 Alessandra Tammaro
Alessandra Tammaro Jesper Kers
Jesper Kers Angelique M. L. Scantlebery1
Angelique M. L. Scantlebery1- 1Department of Pathology, Amsterdam UMC, Amsterdam Infection & Immunity Institute, University of Amsterdam, Amsterdam, Netherlands
- 2Department of Pathology, Leiden University Medical Center, Leiden, Netherlands
- 3Biomolecular Systems Analytics, Van ‘t Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, Amsterdam, Netherlands
Renal ischemia reperfusion injury (IRI), a common event after renal transplantation, causes acute kidney injury (AKI), increases the risk of delayed graft function (DGF), primes the donor kidney for rejection, and contributes to the long-term risk of graft loss. In the last decade, epidemiological studies have linked even mild episodes of AKI to chronic kidney disease (CKD) progression, and innate immunity seems to play a crucial role. The ischemic insult triggers an acute inflammatory reaction that is elicited by Pattern Recognition Receptors (PRRs), expressed on both infiltrating immune cells as well as tubular epithelial cells (TECs). Among the PRRs, Toll-like receptors (TLRs), their synergistic receptors, Nod-like receptors (NLRs), and the inflammasomes, play a pivotal role in shaping inflammation and TEC repair, in response to renal IRI. These receptors represent promising targets to modulate the extent of inflammation, but also function as gatekeepers of tissue repair, protecting against AKI-to-CKD progression. Despite the important considerations on timely use of therapeutics, in the context of IRI, treatment options are limited by a lack of understanding of the intra- and intercellular mechanisms associated with the activation of innate immune receptors and their impact on adaptive tubular repair. Accumulating evidence suggests that TEC-associated innate immunity shapes the tubular response to stress through the regulation of immunometabolism. Engagement of innate immune receptors provides TECs with the metabolic flexibility necessary for their plasticity during injury and repair. This could significantly affect pathogenic processes within TECs, such as cell death, mitochondrial damage, senescence, and pro-fibrotic cytokine secretion, well-known to exacerbate inflammation and fibrosis. This article provides an overview of the past 5 years of research on the role of innate immunity in experimental and human IRI, with a focus on the cascade of events activated by hypoxic damage in TECs: from programmed cell death (PCD) and mitochondrial dysfunction-mediated metabolic rewiring of TECs to maladaptive repair and progression to fibrosis. Finally, we will discuss the important crosstalk between metabolism and innate immunity observed in TECs and their therapeutic potential in both experimental and clinical research.
Introduction
Kidney diseases are a growing health problem, considered to have a direct and indirect impact on morbidity and mortality worldwide, by increasing the risks associated with the major killers, such as cardiovascular diseases, diabetes, hypertension, and infection (1). The timely identification and management of kidney diseases represent the most effective strategy to sustainably address the growing global burden and prevent the progression to end-stage renal disease (ESRD).
In 2010, 2.62 million people received dialysis worldwide, and the need for dialysis was projected to double by 2030. Despite being a life-saving treatment, dialysis is merely a supportive measure. Indeed, the life expectancy and the quality of life in dialysis patients is much lower compared to the general population (2). Basic research studies have identified many targets to delay the progression of kidney disease; however, only few of these promising results can be recapitulated in clinical studies, and yet do not represent a great alternative to dialysis or transplantation.
The life-sustaining job of the kidneys comprises filtering and reabsorbing about 180 liters of fluid from the bloodstream every 24 h. As a consequence, it is the organ with the highest metabolic rate as determined by tubular epithelial cell (TEC) metabolism (3). TECs, the most abundant cell type in the kidney, are densely packed with mitochondria. These cells combust fatty acids to generate adenosine triphosphate (ATP) through oxidative phosphorylation (4). Proper mitochondrial function and metabolism of these cells is crucial for the high transport and reabsorption activities. Given the high metabolic demand, the kidneys are sensitive to decreased blood oxygenation and perfusion. Whenever the kidneys experience extremely low oxygen exposure, or metabolic substrates become inadequate, acute kidney injury (AKI) occurs.
AKI is defined as an abrupt reduction in kidney function that results in disturbances in the milieu intérieur and the retention of uremic toxins that especially influence the cardiovascular, immune, and nervous system. With an incidence of 9–15% of hospital admissions and up to 40% in critically ill patients, AKI is a major cause of morbidity, mortality (40–70% for the critically ill needing dialysis), increased health-care costs, and chronic kidney disease (CKD) (5, 6). Although research has made great efforts to understand the pathogenesis of AKI, the only available therapy is still supportive (dialysis) and has not changed for decades. Additionally, AKI patients have a greater risk to develop chronic complications later in life (7). A systematic review and meta-analysis comprising estimates from more than 2,000,000 individuals identified AKI as a risk factor for new or progressive CKD (hazard ratio [HR] = 2.67), ESRD (HR = 4.81), and mortality (HR = 1.80) (7). These important clinical findings have resulted in a shift toward studies that investigate the link between experimental AKI and progressive kidney fibrosis and failure. Pattern recognition receptors (PRRs), the sensors of the innate immune system, are one of the major players determining short-term outcomes after experimental AKI, a topic of many good reviews (8, 9). In this review, we will discuss recent data showing that danger signals and PRRs are involved in cell fate decisions, metabolism, and mitochondrial function in TECs, thereby determining not only whether a pro- or anti-inflammatory phenotype will emerge, but also the success of regeneration and repair. We specifically intend to highlight the link between metabolism and innate immunity in TECs and the altered cell phenotype that occurs during experimental and human ischemia reperfusion injury (IRI)-induced AKI.
AKI and Experimental Ischemia-Reperfusion Injury
The pathophysiology of AKI is complex. One of the major causes of AKI is IRI (10, 11). IRI is an inevitable event during renal transplantation and is responsible for delayed graft function (DGF), resulting in loss of vital kidney parenchyma and priming of adaptive immune responses that initiate rejection, which altogether lead to graft loss (10, 12). DGF is a clinical syndrome defined as the need for renal replacement therapy in the early phase after transplantation in order to support the function of the newly acquired renal transplant (12). Besides renal transplantation, IRI can also develop in the context of other diseases with low perfusion and/or oxygenation states, including thrombotic diseases, sepsis, trauma, and cardiac surgery (13). Experimental IRI in the mouse is the most widely used preclinical model to mimic human AKI. Despite having many limitations, particularly regarding the immune response (14) it is still the most valuable tool for understanding AKI pathophysiology.
The Early Phase of Experimental IRI Is Characterized by Programmed Cell Death That Activates a Pro-inflammatory Innate Immune Response
In experimental IRI, following the hypoxic event, TECs, especially the proximal TECs located in the S3 segment of the nephron at the cortico-medullary area, are unable to maintain adequate intracellular ATP levels for the essential processes mentioned earlier. Additionally, restoration of blood perfusion, followed by re-oxygenation of the kidney, provokes the production of reactive oxygen species (ROS), eliciting mitochondrial dysfunction. This, together with the ATP-depletion, leads to cell activation and injury, and if severe enough, can lead to programmed cell death (PCD) and secondary necrosis, the hallmark of early IRI (15).
PCD is essential, not only for the maintenance of cellular homeostasis but also in response to irreparable damage caused by injury or disease (15–17). The pathways of cell death execution can present as a diverse morphological pallet in the spectrum from apoptotic to necrotic, with a corresponding degree of danger-associated molecular pattern (DAMP) release and inflammatory potential (18–20). DAMPs are a heterogeneous group of ligands, which are constitutively expressed in different biological compartments of cells hidden from the innate immune system. DAMPs are recognized by a broad spectrum of PRRs, in order to elicit an innate inflammatory response (19).
It has been observed that the severity of the injury determines the path by which cell death is realized, indicating a possible role for PRRs in guiding cell fate decisions by integrating signals from the microenvironment (21). Various currently recognized forms of PCD (18, 22), including necroptosis, pyroptosis, ferroptosis, mitochondrial permeabilization transition (MPT)-mediated regulated necrosis, and parthanatos are initiated as a response to hypoxic injury, either directly or indirectly, and blocking their crucial pathways during the early phase of IRI generally leads to reduced necrotic (tubular) damage, reduced inflammation, preservation of renal function, and reduced mortality (15). PRRs are known inducers of PCD, particularly necroptosis, pyroptosis, and apoptosis, which is why these modalities are discussed in more detail below.
Pattern Recognition Receptors Are Involved in Cell Death Signaling During IRI
In necroptosis, receptor-interacting protein (RIP) kinase-1 and 3 interaction via their RIP–homotypic interacting motif (RHIM) (23) leads to phosphorylation and activation of RIPK3. In turn, RIPK3 catalyzes the phosphorylation and oligomerization of mixed lineage kinase domain–like (MLKL) (24), thereby inducing a molecular switch that leads to plasma membrane rupture and cell death (23–26). Blocking components of the necroptosis pathway in a similar renal phenotype of reduced tubular necrosis, reduced inflammation, better preservation of renal function, and reduced mortality in the first period after reperfusion (27). A recent report by Chen and colleagues showed, in a chimeric bone marrow transplantation model, that in the early phase of IRI, RIPK3 and MLKL in kidney parenchymal cells (including proximal TECs, as shown by in vitro studies) are important for initiation of the vicious inflammatory circle, but that pyroptosis in macrophages is more important in the later stage after reperfusion, suggesting temporal variation in cell death modalities during the course of IRI (28). Pyroptosis is a necrotic form of cell death most often observed in immune cells, such as macrophages and dendritic cells (DCs) (18). During pyroptosis, the presence of DAMPs initiates inflammasome formation, which activates both caspase-1 and caspase-11 (29–31). An effector function of these caspases is to process the inactive precursors of IL-18 and IL-1beta, leading to an intracellular accumulation of pro-inflammatory cytokines (31). These caspases also induce plasma membrane rupture, and essentially cell death, through the cleavage of gasdermin D (GSDMD) (32). The inevitable release of IL-18 and IL-1beta makes this form of cell death highly inflammatory (33). There is some debate as to whether pyroptosis occurs in renal cells as well, however, Yang et al. suggest the occurrence of pyroptosis in TECs based on a significant increase in pyroptosis-related proteins following IRI (34). A recent report by Miao et al. suggests the direct involvement of pyroptosis in IRI and cisplatin toxicity based on Casp11 KO mice (35). In additional experiments they showed that in Gsdmd KO mice, renal tubular damage was less severe, and urinary IL-18 levels were reduced upon cisplatin toxicity (35). Although very suggestive, we do not know whether Gsdmd KO mice have the same phenotype in IRI compared to cisplatin toxicitiy in vivo. Apoptosis is considered a more quiescent form of regulated cell death due to the swift clearance of apoptotic bodies by phagocytes. Apoptosis is initiated through an intrinsic route via mitochondrial outer membrane permeabilization (MOMP) or an extrinsic route via death or dependence receptors (36, 37). Death receptors can initiate apoptosis via cognate ligand binding (e.g., FAS or TNFR1 signaling) whereas dependence receptors initiate apoptosis when there is a lack of ligand binding (i.e., reduced homeostatic survival signaling via e.g., the netrin 1 receptors) (38). After initiation, executioner caspases-3 and−7 are responsible for neat cellular and nuclear fragmentation during apoptosis, releasing “find-me” signals, and flagging apoptotic bodies to be phagocytosed via “eat-me” signals (39). Apoptosis is believed to play a minor role in the early pro-inflammatory phase after reperfusion. Multiple reports have shown that pharmacologically inhibited apoptosis by a pan-caspase inhibitor zVAD or genetic KO of executioner caspase-3 did not reduce but rather exaggerated renal tubular damage and failure (40, 41). In the long-term, Casp3 KO mice appeared to have less peritubular capillary rarefaction, less activated interstitial fibroblasts, less interstitial fibrosis, and evidence of less tubular hypoxia after reperfusion, suggesting a potentially interesting link between late peritubular capillary apoptosis and endothelial-mesenchymal transition and/or pericyte-fibroblast transdifferentiation (41). PRRs can initiate regulated cell death in multiple ways. Toll-like receptor (TLR) signaling via MYD88 results in activation of NFkB, transcriptionally regulating multiple cytokines that can subsequently induce regulated cell death via para- and autocrine signaling to death receptors. However, a more direct route of cell death initiation by TLRs is via Toll/IL-1R domain-containing adaptor-inducing interferon (IFN)-beta (TRIF). TRIF can initiate apoptosis via FADD- and caspase-8-dependent pathways. TRIF also contains a RHIM domain, and could therefore function as a docking site for the RIPK3-MLKL complex during necroptosis initiation (42), as was shown for TLR3 (43). TLR-TRIF-induced active caspase-8 was able to cleave Gasdermin D in macrophages inducing pyroptosis (44), suggesting the bypassing of the inflammasome in these cells.
(mal)Adaptive Repair Responses as a Model for AKI-to-CKD Progression
Tubular regeneration and successful renal repair after an episode of AKI can be observed in the majority of surviving patients, especially in cases of mild injury (45–47). Adaptive tubular repair depends on the presence of an appropriate microenvironment, in which inflammation and tubular response to damage are balanced. In the adaptive repair, surviving TECs undergo dedifferentiation and proliferation in order to restore a functional epithelium. However, in case of severe or repetitive injuries or aged kidneys, maladaptive repair of proximal tubules can occur, which can contribute to progressive renal fibrosis (47). Maladaptive repair of kidney tissue after AKI is characterized by rarefaction of peritubular capillaries, interstitial fibrosis and tubular atrophy, glomerulosclerosis, and vascular remodeling, which interfere with repair and eventually lead to a decline in renal function. Therefore, AKI-to-CKD should be regarded as accelerated renal aging (47, 48). As the determinants of renal aging and CKD overlap (49), identifying patients with premature renal aging could be a strategy to identify AKI survivors at risk for CKD.
Among the culprits in the AKI-to-CKD progression, is the persistence of a senescent state in TECs (45). Senescence describes a proliferative arrest with changes in chromatin organization, gene transcription, and protein secretion, which can occur as a response to cell stress and aging (50). Given the high degree of pro-inflammatory molecules released by senescent TECs, it remains elusive whether progressive accumulation of senescent TECs is causally related to an aberrant innate immune response. Recent results from our group and others (51–53) point toward a role for TEC-mediated inflammation, innate immunity, and mitochondrial metabolism in senescence and fibrosis. Given that these mechanisms fall into the new discipline of immunometabolism, and TECs can be regarded as part of the renal innate immune system, further studies are required to characterize the role of immunometabolism in AKI-to-CKD progression.
Innate Immune Receptors as Gatekeeper of Damage and Repair
The role of the innate immune system was originally to combat infections. However, we now know that its role extends beyond that to include the surveillance of tissue homeostasis, by detecting distinct DAMPs released during tissue injury. Our group pioneered the discovery of TEC-associated innate immune sensors having a crucial role in the initiation of the injury response during IRI, shedding a novel light on the role of TECs as innate immune cells of the kidney (54). Experimental data suggests that the DAMPs released by necrotic cells activate the inflammatory signal initiated through TLRs, and their synergistic receptors, the Nod-like receptors (NLRs) and the NLRP3 inflammasome (55, 56). Initial considerations suggested that pharmaceutical intervention to block TEC-induced innate immune cell signaling could lead to novel therapeutics against renal tissue inflammation and injury after IRI (54, 57). However, in the last decade, seminal studies on the role of innate immune sensors and their ligands in renal IRI have provided an additional prospective: the innate immune sensors translate kidney injury into an immune response, essential for shaping adaptive tubular repair and kidney regeneration. Thus, the tubular innate immune response to IRI seems to be a very well-orchestrated phenomenon (58). After the early inflammatory phase, macrophage populations assume a reparative phenotype that is characterized by the production of numerous growth factors, including Platelet-derived growth factor (PDGF), Transforming growth factor beta 1 (TGF-beta1), Insulin-like Growth Factor I (IGF-1), and Vascular endothelial growth factor A (VEGF-A) that promote tubular regeneration (59–61). Long-term sustained inflammation is detrimental, but the absence of the inflammatory response can also predispose to the development and progression of CKD (8). Whether this is dependent on a faulty inflammatory response or the inability of TECs to regenerate due to extensive damage is still not completely understood. Improving our understanding of the role of innate immune receptors, not only in the early pro-inflammatory phase during IRI, but also in (mal)adaptive tubular repair, is therefore crucial for the development of specific therapies or prevention of AKI and its detrimental sequelae.
Mitochondrial Dysfunction and Metabolic Reprogramming
Mitochondrial injury, fragmentation, and ROS generation induce aberrant tubular inflammation and are central mediators of AKI, as well as the AKI-to-CKD progression (62–66). Oxygen tension in TECs is crucial for proper mitochondrial function. When this becomes inadequate, mitochondrial respiration is inhibited and the kidney undergoes a metabolic rewiring toward glycolysis, thereby decreasing ATP production (67). Mitochondrial number and integrity (through fission and fusion processes) (65, 66) are associated with accelerated tubular repair and improved survival after IRI, suggesting that in order to achieve adaptive tubular repair, restoration of mitochondrial homeostasis is an indispensable event (64). Thus, metabolic flexibility seems to be a crucial ability of the tubular epithelium to quickly adapt to the hypoxic environment. However, in order to promote repair, TECs should be able to return to their primary fuel source, and when this flexibility is impaired and metabolic rewiring persists during repair, this leads to a failed re-differentiation and mesenchymal arrest (68).
Hypoxia is likely not the only cause of reduced oxygen availability in the kidney. The widening of interstitial spaces by edema and inflammation and the regression of capillaries during fibrosis could also be involved. Indeed, pathologic hypoxia persists as fibrosis develops and could thus prevent epithelial recovery through feedback effects, ultimately leading to tubular atrophy (69). Consequent to hypoxia, tubular repair after AKI could be impaired by oxidative stress and growth arrest or senescence, which represents the known adverse effects of hypoxia (51, 52, 70, 71). TECs rely mostly on fatty acid oxidation (FAO) for their functions (72), which requires mitochondria and oxygen for efficient ATP generation. All the enzymes required for FAO reside in the mitochondrial matrix, and proper functioning of the mitochondrial cristae is necessary to provide substrates for the respiratory complex. Recently, several groups have reported that mitochondrial function and energy metabolism are involved in the progression from AKI-to-CKD.
Within mitochondria, the coenzyme Nicotinamide adenine dinucleotide (NAD+) carries high-energy electrons from FAO to the electron transport chain. NAD+ is, therefore, a rate-limiting catalyst for FAO (67). In other terms, decreased NAD+ availability results in impaired energy metabolism in these cells (73). Hypoxia and aging (74, 75) have been known to induce NAD+-consuming enzymes, which lowers NAD+ availability. Tran and colleagues recently showed that renal tubular cell NAD+ levels are suppressed in IRI-induced AKI, and this reduction in NAD+ may impede FAO, reduce ATP generation, and elevate susceptibility to AKI stressors (76). They identified a novel function of the mitochondrial biogenesis regulator PPAR-gamma-coactivator-1alpha (PGC1-alpha) to induce the enzymes that sequentially convert the amino acid tryptophan to NAD+, after IRI. Tubular PGC1-alpha expression protects against hypoxia-related stress and enhancing NAD+ could effectively mimic PGC1-alpha's effects in the tubule (76). Interestingly, biopsies of human AKI showed reduced PGC1-alpha expression (77).
The same group, through a metabolomics study, has demonstrated the elevation of urinary quinolinic acid (uQuin) in murine AKI. Quin becomes NAD+ through the action of quinolinate phosphoribosyltransferase (QPRT) and subsequent enzymes. The elevation of uQuin suggested suppression of QPRT during AKI. Genetic targeting of Qprt showed enhanced susceptibility to IRI and recapitulated the majority of the urinary metabolic changes measured in experimental AKI (78). Additionally, few other studies described that either de novo NAD+ biosynthetic pathway activation (79) or replenishment by means of NAD+ precursors (80) are able to protect the kidney from ischemic damage. This is of great importance given that in mammals, only the kidney and the liver exhibit appreciable de novo NAD+ biosynthesis (79, 81). Taken together, these encouraging studies points toward a therapeutic potential of NAD+ enhancers in AKI and its long-term sequelae.
Apart from PGC1-alpha, the AMP-activated protein kinase (AMPK) is a promising component of a signaling cascade that may modulate the severity of ischemic injury (82). AMPK is a ubiquitously expressed serine-threonine kinase that serves as an important intracellular energy sensor. It is activated by conditions that deplete ATP and alter the AMP:ATP ratio, including ischemia and glucose deprivation. AMPK stimulates FAO, glucose uptake, and glycolysis, while downregulating ATP-utilizing systems. Other targets of AMPK include pathways modulating inflammation, apoptosis, angiogenesis, blood flow, and maintenance of cell polarity, with or without energetic stress (83). Metformin, a widely used drug for the treatment of type II diabetes mellitus, enhances AMPK activity (84). AMPK pre-activation partially ameliorates renal IRI in vivo but also long-term sequelae after IRI (84).
Defective FAO in TECs plays a pivotal role in renal aging (85) and fibrosis (72), but studies directly linking defective FAO to renal IRI are currently missing. The Susztak group showed that the dramatic repression of FAO is induced by TGF-beta signaling (72), which seems to play a role in the early events of renal IRI, therefore, it would be interesting to investigate whether failure to fully return to FAO fuel after IRI might be underlying the AKI-to-CKD transition, or whether defective FAO is associated with a senescent tubular phenotype. Although fatty acid accumulation has been shown to be associated with lipid deposition, this is not per se enough to drive fibrosis.
Interestingly, TGF-beta1 stimulates the Warburg-like metabolic reprogramming in kidney cells, which is relevant because it mirrors the metabolic state during AKI (72). Metabolic reprogramming, toward glycolysis, rapidly generates ATP and involves the enzyme Pyruvate Kinase M2 (PKM2), which is involved in the last step of glycolysis (86). Zhou and colleagues found that disabling PKM2 resulted in a significant increase in cell-repair and a concomitant decrease in energy generation, leading to significant protection against kidney injury in mice (87). A key molecule in this process is nitric oxide (NO), which can be transported to different proteins through Co-enzyme A, thereby, switching off their activity. PKM2 is one such protein. Indeed, adding NO to PKM2 activates repair, suggesting it as an important mechanism that can be used to determine whether kidney cells are using their pathways for energy or repair. The same team found that a protein called AKR1A1 could remove NO from PKM2, thereby switching it on and re-activating a robust energy-generating process. Disabling AKR1A1 protected the kidney from disease by stimulating repair (87).
It has been well-established that mitochondrial dysfunction is a principal mediator of AKI through decreased ATP production, oxidative stress, mitochondrial DNA (mtDNA) release, and cell death. Mitochondria also play a crucial role in maintaining organelle function in cells. Organelle stress and crosstalk in the AKI-to-CKD transition has been recently reviewed (64). The ROS generated upon re-oxygenation and the inability to maintain endogenous antioxidant levels, results in mitochondrial oxidative stress and promotes AKI (88). Enhancement of the antioxidant defense via mitochondria-targeted approaches has been successful in ameliorating IRI-induced AKI. These have been summarized in Table 1. Additionally, a visual representation of the phenotypic changes occurring in TECs after IRI and the majority of the mechanisms described in this chapter can be found in Figures 1, 2.
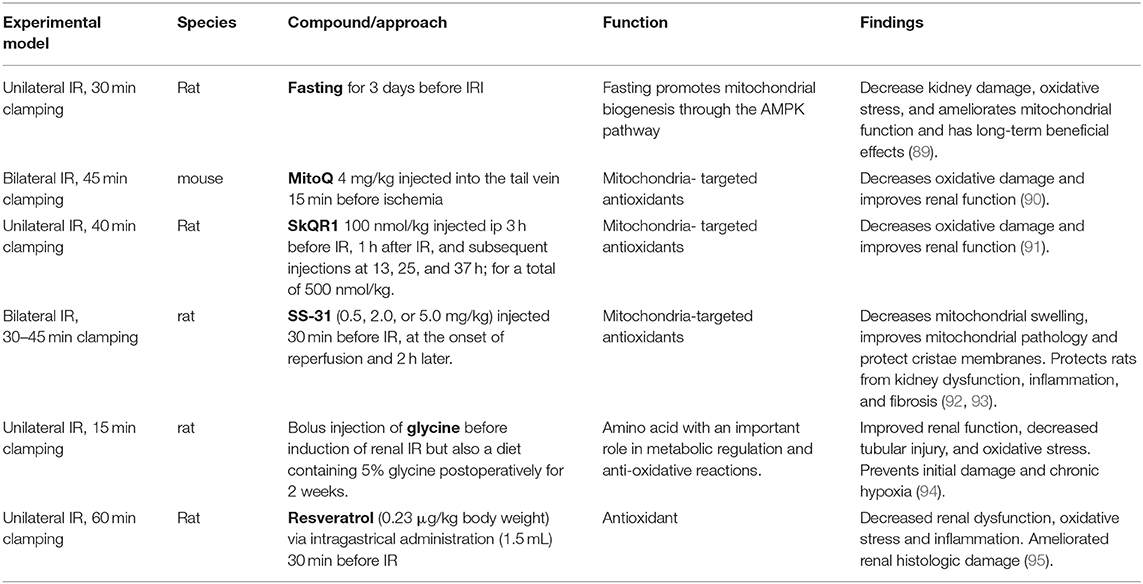
Table 1. Mitochondria-targeted approaches in renal IRI.
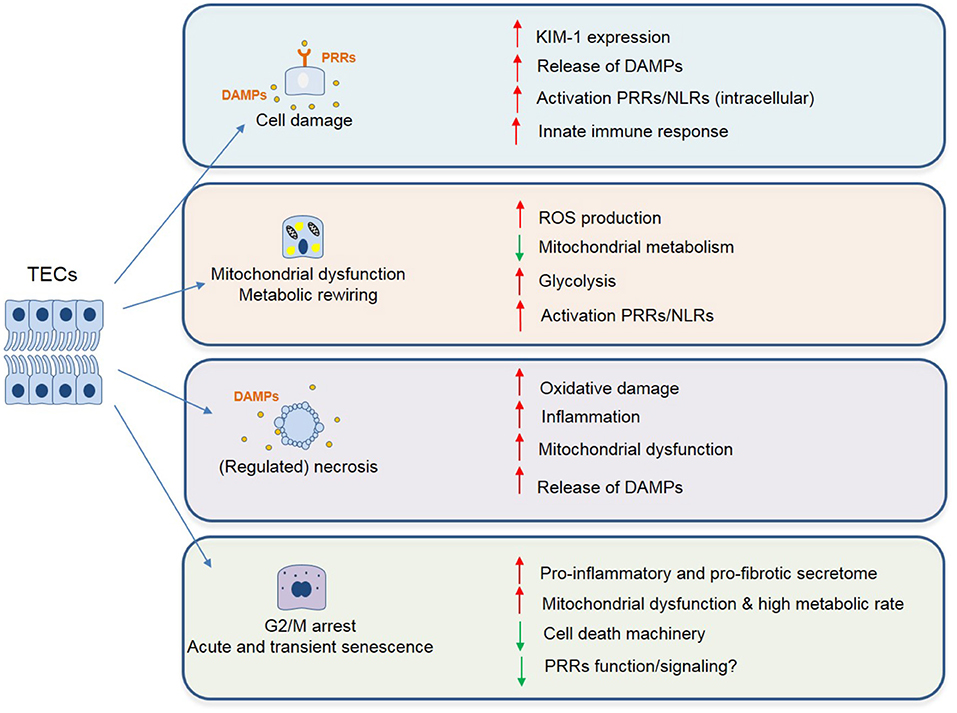
Figure 1. Phenotypic changes described in TECs following IRI associated with maladaptive tubular repair and progressive renal interstitial fibrosis. These changes include but are not limited to: cell damage (chronic inflammation with persistent cytokine production and immune cell infiltrate), mitochondrial dysfunction and cell death (enhanced ROS signaling, metabolic reprogramming, and release of mitochondrial DNA/ROS acting as danger molecules, ultimately leading to cell death). Lastly, as a result of incomplete repair or severe damage, TECs can undergo transient cell cycle arrest, as a protective mechanism to ensure genome stability. However, if persistent, this leads to a pro-inflammatory and profibrotic secretome, ultimately leading to fibrosis.
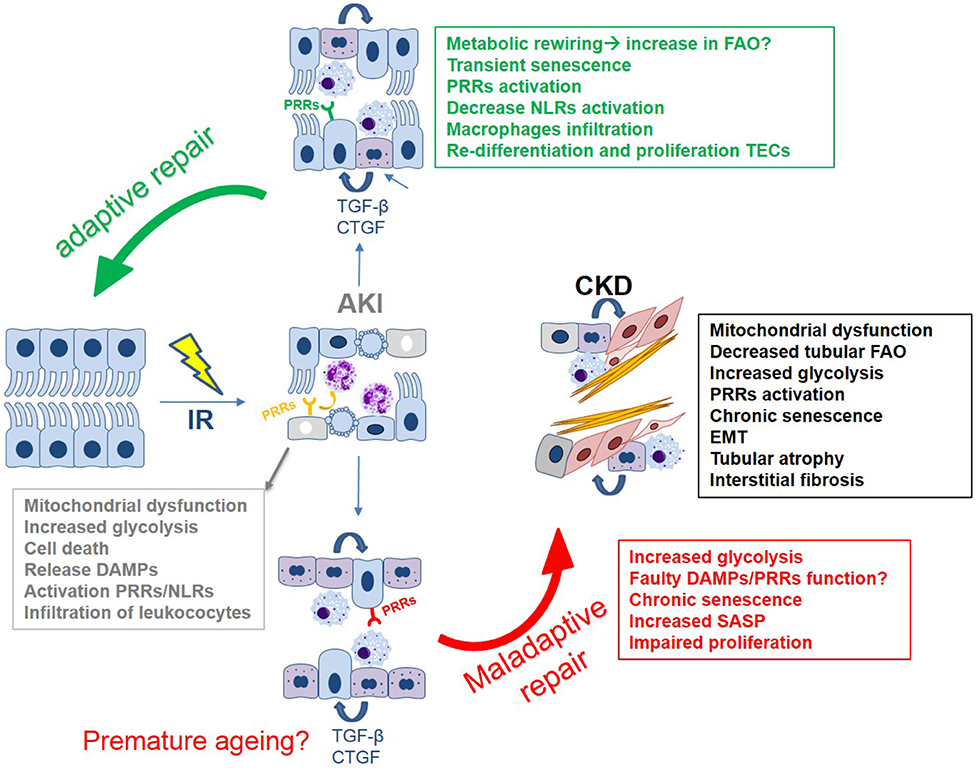
Figure 2. The tubular epithelial response to IR: a simplified overview of the main mechanisms driving (mal)adaptive repair leading to kidney fibrosis. Key mechanisms include: epithelial innate immune response, cell death, mitochondrial dysfunction, metabolic reprogramming, and cell cycle arrest/cellular senescence.
Senescence-Mediated (Mal)adaptive Repair
Kidney regeneration after IRI is accomplished by active tissue repair, in which the role of macrophage phenotype is crucial, as described in recent work (96). A balanced inflammatory response, together with a moderate level of damage, establishes an optimal microenvironment in which surviving TECs fully repair (3, 97). In this scenario of adaptive repair, the kidney regenerates over the course of a few days without scarring. The progression of AKI-to-CKD is the result of maladaptive tubular repair, which can be mediated by TEC senescence.
The hypoxic injury activates a DNA damage response (DDR) in TECs [Ataxia-Telangiectasia, Mutated(ATM)/ATM and Rad3-related(ATR) pathway] which results in cell cycle arrest (71, 98–100). This allows TECs to repair the damage and avoid further amplification of the vicious cycle of injury-induced cell death. ATR, an enzyme involved in DDR activation has been shown to protect against maladaptive tubular repair (100), further demonstrating a crucial role for DDR activation in TEC repair.
One of the consequences of irreparable DNA damage and mitochondrial dysfunction, especially in cells with a high energy demand, is a proliferation arrest termed “senescence” (101). Senescence is generally regarded as an irreversible event (102); however, there are reports that describe senescent cells being able to re-enter the cell cycle, suggesting that more research is needed to understand which scenario determines the fate of these cells (103). Senescent TECs, identified by different markers, with Senescence-associated beta-galactosidase (SA-β-gal) and the anti-proliferative proteins p16 and p21 being the most common, seem to accumulate acutely following experimental and human IRI (53, 104), as a consequence of oxidative stress, the so-called stress-induced senescence. In vivo studies using the renal IRI model showed that elimination of these cells might hinder regeneration (104, 105). Indeed, senescence also plays a key role in the healing of wounds, tissue repair, and during embryonic development (106). Acute and transient senescence, where damaged cells are eliminated, clearly has beneficial effects for an organism and in the adaptive tubular repair after IRI. Paradoxically, subtle accumulation of senescent cells (chronic senescence), instead, impairs the kidney's regenerative capacity, leading to AKI-to-CKD transition (52, 107, 108).
In this scenario, the use of senolytics (aimed at clearing senescent cells) seems a promising therapeutic strategy (49). However, given the non-specificity of current senescence markers and the existence of different senescence programs, elegantly reviewed by the De Maria group, more research is needed for this novel potential therapeutic field (109). The intervention strategies aimed at senescent cell removal or modulation, which seem to be effective in limiting the progression of IRI, have been reviewed previously (104). Nonetheless, having a specific biomarker for tubular senescence burden could be an effective way to stratify patients that could benefit from the use of senolytics.
Despite being arrested in the cell cycle, senescent cells remain metabolically very active and become fibrogenic due to their innate ability to create a pro-inflammatory environment and secrete pro-fibrotic molecules, components of the senescence-associated secretory phenotype (SASP) (110). Activation of SASP in TECs leads to activation and proliferation of fibroblasts and perivascular pericytes, which in turn induce extracellular matrix production and tubulointerstitial inflammation, with impaired M2 macrophage conversion (47, 71, 96). Perhaps redirecting the metabolism of senescent cells could be an effective way to turn their detrimental phenotype and fate. Recent studies have indeed highlighted the plasticity of these cells, being able to re-enter the cell cycle, posing a new challenge to the postulated theory, that cells can instead be re-programmed to change their cell fate (111).
In addition to the common components of the SASP, senescent cells produce exacerbated levels of mitochondrial-derived ROS and might also release mtDNA, which can act as a DAMP, further fueling PRR activation (101, 112). Collectively, they amplify the vicious cycle of inflammation, mitochondrial dysfunction, and senescence, ultimately leading to maladaptive tubular repair. Our group has recently described that the innate immune receptor TREM-1 links mitochondrial dysfunction, tubular senescence and maladaptive repair after AKI. This is the first study linking epithelial immunometabolism to cellular senescence in the context of IRI (52).
Epithelial Immunometabolism
TECs possess an incredible plasticity. By tightly controlling their metabolism, they are able to anticipate and adapt to constantly changing environments in both health and disease. Activation of the innate immune system and metabolic reprogramming are tightly linked (113). The theory of immunometabolism has been increasingly appreciated to drive effector functions in immune cells. Despite TECs being regarded as innate immune cells of the kidney, research on epithelial immunometabolism is still in its infancy, particularly in the context of renal IRI (113). In the last 2 years, our group has found that the metabolic choice of TECs is driven by either membrane-bound or intracellular receptors of the innate immune system, proposing the involvement of epithelial immunometabolism in the pathogenesis of IRI and its detrimental consequences (51, 52).
The effect of renal injury on cellular metabolism has been well-established in various AKI models. The induction of AKI with mercuric chloride results in increased glycolytic activity (114), while FAO has been reported to be reduced in folic acid nephropathy (72). The general consensus is that such metabolic shifts are likely necessary to facilitate the initial recovery process; however, their effects may become deleterious when the altered metabolic state persists. Specifically, numerous studies have reported the damaging effects of increased glycolytic activity in TEC repair following IRI. Increased levels of glycolysis and glycolytic enzymes have been observed in TECs that failed to re-differentiate and underwent atrophy (68). As mentioned earlier, Zhou et al. reported the renoprotective effect of PKM2 inhibition by the S-nitroso-CoA reductase system following bilateral IRI. This obstruction of the glycolytic pathway was accompanied by a shift toward the pentose phosphate pathway (PPP), which generates Nicotinamide adenine dinucleotide phosphate (NADPH) and aids in the supplementation of antioxidant reservoirs, thereby offering protection against ROS-induced injury (87). Similarly, Kim et al. report the renoprotective effect of TP53 Induced Glycolysis Regulatory Phosphatase (TIGAR) activation in IRI, which leads to a metabolic shift away from glycolysis and employs the redox protection of the PPP (115). These studies emphasize the need for 'metabolic fluidity' while simultaneously displaying the need for strict metabolic control during IRI. Results from our group and that of others have shown the effect of TEC-specific expression of PRRs during IRI (8), while our recent reports demonstrate the direct involvement of innate immune signaling (via NLRX1 and TREM1) in regulating TEC metabolism following IRI (51, 52). Altogether these data show the inextricable connection between innate immune signaling and cellular metabolism in TECs, which ultimately defines their fate following an acute insult. It is for these reasons that immunometabolism should be recognized as a crucial mechanism through which TECs respond to, and recover from, IRI.
PRRs, the Inflammasome and Their Ligands in Experimental AKI
Toll-Like Receptors
In addition to their function as gatekeeper, protecting against micro-organism invasion, TLRs can be activated by a wide range of DAMPs released upon tissue injury. Upon IRI, tubular necrosis occurs together with the release of potential DAMPs, including High mobility group box 1 (HMGB1), histones, heat shock proteins, S100A8/A9, hyaluronic acid, biglycan, mtDNA, ROS, and ATP, which can function as ligands for PRRs. The type of ligands recognized by a specific TLR is partially dependent on their intracellular localization. One group can be found on the cellular surface and includes TLR2 and TLR4 and the other group, including TLR3, TLR7, TLR8, and TLR9, is expressed in intracellular vesicles such as endosomes, lysosomes, and the endoplasmic reticulum. Upon ligand recognition, the intracellular TIR-domain of a TLR functions as a scaffold for the recruitment of specific adaptor proteins via homotypic interactions between their TIR-domains.
Signaling pathways activated downstream of these adaptor molecules promote the expression of pro-inflammatory cytokines, chemokines, and type I and type III IFNs. Although TLRs provide protection against a wide variety of pathogens, inappropriate or unregulated activation of TLR signaling can lead to chronic inflammatory and autoimmune disorders (116, 117).
TLR2/4
Tlr2 and Tlr4 mRNA is constitutively expressed by TECs and is upregulated upon IRI in mice (118). Both Tlr2 and Tlr4 overexpression in TECs after IRI induced an exaggerated inflammatory response, resulting in impaired renal function (8, 57, 119). Antisense oligonucleotides targeting Tlr2 protected renal tissue and function after IRI in mice. Compared to the single KO mice, double Tlr2 and Tlr4 KO animals showed no additional protective effects (120). Likewise, mice deficient for Myd88 or Trif were not significantly protected against IRI compared to WT animals (119). This might be the consequence of adaptive mechanisms in these genetically modified animals.
Inhibition of TLRs must take place in the acute phase of injury and cannot be longstanding. Indeed, it has been shown that TLR4 blockade during the recovery phase after IRI slows down the process of tubular repair after IRI in rodents (58). In light of these data and in the context of our review, we believe that inhibiting the endogenous ligands, rather than the receptor itself, might be a safer approach.
TLR2/4 Ligands
One such potential ligand is HMGB1. HMGB1 is a nuclear factor that is highly and ubiquitously expressed in nearly all cell types. Upon renal IRI, HMGB1 is overexpressed, and secondary to cell injury, HMGB1 can be leaked into the circulation and activate TLR2, TLR4, and TLR9 (see below). We and others showed that treatment with anti-HMGB1 antibodies reduced renal injury, inflammation, and dysfunction in a murine model of IRI (121, 122). This protective effect of anti-HMGB1 treatment was confirmed in a model of cold and warm renal IRI in miniature swine (123). Another potential agonist for TLR4 is uromodulin. Uromodulin, or Tamm-Horsfall protein, is a highly glycosylated protein, normally secreted by epithelial cells of the thick ascending limb of Henle's loop in the intraluminal compartment. The function of uromodulin remains elusive, but data suggest that uromodulin might prevent the formation of kidney stones and urinary tract infection (124). In vitro, uromodulin has been shown to activate TLR4 signaling in myeloid DCs and bone marrow-derived macrophages (125). Upon kidney injury, uromodulin can leak into the renal interstitial space, potentially leading to the activation of intrarenal resident macrophages and DCs. In contrast with our expectations, uromodulin-deficient mice have markedly fewer resident macrophages. Upon IRI, these uromodulin-deficient mice exhibit aggravated renal injury and impaired polarization of macrophages toward an M2 healing phenotype (126). The discrepancy between in vivo and in vitro data might eventually be due to the different forms of uromodulin, i.e., full-length or truncated, monomeric or aggregating form.
Histones released from dying cells were also found to exacerbate renal tissue injury in AKI in a TLR2/TLR4 dependent manner, and administration of anti-histone antibody suppressed renal inflammation and injury and improved renal function (127).
Among other DAMPs, the calcium binding proteins (S100A8/A9), ligands of TLR4 and the RAGE receptor, are released following renal IRI and seem to play a pivotal role in orchestrating the repair response after hypoxic damage. During the initial injury phase, immune cells, particularly macrophages, are polarized toward an M1-like phenotype and produce pro-inflammatory cytokines, such as TNF-alpha and IL-6, which exacerbate inflammation. However, in a later phase, they switch their phenotype to a more reparative one, the M2-like phenotype, which secretes anti-inflammatory factors, determining the resolution of inflammation and stimulating tubular regeneration (128). Our group showed that S100A8/A9 proteins play a role in this process of macrophage polarization. By controlling excessive M2 polarization, S100A8/A9 fine-tunes the adaptive response of the kidney to IRI-induced AKI (56).
TLR3
TLR3 is activated upon the binding of single or double-stranded RNA and induces an antiviral immune response, characterized by the production of type I IFNs. TLR3 is constitutively expressed in mouse and human TECs and is activated earlier (few minutes) than TLR2 and TLR4 after IRI. In a model of bilateral IRI, Tlr3-deficient mice were significantly protected against kidney injury as indicated by less inflammation, less tubular apoptosis and necrosis, and preserved renal function 24 h after reperfusion (129). How TLR3 is involved in IRI remains speculative. Since Tlr3 KO mice had diminished TRIF protein expression, reduction of necroptosis might be one explanation. Also, type 1 IFN-mediated auto- and paracrine activation of IFN receptors might contribute, for instance, to pyroptosis via transactivation of caspase-11 (130, 131). The effects of TLR3 in the later phases of reperfusion were not studied in this paper.
TLR3 Ligands
Double-stranded RNA released upon viral replication is the major ligand of TLR3, but TLR3 can also recognize mRNA and mRNA-protein complexes released by necrotic cells (132). Therefore, it is tempting to speculate that both mRNA and mRNA-protein complexes might amplify the pro-inflammatory loop via their interaction with TLR3 upon renal IRI. However, scientific evidences are still lacking.
TLR9
TLR9 is a cytosolic DNA sensing receptor and has evolved to detect unmethylated CpG DNA, commonly found in microbial DNA and DNA viruses, and initiate the production of type I IFN and proinflammatory cytokines. TLR9 is highly expressed in professional innate immune cells, such as plasmacytoid DCs and macrophages but also in the kidney. In a model of moderate renal IRI, TLR9 was not involved in renal dysfunction (133). In contrast, in a model of severe IRI, we showed that Tlr9 deficiency resulted in improved survival in mice but not in the improvement of renal function and kidney damage (134). Improved survival was associated with reduced plasma mtDNA content and a subsequent decrease in hepatic injury. Surprisingly, and somewhat in contrast with our study, a recent study by Han et al. showed that selective intestinal TLR9 deficiency led to increased ischemic AKI and was associated with remote intestinal and hepatic injury. Intestinal Tlr9 deficiency was associated with enlarged Paneth cell granules and increased IL-17A expression (135). We would expect that Tlr9 KO animals would also present with this phenotype, but caution must be applied when interpreting scientific results generated by genetically-modified animals.
The mitochondrial dysfunction, cellular stress, and cell death involved in renal IRI result in the liberation of mtDNA, both in murine models of IRI and after renal transplantation in humans. Recently, our group reported a correlation between urinary mtDNA levels and the occurrence of DGF following renal transplantation (136). Outside of the mitochondrial matrix, mtDNA acts as a DAMP that can elicit neutrophil-mediated injury through TLR9, and other receptors (137). Indeed, the cyclic GMP–AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway is also able to recognize cytosolic DNA. Cytosolic cGAS binds double-stranded DNA and catalyzes the production of the novel second messenger 2′-3′-cyclic AMP-GMP (2′3′-cGAMP) from ATP and GTP. The binding of cGAMP to the ER-resident protein STING releases DNA-triggered signals and activates the innate immune system (138, 139). Recently, Maekawa et al. showed that cytosolic translocation of mtDNA leads to tubular inflammation via the cGAS-STING pathway, linking mitochondrial dysfunction to enhanced inflammation (112). cGAS-STING is activated in the cortex of animals 24 h after renal IRI and Sting KO animals display mild tubular injury and inflammation. Therefore, we cannot exclude the possibility that the absence of reprotection in Tlr9 KO animals, described in (134) after renal IRI, can be explained by an additional contribution of cGAS-STING. As cGAS-STING is activated in other pathogenic processes such as renal fibrosis (140) and senescence (141), further study into its effect on tubular repair will shed more light on the role of immunometabolism in AKI-to-CKD progression. Altogether, these studies strongly suggest that mitochondrial protection and a decrease in oxidative stress can be an elegant way to prevent AKI.
TREM-1
Triggering receptor expressed on myeloid cells-1 (TREM-1) is an activating receptor located primarily on cells of the innate immune system and some parenchymal cells (142). TREM-1 signals through its adapter protein, DNAX-activating protein (DAP12), to activate transcription factors capable of inducing the expression of pro-inflammatory cytokines and chemokines. TREM-1 can initiate inflammation, but can also work synergistically with TLRs to enhance an inflammatory response. Although initially studied in the context of infectious diseases, TREM-1 is also active in sterile inflammation (142). TREM-1 seems to be a hypoxia-inducible gene in myeloid DCs and TECs (52, 143) and might be involved in regulated cell death through amplification of inflammatory signals leading to necroptosis and pyroptosis, as has been shown in brain microglia (144). Interestingly, however, TREM-1 was also shown to mediate an inhibitory effect on necroptosis and pyroptosis in neonatal lung tissue, as suggested in previous work by Syed et al. (145). In our hands, in the early phase of renal IRI, TREM-1 modulation did not affect tubular damage or renal function (146). However, we did find that mice lacking TREM-1 displayed maladaptive repair characterized by persistent tubular damage, inflammation, fibrosis, and mitochondrial dysfunction-induced TEC senescence when exposed to renal IRI (52).
Inflammasomes
NOD1/2
Nucleotide-binding oligomerization domain (NOD) 1 and 2 are part of the NOD-like receptor (NLR) family of cytoplasmic PRRs. NOD1/2 can either initiate immune responses to pathogenic invasion through the recognition of PAMPs or sterile inflammation in response to DAMPs released upon cell stress (8). Both NOD1 and NOD2 contain 3 basic structural units: a NOD region, a caspase recruitment domain (CARD), and a ligand-binding domain. Once activated through ligand binding, the resulting oligomerization leads to the binding of signaling molecules to CARD. NOD1/2 activation can lead to apoptotic signaling through RIP-like interacting caspase-like apoptosis-regulatory protein kinase (RIP2) or the release of inflammatory chemokines and cytokines (147). NOD1/2 are expressed on TECs in both mice and humans (148), and due to their structural resemblance to other NLRs and TLRs that are known to play a role in kidney disease, it is plausible that NOD1/2 may play a role in renal injury. Indeed, a study by Shigeoka et al. found that Nod1/2 double KO led to a reduction in apoptosis following IRI (148). Expression of the pro-inflammatory cytokines IL-6, KC, and TNF-alpha were also reduced in mice deficient for NOD1/2, leading to the general suppression of the inflammatory response to IRI. Interestingly, NOD2-deficient mice were better protected against renal injury than NOD1-deficient mice, indicating that although both NOD proteins are involved in renal injury, the underlying mechanisms may be different (148). Endogenous ligands for NOD1/2 remain unknown, therefore, their discovery would certainly aid in the development of a therapeutic intervention.
NLRP3
NLRP3 (NOD-, LRR-, and pyrin domain-containing 3) is by far the best-characterized inflammasome-forming protein in the kidney. Once activated, the cytosolic innate immune receptor NLRP3 initiates the assembly of an inflammasome, leading to an inflammatory form of cell death (pyroptosis) and the proteolytic activation of the IL-1beta family of pro-inflammatory cytokines. The NLRP3 inflammasome can trigger inflammation by sensing a wide range of stimuli, but the specific mechanisms are still unclear. Among other factors, K+ efflux, ATP released from damaged mitochondria, and ROS production promote NLRP3 inflammasome activation (149).
Nlrp3 gene expression in murine kidneys increased after IRI and peaked 5 days after reperfusion, corresponding to the repair phase. Although the Nlrp3 gene was primarily expressed by leukocytes, TECs also expressed Nlrp3 after hypoxia or LPS stimulation (150). In a murine model of bilateral IRI, we showed that Nlrp3-deficient animals were protected against mortality, renal dysfunction, and displayed a reduced influx of neutrophils into the kidneys despite similar degrees of tubular necrosis. In this study, activation of NLRP3 was triggered, in part, through ATP produced by mitochondria released from necrotic cells (151).
Uromodulin has also been shown to activate the NLRP3 inflammasome in human peripheral blood mononuclear cells, leading to the secretion of IL-1beta. However, as previously mentioned, uromodulin-deficient mice displayed aggravated renal injury upon IRI. This illustrates the complex role of uromodulin in regulating inflammation.
The role of leukocyte- vs. renal-associated Nlrp3 expression has been studied in chimeric mice. In the early phase following IRI (day 1) only renal Nlrp3 contributed to renal dysfunction based on serum creatinine. In contrast, 5 days after reperfusion (repair phase) both renal- and leukocyte-associated Nlrp3 mediated loss of renal function. Interestingly, Nlrp3-deficient TECs showed increased proliferation and a superior repair response both in vivo and in vitro when compared to wildtype TECs (150).
More recently, it was shown that NLRP3 relocalizes from the cytosol to the mitochondria in TECs during hypoxia. The deletion of NLRP3 in TECs resulted in less mitochondrial ROS production, less mitochondrial damage, and less apoptosis in a model of in vitro hypoxia (152). Understanding the mechanisms of NLRP3 inflammasome activation will boost the development of small-molecule inhibitors for the treatment of NLRP3-related diseases (153).
NLRC5
NLR family CARD domain containing 5 (NLRC5) protein is a recently identified member of the NLR family that interferes with the assembly and activity of the NALP3 inflammasome complex by competing with ASC for pro-caspase-1 binding. Although NLRC5 activity leads to caspase-1 activation, induction of pyroptotic cell death dependent on NLRC5 has not yet been shown (154). Nlrc5 is significantly upregulated in the kidney 24 and 48 h after IRI. Nlrc5 deficiency significantly ameliorated renal function, injury, and inflammation 24 and 48 h after IRI. This was associated with less apoptosis in TECs and reduced inflammation in the kidneys (155).
NLRX1
In contrast to the hereinabove studied innate immune receptors, NLR family member X1 (NLRX1) exerts inflammasome independent anti-inflammatory effects by interfering with the canonical NF-kB signaling via inhibition of TRAF6 binding to IkB kinase (156). A unique feature of NLRX1 is its localization in the mitochondria. In 2017, we reported that NLRX1 protects against mortality and renal dysfunction after IRI by preventing excessive oxidative stress. We found that NLRX1 may act as an inhibitor of mitochondrial activity and prevents excessive oxidative stress, thereby preventing apoptosis of TECs during IRI (51).
Contrary to the studies by Zhou and Kim, we discovered renoprotective effects of glycolysis in IRI through our studies of NLRX1 (87, 152). Genetic deletion of Nlrx1 potentiates mitochondrial oxidative phosphorylation in TECs, while glycolysis results in enhanced oxidative stress after ischemia and ultimately increases cell death. Using a KO mouse model for acute renal IRI, we found that NLRX1 deficiency enhanced oxidative stress, thereby profoundly enhancing tubular apoptosis, renal dysfunction, and mortality, in the early days after IRI (51). However, the consequences of this metabolic rewiring toward glycolysis on epithelial repair have not yet been investigated. A proper investigation into our hypothesis on the renoprotective effects of NLRX1 in IRI would require NLRX1 activation. In silico studies have postulated polyunsaturated fatty acids as NLRX1 ligands, although further validation is required (157). As these are naturally occurring lipids, we propose that NLRX1 activation through nutritional supplementation may be a possible therapeutic approach to impede AKI-induced oxidative stress.
Innate Immunity in Human IRI and AKI
Pattern Recognition Receptor Expression and Variation During Human IRI
Little is known about the direct in vivo role for PRRs in human AKI and specific data on the spatiotemporal dynamics of activation are lacking. Instead, most of the studies conducted in humans that investigated PRRs tried to identify single nucleotide variants related to certain outcomes as a weak proxy for a human knock-out or knock-in model, depending on the (often estimated) consequence of the genetic variant. We previously conducted a comprehensive screening of TLR single nucleotide variants (TLR1-8 + SIGIRR) that were of interest because of their estimated effect on protein function. In a cohort of over 1,000 matched donor and recipient DNA samples, neither donor (renal) nor recipient (inflammatory cells) genetic variants were associated with DGF and the calculated effect size was low for individual variants, even after stratification for deceased donor type or analyzing the effect of all TLR gene variants in bulk, as compared to a baseline prediction model of cold ischemia time, donor age, and recipient age (158). A smaller study from Germany was able to find a positive association between a TLR3 gene variant and DGF, although for the other TLR genes they were unable to find a relation with DGF (159). Furthermore, in a Brazilian study by Nogueira et al., again, no association between several TLR4 gene variants and DGF were found (160). In the same cohort, with a similar experimental setup, we were also unable to find NLRP3 and TREM1 gene variants associated with DGF (146, 161). These data altogether suggest that the potential impact of these PRR genetic variants do not determine whether a patient will develop acute tubular necrosis and DGF, even though they were estimated to result in a PRR protein anomaly. Redundancy among these relatively common PRR genetic variants might partly explain this lack of association, since complete knockout of the gene of interest is a very rare event and not covered in the variants tested. At the expression level, polymerase chain reaction on implantation (time = 0) renal transplant biopsies showed a higher expression of TLR4 and MYD88 mRNA in deceased vs. living donor kidneys, but the authors could not find an association with the development of DGF (162). In an interesting study by McGuinness and colleagues from Glasgow, multi-omics analysis on pre- and postperfusion biopies (RNA sequencing, DNA methylation by whole genome bisulphite sequencing, and western blotting) identified a panel of expression markers associated with the development of DGF. They identified a transcriptional panel for DGF that was associated with innate immune signaling, TREM1 signaling, PRRs, and B cell development, with top-ranked networks related to immune system activation, cell death, and survival and cellular fitness. It seemed that cellular stress and restoration of physiological homeostasis (i.e., regeneration) was exacerbated in patients who developed DGF. At the protein level, DGF and perfusion status were associated with a state of cellular senescence (163). Altogether, the data from this study put PRR signaling, cell death and survival signaling, cellular stress and fitness, an exacerbated regenerative response, and cellular senescence at the center stage in the development of human IRI, and the authors suggest that these events probably occur independently of (minor) genetic differences in PRR. These conceptual findings seem to be in line with animal models.
The MABSOT Project and the Human TLR2 Opsona OPN-305 Trial
As mentioned before, within the context of the EU FP7 MABSOT project, the humanized anti-TLR2 monoclonal antibody (OPN-305) was tested for the treatment of DGF after transplantation. In 2016, a phase I/II multicenter, randomized, double-blind placebo-controlled trial (NCT01794663) (164) in selected individuals at high risk for DGF showed that a preferred dose of 0.5 mg/kg (lowest dose tested) resulted in a lower percentage of DGF (primary endpoint; 26.5% in the treatment arm vs. 29.4% in the placebo arm) and functional DGF (failure of serum creatinine to decrease at least 10% daily; 38.2 vs. 52.9%, respectively) (165). Doses of 1.5 and 5 mg/kg appeared to be associated with a higher percentage of functional DGF, possibly in part due to the low number of DGF cases in the placebo group, according to the writers of the preliminary report. Data on long-term follow-up of the enrolled patients is pending, and a follow-up randomized controlled trial specifically looking at the clinical impact of the 0.5 mg/kg in recipients of an extended criteria donor kidney is planned according to the preliminary report. What can we learn thus far from this efficacy MABSOT trial as a first proof-of-concept for PRR blockade during human IRI? An interesting finding from the trial is the efficacy of the lowest antibody dose tested. Mean duration of 100% TLR2 receptor occupancy for this dose was 1 day, whereas for 1.5 mg/kg this was 6 days, and for the highest dose this was even 13 days. The use of OPN-305 in the prevention of IRI is limited by the fact that it does not act specifically on TECs, and the systemic blocking of TLR2 in all cells might result in an increased risk for (severe) infections. It might be the case that TLR blockade in the reparative phase after IRI in humans results in delayed tubular regeneration and even maladaptive repair, increasing the risk of renal fibrosis and infections. Long-term follow-up, with respect to rejection rate and renal function, would be of great interest for further investigation. Efficacy came at the cost of serious infectious and renal side effects in a select number of patients, but these results are the first to suggest the therapeutic potential for early pharmaceutical manipulation of PRRs in the context of human renal IRI.
Concluding Remarks and Future Outlook
This review described the main phenotypic changes occurring during renal IRI in TECs and the role of innate immunity in dictating inflammation and repair. The central dogma on the role of innate immunity in IRI still remains; excessive inflammation is just as detrimental as a faulty response, as both scenarios predispose to maladaptive repair and chronic progression (see Figure 3). We summarized the current evidence that the activation of innate immune receptors, in a very delicate balance (intensity, timing, cell types), is also necessary for adaptive repair. Moreover, here we have provided an additional and novel perspective about the role of epithelial metabolism and cell fate in IRI, which may be controlled by innate immunity.
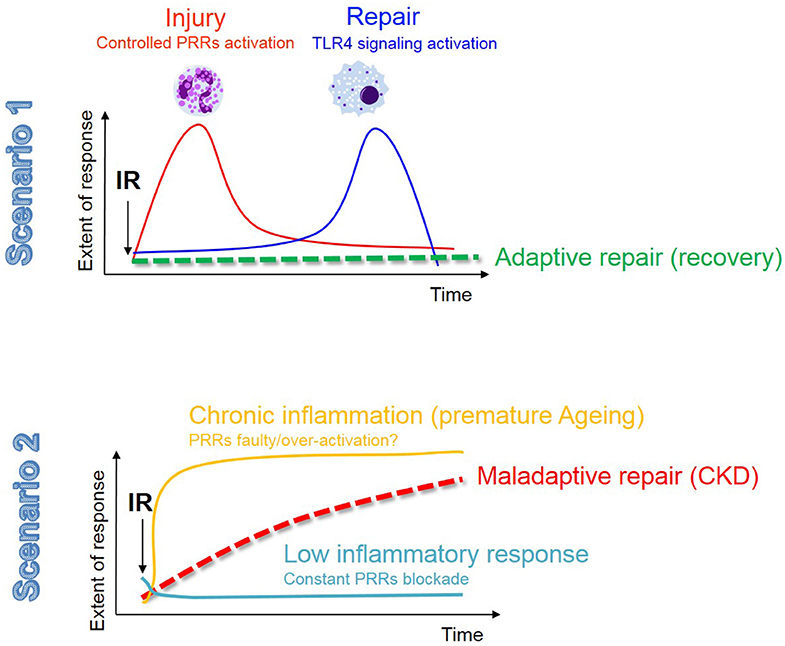
Figure 3. The innate immune response in renal IR: the fine balance between adaptive repair and chronic degeneration. In this figure we describe the 2 possible scenarios that may occur in the kidney following AKI. The fist scenario involves a moderate injury resolved by a balanced and timely activation of the innate immune response, most likely associated with a low and transient senescence burden, resulting in adaptive tubular repair and kidney regeneration. In the second scenario we envision that either a low-grade chronic inflammation (possibly related to an aging phenotype and the associated high senescent burden) or a constant PRRs blockade (to avoid excessive inflammation), contribute to phenotypic changes in TECs (as described in Figure 1) and maladaptive tubular repair.
As TECs have an incredible phenotypic plasticity and are among the most metabolically active cells, they are crucial for kidney function and homeostasis. Consequently, targeting epithelial immunometabolism holds great potential to alter the initiation of acute renal failure, but also progression towards CKD. As a final consideration, we are in a time where longevity is increasing and research on cellular senescence in the kidney, as a strategy to limit the AKI-to-CKD transition, is a fascinating area to explore for therapeutic potential. Unraveling differences between drivers of acute and chronic senescence will be essential to further investigate whether these cells could be trained to change their cell fate and function, in order to slow down the progression of kidney remodeling. Changes in mitochondrial homeostasis, metabolism, and function of innate immune receptors are known pathological mechanisms associated with aging, and thus could also take place in senescent TECs. Detailed studies on the hallmarks of senescent TECs in the different phases of renal IRI may shed light on novel mechanisms that can be targeted to redirect their phenotype and overcome the issue of repair by elimination. We envision that determining the therapeutic window of opportunity to regulate renal homeostasis by targeting innate immunity, immunometabolism, and cellular senescence should be among the future research goals in the field of nephrology and kidney transplantation.
Author Contributions
AT, JK, and SF designed and directed the project. AS contributed to the study. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Luyckx VA, Tonelli M, Stanifer JW. The global burden of kidney disease and the sustainable development goals. Bull World Health Organ. (2018) 96:414–22C. doi: 10.2471/BLT.17.206441
2. Dabrowska-Bender M, Dykowska G, Zuk W, Milewska M, Staniszewska A. The impact on quality of life of dialysis patients with renal insufficiency. Patient Prefer Adherence. (2018) 12:577–83. doi: 10.2147/PPA.S156356
3. Liu B-C, Tang T-T, Lv L-L, Lan H-Y. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. (2018) 93:568–79. doi: 10.1016/j.kint.2017.09.033
4. Ishimoto Y, Inagi R. Mitochondria: a therapeutic target in acute kidney injury. Nephrol Dial Transplant. (2016) 31:1062–9. doi: 10.1093/ndt/gfv317
5. Levey AS, James MT. Acute kidney injury. Ann Intern Med. (2017) 167:ITC66–80. doi: 10.7326/AITC201711070
6. Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. (2014) 371:58–66. doi: 10.1056/NEJMra1214243
7. See EJ, Jayasinghe K, Glassford N, Bailey M, Johnson DW, Polkinghorne KR, et al. Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. (2019) 95:160–72. doi: 10.1016/j.kint.2018.08.036
8. Leemans JC, Kors L, Anders H-J, Florquin S. Pattern recognition receptors and the inflammasome in kidney disease. Nat Rev Nephrol. (2014) 10:398–414. doi: 10.1038/nrneph.2014.91
9. Rabb H, Griffin MD, McKay DiB, Swaminathan S, Pickkers P, Rosner MH, et al. Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrol. (2016) 27:371–9. doi: 10.1681/ASN.2015030261
10. Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. (2011) 7:189–200. doi: 10.1038/nrneph.2011.16
11. Agarwal A, Dong Z, Harris R, Murray P, Parikh SM, Rosner MH, et al. Cellular molecular mechanisms of AKI. J Am Soc Nephrol. (2016) 27:1288–99. doi: 10.1681/ASN.2015070740
12. Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transplant. (2011) 11:2279–96. doi: 10.1111/j.1600-6143.2011.03754.x
13. Wu MY, Yiang GT, Liao WT, Tsai APY, Cheng YL, Cheng PW, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. (2018) 46:1650–67. doi: 10.1159/000489241
14. Shay T, Jojic V, Zuk O, Rothamel K, Puyraimond-Zemmour D, Feng T, et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc Natl Acad Sci USA. (2013) 110:2946–51. doi: 10.1073/pnas.1222738110
15. Kers J, Leemans JC, Linkermann A. An overview of pathways of regulated necrosis in acute kidney injury. Semin Nephrol. (2016) 36:139–52. doi: 10.1016/j.semnephrol.2016.03.002
16. Glücksmann A. Cell deaths in normal vertebrate ontogeny. Biol Rev. (1951) 26:59–86. doi: 10.1111/j.1469-185X.1951.tb00774.x
17. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. (2011) 147:742–58. doi: 10.1016/j.cell.2011.10.033
18. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-018-0102-y
19. Rosin DL, Okusa MD. Dangers within: DAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol. (2011) 22:416–25. doi: 10.1681/ASN.2010040430
20. Todd JL, Palmer SM. Danger signals in regulating the immune response to solid organ transplantation. J Clin Invest. (2017) 127:2464–72. doi: 10.1172/JCI90594
21. Priante G, Gianesello L, Ceol M, Del Prete D, Anglani F. Cell death in the kidney. Int J Mol Sci. (2019) 20:3598. doi: 10.3390/ijms20143598
22. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. (2015) 22:58–73. doi: 10.1038/cdd.2015.54
23. McNamara DE, Quarato G, Guy CS, Green DR, Moldoveanu T. Characterization of MLKL-mediated plasma membrane rupture in necroptosis. J Vis Exp. (2018) 58088. doi: 10.3791/58088
24. Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. (2016) 23:76–88. doi: 10.1038/cdd.2015.70
25. Linkermann A, Kunzendorf U, Krautwald S. Phosphorylated MLKL causes plasma membrane rupture. Mol Cell Oncol. (2014) 1:e29915. doi: 10.4161/mco.29915
26. Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, et al. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol Cell. (2016) 61:589–601. doi: 10.1016/j.molcel.2016.01.011
27. Pefanis A, Ierino FL, Murphy JM, Cowan PJ. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int. (2019) 96:291–301. doi: 10.1016/j.kint.2019.02.009
28. Chen H, Fang Y, Wu J, Chen H, Zou Z, Zhang X, et al. RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis. (2018) 9:878. doi: 10.1038/s41419-018-0936-8
29. Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z, et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. (2008) 117:3216–26. doi: 10.1161/CIRCULATIONAHA.108.769331
30. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
31. Sollberger G, Strittmatter GE, Garstkiewicz M, Sand J, Beer H-D. Caspase-1: the inflammasome and beyond. Innate Immun. (2014) 20:115–25. doi: 10.1177/1753425913484374
32. Aglietti RA, Dueber EC. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. (2017) 38:261–71. doi: 10.1016/j.it.2017.01.003
33. Chen X, He W-T, Hu L, Li J, Fang Y, Wang X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. (2016) 26:1007–20. doi: 10.1038/cr.2016.100
34. Yang J-R, Yao F-H, Zhang J-G, Ji Z-Y, Li K-L, Zhan J, et al. Ischemia-reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am J Physiol Renal Physiol. (2014) 306:F75–84. doi: 10.1152/ajprenal.00117.2013
35. Miao N, Yin F, Xie H, Wang Y, Xu Y, Shen Y, et al. The cleavage of gasdermin D by caspase-11 promotes tubular epithelial cell pyroptosis and urinary IL-18 excretion in acute kidney injury. Kidney Int. (2019) 96:1105–20. doi: 10.1016/j.kint.2019.04.035
36. Kushnareva Y, Newmeyer DD. Bioenergetics and cell death. Ann N Y Acad Sci. (2010) 1201:50–7. doi: 10.1111/j.1749-6632.2010.05633.x
37. Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. (2010) 11:621–32. doi: 10.1038/nrm2952
38. Bredesen DE, Mehlen P, Rabizadeh S. Apoptosis and dependence receptors: a molecular basis for cellular addiction. Physiol Rev. (2004) 84:411–30. doi: 10.1152/physrev.00027.2003
39. Lemke G. How macrophages deal with death. Nat Rev Immunol. (2019) 19:539–49. doi: 10.1038/s41577-019-0167-y
40. Kaushal GP, Shah S V. Non-apoptotic effects of antiapoptotic agent zVAD-fmk in renal injury. Kidney Int. (2013) 83:531. doi: 10.1038/ki.2012.470
41. Yang B, Lan S, Dieudé M, Sabo-Vatasescu J-P, Karakeussian-Rimbaud A, Turgeon J, et al. Caspase-3 is a pivotal regulator of microvascular rarefaction and renal fibrosis after ischemia-reperfusion injury. J Am Soc Nephrol. (2018) 29:1900–16. doi: 10.1681/ASN.2017050581
42. Kaiser WJ, Offermann MK. Apoptosis induced by the toll-like receptor adaptor trif is dependent on its receptor interacting protein homotypic interaction motif. J Immunol. (2005) 174:4942–52. doi: 10.4049/jimmunol.174.8.4942
43. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. (2013) 288:31268–79. doi: 10.1074/jbc.M113.462341
44. Sanjo H, Nakayama J, Yoshizawa T, Fehling HJ, Akira S, Taki S. Cutting edge: TAK1 safeguards macrophages against proinflammatory cell death. J Immunol. (2019) 203:783–8. doi: 10.4049/jimmunol.1900202
45. Ferenbach DA, Bonventre J V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Publ Gr. (2015) 11:264–76. doi: 10.1038/nrneph.2015.3
46. Basile DP, Bonventre J V, Mehta R, Nangaku M, Unwin R, Rosner MH, et al. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol. (2016) 27:687–97. doi: 10.1681/ASN.2015030309
47. Andrade L, Rodrigues CE, Gomes SA, Noronha IL. Acute kidney injury as a condition of renal senescence. Cell Transplant. (2018) 27:739–53. doi: 10.1177/0963689717743512
48. O'Sullivan ED, Hughes J, Ferenbach DA. Renal aging: causes and consequences. J Am Soc Nephrol. (2017) 28:407–20. doi: 10.1681/ASN.2015121308
49. Goligorsky M. Chronic kidney disease: a vicarious relation to premature cell senescence? Am J Pathol. (2020) 190:1164–71. doi: 10.1016/j.ajpath.2020.01.016
50. Docherty MH, O'Sullivan ED, Bonventre JV, Ferenbach DA. Cellular senescence in the kidney. J Am Soc Nephrol. (2019) 30:726–36. doi: 10.1681/ASN.2018121251
51. Stokman G, Kors L, Bakker PJ, Rampanelli E, Claessen N, Teske GJD, et al. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J Exp Med. (2017) 214:2405–20. doi: 10.1084/jem.20161031
52. Tammaro A, Scantlebery AML, Rampanelli E, Borrelli C, Claessen N, Butter LM, et al. TREM1/3 deficiency impairs tissue repair after acute kidney injury and mitochondrial metabolic flexibility in tubular epithelial cells. Front Immunol. (2019) 10:1469. doi: 10.3389/fimmu.2019.01469
53. Jin H, Zhang Y, Ding Q, Wang SS, Rastogi P, Dai D-F, et al. Epithelial innate immunity mediates tubular cell senescence after kidney injury. JCI insight. (2019) 4:e125490. doi: 10.1172/jci.insight.125490
54. Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. (2005) 115:2894–903. doi: 10.1172/JCI22832
55. Anders HJ, Schaefer L. Beyond tissue injury - damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J Am Soc Nephrol. (2014) 25:1387–400. doi: 10.1681/ASN.2014010117
56. Dessing MC, Tammaro A, Pulskens WP, Teske GJ, Butter LM, Claessen N, et al. The calcium-binding protein complex S100A8/A9 has a crucial role in controlling macrophage-mediated renal repair following ischemia/reperfusion. Kidney Int. (2015) 87:85–94. doi: 10.1038/ki.2014.216
57. Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. (2007) 117:2847–59. doi: 10.1172/JCI31008
58. Kulkarni OP, Hartter I, Mulay SR, Hagemann J, Darisipudi MN, Kumar Vr S, et al. Toll-like receptor 4-induced IL-22 accelerates kidney regeneration. J Am Soc Nephrol. (2014) 25:978–89. doi: 10.1681/ASN.2013050528
59. Zhang M-Z, Yao B, Yang S, Jiang L, Wang S, Fan X, et al. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. (2012) 122:4519–32. doi: 10.1172/JCI60363
60. Kinsey GR. Macrophage dynamics in AKI to CKD progression. J Am Soc Nephrol. (2014) 25:209–11. doi: 10.1681/ASN.2013101110
61. Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. (2011) 22:317–26. doi: 10.1681/ASN.2009060615
62. Tábara LC, Poveda J, Martin-Cleary C, Selgas R, Ortiz A, Sanchez-Niño MD. Mitochondria-targeted therapies for acute kidney injury. Expert Rev Mol Med. (2014) 16:e13. doi: 10.1017/erm.2014.14
63. Turrens JF, Beconi M, Barilla J, Chavez UB, McCord JM. Mitochondrial generation of oxygen radicals during reoxygenation of ischemic tissues. Free Radic Res Commun. (1991) 12–13:681–9. doi: 10.3109/10715769109145847
64. Maekawa H, Inagi R. Pathophysiological role of organelle stress/crosstalk in AKI-to-CKD transition. Semin Nephrol. (2019) 39:581–88. doi: 10.1016/j.semnephrol.2019.10.007
65. Gall JM, Wang Z, Bonegio RG, Havasi A, Liesa M, Vemula P, et al. Conditional knockout of proximal tubule mitofusin 2 accelerates recovery and improves survival after renal ischemia. J Am Soc Nephrol. (2015) 26:1092–102. doi: 10.1681/ASN.2014010126
66. Perry HM, Huang L, Wilson RJ, Bajwa A, Sesaki H, Yan Z, et al. Dynamin-related protein 1 deficiency promotes recovery from AKI. J Am Soc Nephrol. (2018) 29:194–206. doi: 10.1681/ASN.2017060659
67. Szeto HH. Pharmacologic approaches to improve mitochondrial function in AKI and CKD. J Am Soc Nephrol. (2017) 28:2856–65. doi: 10.1681/ASN.2017030247
68. Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol. (2016) 27:3356–67. doi: 10.1681/ASN.2015020177
69. Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol. (2015) 26:1765–76. doi: 10.1681/ASN.2015010006
70. He L, Wei Q, Liu J, Yi M, Liu Y, Liu H, et al. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. (2017) 92:1071–83. doi: 10.1016/j.kint.2017.06.030
71. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. (2010) 16:535–43. doi: 10.1038/nm.2144
72. Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. (2015) 21:37–46. doi: 10.1038/nm.3762
73. Zheng J, Devalaraja-Narashimha K, Singaravelu K, Padanilam BJ. Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol. (2005) 288:F387–98. doi: 10.1152/ajprenal.00436.2003
74. Prolla TA, Denu JM. NAD+ deficiency in age-related mitochondrial dysfunction. Cell Metab. (2014) 19:178–80. doi: 10.1016/j.cmet.2014.01.005
75. Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. (2015) 350:1208–13. doi: 10.1126/science.aac4854
76. Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. (2016) 531:528–32. doi: 10.1038/nature17184
77. Drury ER, Zsengeller ZK, Stillman IE, Khankin E V, Pavlakis M, Parikh SM. Renal PGC1α may be associated with recovery after delayed graft function. Nephron. (2018) 138:303–9. doi: 10.1159/000485663
78. Poyan Mehr A, Tran MT, Ralto KM, Leaf DE, Washco V, Messmer J, et al. De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat Med. (2018) 24:1351–9. doi: 10.1038/s41591-018-0138-z
79. Parikh SM. Metabolic stress resistance in acute kidney injury: evidence for a PPAR-Gamma-coactivator-1 alpha-nicotinamide adenine dinucleotide pathway. Nephron. (2019) 143:184–7. doi: 10.1159/000500168
80. Guan Y, Wang SR, Huang XZ, Xie QH, Xu YY, Shang D, et al. Nicotinamide mononucleotide, an NAD+ precursor, rescues age-associated susceptibility to AKI in a sirtuin 1-dependent manner. J Am Soc Nephrol. (2017) 28:2337–52. doi: 10.1681/ASN.2016040385
81. Liu L, Su X, Quinn WJ, Hui S, Krukenberg K, Frederick DW, et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab. (2018) 27:1067–80.e5. doi: 10.1016/j.cmet.2018.03.018
82. Declèves A-E, Sharma K, Satriano J. Beneficial effects of AMP-activated protein kinase agonists in kidney ischemia-reperfusion: autophagy and cellular stress markers. Nephron Exp Nephrol. (2014). doi: 10.1159/000368932. [Epub ahead of print].
83. Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. (2016) 48:e245. doi: 10.1038/emm.2016.81
84. Seo-Mayer PW, Thulin G, Zhang L, Alves DS, Ardito T, Kashgarian M, et al. Preactivation of AMPK by metformin may ameliorate the epithelial cell damage caused by renal ischemia. Am J Physiol Renal Physiol. (2011) 301:F1346–57. doi: 10.1152/ajprenal.00420.2010
85. Chung KW, Lee EK, Lee MK, Oh GT, Yu BP, Chung HY. Impairment of PPARα and the fatty acid oxidation pathway aggravates renal fibrosis during aging. J Am Soc Nephrol. (2018) 29:1223–37. doi: 10.1681/ASN.2017070802
86. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. (2008) 452:230–3. doi: 10.1038/nature06734
87. Zhou H-L, Zhang R, Anand P, Stomberski CT, Qian Z, Hausladen A, et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature. (2019) 565:96–100. doi: 10.1038/s41586-018-0749-z
88. Pan JS, Sheikh-Hamad D. Mitochondrial dysfunction in acute kidney injury and sex-specific implications. Med Res Arch. (2019) 7:10.18103/mra.v7i2.1898. doi: 10.18103/mra.v7i2.1898
89. Rojas-Morales P, León-Contreras JC, Aparicio-Trejo OE, Reyes-Ocampo JG, Medina-Campos ON, Jiménez-Osorio AS, et al. Fasting reduces oxidative stress, mitochondrial dysfunction and fibrosis induced by renal ischemia-reperfusion injury. Free Radic Biol Med. (2019) 135:60–7. doi: 10.1016/j.freeradbiomed.2019.02.018
90. Dare AJ, Bolton EA, Pettigrew GJ, Bradley JA, Saeb-Parsy K, Murphy MP. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. (2015) 5:163–8. doi: 10.1016/j.redox.2015.04.008
91. Plotnikov EY, Chupyrkina AA, Jankauskas SS, Pevzner IB, Silachev DN, Skulachev VP, et al. Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochim Biophys Acta. (2011) 1812:77–86. doi: 10.1016/j.bbadis.2010.09.008
92. Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng F-Y, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. (2011) 22:1041–52. doi: 10.1681/ASN.2010080808
93. Birk A V, Liu S, Soong Y, Mills W, Singh P, Warren JD, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. (2013) 24:1250–61. doi: 10.1681/ASN.2012121216
94. Yin M, Zhong Z, Connor HD, Bunzendahl H, Finn WF, Rusyn I, et al. Protective effect of glycine on renal injury induced by ischemia-reperfusion in vivo. Am J Physiol Renal Physiol. (2002) 282:F417–23. doi: 10.1152/ajprenal.00011.2001
95. Li J, Li L, Wang S, Zhang C, Zheng L, Jia Y, et al. Resveratrol alleviates inflammatory responses and oxidative stress in rat kidney ischemia-reperfusion injury and H2O2-Induced NRK-52E cells via the Nrf2/TLR4/NF-κB pathway. Cell Physiol Biochem. (2018) 45:1677–89. doi: 10.1159/000487735
96. Kim MG, Yang J, Ko YS, Lee HY, Oh SW, Cho WY, et al. Impact of aging on transition of acute kidney injury to chronic kidney disease. Sci Rep. (2019) 9:18445. doi: 10.1038/s41598-019-54585-1
97. Berger K, Bangen J-M, Hammerich L, Liedtke C, Floege J, Smeets B, et al. Origin of regenerating tubular cells after acute kidney injury. Proc Natl Acad Sci USA. (2014) 111:1533–8. doi: 10.1073/pnas.1316177111
98. Koyano T, Namba M, Kobayashi T, Nakakuni K, Nakano D, Fukushima M, et al. The p21 dependent G2 arrest of the cell cycle in epithelial tubular cells links to the early stage of renal fibrosis. Sci Rep. (2019) 9:12059. doi: 10.1038/s41598-019-48557-8
99. Matos DA, Zhang JM, Ouyang J, Nguyen HD, Genois MM, Zou L. ATR protects the genome against R loops through a MUS81-triggered feedback loop. Mol Cell. (2020) 77:514–27.e4. doi: 10.1016/j.molcel.2019.10.010
100. Kishi S, Brooks CR, Taguchi K, Ichimura T, Mori Y, Akinfolarin A, et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J Clin Invest. (2019) 129:4797–816. doi: 10.1172/JCI122313
101. Chapman J, Fielder E, Passos JF. Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett. (2019) 593:1566–79. doi: 10.1002/1873-3468.13498
102. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. (2014) 15:482–96. doi: 10.1038/nrm3823
103. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
104. Valentijn FA, Falke LL, Nguyen TQ, Goldschmeding R. Cellular senescence in the aging and diseased kidney. J Cell Commun Signal. (2018) 12:69–82. doi: 10.1007/s12079-017-0434-2
105. Megyesi J, Price PM, Tamayo E, Safirstein RL. The lack of a functional p21(WAF1/CIP1) gene ameliorates progression to chronic renal failure. Proc Natl Acad Sci USA. (1999) 96:10830–5. doi: 10.1073/pnas.96.19.10830
106. Rhinn M, Ritschka B, Keyes WM. Cellular senescence in development, regeneration and disease. Development. (2019) 146:dev151837. doi: 10.1242/dev.151837
107. Sturmlechner I, Durik M, Sieben CJ, Baker DJ, van Deursen JM. Cellular senescence in renal ageing and disease. Nat Rev Nephrol. (2017) 13:77–89. doi: 10.1038/nrneph.2016.183
108. Luo C, Zhou S, Zhou Z, Liu Y, Yang L, Liu J, et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J Am Soc Nephrol. (2018) 29:1238–56. doi: 10.1681/ASN.2017050574
109. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. (2018) 28:436–53. doi: 10.1016/j.tcb.2018.02.001
110. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. (2016) 23:303–14. doi: 10.1016/j.cmet.2015.11.011
111. Li Y, Zhao H, Huang X, Tang J, Zhang S, Li Y, et al. Embryonic senescent cells re-enter cell cycle and contribute to tissues after birth. Cell Res. (2018) 28:775–8. doi: 10.1038/s41422-018-0050-6
112. Maekawa H, Inoue T, Hirokawa N, Nangaku M, Inagi R. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. (2019) 29:1261–1273.e6. doi: 10.2139/ssrn.3366988
113. O'Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
114. Ash SR, Cuppage FE. Shift toward anaerobic glycolysis in the regenerating rat kidney. Am J Pathol. (1970) 60:385–402.
115. Kim J, Devalaraja-Narashimha K, Padanilam BJ. TIGAR regulates glycolysis in ischemic kidney proximal tubules. Am J Physiol Renal Physiol. (2015) 308:F298–308. doi: 10.1152/ajprenal.00459.2014
116. Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. (2003) 300:1524–5. doi: 10.1126/science.1085536
117. Farrugia M, Baron B. The role of toll-like receptors in autoimmune diseases through failure of the self-recognition mechanism. Int J Inflamm. (2017) 2017:8391230. doi: 10.1155/2017/8391230
118. Wolfs TG, Buurman WA, van Schadewijk A, de Vries B, Daemen MA, Hiemstra PS, et al. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J Immunol. (2002) 168:1286–93. doi: 10.4049/jimmunol.168.3.1286
119. Pulskens WP, Teske GJ, Butter LM, Roelofs JJ, van der Poll T, Florquin S, et al. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS ONE. (2008) 3:e3596. doi: 10.1371/journal.pone.0003596
120. Rusai K, Sollinger D, Baumann M, Wagner B, Strobl M, Schmaderer C, et al. Toll-like receptors 2 and 4 in renal ischemia/reperfusion injury. Pediatr Nephrol. (2010) 25:853–60. doi: 10.1007/s00467-009-1422-4
121. Dessing MC, Pulskens WP, Teske GJ, Butter LM, van der Poll T, Yang H, et al. RAGE does not contribute to renal injury and damage upon ischemia/reperfusion-induced injury. J Innate Immun. (2012) 4:80–5. doi: 10.1159/000334251
122. Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. (2010) 21:1878–90. doi: 10.1681/ASN.2009101048
123. Miura K, Sahara H, Sekijima M, Kawai A, Waki S, Nishimura H, et al. Protective effect of neutralization of the extracellular high-mobility group box 1 on renal ischemia-reperfusion injury in miniature swine. Transplantation. (2014) 98:937–43. doi: 10.1097/TP.0000000000000358
124. Devuyst O, Bochud M. Uromodulin, kidney function, cardiovascular disease, and mortality. Kidney Int. (2015) 88:944–6. doi: 10.1038/ki.2015.267
125. Säemann MD, Weichhart T, Zeyda M, Staffler G, Schunn M, Stuhlmeier KM, et al. Tamm-Horsfall glycoprotein links innate immune cell activation with adaptive immunity via a Toll-like receptor-4-dependent mechanism. J Clin Invest. (2005) 115:468–75. doi: 10.1172/JCI200522720
126. Micanovic R, Khan S, Janosevic D, Lee ME, Hato T, Srour EF, et al. Tamm-horsfall protein regulates mononuclear phagocytes in the kidney. J Am Soc Nephrol. (2018) 29:841–56. doi: 10.1681/ASN.2017040409
127. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. (2012) 23:1375–88. doi: 10.1681/ASN.2011111077
128. Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. (2008) 118:3522–30. doi: 10.1172/JCI36150
129. Paulus P, Rupprecht K, Baer P, Obermüller N, Penzkofer D, Reissig C, et al. The early activation of toll-like receptor (TLR)-3 initiates kidney injury after ischemia and reperfusion. PLoS ONE. (2014) 9:e94366. doi: 10.1371/journal.pone.0094366
130. Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JP-Y, Coers J, et al. Canonical inflammasomes drive IFN-γ to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe. (2015) 18:320–32. doi: 10.1016/j.chom.2015.07.016
131. Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. (2012) 150:606–19. doi: 10.1016/j.cell.2012.07.007
132. Karikó K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for toll-like receptor 3. J Biol Chem. (2004) 279:12542–50. doi: 10.1074/jbc.M310175200
133. Li X, Yun Z, Tan Z, Li S, Wang D, Ma K, et al. The role of toll-like receptor (TLR) 2 and 9 in renal ischemia and reperfusion injury. Urology. (2013) 81:1379.e15–20. doi: 10.1016/j.urology.2013.02.016
134. Bakker PJ, Scantlebery AM, Butter LM, Claessen N, Teske GJD, van Der Poll T, et al. TLR9 mediates remote liver injury following severe renal ischemia reperfusion. PLoS ONE. (2015) 10:e0137511. doi: 10.1371/journal.pone.0137511
135. Han SJ, Li H, Kim M, D'Agati V, Lee HT. Intestinal toll-like receptor 9 deficiency leads to paneth cell hyperplasia and exacerbates kidney, intestine, and liver injury after ischemia/reperfusion injury. Kidney Int. (2019) 95:859–79. doi: 10.1016/j.kint.2018.10.035
136. Jansen MPB, Pulskens WPC, Uil M, Claessen N, Nieuwenhuizen G, Standaar D, et al. Urinary mitochondrial deoxyribonucleic acid associates with delayed graft function following renal transplantation. Nephrol Dial Transplant. (2018). doi: 10.1093/ndt/gfy372. [Epub ahead of print].
137. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via P38 map kinase. Shock. (2010) 34:55–9. doi: 10.1097/SHK.0b013e3181cd8c08
138. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. (2013) 153:1094–107. doi: 10.1016/j.cell.2013.04.046
139. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. (2011) 478:515–8. doi: 10.1038/nature10429
140. Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. (2019) 30:784–99.e5. doi: 10.1016/j.cmet.2019.08.003
141. Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. (2017) 19:1061–70. doi: 10.1038/ncb3586
142. Tammaro A, Derive M, Gibot S, Leemans JC, Florquin S, Dessing MC. TREM-1 and its potential ligands in non-infectious diseases: from biology to clinical perspectives. Pharmacol Ther. (2017) 177:81–95. doi: 10.1016/j.pharmthera.2017.02.043
143. Bosco MC, Pierobon D, Blengio F, Raggi F, Vanni C, Gattorno M, et al. Hypoxia modulates the gene expression profile of immunoregulatory receptors in human mature dendritic cells: identification of TREM-1 as a novel hypoxic marker in vitro and in vivo. Blood. (2011) 117:2625–39. doi: 10.1182/blood-2010-06-292136
144. Xu P, Zhang X, Liu Q, Xie Y, Shi X, Chen J, et al. Microglial TREM-1 receptor mediates neuroinflammatory injury via interaction with SYK in experimental ischemic stroke. Cell Death Dis. (2019) 10:555. doi: 10.1038/s41419-019-1777-9
145. Syed MA, Shah D, Das P, Andersson S, Pryhuber G, Bhandari V. TREM-1 attenuates RIPK3-mediated necroptosis in hyperoxia-induced lung injury in neonatal mice. Am J Respir Cell Mol Biol. (2019) 60:308–22. doi: 10.1165/rcmb.2018-0219OC
146. Tammaro A, Kers J, Emal D, Stroo I, Teske GJD, Butter LM, et al. Effect of TREM-1 blockade and single nucleotide variants in experimental renal injury and kidney transplantation. Sci Rep. (2016) 6:38275. doi: 10.1038/srep38275
147. Masumoto J, Yang K, Varambally S, Hasegawa M, Tomlins SA, Qiu S, et al. Nod1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J Exp Med. (2006) 203:203–13. doi: 10.1084/jem.20051229
148. Shigeoka AA, Kambo A, Mathison JC, King AJ, Hall WF, da Silva Correia J, et al. Nod1 and nod2 are expressed in human and murine renal tubular epithelial cells and participate in renal ischemia reperfusion injury. J Immunol. (2010) 184:2297–304. doi: 10.4049/jimmunol.0903065
149. Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
150. Bakker PJ, Butter LM, Claessen N, Teske GJD, Sutterwala FS, Florquin S, et al. A tissue-specific role for Nlrp3 in tubular epithelial repair after renal ischemia/reperfusion. Am J Pathol. (2014) 184:2013–22. doi: 10.1016/j.ajpath.2014.04.005
151. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. (2009) 106:20388–93. doi: 10.1073/pnas.0908698106
152. Kim SM, Kim YG, Kim DJ, Park SH, Jeong KH, Lee YH, et al. Inflammasome-independent role of NLRP3 mediates mitochondrial regulation in renal injury. Front Immunol. (2018) 9:2563. doi: 10.3389/fimmu.2018.02563
153. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. (2019) 10:128. doi: 10.1038/s41419-019-1413-8
154. Wu Y, Shi T, Li J. NLRC5: A paradigm for NLRs in immunological and inflammatory reaction. Cancer Lett. (2019) 451:92–9. doi: 10.1016/j.canlet.2019.03.005
155. Li Q, Wang Z, Zhang Y, Zhu J, Li L, Wang X, et al. NLRC5 deficiency protects against acute kidney injury in mice by mediating carcinoembryonic antigen–related cell adhesion molecule 1 signaling. Kidney Int. (2018) 94:551–66. doi: 10.1016/j.kint.2018.02.031
156. Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity. (2011) 34:843–53. doi: 10.1016/j.immuni.2011.02.022
157. Lu P, Hontecillas R, Abedi V, Kale S, Leber A, Heltzel C, et al. Modeling-enabled characterization of novel NLRX1 ligands. PLoS ONE. (2015) 10:e0145420. doi: 10.1371/journal.pone.0145420
158. Dessing MC, Kers J, Damman J, Leuvenink HGD, van Goor H, Hillebrands J-L, et al. Toll-like receptor family polymorphisms are associated with primary renal diseases but not with renal outcomes following kidney transplantation. PLoS ONE. (2015) 10:e0139769. doi: 10.1371/journal.pone.0139769
159. Krüger B, Banas MC, Walberer A, Böger CA, Farkas S, Hoffmann U, et al. A comprehensive genotype-phenotype interaction of different toll-like receptor variations in a renal transplant cohort. Clin Sci. (2010) 119:535–44. doi: 10.1042/CS20100190
160. Nogueira E, Ozaki KS, Macusso GD, Quarim RF, Camara NO, Pacheco-Silva A. Incidence of donor and recipient toll-like receptor-4 polymorphisms in kidney transplantation. Transpl Proc. (2007) 39:412–4. doi: 10.1016/j.transproceed.2007.01.026
161. Dessing MC, Kers J, Damman J, Navis GJ, Florquin S, Leemans JC. Donor and recipient genetic variants in NLRP3 associate with early acute rejection following kidney transplantation. Sci Rep. (2016) 6:36315. doi: 10.1038/srep36315
162. Andrade-Oliveira V, Campos EF, Goncalves-Primo A, Grenzi PC, Medina-Pestana JO, Tedesco-Silva H, et al. TLR4 mRNA levels as tools to estimate risk for early posttransplantation kidney graft dysfunction. Transplantation. (2012) 94:589–95. doi: 10.1097/TP.0b013e31825db680
163. McGuinness D, Mohammed S, Monaghan L, Wilson PA, Kingsmore DB, Shapter O, et al. A molecular signature for delayed graft function. Aging Cell. (2018) 17:e12825. doi: 10.1111/acel.12825
164. Placebo-Controlled Study to Evaluate the Safety Efficacy of OPN-305 in Preventing Delayed Renal Graft Function - Full Text View - ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/ct2/show/NCT01794663 (accessed November 1, 2019)
165. Final Report Summary - MABSOT (Development of OPN-305 as An Orphan Drug For The Treatment of Delayed Graft Function Post Solid Organ Transplantation) | Report Summary | MABSOT | FP7 | CORDIS | European Commission. Available online at: https://cordis.europa.eu/project/rcn/98249/reporting/en (accessed November 1, 2019)
Keywords: kidney transplantation, tubular repair, innate immunity, cell death, mitochondria, senescence
Citation: Tammaro A, Kers J, Scantlebery AML and Florquin S (2020) Metabolic Flexibility and Innate Immunity in Renal Ischemia Reperfusion Injury: The Fine Balance Between Adaptive Repair and Tissue Degeneration. Front. Immunol. 11:1346. doi: 10.3389/fimmu.2020.01346
Received: 02 November 2019; Accepted: 27 May 2020;
Published: 07 July 2020.
Edited by:
Cees Van Kooten, Leiden University, NetherlandsReviewed by:
Philippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique, FranceKaren Bieback, Heidelberg University, Germany
Copyright © 2020 Tammaro, Kers, Scantlebery and Florquin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Tammaro, YS50YW1tYXJvQGFtc3RlcmRhbXVtYy5ubA==
†These authors have contributed equally to this work