 Micah D. Dunlap1,2†
Micah D. Dunlap1,2† Oliver A. Prince2†
Oliver A. Prince2† Javier Rangel-Moreno3
Javier Rangel-Moreno3 Kimberly A. Thomas2Julia M. Scordo4
Kimberly A. Thomas2Julia M. Scordo4 Jordi B. Torrelles4Jeffery Cox5
Jordi B. Torrelles4Jeffery Cox5 Adrie J. C. Steyn6,7
Adrie J. C. Steyn6,7 Joaquín Zúñiga8
Joaquín Zúñiga8 Deepak Kaushal4,9,10*
Deepak Kaushal4,9,10* Shabaana A. Khader1,2*
Shabaana A. Khader1,2*- 1Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO, United States
- 2Department of Molecular Microbiology, Washington University School of Medicine, St. Louis, MO, United States
- 3University of Rochester Medical Center, Rochester, NY, United States
- 4Texas Biomedical Research Institute, San Antonio, TX, United States
- 5Department of Molecular and Cell Biology, University of California, Berkeley, Berkeley, CA, United States
- 6Department of Microbiology, Centers for AIDS Research and Free Radical Biology, University of Alabama at Alabama, Birmingham, AL, United States
- 7African Health Research Institute (AHRI), Durban, South Africa
- 8Instituto Nacional de Enfermedades Respiratorias, Mexico City, Mexico
- 9Division of Bacteriology, Tulane National Primate Research Center, Covington, LA, United States
- 10Department of Microbiology and Immunology, Tulane University School of Medicine, New Orleans, LA, United States
Mycobacterium tuberculosis (Mtb) is the causative agent of the infectious disease tuberculosis (TB), which is a leading cause of death worldwide. Approximately one fourth of the world's population is infected with Mtb. A major unresolved question is delineating the inducers of protective long-lasting immune response without inducing overt, lung inflammation. Previous studies have shown that the presence of inducible Bronchus-Associated Lymphoid Tissue (iBALT) correlate with protection from Mtb infection. In this study, we hypothesized that specific Mtb factors could influence the formation of iBALT, thus skewing the outcome of TB disease. We infected non-human primates (NHPs) with a transposon mutant library of Mtb, and identified specific Mtb mutants that were over-represented within iBALT-containing granulomas. A major pathway reflected in these mutants was Mtb cell wall lipid transport and metabolism. We mechanistically addressed the function of one such Mtb mutant lacking mycobacteria membrane protein large 7 (MmpL7), which transports phthiocerol dimycocerosate (PDIM) to the mycobacterial outer membrane (MOM). Accordingly, murine aerosol infection with the Mtb mutant Δmmpl7 correlated with increased iBALT-containing granulomas. Our studies showed that the Δmmpl7 mutant lacking PDIMs on the surface overexpressed diacyl trehaloses (DATs) in the cell wall, which altered the cytokine/chemokine production of epithelial and myeloid cells, thus leading to a dampened inflammatory response. Thus, this study describes an Mtb specific factor that participates in the induction of iBALT formation during TB by directly modulating cytokine and chemokine production in host cells.
Introduction
Mycobacterium tuberculosis (Mtb) is the causative agent of pulmonary tuberculosis (TB). TB is caused by inhalation of Mtb bacilli and it is currently estimated that approximately one fourth of the world's population is infected with Mtb (1). While most individuals infected with Mtb are asymptomatic and do not exhibit clinical symptoms, 5–10% of infected individuals can progress to pulmonary clinical TB. The immune parameters that distinguish latent TB from active pulmonary TB are not as yet clearly defined.
The first interactions between Mtb and the host happen in the lung airways immediately after inhalation of Mtb bacilli. These early interactions involve alveolar macrophages (2) (AMs), dendritic cells (3, 4) (DCs), and epithelial cells lining the airway (5–7) and have the potential to drive immune responses. As a result of initiation of immune responses, the tubercle granuloma is formed, which is a hallmark immune structure formed during TB (8). We have previously shown that protective granulomas that are formed during latent TB are associated with the formation of B-cell containing lymphoid follicles (9). During severe active TB, granulomas that do not effectively contain Mtb are comprised predominately of neutrophils (10, 11) and permissive monocytes (12), which have been implicated in general tissue destruction, thus skewing responses toward disease progression. Non-protective granulomas are hallmarked by the formation of hypoxic, necrotic cores that do not prevent Mtb growth and eventually lead to dissemination to other organs and tissues (10). The earliest mechanism(s) which determine the nature and outcome of granulomas during infection remains elusive, and are a focus of this work.
In this study, we aimed to determine the Mtb and host specific factors that drive the formation of inducible bronchus associated lymphoid tissue (iBALT) during TB. To examine the Mtb specific factors involved, we utilized an Mtb transposon mutant library to screen the induction of lymphoid follicles using the Non-Human primate (NHP) model of pulmonary TB. This screen allowed us to identify Mtb genes which when mutated led to increased induction of iBALT within granulomas. The NHP model exhibits characteristics associated with human TB including the pulmonary cellular and acellular lesions (13, 14). We found an over-representation of Mtb cell wall mutants within iBALT containing granulomas and further characterized the role of one such mutant using the mouse model of TB. The mycobacteria membrane protein large 7 (Mmpl7) inner membrane protein is best known for the transport of phthiocerol dimycocerosate (PDIM) to the free fatty acid layer of the mycobacterial outer membrane (MOM) (15, 16). The role of lipid presentation in the MOM is well-documented in modulating various host responses (17–19), suggesting that MOM lipids contact with host cells in the lung plays a role in disease progression. In light of these observations, we aimed to understand how the Mtb Δmmpl7 cell wall mutant identified in our screen modulates early epithelial signaling and myeloid recruitment in order to coordinate granuloma structure and the formation of iBALT. In this study we found that in the mouse model, infection with the Δmmpl7 mutant drives decreased bacterial burden and increased formation of iBALT, as was observed in NHPs. Furthermore, the Δmmpl7 mutant also drove decreased inflammatory cytokine and chemokine production in vitro and in vivo, along with decreased accumulation of myeloid and lymphoid immune cells. We demonstrate that the Δmmpl7 mutant overexpresses diacyl trehaloses (DATs), a Mtb cell wall lipid, which can drive the observed decreased inflammatory cytokines and chemokine production by in vitro macrophages and also yields increased production of IL-10.
Results
Identification of Mtb Genes Associated With Formation of Protective Lymphoid Follicles
TB granulomas contain distinct iBALT structures which are protective in mice and macaques (9). NHPs infected with Mtb exhibit the spectrum of disease severity observed clinically during human TB, with a diverse array of granuloma structures reflected by differences in immune cell recruitment and disease outcome (14, 20). Thus, we used the NHP model to probe the early host-Mtb interactions that mediate the signaling events that initiate the induction of iBALT within TB granulomas. NHPs were infected with 1 × 105 CFU of Mtb (H37Rv) mutants from the himar1 transposon mutagenesis site hybridization (TraSH) library (21, 22). Four-six weeks post-infection, the animals were humanely euthanized due to TB disease. At this time, macaque lungs demonstrated a wide distribution of caseous and follicular granulomatous structures (Figure 1), with follicular granulomas featuring prominent cellular structure. In combination with mesodissection, DNA sequencing analysis of separate B cell follicle containing granulomas, seeded by Mtb mutants (23, 24), identified nine Mtb single mutants that were highly represented within distinct iBALT containing TB granulomas in macaques (Figure 1 and Table 1). Of the Mtb mutants represented within these “protective” granulomas, two members of the mycobacterial membrane protein large family (Mmpl2 and Mmpl7), were well-reflected. MmpL2 is a putative inner membrane transporter protein with unknown substrate, while MmpL7 is an inner membrane protein associated with phthiocerol dimycocerosate (PDIM) transport to the MOM (15, 16). The NAD dehydrogenase, NdhA is a non-essential protein found in the inner mycobacterial membrane associated with NADH mediated electron transfer to the electron transport chain (25). EccD5 is an essential inner membrane protein that makes up part of the type VII secretion system, the absence of which results in sensitivity to detergents and killing by macrophage (26). ClpB and Acr2 are both heat stress molecular chaperones regulated by the alternative sigma factors SigH and SigE, respectively (27). Of the two, only ClpB is essential for in vitro growth. The non-redundant glutamine synthetases GlnA2 and GlnA4 are involved in nitrogen metabolism but are not associated with any loss of virulence in vivo (28). Cmtr is a cytosolic sensor of cadmium Cd(II) and lead Pb(II) (29) which are trace metal contaminants found in the lung (Table 1).
