
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 08 July 2020
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01444
This article is part of the Research Topic Purinergic Signaling and Inflammation View all 10 articles
 Shaima'a Hamarsheh1,2
Shaima'a Hamarsheh1,2 Robert Zeiser1,3,4,5*
Robert Zeiser1,3,4,5*Inflammation is involved in tumor development and progression as well as antitumor response to therapy. In the past decade, the crosstalk between inflammation, immunity, and cancer has been investigated extensively, which led to the identification of several underlying mechanisms and cells involved. The formation of inflammasome complexes leads to the activation of caspase-1, production of interleukin (IL)-1β, and IL-18 and pyroptosis. Multiple studies have shown the involvement of NLRP3 inflammasome in tumorigenesis. Conversely, other reports have indicated a protective role in certain cancers. In this review, we summarize these contradictory roles of NLRP3 inflammasome in cancer, shed the light on oncogenic signaling leading to NLRP3 activation and IL-1β production and outline the current knowledge on therapeutic approaches.
It is well-established that inflammation caused by viral or microbial infections contributes to tumorigenesis. However, emerging evidence have shown that it as well has a pivotal role in most stages of cancer development, besides interfering with the ability of immune system to counteract tumor cells and affecting response to treatment. These mechanisms are mainly driven by innate and adaptive immune cells, such as dendritic cells, macrophages, natural killer (NK) cells, neutrophils, and lymphocytes (1, 2).
One of the central mechanisms contributing to inflammation in immune cells is mediated by special cytoplasmic protein complexes known as inflammasomes. They are divided based on their structural features into nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) and absent in melanoma 2 (AIM2)-like receptors (ALRs). In addition, inflammasomes belong to a larger family of receptors known as pattern recognition receptors (PRRs), where their function is the recognition of pathogen- or danger-associated molecular patterns (PAMPs or DAMPs), causing the activation, maturation, and production of pro-inflammatory cytokines (3). Besides, emerging evidence has proposed that inflammasomes act as a “signal integrator” detecting changes in cytoplasmic homeostasis. These perturbations, named as homeostasis-altering molecular processes (HAMPs), are induced by the functional consequences of cellular processes, where the inflammasome responds to a cellular imbalance rather than a molecular pattern, triggering inflammation in a sterile context. This provides hints that inflammasome activation via the HAMP detection pathway might also be involved in disease pathogenesis (4, 5).
Amongst the inflammasomes family, NLRP3 inflammasome is the most characterized. Mutations in NLRP3 are associated with several autoimmune and inflammatory diseases, particularly a group known as cold-induced auto-inflammatory syndrome (CAPS). In addition, NLRP3 has been implicated in several other diseases including inflammatory bowel disease (IBD), rheumatoid arthritis, and Parkinson's disease (6). In cancer, analysis of copy number alterations in tumor samples has shown NLRP3 with a high frequency of copy gains, thus acting more as an oncogene (7). However, different roles of inflammasomes in tumorigenesis and antitumor immunity have emerged in the past decade (8), without overlooking the well-established role of cytokines in cancer pathogenesis (9).
Here, we discuss the structure and activation pathways of NLRP3, and provide a brief updated review on the most recent research investigating its opposing roles in cancer. Lastly, we list the potential therapeutic targets and the latest reports and clinical trials investigating them.
Since the cloning of IL-1β in 1984 (10, 11) and the characterization of its various immunological activities, enormous research has been conducted to further explore the biology of cytokines and their effects on inflammation and other physiological roles. The first major contribution following this, was the identification of IL-1-converting enzyme (ICE), now named as caspase-1 (12, 13). Despite that, the underlying mechanisms causing the processing and release of IL-1ß remained unclear. It was only until 2002, when Martinon et al. (14) identified a caspase-activating complex, which leads to the maturation and secretion of IL-1β, now known as the inflammasome. They continued their pioneering work in this field (15), which led to discovering the association of inflammasomes with CAPS (16), as well as gout and type 2 diabetes. Additionally, they reported several inflammasome agonists, PAMPs including muramyl dipeptide (MDP) (17), viral DNA (18) and malaria-associated hemozoin (19); DAMPs such as monosodium urate (MSU) crystals (20); and environment-derived factors like asbestos, silica (21) and alum (22). A number of different clinical trials for inflammasome-related inflammatory diseases were conducted which led to the development of a therapy for CAPS patients in the clinic (23), in addition to promising results in several clinical studies involving gouty arthritis patients treated with anakinra (24, 25). These revolutionary discoveries paved a new path in the fields of inflammasome activation, innate immunity cytokines production, and their involvement in health and disease.
Inflammasomes are danger-sensing, multimeric protein complexes that are part of the innate immune response. The most widely studied and well-characterized inflammasome is NLRP3, which is characterized by the presence of a central nucleotide-binding and oligomerization (NACHT) domain, which is usually flanked by C-terminal leucine-rich repeat (LRR), and N-terminal pyrin domain (PYD) (Figure 1A) (3). In brief, a danger signal sensed leads to a conformational change of NLRP3 causing the exposure of NACHT domain. NLRP3 undergoes oligomerization by homotypic interactions between NACHT domains. As a result, the PYD domain of NLRP3 becomes exposed, recruit the adaptor apoptosis speck protein (ASC, also known as PYCARD) and bind through their shared PYD domains (Figure 1A). Following, ASC converts to a prion-like form and generates long ASC filaments. This interaction recruits the CARD of pro-caspase-1 facilitating its binding to the complex. Additionally, the clustering of pro-caspase-1 forms its own prion-like filaments that separates from the ASC filaments allowing the auto-cleavage and formation of the active caspase-1 p10/p20 tetramer, which then processes cytokine pro-forms into active molecules. Therefore, the cluster of oligomerized NLRP3-ASC-pro-caspase-1 complex results in the assembly of the multi-subunit wheel-shaped inflammasome complex (Figure 1B) (3, 14, 26–29). The activation of NLRP3 inflammasome causes two main effects, the induction of programmed cell death known as pyroptosis, and/or a pro-inflammatory response caused by the release of inflammatory cytokines IL-1β and IL-18.
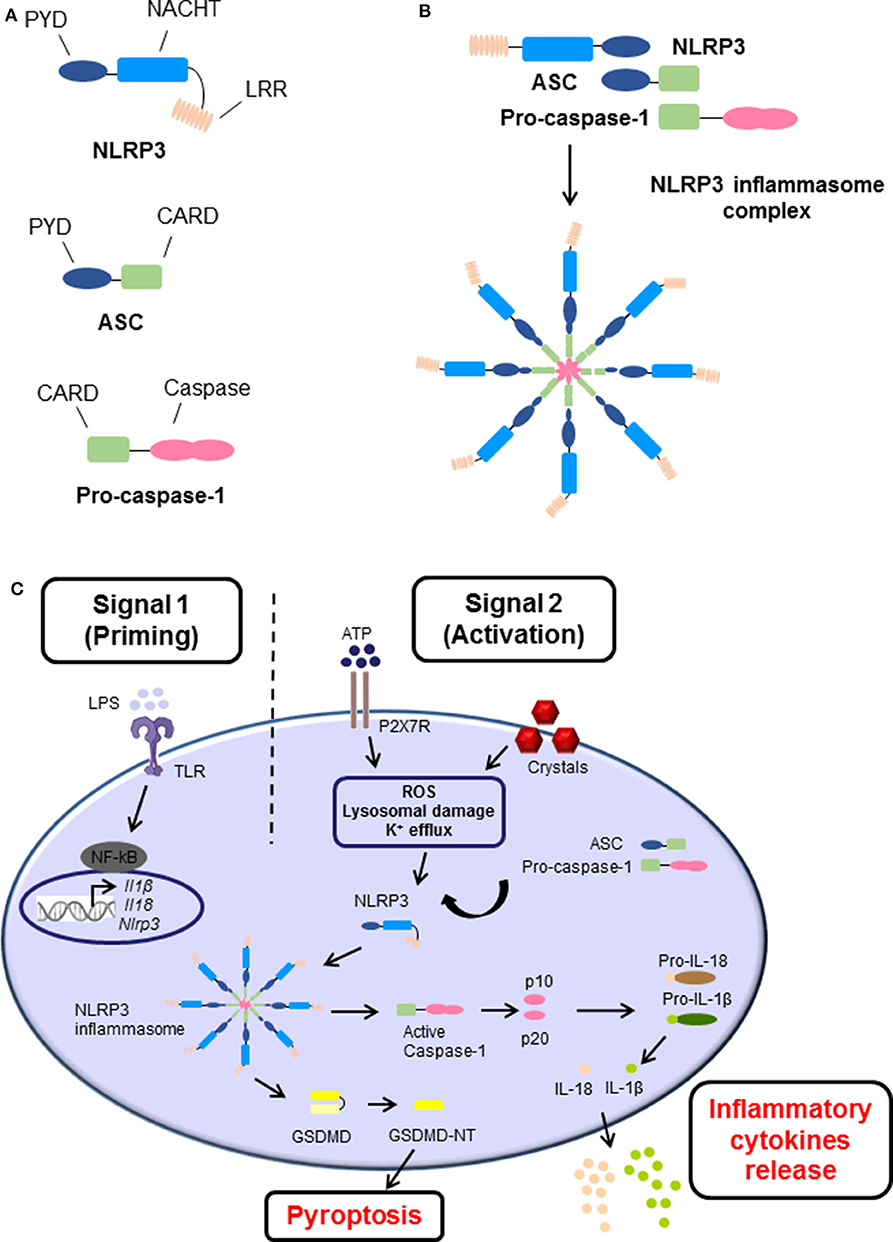
Figure 1. The structure and canonical activation of the NLRP3 inflammasome complex. (A) The structure of NLRP3 is comprised of three main domains: (i) NLRP3, containing an N-terminal pyrin domain (PYD), a central NACHT domain, and a C-terminal leucine-rich repeat (LRR) domain; (ii) adaptor apoptosis speck (ASC) which contains PYD and CARD domains; and (iii) pro-caspase-1 which contains caspase-1 and CARD domains. (B) Upon activation, NLRP3 undergoes oligomerization, recruits, and binds ASC, which subsequently recruits and binds pro-caspase-1 via their shared domains. The formation of this NLRP3 inflammasome cluster results in a prion-like assembly of the complex. (C) The activation process of NLRP3 inflammasome consists of two main signals: (i) Signal 1 (Priming), which is induced by pathogen recognition receptors (PRRs) such as toll-like receptors (TLRs) activated by pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS), or other endogenous factors and mechanisms such as reactive oxygen species (ROS), hypoxia, metabolites, oxidized low-density lipoprotein (oxLDL), amyloids, and complement (not shown). This leads to the transcriptional upregulation of Nlrp3, Ill1b, and Il18 via transcription factors such as NF-κB. (ii) Signal 2 (Activation), is provided by PAMPs or damage-associated molecular patterns (DAMPs), such as adenosine triphosphate (ATP) and crystals which activate different signaling events including ROS, lysosomal damage and K+ efflux, leading to activation and recruitment of NLRP3, oligomerization, and formation of NLRP3 inflammasome complex. The activation and formation of NLRP3 inflammasome has two main consequences: (i) cleavage of Gasdermin D GSDMD and inducing pyroptosis and/or (ii) auto-cleavage and formation of the active caspase-1 and p10/p20 tetramer which then proteolytically cleaves pro-IL-1β and pro-IL-18 into their bioactive forms IL-1β and IL-18 prior to their release.
The canonical activation process requires two main steps known as priming signal and activating signal (Figure 1C). The first step is provided by inflammatory stimuli from toll-like receptors (TLR) ligands or endogenous molecules, which induce the expression of NF-κB. Additionally, other endogenous factors and mechanisms have been identified to prime the inflammasome in sterile inflammatory diseases, such as reactive oxygen species (ROS), hypoxia, metabolites, oxidized low-density lipoprotein (oxLDL), amyloids, and complement. The second step is usually promoted by PAMPs and DAMPs, which cause potassium ion (K+) efflux, calcium (Ca+2) flux, lysosomal damage or ROS production leading to NLRP3 inflammasome assembly, caspase-1 cleavage, and thus the maturation and secretion of IL-1β and IL-18 (27, 28, 30).
On the other hand, other pathways for NLRP3 inflammasome activation were described (reviewed elsewhere (31, 32). The non-canonical NLRP3 inflammasome pathway is activated by most Gram-negative bacteria, and requires capase-11 (33) as well as vacuolar rupture mediated by interferon-inducible guanylate-binding proteins (GBPs). Also, an alternative NLRP3 inflammasome pathway is activated in human monocytes induced by LPS and requires the molecules RIPK1, FAS-associated death domain protein (FADD), and caspase-8 (34).
The function of NLRP3 inflammasome in human cancers is rather a conflicting topic (8, 35), where there is evidence of a protective anti-tumorigenic effect as well as a pro-tumorigenic role in different types of cancer (summarized in Table 1). Here, we discuss both roles shown in murine and human studies and introduce new insights for the effect of oncogenic mutations in inducing NLRP3 inflammasome activation in leukemias.

Table 1. The dual effect of NLRP3 inflammasome in cancers.
NLRP3 inflammasome have been shown to promote the development of several cancers, where most studies were focused on proliferation, survival, metastasis, angiogenesis, and immunosuppression. In breast cancer, NLRP3 inflammasome, and IL-1β production promote the infiltration of myeloid cells such as myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs), providing an inflammatory microenvironment thus promoting breast cancer progression (36). In addition, NLRP3 inflammasome in fibroblasts is further linked with progression and metastasis (37), and IL-1β was found to have an immunosuppressive, pro-tumorigenic in the tumor microenvironment (38). Besides, the NLRP3 inflammasome seems to be an effector for promoting metastasis via the lymphatic system and favoring mammary carcinoma development (39). Interestingly, Huber et al. (42) demonstrated that IL-18 induced by NLRP3 causes the downregulation of the soluble IL-22 receptor, IL-22-binding protein (IL-22BP), leading to an increase in the ratio of IL-22/IL-22BP, which at later stages promotes tumor development (42). Additionally, NLRP3 deficiency leads to suppression of metastases and methylcholanthrene (MCA)-induced sarcomas in mouse models, which were dependent on NK cell and IFN-γ (44). In epithelial skin cancer, mice deficient for IL-1R and caspase-1 showed partial protection against skin cancer development (43). Besides, the roles of inflammasomes in melanoma pathogenesis is established (65). In particular, NLRP3 inflammasome was shown to be constitutively expressed and activated in human melanoma cells. However, these cells secrete biologically active IL-1β in an autonomous way without the presence of exogenous stimuli at late stages of the disease (54). In HNSCC, NLRP3 inflammasome is found upregulated in carcinoma tissues and associated with carcinogenesis and cancer stem cells (CSCs) self-renewal activation (46–48). Also, NLRP3 signaling seems to drive immunosuppression in pancreatic carcinoma, by promoting tolerogenic T cell differentiation and adaptive immune suppression via IL-10 (56).
In hematological malignancies, the role of NLRP3 inflammasome in normal and malignant hematopoiesis has been lately reviewed (66). We have recently reported a novel function of the NLRP3 inflammasome in the pathogenesis of hematological malignancies, particularly myeloproliferation in leukemias. Interestingly, and despite the manifestation of oncogenic KRAS in hematopoietic cells, we could show that the NLRP3 inflammasome has a key role in the development of several myeloid leukemias features in vivo, including cytopenias, splenomegaly, and myeloproliferation. These phenotypes are often seen in chronic myelomonocytic leukemia (CMML), juvenile myelomonocytic leukemia (JMML) and more rarely acute myeloid leukemia (AML) patients harboring KRAS mutations. Additionally, we found evidence of NLRP3 inflammasome activation upon analyzing JMML, CMML, and AML patient samples harboring KRAS mutations, providing a stronger evidence of the participation of NLRP3 inflammasome in the disease development (49). An open question remains how the NLRP3 inflammasome activation drives hematological malignancies, whether by a cell-autonomous signal that promotes cell proliferation directly or via a modification of the TME or both.
Conversely, NLRP3 inflammasome was also shown to have an anti-tumorigenic role. Previously, Ghiringhelli et al. (67) have proposed that NLRP3 inflammasome is required for dendritic cell-mediated priming of IFN-γ-producing T lymphocytes against tumor cells. NLRP3 inflammasome seems to act as a negative modulator of tumorigenesis in colitis-associated cancer (59), which is confirmatory to the study emphasizing the role of NLRP3 inflammasome in the regulation of intestinal homeostasis and thus protection against colitis (60). In addition, NLRP3 inflammasome deficiency seems to cause increased tumor burdens in colorectal cancer. Moreover, Dupaul-Chicoine et al. (62) reported that NLRP3 inflammasome-mediated IL-18 production suppresses colorectal cancer metastatic growth in the liver. In contrast to the tumor-promoting function of IL-22 discussed above, NLRP3/IL-18-mediated downregulation of IL-22BP under controlled production can also provide protective roles against intestinal tissue damage during the inflammation peak (42). In melanoma, it was shown that NLRP3 in the TME weakens the anti-tumor immune response to a cancer vaccine, by assisting the migration of myeloid-derived suppressor cells (MDSCs), thus suppressing the T cell response (64).
NLRP3 inflammasome signaling in humans is controlled by a variety of factors, such as genetic polymorphisms and mutations that can affect gene expression and ultimately lead to its activation. These effects were seen in patients with inflammatory diseases (68–71). Similarly, genetic polymorphisms involved with NLRP3 inflammasome have also been linked to cancer. For instance, a single-nucleotide polymorphism (SNP) in the NLRP3 gene, Q705K (rs35829419), was correlated with poorer survival in patients with invasive colorectal cancer (41), postulated as a risk allele for sporadic metastatic melanoma in Swedish males (72), and also occurs at high frequency in pancreatic cancer patients (73). Additionally, those with NLRP3 polymorphisms (rs10754558 and rs4612666) are more susceptible to gastric cancer when infected with Helicobacter pylori (74). In hematological malignancies, polymorphisms restricted only to IL-1β and IL-18 were associated with clinical and pathophysiological characteristics in AML and chronic myeloid leukemia (CML) (75, 76). Besides, studies utilizing gene expression profiling have also implicated the upregulation of NLRP3 inflammasome in several cancers. For example, NLRP3 is overexpressed in HNSCC, LSCC, and squamous cell carcinoma tissues compared to normal tissues, and often correlated with poor prognosis and worse pathology (48, 50, 77). In bladder cancer, high expression of NLRP3 inflammasome is also found, making it a potential biomarker for its diagnosis (78). Further studies will be required to understand the association between genetic polymorphisms or differential expression of NLRP3 inflammasome and clinical features of cancer.
The understanding of this crosstalk between immunity, inflammasomes, inflammation, and cancer is the foundation for implementing anti-inflammatory therapeutic options in cancer prevention and treatment.
The involvement of the NLRP3 inflammasome in several inflammation-related diseases, including cancer, provided it as an attractive potential target in designing new drugs for treatment. Several reported molecules and drugs were shown to regulate the inflammasome activity. However, many indirectly affect the inflammasome effector functions by targeting other molecules. Until today, current treatment of NLRP3 inflammasome-related diseases in the clinic involve targeting IL-1β or IL-1β receptor by monoclonal IL-1β antibodies or recombinant IL-1 receptor antagonists. Nevertheless, several specific small-molecule compounds have been shown to have anti-inflammatory effects. Here, we review the variety of NLRP3 inflammasome inhibitors which either target components of its canonical signaling pathway or are specific to NLRP3 protein (summarized in Table 2).
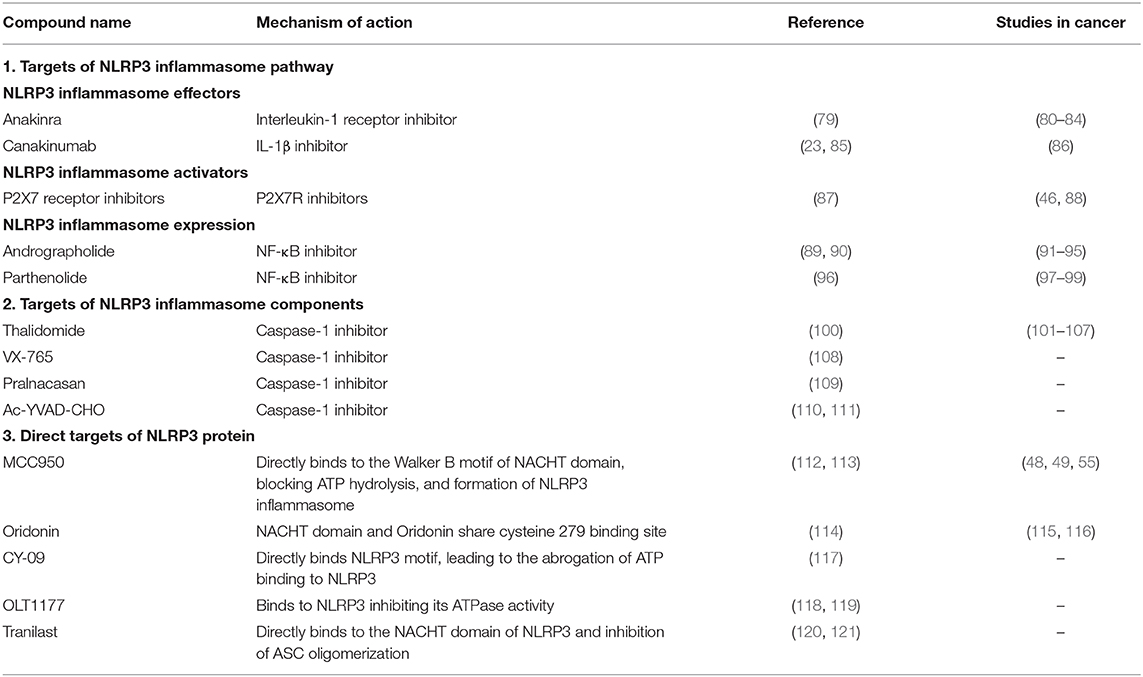
Table 2. A list of compounds targeting NLRP3 inflammasome either indirectly or directly and their therapeutic potential in cancers.
Anakinra is a recombinant form of interleukin-1 receptor antagonist (IL-1Ra) (79), which was approved by the US Food and Drug Administration (FDA) for the treatment of rheumatoid arthritis patients and autoinflammatory disorders (122, 123). We have recently reported that treating KrasG12D-mutant leukemia mouse models with anakinra improves myeloproliferation and cytopenia phenotypes (49). Due to its clinical safety record and short life, anakinra is an ideal drug to be used in conjugation with chemotherapy. Indeed, one clinical trial on metastatic colorectal cancer reported that the treatment of anakinra besides fluorouracil (5-FU) plus bevacizumab showed survival benefit (80), while another showed improved outcome in PDAC patients when combining anakinra with gemcitabine, nab-paclitaxel, and cisplatin (AGAP) (81). Although older reports indicated that anakinra alone was not able to induce myeloma cell death, a study involving multiple myeloma patients used anakinra in combination with low-dose weekly dexamethasone, showed an improved survival for over 10 years compared to the controls (82). In breast cancer, the use of pre-clinical mouse models indicated that anakinra treatment decreased tumor growth and bone metastasis (83). Besides, a clinical pilot study investigated the administration of anakinra prior to standard chemotherapy in HER2-negative metastatic breast cancer female patients. The study revealed that 2-weeks of anakinra treatment alone could downregulate the expression of several genes for TLR and IL-1β families, but upregulate the expression of tumor lysis-associated genes like NK and CD8+ T-cells (84). These results indicate a promising outlook for the use of anakinra combined with standard chemotherapy in difference cancers. However, the effectiveness of anakinra in antitumor applications needs further investigation through in vivo models and later in clinical trials.
Canakinumab is a human anti–IL-1β monoclonal antibody, known for its high specificity to block IL-1β without interference or cross-reactivity with other IL-1 family members. It was approved by the US FDA and European Medicines Agency for treating CAPS (23, 85). Canakinumab has a half-life of a typical IgG1 antibody (124), which gives it an advantage over recombinant IL-1Ra by ensuring the full inhibition of IL-1β over a lengthier period. Interestingly, Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS), a randomized, double-blinded clinical trial of 10,061 lung cancer and atherosclerosis patients implemented the use of canakinumab, and resulted in a significant reduction of lung cancer-caused mortality. This antitumor effect was evident in lung adenocarcinoma or poorly differentiated large cell cancer due to the few cases of small-cell lung cancers or squamous cell carcinomas (86). Currently, canakinumab is being applied in clinical trials focusing on non-small cell lung cancer (NSCLC), Triple Negative Breast Cancer (TNBC), colorectal cancer and metastatic melanoma. In particular, two ongoing Phase III clinical trials conducted by Novartis pharmaceuticals (CANOPY-1 and CANOPY-2) are currently investigating pembrolizumab plus chemotherapy with or without canakinumab, or docetaxel with canakinumab in NSCLC (ClinicalTrials.gov Identifier: NCT03626545, NCT03631199). The forthcoming results will provide a better insight on in safety and efficacy of using it as combination treatment. However, investigating the use of canakinumab in other cancers remain less prominent, and relatively requires more recognition.
P2X7R mediates NLRP3 inflammasome activation and cytokine release. However, the role of P2X7R in tumor cells is shown to be either pro-tumorigenic or anti-tumorigenic [reviewed in Savio et al. (87)]. Nevertheless, several reports have evaluated the potential of P2X7R antagonists in different cancers and suggested their efficacy in altering tumor cells and suppressing cancer progression. For instance, inhibition of P2X7R caused attenuated tumor proliferation and invasion in PDAC (88), and decreased invasiveness of A253 cells derived from epidermoid carcinoma (46).
Thalidomide, a sedative or hypnotic drug, was used particularly for morning sickness in pregnant women (100). However, it was shown to have an anti-tumor activity due to its antiangiogenic properties (125, 126), and later be an inhibitor of caspase-1 (127). It has been approved as a first-line therapeutic option in patients with advanced multiple myeloma in combination with other chemotherapy drugs because of its anti-tumor activities, resulting in improved response (101, 102). In prostate cancer, the administration of thalidomide alone or in combination with docetaxel resulted in improved response and overall median survival (103, 104). However, its application in other cancer types, such as metastatic melanoma, NSCLC and hepatocellular carcinoma (105–107), did not show significant usefulness.
In addition, VX-765 (108), Pralnacasan (109), and Ac-YVAD-CHO (110, 111) are other caspase-1 inhibitors which have shown few but promising results in their potential in NLRP3-related diseases. However, their potential as therapeutic targets in cancer was not investigated.
Other compounds include Andrographolide (89, 90) and Parthenolide (96), which mainly target NF-κB signaling pathway, but the later was also shown to directly inhibit NLRP3 inflammasome by interfering with its ATPase activity (128), have also shown promising results in several cancers (129). For instance, andrographolide was shown to suppress cancer cell proliferation, promote apoptosis in colon cancer (91), breast cancer (92, 93), multiple myeloma (94), and enhance the antitumor effect of 5-FU in colorectal cancer (95). Besides, parthenolide have shown positive results in inhibiting tumor cell proliferation in gastric cancer (97), pancreatic adenocarcinoma (98), colorectal cancer (99). However, these two compounds have not been taken further beyond pre-clinical studies.
A number of small-molecule compounds were proposed to show specific inhibitory effects on NLRP3 activation [reviewed further in detail elsewhere (130, 131)]. One example is MCC950, which prevents NLRP3-induced ASC oligomerization, leading to the inhibition of both canonical and non-canonical NLRP3 inflammasome activation as well as IL-1β secretion, presenting it as a promising agent in NLRP3-related diseases (112). Mechanistic studies have revealed that MCC950 directly binds to the Walker B motif of the NLRP3 central NACHT domain, blocking the hydrolysis of ATP and thus the formation of NLRP3 inflammasome. This action is independent of K+ efflux, Ca2+ flux, or NLRP3–ASC interactions, and occurs without interfering with TLR signaling or the priming step of NLRP3 activation (112, 113, 130). The use of MCC950 in head and neck squamous cell carcinoma was shown to delay tumorigenesis and improve the antitumor response by reducing the numbers of MDSCs; regulatory T cells (Tregs) and TAMs (48). Besides, MCC950 treatment in MDS was sufficient to halt restore effective hematopoiesis by inhibition of pyroptosis (55). Furthermore, we have recently reported that the use of MCC950 in KrasG12D-mutant leukemia mouse models improves myeloproliferation and cytopenia phenotypes, by attenuating NLRP3 inflammasome (49). However, despite its promising potential in Parkinson's disease (132), preclinical and clinical reports studying MCC950 in cancer remain rather limited.
Oridonin is a major bioactive component of herbal plant Rabdosia rubescens, and is widely used as an over-the-counter (OTC) herbal medicine for the treatment of inflammatory diseases (130). Studies have shown that Oridonin can specifically inhibit NLRP3 inflammasome activation, where NACHT domain and Oridonin share cysteine 279 binding site (114). The ability of Oridonin to suppress cell proliferation was previously demonstrated in breast (133) ovarian (115) and esophageal (116) cancers. On the other hand, CY-09 (117), OLT1177(118, 119), Tranilast (120, 121) present as promising specific NLRP3 inhibitors. However, their potential in NLRP3-related cancers has not been investigated yet.
In conclusion, despite the promising prospective of the compounds mentioned above, further studies are still needed to fully understand their therapeutic potential in NLRP3-related diseases, especially in cancers.
Despite the well-characterized crucial functions for NLRP3 inflammasome in the immune system, their roles in cancer remain rather complicated and elusive. The double-edged sword effect of NLRP3 inflammasome in cancer appears to be dependent on several factors, including its levels of expression, downstream effector molecules (i.e., IL-1β or IL-18), cancer type, stages of tumorigenesis as well as the potential presence of mutations affecting NLRP3 expression. Therefore, in order to further understand these roles, future research needs to address several points: (i) driving factors of NLRP3 inflammasome activation in tumors, such as oncogenic mutations or mutations of inflammasome components, (ii) possible cross-talk pathways and molecules interacting and affecting the regulation of NLRP3 inflammasome, (iii) effects of TME and its components on NLRP3 inflammasome activation and vice versa, (iv) effect of NLRP3 inflammasome on the regulation of immune cells, antitumor immunity and efficiency of immunotherapy. In summary, targeting the NLRP3 inflammasome or its downstream pathways, either solely or in combination with chemotherapy or other immunotherapeutic approaches, hold a promising potential in cancers.
SH and RZ performed literature research together, discussed the articles, and wrote the manuscript together. All authors contributed to the article and approved the submitted version.
This study was supported by the Deutsche Forschungsgemeinschaft, Germany, SFB TRR167 (to RZ), SFB1160 TP B09 to RZ, SFB 850 (RZ), DFG individual grant 872/4-1 to RZ, the European Union: GVHDCure (ERC consolidator grant to RZ), the Deutsche Krebshilfe (grant number 70113473) and the Jose-Carreras Leukemia foundation grant number DJCLS 01R/2019 (RZ) and by the Germany's Excellence Strategy (CIBSS—EXC-2189—Project ID 390939984 to RZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
3. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
4. Liston A, Masters SL. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol. (2017) 17:208–14. doi: 10.1038/nri.2016.151
5. Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. (2019) 40:1035–52. doi: 10.1016/j.it.2019.09.005
6. Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol. (2011) 166:1–5. doi: 10.1111/j.1365-2249.2011.04440.x
7. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. (2010) 466:869–73. doi: 10.1038/nature09208
8. Karki R, Kanneganti TD. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer. (2019) 19:197–214. doi: 10.1038/s41568-019-0123-y
9. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. (2004) 4:11–22. doi: 10.1038/nrc1252
10. Auron PE, Webb AC, Rosenwasser LJ, Mucci SF, Rich A, Wolff SM, et al. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc Natl Acad Sci U S A. (1984) 81:7907–11. doi: 10.1073/pnas.81.24.7907
11. Kasaian M, Collins M. Interleukin-13. Contemp Target Ther Rheumatol. (2007) 6:159–74. doi: 10.3109/9780203694145-14
12. Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, et al. Molecular cloning of the interleukin-1β converting enzyme. Science. (1992) 256:97–100. doi: 10.1126/science.1373520
13. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. (1992) 356:768–74. doi: 10.1038/356768a0
14. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
15. Dagenais M, Skeldon A, Saleh M. The inflammasome: in memory of Dr. Jurg Tschopp. Cell Death Differ. (2012) 19:5–12. doi: 10.1038/cdd.2011.159
16. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. (2004) 20:319–25. doi: 10.1016/S1074-7613(04)00046-9
17. Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/Cryopyrin inflammasome. Curr Biol. (2004) 14:1929–34. doi: 10.1016/j.cub.2004.10.027
18. Muruve DA, Pétrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. (2008) 452:103–7. doi: 10.1038/nature06664
19. Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One. (2009) 4:e6510. doi: 10.1371/journal.pone.0006510
20. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. (2006) 440:237–41. doi: 10.1038/nature04516
21. Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. (2008) 320:674–7. doi: 10.1126/science.1156995
22. Kool M, Pétrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. (2008) 181:3755–9. doi: 10.4049/jimmunol.181.6.3755
23. Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. (2009) 360:2416–25. doi: 10.1056/NEJMoa0810787
24. So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. (2007) 9:R28. doi: 10.1186/ar2143
25. Ottaviani S, Moltó A, Ea HK, Neveu S, Gill G, Brunier L, et al. Efficacy of anakinra in gouty arthritis: a retrospective study of 40 cases. Arthritis Res Ther. (2013) 15:R123. doi: 10.1186/ar4303
26. Guo H, Callaway JB, Ting JPY. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
27. Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. (2010) 327:296–300. doi: 10.1126/science.1184003
28. Malik A, Kanneganti T-D. Inflammasome activation and assembly at a glance. J Cell Sci. (2017) 130:3955–63. doi: 10.1242/jcs.207365
29. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
30. Patel MN, Carroll RG, Galván-Peña S, Mills EL, Olden R, Triantafilou M, et al. Inflammasome priming in sterile inflammatory disease. Trends Mol Med. (2017) 23:165–80. doi: 10.1016/j.molmed.2016.12.007
31. Man SM, Kanneganti T-D. Regulation of inflammasome activation. Immunol Rev. (2015) 265:6–21. doi: 10.1111/imr.12296
32. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
33. Kayagaki N, Warming S, Lamkanfi M, Walle L, Vande Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. (2011) 479:117–21. doi: 10.1038/nature10558
34. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
35. Kantono M, Guo B. Inflammasomes and cancer: the dynamic role of the inflammasome in tumor development. Front Immunol. (2017) 8:1132. doi: 10.3389/fimmu.2017.01132
36. Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep. (2016) 6:1–2. doi: 10.1038/srep36107
37. Ershaid N, Sharon Y, Doron H, Raz Y, Shani O, Cohen N, et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat Commun. (2019) 10:4375. doi: 10.1038/s41467-019-12370-8
38. Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, et al. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci U S A. (2019) 116:1361–9. doi: 10.1073/pnas.1812266115
39. Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J Exp Med. (2017) 214:2695–713. doi: 10.1084/jem.20160392
40. Wang H, Wang Y, Du Q, Lu P, Fan H, Lu J, et al. Inflammasome-independent NLRP3 is required for epithelial-mesenchymal transition in colon cancer cells. Exp Cell Res. (2016) 342:184–92. doi: 10.1016/j.yexcr.2016.03.009
41. Ungerbäck J, Belenki D, Jawad ul-Hassan A, Fredrikson M, Fransén K, Elander N, et al. Genetic variation and alterations of genes involved in NFκB/TNFAIP3- and NLRP3-inflammasome signaling affect susceptibility and outcome of colorectal cancer. Carcinogenesis. (2012) 33:2126–34. doi: 10.1093/carcin/bgs256
42. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. (2012) 491:259–63. doi: 10.1038/nature11535
43. Drexler SK, Bonsignore L, Masin M, Tardivel A, Jackstadt R, Hermeking H, et al. Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc Natl Acad Sci U S A. (2012) 109:18384–9. doi: 10.1073/pnas.1209171109
44. Chow MT, Sceneay J, Paget C, Wong CSF, Duret H, Tschopp J, et al. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. (2012) 72:5721–32. doi: 10.1158/0008-5472.CAN-12-0509
45. Li S, Liang X, Ma L, Shen L, Li T, Zheng L, et al. MiR-22 sustains NLRP3 expression and attenuates H. Pyloriinduced gastric carcinogenesis. Oncogene. (2018) 37:884–96. doi: 10.1038/onc.2017.381
46. Bae JY, Lee S-W, Shin Y-H, Lee J-H, Jahng JW, Park K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget. (2017) 8:48972–82. doi: 10.18632/oncotarget.16903
47. Huang CF, Chen L, Li YC, Wu L, Yu GT, Zhang WF, et al. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. (2017) 36:116. doi: 10.1186/s13046-017-0589-y
48. Chen L, Huang CF, Li YC, Deng WW, Mao L, Wu L, et al. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell Mol Life Sci. (2018) 75:2045–58. doi: 10.1007/s00018-017-2720-9
49. Hamarsheh S, Osswald L, Saller BS, Unger S, De Feo D, Vinnakota JM, et al. Oncogenic KrasG12D causes myeloproliferation via NLRP3 inflammasome activation. Nat Commun. (2020) 11:1659. doi: 10.1038/s41467-020-15497-1
50. Xue Y, Du H-D, Tang D, Zhang D, Zhou J, Zhai C-W, et al. Correlation between the NLRP3 inflammasome and the prognosis of patients with LSCC. Front Oncol. (2019) 9:588. doi: 10.3389/fonc.2019.00588
51. Wang Y, Kong H, Zeng X, Liu W, Wang Z, Yan X, et al. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol Rep. (2016) 35:2053–64. doi: 10.3892/or.2016.4569
52. Zhao X, Zhang C, Hua M, Wang R, Zhong C, Yu J, et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget. (2017) 8:108571–83. doi: 10.18632/oncotarget.21010
53. Ahmad I, Muneer KM, Tamimi IA, Chang ME, Ata MO, Yusuf N. Thymoquinone suppresses metastasis of melanoma cells by inhibition of NLRP3 inflammasome. Toxicol Appl Pharmacol. (2013) 270:70–6. doi: 10.1016/j.taap.2013.03.027
54. Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, et al. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1β. J Biol Chem. (2010) 285:6477–88. doi: 10.1074/jbc.M109.064907
55. Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood. (2016) 128:2960–75. doi: 10.1182/blood-2016-07-730556
56. Daley D, Mani VR, Mohan N, Akkad N, Pandian GSDB, Savadkar S, et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med. (2017) 214:1711–24. doi: 10.1084/jem.20161707
57. Veeranki S. Role of inflammasomes and their regulators in prostate cancer initiation, progression and metastasis. Cell Mol Biol Lett. (2013) 18:355–67. doi: 10.2478/s11658-013-0095-y
58. Panchanathan R, Liu H, Choubey D. Hypoxia primes human normal prostate epithelial cells and cancer cell lines for the NLRP3 and AIM2 inflammasome activation. Oncotarget. (2016) 7:28183–94. doi: 10.18632/oncotarget.8594
59. Allen IC, TeKippe EM, Woodford R-MT, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. (2010) 207:1045–56. doi: 10.1084/jem.20100050
60. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti T-D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. (2010) 32:379–91. doi: 10.1016/j.immuni.2010.03.003
61. Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti T-D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. (2010) 185:4912–20. doi: 10.4049/jimmunol.1002046
62. Dupaul-Chicoine J, Arabzadeh A, Dagenais M, Douglas T, Champagne C, Morizot A, et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity. (2015) 43:751–63. doi: 10.1016/j.immuni.2015.08.013
63. Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, et al. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Investig. (2014) 94:52–62. doi: 10.1038/labinvest.2013.126
64. Van Deventer HW, Burgents JE, Wu QP, Woodford RMT, Brickey WJ, Allen IC, et al. The inflammasome component Nlrp3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. (2010) 70:10161–9. doi: 10.1158/0008-5472.CAN-10-1921
65. Dunn JH, Ellis LZ, Fujita M. Inflammasomes as molecular mediators of inflammation and cancer: potential role in melanoma. Cancer Lett. (2012) 314:24–33. doi: 10.1016/j.canlet.2011.10.001
66. Ratajczak MZ, Bujko K, Cymer M, Thapa A, Adamiak M, Ratajczak J, et al. The Nlrp3 inflammasome as a “rising star” in studies of normal and malignant hematopoiesis. Leukemia. (2020) 34:1512–23. doi: 10.1038/s41375-020-0827-8
67. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1?-dependent adaptive immunity against tumors. Nat Med. (2009) 15:1170–8. doi: 10.1038/nm.2028
68. Verma D, Särndahl E, Andersson H, Eriksson P, Fredrikson M, Jönsson JI, et al. The Q705K polymorphism in NLRP3 is a gain-of-function alteration leading to excessive interleukin-1β and IL-18 production. PLoS One. (2012) 7:e34977. doi: 10.1371/journal.pone.0034977
69. Touitou I, Lesage S, McDermott M, Cuisset L, Hoffman H, Dode C, et al. Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat. (2004) 24:194–8. doi: 10.1002/humu.20080
70. Levy R, Gérard L, Kuemmerle-Deschner J, Lachmann HJ, Koné-Paut I, Cantarini L, et al. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: a series of 136 patients from the Eurofever Registry. Ann Rheum Dis. (2015) 74:2043–9. doi: 10.1136/annrheumdis-2013-204991
71. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. (2001) 29:301–5. doi: 10.1038/ng756
72. Verma D, Bivik C, Farahani E, Synnerstad I, Fredrikson M, Enerbäck C, et al. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res. (2012) 25:506–13. doi: 10.1111/j.1755-148X.2012.01008.x
73. Miskiewicz A, Szparecki G, Durlik M, Rydzewska G, Ziobrowski I, Górska R. The Q705K and F359L single-nucleotide polymorphisms of NOD-like receptor signaling pathway: association with chronic pancreatitis, pancreatic cancer, and periodontitis. Arch Immunol Ther Exp (Warsz). (2015) 63:485–94. doi: 10.1007/s00005-015-0355-9
74. Castaño-Rodríguez N, Kaakoush NO, Goh KL, Fock KM, Mitchell HM. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: a case-control study and gene expression analyses. PLoS One. (2014) 9:e98899. doi: 10.1371/journal.pone.0098899
75. Zhang A, Yu J, Yan S, Zhao X, Chen C, Zhou Y, et al. The genetic polymorphism and expression profiles of NLRP3 inflammasome in patients with chronic myeloid leukemia. Hum Immunol. (2018) 79:57–62. doi: 10.1016/j.humimm.2017.10.013
76. Wang H, Hua M, Wang S, Yu J, Chen C, Zhao X, et al. Genetic polymorphisms of IL-18 rs1946518 and IL-1β rs16944 are associated with prognosis and survival of acute myeloid leukemia. Inflamm Res. (2017) 66:249–58. doi: 10.1007/s00011-016-1012-4
77. Wu CS, Chang KP, OuYang CN, Kao HK, Hsueh C, Chen LC, et al. ASC contributes to metastasis of oral cavity squamous cell carcinoma. Oncotarget. (2016) 7:50074–85. doi: 10.18632/oncotarget.10317
78. Poli G, Brancorsini S, Cochetti G, Barillaro F, Egidi MG, Mearini E. Expression of inflammasome-related genes in bladder cancer and their association with cytokeratin 20 messenger RNA. Urol Oncol Semin Orig Investig. (2015) 33:505.e1–e7. doi: 10.1016/j.urolonc.2015.07.012
79. Arend WP. Interleukin 1 receptor antagonist. A new member of the interleukin 1 family. J Clin Invest. (1991) 88:1445–51. doi: 10.1172/JCI115453
80. Isambert N, Hervieu A, Rébé C, Hennequin A, Borg C, Zanetta S, et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): a single-arm phase 2 study. Oncoimmunology. (2018) 7:e1474319. doi: 10.1080/2162402X.2018.1474319
81. Becerra C, Paulson AS, Cavaness KM, Celinski SA. Gemcitabine, nab-paclitaxel, cisplatin, and anakinra (AGAP) treatment in patients with localized pancreatic ductal adenocarcinoma (PDAC). J Clin Oncol. (2018) 36:449–9. doi: 10.1200/JCO.2018.36.4_suppl.449
82. Lust JA, Lacy MQ, Zeldenrust SR, Witzig TE, Moon-Tasson LL, Dinarello CA, et al. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am J Hematol. (2016) 91:571–4. doi: 10.1002/ajh.24352
83. Holen I, Lefley D V, Francis SE, Rennicks S, Bradbury S, Coleman RE, et al. IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget. (2016) 7:75571–84. doi: 10.18632/oncotarget.12289
84. Wu T-C, Xu K, Martinek J, Young RR, Banchereau R, George J, et al. IL1 receptor antagonist controls transcriptional signature of inflammation in patients with metastatic breast cancer. Cancer Res. (2018) 78:5243–58. doi: 10.1158/0008-5472.CAN-18-0413
85. Alten R, Gram H, Joosten LA, van den Berg WB, Sieper J, Wassenberg S, et al. The human anti-IL-1 beta monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof-of-concept study in patients with rheumatoid arthritis. Arthritis Res Ther. (2008) 10:R67. doi: 10.1186/ar2438
86. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:1833–42. doi: 10.1016/S0140-6736(17)32247-X
87. Savio LEB, de Andrade Mello P, da Silva CG, Coutinho-Silva R. The P2X7 receptor in inflammatory diseases: angel or demon? Front Pharmacol. (2018) 9:52. doi: 10.3389/fphar.2018.00052
88. Giannuzzo A, Pedersen SF, Novak I. The P2X7 receptor regulates cell survival, migration and invasion of pancreatic ductal adenocarcinoma cells. Mol Cancer. (2015) 14:203. doi: 10.1186/s12943-015-0472-4
89. Guo W, Sun Y, Liu W, Wu X, Guo L, Cai P, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. (2014) 10:972–85. doi: 10.4161/auto.28374
90. Bao Z, Guan S, Cheng C, Wu S, Wong SH, Michael Kemeny D, et al. A novel antiinflammatory role for andrographolide in asthma via inhibition of the nuclear factor-κb pathway. Am J Respir Crit Care Med. (2009) 179:657–65. doi: 10.1164/rccm.200809-1516OC
91. Zhang R, Zhao J, Xu J, Jiao DX, Wang J, Gong ZQ, et al. Andrographolide suppresses proliferation of human colon cancer SW620 cells through the TLR4/NF-κB/MMP-9 signaling pathway. Oncol Lett. (2017) 14:4305–10. doi: 10.3892/ol.2017.6669
92. Zhai Z, Qu X, Yan W, Li H, Liu G, Liu X, et al. Andrographolide prevents human breast cancer-induced osteoclastic bone loss via attenuated RANKL signaling. Breast Cancer Res Treat. (2014) 144:33–45. doi: 10.1007/s10549-014-2844-7
93. Zhai Z, Qu X, Li H, Ouyang Z, Yan W, Liu G, et al. Inhibition of MDA-MB-231 breast cancer cell migration and invasion activity by andrographolide via suppression of nuclear factor-κB-dependent matrix metalloproteinase-9 expression. Mol Med Rep. (2015) 11:1139–45. doi: 10.3892/mmr.2014.2872
94. Gao H, Wang J. Andrographolide inhibits multiple myeloma cells by inhibiting the TLR4/NF-κB signaling pathway. Mol Med Rep. (2016) 13:1827–32. doi: 10.3892/mmr.2015.4703
95. Su M, Qin B, Liu F, Chen Y, Zhang R. Andrographolide enhanced 5-fluorouracil-induced antitumor effect in colorectal cancer via inhibition of c-MET pathway. Drug Des Devel Ther. (2017) 11:3333–41. doi: 10.2147/DDDT.S140354
96. Zhang D, Qiu L, Jin X, Guo Z, Guo C. Nuclear factor-kappaB inhibition by parthenolide potentiates the efficacy of Taxol in non-small cell lung cancer in vitro and in vivo. Mol Cancer Res. (2009) 7:1139–49. doi: 10.1158/1541-7786.MCR-08-0410
97. Sohma I, Fujiwara Y, Sugita Y, Yoshioka A, Shirakawa M, Moon JH, et al. Parthenolide, an NF-κB inhibitor, suppresses tumor growth and enhances response to chemotherapy in gastric cancer. Cancer Genomics Proteomics. (2011) 8:39–47.
98. Liu JW, Cai MX, Xin Y, Wu QS, Ma J, Yang P, et al. Parthenolide induces proliferation inhibition and apoptosis of pancreatic cancer cells in vitro. J Exp Clin Cancer Res. (2010) 29:108. doi: 10.1186/1756-9966-29-108
99. Kim SL, Trang KTT, Kim SH, Kim IH, Lee SO, Lee ST, et al. Parthenolide suppresses tumor growth in a xenograft model of colorectal cancer cells by inducing mitochondrial dysfunction and apoptosis. Int J Oncol. (2012) 41:1547–53. doi: 10.3892/ijo.2012.1587
100. Franks ME, Macpherson GR, Figg WD. Thalidomide. Lancet. (2004) 363:1802–11. doi: 10.1016/S0140-6736(04)16308-3
101. Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. (1999) 341:1565–71. doi: 10.1056/NEJM199911183412102
102. Palumbo A, Facon T, Sonneveld P, Bladè J, Offidani M, Gay F, et al. Thalidomide for treatment of multiple myeloma: 10 years later. Blood. (2008) 111:3968–77. doi: 10.1182/blood-2007-10-117457
103. Dahut WL, Gulley JL, Arlen PM, Liu Y, Fedenko KM, Steinberg SM, et al. Randomized phase II trial of docetaxel plus thalidomide in androgen-independent prostate cancer. J Clin Oncol. (2004) 22:2532–9. doi: 10.1200/JCO.2004.05.074
104. Figg WD, Dahut W, Duray P, Hamilton M, Tompkins A, Steinberg SM, et al. A randomized Phase II trial of thalidomide, an angiogenesis inhibitor, in patients with androgen-independent prostate cancer. Clin Cancer Res. (2001) 7:1888–93.
105. Lin AY, Brophy N, Fisher GA, So S, Biggs C, Yock TI, et al. Phase II study of thalidomide in patients with unresectable hepatocellular carcinoma. Cancer. (2005) 103:119–25. doi: 10.1002/cncr.20732
106. Lee SM, Rudd R, Woll PJ, Ottensmeier C, Gilligan D, Price A, et al. Randomized double-blind placebo-controlled trial of thalidomide in combination with gemcitabine and Carboplatin in advanced non-small-cell lung cancer. J Clin Oncol. (2009) 27:5248–4. doi: 10.1200/JCO.2009.21.9733
107. Ott PA, Chang JL, Oratz R, Jones A, Farrell K, Muggia F, et al. Phase II trial of dacarbazine and thalidomide for the treatment of metastatic melanoma. Chemotherapy. (2009) 55:221–7. doi: 10.1159/000219435
108. Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inh. J Pharmacol Exp Ther. (2007) 321:509–16. doi: 10.1124/jpet.106.111344
109. Randle JC, Harding MW, Ku G, Schönharting M, Kurrle R. ICE/Caspase-1 inhibitors as novel anti-inflammatory drugs. Expert Opin Investig Drugs. (2001) 10:1207–9. doi: 10.1517/13543784.10.7.1207
110. Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B: functional relationships established for key mediators of apoptosis. J Biol Chem. (1997) 272:17907–11. doi: 10.1074/jbc.272.29.17907
111. Talanian R V, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, et al. Substrate specificities of caspase family proteases. J Biol Chem. (1997) 272:9677–82. doi: 10.1074/jbc.272.15.9677
112. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. (2015) 21:248–57. doi: 10.1038/nm.3806
113. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
114. He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. (2018) 9:2550. doi: 10.1038/s41467-018-04947-6
115. Xia R, Chen SX, Qin Q, Chen Y, Zhang WW, Zhu RR, et al. Oridonin suppresses proliferation of human ovarian cancer cells via blockage of mTOR signaling. Asian Pacific J Cancer Prev. (2016) 17:667–71. doi: 10.7314/APJCP.2016.17.2.667
116. Song M, Liu X, Liu K, Zhao R, Huang H, Shi Y, et al. Targeting AKT with oridonin inhibits growth of esophageal squamous cell carcinoma in vitro and patient-derived xenografts in vivo. Mol Cancer Ther. (2018) 17:1540–53. doi: 10.1158/1535-7163.MCT-17-0823
117. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. (2017) 214:3219–38. doi: 10.1084/jem.20171419
118. Jansen TL, Klück V, Janssen M, Comarniceanu A, Efdé M, Scribner CL, et al. P160 The first phase 2A proof-of-concept study of a selective NLRP3 inflammasome inhibitor, dapansutrileTM (OLT1177TM), in acute gout. Ann Rheum Dis. (2019) 78(Suppl 1):A70–A1. doi: 10.1136/annrheumdis-2018-EWRR2019.142
119. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U S A. (2018) 115:E1530–E9. doi: 10.1073/pnas.1716095115
120. Darakhshan S, Pour AB. Tranilast: a review of its therapeutic applications. Pharmacol Res. (2015) 91:15–28. doi: 10.1016/j.phrs.2014.10.009
121. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. (2018) 10:e8689. doi: 10.15252/emmm.201708689
122. Dinarello CA. The role of the interleukin-1-receptor antagonist in blocking inflammation mediated by interleukin-1. N Engl J Med. (2000) 343:732–4. doi: 10.1056/NEJM200009073431011
123. Furst DE. Anakinra: review of recombinant human interleukin-I receptor antagonist in the treatment of rheumatoid arthritis. Clin Ther. (2004) 26:1960–75. doi: 10.1016/j.clinthera.2004.12.019
125. D'Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. (1994) 91:4082–5. doi: 10.1073/pnas.91.9.4082
126. Wu JJ, Huang DB, Pang KR, Hsu S, Tyring SK. Thalidomide: dermatological indications, mechanisms of action and side-effects. Br J Dermatol. (2005) 153:254–73. doi: 10.1111/j.1365-2133.2005.06747.x
127. Keller M, Sollberger G, Beer H-D. Thalidomide inhibits activation of caspase-1. J Immunol. (2009) 183:5593–9. doi: 10.4049/jimmunol.0900476
128. Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu J-W, et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem. (2010) 285:9792–802. doi: 10.1074/jbc.M109.082305
129. Xu S, Li X, Liu Y, Xia Y, Chang R, Zhang C. Inflammasome inhibitors: promising therapeutic approaches against cancer. J Hematol Oncol. (2019) 12:64. doi: 10.1186/s13045-019-0755-0
130. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. (2019) 10:128. doi: 10.1038/s41419-019-1413-8
131. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological inhibitors of the nlrp3 inflammasome. Front Immunol. (2019) 10:2538. doi: 10.3389/fimmu.2019.02538
132. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med. (2018) 10:eaah4066 doi: 10.1126/scitranslmed.aah4066
Keywords: NLRP3, inflammasome, cancer, therapeutic targets, activation
Citation: Hamarsheh S and Zeiser R (2020) NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front. Immunol. 11:1444. doi: 10.3389/fimmu.2020.01444
Received: 26 February 2020; Accepted: 03 June 2020;
Published: 08 July 2020.
Edited by:
Davide Ferrari, University of Ferrara, ItalyReviewed by:
Angela Bonura, National Research Council (CNR), ItalyCopyright © 2020 Hamarsheh and Zeiser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Zeiser, cm9iZXJ0LnplaXNlckB1bmlrbGluaWstZnJlaWJ1cmcuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.