 Verena Harpf
Verena Harpf Günter Rambach
Günter Rambach Reinhard Würzner
Reinhard Würzner Cornelia Lass-Flörl
Cornelia Lass-Flörl Cornelia Speth
Cornelia Speth
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 July 2020
Sec. Microbial Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01471
This article is part of the Research Topic The Role of Complement in Microbial Infections View all 15 articles
Candida is a dominant fungal pathogen in immunocompromised hosts, leading to opportunistic infections. Complement with its multifaceted functions is involved in the immune defense against this yeast, and recently several novel aspects have emerged in this old battle. It is clear that Candida can adopt both roles as a colonizer or as a pathogen. In our article, we focus on the molecular mechanisms of the Candida-complement interplay, which occur in disseminated disease as well as locally on skin or on mucous membranes in mouth and vagina; the mechanisms can be supposed to be the same. Activation of the complement system by Candida is facilitated by directly triggering the three dominant pathways, but also indirectly via the coagulation and fibrinolysis systems. The complement-mediated anti-Candida effects induced thereby clearly extend chemotaxis, opsonization, and phagocytosis, and even the membrane attack complex formed on the fungal surface plays a modulatory role, although lysis of the yeast per se cannot be induced due to the thick fungal cell wall. In order to avoid the hostile action of complement, several evasion mechanisms have evolved during co-evolution, comprising the avoidance of recognition, and destruction. The latter comes in many flavors, in particular the cleavage of complement proteins by yeast enzymes and the exploitation of regulatory proteins by recruiting them on the cell wall, such as factor H. The rationale behind that is that the fluid phase regulators on the fungal cell surface down-regulate complement locally. Interestingly, however, evasion protein knockout strains do not necessarily lead to an attenuated disease, so it is likely more complex in vivo than initially thought. The interactions between complement and non-albicans species also deserve attention, especially Candida auris, a recently identified drug-resistant species of medical importance. This is in particular worth investigating, as deciphering of these interactions may lead to alternative anti-fungal therapies directly targeting the molecular mechanisms.
The complement system is an ancient and effective multicomponent bodily system for antimicrobial defense, homeostasis, and immunomodulation [reviewed in (1, 2)]. These functions are elicited by a system of soluble and membrane-bound effector, regulator, and receptor molecules, and form a tightly regulated cascade. Complement detects invading microbes and modified self-surfaces (e.g., on apoptotic cells) using various pattern recognition molecules. Via different effector molecules of the cascade, this detection results in opsonization with subsequent phagocytic clearance, formation of the lytic C5b-9 complex of the terminal pathway, induction of inflammation, and modulation of other innate, and adaptive immune weapons.
Three activation pathways can push this system: the classical pathway (mainly induced by binding of immunoglobulins on target surfaces), the alternative pathway (triggered by deposition of spontaneously formed C3b on foreign surfaces with subsequent amplification loop), and the lectin pathway (elicited by foreign or aberrant carbohydrate structures on surfaces). The consequence of all three pathways is the covalent deposition of complement on the foreign surface and the generation of a C3-convertase that cleaves complement factor C3 into the larger C3b fragment and the smaller C3a anaphylatoxin. The composition of the C3-convertase varies upon the activation pathway (C4b2b for the classical and lectin pathway and C3bBb for the alternative pathway). The newly formed C3b either can amplify the alternative pathway by generating new C3-convertase molecules or can form a complex with the existing C3-convertases to generate a C5-convertase. The enzymatic function of the C5-convertases (cleavage of complement protein C5 into C5a anaphylatoxin and the C5b fragment) initiates the sequence of the terminal pathway. Several aggregation events finally result in generation of the C5b-9 complex. Inserted into the target membrane, C5b-9 is called membrane attack complex (MAC) and induces lysis of the pathogen or altered cell (3). If soluble, C5b-9 (called terminal complement complex TCC) harbors further functions such as cell activation and pro-inflammatory immune activation (4, 5).
The presence of such a powerful tool in the body requires the tight control by numerous regulators. Major soluble controller of the alternative pathway is factor H (FH), which is a single chained 150 kDa plasma glycoprotein (6). This protein is composed of 20 homologous domains known as short consensus repeats (SCR) or complement control protein (CCP) modules, each of which is made of 60 amino acids and stabilized by two internal disulfide-bonds (7, 8). The SCRs bear different functions. While the C-terminal domains SCRs 19 and 20 are essential for the target recognition, the N-terminal domains SCRs 1–4 mediate the diverse complement regulator functions (9–11) (Figure 1). Complement regulation by FH can be divided into three parts: First, FH competes with factor B for the C3b binding and therefore inhibits the assembly of the alternative pathway C3- and C5-convertases. Second, the protein exhibits a “decay accelerating activity,” as it enhances the disassembly of these convertases by displacing bound factor Bb; and third, FH is used as a cofactor for the serine protease factor I in cleaving and inactivating C3b (8, 12, 13). An alternatively spliced transcript of the FH gene is factor H-like protein 1 (FHL-1) (14, 15). This 43 kDa plasma glycoprotein consists of the N-terminal domains SCRs 1–7 with four additional amino acids added at the C-terminus (16) (Figure 1). Compared to FH, the truncated form FHL-1 is less abundant in plasma (500 vs. 50 μg/ml, respectively) (17). FHL-1 still bears the same complement regulatory functions as FH and is therefore an important protein of the alternative pathway (8, 12, 13). The classical and lectin pathway, however, are mainly controlled by C4b binding protein (C4BP) [reviewed in (18)]. It binds to and inhibits the function of the activated complement compound C4b. C4BP acts as a cofactor of factor I (FI)-mediated cleavage and inactivation of soluble and cell-bound C4b, thus preventing the assembly of the C3-convertase C4b2b. In addition, C4BP accelerates the decay of functional C4b2b. Although FH is the main controller of the alternative pathway, C4BP also affects this pathway since it acts as a cofactor of FI in the proteolytic cleavage of C3b (18, 19).
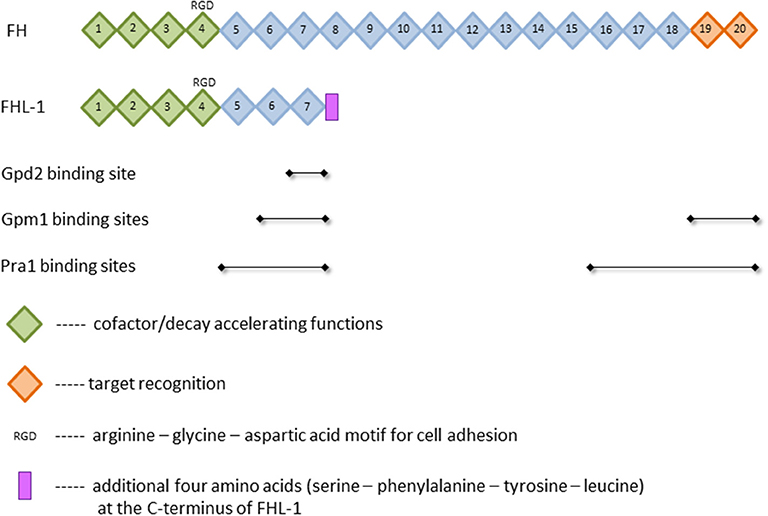
Figure 1. Structure of factor H (FH) and factor H-like protein 1 (FHL-1). Depicted are selected functions of short consensus repeats (SCR), illustrated as squares, and the known binding sites of the FH-binding proteins phosphoglycerate mutase 1 (Gpm1), pH-regulated antigen 1 (Pra1), and glycerol-3-phosphate dehydrogenase 2 (Gpd2). For further details and references, see text.
A similar cofactor activity is attributed to the cell surface-expressed regulator molecule membrane cofactor protein (MCP; CD46). MCP and a further surface regulator, decay accelerating factor (DAF, CD55), accelerate the decay of both C3 convertases (3). Important regulators of the terminal pathway are CD59 (protectin) and vitronectin. Both molecules interfere with assembly of the C5b-9 complex and thus prevent its insertion into the membrane to form a lytic pore (20, 21).
Candida spp. are common members of the human mycobiome, but also opportunistic fungal pathogens [reviewed in (22, 23)]. In healthy individuals, Candida resides on the skin or as colonizer of the oral cavity, the gastrointestinal and the urogenital tract. When shifting from a colonizer to a pathogen, it causes cutaneous and mucocutaneous candidiasis as well as life-threatening invasive infections of inner organs and the bloodstream. The shift is enabled by changes in the host microbiota (e.g., by antibiotics), impairment of the host immune response (e.g., by immunosuppressive therapy), or alterations in the local conditions (e.g., shifts in nutrients or pH) (23, 24). The most common manifestation is vulvovaginal candidiasis affecting millions of women worldwide (25). The growing number of immunocompromised patients with intravenous catheters, cancer chemotherapy, or organ transplantations contributes to the boost in Candida-induced bloodstream infections (26).
The genus Candida includes at least 30 species of clinical importance; the most frequent one is Candida albicans, but non-albicans species become more and more common and are often associated with reduced antifungal susceptibility and outbreaks (22). Candida auris is a recently discovered emerging multidrug-resistant species that is responsible for an increasing number of nosocomial outbreaks.
Understanding of anti-Candida host defense mechanisms remains an urgent need. One focus is the interaction with the complement system as a universal and fast-acting immune weapon. The relevance of this interaction is accentuated by the fact that local production of complement is revealed for most organs, indicating a role in both superficial candidiasis and in Candida sepsis. The picture of Candida—complement interplay is complex and multifaceted and will be depicted in the following. Most knowledge exists about Candida albicans, but also non-albicans species are included, particularly Candida auris.
Various pathways are capable to mediate complement activation by Candida, underlining the relevance of this innate immune system in the case of infection.
The direct mechanisms of complement activation include the classical, lectin, and alternative pathway, which all were described to be triggered by Candida (27–29). Early experiments already showed that classical and alternative pathway are activated with different kinetics (30). The classical pathway might not only be triggered by interaction between the pattern recognition molecule C1q and specific anti-Candida antibodies: a recent report described that serum amyloid P (SAP) component, a member of the pentraxin family, binds to the Candida albicans surface (31). Although this report shows reduced phagocytosis after SAP binding, other authors described that SAP is able to activate the classical complement pathway (32, 33).
The efficiency of complement to react on the presence of Candida strongly depends on the surface composition of the yeast. On intact Candida albicans cells, β-glucan is located, together with chitin, in the inner fungal cell wall. However, during C. albicans infection or by treatment with caspofungin, the inner β-glucan components become exposed (34) and can initiate the alternative pathway of the complement system (35). Similarly, purified β-glucan triggered the alternative pathway when co-incubated with the purified AP proteins (35).
The lectin pathway is an evident candidate for Candida-triggered complement activation, since mannans represent about 40% of the total polysaccharide yeast cell wall content (36). Neth et al. (29) found diverse Candida species to strongly bind MBL (mannan-binding lectin), a starter molecule of the lectin pathway, with subsequent C4 deposition on the yeast surface. Another pattern recognition molecule of the lectin pathway, collectin-11, also bound to carbohydrate residues on Candida albicans and triggered complement activation with C4b and C3b deposition on the surface (37). Blocking of MBL in a mouse model increased C. albicans colonization, and MBL-deficient animals had a higher level of colonization than wild-type mice (38).
More recent findings imply that Candida activates the complement system not only via classical, lectin, or alternative pathway. More and more indirect mechanisms were discovered, involving molecules of the contact system, the fibrinolysis system, and the coagulation system. This complex pattern of Candida-induced complement activation is summarized in Figure 2.
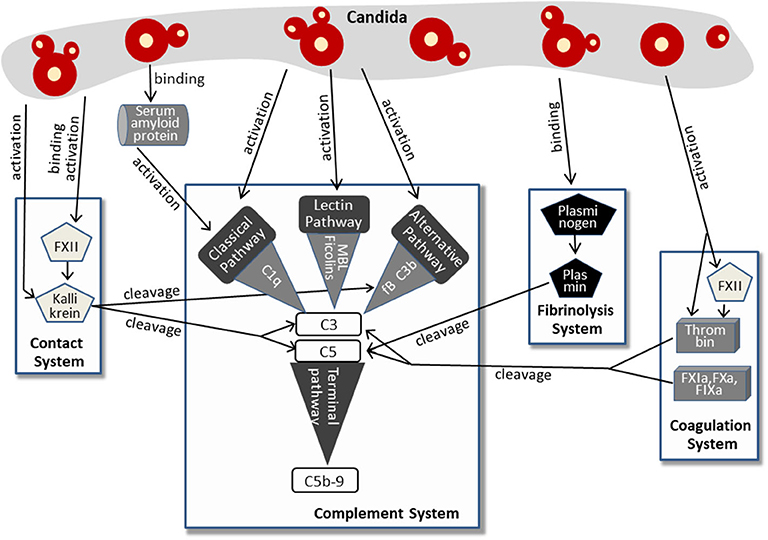
Figure 2. Pathways of complement activation by Candida, either directly or by affecting the contact system, fibrinolysis system, or coagulation system. For further details, see text.
The human plasma contact system represents a powerful immune surveillance tool that is activated by negatively charged surfaces, (e.g., on fungi). The contact of factor XII (FXII) to these surfaces triggers autoactivation with subsequent conversion to the active serine protease FXIIa. This process is also induced by contact with the cell walls of Candida albicans and C. tropicalis (39). In addition, previous work revealed that Candida-derived proteinases also activate factor XII (40), which appears to be a multifunctional molecule in innate immunity (41). Active FXIIa, either generated by Candida surface contact or by Candida proteases, cleaves prekallikrein to form kallikrein. Kallikrein reciprocally activates FXIIa, thus increasing rapidly the plasma level of both enzymes (41, 42). Furthermore, a Candida albicans proteinase directly converts plasma prekallikrein to active kallikrein (43). Kallikrein was shown to cleave the central complement protein C3 to yield the active fragments C3b and C3a. The proteolytic cleavage site used by kallikrein corresponds to that used by the C3 convertase, and the kallikrein-generated fragment C3b forms C3-convertase, thus triggering the C3 amplification loop. In addition, the C3a fragment resulting from kallikrein cleavage was determined to be biologically active and to harbor antimicrobial activity (44). The link from the contact system to complement activation in Candida infection is strengthened by findings that kallikrein also cleaves factor B, yielding Bb and Ba and thereby triggering the alternative pathway (44). Furthermore, contact system and complement system share regulator mechanisms, since C1INH, the regulator of the proteases of all three complement pathways, also represents the primary plasma control for FXIIa and kallikrein [reviewed in (45, 46)].
A second system that is activated by Candida and that cross-triggers complement activation is the coagulation system (Figure 2), a complex set of enzymes that mediates blood clotting. FXII, described above to be activated by Candida surfaces or Candida proteinases, also plays a central role in this system and promotes the generation of active thrombin (47), a key enzyme of coagulation (48). Active thrombin also results directly from the enzymatic function of Candida proteases (40). Thrombin effectively cleaves C3 to generate C3a that was capable to induce a chemotactic response of neutrophils (49, 50). In addition, thrombin is able to cleave C5 and thereby generates biologically active C5a and a functional C5-convertase even in the absence of C3 (51). Thrombin-generated C5 products also support triggering of the terminal complement pathway (52). Other components of the coagulation system, the factors IXa, Xa, and XIa, were also capable to cleave C3 and C5 (49).
The third system triggered by Candida and contributing to Candida-induced complement activation is the fibrinolysis system (Figure 2). Here, plasminogen/plasmin represent the central molecules. Urokinase-type plasminogen activator cleaves the plasma protein plasminogen to form plasmin, a serine protease that lyses fibrin clots and promotes degradation of the extracellular matrix. Candida albicans binds plasminogen to its surface via the proteins Gpm1 and Pra1 (53, 54). After conversion into proteolytically active plasmin, the subsequent degradation of extracellular matrix proteins and cell junction proteins might favor dissemination of the yeast. However, plasmin also mediates complement activation by cleaving C3 and C5 (49), and the plasmin-activated C5 was capable to yield a functional membrane attack complex (MAC) (55, 56). It must be mentioned that the role of plasminogen/plasmin in complement activation seems to be ambiguous, since other reports also describe an inhibitory role and list plasminogen as a complement inhibitor (see below).
The infection-induced complement activation leads in general to opsonization and lysis of invading pathogens, initiation of inflammation, guidance of immune cells to the site of infection, and last but not least to stimulation of the adaptive immune response (57). The central role of complement in the host defense against Candida infection has been clearly demonstrated in a variety of experimental models with various deficiencies for complement proteins. C3-depleted guinea pigs showed higher susceptibility to lethal Candida infections than untreated control animals (58). Tsoni et al. revealed a higher susceptibility to develop systemic and lethal infections with C. albicans and C. glabrata in C3 deficient mice compared to immunocompetent animals (59). Mice congenitally deficient in C5, such as DBA/2 or A/J, were highly susceptible to C. albicans infection (60–62). Furthermore, mice deficient in MBL, or double knockout mice lacking factor B and C2, showed highly increased mortality and elevated fungal load in the tissues after infection with C. albicans (63). The central role of complement in Candida infection has also been investigated in vaginal candidiasis. Notably, MBL as pattern recognition molecule plays a predominant role to determine severity and recurrence of disease. MBL was demonstrated to bind to Candida colonizing the vaginal cavity (64). The gene coding for MBL protein is polymorphic. A polymorphism associated with unstable MBL protein and thus decreased MBL concentrations is more prevalent in women developing primary vulvar vestibulitis syndrome than in control groups (65). Other reports confirm the relation of MBL polymorphism with recurrent vulvovaginal candidiasis and therapy resistance (36, 66–68). Similarly, cutaneous candidiasis was described to be strongly associated with complement. Mice with deficiency in C5 were less efficient in clearing experimental cutaneous Candida infection (69). Complement activation and deposition of C3 and factor B on Candida surface was demonstrated in an in vitro model of cutaneous candidiasis and were suggested to inhibit Candida adherence to corneocytes (70).
One predominant role of complement factor C3 is the opsonization of foreign structures to facilitate their uptake by complement receptor-bearing phagocytes (71). Opsonization of the Candida surface with C3 fragments is a very fast and efficient process, underlining the relevance of this mechanism particularly in early stages of infection (28). The most prominent receptor for C3b and iC3b deposited on the Candida surface is complement receptor 3 (CR3), widely expressed in professional phagocytes. Interestingly, Candida exposes a CR3-like molecule on its surface (72) that binds the negative complement regulator factor H and thus contributes to complement evasion, as described below.
Phagocytosis of Candida is not only facilitated by deposition of C3 fragments, but is further supported by an MBL-mediated “shortcut.” Besides being the main pattern recognition molecule of the lectin pathway, MBL was shown to possess opsonic functions itself to bridge the Candida surface to the cellular complement receptor CR1 (CD35) expressed on monocytes and neutrophils (73). MBL-mediated opsonophagocytosis of Candida albicans by neutrophils is independent from complement activation and stimulates the intracellular expression of Dectin-1 with subsequent production of reactive oxygen species (ROS) (74). Since generation of toxic ROS represents an important fungicidal mechanism employed by innate immune cells, MBL acts as a dual complement effector molecule that utilizes both complement activation and activation-independent techniques for antifungal protection.
The pro-inflammatory anaphylatoxins C3a and C5a, which are generated by cleavage of complement factors C3 and C5, support these anti-Candida effects by chemotactically attracting phagocytes to the site of infection (75). C5a is particularly potent to attract neutrophils and monocytes; by binding to the corresponding receptors on neutrophils, C5a also increases their antimicrobial effector functions toward Candida albicans such as phagocytosis, oxidative burst, and degranulation (76). The induction of the release of the pro-inflammatory cytokines IL-6 and IL-1β by human peripheral blood mononuclear cells (PBMCs) completes the antifungal armamentarium of C5a (75, 77).
Similarly, the anaphylatoxin C3a exerts a broad spectrum of antimicrobial effects including chemotaxis and immune cell activation (78). However, C3a-like peptides were also shown to act directly as antifungal weapons by binding to the Candida surface and inducing membrane perturbations (79).
In contrast, the influence of the terminal complement cascade on host defense against yeasts is less clear. In any case, the progress of the terminal pathway will lead to formation of the C5b-9 complex on the yeast cell surface. Although C5b-9 is also termed membrane attack complex (MAC), the thick fungal cell wall inhibits its lytic activity against fungal cells (unlike for gram-negative bacteria or erythrocytes) (3). However, deposited MAC may cause immune modulatory effects, as it induces a higher fungal mitochondrial activation and augments phagocytosis (80).
As described above, β-glucan molecules from the inner part of the Candida cell wall become exposed during infection or by treatment with the echinocandin caspofungin (34). Triggering of the alternative pathway of the complement system is a consequence of β-glucan exposure on the surface (35). Intact Candida albicans cells, however, are covered by a sheath of mannan that masks the complement-activating β-glucan layer (81) (Table 1). This mannan sheath provides resistance against alternative pathway activation (see Figure 3); conversion of mannose polysaccharides to polyalcohols or chemical removal of the surface mannan was shown to overcome this resistance (35). Specific antibodies against C. albicans mannan are a further tool to overcome this masking mechanism and to improve the outcome in experimental disseminated candidiasis (97). Interestingly, antibodies specifically recognizing β-mannans were protective against candidiasis, whereas α-mannan antibodies turned out to be non-protective. This difference perfectly correlated with the capacity of the antibodies to bind complement factor C3 on the yeast surface, a process that is much more efficient for the protective antibodies (97). The potency of mannan antibodies to overcome complement resistance is of particular interest, as most individuals have naturally occurring antibodies reactive with Candida mannan epitopes. However, these antibodies vary considerably regarding quantity and epitope specificity (98).
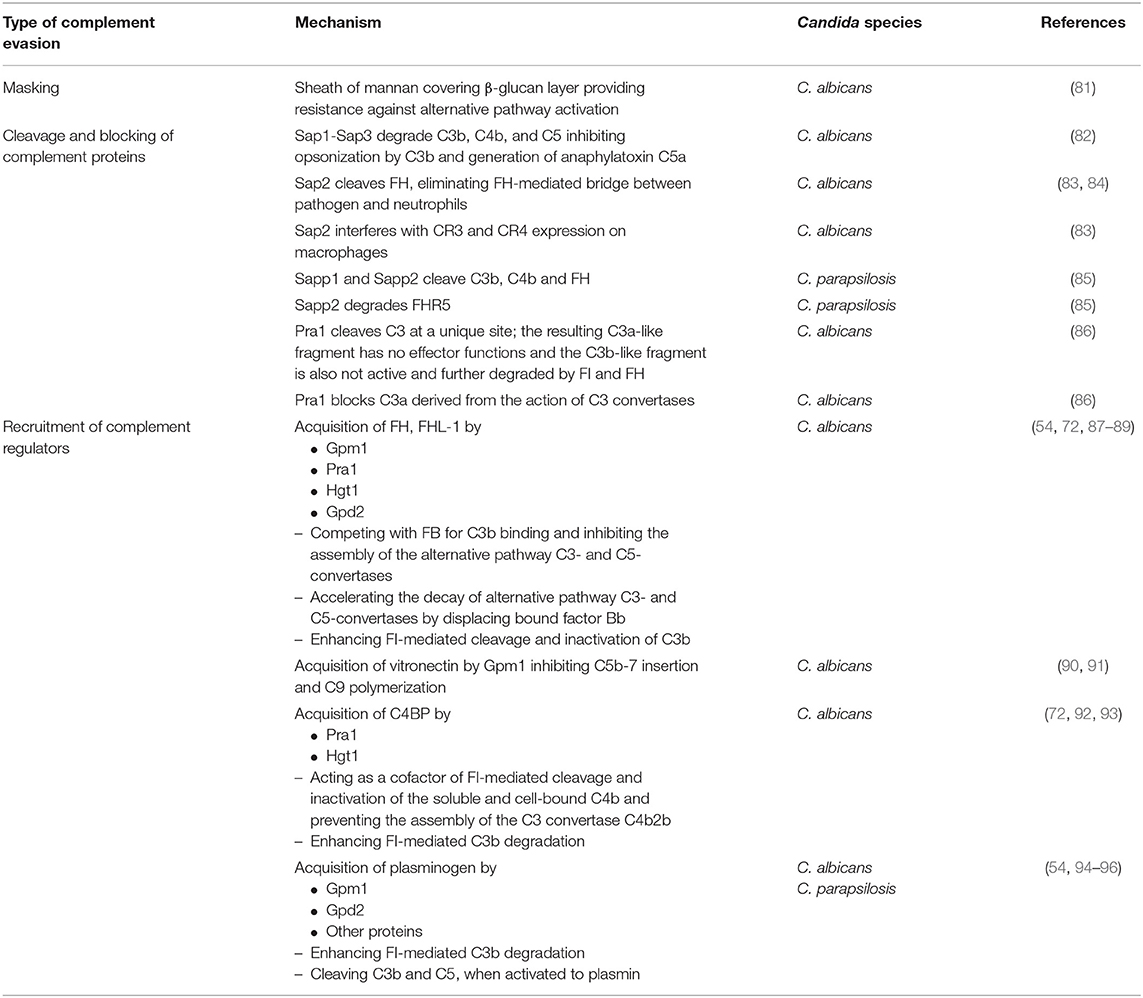
Table 1. Overview of the different types of complement evasion performed by Candida species and the underlying mechanisms. For further details, see text.
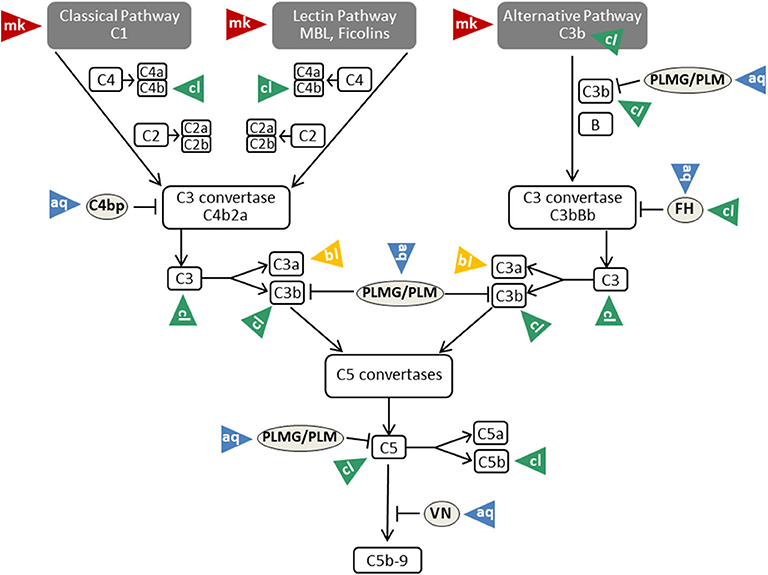
Figure 3. Mechanisms of complement evasion by Candida. The different evasion mechanisms affecting the complete flow of the complement cascade are indicated by triangles and labeled with mk (masking; red triangles), aq (acquisition; blue triangles), cl (cleavage; green triangles), and bl (blocking; yellow triangles). VN, vitronectin; PLMG, Plasminogen; PLM, Plasmin; FH, factor H. For further details and references, see text.
Candida albicans produces a family of 10 related secreted aspartyl proteases (Sap proteins) that are considered to contribute substantially to pathogenicity by favoring adhesion, invasion, and tissue damage (99, 100). Furthermore, Sap proteins are of central relevance for pathogen survival in the host, since they destroy molecules of the immune system (antibodies, complement factors, cytokines) and thus limit microbicidal attacks (101). In this respect, elimination of an effective complement cascade as first-line defense is of special importance (Figure 3). Three members of the Sap family (Sap1-Sap3) were proven to bind and proteolytically degrade the complement proteins C3b, C4b, and C5 (82) and thereby subsequently inhibit complement activation (Table 1).
The functionality of this process was demonstrated by the findings that Sap proteins inhibit the opsonization of C. albicans by C3b and the generation of the anaphylatoxin C5a (82). A second complement-evading mechanism could be attributed to Sap2. It cleaves factor H (FH), a regulator of complement activity and, in addition, a putative bridging molecule between the pathogen and complement receptor CR3 (83) (Table 1). Losse et al. (84) revealed that FH, factor H-like protein (FHL-1) and factor H-related protein-1 (FHR-1) bind to C. albicans as well as to neutrophils. CR3 (CD11b/CD18) was identified to be the major receptor on neutrophils and thus facilitates the attachment of neutrophils to FH-bound Candida. When attached to Candida surface, FH and FHR1 enhanced the antimicrobial activity of the granulocytes and increased the killing of the pathogen (84). Secretion of Sap2 by Candida thus can contribute to immune evasion by proteolytically eliminating the FH-mediated bridge between pathogen and neutrophils. Furthermore, Sap2 interferes with CR3 and CR4 expression on macrophages (83) (Table 1).
The relevance of Sap proteins as virulence factors is not limited to C. albicans. A recent paper describes the functional characterization of Saps produced by Candida parapsilosis (85). C. parapsilosis possesses three secreted aspartyl protease-encoding genes (Sapp1-Sapp3). The purified enzymes Sapp1 and Sapp2 were demonstrated to efficiently cleave the complement components C4b, C3b, and FH; Sapp2 additionally degraded factor H-related protein 5 (FHR5) (Table 1). Macrophages phagocytosed and killed C. parapsilosis sapp1/2/3-/- deletion mutants more efficiently than wild type strains, indicating that Sap-mediated complement evasion is not limited to C. albicans (85). Two additional species that possess Sap genes, namely C. tropicalis (Sapt1-Sapt4) and C. dubliniensis (Sapcd1-Sapcd4, Sapcd7-Sapcd10), might also utilize Saps to modulate the complement system due to the high similarity of their Sap genes with those of C. albicans and C. parapsilosis (102, 103).
Saps are not the only Candida-derived proteases that contribute to virulence. The pH-regulated antigen 1 (Pra1) is described to interfere with complement-mediated antimicrobial functions by several mechanisms. Besides recruiting in its surface-bound form the complement inhibitors FH and C4BP to the Candida surface (see below), it is also released by C. albicans yeast and hyphal forms (87). Recombinant Pra1 was identified as a C3-degrading protease, cleaving C3 at a unique site (Table 1). Pra1 blocks effector functions of the resulting C3a fragment, whereas the Pra1-generated C3b-like fragment is processed and thus inactivated by the complement regulators factor I and factor H. Furthermore, Pra1 can also block C3a derived from the action of the C3 convertases (86) (Table 1).
Although factor H (FH) and factor H-like protein 1 (FHL-1), beside others, should prevent the detrimental effect of the complement system on endogenous cells, such proteins are frequently used by pathogens to avoid recognition or destruction by complement. Regarding FH and FHL-1, it is known so far that Candida albicans expresses four different so-called FH-binding proteins, namely phosphoglycerate mutase 1 (Gpm1), pH-regulated antigen 1 (Pra1), high affinity glucose transporter 1 (Hgt1), and glycerol-3-phosphate dehydrogenase 2 (Gpd2) (53, 54, 72, 88) (Table 1).
Gpm1 was the first protein shown to bind FH (54). The native protein has a mass of 27.5 kDa and regulates the catalytic conversion of 3-phosphoglycerate to 2-phosphoglycerate in glycolysis and the reverse reaction in gluconeogenesis in the cytoplasm (54, 90). Beside the localization in the cytoplasm, Gpm1 is present in the cell wall of yeast cells and hyphae, latter especially on the tip of the hyphae. The lack of classical signal and transport sequences implies that export from the cytoplasm to the yeast surface circumvents endoplasmic reticulum and Golgi, but rather occurs via a non-conventional transport mechanism (54). When located on the cell wall, Gpm1 binds different complement regulators, namely FH, FHL-1 as well as vitronectin and plasminogen (see below). Regarding FH, Gpm1 bears two binding sites for this protein, which are located at SCRs 6-7 and SCRs 19–20 (Figure 1), the former is shared with FHL-1. These regulators stay active when Gpm1 binds them to Candida (54). Even though this moonlighting protein was detected on the cell surface of different Candida species, namely C. albicans, C. parapsilosis, and C. glabrata, only little is known on the functions of Gpm1 in non-albicans species (104).
In 2009, 2 years after the first FH-binding molecule was described, Pra1 was discovered to bind FH to Candida albicans. In its monomeric form, Pra1 has a mass of 68 kDa and is composed of 299 amino acids (53, 105). It was initially described as a fibrinogen binding protein, which is localized on the cell wall of yeast and hyphae (105). The concentration is enhanced on the hyphal surface and notably high on the tips (53). Pra1 can either act as a pH-dependent zincophore, or manipulate complement to Candida's favor, as described above (53, 87, 106). Additionally, Pra1 binds directly or indirectly to different immune cells, resulting in contradictory effects. On the one hand, it can bind to CD4+ T-cells and inhibit cytokine secretion; on the other hand, it serves as a ligand for the integrin αMβ2 (CD11b/CD18, CR3) expressed on neutrophilic granulocytes and therefore enables enhanced phagocytosis (107–109). FH is bound to Pra1 by two binding sites, namely SCRs 16–20, distinct for FH, and SCRs 5–7 (Figure 1), which is also a binding site for FHL-1. As the first four SCRs, mediating the complement regulator functions, are not affected by these different binding sites, pathogen-bound FH and FHL-1 stay functionally active (53).
In 2011, another FH-binding molecule named Hgt1 was discovered (72). This protein belongs to a large gene family of glucose transporters (20 proteins are described by now) and consists of 12 transmembrane domains, 545 amino acids, and weighs 60.67 kDa (110). Hgt1 can also be found in the cell wall of yeast cells and hyphae, where it can serve, in addition to its function as glucose transporter, as a binding molecule for the complement regulators FH and C4b-binding protein (C4BP) (72, 89). With binding of FH as main regulator of alternative pathway and of C4BP as main inhibitor of the classical and lectin pathway, Hgt1 has the capacity to comprehensively interfere with complement activation.
Gpd2, the fourth and by now last protein realized to bind FH, is made up of 371 amino acids, weighs 52 kDa and is localized on the surface of both yeast cells and hyphae (88). Initially, this protein was described as a nicotinamide adenine dinucleotide (NAD+)-dependent enzyme contributing to the degradation of glycerol (111). Gpd2 on Candida was shown to bind to human epithelial and endothelial cells, which might support Candida infection and dissemination in the host. In addition, Gpd2 controls the complement system by binding plasminogen via lysine residues (like Gpm1) and FH/FHL-1 by an amino acid sequence in SCR 7 (88) (Figure 1). FH/FHL-1 derived from individuals with a known human polymorphism on SCR7 (112, 113) may be unable to attach to Gpd2; it is interesting to speculate if these individuals show a better outcome in Candida infections.
Factor H binding by Candida might, however, also represent a double-edged sword for the pathogen. Since FH was described to also serve as a ligand for CR3 (114), Candida-bound FH might interact with CR3 on neutrophils and macrophages, thus bridging the pathogen to professional phagocytes and supporting clearance of the invader.
The family of factor H, factor H-like proteins, and factor H-related proteins are not the only complement regulators that are exploited by Candida to evade from complement. The soluble plasma proteins C4BP, plasminogen and vitronectin were also described to attach to Candida surface, to retain their complement regulatory functions and to assist in complement evasion (19, 87, 115) (Figure 3).
Acquisition of vitronectin on the surfaces of pathogens is a common complement evasion mechanism that was described for bacterial and viral pathogens (116, 117). As mentioned above, vitronectin is an inhibitor of the terminal pathway of complement; by inhibiting the membrane insertion of C5b-7 and the polymerization of C9, it interferes with assembly of the C5b-9 complex and formation of a lytic pore [reviewed in (19, 118)]. Vitronectin was shown to be a ligand for the Candida albicans surface protein Gpm1 (90). Acquisition of vitronectin to Candida leaves its terminal pathway regulatory domain accessible, thus enabling inhibition of C5b-7 insertion and C9 polymerization (91) (Table 1). Furthermore, vitronectin also binds to C. parapsilosis and C. tropicalis pseudohyphae via various surface compounds (119), implying that a similar complement evasion mechanism might be possible for these Candida species.
To limit complement activation for survival in the presence of innate immunity, Candida exploits the efficacy of the main regulator of the classical and lectin pathway, C4BP. For this purpose, both the yeast and the hyphal forms of Candida albicans are capable of binding C4BP to the fungal surface (92, 120). Preferential binding sites are located on the tips of the germ tubes. The surface-attached C4BP retains its regulatory capacity, inactivates C4b and thus limits the flow of the complement cascade (92) (Table 1). The pH-regulated antigen 1 (Pra1) and high affinity glucose transporter 1 (Hgt1) of Candida albicans were described to be the corresponding C4BP-binding molecules that enable this mechanism of fungal complement evasion (72, 87). Pra1 expression levels of different clinical Candida albicans isolates correlated with C4BP binding activity, increased fungal virulence and enhanced survival of the pathogen in the presence of complement (93). The acquisition of C4BP on the fungal surface is not limited to Candida albicans. Meri et al. also proved the binding of this protein to the yeast form of C. tropicalis, C. glabrata, and C. krusei, but it is not known whether the bound C4BP remains regulatory active in these species (92).
The third usage of complement regulators for Candida albicans survival concerns plasminogen. C. albicans acquires plasminogen on its surface via attachment to Gpm1, and bound plasminogen can be converted into the active protease plasmin by urokinase-type plasminogen activator (54). A recent report even lists eight plasminogen-binding proteins on Candida albicans and four proteins in Candida parapsilosis (94, 95). As mentioned above, some reports describe plasminogen/plasmin as a trigger for complement activation with generation of active C5 fragments. However, various reports show opposing results and picture plasminogen as complement inhibitor. Barthel et al. mention plasminogen as a complement inhibitor that enhances FI-mediated C3b degradation and, activated to plasmin, cleaves C3b and C5 (96) (Table 1).
In the last decade, Candida auris emerged as a multidrug-resistant yeast with severe cases of nosocomial infections. Outbreaks were observed all around the world in more than 30 countries (121, 122). The new species is intrinsically resistant to fluconazole and shows variable susceptibility to other azoles, echinocandins, and amphotericin B (122, 123). The genetic relation is closer to rarer Candida species such as C. haemulonii than to common species such as C. albicans and C. glabrata (123). Meanwhile, four distinct clades were distinguished with considerable inter-clade variation, implying independent emergence in multiple geographic regions.
The multidrug resistance strongly asks for deeper insights as a basis to develop immune-based supporting therapies. However, the knowledge about the interaction of C. auris with innate immunity is still limited. Xin et al. described that a mouse strain deficient in complement protein C5 (ΔC5) showed a high susceptibility for disseminated C. auris infection, compared to strains expressing C5. Immunosuppression with cyclophosphamide further substantially increased this susceptibility in ΔC5 mice, whereas treatment of animals with intact C5 gene had only minor effect. Detailed analysis of fungal burden in different organs revealed that expression of C5 is essential to limit the dissemination of C. auris into kidney, heart, and brain in infected mice (124).
Mimicry of complement receptors on the fungal surface is another link between C. auris and the complement system. The Candida complement receptor 3-related protein (CR3-RP) is a surface protein expressed by various Candida species during biofilm formation. CR3-RP is functionally and structurally related to the human CR3 (Mac-1, CD11b/CD18) expressed on neutrophils, monocytes, and macrophages. CR3-RP binds the human complement fragment iC3b and plays an important role in adherence to epithelial cells and in biofilm formation of different Candida species (125). Recent analysis confirmed the presence of CR3-RP also on the surface of C. auris. Blocking of CR3-RP with an antibody in the adherence phase inhibited C. auris biofilm formation. Furthermore, a polyclonal anti-CR3-RP antibody decreased the metabolic activity of pre-formed C. auris biofilms (126). Future work concerning C. auris-triggered complement activation, opsonization, and evasion mechanisms are necessary to get deeper insight into the interplay between this multidrug-resistant pathogen and the complement cascade.
As described in our review, complement as a fast-acting immune weapon with nearly overall presence in the body and a broad spectrum of pathogen recognition is of central relevance for local and invasive Candida infections.
Although comprehensive views on this interesting mutual interplay between a pathogen and the corresponding innate immune response already exist, some aspects ask for further studies. Only limited precise data are available on complement and local Candida lesions in mouth, gut, skin, and vagina. Since many complement proteins are acute-phase proteins, the extent of the topical synthesis of single complement proteins in different stages of disease would be interesting. Furthermore, the contribution of excessive complement activation to development and severity of local lesions is a relevant aspect to be clarified.
Another emphasis should be a broader insight into non-albicans species. Although some data are available on the expression of Saps or plasminogen-binding proteins for non-albicans, (e.g., for C. parapsilosis or C. glabrata, an overview about the relevance of single complement aspects for the different species is lacking). A special focus in this respect should be laid on Candida auris. The relevance of complement for the defense against this species is emphasized by the susceptibility of complement-deficient animals. However, it is unclear which activation pathway has the highest impact to trigger the cascade. The rather small number of patients makes it difficult to weight the role of MBL polymorphism for the susceptibility against C. auris. Furthermore, the evasion mechanisms of C. auris are still completely unknown. Here, the acquisition of factor H, a central evasion mechanism for other Candida species, deserves particular attention.
Exploitation of complement molecules by Candida to down-regulate inflammatory immune responses is another aspect that should be expanded to non-albicans species. C. albicans was described to secrete soluble β-glucan that binds to complement receptor CR3 on host monocytes. Consequently, monocytes form and release vesicles that transport TGF-β1. TGF-β1-transporting extracellular vesicles down-modulate inflammation in whole-blood cells and amplify the anti-inflammatory reaction of endothelial cells (127). This fascinating mechanism might also play a role for other Candida species. The battle between Candida and complement is old, but still reveals many new aspects.
VH wrote the section Recruitment of Complement Regulators FH and FHL-1 on the Candida Surface. GR wrote chapter AN EMERGING FIELD: CANDIDA AURIS AND COMPLEMENT and references. RW wrote section COMPLEMENT-MEDIATED ANTI-CANDIDA EFFECTS. CS wrote the other chapters. CL-F did proofreading and corrections. All authors contributed to the article and approved the submitted version.
RW, VH, CS, and CL-F were supported by the Austrian Science Fund (FWF) Doctoral Program of Excellence HOROS (W1253).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bordron A, Bagacean C, Tempescul A, Berthou C, Bettacchioli E, Hillion S, et al. Complement system: a neglected pathway in immunotherapy. Clin Rev Allergy Immunol. (2019) 58:155–71. doi: 10.1007/s12016-019-08741-0
2. Vignesh P, Rawat A, Sharma M, Singh S. Complement in autoimmune diseases. Clin Chim Acta. (2017) 465:123–30. doi: 10.1016/j.cca.2016.12.017
3. Speth C, Prodinger W, Würzner R, Stoiber H, Dierich MP. Complement. In: Paul WE, editor. Fundamental Immunology. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins (2008). p. 1047–78.
4. Chen Y, Yang C, Jin N, Xie Z, Tang Y, Fei L, et al. Terminal complement complex C5b-9-treated human monocyte-derived dendritic cells undergo maturation and induce Th1 polarization. Eur J Immunol. (2007) 37:167–76. doi: 10.1002/eji.200636285
5. Nicholson-Weller A, Halperin JA. Membrane signaling by complement C5b-9, the membrane attack complex. Immunol Res. (1993) 12:244–57. doi: 10.1007/BF02918256
6. Rodriguez de Cordoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sanchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. (2004) 41:355–67. doi: 10.1016/j.molimm.2004.02.005
7. Klickstein LB, Wong WW, Smith JA, Weis JH, Wilson JG, Fearon DT. Human C3b/C4b receptor (CR1). Demonstration of long homologous repeating domains that are composed of the short consensus repeats characteristics of C3/C4 binding proteins. J Exp Med. (1987) 165:1095–112. doi: 10.1084/jem.165.4.1095
8. Kopp A, Hebecker M, Svobodova E, Jozsi M. Factor h: a complement regulator in health and disease, and a mediator of cellular interactions. Biomolecules. (2012) 2:46–75. doi: 10.3390/biom2010046
9. Gordon DL, Kaufman RM, Blackmore TK, Kwong J, Lublin DM. Identification of complement regulatory domains in human factor H. J Immunol. (1995) 155:348–56.
10. Kuhn S, Skerka C, Zipfel PF. Mapping of the complement regulatory domains in the human factor H-like protein 1 and in factor H1. J Immunol. (1995) 155:5663–70.
11. Pangburn MK. Cutting edge: localization of the host recognition functions of complement factor H at the carboxyl-terminal: implications for hemolytic uremic syndrome. J Immunol. (2002) 169:4702–6. doi: 10.4049/jimmunol.169.9.4702
12. Farries TC, Seya T, Harrison RA, Atkinson JP. Competition for binding sites on C3b by CR1, CR2, MCP, factor B and factor H. Complement Inflamm. (1990) 7:30–41. doi: 10.1159/000463124
13. Meri T, Hartmann A, Lenk D, Eck R, Wurzner R, Hellwage J, et al. The yeast Candida albicans binds complement regulators factor H and FHL-1. Infect Immun. (2002) 70:5185–92. doi: 10.1128/IAI.70.9.5185-5192.2002
14. Zipfel PF, Hellwage J, Friese MA, Hegasy G, Jokiranta ST, Meri S. Factor H and disease: a complement regulator affects vital body functions. Mol Immunol. (1999) 36:241–8. doi: 10.1016/S0161-5890(99)00038-3
15. Zipfel PF, Jokiranta TS, Hellwage J, Koistinen V, Meri S. The factor H protein family. Immunopharmacology. (1999) 42:53–60. doi: 10.1016/S0162-3109(99)00015-6
16. Schwaeble W, Zwirner J, Schulz TF, Linke RP, Dierich MP, Weiss EH. Human complement factor H: expression of an additional truncated gene product of 43 kDa in human liver. Eur J Immunol. (1987) 17:1485–9. doi: 10.1002/eji.1830171015
17. Friese MA, Hellwage J, Jokiranta TS, Meri S, Peter HH, Eibel H, et al. FHL-1/reconectin and factor H: two human complement regulators which are encoded by the same gene are differently expressed and regulated. Mol Immunol. (1999) 36:809–18. doi: 10.1016/S0161-5890(99)00101-7
18. Ermert D, Blom AM. C4b-binding protein: the good, the bad and the deadly. Novel functions of an old friend. Immunol Lett. (2016) 169:82–92. doi: 10.1016/j.imlet.2015.11.014
19. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. (2009) 9:729–40. doi: 10.1038/nri2620
20. Defendi F, Thielens NM, Clavarino G, Cesbron JY, Dumestre-Perard C. The immunopathology of complement proteins and innate immunity in autoimmune disease. Clin Rev Allergy Immunol. (2020) 58:229–51. doi: 10.1007/s12016-019-08774-5
21. Mathern DR, Heeger PS. Molecules great and small: the complement system. Clin J Am Soc Nephrol. (2015) 10:1636–50. doi: 10.2215/CJN.06230614
22. Eggimann P, Garbino J, Pittet D. Epidemiology of Candida species infections in critically ill non-immunosuppressed patients. Lancet Infect Dis. (2003) 3:685–702. doi: 10.1016/S1473-3099(03)00801-6
23. Nobile CJ, Johnson AD. Candida albicans biofilms and human disease. Annu Rev Microbiol. (2015) 69:71–92. doi: 10.1146/annurev-micro-091014-104330
24. Romo JA, Kumamoto CA. On commensalism of Candida. J Fungi. (2020) 6:e10016. doi: 10.3390/jof6010016
25. Sobel JD. Vulvovaginal candidosis. Lancet. (2007) 369:1961–71. doi: 10.1016/S0140-6736(07)60917-9
26. Sardi JC, Scorzoni L, Bernardi T, Fusco-Almeida AM, Mendes Giannini MJ. Candida species: current epidemiology, pathogenicity, biofilm formation, natural antifungal products and new therapeutic options. J Med Microbiol. (2013) 62:10–24. doi: 10.1099/jmm.0.045054-0
27. Cheng SC, Joosten LA, Kullberg BJ, Netea MG. Interplay between Candida albicans and the mammalian innate host defense. Infect Immun. (2012) 80:1304–13. doi: 10.1128/IAI.06146-11
28. Kozel TR. Activation of the complement system by pathogenic fungi. Clin Microbiol Rev. (1996) 9:34–46. doi: 10.1128/CMR.9.1.34
29. Neth O, Jack DL, Dodds AW, Holzel H, Klein NJ, Turner MW. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect Immun. (2000) 68:688–93. doi: 10.1128/IAI.68.2.688-693.2000
30. Kozel TR, Weinhold LC, Lupan DM. Distinct characteristics of initiation of the classical and alternative complement pathways by Candida albicans. Infect Immun. (1996) 64:3360–8. doi: 10.1128/IAI.64.8.3360-3368.1996
31. Behrens NE, Lipke PN, Pilling D, Gomer RH, Klotz SA. Serum amyloid P component binds fungal surface amyloid and decreases human macrophage phagocytosis and secretion of inflammatory cytokines. MBio. (2019) 10:19. doi: 10.1128/mBio.00218-19
32. Roy N, Ohtani K, Hidaka Y, Amano Y, Matsuda Y, Mori K, et al. Three pentraxins C-reactive protein, serum amyloid p component and pentraxin 3 mediate complement activation using Collectin CL-P1. Biochim Biophys Acta General Subjects. (2017) 1861:1–14. doi: 10.1016/j.bbagen.2016.11.023
33. Ying SC, Gewurz AT, Jiang H, Gewurz H. Human serum amyloid P component oligomers bind and activate the classical complement pathway via residues 14–26 and 76–92 of the A chain collagen-like region of C1q. J Immunol. (1993) 150:169–76.
34. Wheeler RT, Kombe D, Agarwala SD, Fink GR. Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog. (2008) 4:e1000227. doi: 10.1371/journal.ppat.1000227
35. Boxx GM, Kozel TR, Nishiya CT, Zhang MX. Influence of mannan and glucan on complement activation and C3 binding by Candida albicans. Infect Immun. (2010) 78:1250–9. doi: 10.1128/IAI.00744-09
36. Hammad NM, El Badawy NE, Ghramh HA, Al Kady LM. Mannose-binding lectin: a potential therapeutic candidate against Candida infection. Biomed Res Int. (2018) 2018:2813737. doi: 10.1155/2018/2813737
37. Ma YJ, Skjoedt MO, Garred P. Collectin-11/MASP complex formation triggers activation of the lectin complement pathway–the fifth lectin pathway initiation complex. J Innate Immun. (2013) 5:242–50. doi: 10.1159/000345356
38. Choteau L, Parny M, Francois N, Bertin B, Fumery M, Dubuquoy L, et al. Role of mannose-binding lectin in intestinal homeostasis and fungal elimination. Mucosal Immunol. (2016) 9:767–76. doi: 10.1038/mi.2015.100
39. Karkowska-Kuleta J, Kozik A, Rapala-Kozik M. Binding and activation of the human plasma kinin-forming system on the cell walls of Candida albicans and Candida tropicalis. Biol Chem. (2010) 391:97–103. doi: 10.1515/bc.2009.145
40. Kaminishi H, Hamatake H, Cho T, Tamaki T, Suenaga N, Fujii T, et al. Activation of blood clotting factors by microbial proteinases. FEMS Microbiol Lett. (1994) 121:327–32. doi: 10.1111/j.1574-6968.1994.tb07121.x
41. Renne T, Stavrou EX. Roles of factor XII in innate immunity. Front Immunol. (2019) 10:2011. doi: 10.3389/fimmu.2019.02011
42. Raghunathan V, Zilberman-Rudenko J, Olson SR, Lupu F, McCarty OJT, Shatzel JJ. The contact pathway and sepsis. Res Practice Thrombosis Haemostasis. (2019) 3:331–9. doi: 10.1002/rth2.12217
43. Kaminishi H, Tanaka M, Cho T, Maeda H, Hagihara Y. Activation of the plasma kallikrein-kinin system by Candida albicans proteinase. Infect Immun. (1990) 58:2139–43. doi: 10.1128/IAI.58.7.2139-2143.1990
44. Irmscher S, Doring N, Halder LD, Jo EA H., Kopka I, Dunker C, et al. Kallikrein cleaves C3 and activates complement. J Innate Immun. (2018) 10:94–105. doi: 10.1159/000484257
45. Davis AE 3rd. The pathophysiology of hereditary angioedema. Clin Immunol. (2005) 114:3–9. doi: 10.1016/j.clim.2004.05.007
46. Longhurst HJ, Bork K. Hereditary angioedema: an update on causes, manifestations and treatment. Br J Hosp Med. (2019) 80:391–8. doi: 10.12968/hmed.2019.80.7.391
47. Gailani D, Bane CE, Gruber A. Factor XI and contact activation as targets for antithrombotic therapy. J Thromb Haemost. (2015) 13:1383–95. doi: 10.1111/jth.13005
48. Negrier C, Shima M, Hoffman M. The central role of thrombin in bleeding disorders. Blood Rev. (2019) 38:100582. doi: 10.1016/j.blre.2019.05.006
49. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
50. Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol. (2008) 632:71–9. doi: 10.1007/978-0-387-78952-1_6
51. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. (2006) 12:682–7. doi: 10.1038/nm1419
52. Krisinger MJ, Goebeler V, Lu Z, Meixner SC, Myles T, Pryzdial EL, et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood. (2012) 120:1717–25. doi: 10.1182/blood-2012-02-412080
53. Luo S, Poltermann S, Kunert A, Rupp S, Zipfel PF. Immune evasion of the human pathogenic yeast Candida albicans: Pra1 is a Factor H, FHL-1 and plasminogen binding surface protein. Mol Immunol. (2009) 47:541–50. doi: 10.1016/j.molimm.2009.07.017
54. Poltermann S, Kunert A, von der Heide M, Eck R, Hartmann A, Zipfel PF. Gpm1p is a factor H-, FHL-1-, and plasminogen-binding surface protein of Candida albicans. J Biol Chem. (2007) 282:37537–44. doi: 10.1074/jbc.M707280200
55. Foley JH, Walton BL, Aleman MM, O'Byrne AM, Lei V, Harrasser M, et al. Complement activation in arterial and venous thrombosis is mediated by plasmin. EBioMedicine. (2016) 5:175–82. doi: 10.1016/j.ebiom.2016.02.011
56. Leung LL, Morser J. Plasmin as a complement C5 convertase. EBioMedicine. (2016) 5:20–1. doi: 10.1016/j.ebiom.2016.03.015
57. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
58. Bottger EC, Metzger S, Bitter-Suermann D, Stevenson G, Kleindienst S, Burger R. Impaired humoral immune response in complement C3-deficient guinea pigs: absence of secondary antibody response. Eur J Immunol. (1986) 16:1231–5. doi: 10.1002/eji.1830161008
59. Tsoni SV, Kerrigan AM, Marakalala MJ, Srinivasan N, Duffield M, Taylor PR, et al. Complement C3 plays an essential role in the control of opportunistic fungal infections. Infect Immun. (2009) 77:3679–85. doi: 10.1128/IAI.00233-09
60. Ashman RB, Fulurija A, Papadimitriou JM. Strain-dependent differences in host response to Candida albicans infection in mice are related to organ susceptibility and infectious load. Infect Immun. (1996) 64:1866–9. doi: 10.1128/IAI.64.5.1866-1869.1996
61. Ashman RB, Papadimitriou JM, Fulurija A, Drysdale KE, Farah CS, Naidoo O, et al. Role of complement C5 and T lymphocytes in pathogenesis of disseminated and mucosal candidiasis in susceptible DBA/2 mice. Microb Pathog. (2003) 34:103–13. doi: 10.1016/S0882-4010(02)00211-5
62. Hector RF, Domer JE, Carrow EW. Immune responses to Candida albicans in genetically distinct mice. Infect Immun. (1982) 38:1020–8. doi: 10.1128/IAI.38.3.1020-1028.1982
63. Held K, Thiel S, Loos M, Petry F. Increased susceptibility of complement factor B/C2 double knockout mice and mannan-binding lectin knockout mice to systemic infection with Candida albicans. Mol Immunol. (2008) 45:3934–41. doi: 10.1016/j.molimm.2008.06.021
64. Pellis V, De Seta F, Crovella S, Bossi F, Bulla R, Guaschino S, et al. Mannose binding lectin and C3 act as recognition molecules for infectious agents in the vagina. Clin Exp Immunol. (2005) 139:120–6. doi: 10.1111/j.1365-2249.2005.02660.x
65. Babula O, Linhares IM, Bongiovanni AM, Ledger WJ, Witkin SS. Association between primary vulvar vestibulitis syndrome, defective induction of tumor necrosis factor-alpha, and carriage of the mannose-binding lectin codon 54 gene polymorphism. Am J Obstet Gynecol. (2008) 198:101.e101–104. doi: 10.1016/j.ajog.2007.05.037
66. Babula O, Lazdane G, Kroica J, Ledger WJ, Witkin SS. Relation between recurrent vulvovaginal candidiasis, vaginal concentrations of mannose-binding lectin, and a mannose-binding lectin gene polymorphism in Latvian women. Clin Infect Dis. (2003) 37:733–7. doi: 10.1086/377234
67. Donders GG, Babula O, Bellen G, Linhares IM, Witkin SS. Mannose-binding lectin gene polymorphism and resistance to therapy in women with recurrent vulvovaginal candidiasis. BJOG. (2008) 115:1225–31. doi: 10.1111/j.1471-0528.2008.01830.x
68. Hammad NM, El Badawy NE, Nasr AM, Ghramh HA, Al Kady LM. Mannose-binding lectin gene polymorphism and its association with susceptibility to recurrent vulvovaginal candidiasis. Biomed Res Int. (2018) 2018:7648152. doi: 10.1155/2018/7648152
69. Wilson BD, Sohnle PG. Neutrophil accumulation and cutaneous responses in experimental cutaneous candidiasis of genetically complement-deficient mice. Clin Immunol Immunopathol. (1988) 46:284–93. doi: 10.1016/0090-1229(88)90190-0
70. Ray TL, Digre KB, Payne CD. Adherence of Candida species to human epidermal corneocytes and buccal mucosal cells: correlation with cutaneous pathogenicity. J Invest Dermatol. (1984) 83:37–41. doi: 10.1111/1523-1747.ep12261661
71. Densen P. Human complement deficiency states and infection. In: Whaley K, Loos M, Weiler JM, editors. Complement in Health and Disease. Dordrecht: Springer Netherlands (1993). p. 173–97. doi: 10.1007/978-94-011-2214-6_6
72. Lesiak-Markowicz I, Vogl G, Schwarzmuller T, Speth C, Lass-Florl C, Dierich MP, et al. Candida albicans Hgt1p, a multifunctional evasion molecule: complement inhibitor, CR3 analogue, and human immunodeficiency virus-binding molecule. J Infect Dis. (2011) 204:802–9. doi: 10.1093/infdis/jir455
73. Ghiran I, Barbashov SF, Klickstein LB, Tas SW, Jensenius JC, Nicholson-Weller A. Complement receptor 1/CD35 is a receptor for mannan-binding lectin. J Exp Med. (2000) 192:1797–808. doi: 10.1084/jem.192.12.1797
74. Li D, Dong B, Tong Z, Wang Q, Liu W, Wang Y, et al. MBL-mediated opsonophagocytosis of Candida albicans by human neutrophils is coupled with intracellular Dectin-1-triggered ROS production. PLoS ONE. (2012) 7:e50589. doi: 10.1371/journal.pone.0050589
75. Zipfel PF, Skerka C. Complement, Candida, and cytokines: the role of C5a in host response to fungi. Eur J Immunol. (2012) 42:822–5. doi: 10.1002/eji.201242466
76. Hunniger K, Bieber K, Martin R, Lehnert T, Figge MT, Loffler J, et al. A second stimulus required for enhanced antifungal activity of human neutrophils in blood is provided by anaphylatoxin C5a. J Immunol. (2015) 194:1199–210. doi: 10.4049/jimmunol.1401845
77. Cheng SC, Sprong T, Joosten LA, van der Meer JW, Kullberg BJ, Hube B, et al. Complement plays a central role in Candida albicans-induced cytokine production by human PBMCs. Eur J Immunol. (2012) 42:993–1004. doi: 10.1002/eji.201142057
78. Nordahl EA, Rydengard V, Nyberg P, Nitsche DP, Morgelin M, Malmsten M, et al. Activation of the complement system generates antibacterial peptides. Proc Natl Acad Sci USA. (2004) 101:16879–84. doi: 10.1073/pnas.0406678101
79. Sonesson A, Ringstad L, Nordahl EA, Malmsten M, Morgelin M, Schmidtchen A. Antifungal activity of C3a and C3a-derived peptides against Candida. Biochim Biophys Acta. (2007) 1768:346–53. doi: 10.1016/j.bbamem.2006.10.017
80. Triebel T, Grillhosl B, Kacani L, Lell CP, Fuchs A, Speth C, et al. Importance of the terminal complement components for immune defence against Candida. Int J Med Microbiol. (2003) 292:527–36. doi: 10.1078/1438-4221-00211
81. Suzuki S. Immunochemical study on mannans of genus Candida. I. Structural investigation of antigenic factors 1, 4, 5, 6, 8, 9, 11, 13, 13b, and 34. Curr Top Med Mycol. (1997) 8:57–70.
82. Gropp K, Schild L, Schindler S, Hube B, Zipfel PF, Skerka C. The yeast Candida albicans evades human complement attack by secretion of aspartic proteases. Mol Immunol. (2009) 47:465–75. doi: 10.1016/j.molimm.2009.08.019
83. Svoboda E, Schneider AE, Sandor N, Lermann U, Staib P, Kremlitzka M, et al. Secreted aspartic protease 2 of Candida albicans inactivates factor H and the macrophage factor H-receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18). Immunol Lett. (2015) 168:13–21. doi: 10.1016/j.imlet.2015.08.009
84. Losse J, Zipfel PF, Jozsi M. Factor H and factor H-related protein 1 bind to human neutrophils via complement receptor 3, mediate attachment to Candida albicans, and enhance neutrophil antimicrobial activity. J Immunol. (2010) 184:912–21. doi: 10.4049/jimmunol.0901702
85. Singh DK, Nemeth T, Papp A, Toth R, Lukacsi S, Heidingsfeld O, et al. Functional characterization of secreted aspartyl proteases in Candida parapsilosis. mSphere. (2019) 4:19. doi: 10.1128/mSphere.00484-19
86. Luo S, Dasari P, Reiher N, Hartmann A, Jacksch S, Wende E, et al. The secreted Candida albicans protein Pra1 disrupts host defense by broadly targeting and blocking complement C3 and C3 activation fragments. Mol Immunol. (2018) 93:266–77. doi: 10.1016/j.molimm.2017.07.010
87. Luo S, Blom AM, Rupp S, Hipler UC, Hube B, Skerka C, et al. The pH-regulated antigen 1 of Candida albicans binds the human complement inhibitor C4b-binding protein and mediates fungal complement evasion. J Biol Chem. (2011) 286:8021–9. doi: 10.1074/jbc.M110.130138
88. Luo S, Hoffmann R, Skerka C, Zipfel PF. Glycerol-3-phosphate dehydrogenase 2 is a novel factor H-, factor H-like protein 1-, and plasminogen-binding surface protein of Candida albicans. J Infect Dis. (2013) 207:594–603. doi: 10.1093/infdis/jis718
89. Kenno S, Speth C, Rambach G, Binder U, Chatterjee S, Caramalho R, et al. Candida albicans factor H binding molecule Hgt1p—a low glucose-induced transmembrane protein is trafficked to the cell wall and impairs phagocytosis and killing by human neutrophils. Front Microbiol. (2018) 9:3319. doi: 10.3389/fmicb.2018.03319
90. Lopez CM, Wallich R, Riesbeck K, Skerka C, Zipfel PF. Candida albicans uses the surface protein Gpm1 to attach to human endothelial cells and to keratinocytes via the adhesive protein vitronectin. PLoS ONE. (2014) 9:e90796. doi: 10.1371/journal.pone.0090796
91. Hallstrom T, Singh B, Kraiczy P, Hammerschmidt S, Skerka C, Zipfel PF, et al. Conserved patterns of microbial immune escape: pathogenic microbes of diverse origin target the human terminal complement inhibitor vitronectin via a single common motif. PLoS ONE. (2016) 11:e0147709. doi: 10.1371/journal.pone.0147709
92. Meri T, Blom AM, Hartmann A, Lenk D, Meri S, Zipfel PF. The hyphal and yeast forms of Candida albicans bind the complement regulator C4b-binding protein. Infect Immun. (2004) 72:6633–41. doi: 10.1128/IAI.72.11.6633-6641.2004
93. Luo S, Hipler UC, Munzberg C, Skerka C, Zipfel PF. Sequence variations and protein expression levels of the two immune evasion proteins Gpm1 and Pra1 influence virulence of clinical Candida albicans isolates. PLoS ONE. (2015) 10:e0113192. doi: 10.1371/journal.pone.0113192
94. Crowe JD, Sievwright IK, Auld GC, Moore NR, Gow NA, Booth NA. Candida albicans binds human plasminogen: identification of eight plasminogen-binding proteins. Mol Microbiol. (2003) 47:1637–51. doi: 10.1046/j.1365-2958.2003.03390.x
95. Karkowska-Kuleta J, Zajac D, Bras G, Bochenska O, Rapala-Kozik M, Kozik A. Binding of human plasminogen and high-molecular-mass kininogen by cell surface-exposed proteins of Candida parapsilosis. Acta Biochim Pol. (2017) 64:391–400. doi: 10.18388/abp.2017_1609
96. Barthel D, Schindler S, Zipfel PF. Plasminogen is a complement inhibitor. J Biol Chem. (2012) 287:18831–42. doi: 10.1074/jbc.M111.323287
97. Han Y, Kozel TR, Zhang MX, MacGill RS, Carroll MC, Cutler JE. Complement is essential for protection by an IgM and an IgG3 monoclonal antibody against experimental, hematogenously disseminated candidiasis. J Immunol. (2001) 167:1550–7. doi: 10.4049/jimmunol.167.3.1550
98. Zhang MX, Bohlman MC, Itatani C, Burton DR, Parren PW, St. Jeor SC, et al. Human recombinant antimannan immunoglobulin G1 antibody confers resistance to hematogenously disseminated candidiasis in mice. Infect Immun. (2006) 74:362–9. doi: 10.1128/IAI.74.1.362-369.2006
99. Naglik J, Albrecht A, Bader O, Hube B. Candida albicans proteinases and host/pathogen interactions. Cell Microbiol. (2004) 6:915–26. doi: 10.1111/j.1462-5822.2004.00439.x
100. Rapala-Kozik M, Bochenska O, Zajac D, Karkowska-Kuleta J, Gogol M, Zawrotniak M, et al. Extracellular proteinases of Candida species pathogenic yeasts. Mol Oral Microbiol. (2018) 33:113–24. doi: 10.1111/omi.12206
101. Monika S, Malgorzata B, Zbigniew O. Contribution of aspartic proteases in Candida virulence. Protease inhibitors against Candida infections. Curr Protein Peptide Sci. (2017) 18:1050–62. doi: 10.2174/1389203717666160809155749
102. Parra-Ortega B, Cruz-Torres H, Villa-Tanaca L, Hernández-Rodríguez C. Phylogeny and evolution of the aspartyl protease family from clinically relevant Candida species. Mem Inst Oswaldo Cruz. (2009) 104:505–12. doi: 10.1590/S0074-02762009000300018
103. Zaugg C, Borg-Von Zepelin M, Reichard U, Sanglard D, Monod M. Secreted aspartic proteinase family of Candida tropicalis. Infect Immun. (2001) 69:405–12. doi: 10.1128/IAI.69.1.405-412.2001
104. Karkowska-Kuleta J, Satala D, Bochenska O, Rapala-Kozik M, Kozik A. Moonlighting proteins are variably exposed at the cell surfaces of Candida glabrata, Candida parapsilosis and Candida tropicalis under certain growth conditions. BMC Microbiol. (2019) 19:149. doi: 10.1186/s12866-019-1524-5
105. Sentandreu M, Elorza MV, Sentandreu R, Fonzi WA. Cloning and characterization of PRA1, a gene encoding a novel pH-regulated antigen of Candida albicans. J Bacteriol. (1998) 180:282–9. doi: 10.1128/JB.180.2.282-289.1998
106. Loboda D, Rowinska-Zyrek M. Zinc binding sites in Pra1, a zincophore from Candida albicans. Dalton Trans. (2017) 46:13695–703. doi: 10.1039/C7DT01675A
107. Bergfeld A, Dasari P, Werner S, Hughes TR, Song WC, Hortschansky P, et al. Direct binding of the pH-regulated protein 1 (Pra1) from Candida albicans inhibits cytokine secretion by mouse CD4(+) T cells. Front Microbiol. (2017) 8:844. doi: 10.3389/fmicb.2017.00844
108. Soloviev DA, Fonzi WA, Sentandreu R, Pluskota E, Forsyth CB, Yadav S, et al. Identification of pH-regulated antigen 1 released from Candida albicans as the major ligand for leukocyte integrin alphaMbeta2. J Immunol. (2007) 178:2038–46. doi: 10.4049/jimmunol.178.4.2038
109. Soloviev DA, Jawhara S, Fonzi WA. Regulation of innate immune response to Candida albicans infections by alphaMbeta2-Pra1p interaction. Infect Immun. (2011) 79:1546–58. doi: 10.1128/IAI.00650-10
110. Fan J, Chaturvedi V, Shen SH. Identification and phylogenetic analysis of a glucose transporter gene family from the human pathogenic yeast Candida albicans. J Mol Evol. (2002) 55:336–46. doi: 10.1007/s00239-002-2330-4
111. Ansell R, Granath K, Hohmann S, Thevelein JM, Adler L. The two isoenzymes for yeast NAD+-dependent glycerol 3-phosphate dehydrogenase encoded by GPD1 and GPD2 have distinct roles in osmoadaptation and redox regulation. EMBO J. (1997) 16:2179–87. doi: 10.1093/emboj/16.9.2179
112. Ormsby RJ, Ranganathan S, Tong JC, Griggs KM, Dimasi DP, Hewitt AW, et al. Functional and structural implications of the complement factor H Y402H polymorphism associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. (2008) 49:1763–70. doi: 10.1167/iovs.07-1297
113. Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. (2011) 478:76–81. doi: 10.1038/nature10449
114. DiScipio RG, Daffern PJ, Schraufstatter IU, Sriramarao P. Human polymorphonuclear leukocytes adhere to complement factor H through an interaction that involves alphaMbeta2 (CD11b/CD18). J Immunol. (1998) 160:4057–66.
115. Behnsen J, Hartmann A, Schmaler J, Gehrke A, Brakhage AA, Zipfel PF. The opportunistic human pathogenic fungus Aspergillus fumigatus evades the host complement system. Infect Immun. (2008) 76:820–7. doi: 10.1128/IAI.01037-07
116. Conde JN, da Silva EM, Allonso D, Coelho DR, Andrade IDS, de Medeiros LN, et al. Inhibition of the membrane attack complex by dengue virus NS1 through Interaction with vitronectin and terminal complement proteins. J Virol. (2016) 90:9570–81. doi: 10.1128/JVI.00912-16
117. Singh B, Su Y.-C., Riesbeck K. Vitronectin in bacterial pathogenesis: a host protein used in complement escape and cellular invasion. Mol Microbiol. (2010) 78:545–60. doi: 10.1111/j.1365-2958.2010.07373.x
118. Blom AM, Hallstrom T, Riesbeck K. Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Mol Immunol. (2009) 46:2808–17. doi: 10.1016/j.molimm.2009.04.025
119. Kozik A, Karkowska-Kuleta J, Zajac D, Bochenska O, Kedracka-Krok S, Jankowska U, et al. Fibronectin-, vitronectin- and laminin-binding proteins at the cell walls of Candida parapsilosis and Candida tropicalis pathogenic yeasts. BMC Microbiol. (2015) 15:197. doi: 10.1186/s12866-015-0531-4
120. Blom AM, Villoutreix BO, Dahlback B. Complement inhibitor C4b-binding protein-friend or foe in the innate immune system? Mol Immunol. (2004) 40:1333–46. doi: 10.1016/j.molimm.2003.12.002
121. Chaabane F, Graf A, Jequier L, Coste AT. Review on antifungal resistance mechanisms in the emerging pathogen Candida auris. Front Microbiol. (2019) 10:2788. doi: 10.3389/fmicb.2019.02788
122. Rhodes J, Fisher MC. Global epidemiology of emerging Candida auris. Curr Opin Microbiol. (2019) 52:84–9. doi: 10.1016/j.mib.2019.05.008
123. Snyder GM, Wright SB. The epidemiology and prevention of Candida auris. Curr Infect Dis Rep. (2019) 21:19. doi: 10.1007/s11908-019-0675-8
124. Xin H, Mohiuddin F, Tran J, Adams A, Eberle K. Experimental mouse models of disseminated Candida auris infection. mSphere. (2019) 4:19. doi: 10.1128/mSphere.00339-19
125. Bujdakova H, Paulovicova E, Paulovicova L, Simova Z. Participation of the Candida albicans surface antigen in adhesion, the first phase of biofilm development. FEMS Immunol Med Microbiol. (2010) 59:485–92. doi: 10.1111/j.1574-695X.2010.00713.x
126. Dekkerova J, Lopez-Ribot JL, Bujdakova H. Activity of anti-CR3-RP polyclonal antibody against biofilms formed by Candida auris, a multidrug-resistant emerging fungal pathogen. Eur J Clin Microbiol Infect Dis. (2019) 38:101–8. doi: 10.1007/s10096-018-3400-x
Keywords: Candida, complement, evasion, innate immunity, factor H, invasive fungal infections
Citation: Harpf V, Rambach G, Würzner R, Lass-Flörl C and Speth C (2020) Candida and Complement: New Aspects in an Old Battle. Front. Immunol. 11:1471. doi: 10.3389/fimmu.2020.01471
Received: 09 April 2020; Accepted: 05 June 2020;
Published: 14 July 2020.
Edited by:
Markus M. Heimesaat, Charité—Universitätsmedizin Berlin, GermanyReviewed by:
Shanshan Luo, Huazhong University of Science and Technology, ChinaCopyright © 2020 Harpf, Rambach, Würzner, Lass-Flörl and Speth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cornelia Speth, Y29ybmVsaWEuc3BldGhAaS1tZWQuYWMuYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.