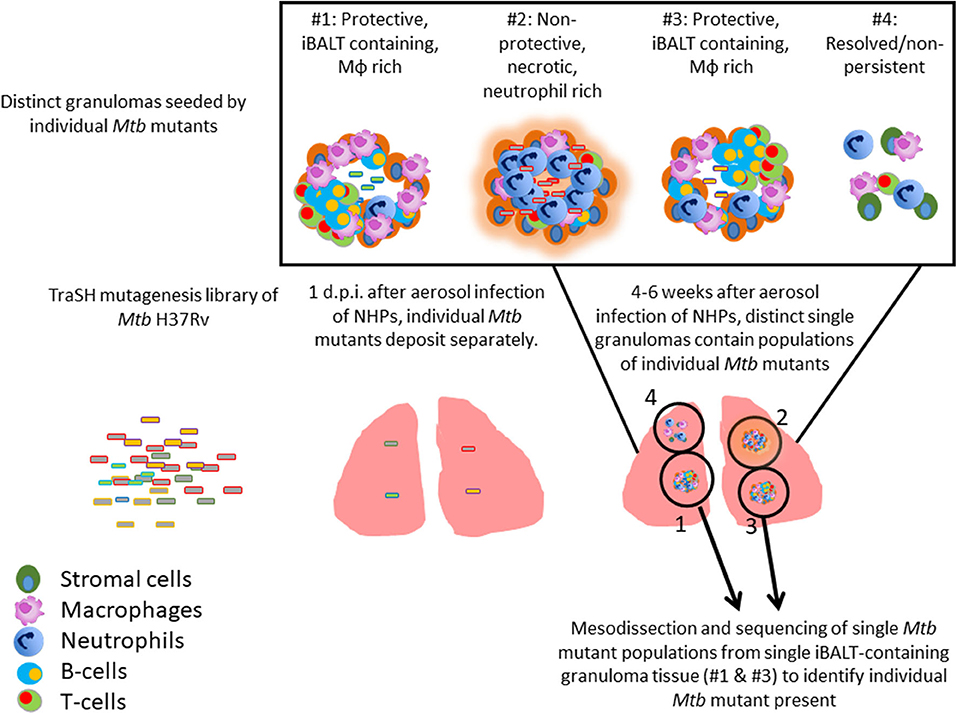
Figure 1. Identification of Mtb genes associated with formation of protective lymphoid follicles: Transposon site hybridization library (TraSH) using the himar1 transposon mutagenesis strategy was employed to create a Mtb mutant library for the specific discovery of Mtb genes influencing the progression of lymphoid follicle containing granulomas in NHPs. NHPs were infected with 100,000 CFU of Mtb mutants and lung sections were harvested 4–6 weeks after infection. In combination with mesodissection, the analysis of >1,200 separate lesions identified nine Mtb single mutants that were highly represented within protective iBALT containing TB granulomas in macaques. Single Mtb mutants were isolated from single granulomas.
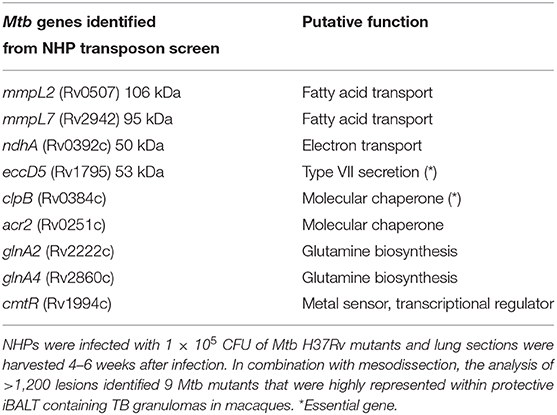
Table 1. Identification of Mtb genes associated with formation of protective lymphoid follicles: Transposon site hybridization library (TraSH) using the himar1 transposon mutagenesis strategy was employed to create a Mtb mutant library for the specific discovery of Mtb genes influencing the progression of lymphoid follicle containing granulomas in NHPs.
Taken together, the Mtb transposon screen conducted in NHPs has identified a novel set of Mtb genes involved in generation of lymphoid follciles within the lung, that appear to control Mtb metabolism and inner membrane lipid transport. In this study, we chose to mainly focus on the interaction driven by Mtb Mmpl7 that mediates formation of the granuloma.
Δmmpl7 Mutant Drives Mtb Enhanced B Cell Follicle Containing iBALT Formation in Mouse Model
To determine the functional role of Mtb Mmpl7 in modulating iBALT generation in vivo, we infected C57BL/6 mice (B6) with 100 CFU Mtb Erdman or the Δmmpl7 mutant Erdman strain and characterized immune recruitment and induction of iBALT features in the lung. As previously shown, infection with the Δmmpl7 mutant resulted in early decreased Mtb CFU which recovered by 40 days post-infection (d.p.i.) in the lung and spleen (Figure 2A). At a time when Mtb CFU was comparable between the WT and mutant strains, we observed that the total inflammation observed in the lungs of the Δmmpl7 mutant infected mice was significantly decreased (Figures 2B,C). However, despite significantly decreased overall inflammation, infection with the Δmmpl7 mutant resulted in increased formation of B cell lymphoid follicle containing area, reflecting iBALT areas (Figures 2D–E). Furthermore, by 40 d.p.i., T cells were well able to infiltrate within iBALT containing granulomas, as the area of T cell cuffing around the granulomas was decreased in Δmmpl7 mutant infected mice (Figures 2D,E). Together, these findings suggest that Mtb lacking the Mmpl7 gene induces improved induction of iBALT and decreased inflammation, suggesting that Mtb actively shuts down iBALT formation utilizing virulence factors such as PDIM, or alternatively, the reduced early Mtb growth in the Mmpl7 mutant in vivo propagates iBALT containing granulomas.
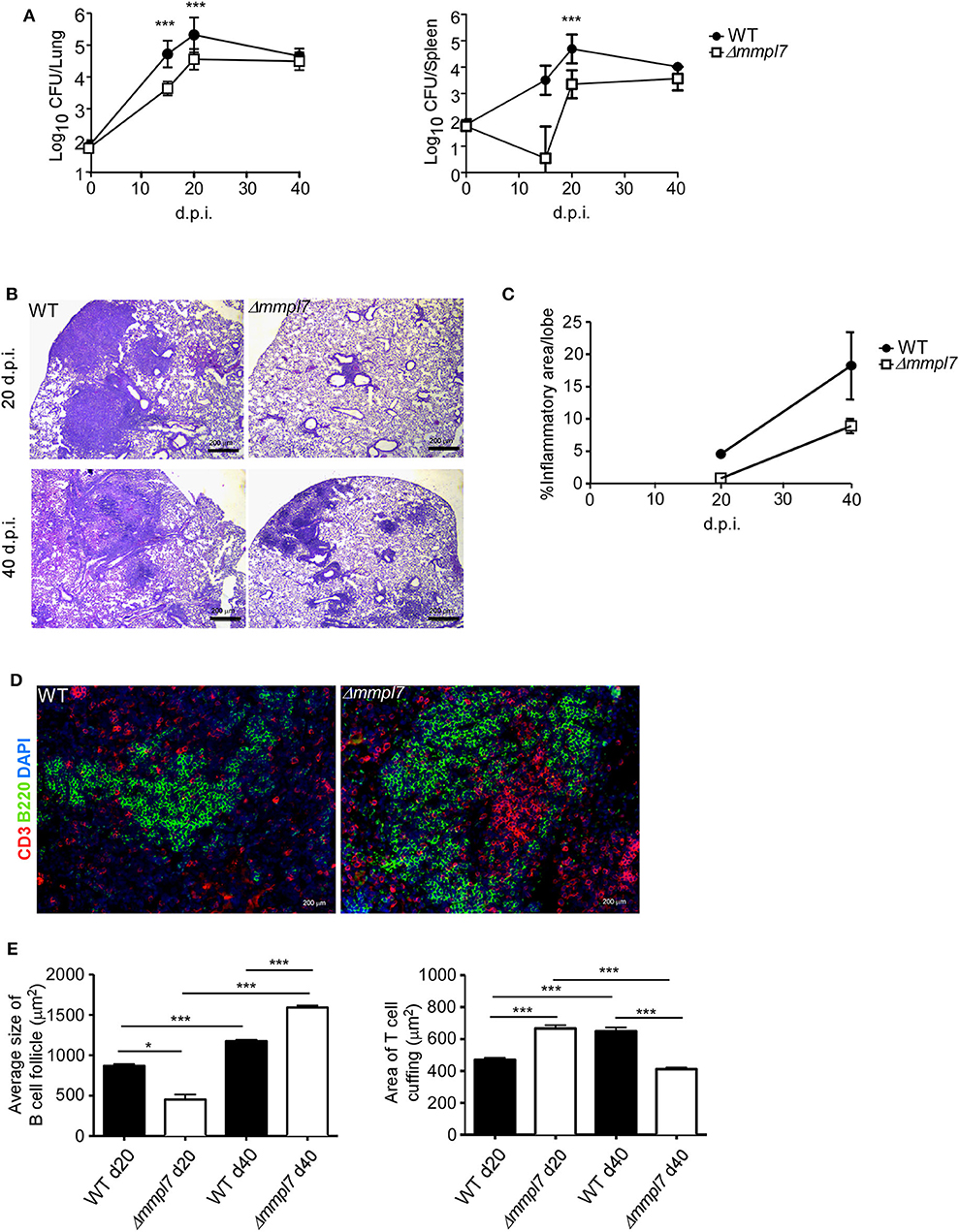
Figure 2. Δmmpl7 mutant drives Mtb enhanced B cell follicle formation in mouse model: Sex and age matched C57BL/6 mice were infected with 100 CFU of Mtb Erdman WT or Δmmpl7. (A) Lung and spleen homogenates were used to determine Mtb CFU counts at indicated time points. (B,C) Lungs from Mtb-infected mice at 20 (top) and 40 (bottom) d.p.i. were formalin fixed, embedded in paraffin and used for H&E staining and inflammation was quantified by tracing areas. (D,E) B cell follicles present within lung sections were visualized by confocal microscopy. Slides were visualized and quantified by outlining the lesions using the automated tool of the Zeiss Axioplan 2 microscope. Infected groups (n ≥ 5) comparing WT Erdman and Δmmpl7 Mtb strains at individual time points were compared using Student's t-test. Mean and standard deviation (SD) were plotted for each group at each indicated time point. *p < 0.05, **p < 0.01, ***p < 0.001.
It has been shown previously that the Δmmpl7 mutant lacks the ability to transport PDIMs, an important Mtb structural lipid and virulence factor, to the MOM, leading to an early growth defect in vivo when mice are infected intravenously (15, 16). Thus, to specifically address if the early decrease in Mtb CFU burden in the mutant infection was responsible for increased iBALT formation, we infected mice with a 5-fold higher dose (500 CFU) of Mtb Δmmpl7 to directly compare the ability of Δmmpl7 mutant to induce iBALT when compared to low dose (100 CFU) infection with wildtype (WT) Mtb Erdman (Supplemental Figure 1). Using the higher dose model, our data show that Δmmpl7 mutant still had early defects in Mtb CFU in vivo in the lung and spleen as previously shown, but at 40 d.p.i. showed comparable CFU to WT Mtb and similar overall inflammation (Supplemental Figures 1A,B). Importantly, the higher dose model maintained the observed increased iBALT formation (Supplemental Figure 1C). Whether this phenotype was due partially or entirely to the lack of PDIMs on the MOM or due to other changes in the Δmmpl7 mutant was further evaluated in this study.
Furthermore, by 40 d.p.i., no differences in bacterial burden were observed in the lung or spleen between WT Erdman and Δmmpl7 mutant, which is more likely due to increased control of WT Erdman while Δmmpl7 mutant CFU levels were maintained. These findings demonstrate that our 5-fold higher CFU dose aerosol infection used to compensate for the growth defect in the Δmmpl7 mutant still recapitulates the in vivo phenotype observed with low dose infection. These data suggest that the observed phenotype of increased iBALT formation in the Δmmpl7 mutant infection is likely not an artifact of abrogated Mtb growth.
Δmmpl7 Mutant Mtb Limits Early Myeloid Cell Accumulation in the Mtb-Infected Lung
To examine the early immune events that occur in the lung that mediate differences in iBALT formation, we infected mice with 100 CFU wildtype Mtb Erdman or 5-fold higher CFU (500 CFU) of the Δmmpl7 mutant and characterized myeloid and T cell accumulation in the lung. Upon infection with the Δmmpl7 mutant, we observed decreased neutrophils, monocytes and recruited macrophages at early time points as well as decreased accumulation of alveolar macrophages (AMs) and myeloid DCs (mDCs) when compared with accumulation in WT Mtb infected lungs (Figures 3A,B). We saw similar decreases in accumulation of total CD3+ T cells (Figure 3C), activated and IFN-γ producing CD4+ (Figure 3D) and CD8+ T cells (Figure 3E) at later time points.
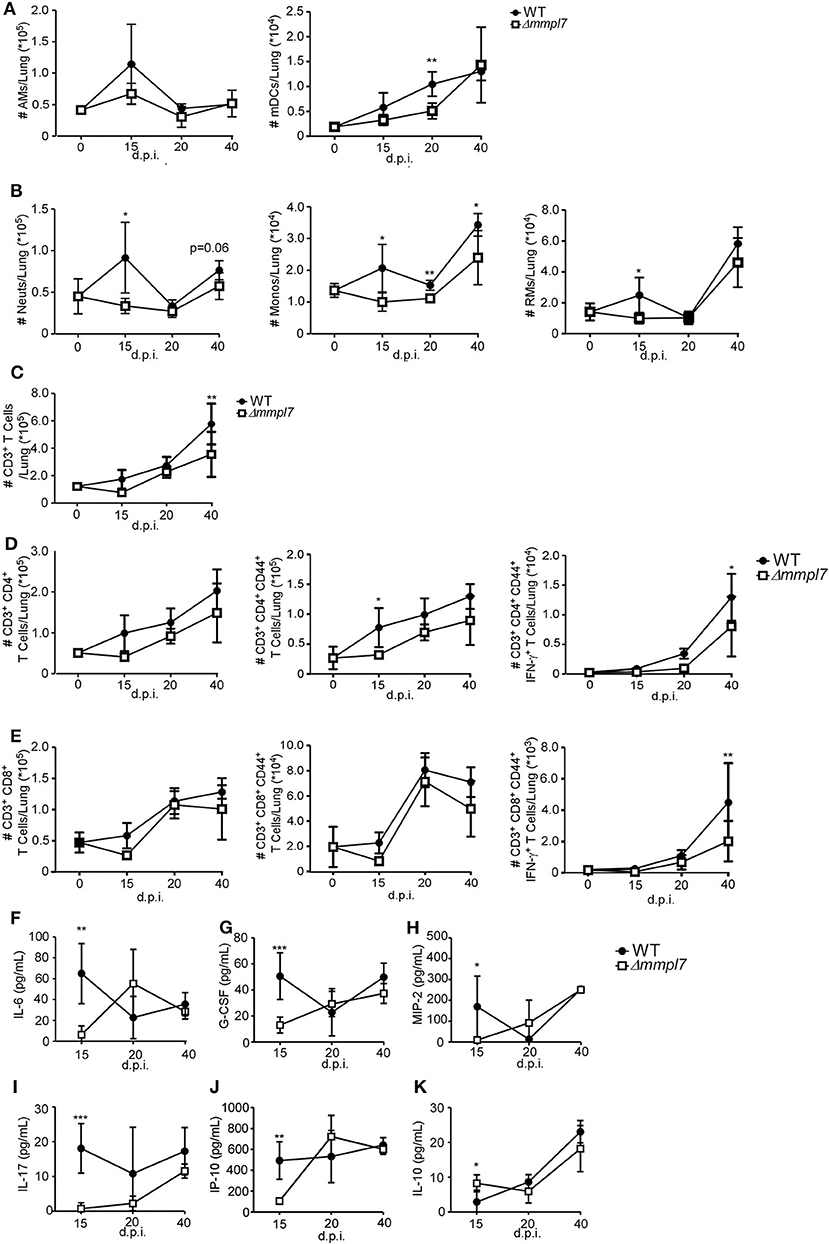
Figure 3. Δmmpl7 mutant Mtb limits myeloid cell accumulation in the lung: Sex and age matched C57BL/6 mice were infected with 100 CFU Mtb Erdman WT or 500 CFU Δmmpl7. Single cell suspensions were processed from infected lungs and immune cell subsets were analyzed by flow cytometry. (A) tissue resident myeloid and (B) recruited myeloid cell accumulation in the Mtb-infected lungs was determined at indicated time points. (C) CD3+ lymphocytes, (D) CD4+ or (E) CD8+ T cells were determined using flow cytometry. (F–K) Protein levels of inflammatory cytokine and chemokines IL-6, G-CSF, MIP-2(CXCL2), IL-17, IP-10 (CXCL10), and IL-10 were determined in lung homogenates at indicated time points. Infected groups (n = 5) comparing WT Erdman and Δmmpl7 Mtb strains at individual time points were compared using Student's t-test. Individual time points within the same group were compared using 2-way ANOVA with Bonferroni post-tests. Mean and standard deviation (SD) were plotted for each group at each indicated time point. *p < 0.05, **p < 0.01, ***p < 0.001.
These cellular changes coincided with early decreased production of inflammatory cytokines, such as IL-6, G-CSF, and IL-17, and chemokines, such as MIP-2 (CXCL2), and IP-10 (CXCL10) that are associated with myeloid recruitment in Mtb Δmmpl7 infected mice (Figures 3F–J). Interestingly, this coincided with an early increased induction of IL-10 at 15 d.p.i., though this effect was abolished by the later timepoints (Figure 3K). As IL-10 is a known mediator of anti-inflammatory responses (30, 31), these findings suggest a potential mechanism for the decrease in pro-inflammatory cytokines and lung inflammation. Taken together, these data demonstrate an overall “dampening” of the immune response during early infection with the Δmmpl7 mutant, suggesting that decreased inflammation and decreased neutrophilia may improve the formation of protective iBALT containing granulomas, thus imporving TB disease.
Δmmpl7 Mutant Mtb Overexpresses DATs and Limits Cytokine Production in Lung Epithelial Cells
Having observed abrogated recruitment of myeloid cells after infection with the Mtb Δmmpl7 mutant and decreased cytokine/chemokine production in vivo (Figure 3), we hypothesized that the Δmmpl7 mutant was specifically modulating inflammatory molecule production and subsequent recruitment of immune cells. Increased early IL-10 production and decreased neutrophilia and inflammation observed after infection with the Δmmpl7 mutant likely skewed the immune response toward protective iBALT containing granulomas. Alternatively, increased neutrophil accumulation and increased pro-inflammatory cytokines, including G-CSF and IL-6, are induced in response to WT Mtb infection and drive non-protective, necrotic granulomas (10, 11, 32). To address if G-CSF was a marker of active TB disease in humans, we measured levels in individuals with active TB disease (ATB) or latent Mtb infection (LTBI), in two human cohorts. We observed that G-CSF production was increased in ATB individuals when compared with LTBI or house contacts (HCs) (Figure 4A). Furthermore, it was previously demonstrated that individuals with ATB also have increased serum levels of IL-6 (33, 34). As such, both G-CSF and IL-6 were used as cytokine indicators of inflammatory response severity for the remainder of this study.
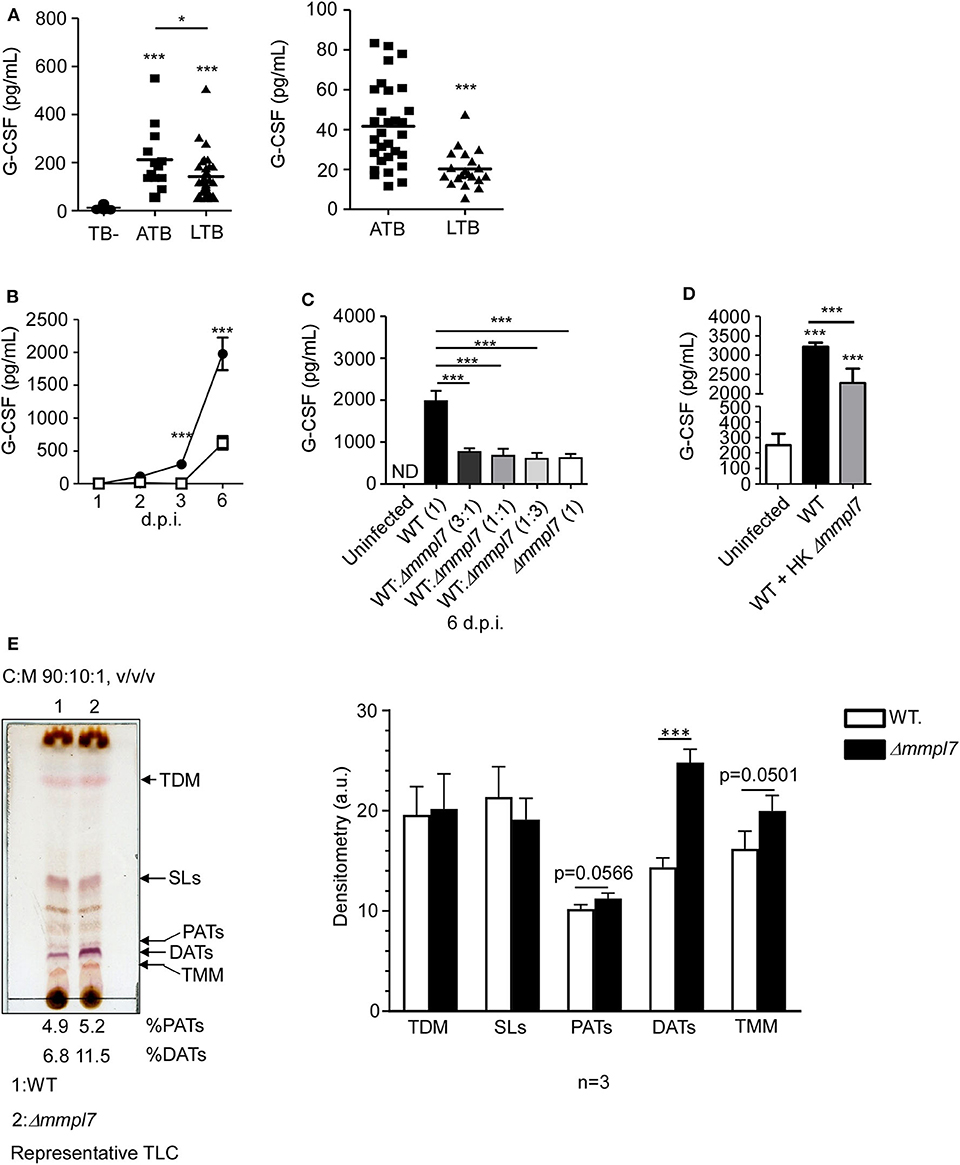
Figure 4. Δmmpl7 mutant Mtb overexpresses DATs and limits cytokine production in lung epithelial cells: (A) Serum samples from human active TB patients and controls (Left) Panel: Mexico (TB- n = 30; ATB n = 13; LTB n = 33), Right Panel: South Africa (ATB n = 30; LTB n = 20) were analyzed for G-CSF levels. Mouse lung epithelial cells (n ≥ 4 per group) were infected with Mtb Erdman WT or Δmmpl7 at (B) MOI = 1 or MOI = 5, respectively, for indicated time points. (C) Mouse lung epithelial cells (n = 4) were infected with Mtb Erdman WT and/or Δmmpl7 at indicated ratios for a constant total MOI = 1. (D) Mouse lung epithelial cells (n = 4) were infected with Mtb Erdman WT with or without the addition of heat-killed (HK) Δmmpl7. (A–D) Supernatants were collected and G-CSF concentration was quantified by ELISA. (E) Total lipids from Mtb Erdman WT or Δmmpl7 were obtained from freshly grown bacteria (3 independent times) and extracted with chloroform:methanol (2:1, v/v/) after normalization by total bacterial number. Total lipids (100 μg) were analyzed by TLC using as a solvent system (chloroform:methanol:water, 90:10:1, v/v/v) and 10% sulfuric acid in ethanol as a developer to visualize total lipid contents (representative experiment shown). Densitometry analyses of the observed lipids were performed using the NIH software ImageJ, n = 3 (where each value corresponds to independent extractions from freshly grown bacteria and TLCs). Human cohort study groups were compared by 1-way ANOVA with Tukey's post-test, or unpaired Student's t-test, respectively. Infected groups comparing WT Erdman and Δmmpl7 Mtb strains under indicated conditions at individual time points were compared using 1-way ANOVA with Tukey's post-test or unpaired Student's t-test. TLC densitometry was analyzed by unpaired Student's t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
To further test this hypothesis in our model, we examined mouse lung epithelial cells in vitro, which are a key source of G-CSF, and other inflammatory cytokines in the context of lung infection (5, 35–39). We assayed epithelial cell derived G-CSF production in vitro after infection with either wildtype Mtb Erdman or Δmmpl7 mutant. Firstly, we found that epithelial cells infected with WT Mtb induced significant G-CSF in a time-dependent manner. In contrast, infection of epithelial cells with Δmmpl7 mutant resulted in significantly decreased G-CSF production when compared to wildtype Erdman infection (Figure 4B). When performing coinfection with varying ratios of wildtype Erdman and Δmmpl7 mutant, we observed that even 3 times the presence of WT Mtb did not reverse the abrogated G-CSF production observed upon Δmmpl7 mutant infection (Figure 4C). Furthermore, we observed a significant decrease in G-CSF production even when wildtype Erdman infected epithelial cells were co-administered heat-killed (HK) Δmmpl7 mutant, suggesting that the inhibition of G-CSF was dependent on a factor expressed by Mtb Δmmpl7 and did not depend on the viability or infectivity of the bacteria (Figure 4D). These data together suggest that a molecular factor present in the Δmmpl7 mutant is likely limiting G-CSF expression in vitro and in vivo.
From these data, we hypothesized that a lack of Mmpl7 in Mtb led to changes in the lipid profile of the MOM beyond the reported loss of PDIMs (15, 16, 40), thus inducing the presence of other lipids and virulence factors than those present on wildtype Mtb Erdman (41). We hypothesized that these lipids may decrease the production of G-CSF, IL-6 and other inflammatory cytokines, thus driving decreased myeloid recruitment and increased iBALT formation upon infection. To test this, we freshly grew Mtb WT and Δmmpl7 mutant bacteria three independent times, normalized by bacteria weight, and extracted and characterized their total lipid content by 1 and 2 dimensional thin layer chromatography (1D or 2D TLC) (Figure 4E and Supplemental Figure 2), using different solvents systems allowing us to fully screen the wide range of lipids present on the mycobacterial cell envelope. We found that while sulpholipids (SLs), trehalose dimycolates (TDMs) and trehalose monomycolates (TMMs) were present at similar levels, diacyl trehaloses (DATs) were expressed at higher levels in the cell envelope of the Δmmpl7 mutant Mtb (11.5% of total lipid) when compared to WT Erdman (6.8% of total lipid) (Figure 4E and Supplemental Figure 2, black arrows). DATs are heterogeneous glycolipids (>30 molecular species described) composed of a trehalose sugar structure with two acyl chains, where the trehalose moiety is thought to bind to the macrophage-inducible C-type lectin Mincle (42, 43). DATs have been previously shown to negatively regulate the pro-inflammatory response of the host, thus promoting Mtb survival (18, 19). Thus, these findings suggested that increased presence of DATs in the Δmmpl7 mutant could be linked to the decreased production of inflammatory molecules like G-CSF and IL-6 and the dampened pulmonary inflammatory response observed in vitro and in vivo (19).
DAT Administration Drives Decreased Inflammatory Molecule Production in Epithelial and Myeloid Cells
We next hypothesized that the increased expression of DATs could abrogate production of cytokines, especially G-CSF, in epithelial cells (5, 7), similar to what we observed with infection with the Δmmpl7 mutant. To demonstrate this, we infected lung epithelial cells with Mtb Erdman and co-treated them with DATs. Our results show that co-treatment with DATs limited the production of G-CSF and IL-6 by WT Mtb infected epithelial cells (Figure 5A), similar to the responses observed when Δmmpl7 mutant single infection and coinfections were carried out (Figure 4). Additionally, the inhibitory effect observed was dose dependent, thus confirming that the overexpression of DATs was one factor that was skewing the induction of cytokines upon infection with the Δmmpl7 mutant. This effect was likely not due to DAT-induced cell death, as levels of total live, apoptotic, and dead cells were only slightly altered after addition of DATs (Supplemental Figure 3A). IL-10 production within this culture was below the limits of detection of the assay for all groups, even with addition of DATs, thus suggesting that epithelial cells are likely not a significant source of anti-inflammatory signals in this phenomenon.
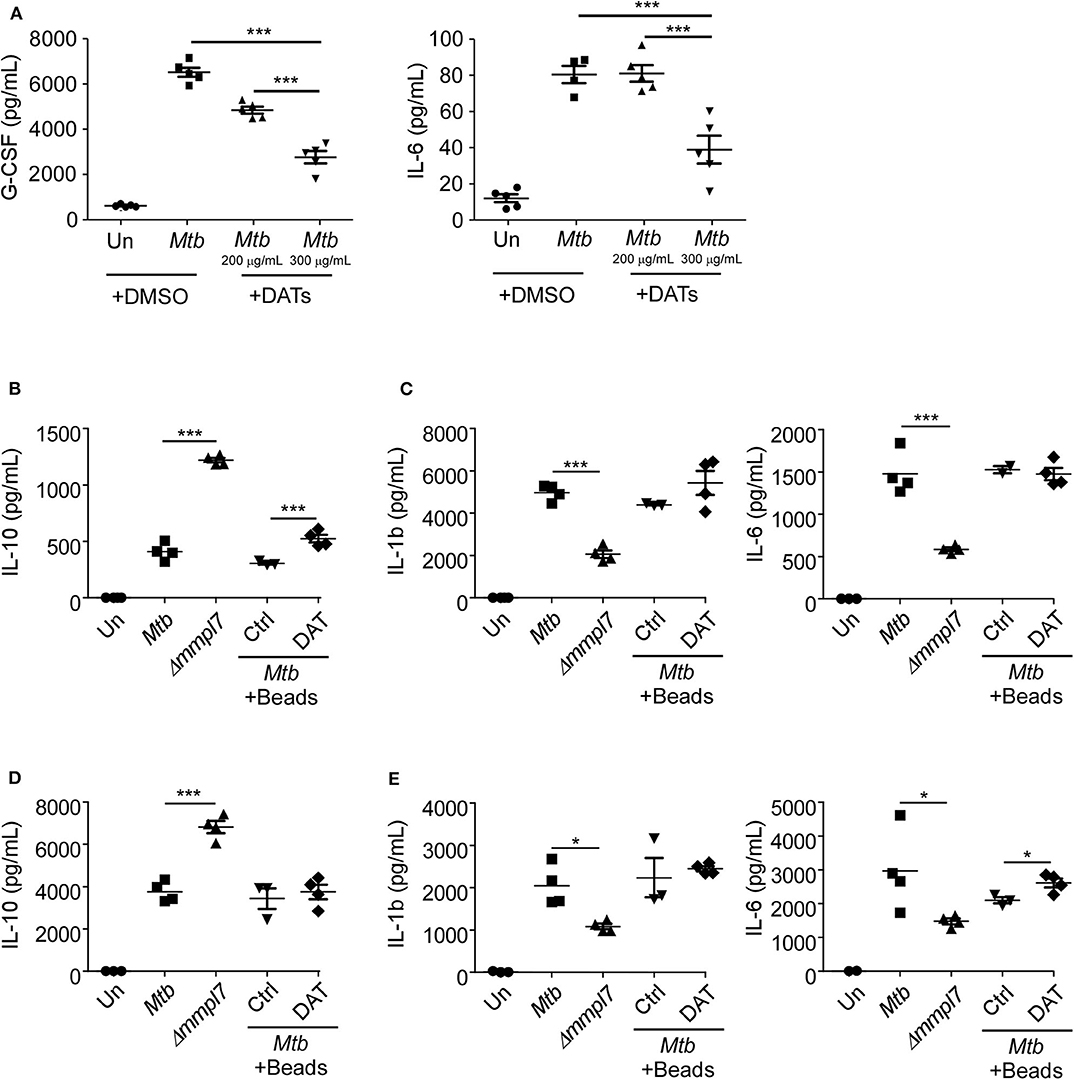
Figure 5. (A) DAT administration drives decreased inflammatory molecule production in epithelial and myeloid cells: Mouse lung epithelial cells (n = 5) were infected with Mtb Erdman WT with or without the addition of raw DATs at MOI = 1 for 6 days. Mouse bone-marrow derived (B,C) macrophages (BMDMs) (n = 5) or (D,E) dendritic cells (BMDCs) (n = 5) were infected with Mtb Erdman WT with or without the addition of DAT coated agarose beads, or Δmmpl7 for 3 days. Supernatants were collected and the concentrations of cytokines G-CSF, IL-6, IL-1β, and IL-10 were quantified by multiplex assays. Multiple groups were compared by 1-way ANOVA with Tukey's post-tests. *p < 0.05, **p < 0.01, ***p < 0.001.
We next aimed to determine if this phenotype was specific to epithelial cells or if the Δmmpl7 mutant also regulated cytokine expression in myeloid phagocytes, and if the presence of DATs modulated these responses as previously shown (19). Therefore, we infected macrophages and DCs with WT and Δmmpl7 mutant (Figures 5B–E). We found that while Δmmpl7 mutant abrogated IL-1β and IL-6 expression in both macrophages and DCs (Figures 5C,E), the Δmmpl7 mutant surprisingly induced increased IL-10 expression in both cell types when compared to WT Mtb (Figures 5B,D). Furthermore, no differences in cell death were observed between WT Mtb and the Δmmpl7 mutant (Supplemental Figures 3B,C), thus suggesting that this finding is due to specific regulation of cytokine production, not apoptosis induced abrogation. The decreased production of IL-1β and IL-6 in phagocytes was likely not only due to active inhibition by DAT alone, as co-treatment with DAT-coated beads did not decrease production of these cytokines during WT infection when compared to control beads. However, in BMDMs the increased induction of IL-10 driven by the Δmmpl7 mutant Mtb over WT Erdman was recapitulated by addition of DATs to WT Erdman infection (Figure 5B), thus suggesting that DAT overexpression on Δmmpl7 mutant Mtb actively induced expression of anti-inflammatory IL-10 produced by macrophages. The increased production of anti-inflammatory IL-10 by lung macrophages may explain the early increased IL-10 observed in vivo in lung homogenates. These early anti-inflammatory signals may abrogate the production of inflammatory cytokines and pulmonary neutrophilia, thus skewing the immune response away from neutrophil rich, non-protective granulomas, and toward iBALT containing, protective granulomas.
Discussion
Although pulmonary Mtb infection is the leading cause of death by an infectious agent worldwide, the initial steps in pathogenesis that govern the induction of iBALT formation within granulomas remain unknown. In this study, using a transposon library in the NHP pulmonary infection model, we have identified Mtb drivers of B cell-containing iBALT formation. Our work here demonstrates that murine infection with the Δmmpl7 mutant induces increased iBALT formation by dampening early innate immune responses. The Δmmpl7 mutant overexpressed DATs in the cell wall, which drove abrogated inflammatory molecule production and increased IL-10 in the lung, thus leading to decreased cellular recruitment. This study provides novel evidence for a critical role for Mtb specific factors in skewing the earliest host pathogen interactions that drive protective or detrimental disease outcomes.
Although significant advances have been made in understanding the factors that aid in the intracellular survival of Mtb and their potential as therapeutic targets using in vitro approaches and the mouse model, it is unclear if these factors play important roles in human tubercle formation. Furthermore, most physiological events cannot be modeled by targeting a single gene or its substrates, as interaction and/or compensation among several genes/pathways underpin complex outcomes. Thus, in this study we have examined the Mtb determinants that mediate granulomas by using the well-established preclinical model of NHP TB infection. The macaque model replicates many facets of clinically observed Mtb infection, thus providing relevance to identification of Mtb determinants that drive or limit granuloma and iBALT formation. By infecting NHPs with an Mtb mutant transposon library we have identified a novel subset of Mtb genes associated with protective iBALT containing granulomas that correspond with effective Mtb control. Although the gene subsets identified using this approach did not fall into any specific pathway, they were interrelated in that they were predominantly involved in mycobacterial growth and survival via responses to stress (Acr2, ClpB, Gln4, NdhA, and Cmtr) or the synthesis/translocation of virulence components including lipids & proteins (Mmpl2, Mmpl7, EccD5).
Among the two heat stress molecular chaperones identified in our study ClpB has already been established as essential for the in vitro growth of Mtb (27), however the importance of Acr2 is yet to be established. Acr2 is the most up-regulated gene following phagocytosis of Mtb by macrophages (27, 44) and is under the control of the master regulator PhoP (45). Wilkinson et al. found that Acr2 was strongly expressed in response to heat shock protein (HSP) Rv0251c and appears to play a role in early immune responses (46). HSPs assist in Mtb survival but also act as signaling agents to the host inflammatory mechanisms. While the role of Acr2 in Mtb virulence in humans is not clear, it remains essential for virulence in a murine model of TB (46). To our knowledge this is the first study to report on the association of absence of Acr2 with protective granulomas in the NHP model.
Until recently NdhA was considered a non-essential protein found in the inner mycobacterial membrane associated with nicotinamide adenine dinucleotide (NADH) mediated electron transfer to the electron transport chain (25). Recent studies by Vilcheze et al. concluded that when NdhA is the only type I NADH dehydrogenase present in Mtb it affects Mtb growth and renders it susceptible to oxidative stress (47). Likewise, while GlnA2 and GlnA4 are not associated with Mtb virulence in vivo another member of this family GlnA1 was found to contribute to Mtb virulence in a guinea pig model (48). Also, while a direct role for Cmtr in Mtb virulence is yet to be established, a transcriptional survey of intracellular mycobacteria and their host macrophages revealed signatures of heavy metal poisoning and an associated strong induction of Cmtr and Csor which are known to encode metal responsive transcriptional regulators (49).
The Mtb cell wall expresses a variety of virulence factors that contribute to bacterial survival and intrinsic drug resistance. Identification of genes that regulate uptake and secretion machinery across the membrane is critical to characterizing the corresponding secretory products and thus the pathogenesis of Mtb. EccD5 has been identified as being associated with protective granulomas. EccD5 as part of the ESX-5 system is involved in the translocation of Proline-Proline Glutamate (PPE) proteins and its disruption affects cell wall integrity leading to strong attenuation of the pathogen in a mouse model (26). But the impact of EccD5 in human Mtb infection or in NHPs is not known. Lastly, we also identified Mmpl2 and Mmpl7 to be associated with protective granulomas in the NHP model. Sequencing of the Mtb genome revealed 12 membrane proteins that were primarily involved in transport of Mtb lipids. MmpL-mediated lipid secretion impacts both the innate ability of the pathogen to survive intracellularly and also the host-pathogen interactions that determine the disease outcome. It has been previously established that only Mmpl4 and the well-characterized Mmpl7, which transports PDIM to the MOM, have both impaired growth kinetics and impaired lethality (50). In summary, our Mtb mutant transposon library infection model in the NHPs has identified for the first time several unique Mtb genes whose roles in Mtb virulence were previously considered as redundant.
Since Mmpl7 is the most well-characterized of all Mmpl genes being the known transporter of the virulence lipid PDIM, and as the substrate for Mmpl2 is currently not known (51), we decided to focus our further studies utilizing the Mmpl7 mutant strain (Δmmpl7). Beyond reduced expression of PDIM, the Δmmpl7 mutant also has an established in vivo growth defect in the lung, spleen, and liver of mice, which we compensated for in our study by infecting with higher doses of the mutant compared to wild type. The decreased CFU in the lungs of Δmmpl7 mutant infected mice that we observed is similar to previous findings by Cox et al. and others (52), but the decreased dissemination to the spleen was not previously observed. This is likely due to the use of different background Mtb strains and routes of infection in our study. Furthermore, our higher dose aerosol infection recapitulates a similar phenotype in increased iBALT formation as the standard low dose aerosol infection. Thus, our findings suggest that the improved TB disease outcomes observed is likely not simply due to the absence of PDIMs or a growth defect of the mutant, but that the observed increased iBALT formation is likely driven specifically by the Δmmpl7 mutant and overpression of DATs.
Despite using a higher dose of the Δmmpl7 mutant in our infection studies, we observed enhanced iBALT formation and abrogated inflammatory molecule production and cellular recruitment, including both myeloid and lymphoid cell types. As T cells are required for the formation of iBALT, it would seem that slightly decreased T cell accumulation should also yield decreased iBALT, which was not the case. However, our main observed differences in cellular recruitment involved decreased early neutrophil associated responses, specifically abrogated cytokines like IL-6 and G-CSF, and increased IL-10. It is known that neutrophils are overrepresented in non-protective, necrotic granulomas that do not control or contain Mtb, and that neutrophil counts in peripheral blood, as well as IL-6 levels, correlate with ATB disease (10, 32, 33).
Our study confirms these findings with heightened blood levels of G-CSF in people with active TB when compared to LTBI, and as such we used these two inflammatory cytokines as readouts for response severity for our in vitro experiments. Thus, these data suggest that a lack of IL-6 and G-CSF and decreased neutrophils may be skewing toward a dampened immune response, potentially mediated by increased early IL-10, that allows for the enhanced formation of iBALT containing granulomas as opposed to necrotic, neutrophil containing lesions. This hypothesis is also supported by our findings in vitro, which show decreased IL-6 and G-CSF in lung epithelial cells, a key source of inflammatory molecules, after Δmmpl7 mutant infection.
Importantly, we observed increased IL-10 production in macrophages and DCs after Δmmpl7 mutant infection. It is known that IL-10 antagonizes IL-17 production, and thus downstream G-CSF production (53), suggesting a potential mechanism for our observed dampened immune responses and improved outcomes. Indeed, Mtb is capable of dampening immune responses mediated by pattern recognition receptors (PRRs), and not just stimulating heightened inflammatory responses by interactions with these receptors.
MmpL7 is colocalized with genes involved in polyketide biosynthesis (pks genes) and genes involved in lipid metabolism (PapA, FadD) suggesting that it is involved in complex lipid transport involving more than one substrate in Mtb (54, 55). Therefore, we aimed to determine if there were any other differences in the lipid profile of Mtb beyond the loss of PDIM that could explain the observed abrogated cytokine responses in vivo and in vitro. We found that DATs are overrepresented in the Δmmpl7 mutant, validated by both 1D and 2D thin layer chromatography (TLC). MmpL10 is considered to be the putative agent responsible for the transport of DAT across the plasma membrane (51). Hence, it is likely that the absence of MmpL7 during infection could be compensated by MmpL10 function accounting for the observed accumulation of DATs. The addition of DATs during WT Mtb infection recapitulated our findings of decreased inflammatory cytokine production in epithelial cells and yielded increased IL-10 production by macrophages, both supporting what was observed in Δmmpl7 mutant infection in vivo. These findings suggest distinct mechanisms for driving dampened immune responses depending on cell types, with likely interactions of IL-10 sourced from macrophages driving decreased production of inflammatory cytokines by epithelial cells. DATs likely do not act in isolation, as addition of DATs to WT Mtb Erdman infection did not fully recapitulate all aspects of Δmmpl7 mutant infection in vitro in these model systems.
Mycobacterial lipid virulence factors such as PDIMs are known to mask pathogen associated molecular patterns (PAMPs) and the resultant downstream PRR signaling (40). Lipid virulence factors are also known to directly act as toll-like receptor (TLR) antagonists (56). Apart from TLRs, other surface receptors including Mincle, that recognizes trehalose moieties (42, 43), are likely involved in this phenomenon as BMDMs from Mincle deficient mice reportedly produce less G-CSF and TNF in response to Mtb infection (57). Moreover, the expression of TLRs is also known to vary between different phagocytic cell types (58). Hence these separate responses are likely due to differences in PRR expression and signaling by these cell types. Interestingly, previous studies have shown DAT dependent abrogation of inflammatory cytokine production in monocytes and macrophages in vitro (19). This study was performed using several different human phagocyte models without abrogating the expression of Mincle. While Mincle is established to interact with trehalose moieties, this study validates our claim that DATs can drive abrogated inflammatory cytokine production, independent of any manipulation of the expression of Mincle, suggesting a separate mechanism. It is likely that the interactions and modulated cytokine/chemokine production we have observed involves other PRRs and Mtb lipid factors, beyond the singular interaction between DATs and Mincle (43).
While the various immune cells and cytokine/chemokine signals that contribute to iBALT formation are well-established in a variety of disease contexts (59), the Mtb specific factors that drive these protective responses, and the mechanism of those interactions, are poorly understood. This study demonstrates that a single Mtb specific lipid factor can differentially affect the ability of various cell types to produce inflammatory and anti-inflammatory cytokines, all of which contribute in different aspects to the milieu that fosters conditions beneficial to iBALT formation. In the context of an immune structure as complex as iBALT, where multiple coordinated and regulated signals and cell types are required, future work will likely need to use a similar approach across several in vivo and in vitro model systems examining multiple cell types in order to elucidate the concise contribution of a specific Mtb factor and PRR interactions. Future work may also need to examine not only multiple cell types, but also locational and temporal cellular interactions throughout the course of infection, as a single Mtb factor is likely neither necessary nor sufficient in isolation to drive iBALT by interactions with a single cell type at one specific time point.
While this study was not exhaustive in analyzing all potential lipid factors involved, it provides a framework for determining the contributions of the other Mtb genes and potential candidate lipids in skewing toward protective outcomes. We acknowledge that the role of any specific lipid in skewing iBALT formation is not an isolated phenomenon, as Mtb mutants knocking out certain lipids inevitably drive compensatory overexpression of others. Furthermore, we expect that the increased iBALT formation we have observed is likely due to a combination of a lack of PDIM along with increased DATs, with the possibility of other lipids being involved that we did not identify or assay for.
Our novel findings demonstrate that genes associated with Mtb cell wall lipids are critical to the initial interactions between Mtb and the host and suggest that Mtb specific lipids are key determinants of the early immune response that skews toward formation of iBALT. Our findings further provide a list of Mtb gene candidates for future work examining conditions needed for protective iBALT formation, specifically aimed at early responses that determine these outcomes. We have also demonstrated for the first time a panel of novel Mtb genes that are associated with enhanced formation of protective iBALT containing granulomas and improved disease outcomes, while most work on this topic examines genes involved in detrimental host outcomes and loss of protection. This study thus provides a framework for future attenuated vaccine candidates and mechanistic studies across model systems.
Materials and Methods
Non-human Primate Infection
An Mtb (H37Rv) transposon site hybridization mutants (TraSH) library (kind gift of Dr. Chris Sassetti) was delivered high dose (≥100,000) CFU via bronchoscopic procedure into the lungs of triplicate Indian rhesus macaques. Delivery of bacteria to the lower lungs was confirmed via dilution plating and Mtb infection was confirmed by direct observation of the clinical signs of TB and concurrent positive tuberculin skin test. NHPs were humanely euthanized after being held 4–6 weeks due to development of disease using prespecified criteria (60–62). Lungs from infected animals were harvested and processed for mesodissection as previously described (63, 64). Briefly, lung sections were removed, formalin fixed and paraffin embedded. A series of unstained and H&E slides were then made and used with the mesodissection instrument and 2iD Imaging software. The H&E guide slide is used to direct the consumable mill bit to dissect the unstained slide and harvest the nuclear material for downstream upscaling and identification. By staining of B cells, iBALT associated lesions were first identified. The appropriate sections from H&E slides were then dissected and used for sequencing as previously described (65) to identify the single Mtb mutant associated with each individual lesion (23, 24).
Mice
C57Bl/6 (B6) mice used were 6–8 weeks old and sex matched. All treatments and conditions used to handle study animals are in accordance with the approved Institutional Animal Care and Use Committee (IACUC) guidelines at Washington University in St. Louis.
Bacterial Strains, Infection and Instillation
Mtb (Erdman) and ΔmmpL7 mutant (MJM39) (16) were stocked in Proskauer-Beck with 0.05% tween 80 and stored at −80°C. Prior to infection, frozen stocks were thawed and placed in a solution of sterile PBS for loading into the Glasscol nebulizer. Mice were infected with aerosolized Mtb in the above Glasscol nebulizer at tested doses of ~100 CFU (low) or ~500 CFU (5-fold higher) of Mtb per mouse. At the indicated time points mice were sacrificed via carbon dioxide narcosis and lungs and spleens were harvested.
Microscopy and Inflammation, B Cell Quantification
Lung pathology from formalin fixed paraffin embedded lungs was assessed digitally using the automated tool of the Zeiss Axioplan 2 microscope (Carl Zeiss) of H&E stained slides to quantify inflammation and immunofluorescence labeled slides B220, CD3 and counter stained with DAPI in order to assess lymphoid follicle area.
Bacterial Culture and Cytokine Analysis
Bacterial burden was assessed using serial 10-fold dilutions of lung or spleen and plated on 7H11 supplemented with OADC (oleic acid, bovine albumin, dextrose, and catalase). After 2–3 weeks incubation colonies were counted visually. Cytokine/chemokine expression was analyzed in lung homogenates from infected mice via Luminex (Millipore-Sigma) or ELISA (R&D).
Flow Cytometry
Flow cytometry was conducted on single cell preparations derived from infected and uninfected lungs using fluorochrome conjugated antibodies:
Myeloid antigen presenting cell panel (66): CD11b (M1/70), CD11c (HL3), Gr-1 (RB6-8C5), Siglec-F (E50-2440), Ly6G (1A8), Ly6C (AL-21), CD64 (X54-5/7.1), MHC-II (M5/114.15.2). Cells were defined as: AMs: CD11b−, CD11c+, Siglec-F+, CD64+. mDCs: CD11b+CD11c+, MHC-II+. Neutrophils: CD11b+, CD11c−, Gr-1hi. Monocytes: CD11b+ CD11c− Gr-1int and/or Ly6C+. Recruited macrophages: CD11b+ CD11c−, Gr-1lo, CD64+/−, Ly6C+/−, MHC-II+/−.
Activated lymphocyte panel (9, 67): CD3ε (500A2), CD4 (RM4-5), CB8α (53-6.7), CD44 (IM7), IFN-γ (XMG1.2).
Live, apoptotic, or dead cell panel (66): Annexin V (PE) and 7-AAD (PerCP-Cy5.5) from the BD Pharmingen Apoptosis Detection kit were utilized and cell percentages were quantified by flow cytometry as per manufacturer's suggested protocol (BD Biosciences). The percentage of cells were defined as live (Annexin V−, 7-AAD−), apoptotic (Annexin V+, 7-AAD−), and dead (Annexin V+, 7-AAD+).
Data collection and analysis were conducted on the BD LSR Fortessa, Fortessa-X20 Cytometers or the FACSJazz Cell Sorter, all with FACS Diva software and post-acquisition analysis conducted on FlowJo.
Human Serum Collection and Analysis
Human samples were collected on approval from the Ethics Committee of the National Institute for Respiratory Diseases, KwaZulu-Natal Research Institute for TB and HIV, Durban, South Africa; and The American British Cowdray Medical Center, Mexico City, Mexico on a protocol approved by the Ethics Committee of the National Institute for Respiratory Diseases (INER), KwaZulu-Natal Research Institute for TB and HIV (Currently renamed AHRI), South Africa and The American British Cowdray Medical Center. All subjects were of similar socioeconomic status and unrelated to the third generation as determined by a questionnaire. TB cases had symptoms (weight loss >10 kg, cough, fever, night sweats for >1 month, or cervical or axillary lymphadenopathy) and chest radiographic findings consistent with recent pulmonary TB, a positive sputum acid-fast smear and culture confirmed for Mtb. Fresh blood samples from active pulmonary TB patients were obtained from patients recruited to the Tuberculosis Outpatient Clinic, INER, Mexico, or the KwaZulu-Natal Research Institute for TB and HIV, South Africa. Serum samples from all patients with active TB were collected prior to anti-Mtb treatment and did not presented comorbidities such as diabetes, HIV, cancer, and COPD. LTBI patients were defined as asymptomatic individuals who had positive IGRA test results as previously described (10). We simultaneously recruited a group of HCs and these individuals were negative for IGRA tests at collection sites.
In vitro Culture, Isolation, Stimulation, and Infection
Mouse lung epithelial cell line C10 cells were cultured to confluence in 24 well plates in DMEM. Prior to in vitro infection hemocytometer counts from a representative well were used to calculate multiplicity of infection (MOI). C10 cells were washed once with sterile PBS and then infected with Mtb or Mtb mutant at an MOI of 1 in DMEM without antibiotics and incubated at 37°C, 7.5% CO2. Supernatants from the infection was collected and stored at −80°C. Cells were collected by tryspinization and stained as per manufacturer's protocol.
Primary B6 mouse bone marrow derived macrophages (BMDMs) and dendritic cells (BMDC) were prepared as previously described (66). Briefly, bone marrow was harvested in a solution of serum free DMEM and then passed through a 70 μm screen, spun down and then resuspended in red blood cell lysis solution. Equal volume of cDMEM was added and again spun down. The single cell suspension was plated at 1 × 106 cells per ml and supplemented with 4% GM-CSF for 7 days. At harvest, adherent cells were collected as BMDMs, and floating cells were collected as BMDCs, gently washed with CDMEM via centrifugation (1,200 rpm, 6 min at 4°C), resuspended and then plated at 1 × 106 cells per ml.
Indicated cell types were stimulated with either DATs resuspended in DMSO, or BSA- control or DAT-coated beads suspended in PBS as indicated at 200 μg/mL. Briefly, beads (1.5 × 109 Polybead polystyrene beads) were washed twice in 0.05 M carbonate-bicarbonate buffer (pH 9.6) and then incubated with 50 μg of DATs or buffer alone for 1 h at 37°C. Beads were then blocked with 5% BSA, washed repeatedly in 0.5% BSA, and finally adjusted to 4.0 × 108/ml in 0.5% BSA before being used (68).
Lipid Extraction and Analysis
Mtb bacterial pellets normalized by bacterial numbers (counted by microscopy, 1 × 1010) were extracted with chloroform:methanol (2:1, v/v) following the previously published method (17). Dried total lipids extracts were analyzed (loaded 100 μg by weight) by thin layer chromatography using chloroform:methanol:water (90:10:1, v/v/v) as the mobile phase, and 10% sulfuric acid in ethanol as a developer as described (68). Densitometry using NIH ImageJ software was used to quantify all the spots/lipids per lane in each TLC, and to calculate the percentage of total DATs and total polyacyl trehaloses (PATs) per lane. Densitometry analyses of each spot/lipids were also calculated as using NIH ImageJ software (n = 3, where each value corresponds to independent extractions and TLCs). DATs were purified by preparative TLC as previously described for other Mtb cell envelope lipids (68), and used for in vitro studies at the indicated concentrations, resuspended in DMSO.
Statistical Analysis
Data analysis was conducted in GraphPad Prism 5 (La Jolla, CA) using unpaired two tailed Student's t-test for comparison between two groups or one-way analysis of variance for multiple comparisons. Significance is denoted as: *p < 0.05, **p < 0.01, ***p < 0.001, not detected, ND.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by AHRI and INER ethics board. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Washington University in St. Louis, Tulane Primate Center.
Author Contributions
MD and OP designed and performed experiments, analyzed data, and prepared the manuscript. KT, JR-M, JS, and JT performed experiments, provided samples, and generated data. JC, JZ, and AS provided samples or reagents. DK and SK designed experiments and provided guidance and funding. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Washington University in St. Louis, NIH grant HL105427, AI111914-02 to SK, and AI123780 and AI134236-02 to SK and DK, NIH/NHLBI T32 AI007172 and T32 AI007163 to MD, T32 HL007317 to OP and KT.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01325/full#supplementary-material
References
2. LeibundGut-Landmann S, Weidner K, Hilbi H, Oxenius A. Nonhematopoietic cells are key players in innate control of bacterial airway infection. J Immunol. (2011) 186:3130–7. doi: 10.4049/jimmunol.1003565
3. Tian T, Woodworth J, Skold M, Behar SM. In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. J Immunol. (2005) 175:3268–72. doi: 10.4049/jimmunol.175.5.3268
4. Mayer-Barber KD, Barber DL. Innate and adaptive cellular immune responses to Mycobacterium tuberculosis infection. Cold Spring Harb Perspect Med. (2015) 5:a018424. doi: 10.1101/cshperspect.a018424
5. Arcos J, Sasindran SJ, Moliva JI, Scordo JM, Sidiki S, Guo H, et al. Mycobacterium tuberculosis cell wall released fragments by the action of the human lung mucosa modulate macrophages to control infection in an IL-10-dependent manner. Mucosal Immunol. (2017) 10:1248–58. doi: 10.1038/mi.2016.115
6. Reuschl AK, Edwards MR, Parker R, Connell DW, Hoang L, Halliday A, et al. Innate activation of human primary epithelial cells broadens the host response to Mycobacterium tuberculosis in the airways. PLoS Pathog. (2017) 13:e1006577. doi: 10.1371/journal.ppat.1006577
7. Scordo JM, Arcos J, Kelley HV, Diangelo L, Sasindran SJ, Youngmin E, et al. Mycobacterium tuberculosis cell wall fragments released upon bacterial contact with the human lung mucosa alter the neutrophil response to infection. Front Immunol. (2017) 8:307. doi: 10.3389/fimmu.2017.00307
8. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol. (2012) 3:411. doi: 10.3389/fimmu.2012.00411
9. Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest. (2013) 123:712–26. doi: 10.1172/JCI65728
10. Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med. (2013) 188:1137–46. doi: 10.1164/rccm.201304-0803OC
11. Nouailles G, Dorhoi A, Koch M, Zerrahn J, Weiner J, Fae KC, et al. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J Clin Invest. (2014) 124:1268–82. doi: 10.1172/JCI72030
12. Lastrucci C, Bénard A, Balboa L, Pingris K, Souriant S, Poincloux R, et al. Tuberculosis is associated with expansion of a motile, permissive and immunomodulatory CD16(+) monocyte population via the IL-10/STAT3 axis. Cell Res. (2015) 25:1333–51. doi: 10.1038/cr.2015.123
13. Flynn JL, Capuano SV, Croix D, Pawar S, Myers A, Zinovik A, et al. Non-human primates: a model for tuberculosis research. Tuberculosis. (2003) 83:116–8. doi: 10.1016/S1472-9792(02)00059-8
14. Kaushal D, Mehra S, Didier PJ, Lackner AA. The non-human primate model of tuberculosis. J Med Primatol. (2012) 41:191–201. doi: 10.1111/j.1600-0684.2012.00536.x
15. Cox JS, Chen B, McNeil M, Jacobs WR Jr. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. (1999) 402:79–83. doi: 10.1038/47042
16. Jain M, Cox JS. Interaction between polyketide synthase and transporter suggests coupled synthesis and export of virulence lipid in M. tuberculosis PLoS Pathog. (2005) 1:e2. doi: 10.1371/journal.ppat.0010002
17. Muñoz M, Lanéelle MA, Luquin M, Torrelles J, Julián E, Ausina V, et al. Occurrence of an antigenic triacyl trehalose in clinical isolates and reference strains of Mycobacterium tuberculosis. FEMS Microbiol Lett. (1997) 157:251–9. doi: 10.1016/S0378-1097(97)00483-7
18. Saavedra R, Segura E, Leyva R, Esparza LA, Lopez-Marin LM. Mycobacterial di-O-acyl-trehalose inhibits mitogen- and antigen-induced proliferation of murine T cells in vitro. Clin Diagn Lab Immunol. (2001) 8:1081–8. doi: 10.1128/CDLI.8.6.1-91-1088.2001
19. Lee K-S, Dubey VS, Kolattukudy PE, Song C-H, Shin AR, Jung S-B, et al. Diacyltrehalose of Mycobacterium tuberculosis inhibits lipopolysaccharide- and mycobacteria-induced proinflammatory cytokine production in human monocytic cells. FEMS Microbiol Lett. (2007) 267:121–8. doi: 10.1111/j.1574-6968.2006.00553.x
20. Scanga CA, Flynn JL. Modeling tuberculosis in nonhuman primates. Cold Spring Harb Perspect Med. (2014) 4:a018564. doi: 10.1101/cshperspect.a018564
21. Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci USA. (2001) 98:12712–7. doi: 10.1073/pnas.231275498
22. Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. (2003) 48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x
23. Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genet. (2011) 43:482–6. doi: 10.1038/ng.811
24. Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med. (2014) 20:75–9. doi: 10.1038/nm.3412
25. Awasthy D, Ambady A, Narayana A, Morayya S, Sharma U. Roles of the two type II NADH dehydrogenases in the survival of Mycobacterium tuberculosis in vitro. Gene. (2014) 550:110–6. doi: 10.1016/j.gene.2014.08.024
26. Bottai D, Di Luca M, Majlessi L, Frigui W, Simeone R, Sayes F, et al. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol Microbiol. (2012) 83:1195–209. doi: 10.1111/j.1365-2958.2012.08001.x
27. Stewart GR, Mangan JA, Laing KG, Hinds J, Young DB, Butcher PD, et al. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays a. Microbiology. (2002) 148:3129–38. doi: 10.1099/00221287-148-10-3129
28. Lee S, Jeon BY, Bardarov S, Chen M, Morris SL, Jacobs WR. Protection elicited by two glutamine auxotrophs of Mycobacterium tuberculosis and in vivo growth phenotypes of the four unique glutamine synthetase mutants in a murine model. Infect Immun. (2006) 74:6491–5. doi: 10.1128/IAI.00531-06
29. Cavet JS, Graham AI, Meng W, Robinson NJ. A cadmium-lead-sensing ArsR-SmtB repressor with novel sensory sites. Complementary metal discrimination by NmtR AND CmtR in a common cytosol. J Biol Chem. (2003) 278:44560–6. doi: 10.1074/jbc.M307877200
30. Wu J, Wang S, Lu C, Shao L, Gao Y, Zhou Z, et al. Multiple cytokine responses in discriminating between active tuberculosis and latent tuberculosis infection. Tuberculosis. (2017) 102:68–75. doi: 10.1016/j.tube.2016.06.001
31. Ouyang W, O'Garra A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity. (2019) 50:871–91. doi: 10.1016/j.immuni.2019.03.020
32. Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. (2010) 466:973–7. doi: 10.1038/nature09247
33. Verbon A, Juffermans N, Van Deventer SJ, Speelman P, Van Deutekom H, Van Der Poll T. Serum concentrations of cytokines in patients with active tuberculosis and after treatment. Clin Exp Immunol. (1999) 115:110–3. doi: 10.1046/j.1365-2249.1999.00783.x
34. Chandrashekara S, Anupama KR, Sambarey A, Chandra N. High IL-6 and low IL-15 levels mark the presence of TB infection: a preliminary study. Cytokine. (2016) 81:57–62. doi: 10.1016/j.cyto.2016.02.003
35. Bermudez LE, Goodman J. Mycobacterium tuberculosis invades and replicates within type II alveolar cells. Infect Immun. (1996) 64:1400–6. doi: 10.1128/IAI.64.4.1400-1406.1996
36. Kube D, Sontich U, Fletcher D, Davis PB. Proinflammatory cytokine responses to P. aeruginosa infection in human airway epithelial cell lines. Am J Physiol Lung Cell Mol Physiol. (2001) 280:L493–502. doi: 10.1152/ajplung.2001.280.3.L493
37. Arcos J, Diangelo LE, Scordo JM, Sasindran SJ, Moliva JI, Turner J, et al. Lung mucosa lining fluid modification of Mycobacterium tuberculosis to reprogram human neutrophil killing mechanisms. J Infect Dis. (2015) 212:948–58. doi: 10.1093/infdis/jiv146
38. Scordo JM, Knoell DL, Torrelles JB. Alveolar epithelial cells in Mycobacterium tuberculosis infection: active players or innocent bystanders? J Innate Immun. (2016) 8:3–14. doi: 10.1159/000439275
39. Dudek M, Puttur F, Arnold-Schrauf C, Kühl AA, Holzmann B, Henriques-Normark B, et al. Lung epithelium and myeloid cells cooperate to clear acute pneumococcal infection. Mucosal Immunol. (2016) 9:1288–302. doi: 10.1038/mi.2015.128
40. Quigley J, Hughitt VK, Velikovsky CA, Mariuzza RA, El-Sayed NM, Briken V. The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of Mycobacterium tuberculosis. MBio. (2017) 8:e00148-17. doi: 10.1128/mBio.00148-17
41. Jackson M. The mycobacterial cell envelope-lipids. Cold Spring Harb Perspect Med. (2014) 4:a021105. doi: 10.1101/cshperspect.a021105
42. Feinberg H, Rambaruth ND, Jegouzo SA, Jacobsen KM, Djurhuus R, Poulsen TB, et al. Binding sites for acylated trehalose analogs of glycolipid ligands on an extended carbohydrate recognition domain of the macrophage receptor mincle. J Biol Chem. (2016) 291:21222–33. doi: 10.1074/jbc.M116.749515
43. Decout A, Silva-Gomes S, Drocourt D, Barbe S, Andre I, Cueto FJ, et al. Rational design of adjuvants targeting the C-type lectin Mincle. Proc Natl Acad Sci USA. (2017) 114:2675–80. doi: 10.1073/pnas.1612421114
44. Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med. (2003) 198:693–704. doi: 10.1084/jem.20030846
45. Sevalkar RR, Arora D, Singh PR, Singh R, Nandicoori VK, Karthikeyan S, et al. Functioning of mycobacterial heat shock repressors requires the master virulence regulator PhoP. J Bacteriol. (2019) 201:e00013-19. doi: 10.1128/JB.00013-19
46. Wilkinson KA, Stewart GR, Newton SM, Vordermeier HM, Wain JR, Murphy HN, et al. Infection biology of a novel alpha-crystallin of Mycobacterium tuberculosis: Acr2. J Immunol. (2005) 174:4237–43. doi: 10.4049/jimmunol.174.7.4237
47. Vilcheze C, Weinrick B, Leung LW, Jacobs WR Jr. Plasticity of Mycobacterium tuberculosis NADH dehydrogenases and their role in virulence. Proc Natl Acad Sci USA. (2018) 115:1599–604. doi: 10.1073/pnas.1721545115
48. Tullius MV, Harth G, Horwitz MA. Glutamine synthetase GlnA1 is essential for growth of Mycobacterium tuberculosis in human THP-1 macrophages and guinea pigs. Infect Immun. (2003) 71:3927–36. doi: 10.1128/IAI.71.7.3927-3936.2003
49. Botella H, Peyron P, Levillain F, Poincloux R, Poquet Y, Brandli I, et al. Mycobacterial p(1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe. (2011) 10:248–59. doi: 10.1016/j.chom.2011.08.006
50. Domenech P, Reed MB, Barry CE. Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect Immun. (2005) 73:3492–501. doi: 10.1128/IAI.73.6.3492-3501.2005
51. Melly G, Purdy GE. MmpL proteins in physiology and pathogenesis of M. tuberculosis. Microorganisms. (2019) 7:70. doi: 10.3390/microorganisms7030070
52. Rousseau C, Winter N, Pivert E, Bordat Y, Neyrolles O, Ave P, et al. Production of phthiocerol dimycocerosates protects Mycobacterium tuberculosis from the cidal activity of reactive nitrogen intermediates produced by macrophages and modulates the early immune response to infection. Cell Microbiol. (2004) 6:277–87. doi: 10.1046/j.1462-5822.2004.00368.x
53. Monin L, Khader SA. Chemokines in tuberculosis: the good, the bad and the ugly. Semin Immunol. (2014) 26:552–8. doi: 10.1016/j.smim.2014.09.004
54. Tekaia F, Gordon SV, Garnier T, Brosch R, Barrell BG, Cole ST. Analysis of the proteome of Mycobacterium tuberculosis in silico. Tuber Lung Dis. (1999) 79:329–42. doi: 10.1054/tuld.1999.0220
55. Minnikin DE, Kremer L, Dover LG, Besra GS. The methyl-branched fortifications of Mycobacterium tuberculosis. Chem Biol. (2002) 9:545–53. doi: 10.1016/S1074-5521(02)00142-4
56. Blanc L, Gilleron M, