inducible NO synthase (iNOS) and arginase-1 (ARG1) (20–22). These enzymes are responsible for metabolism of L-arginine, which is essential for the proliferation and proper functioning of T cells (23). Moreover, NO and ROS produced in these reactions are involved in the inactivation of the T-cell receptor (TCR), causing a decrease in the expression of CD3ζ chain and inducing T-cell apoptosis (18, 19, 22).
It is widely accepted that the level of circulating MDSCs increases in the late stage of cancer, correlating with disease progression and formation of metastases (15, 24–26). However, recently, Ma et al. showed that the MDSC level in circulation also increases in premalignant states, such as colon polyposis (27).
The development of MDSCs is caused by various mediators released under chronic inflammatory conditions, including the release of chemokines (11, 15, 28, 29). One of them that is particularly relevant is CCL2, which contributes to tumor growth, progression, and metastasis development in many tumors, including breast, ovarian, prostate, and CRC (30–33). Previous studies in mice showed that CRC growth could be supported by myeloid cells recruited by the CCL2-CCR2 signaling pathway (33). CCL2 caused accumulation of MDSCs and enhanced their immunosuppressive function during colorectal carcinogenesis (34). It was also shown that the level of CCL2 increased simultaneously with the progression of CRC (humans), while the deletion of CCL2 led to the reduction of the MDSC level (mouse model) (34). Further, RNS produced by MDSCs may nitrite chemokines, e.g., CCL2 to N-CCL2, which do not attract CD8+ T cells (like unmodified CCL2 does) but instead recruit myeloid cells, e.g., monocytes (35). On the other hand, several studies documented that CXCL1 is elevated in human CRC (36–38). Further data indicated that CXCR2-positive MDSCs are recruited through CXCR2 ligands, e.g., CXCL1 and CXCl2 are essential for chronic colonic inflammation and colitis-associated tumorigenesis (39).
In addition to chemokines, an important role in the regulation of MDSC activity is attributed to other inflammatory mediators such as histamine and prostaglandins. It has been documented that histamine induces MDSC proliferation and promotes ARG1 and iNOS expression in Mo-MDSCs. At the same time, histamine inhibits the expression of ARG1 and iNOS in PMN-MDSCs, promoting the production of IL-13 and IL-4 (40). Thus, histamine may activate Mo-MDSCs and PMN-MDSCs in different ways (40, 41). Prostaglandin E2 (PGE2), on the other hand, is a strong proinflammatory mediator produced by COX-2 (42) and may activate MDSCs through STAT3 phosphorylation (43, 44). In CRC, persistent STAT3 activation is associated with tumor growth (45, 46) and activation of MDSCs (47, 48). These observations are consistent with the results showing effectiveness of COX-2 inhibitors in the reduction of the MDSC level through blocking COX-2 and subsequent inhibition of the STAT3 pathway (43, 44, 49, 50). Another arachidonic acid metabolite, leukotriene B4 (LTB4), a product of 5-lipoxygenase (5LO), acts as a chemoattractant for MDSCs, leading to their accumulation. Deficiency of 5LO is associated not only with a lowered circulation level of MDSCs but also with decreased activity of ARG1 and iNOS (51).
The tumor microenvironment stimulates MDSCs also by other factors induced by local hypoxia and low pH (52, 53). One of them is hypoxia-inducible factor (HIF). Over-expression of HIF-1α and also HIF-2α is associated with poor prognosis in the majority of cancers, including CRC (54). HIF-1α is associated with increased activity of ARG1 and iNOS in MDSCs, leading to stronger inhibition of T-cell functions (55). Moreover, HIF-1α can also enhance the suppressive nature of MDSCs by inducing expression of programmed death-ligand 1 (PD-L1) (56), a ligand for PD-1, leading to inhibition of IL-2 production and decreased proliferation of cytotoxic T cells (56, 57). Additionally, HIF-1α, by binding to a conserved hypoxia response element in the V-domain of Ig suppressor of T-cell activation (VISTA) promoter, upregulates VISTA expression on MDSCs, thereby inducing their suppressive activity in the tumor microenvironment (58).
Many studies have shown that not only soluble mediators but also extracellular vesicles, e.g., exosomes secreted by tumor cells, may directly induce MDSC development and modulate their activity (59). This was demonstrated for many malignancies, including melanoma, breast, lung, and CRC (60). The role of cancer exosomes in CRC is complex, based on the type of cargo material transferred from cancer cells to the cells of the immune system, including MDSCs. This may occur through the delivery of tumor proteins, e.g., FasL (61) and Hsp72 (62), mRNA (63), and non-coding microRNAs (miRNA) (64). The role of miRNA in CRC, in particular, has been documented recently, with an elevated level of miRNA-21 in patients' sera correlating with poor prognosis (65, 66).
The suppressive function of MDSCs in CRC is mainly associated with their ability to inhibit T-cell proliferation and to stimulate Treg development (15). One of the important factors involved in interactions between T cells and MDSCs is L-arginine, an amino acid that is essential for T-cell proliferation and proper functioning. MDSCs highly express ARG1, which uses L-arginine, causing its depletion from the microenvironment (21, 22), which in turn affects T-cell functionality. Lack of L-arginine blocks T-cell proliferation and decreases expression of CD3ζ chain and IFNγ production (67–69). Studies on CRC have shown that MDSCs impair T-cell activation through production and iNOS activity (70, 71), which can be reversed by MDSC depletion or the use of iNOS and inhibitors (72). The mechanism of ARG1- and iNOS-dependent T-cell suppression has been explained by studies showing that, under conditions where the L-arginine level is reduced due to ARG1 activity, L-arginine is preferentially used by iNOS for and NO production, while under normal conditions, where the L-arginine level is high, only NO is produced (73). After mutual reaction of NO with , a strongly reactive oxidizing agent, peroxynitrite (ONOO−), is formed. It can cause nitration of proteins (74, 75) as well as the induction of T-cell apoptosis through the TCR/CD3 complex tyrosine phosphorylation pathway (22, 76, 77). Recent results have also shown that MDSC level correlates with reduction in the adaptive immune response to tumor antigens, e.g., MUC-1, both by lowering the production of specific antibodies and activation of tumor-specific T cells (27).
The interactions between MDSCs and Tregs in cancer are well-documented. Mainly, the activation of Tregs by MDSCs is caused by cytokines, including IL-10 and TGF-β, where the latter is also associated with MDSC induction (78). However, the relationship between MDSCs and Tregs in CRC is questionable. Some authors indicate that MDSCs in CRC do not induce Tregs development in vitro (70). On the other hand, mouse MDSCs were able to induce Tregs in vitro and in vivo through the IL-10- and IFN-γ-dependent pathway (79).
In addition to the role of MDSCs in immunosuppression that is observed during tumor progression, they may also directly stimulate tumor growth and metastases, inducing, in cooperation with VEGF, angiogenesis. Furthermore, MDSCs may introduce high levels of MMP9 and pro-MMP9 into the extracellular milieu, regulating VEGF bioavailability for colorectal cancer cells (80, 81). At the initial stage of cancer, MDSCs, through TGF-β, can also induce the epithelial to mesenchymal cell transition (EMT) process, which is essential for metastases at the late stage. These cells participate in extracellular matrix degradation in order to prepare distant tissue for receiving metastatic cells (82, 83). The latest findings reveal that PMN-MDSCs also enhance CRC growth by exosomes and exosomal protein S100A9 in the tumor microenvironment, especially under hypoxic conditions (84).
Both populations of MDSCs can effectively inhibit T-cell activity but using different mechanisms (85, 86). Some authors suggest that Mo-MDSCs are more suppressive than PMN-MDSCs (87), while others show the opposite result (88, 89). PMN-MDSCs are mainly responsible for ROS production, while Mo-MDSCs have high expression of iNOS, producing large amounts of NO, which has a longer activity than ROS. Thus, PMN-MDSCs, in contrast to Mo-MDSCs, need direct cell-to-cell contact to suppress T cells (85, 90). In this context, it has been documented that PMN-MDSCs preferentially settle the peripheral lymphoid organs, while Mo-MDSCs mainly persist in the tumor bed (85). In addition, MDSCs can also downregulate innate immune response, e.g., affecting the activity of NK cells (91). The crosstalk between MDSCs and cells in the CRC microenvironment is summarized in Figure 1. According to some authors, in human CRC, a major proportion of the MDSCs in peripheral blood are PMN-MDSCs (86). However, there are also studies showing an increased level of both populations (92–95). Additionally, an e-MDSC population was also detected in CRC patients (27, 96).
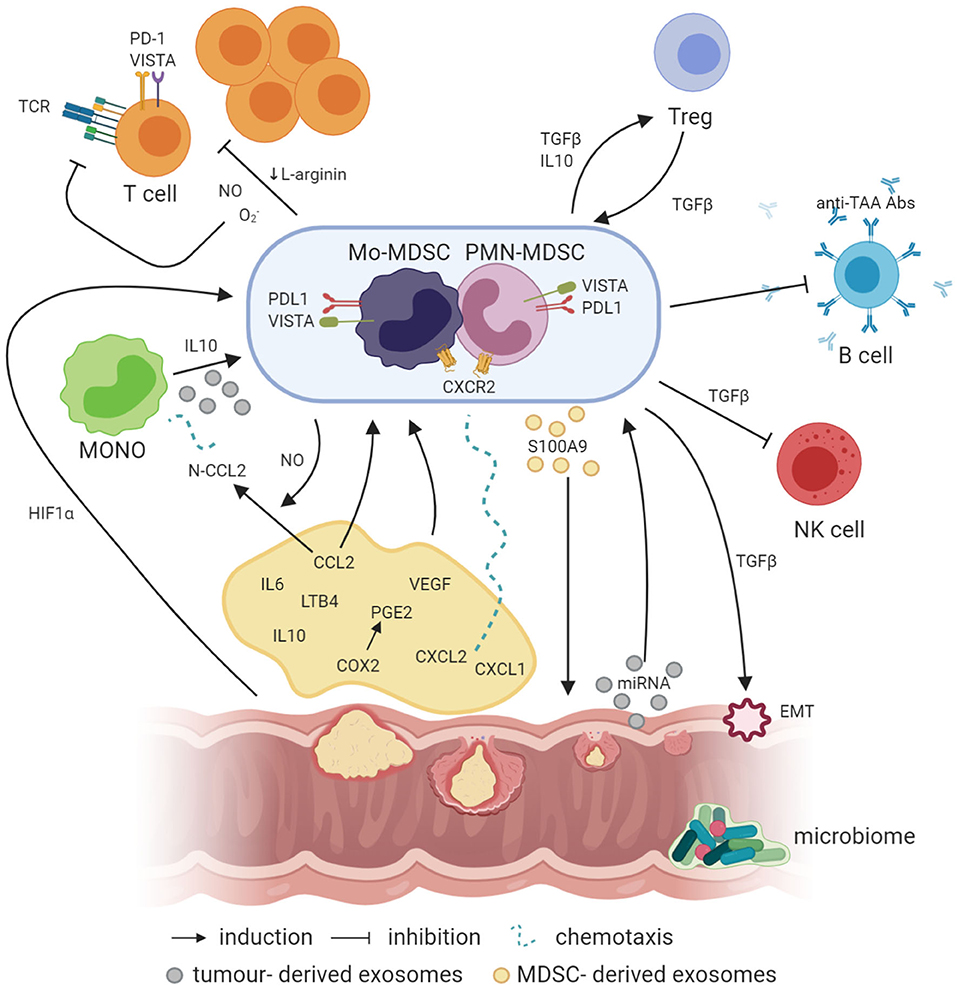
Figure 1. Crosstalk between MDSCs and other cells in the cancer microenvironment (created with BioRender.com). Factors like PGE2, IL-6, IL-10, and LTB4 are involved in the induction of MDSCs, where IL-10 can also be involved in the generation of Mo-MDSCs from circulating blood monocytes. In addition, NO produced by iNOS is required for the production of N-CCL2 from CCL2, acting as a chemoattractant for monocytes. In a similar manner, CXCL1 and CXCL2 binding to CXCR2 may recruit MDSCs to the tumor bed. Simultaneously, exosomes containing exosomal S100A9 protein are released by PMN-MDSCs, supporting the tumor growth. On the other hand, EVs generated by the tumor transfer biologically active tumor-related factors, e.g., proteins and miRNAs, which may also be involved in the induction of MDSCs from infiltrating monocytes. Moreover, hypoxia per se and hypoxia-related factors, including HIF1a, are also responsible for the induction of the expression of suppressive molecules such as VISTA or PD-L1 on the surface of MDSCs, which act through VISTA receptor and PD-1 on the T cells, respectively. TGFβ produced by MDSCs has a number of suppressive actions, e.g., MDSCs, through TGFβ, can induce the epithelial to mesenchymal cell transition (EMT) process, which is essential for metastasis formation, or inhibit NK cells. Moreover, TGFβ has a great influence, together with IL-10, on the induction of Tregs, while Tregs, producing TGFβ, induce in return MDSCs as a result of a positive feedback loop. In addition, MDSCs may also inhibit the production of antibodies and T cells directed against tumor-associated antigens (TAA), such as MUC1. Additionally, NO, O2-, and a reduced concentration of L-arginine, which are associated with MDSC activity in the tumor microenvironment, inhibit T-cell proliferation. Moreover, NO by itself can modify TCR structure and induce T-cell apoptosis.
The composition of phenotype markers used for MDSC detection and characterization in CRC quite often differs between studies. The phenotype markers and functional characteristics of MDSCs from various studies on human CRC are presented in Table 1. While the majority of the authors agree that the general phenotype of MDSCs is CD11b+ HLA-DR− Lin− CD33+ or functional markers, e.g., iNOS+ and ARG1+, there is no consensus with respect to more specific markers such as CD14, CD15, PD-L1, or CD124 (IL-4αR). The recent recommendations of the COST-Mye-EUNITER consortium provide the minimal phenotype characteristics necessary to identify cells as MDSCs: CD14−CD11b+CD15+(or CD66b+) for PMN-MDSCs; CD11b+CD14+HLA-DRlow/− CD15− for Mo-MDSCs, and Lin−(CD3/14/15/19/56)/HLA-DR−/CD33+ for e-MDSCs (17).

Table 1. The phenotype markers and functional characteristics of MDSCs as published in various studies on human CRC.
Despite the availability of chemo- and immunotherapy, surgery is still the primary method of CRC treatment. However, in a mouse model, it was shown that surgical removal of tumor mass recruits MDSCs to the peritoneal cavity and promotes tumor progression due to the surgical trauma, downregulating the CXCL4 expression. CXCL4 inhibits tumor growth and angiogenesis, which might be due to its inhibitive impact on the recruitment of MDSCs (97). In this context, it seems that MDSC-targeted therapy is urgently required for this type of cancer.
There are numerous studies concerning different small-molecule compounds that are able to inhibit the suppressive activity of MDSCs. In this section, however, the compounds with potential for CRC treatment are mainly being discussed. One such is AT38, an inhibitor of RNS, which was used in a mouse model of CRC where it proved to effectively reduce nitration of chemokines, including CCL2. Administration of AT38 also decreased the level of iNOS and ARG1 (35). Another example is nitroaspirine, which, in a mouse model, increased the number of tumor antigen-specific T cells and reduced both ARG1 and iNOS activity in MDSCs (98). Triterpenoids were also shown to reduce the suppressive functions of MDSCs through downregulation of ROS and inhibition of STAT3. However, they did not exert any effects on ARG1 activity, on NO production, or on the frequency of MDSCs (99). In the human CRC, amiloride, normally used to reduce high blood pressure, can also inhibit tumor exosome formation, which has been shown to induce suppressive functions in MDSCs (62). It was also reported that H2 blockers, e.g., cimetidine, appear to induce apoptosis of MDSCs through a Fas-FasL-dependent pathway (100).
Another therapeutic approach involves the reduction of MDSC expansion by using COX2 or PGE2 inhibitors, as PGE2 production could be associated with MDSC expansion in cancer (43). Such inhibitors, e.g., indomethacin, celocoxib, melocoxib, and acethylosalicylo acid (ASA), were able to reduce tumor growth in various tumor models, including CRC (101–103). This treatment could also modulate MDSC functions by inhibiting ARG1 expression and ROS and NO production (104, 105). ASA also reduced the level of chemokines, including CCL2, a potent chemoattractant for MDSCs (106). Another way to block MDSC accumulation is the inhibition of stem cell factor (SCF), which causes MDSC recruitment when produced in the tumor environment (107).
Another option for targeting MDSCs is inducing their differentiation. For example, curcumin used in a mouse model of CRC was able to decrease the level of PMN-MDSCs and to induce differentiation of Mo-MDSCs into cells with M1-like phenotype (108).
Another strategy for potential MDSC-targeted therapy was suggested by Condamine et al. who pointed to a shorter lifespan for MDSCs compared with neutrophils and monocytes (109). This was associated with their increased apoptosis rate in the periphery, related to high expression of TNF-related apoptosis–induced ligand receptors (TRAIL-Rs) due to the stress in endoplasmic reticulum (ER) occurring under pathophysiological conditions like cancer. Thus, targeting TRAIL-Rs by selective agonists can be considered as a future therapy for reducing MDSC activity and number (109).
Immunotherapy designed to target the checkpoint inhibitors of the PD-1–PD-L1 pathway is currently one of the most promising possibilities for reducing MDSC activity. Currently, four monoclonal antibodies are already approved by the FDA for the inhibition of this pathway: anti-PD-1 nivolumab and pembrolizumab, and anti-PD-L1 atezolizumab and avelumab. These inhibitors and several other checkpoint modulators are under clinical investigation for CRC treatment (110). In the clinical studies, nivolumab and pembrolizumab showed good response rates of 26 and 57%, respectively (111). Better results were obtained in the case of nivolumab combined with ipilimumab (anti-CTLA-4) (111–113). However, in the context of MDSCs, more satisfactory results were obtained where the PD-L1 inhibitor was used (56). Recently, several chemotherapeutic agents, e.g., gemcitabine, 5-fluorouracil, and doxorubicin, which are used in conventional cancer chemotherapy have been found to reduce MDSC numbers through the induction of apoptosis in tumor tissues as well as in the peripheral lymphoid organs (114–116), and combining these agents with immunotherapy improved survival of tumor-bearing hosts. In keeping with this, Limagne et al. in their study, provided a clinical rationale for combining chemotherapy with anti-PD-1/PD-L1 antibodies for more effective reduction of the immunosuppression caused by PMN-MDSCs in metastatic CRC (89). In this context, FOLFOX (5-fluorouracil + oxaliplatin) chemotherapy was shown to act synergistically with anti-PD-1 (117).
In the context of immunotherapy, it is worth mentioning the heterogenic genetic composition of CRC, which has important therapeutic implications. The effectiveness of immunotherapy, particularly immune checkpoint inhibition therapy, such as CTLA-4 and PD-1, has been confirmed in mismatch-repair-deficient (dMMR) and microsatellite instability-high (MSI-H) (dMMR-MSI-H) tumors, while it was ineffective in mismatch-repair-proficient (pMMR) and microsatellite instability-low (MSI-L) (pMMR-MSI-L) tumors (118). This resistance for immunotherapy of MMR-MSI-L tumors results from the inability of immune cells to recognize MSI-L mutated tumor cells and thereby reduced T-cell infiltration (119). However, it was noticed that pMMR-MSI-L tumors are more extensively infiltrated by Tregs and MDSCs than dMMR-MSI-H, which may also explain the poor immune response (120). Thus, to use of MDSC-targeted therapy seems to be a beneficial opportunity to assist the effectiveness of surgery in patients with pMMR-MSI-L cancer.
Tumor develops a variety of mechanisms to escape from immune system surveillance, including the generation of MDSCs. There is substantial evidence that MDSCs are involved in CRC development and progression. MDSCs can be detected both in the peripheral blood and tumor tissue; however, it is not known if both or one of them are relevant for predicting the prognosis for patients in the clinic. Therefore, more in-depth investigation of the mechanisms of MDSC actions in the tumor bed is still needed. Finally, more advanced pharmacological data on specific treatments targeting MDSCs are required. This could significantly improve the effectiveness of the treatment of CRC patients, and also those with pMMR-MSI-L tumors, who respond poorly to current forms of immunotherapy.
IS wrote the paper. JB critically revised the paper. All authors contributed to the article and approved the submitted version.
The authors declare and acknowledge financial support from project grant from the EU Program H2020-MSCA-RISE-2017 CANCER (777682).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. De la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer. (2004) 4:769–80. doi: 10.1038/nrc1453
3. Jia Y, Guo M. Epigenetic changes in colorectal cancer. Chin J Cancer. (2013) 32:21. doi: 10.5732/cjc.011.10245
4. Labianca R, Beretta G, Gatta G, de Braud F, Wils J. Colon cancer. Crit Rev Oncol Hematol. (2004) 51:145–70. doi: 10.1016/j.critrevonc.2004.03.003
5. De Almeida CV, de Camargo MR, Russo E, Amedei A. Role of diet and gut microbiota on colorectal cancer immunomodulation. World J Gastroentero. (2019) 25:151. doi: 10.3748/wjg.v25.i2.151
6. Gillen CD, Andrews HA, Prior P, Allan RN. Crohn's disease and colorectal cancer. Gut. (1994) 35:651–5. doi: 10.1136/gut.35.5.651
7. Cole BF, Logan RF, Halabi S, Benamouzig R, Sandler RS, Grainge MJ, et al. Aspirin for the chemoprevention of colorectal adenomas: meta-analysis of the randomized trials. JNCI J Natl Cancer Inst. (2009) 101:256–66. doi: 10.1093/jnci/djn485
8. Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. (2010) 376:1741–50. doi: 10.1016/S0140-6736(10)61543-7
9. Mei Z, Liu Y, Liu C, Cui A, Liang Z, Wang G, et al. Tumour-infiltrating inflammation and prognosis in colorectal cancer: systematic review and meta-analysis. Br J Cancer. (2014) 110:1595–605. doi: 10.1038/bjc.2014.46
10. Anitei MG, Zeitoun G, Mlecnik B, Marliot B, Haicheur N, Todosi AM, et al. Prognostic and predictive values of the immunoscore in patients with rectal cancer. Clin Cancer Res. (2014) 20:1891–9. doi: 10.1158/1078-0432.CCR-13-2830
11. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
13. Gabrilovich DI, Bronte V, Chen SH, Colombo SM, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. (2007) 67:425. doi: 10.1158/0008-5472.CAN-06-3037
14. Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S. Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood. (2011) 117:5381–90. doi: 10.1182/blood-2010-11-321752
15. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
16. Umansky V, Adema GJ, Baran J, Brandau S, Van Ginderachter JA, Hu X, et al. Interactions among myeloid regulatory cells in cancer. Cancer Immunol Immunother. (2019) 68:645–60. doi: 10.1007/s00262-018-2200-6
17. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat comm. (2016) 7:12150. doi: 10.1038/ncomms12150
18. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells. Advanc Cancer Res. (2015) 128:95–139. doi: 10.1016/bs.acr.2015.04.002
19. Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann Ny Acad Sci. (2014) 1319:47–65. doi: 10.1111/nyas.12469
20. Highfill L, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. (2010) 116:5738–47. doi: 10.1182/blood-2010-06-287839
21. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. (2005) 65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505
22. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. (2005) 5:641–54. doi: 10.1038/nri1668
23. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–42. doi: 10.1016/j.cell.2016.09.031
24. Wang PF, Song SY, Wang TJ, Ji WJ, Li SW, Liu N, et al. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: a meta-analysis of 40 studies. Oncoimmunology. (2018) 7:1494113. doi: 10.1080/2162402X.2018.1494113
25. Brandau S, Moses K, Lang S. The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer : cousins, siblings or twins? Semin Cancer Biol. (2013) 23:171–82. doi: 10.1016/j.semcancer.2013.02.007
26. Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS ONE. (2013) 8:57114. doi: 10.1371/journal.pone.0057114
27. Ma P, Beatty PL, McKolanis J, Brand R, Schoen RE, Finn OJ. Circulating myeloid derived suppressor cells (MDSC) that accumulate in premalignancy share phenotypic and functional characteristics with MDSC in cancer. Front Immunol. (2019) 10:1401. doi: 10.3389/fimmu.2019.01401
28. Kusmartsev S, Gabrilovich DI. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cance Metastasis Rev. (2006) 25:323–31. doi: 10.1007/s10555-006-9002-6
29. Meirow Y, Kanterman J, Baniyash M. Paving the road to tumor development and spreading: myeloid-derived suppressor cells are ruling the fate. Front Immunol. (2015) 6:523. doi: 10.3389/fimmu.2015.00523
30. Zhang J, Lu Y, Pienta KI. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst. (2010) 102:522–8. doi: 10.1093/jnci/djq044
31. Qian BZ, Li L, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
32. Fader AN, Rasool N, Vaziri SAJ, Kozuki T, Faber PW, Elson P, et al. CCL2 expression in primary ovarian carcinoma is correlated with chemotherapy response and survival outcomes. Anticancer Res. (2010) 30:4791–8.
33. McClellan JL, Davis JM, Steiner JL, Enos RT, Jung SH, Carson JA, et al. Linking tumor-associated macrophages, inflammation, and intestinal tumorigenesis: role of MCP-1. AJP Gastrointest Liver Physiol. (2012) 303:1087–95. doi: 10.1152/ajpgi.00252.2012
34. Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, et al. CCL2 Promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. (2015) 12:244–57. doi: 10.1016/j.celrep.2015.06.024
35. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
36. Rubie C, Frick VO, Wagner M, Schuld J, Graber S, Brittner B, et al. ELR+ CXC chemokine expression in benign and malignant colorectal conditions. BMC Cancer. (2008) 8:178. doi: 10.1186/1471-2407-8-178
37. Wang D, Wang H, Brown J, Daikoku T, Ning W, Shi Q, et al. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med. (2006) 203:941–51. doi: 10.1084/jem.20052124
38. Wen Y, Giardina SF, Hamming D, Greenman J, Zachariah E, Bacolod MD, et al. GROalpha is highly expressed in adenocarcinoma of the colon and down-regulates fibulin-1. Clin Cancer Res. (2006) 12:5951–9. doi: 10.1158/1078-0432.CCR-06-0736
39. Katoh H, Wang D, Daikoku T, Sun H, Dey SK, DuBois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell. (2013) 24:631–44. doi: 10.1016/j.ccr.2013.10.009
40. Martin RK, Saleem SJ, Folgosa L, Zellner HB, Damle SR, Nguyen GT et al. Mast cell histamine promotes the immunoregulatory activity of myeloid-derived suppressor cells. J Lekoc Biol. (2014) 96:151–9. doi: 10.1189/jlb.5A1213-644R
41. Saleem SJ, Martin RK, Morales JK, Sturgill JL, Gibb DR, Graham L, et al. Mast cells critically augment myeloid-derived suppressor cell activity. J Immunol. (2012) 189:511–5. doi: 10.4049/jimmunol.1200647
42. Burdan F, Chałas A, Szumiło J. Cyclooxygenase and prostanoids – biological implications. Postepy Hig Med Dosw. (2006) 3:129–41.
43. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. (2011) 118:5498–505. doi: 10.1182/blood-2011-07-365825
44. Han C, Demetris AJ, Stolz DB, Xu L, Lim K, Wu T. Modulation of Stat3 Activation by the cytosolic phospholipase a 2 α and cyclooxygenase-2-controlled prostaglandin E2 signaling pathway. J Biol Chem. (2006) 281:24831–46. doi: 10.1074/jbc.M602201200
45. Corvinu FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. (2005) 7:545. doi: 10.1593/neo.04571
46. Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, et al. Persistently activated stat3 maintains constitutive NF-κB activity in tumors. Cancer Cell. (2009) 15:283–93. doi: 10.1016/j.ccr.2009.02.015
47. Panni RZ, Sanford DE, Belt BA, Mitchem JB, Worley LA, Goetz BD, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother. (2014) 63:513–28. doi: 10.1007/s00262-014-1527-x
48. Su YL, Banerjee S, White SV, Kortylewski M. STAT3 in tumor-associated myeloid cells: multitasking to disrupt immunity. In J Mol Sci. (2018) 19:1803. doi: 10.3390/ijms19061803
49. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. (2007) 67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174
50. Serafini P. PGE2-producing MDSC: a role in tumor progression? J Leukocyte Biol. (2010) 88:827–9. doi: 10.1189/jlb.0510303
51. Cheon EC, Khazaie K, Khan MW, Strouch MJ, Krantz SB, Phillips J, et al. Mast cell 5-lipoxygenase activity promotes intestinal polyposis in APC 468 Mice. Cancer Res. (2011) 71:1627–36. doi: 10.1158/0008-5472.CAN-10-1923
52. Chouaib S, Umansky V, Kieda C. The role of hypoxia in shaping the recruitment of proangiogenic and immunosuppressive cells in the tumor microenvironment. Contemp Oncol. (2018) 22:7–13. doi: 10.5114/wo.2018.73874
53. Won WJ, Deshane JS, Leavenworth JW, Oliva CR, Griguer CE. Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress. (2019) 3:47. doi: 10.15698/cst2019.02.176
54. Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. (2011) 12:9–22. doi: 10.1038/nrc3183
55. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
56. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
57. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J Immunother Cancer. (2014) 2:3. doi: 10.1186/2051-1426-2-3
58. Deng J, Li J, Sarde A, Lines JL, Lee YC, Qian DC, et al. Hypoxia-induced VISTA promotes the suppressive function of myeloid-derived suppressor cells in tumor microenvironment. Cancer Immunol Res. (2019) 7:1079–90. doi: 10.1158/2326-6066.CIR-18-0507
59. Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. (2009) 124:2621–33. doi: 10.1002/ijc.24249
60. Fleming V, Hu X, Weller C, Weber R, Groth C, Riester Z, et al. Melanoma extracellular vesicles generate immunosuppressive myeloid cells by upregulating PD-L1 via TLR4 signaling. Cancer Res. (2019) 79:4715–28. doi: 10.1158/0008-5472.CAN-19-0053
61. Huber V, Fais S, Valenti R, Canese P, Cova A, Sovena G, et al. Human colon carcinoma cells release microvesicles bearing active FasL and TRAIL and affecting tumor/immune interactions. AACR. (2004) 45:504.
62. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. (2010) 120:457–71. doi: 10.1172/JCI40483
63. Baj-Krzyworzeka M, Szatanek R, Weglarczyk K, Baran J, Urbanowicz B, Branski P, et al. Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol Immunother. (2006) 55:808–18. doi: 10.1007/s00262-005-0075-9
64. Francavilla A, Turoczi S, Tarallo S, Vodicka P, Pardini B, Naccarati A. Exosomal microRNAs and other non-coding RNAs as colorectal cancer biomarkers: a review. Mutagenesis. (2019) 35:243–60. doi: 10.1093/mutage/gez038
65. Ogata-Kawata H, Izumiya M, Kurioka D, Honma Y, Yamada Y, Furuta K, et al. Circulating exosomal microRNAs as biomarkers of colon cancer. PLoS ONE. (2014) 9:92921. doi: 10.1371/journal.pone.0092921
66. Nielsen BS, Jørgensen S, Fog JU, Søkilde R, Christensen IJ, Hansen U, et al. High levels of microRNA-21 in the stroma of colorectal cancers predict short disease-free survival in stage II colon cancer patients. Clin Exp Metas. (2011) 28:27–38. doi: 10.1007/s10585-010-9355-7
67. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3ζ chain expression byl-arginine. J Biol Chem. (2002) 277:21123–9. doi: 10.1074/jbc.M110675200
68. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-Cell receptor expression and antigen-specific T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
69. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. (2007) 109:1568–73. doi: 10.1182/blood-2006-06-031856
70. OuYang LY, Wu XJ, Ye SB, Zhang R, Li RL, Liao W, et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Transl Med. (2015) 13:47. doi: 10.1186/s12967-015-0410-7
71. Zhao F, Hoechst B, Duffy A, Gamrekelashvili J, Fioravanti S, Manns MP, et al. S100A9a new marker for monocytic human myeloid-derived suppressor cells. Immunology. (2012) 136:176–83. doi: 10.1111/j.1365-2567.2012.03566.x
72. Kusmartsev SA, Li Y, Chen SH. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immuno. (2000) l:779–85. doi: 10.4049/jimmunol.165.2.779
73. Mondanelli G, Ugel S, Grohmann U, Bronte V. The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr Opin Pharmacol. (2017) 35:30–9. doi: 10.1016/j.coph.2017.05.002
74. Pall ML. The NO/ONOO-cycle as the central cause of heart failure. Int J Mol Sci. (2013) 14:22274–330. doi: 10.3390/ijms141122274
75. Bogdan C. Nitric oxide and the immune response. Nat Immunol. (2001) 2:907–16. doi: 10.1038/ni1001-907
76. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. (2007) 13:828–35. doi: 10.1038/nm1609
77. Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. (2010) 184:3106–16. doi: 10.4049/jimmunol.0902661
78. Lee CR, Kwak Y, Yang T, Han JH, Park SH, Michael BY, et al. Myeloid-derived suppressorcells are controlled by regulatory T cells via TGF-β during murine colitis. Cell Rep. (2016) 17:3219–32. doi: 10.1016/j.celrep.2016.11.062
79. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1 + CD115 + Immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in Tumor-Bearing Host. Cancer Res. (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
80. Yang L, DeBusk M, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. (2004) 6:409–21. doi: 10.1016/j.ccr.2004.08.031
81. Mira E, Lacalle RA, Buesa JM, de Buitrago GG, Jiménez-Baranda S, Gómez-Moutón C, et al. Secreted MMP9 promotes angiogenesis more efficiently than constitutive active MMP9 bound to the tumor cell surface. J Cell Sci. (2004) 117:1847–57. doi: 10.1242/jcs.01035
82. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGFβ signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. (2008) 13:23–35. doi: 10.1016/j.ccr.2007.12.004
83. Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. (2011) 9:1001162. doi: 10.1371/journal.pbio.1001162
84. Wang Y, Yin K, Tian J, Xia X, Ma J, Tang X, et al. Granulocytic myeloid-derived suppressor cells promote the stemness of colorectal cancer cells through exosomal S100A9. Adv Sci. (2019) 6:1901278. doi: 10.1002/advs.201901278
85. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
86. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. (2015) 3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036
87. Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. (2010) 40:22–35. doi: 10.1002/eji.200939903
88. Lang S, Bruderek K, Kaspar C, Höing B, Kanaan O, Dominas N, et al. Clinical relevance and suppressive capacity of human myeloid-derived suppressor cell subsets. Clin Cancer Res. (2018) 24:4834–44. doi: 10.1158/1078-0432.CCR-17-3726
89. Limagne E, Euvrard R, Thibaudin M, Rébé C, Derangère V, Chevriaux A, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX–bevacizumab drug treatment regimen. Cancer Res. (2016) 76:5241–52. doi: 10.1158/0008-5472.CAN-15-3164
90. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer. (2014) 134:2853–64. doi: 10.1002/ijc.28622
91. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloidderived suppressor cells induce anergy of NK cells through membrane-bound TGF- 1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
92. Karakasheva TA, Dominguez GA, Hashimoto A, Lin EW, Chiu C, Sasser K, et al. CD38+ M-MDSC expansion characterizes a subset of advanced colorectal cancer patients. JCI Insight. (2018) 3:e97022. doi: 10.1172/jci.insight.97022
93. Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, et al. IL4Rα+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. (2009) 182:6562–8. doi: 10.4049/jimmunol.0803831
94. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. γδT17 Cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. (2014) 40:785–800. doi: 10.1016/j.immuni.2014.03.013
95. Mundy-Bosse BL, James Cancer Hospital AG, Young GS, James AG, Bauer T, Binkley E, et al. Distinct myeloid suppressor cell subsets correlate with plasma IL-6 and IL-10 and reduced interferon-alpha signaling in CD4 + T cells from patients with GI malignancy. Cancer Immunol Immun. (2011) 60:1269–79. doi: 10.1007/s00262-011-1029-z
96. Toor SM, Syed Khaja AS, El Salhat H, Bekdache O, Kanbar J, Jaloudi M, et al. Increased levels of circulating and tumor-infiltrating granulocytic myeloid cells in colorectal cancer patients. Front Immunol. (2016) 7:560. doi: 10.3389/fimmu.2016.00560
97. Xu P, He H, Gu Y, Wang Y, Sun Z, Yang L, et al. Surgical trauma contributes to progression of colon cancer by downregulating CXCL4 and recruiting MDSCs. Exp Cell Res. (2018) 370:692–8. doi: 10.1016/j.yexcr.2018.07.035
98. De Santo C, Serafini P, Marigo I, Dolcetti L. M, Bolla M, Del Soldato P, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci USA. (2005) 102:4185–90. doi: 10.1073/pnas.0409783102
99. Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune responsein cancer. Clin Cancer Res. (2010) 16:1812–23. doi: 10.1158/1078-0432.CCR-09-3272
100. Zheng Y, Xu M, Li X, Jia J, Fan K, Lai G. Cimetidine suppresses lung tumor growth in mice through proapoptosis of myeloid-derived suppressor cells. Mol Immunol. (2013) 54:74–83. doi: 10.1016/j.molimm.2012.10.035
101. Levin G, Kariv N, Khomiak E, Raz A. Indomethacin inhibits the accumulation of tumor cells in mouse lungs and subsequent growth of lung metastases. Chemotherapy. (2000) 46:429–37. doi: 10.1159/000007323
102. Leahy KM, Ornberg RL, Wang Y, Zweifel BS, Koki AT, Masferrer JL. Cyclooxygenase-2 inhibition by celecoxib reduces proliferation and induces apoptosis in angiogenic endothelial cells in vivo. Cancer Res. (2002) 62:625–31.
103. Tsubouchi Y, Mukai S, Kawahito Y, Yamada R, Kohno M, Inoue K, et al. Meloxicam inhibits the growth of non-small cell lung cancer. Anticancer Res. (2000) 20:2867−72.
104. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer. (2010) 10:464. doi: 10.1186/1471-2407-10-464
105. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. (2005) 202:931–9. doi: 10.1084/jem.20050715
106. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. (2011) 71:2664–74. doi: 10.1158/0008-5472.CAN-10-3055
107. Pan PY, Wang GX, Yin B, Ozao J, Ku T, Divino CM, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. (2008) 111:219–28. doi: 10.1182/blood-2007-04-086835
108. Tu SP, Jin H, Shi JD, Zhu LM, Suo Y, Lu G, et al. Curcumin induces the differentiation of myeloid-derived suppressor cells and inhibits their interaction with cancer cells and related tumor growth. Cancer Prev Res (Phila). (2012) 5:205–15. doi: 10.1158/1940-6207.CAPR-11-0247
109. Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R–mediated apoptosis. J Clin Invest. (2014) 124:2626–39. doi: 10.1172/JCI74056
110. Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Signal transduct Targ Ther. (2020) 5:1–30. doi: 10.1038/s41392-020-0116-z
111. Overman M, Kopetz S, McDermott R, Leach J. Nivolumab±ipilimumab in treatment (tx) of patients (pts) with metastatic colorectal cancer (mCRC) with and without high microsatellite instability (MSI-H): checkMate-142 interim results. J Clin Oncol. (2016) 34:3501. doi: 10.1200/JCO.2016.34.15_suppl.3501
112. Boland P, Ma W. Immunotherapy for colorectal cancer. Cancers. (2017) 9:50. doi: 10.3390/cancers9050050
113. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
114. Wang Z, Till B, Gao Q. Chemotherapeutic agent-mediated elimination of myeloid-derived suppressor cells. Oncoimmunology. (2017) 7:1331807. doi: 10.1080/2162402X.2017.1331807
115. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell–dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690
116. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. (2018) 9:398. doi: 10.3389/fimmu.2018.00398
117. Dosset M, Vargas TR, Lagrange A, Boidot R, Végran F, Roussey A, et al. PD-1/PD-L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology. (2018) 7:1433981. doi: 10.1080/2162402X.2018.1433981
118. Yamagishi H. Kuroda H. Imai Y. Hiraishi H. Molecular pathogenesis of sporadic colorectal cancers. Chin J Cancer. (2016) 35:4. doi: 10.1186/s40880-015-0066-y
119. Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. (2015) 16:64. doi: 10.1186/s13059-015-0620-6
Keywords: colorectal cancer (CRC), myeloid-derived suppressor cells (MDSCs), inducible NO synthase (iNOS), arginase-1 (ARG1), T regulatory cells (Tregs)
Citation: Sieminska I and Baran J (2020) Myeloid-Derived Suppressor Cells in Colorectal Cancer. Front. Immunol. 11:1526. doi: 10.3389/fimmu.2020.01526
Received: 16 April 2020; Accepted: 10 June 2020;
Published: 07 August 2020.
Edited by:
Sergei Kusmartsev, University of Florida, United StatesReviewed by:
Kebin Liu, Augusta University, United StatesCopyright © 2020 Sieminska and Baran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jarek Baran, bWliYXJhbkBjeWYta3IuZWR1LnBs
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.