 Tracey L. Freeman
Tracey L. Freeman Talia H. Swartz
Talia H. Swartz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 23 June 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01518
This article is part of the Research Topic Coronavirus Disease (COVID-19): Pathophysiology, Epidemiology, Clinical Management and Public Health Response View all 400 articles
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a member of the genus Betacoronavirus within the family Coronaviridae. It is an enveloped single-stranded positive-sense RNA virus. Since December of 2019, a global expansion of the infection has occurred with widespread dissemination of coronavirus disease 2019 (COVID-19). COVID-19 often manifests as only mild cold-like symptomatology, but severe disease with complications occurs in 15% of cases. Respiratory failure occurs in severe disease that can be accompanied by a systemic inflammatory reaction characterized by inflammatory cytokine release. In severe cases, fatality is caused by the rapid development of severe lung injury characteristic of acute respiratory distress syndrome (ARDS). Although ARDS is a complication of SARS-CoV-2 infection, it is not viral replication or infection that causes tissue injury; rather, it is the result of dysregulated hyperinflammation in response to viral infection. This pathology is characterized by intense, rapid stimulation of the innate immune response that triggers activation of the Nod-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome pathway and release of its products including the proinflammatory cytokines IL-6 and IL-1β. Here we review the literature that describes the pathogenesis of severe COVID-19 and NLRP3 activation and describe an important role in targeting this pathway for the treatment of severe COVID-19.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a member of the genus Betacoronavirus within the family Coronaviridae. It is an enveloped single-stranded positive-sense RNA virus (1). In December of 2019, the first cases of an atypical viral pneumonia were reported in Wuhan, China. Since that time, a global expansion of the infection has occurred with widespread dissemination of coronavirus disease 2019 (COVID-19) (2, 3). For most, the infection is mild with low-grade fever and cough, but 15% are associated with respiratory compromise. Severe cases result in acute respiratory distress syndrome (ARDS) with systemic inflammation in which lung injury is associated with release of inflammatory cytokines IL-6 and IL-1β (2, 4). The systemic inflammatory syndrome is characterized by dysregulated proinflammatory cytokine cascades triggered by an intense, rapid activation of the innate immune response. COVID-19 severity is associated with increased proinflammatory cytokines and chemokines and IL-6, specifically, is predictive of COVID-19 fatality (5). High levels of interleukin IL-1β and IL-6 were detected in autopsy tissues from SARS-CoV patients (6) and single cell RNA-seq analysis of peripheral blood in COVID-19 patients show increased subsets of CD14+ IL-1β-producing monocytes (7). A clear mechanism is not yet understood. The inflammatory basis underlying COVID-19 fatality renders development of immunoregulatory agents of paramount importance (8). There is significant literature implicating the Nod-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome, and cytokine release syndrome or cytokine storm in this pathogenesis (9–12). The NLRP3 inflammasome is an important cause of activation of the innate immune system to recognize pathogens, including viral infections (13, 14). SARS-CoV 3a protein activates the NLRP3 inflammasome in lipopolysaccharide-primed macrophages with 3a-mediated IL-1β secretion associated with K+ efflux and mitochondrial reactive oxygen species (15).
Individuals at risk for this inflammatory syndrome include those with hypertension, diabetes, cardiovascular disease, respiratory disease, and cancer (16, 17). It is not clear why individuals at risk include those with cardiovascular risk factors but may relate to the virology of SARS-CoV-2 infection. SARS-CoV uses the spike glycoprotein (S protein) on the surface of the virion to mediate viral membrane fusion (18). The S protein is a trimer that is cleaved into S1 and S2 subunits; S1 binds directly to the peptidase domain of angiotensin-converting enzyme 2 (ACE2) (19) to expose S2 to cleavage that enables fusion and entry (20). The physiological function of ACE2 in the cell is the maturation of angiotensin (Ang) which regulates blood pressure through vasoconstriction. Clinical literature based on the 2003 SARS-CoV epidemic suggested that the virus caused ACE2 downregulation and that lung injury may be improved by Angiotensive II Receptor Blocker (ARB) treatment (21, 22). Further literature implicates ACE2 signaling in NLRP3 activation in multiple settings. AngII can induce NLRP3 inflammasome activation in renal tubular epithelial cells (23), AngII induces pulmonary fibrosis which is attenuated by ACE2 (24), and NLRP3 inflammasome activation drives Ang II-induced vascular smooth muscle cell (VSMC) proliferation and vascular remodeling and hypertension (25, 26).
Individuals infected with SARS-CoV-2 can present with an array of clinical severity from asymptomatic through severe disease characterized by pneumonia requiring supplemental oxygen, and progression to acute respiratory distress syndrome (ARDS) with systemic inflammatory response syndrome (SIRS), shock and multiorgan dysfunction, coagulopathy, and death (27). Early symptoms can include shortness of breath, fever, and cough with increasing reports of loss of taste and smell (4, 17, 28–30). Individuals demonstrated to be at high risk of severe outcomes include those with advanced age, hypertension, cardiovascular disease, and diabetes mellitus (4, 29, 31, 32). Severe COVID-19 is associated with increased serum inflammatory cytokine levels including IL-1, IL-6, granulocyte-colony stimulating factor (G-CSF), interferon-γ inducible protein 10 (IP-10), and tumor necrosis factor-α (TNF-α) (5, 17, 33–36).
Overwhelming inflammatory cytokine secretion can result in ARDS through massive recruitment of immune cells leading to vascular leakage, fluid accumulation causing pulmonary edema, and resulting hypoxemia (37–39). Reports of patients with severe COVID-19 indicate that elevated levels of IL-1β and IL-6 are associated with elevated immune exhaustion and reduced T cell functional diversity (40). By contrast, individuals with COVID-19 who experience more mild disease have lower levels of IL-6, together with activated T lymphocytes and IgM SARS-CoV-2-binding antibodies (41). These observations indicate that a robust inflammatory cytokine response mediates severe disease while low inflammatory cytokine responses may be associated with an adaptive response that favors disease resolution. IL-1β is a key regulator of many chronic inflammatory diseases (42–49). Therefore, probing the role of IL-1β and its inhibition might lead to reduced inflammatory signaling, thus reducing lung injury in ARDS associated with severe COVID-19.
The NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) inflammasome consists of a sensor (NLRP3), an adaptor (ASC; also known as PYCARD), and an effector (caspase 1) (50). NLRP3 contains an amino-terminal pyrin domain (PYD), a central NACHT domain (domain present in NAIP, CIITA, HET-E, and TP1) and a carboxy-terminal leucine-rich repeat domain (LRR domain). The NACHT domain mediates ATPase function that is vital for NLRP3 self-association and function (51) and the LRR domains autoregulate through folding back onto the NACHT domain. ASC has two protein binding domains, an amino-terminal PYD and a carboxy-terminal caspase recruitment domain (CARD). NLRP3 can oligomerize between NACHT domains upon stimulation which leads to ASC recruitment through PYD–PYD interactions. The formation of multiple ASC filaments is referred to as an ASC speck (52–54). The assembled ASC complex can recruit caspase 1 to facilitate cleavage and activation.
Activation of the inflammasome is highly regulated and mediated by a two-step process in which first priming occurs and then activation occurs. Priming allows for transcription upregulation of the NLRP3 genes in response to recognition of pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharides and viral RNA, or damage-associated molecular patterns (DAMPs), such as ATP and reactive oxygen species, through purine sensing receptors including P2RX7 (13, 14, 54–56). Engagement of PAMPS and/or DAMPS can activate pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) or nucleotide-binding oligomerization domain-containing protein 2 (NOD2). This leads to activation of nuclear factor-κB (NF-κB) activation and gene transcription (57). Priming also shifts oxidative phosphorylation to glycolysis in macrophages, resulting in stabilization of hypoxia-inducible factor 1α (HIF1α) and increase in IL1B gene transcription (58). Priming additionally induces post-translational modifications of the NLRP3 inflammasome which include ubiquitylation, phosphorylation, and sumoylation that stabilize the NLRP3 inflammasome in an auto-suppressed inactive, signal-competent, state (59).
After priming, NLRP3 inflammasome activation can occur in response to an array of pathogens or endogenous DAMPs. Multiple cellular signaling events can result in NLRP3 activation at the membrane, including efflux of potassium (K+) or chloride ions (Cl−), and flux of calcium ions (Ca2+) (60–70) as well as other cellular functions including lysosomal disruption, mitochondrial dysfunction, metabolic changes, and trans-Golgi disassembly (50).
NLRP3 activation can lead to pyroptosis, an inflammatory programmed cell death pathway that takes place in T lymphocytes (71). This inflammatory cell death is activated through gasdermin D (GSDMD) cleavage by caspase 1, 4, 5, and/or 11 and results in a series of cellular events including swelling of the cytoplasm, plasma membrane rupture, and consolidation of the nucleus with release of cytoplasmic contents into the extracellular space (72, 73). GSDMD contains an amino-terminal cell death domain (GSDMDNterm) which is exposed through caspase cleavage to bind phosphatidylinositol phosphates and phosphatidylserine in the cell membrane, inserting into the plasma membrane and forming a pore that kills the cell from within (74, 75). Additionally, GSDMD can mediate IL-1β and IL-18 secretion (76, 77) and this occurs both through pathways dependent and independent of NLRP3 signaling.
Cell death is an important cause of pathogenesis in viral infections. HIV-1 infection is associated with programmed cell death through pyroptosis in bystander cells (78–82) and represents an important mechanism of NLRP3 inflammasome-mediated immune cell depletion. Programmed cell death through multiple mechanisms has been reported in coronavirus infections as an important mechanism of viral pathogenesis (83–88).
There are numerous studies that implicate the NLRP3 inflammasome and IL-1β in mediating inflammation during lung injury and ARDS (39, 89, 90). Bronchoalveolar fluid and plasma in patients with ARDS have elevated IL-1β levels compared to healthy controls (91–94) and is associated with worse clinical outcomes. In other coronavirus infections including MERS-CoV and SARS-CoV, patients with ARDS had high levels of IL-1β, IL-6, and IL-8 (6, 95–97). In other respiratory viral infections such as influenza, high levels of IL-1β have been detected in bronchoalveolar fluid and plasma from patients with lung injury (91–94, 98–101). Furthermore, animal studies in which mice deficient in components of the inflammasome have reduced lung injury and enhanced survival with influenza infection (45, 102). In pharmacologic studies in which IL-1β or IL-1R was antagonized, influenza associated lung injury was reduced (103, 104). Taken together, IL-1β appears to play a key role in acute lung injury with respiratory viral infections and pharmacologic targeting of this pathway represents an important area of intervention.
Injury of type II alveolar epithelial cells expressing ACE2 leads to NLRP3 inflammasome activation (14, 15, 105). The acute immune response to SARS-CoV-2 infection is largely driven by inflammatory alveolar and monocyte-derived macrophages that are activated by PAMPs and DAMPs released by infected, apoptotic pneumocytes (11, 106–108). TNF-α and IL-1β secreted by alveolar macrophages initiate the acute proinflammatory cascade immediately following infection. The secretion of these cytokines induces cell death and damage, PAMP/DAMP production, immune cell recruitment, and widespread NLRP3 activation, establishing a proinflammatory positive feedback cascade (11, 106, 108–110). More recently, Blanco-Melo et al. demonstrated that SARS-CoV-2 infection of primary human bronchial epithelial cells resulted in expression of multiple cytokines and chemokines including TNF-α, IL-6, and IL-1β (111).
This localized inflammatory cell death extends to the vasculature, inducing the leakage, edema, and pneumonia characteristic of COVID-19 (11, 108, 109). It is important to note that the onset of this pathological immune response is characterized not by systemic inflammation, but by a hyperinflammatory microenvironment localized to the site of tissue injury. As the inflammatory cascade progresses, IL-1β, and TNF-α induce the secretion of additional NLRP3 cytokines such as IL-6 which can subsequently be observed in the peripheral blood due to the loss of vascular integrity (11, 107–110, 112, 113). The kinetics of the inflammatory response are essential to effective clinical practice—circulating biomarkers such as IL-6 may prove useful to predicting outcomes and informing immunomodulatory treatment decisions (31, 33, 114–116).
The rapid decline of COVID-19 patients coincides with an abrupt shift from the NLRP3 cytokine storm to a compensatory immunosuppressive state (5, 107). This repair and recovery-oriented phase is characterized by production of IL-10, polarization of macrophages to the anti-inflammatory M2 state, suppression of NLRP3, and recruitment of fibroblasts and platelets. The accumulation of fibroblasts and M2 macrophages in the lung initiates the deposition of collagen and construction of the extracellular matrices that characterize ARDS fibrosis (11, 108, 117). M2 macrophages and other markers of this pro-fibrotic, anti-inflammatory environment have been detected in the bronchioalveolar fluid of severe COVID-19 patients (117, 118).
Unique to SARS-CoV and SARS-CoV-2 is the downmodulation of the ACE2 receptor. SARS-CoV entry has been reported to be dependent on TNF-α converting enzyme and coupled to the release of TNF-α from the cell membrane (110). TNF-α, specifically, has been shown to act as an alternative toll-like receptor (TLR) agonist that may increase the sensitivity and longevity of NLRP3 activation (113, 119). Downregulation of ACE2 is associated with both SARS-CoV and SARS-CoV-2 disease severity (21, 120, 121); this contrasts with a minimally symptomatic coronavirus strain, HCoV-NL63, that utilizes but does not cleave or downmodulate the ACE2 receptor (122). The overproduction of TNF-α in COVID-19 may preferentially activate the NLRP3 inflammasome relative to other immunological pathways. These observations warrant further investigation into the mechanisms by and extent to which TNF-α acts as a significant modulator of severe COVID-19.
The SARS-CoV genome encodes 3 ion channel proteins: E, open reading frame 3a (ORF3a), and ORF8a in which E and ORF3a are required for both replication and virulence (87, 109, 123–126). In addition to the canonical NLRP3 activation pathway by PAMPs and DAMPs, the E, 3a, and 8b proteins of SARS-CoV function as NLRP3 agonists (84, 107, 109, 123, 127); many of these sequences are conserved in SARS-CoV-2 and likely play a role in inflammatory pathogenesis (107, 128). The SARS-CoV E, 3a, and 8b proteins are all reported to induce NLRP3 activation and IL-1β release in LPS-primed macrophage models (15, 127). A wide variety of mechanisms have been proposed for this NLRP3 agonism including E-, 3a-, and 8b-induced viroporin activity, interferon antagonism, membrane-bound organelle stress, reactive oxygen species production, and direct binding to and regulation of inflammasome components such as caspase 1, NLRP3, and NF-κB (15, 86, 107, 109, 112, 123, 127). There are multiple pathways by which SARS-CoV triggers NLRP3 activation which have yet to be characterized and are likely influenced by cell type and the extracellular microenvironment (15, 84, 86, 88, 107).
Notably, the NLRP3-implicated ORFs 3a and 8 are the primary sites driving genetic diversification of SARS-CoV-2. ORF3a, specifically, is the only gene undergoing diversifying mutations that are predicted to exhibit altered phenotypes (84, 113, 127, 129). Ongoing mutations in ORF8 are particularly concerning, as a 29-nt deletion of the SARS-CoV genome is suspected to have increased the pathogenicity of the virus during the SARS-CoV epidemic by antagonizing interferon, increasing viral titers, and agonizing NLRP3 (127, 130). The uniquely low homology between SARS-CoV-2 and SARS-CoV ORFs 3a and 8 may play a role in the differences in virulence and pathogenesis between these two related viral infections (107, 131). Defining the inflammatory activities of these two proteins is therefore critical to predictive monitoring and modeling of novel SARS-CoV-2 strain emergence.
Genetic variations in host inflammasome pathways may also influence disease outcome. Mutations in the LRR domain of bat NLRP3 mediate an overall dampened NLRP3 response to agonists (85). In the context of coronavirus infections, MERS-CoV does not induce clinical disease in bats despite high viral titers; this appears to be mediated by NLRP3 (85). Interestingly, SARS-CoV ORF8b is reported to activate NLRP3 via direct binding to the LRR domain, suggesting a mechanism of coronavirus-induced NLRP3 activation and further indicating therapeutic potential for NLRP3 immunomodulatory agents (127). Defining these mechanisms should be a focus of SARS-CoV-2 research so as to identify targeted therapeutics such as those summarized in Table 1.
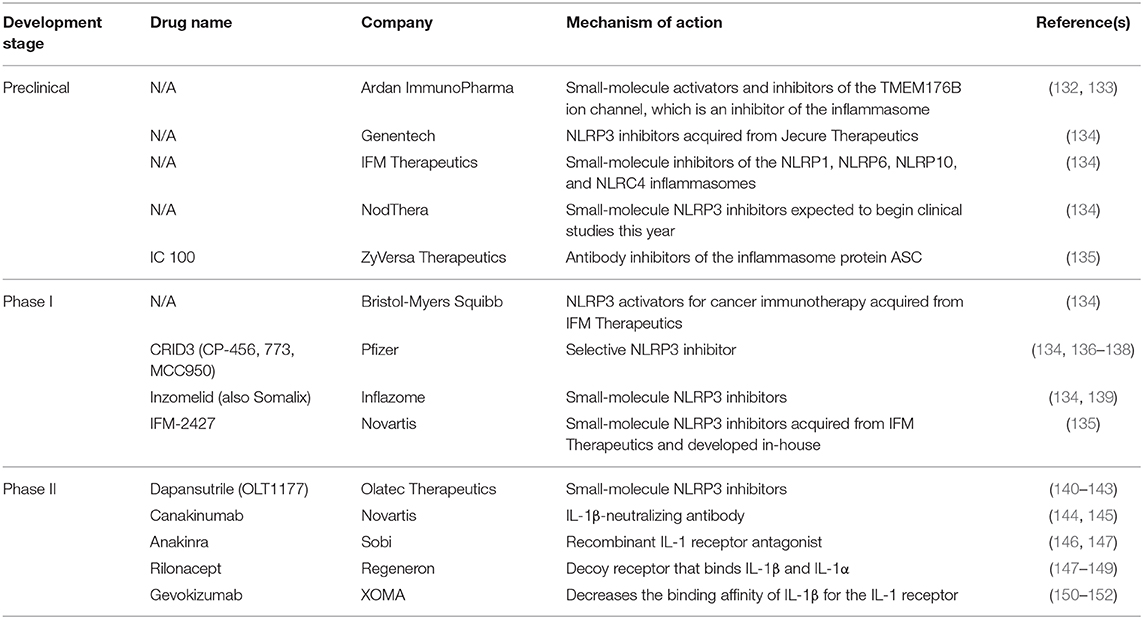
Table 1. NLRP3 inflammasome-targeted therapeutics in development.
Cytokine release syndrome (CRS) is a systemic inflammatory response that can be triggered by a number of stimuli including drugs and infections (153, 154). The term was originally coined in response to administration of anti-T-cell antibody muromonab-CD3 (OKT3) to solid organ transplant patients who experienced an idiosyncratic cytokine storm following treatment (155, 156). A number of other drugs have stimulated similar infusion reactions including antibody-based therapies (157–164) and cancer therapeutics (165, 166). Other reported stimuli for the development of CRS include haploidentical donor stem cell transplantation, graft-vs.-host disease (167, 168), and respiratory viral infections including influenza (11, 169). Most recently, new classes of immunotherapeutic agents are used in a variety of hematologic malignancies including bispecific antibody constructs and chimeric antigen receptor (CAR) T cell therapies.
In response to these stimuli, patients experience robust cytokine-mediated response that is associated with fever, hypotension and hypoxemia. The syndrome can be mild and resolve spontaneously or can progress to persistent high-grade fevers, vasodilatory shock with hemodynamic instability, severe hypoxemia requiring mechanical ventilation. This can be associated with end-organ damage including liver injury, cardiac ischemia, clotting dysfunction, kidney dysfunction, and hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) (154). The timing of onset is unpredictable, between 1 day to 2 months after exposure (170).
In SARS-CoV-2 infection, a cytokine storm occurs that has similar features to CRS as described above. Individuals with severe COVID-19 with cytokine storm have elevated systemic inflammatory biomarkers including C-reactive protein, D-dimer, ferritin (3, 115, 171–173). Patients experience a dysfunctional immune response characterized by high levels of plasma cytokines including IL-6, TNF-α, IL-8, IL-10, IL-1RA, and CXCL10 (4, 117). IL-6 levels increase over time higher in those who die of the infection compared to those who survive (27). The stimulation of inflammatory cytokines, largely through activated macrophages, leads to acute lung injury, acute respiratory distress syndrome, systemic inflammatory response syndrome (SIRS), shock and multiorgan dysfunction, and coagulopathy (117). This is described in Figure 1.
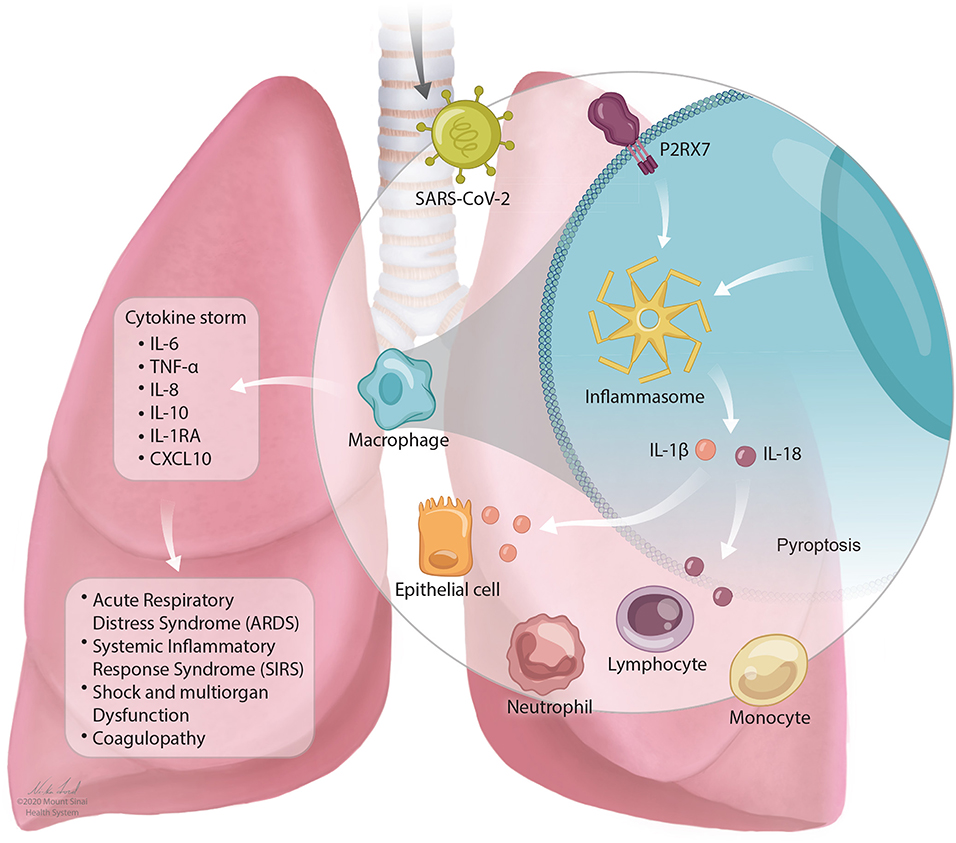
Figure 1. The NLRP3 inflammasome mediates lung inflammation in SARS-CoV-2 infection. SARS-CoV-2 is inhaled into the airway and mediates activation of the P2RX7 receptor by release of extracellular ATP. P2RX7 signaling can lead to NLRP3 activation through direct or indirect activation in activated macrophages. Activation of the NLRP3 inflammasome drives the secretion of IL-1β and IL-18 which can result in pyroptosis (programmed cell death). Activation of immune cell subsets, largely through activated macrophages, results in a cascade of massive inflammatory cytokine activation including IL-6, TNF-α, IL-8, IL-10, IL-1RA, and CXCL10 that lead to acute lung injury with acute respiratory distress syndrome, systemic inflammatory response syndrome (SIRS), shock and multiorgan dysfunction, and coagulopathy.
Individuals with severe COVID-19 have developed a coagulopathy which is associated with reduced platelet count, increased levels of fibrin degradation productions (D-dimer), and increased microthrombi in lungs, brain, kidney, and extremities (174–176). The NLRP3 inflammasome may play a key role in mediating this coagulopathy. Activated macrophages undergoing NLRP3 inflammasome activation release tissue factor which initiates coagulation (177, 178), regulation of platelet integrins (179, 180), and through hypoxia-inducible factor 1-alpha (HIF-1α) (181). Whether NLRP3 inflammasome activation as a mediator of coagulopathy is an area of great interest for future investigation.
Experimental therapeutics assessed in vitro and in vivo have provided further insight into the role of NLRP3 in mediating SARS-CoV pathogenicity. In bone marrow-derived macrophages, a mitochondrial antioxidant reduced IL-1β secretion induced by SARS-CoV 3a and E proteins (15). In SARS-CoV-infected mice, the NF-κB antagonists CAPE, resveratrol, Bay11-7082, and parthenolide improved survival and reduced proinflammatory cytokine levels in the lungs (182). Depletion of inflammatory macrophages also mitigated SARS-CoV-associated inflammatory lung pathology in mice without impacting viral load (108). These reports elucidate molecular and clinical inflammatory phenotypes that appear to parallel those seen in COVID-19 and should be used to inform novel therapeutic development and pathogenesis studies.
Cross-regulation between type I interferon (IFN-I) and the NLRP3 inflammasome is implicated in the abrupt proinflammatory response to immunosuppressive switch characteristic of SARS and COVID-19 ARDS through an undefined mechanism (5, 107). Early IFN-I administration may therapeutically regulate NLRP3 and has been shown to abrogate clinical symptomatology in SARS-CoV-infected macaques (112) and mice (108). Dual corticosteroid-IFN-I treatment appeared to improve outcomes in a small-cohort SARS-CoV trial (183, 184). The therapeutic impact observed in mice, macaques, and humans in each setting occurred despite unchanged viral loads (108, 112, 183, 184).
Both IL-6R and IL-1 receptor blocking agents have been used for the treatment of CRS (185, 186). Tocilizumab, an IL-6R blocking antibody has been used to treat severe CRS (187, 188) in the setting of CAR-T cell therapy and in the setting of SARS-CoV-2 infection (5). Similarly, the IL-1 receptor antagonist anakinra improves CAR-T cell therapy CRS outcomes and also significantly increases survival of SARS-CoV-infected mice with hyperactive NLRP3 inflammasomes (186, 189, 190). In a retrospective cohort analysis, intravenous administration of high-dose anakinra increased survival and clinical improvement in COVID-19 patients with ARDS (191). Evidence from CAR-T-induced CRS suggests parallels to the COVID-19 inflammatory response that would suggest that targeting IL-1β would reduce the inflammatory signaling that mediates lung injury, ARDS, and mortality. Table 1 shows a list of agents in various stages of development that target the NLRP3 inflammasome.
Therapeutics targeting IL-1β and the NLRP3 inflammasome pathway have similarly been employed and efficacious in the context of cardiovascular disease. The NLRP3 inhibitors arglabin and MCC950 reduced IL-1β plasma levels and decreased atherosclerotic lesion size (48, 192). IL-1β neutralizing antibodies and anakinra showed reduced cardiac hypertrophy and myocardial dysfunction post-MI (193–195). The CANTOS trial randomized patients with past MI and elevated hsCRP to receive canakinumab, a monoclonal antibody targeting IL-1β and found a 15% reduction in major CV events (144).
In sum, COVID-19 causes an array of disease manifestations, the most severe of which is mediated by a massive inflammatory response that appears to occur through stimulation of the NLRP3 inflammasome. Direct data linking the NLRP3 inflammasome and SARS-CoV-2 infection are limited given the recent onset of this new pathogen and its global impact. The pathogenesis of this infection and cytokine storm, mirrors many of those features observed in cardiovascular disease, HIV-1 pathogenesis, and SARS-CoV. For this reason, it is of value to contextualize what is already known about the NLRP3 as a mediator of inflammatory signaling to inform future studies of pathogenesis and therapeutic development given the urgent need for drug discovery.
Significant evidence supports the role of IL-1β and NLRP3-dependent inflammasome activation in the pathogenesis of acute lung injury. An abundance of literature supports targeting this pathway in the development of therapeutic strategies. In consideration of direct acting anti-viral agents, viral load appears non- or minimally consequential in determining SARS-CoV and SARS-CoV-2 disease outcomes. When tested in the context of SARS-CoV infection, treatments targeting NLRP3 pathway components including NF-κB, inflammatory macrophages, and IFN-I all demonstrated significant efficacy despite unchanged viral titers their respective human, murine, macaque, and/or in vitro models (5, 35, 106, 107, 196). In COVID-19 clinical trials, hydroxychloroquine demonstrated antiviral activity (197, 198), yet without demonstrated clinical benefit (199–201). The known role of NLRP3 in hyperinflammatory ARDS and CRS, documented NLRP3 involvement in MERS-CoV and SARS-CoV severity, and apparent efficacy of anti-NLRP3 therapeutics in SARS-CoV and SARS-CoV-2 clinical trials and animal models strongly indicate that NLRP3 is a central mediator of severe COVID-19. The potential central role of NLRP3 in severe COVID-19 necessitates investigation into the therapeutic targeting of the NLRP3 inflammasome.
Timing of therapy is critical as once individuals develop ARDS, the chances of improved outcomes with therapy are severely reduced. Targeted therapy for individuals with moderate disease before the development of respiratory failure will be critical. There is an urgent need to develop therapeutics that improve patient outcomes in severe COVID-19. Therefore, targeting this pathway through existing available therapeutic options would represent an important and viable approach to reducing SARS-CoV-2-induced inflammatory cytokine signaling and immediately improve patient outcomes.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was funded by NIH/NIAID 1K08AI120806 to TS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. (2020) 5:536–44. doi: 10.1038/s41564-020-0695-z
2. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China 2019. N Engl J Med. (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
3. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
4. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
5. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. (2020) 395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0
6. He L, Ding Y, Zhang Q, Che X, He Y, Shen H, et al. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J Pathol. (2006) 210:288–97. doi: 10.1002/path.2067
7. Wen W, Su W, Tang H, Le W, Zhang X, Zheng Y, et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. (2020) 6:31. doi: 10.1038/s41421-020-0168-9
8. Vabret N, Samstein R, Fernandez N, Merad M, Project SIR, The Sinai Immunology Review Project, Trainees & Faculty. Advancing scientific knowledge in times of pandemics. Nat Rev Immunol. (2020) 20:338. doi: 10.1038/s41577-020-0319-0
9. Lin L, Xu L, Lv W, Han L, Xiang Y, Fu L, et al. An NLRP3 inflammasome-triggered cytokine storm contributes to Streptococcal toxic shock-like syndrome (STSLS). PLoS Pathog. (2019) 15:e1007795. doi: 10.1371/journal.ppat.1007795
10. Sendler M, van den Brandt C, Glaubitz J, Wilden A, Golchert J, Weiss FU, et al. NLRP3 inflammasome regulates development of systemic inflammatory response and compensatory anti-inflammatory response syndromes in mice with acute pancreatitis. Gastroenterology. (2020) 158:253–69.e14. doi: 10.1053/j.gastro.2019.09.040
11. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. (2012) 76:16–32. doi: 10.1128/MMBR.05015-11
12. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. (2017) 39:517–28. doi: 10.1007/s00281-017-0639-8
13. Bauernfeind F, Ablasser A, Bartok E, Kim S, Schmid-Burgk J, Cavlar T, et al. Inflammasomes: current understanding and open questions. Cell Mol Life Sci. (2011) 68:765–83. doi: 10.1007/s00018-010-0567-4
14. Zhao C, Zhao W. NLRP3 inflammasome-a key player in antiviral responses. Front Immunol. (2020) 11:211. doi: 10.3389/fimmu.2020.00211
15. Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. (2019) 10:50. doi: 10.3389/fmicb.2019.00050
16. Li JY, You Z, Wang Q, Zhou ZJ, Qiu Y, Luo R, et al. The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microbes Infect. (2020) 22:80–5. doi: 10.1016/j.micinf.2020.02.002
17. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
18. Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc Natl Acad Sci USA. (2004) 101:4240–5. doi: 10.1073/pnas.0306446101
19. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. (2005) 309:1864–8. doi: 10.1126/science.1116480
20. Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. (2015) 202:120–34. doi: 10.1016/j.virusres.2014.11.021
21. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. (2005) 11:875–9. doi: 10.1038/nm1267
22. Patel AB, Verma A. COVID-19 and angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: what is the evidence? JAMA. (2020) 323:1769–70. doi: 10.1001/jama.2020.4812
23. Wen Y, Liu Y, Tang T, Lv L, Liu H, Ma K, et al. NLRP3 inflammasome activation is involved in Ang II-induced kidney damage via mitochondrial dysfunction. Oncotarget. (2016) 7:54290–302. doi: 10.18632/oncotarget.11091
24. Sun NN, Yu CH, Pan MX, Zhang Y, Zheng BJ, Yang QJ, et al. Mir-21 mediates the inhibitory effect of Ang (1-7) on AngII-induced NLRP3 inflammasome activation by targeting Spry1 in lung fibroblasts. Sci Rep. (2017) 7:14369. doi: 10.1038/s41598-017-13305-3
25. Ren XS, Tong Y, Ling L, Chen D, Sun HJ, Zhou H, et al. NLRP3 gene deletion attenuates angiotensin ii-induced phenotypic transformation of vascular smooth muscle cells and vascular remodeling. Cell Physiol Biochem. (2017) 44:2269–80. doi: 10.1159/000486061
26. Sun HJ, Ren XS, Xiong XQ, Chen YZ, Zhao MX, Wang JJ, et al. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. (2017) 8:e3074. doi: 10.1038/cddis.2017.470
27. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
28. Team CC-R. Characteristics of health care personnel with COVID-19 - United States, February 12-April 9, 2020. MMWR Morb Mortal Wkly Rep. (2020) 69:477–81. doi: 10.15585/mmwr.mm6915e6
29. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
30. Zhao D, Yao F, Wang L, Zheng L, Gao Y, Ye J, et al. A comparative study on the clinical features of COVID-19 pneumonia to other pneumonias. Clin Infect Dis. (2020) ciaa247. doi: 10.1093/cid/ciaa247
31. Zhu Z, Cai T, Fan L, Lou K, Hua X, Huang Z, et al. Clinical value of immune-inflammatory parameters to assess the severity of coronavirus disease 2019. Int J Infect Dis. (2020)95:332–9. doi: 10.1016/j.ijid.2020.04.041
32. Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. (2020) 368:m1091. doi: 10.1136/bmj.m1091
33. Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. (2020) 34:1. doi: 10.23812/CONTI-E
34. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. (2020) 27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009
35. Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int J Antimicrob Agents. (2020) 55:105954. doi: 10.1016/j.ijantimicag.2020.105954
36. Amlani A, Barber C, Fifi-Mah A, Monzon J. Successful treatment of cytokine release syndrome with IL-6 blockade in a patient transitioning from immune-checkpoint to MEK/BRAF inhibition: a case report and review of literature. Oncologist. (2020) 25:1–4. doi: 10.1634/theoncologist.2020-0194
37. Leff JA, Baer JW, Bodman ME, Kirkman JM, Shanley PF, Patton LM, et al. Interleukin-1-induced lung neutrophil accumulation and oxygen metabolite-mediated lung leak in rats. Am J Physiol. (1994) 266:L2–8. doi: 10.1152/ajplung.1994.266.1.L2
38. Olman MA, White KE, Ware LB, Simmons WL, Benveniste EN, Zhu S, et al. Pulmonary edema fluid from patients with early lung injury stimulates fibroblast proliferation through IL-1 beta-induced IL-6 expression. J Immunol. (2004) 172:2668–77. doi: 10.4049/jimmunol.172.4.2668
39. Ganter MT, Roux J, Miyazawa B, Howard M, Frank JA, Su G, et al. Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ Res. (2008) 102:804–12. doi: 10.1161/CIRCRESAHA.107.161067
40. Zheng HY, Zhang M, Yang CX, Zhang N, Wang XC, Yang XP, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol Immunol. (2020) 17:541–3. doi: 10.1038/s41423-020-0401-3
41. Thevarajan I, Nguyen THO, Koutsakos M, Druce J, Caly L, van de Sandt CE, et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat Med. (2020) 26:453–5. doi: 10.1038/s41591-020-0819-2
42. Toldo S, Mezzaroma E, Bressi E, Marchetti C, Carbone S, Sonnino C, et al. Interleukin-1β blockade improves left ventricular systolic/diastolic function and restores contractility reserve in severe ischemic cardiomyopathy in the mouse. J Cardiovasc Pharmacol. (2014) 64:1–6. doi: 10.1097/FJC.0000000000000106
43. Mantovani A, Dinarello CA, Molgora M, Garlanda C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity. (2019) 50:778–95. doi: 10.1016/j.immuni.2019.03.012
44. Zhao C, Gu Y, Zeng X, Wang J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clin Immunol. (2018) 197:154–60. doi: 10.1016/j.clim.2018.09.007
45. Zhang H, Luo J, Alcorn JF, Chen K, Fan S, Pilewski J, et al. AIM2 inflammasome is critical for influenza-induced lung injury and mortality. J Immunol. (2017) 198:4383–93. doi: 10.4049/jimmunol.1600714
46. Yang CA, Huang ST, Chiang BL. Association of NLRP3 and CARD8 genetic polymorphisms with juvenile idiopathic arthritis in a Taiwanese population. Scand J Rheumatol. (2014) 43:146–52. doi: 10.3109/03009742.2013.834962
47. Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG, et al. Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology. (2014) 11:35. doi: 10.1186/1742-4690-11-35
48. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, et al. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol. (2017) 37:1457–61. doi: 10.1161/ATVBAHA.117.309575
49. Tan HY, Yong YK, Shankar EM, Paukovics G, Ellegard R, Larsson M, et al. Aberrant inflammasome activation characterizes tuberculosis-associated immune reconstitution inflammatory syndrome. J Immunol. (2016) 196:4052–63. doi: 10.4049/jimmunol.1502203
50. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
51. Duncan JA, Bergstralh DT, Wang Y, Willingham SB, Ye Z, Zimmermann AG, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci USA. (2007) 104:8041–6. doi: 10.1073/pnas.0611496104
52. Schmidt FI, Lu A, Chen JW, Ruan J, Tang C, Wu H, et al. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J Exp Med. (2016) 213:771–90. doi: 10.1084/jem.20151790
53. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. (2014) 156:1193–206. doi: 10.1016/j.cell.2014.02.008
54. Ruland J. Inflammasome: putting the pieces together. Cell. (2014) 156:1127–9. doi: 10.1016/j.cell.2014.02.038
55. Skeldon AM, Faraj M, Saleh M. Caspases and inflammasomes in metabolic inflammation. Immunol Cell Biol. (2014) 92:304–13. doi: 10.1038/icb.2014.5
56. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. (2017) 47:15–31. doi: 10.1016/j.immuni.2017.06.020
57. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
58. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
59. Shim DW, Lee KH. Posttranslational regulation of the NLR family pyrin domain-containing 3 inflammasome. Front Immunol. (2018) 9:1054. doi: 10.3389/fimmu.2018.01054
60. Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem. (1994) 269:15195–203.
61. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science. (1996) 272:735–8. doi: 10.1126/science.272.5262.735
62. Samways DS, Li Z, Egan TM. Principles and properties of ion flow in P2X receptors. Front Cell Neurosci. (2014) 8:6. doi: 10.3389/fncel.2014.00006
63. Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, et al. The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity. (2018) 49:56–65.e4. doi: 10.1016/j.immuni.2018.04.032
64. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. (2013) 126:2903–13. doi: 10.1242/jcs.124388
65. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
66. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci USA. (2012) 109:11282–7. doi: 10.1073/pnas.1117765109
67. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. (2012) 492:123–7. doi: 10.1038/nature11588
68. Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F, et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis. (2015) 6:e1954. doi: 10.1038/cddis.2015.277
69. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun. (2017) 8:202. doi: 10.1038/s41467-017-00227-x
70. Domingo-Fernández R, Coll RC, Kearney J, Breit S, O'Neill LAJ. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1β transcription and activate the NLRP3 inflammasome. J Biol Chem. (2017) 292:12077–87. doi: 10.1074/jbc.M117.797126
71. de Gassart A, Martinon F. Pyroptosis: caspase-11 unlocks the gates of death. Immunity. (2015) 43:835–7. doi: 10.1016/j.immuni.2015.10.024
72. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
73. McIntire CR, Yeretssian G, Saleh M. Inflammasomes in infection and inflammation. Apoptosis. (2009) 14:522–35. doi: 10.1007/s10495-009-0312-3
74. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
75. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. (2015) 25:1285–98. doi: 10.1038/cr.2015.139
76. Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, et al. Interleukin-1β maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep. (2018) 24:1425–33. doi: 10.1016/j.celrep.2018.07.027
77. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. (2018) 48:35–44.e6. doi: 10.1016/j.immuni.2017.11.013
78. Doitsh G, Greene WC. Dissecting how CD4 T cells are lost during HIV infection. Cell Host Microbe. (2016) 19:280–91. doi: 10.1016/j.chom.2016.02.012
79. Galloway NL, Doitsh G, Monroe KM, Yang Z, Muñoz-Arias I, Levy DN, et al. Cell-to-cell transmission of HIV-1 is required to trigger pyroptotic death of lymphoid-tissue-derived CD4 T cells. Cell Rep. (2015) 12:1555–63. doi: 10.1016/j.celrep.2015.08.011
80. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. (2014) 505:509–14. doi: 10.1038/nature12940
81. Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science. (2014) 343:428–32. doi: 10.1126/science.1243640
82. Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, et al. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell. (2010) 143:789–801. doi: 10.1016/j.cell.2010.11.001
83. Zhang J, Han Y, Shi H, Chen J, Zhang X, Wang X, et al. Swine acute diarrhea syndrome coronavirus-induced apoptosis is caspase- and cyclophilin D- dependent. Emerg Microbes Infect. (2020) 9:439–56. doi: 10.1080/22221751.2020.1722758
84. Siu KL, Yuen KS, Castaño-Rodriguez C, Ye ZW, Yeung ML, Fung SY, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. (2019) 33:8865–77. doi: 10.1096/fj.201802418R
85. Ahn M, Anderson DE, Zhang Q, Tan CW, Lim BL, Luko K, et al. Dampened NLRP3-mediated inflammation in bats and implications for a special viral reservoir host. Nat Microbiol. (2019) 4:789–99. doi: 10.1038/s41564-019-0371-3
86. Yue Y, Nabar NR, Shi CS, Kamenyeva O, Xiao X, Hwang IY, et al. SARS-coronavirus open reading frame-3a drives multimodal necrotic cell death. Cell Death Dis. (2018) 9:904. doi: 10.1038/s41419-018-0917-y
87. DeDiego ML, Nieto-Torres JL, Jimenez-Guardeño JM, Regla-Nava JA, Castaño-Rodriguez C, Fernandez-Delgado R, et al. Coronavirus virulence genes with main focus on SARS-CoV envelope gene. Virus Res. (2014) 194:124–37. doi: 10.1016/j.virusres.2014.07.024
88. Tan YJ, Lim SG, Hong W. Regulation of cell death during infection by the severe acute respiratory syndrome coronavirus and other coronaviruses. Cell Microbiol. (2007) 9:2552–61. doi: 10.1111/j.1462-5822.2007.01034.x
89. Patton LM, Saggart BS, Ahmed NK, Leff JA, Repine JE. Interleukin-1 beta-induced neutrophil recruitment and acute lung injury in hamsters. Inflammation. (1995) 19:23–9. doi: 10.1007/BF01534377
90. Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. (2001) 107:1529–36. doi: 10.1172/JCI12568
91. Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, et al. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. (1995) 107:1062–73. doi: 10.1378/chest.107.4.1062
92. Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A. Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest. (1995) 108:1303–14. doi: 10.1378/chest.108.5.1303
93. Park WY, Goodman RB, Steinberg KP, Ruzinski JT, Radella F, Park DR, et al. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. (2001) 164:1896–903. doi: 10.1164/ajrccm.164.10.2104013
94. Bouros D, Alexandrakis MG, Antoniou KM, Agouridakis P, Pneumatikos I, Anevlavis S, et al. The clinical significance of serum and bronchoalveolar lavage inflammatory cytokines in patients at risk for acute respiratory distress syndrome. BMC Pulm Med. (2004) 4:6. doi: 10.1186/1471-2466-4-6
95. Lau SKP, Lau CCY, Chan KH, Li CPY, Chen H, Jin DY, et al. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel middle east respiratory syndrome coronavirus: implications for pathogenesis and treatment. J Gen Virol. (2013) 94:2679–90. doi: 10.1099/vir.0.055533-0
96. Min CK, Cheon S, Ha NY, Sohn KM, Kim Y, Aigerim A, et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci Rep. (2016) 6:25359. doi: 10.1038/srep25359
97. Alosaimi B, Hamed ME, Naeem A, Alsharef AA, AlQahtani SY, AlDosari KM, et al. MERS-CoV infection is associated with downregulation of genes encoding Th1 and Th2 cytokines/chemokines and elevated inflammatory innate immune response in the lower respiratory tract. Cytokine. (2020) 126:154895. doi: 10.1016/j.cyto.2019.154895
98. Beigel JH, Farrar J, Han AM, Hayden FG, Hyer R, de Jong MD, et al. Avian influenza A (H5N1) infection in humans. N Engl J Med. (2005) 353:1374–85. doi: 10.1056/NEJMra052211
99. Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solórzano A, Swayne DE, et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. (2005) 310:77–80. doi: 10.1126/science.1119392
100. Perrone LA, Plowden JK, García-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. (2008) 4:e1000115. doi: 10.1371/journal.ppat.1000115
101. Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. (2007) 445:319–23. doi: 10.1038/nature05495
102. Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol. (2005) 79:6441–8. doi: 10.1128/JVI.79.10.6441-6448.2005
103. Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. (2007) 117:3786–99. doi: 10.1172/JCI32285
104. Kim KS, Jung H, Shin IK, Choi BR, Kim DH. Induction of interleukin-1 beta (IL-1β) is a critical component of lung inflammation during influenza A (H1N1) virus infection. J Med Virol. (2015) 87:1104–12. doi: 10.1002/jmv.24138
105. Xu H, Zhong L, Deng J, Peng J, Dan H, Zeng X, et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. (2020) 12:8. doi: 10.1038/s41368-020-0074-x
106. Fu B, Xu X, Wei H. Why tocilizumab could be an effective treatment for severe COVID-19? J Transl Med. (2020) 18:164. doi: 10.1186/s12967-020-02339-3
107. Fung SY, Yuen KS, Ye ZW, Chan CP, Jin DY. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microbes Infect. (2020) 9:558–70. doi: 10.1080/22221751.2020.1736644
108. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
109. Nieto-Torres JL, DeDiego ML, Verdiá-Báguena C, Jimenez-Guardeño JM, Regla-Nava JA, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. (2014) 10:e1004077. doi: 10.1371/journal.ppat.1004077
110. Fu Y, Cheng Y, Wu Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol Sin. (2020). doi: 10.1007/s12250-020-00207-4. [Epub ahead of print].
111. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. (2020) 181:1036–45.e9. doi: 10.1016/j.cell.2020.04.026
112. Smits SL, de Lang A, van den Brand JM, Leijten LM, van IJcken WF, Eijkemans MJ, et al. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. (2010) 6:e1000756. doi: 10.1371/journal.ppat.1000756
113. Barker BR, Taxman DJ, Ting JP. Cross-regulation between the IL-1β/IL-18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol. (2011) 23:591–7. doi: 10.1016/j.coi.2011.07.005
114. Zhang S, Li L, Shen A, Chen Y, Qi Z. Rational use of tocilizumab in the treatment of novel coronavirus pneumonia. Clin Drug Investig. (2020) 40:511–8. doi: 10.1007/s40261-020-00917-3
115. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. (2020) 130:2620–9. doi: 10.1172/JCI137244
116. An PJ, Yi ZZ, Yang LP. Biochemical indicators of coronavirus disease 2019 exacerbation and the clinical implications. Pharmacol Res. (2020) 159:104946. doi: 10.1016/j.phrs.2020.104946
117. Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. (2020) 20:355–62. doi: 10.1038/s41577-020-0331-4
118. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. (2020). doi: 10.1038/s41591-020-0901-9
119. Franchi L, Eigenbrod T, Núñez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol. (2009) 183:792–6. doi: 10.4049/jimmunol.0900173
120. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. (2020) 46:586–90. doi: 10.1007/s00134-020-05985-9
121. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. (2020) 76:14–20. doi: 10.1016/j.ejim.2020.04.037
122. Glowacka I, Bertram S, Herzog P, Pfefferle S, Steffen I, Muench MO, et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol. (2010) 84:1198–205. doi: 10.1128/JVI.01248-09
123. Nieto-Torres JL, Verdiá-Báguena C, Jimenez-Guardeño JM, Regla-Nava JA, Castaño-Rodriguez C, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. (2015) 485:330–9. doi: 10.1016/j.virol.2015.08.010
124. Lu W, Zheng BJ, Xu K, Schwarz W, Du L, Wong CK, et al. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc Natl Acad Sci USA. (2006) 103:12540–5. doi: 10.1073/pnas.0605402103
125. Chen CC, Krüger J, Sramala I, Hsu HJ, Henklein P, Chen YM, et al. ORF8a of SARS-CoV forms an ion channel: experiments and molecular dynamics simulations. Biochim Biophys Acta. (2011) 1808:572–9. doi: 10.1016/j.bbamem.2010.08.004
126. Castaño-Rodriguez C, Honrubia JM, Gutiérrez-Álvarez J, DeDiego ML, Nieto-Torres JL, Jimenez-Guardeño JM, et al. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. mBio. (2018) 9:e02325-17. doi: 10.1128/mBio.02325-17
127. Shi CS, Nabar NR, Huang NN, Kehrl JH. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. (2019) 5:101. doi: 10.1038/s41420-019-0181-7
128. Astuti I, Ysrafil. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diabetes Metab Syndr. (2020) 14:407–12. doi: 10.1016/j.dsx.2020.04.020
129. Velazquez-Salinas L, Zarate S, Eberl S, Gladue DP, Novella I, Borca MV. Positive selection of ORF3a and ORF8 genes drives the evolution of SARS-CoV-2 during the 2020 COVID-19 pandemic. bioRxiv [preprint]. (2020). doi: 10.1101/2020.04.10.035964
130. Lau SK, Feng Y, Chen H, Luk HK, Yang WH, Li KS, et al. Severe acute respiratory syndrome (SARS) Coronavirus ORF8 protein is acquired from SARS-related coronavirus from greater horseshoe bats through recombination. J Virol. (2015) 89:10532–47. doi: 10.1128/JVI.01048-15
131. Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. (2020) 18:179. doi: 10.1186/s12967-020-02344-6
132. Segovia M, Russo S, Jeldres M, Mahmoud YD, Perez V, Duhalde M, et al. Targeting TMEM176B enhances antitumor immunity and augments the efficacy of immune checkpoint blockers by unleashing inflammasome activation. Cancer Cell. (2019) 35:767–81.e6. doi: 10.1016/j.ccell.2019.04.003
133. Segovia M, Russo S, Girotti MR, Rabinovich GA, Hill M. Role of inflammasome activation in tumor immunity triggered by immune checkpoint blockers. Clin Exp Immunol. (2020) 200:155–62. doi: 10.1111/cei.13433
134. Mullard A. NLRP3 inhibitors stoke anti-inflammatory ambitions. Nat Rev Drug Discov. (2019) 18:405–7. doi: 10.1038/d41573-019-00086-9
135. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. (2019) 10:128. doi: 10.1038/s41419-019-1413-8
136. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. (2018) 17:688. doi: 10.1038/nrd.2018.97
137. Xu L, Zhang C, Jiang N, He D, Bai Y, Xin Y. Rapamycin combined with MCC950 to treat multiple sclerosis in experimental autoimmune encephalomyelitis. J Cell Biochem. (2019) 120:5160–8. doi: 10.1002/jcb.27792
138. Perregaux DG, McNiff P, Laliberte R, Hawryluk N, Peurano H, Stam E, et al. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J Pharmacol Exp Ther. (2001) 299:187–97.
139. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med. (2018) 10:eaah4066. doi: 10.1126/scitranslmed.aah4066
140. Toldo S, Mauro AG, Cutter Z, Van Tassell BW, Mezzaroma E, Del Buono MG, et al. The NLRP3 inflammasome inhibitor, OLT1177 (Dapansutrile), reduces infarct size and preserves contractile function after ischemia reperfusion injury in the mouse. J Cardiovasc Pharmacol. (2019) 73:215–22. doi: 10.1097/FJC.0000000000000658
141. Sánchez-Fernández A, Skouras DB, Dinarello CA, López-Vales R. OLT1177 (Dapansutrile), a selective NLRP3 inflammasome inhibitor, ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front Immunol. (2019) 10:2578. doi: 10.3389/fimmu.2019.02578
142. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci USA. (2018) 115:E1530–9. doi: 10.1073/pnas.1716095115
143. Marchetti C, Swartzwelter B, Koenders MI, Azam T, Tengesdal IW, Powers N, et al. NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Res Ther. (2018) 20:169. doi: 10.1186/s13075-018-1664-2
144. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
145. Zheng ZH, Zeng X, Nie XY, Cheng YJ, Liu J, Lin XX, et al. Interleukin-1 blockade treatment decreasing cardiovascular risk. Clin Cardiol. (2019) 42:942–51. doi: 10.1002/clc.23246
146. Ikonomidis I, Tzortzis S, Lekakis J, Paraskevaidis I, Andreadou I, Nikolaou M, et al. Lowering interleukin-1 activity with anakinra improves myocardial deformation in rheumatoid arthritis. Heart. (2009) 95:1502–7. doi: 10.1136/hrt.2009.168971
147. Gómez-García F, Sanz-Cabanillas JL, Viguera-Guerra I, Isla-Tejera B, Nieto AV, Ruano J. Scoping review on use of drugs targeting interleukin 1 pathway in DIRA and DITRA. Dermatol Ther. (2018) 8:539–56. doi: 10.1007/s13555-018-0269-7
148. Schumacher HR, Sundy JS, Terkeltaub R, Knapp HR, Mellis SJ, Stahl N, et al. Rilonacept (interleukin-1 trap) in the prevention of acute gout flares during initiation of urate-lowering therapy: results of a phase II randomized, double-blind, placebo-controlled trial. Arthritis Rheum. (2012) 64:876–84. doi: 10.1002/art.33412
149. Kapur S, Bonk ME. Rilonacept (arcalyst), an interleukin-1 trap for the treatment of cryopyrin-associated periodic syndromes. P T. (2009) 34:138–41.
150. Knickelbein JE, Tucker WR, Bhatt N, Armbrust K, Valent D, Obiyor D, et al. Gevokizumab in the treatment of autoimmune non-necrotizing anterior scleritis: results of a phase I/II clinical trial. Am J Ophthalmol. (2016) 172:104–10. doi: 10.1016/j.ajo.2016.09.017
151. Owyang AM, Issafras H, Corbin J, Ahluwalia K, Larsen P, Pongo E, et al. XOMA 052, a potent, high-affinity monoclonal antibody for the treatment of IL-1β-mediated diseases. MAbs. (2011) 3:49–60. doi: 10.4161/mabs.3.1.13989
152. Blech M, Peter D, Fischer P, Bauer MM, Hafner M, Zeeb M, et al. One target-two different binding modes: structural insights into gevokizumab and canakinumab interactions to interleukin-1β. J Mol Biol. (2013) 425:94–111. doi: 10.1016/j.jmb.2012.09.021
153. Murthy H, Iqbal M, Chavez JC, Kharfan-Dabaja MA. Cytokine release syndrome: current perspectives. Immunotargets Ther. (2019) 8:43–52. doi: 10.2147/ITT.S202015
154. Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. (2018) 6:56. doi: 10.1186/s40425-018-0343-9
155. Chatenoud L, Ferran C, Reuter A, Legendre C, Gevaert Y, Kreis H, et al. Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma [corrected]. N Engl J Med. (1989) 320:1420–1. doi: 10.1056/NEJM198905253202117
156. Chatenoud L, Ferran C, Legendre C, Thouard I, Merite S, Reuter A, et al. In vivo cell activation following OKT3 administration. Systemic cytokine release and modulation by corticosteroids. Transplantation. (1990) 49:697–702. doi: 10.1097/00007890-199004000-00009
157. Pihusch R, Holler E, Mühlbayer D, Göhring P, Stötzer O, Pihusch M, et al. The impact of antithymocyte globulin on short-term toxicity after allogeneic stem cell transplantation. Bone Marrow Transplant. (2002) 30:347–54. doi: 10.1038/sj.bmt.1703640
158. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. (2006) 355:1018–28. doi: 10.1056/NEJMoa063842
159. Winkler U, Jensen M, Manzke O, Schulz H, Diehl V, Engert A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood. (1999) 94:2217–24. doi: 10.1182/blood.V94.7.2217.419k02_2217_2224
160. Freeman CL, Morschhauser F, Sehn L, Dixon M, Houghton R, Lamy T, et al. Cytokine release in patients with CLL treated with obinutuzumab and possible relationship with infusion-related reactions. Blood. (2015) 126:2646–9. doi: 10.1182/blood-2015-09-670802
161. Wing MG, Moreau T, Greenwood J, Smith RM, Hale G, Isaacs J, et al. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J Clin Invest. (1996) 98:2819–26. doi: 10.1172/JCI119110
162. Alig SK, Dreyling M, Seppi B, Aulinger B, Witkowski L, Rieger CT. Severe cytokine release syndrome after the first dose of Brentuximab Vedotin in a patient with relapsed systemic anaplastic large cell lymphoma (sALCL): a case report and review of literature. Eur J Haematol. (2015) 94:554–7. doi: 10.1111/ejh.12396
163. de Vos S, Forero-Torres A, Ansell SM, Kahl B, Cheson BD, Bartlett NL, et al. A phase II study of dacetuzumab (SGN-40) in patients with relapsed diffuse large B-cell lymphoma (DLBCL) and correlative analyses of patient-specific factors. J Hematol Oncol. (2014) 7:44. doi: 10.1186/1756-8722-7-44
164. Rotz SJ, Leino D, Szabo S, Mangino JL, Turpin BK, Pressey JG. Severe cytokine release syndrome in a patient receiving PD-1-directed therapy. Pediatr Blood Cancer. (2017) 64. doi: 10.1002/pbc.26642
165. Tonini G, Santini D, Vincenzi B, Borzomati D, Dicuonzo G, La Cesa A, et al. Oxaliplatin may induce cytokine-release syndrome in colorectal cancer patients. J Biol Regul Homeost Agents. (2002) 16:105–9.
166. Aue G, Njuguna N, Tian X, Soto S, Hughes T, Vire B, et al. Lenalidomide-induced upregulation of CD80 on tumor cells correlates with T-cell activation, the rapid onset of a cytokine release syndrome and leukemic cell clearance in chronic lymphocytic leukemia. Haematologica. (2009) 94:1266–73. doi: 10.3324/haematol.2009.005835
167. Abboud R, Keller J, Slade M, DiPersio JF, Westervelt P, Rettig MP, et al. Severe cytokine-release syndrome after T cell-replete peripheral blood haploidentical donor transplantation is associated with poor survival and anti-IL-6 therapy is safe and well tolerated. Biol Blood Marrow Transplant. (2016) 22:1851–60. doi: 10.1016/j.bbmt.2016.06.010
168. Cho C, Perales MA. Rapid identification of cytokine release syndrome after haploidentical PBSC transplantation and successful therapy with tocilizumab. Bone Marrow Transplant. (2016) 51:1620–1. doi: 10.1038/bmt.2016.229
169. Lundemose JB, Smith H, Sweet C. Cytokine release from human peripheral blood leucocytes incubated with endotoxin with and without prior infection with influenza virus: relevance to the sudden infant death syndrome. Int J Exp Pathol. (1993) 74:291–7.
170. Porter D, Frey N, Wood PA, Weng Y, Grupp SA. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol. (2018) 11:35. doi: 10.1186/s13045-018-0571-y
171. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in china: summary of a report of 72 314 cases from the chinese center for disease control and prevention. JAMA. (2020) 323:1239–42. doi: 10.1001/jama.2020.2648
172. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. (2020) 46:846–8. doi: 10.1007/s00134-020-05991-x
173. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. (2020). doi: 10.2139/ssrn.3541136. [Epub ahead of print].
174. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. (2020) 135:2033–40. doi: 10.1182/blood.2020006000
175. Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. (2020) 46:1089–98. doi: 10.1007/s00134-020-06062-x
176. Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med. (2020) 382:e38. doi: 10.1056/NEJMc2007575
177. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity. (2019) 50:1401–1411.e4. doi: 10.1016/j.immuni.2019.04.003
178. Bortolotti P, Faure E, Kipnis E. Inflammasomes in tissue damages and immune disorders after trauma. Front Immunol. (2018) 9:1900. doi: 10.3389/fimmu.2018.01900
179. Qiao J, Wu X, Luo Q, Wei G, Xu M, Wu Y, et al. NLRP3 regulates platelet integrin αIIbβ3 outside-in signaling, hemostasis and arterial thrombosis. Haematologica. (2018) 103:1568–76. doi: 10.3324/haematol.2018.191700
180. Murthy P, Durco F, Miller-Ocuin JL, Takedai T, Shankar S, Liang X, et al. The NLRP3 inflammasome and bruton's tyrosine kinase in platelets co-regulate platelet activation, aggregation, and in vitro thrombus formation. Biochem Biophys Res Commun. (2017) 483:230–6. doi: 10.1016/j.bbrc.2016.12.161
181. Gupta N, Sahu A, Prabhakar A, Chatterjee T, Tyagi T, Kumari B, et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc Natl Acad Sci USA. (2017) 114:4763–8. doi: 10.1073/pnas.1620458114
182. DeDiego ML, Nieto-Torres JL, Regla-Nava JA, Jimenez-Guardeño JM, Fernandez-Delgado R, Fett C, et al. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J Virol. (2014) 88:913–24. doi: 10.1128/JVI.02576-13
183. Ward SE, Loutfy MR, Blatt LM, Siminovitch KA, Chen J, Hinek A, et al. Dynamic changes in clinical features and cytokine/chemokine responses in SARS patients treated with interferon alfacon-1 plus corticosteroids. Antivir Ther. (2005) 10:263–75.
184. Loutfy MR, Blatt LM, Siminovitch KA, Ward S, Wolff B, Lho H, et al. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. JAMA. (2003) 290:3222–8. doi: 10.1001/jama.290.24.3222
185. Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med. (2016) 44:275–81. doi: 10.1097/CCM.0000000000001402
186. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
187. Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, et al. FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. Oncologist. (2018) 23:943–7. doi: 10.1634/theoncologist.2018-0028
188. Kotch C, Barrett D, Teachey DT. Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev Clin Immunol. (2019) 15:813–22. doi: 10.1080/1744666X.2019.1629904
189. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
190. Vijay R, Fehr AR, Janowski AM, Athmer J, Wheeler DL, Grunewald M, et al. Virus-induced inflammasome activation is suppressed by prostaglandin D. Proc Natl Acad Sci USA. (2017) 114:E5444–E5453. doi: 10.1073/pnas.1704099114
191. Cavalli G, Luca GD, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. (2020) 2:e310-1. doi: 10.1016/S2665-9913.(20)30127-2
192. Abderrazak A, Couchie D, Mahmood DF, Elhage R, Vindis C, Laffargue M, et al. Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation. (2015) 131:1061–70. doi: 10.1161/CIRCULATIONAHA.114.013730
193. Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am J Cardiol. (2013) 111:1394–400. doi: 10.1016/j.amjcard.2013.01.287
194. Abbate A, Van Tassell BW, Seropian IM, Toldo S, Robati R, Varma A, et al. Interleukin-1beta modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur J Heart Fail. (2010) 12:319–22. doi: 10.1093/eurjhf/hfq017
195. Liu D, Zeng X, Li X, Mehta JL, Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. (2017) 113:5. doi: 10.1007/s00395-017-0663-9
196. Zhao M. Cytokine storm and immunomodulatory therapy in COVID-19: role of chloroquine and anti-IL-6 monoclonal antibodies. Int J Antimicrob Agents. (2020) 55:105982. doi: 10.1016/j.ijantimicag.2020.105982
197. Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. (2020) 6:16. doi: 10.1038/s41421-020-0156-0
198. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. (2020) 30:269–71. doi: 10.1038/s41422-020-0282-0
199. Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Mailhe M, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. (2020) 2020:105949. doi: 10.1016/j.ijantimicag.2020.105949
200. Geleris J, Sun Y, Platt J, Zucker J, Baldwin M, Hripcsak G, et al. Observational study of hydroxychloroquine in hospitalized patients with Covid-19. N Engl J Med. (2020). doi: 10.1056/NEJMoa2012410. [Epub ahead of print].
Keywords: NLRP3 inflammasome, COVID-19, SARS-CoV-2, IL-1β, cytokine release syndrome (CRS), cytokine storm, coronavirus, acute respiratory distress syndrome (ARDS)
Citation: Freeman TL and Swartz TH (2020) Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 11:1518. doi: 10.3389/fimmu.2020.01518
Received: 08 May 2020; Accepted: 09 June 2020;
Published: 23 June 2020.
Edited by:
Linda F. Van Dyk, University of Colorado Denver, United StatesReviewed by:
Kian Fan Chung, Imperial College London, United KingdomCopyright © 2020 Freeman and Swartz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Talia H. Swartz, dGFsaWEuc3dhcnR6QG1zc20uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.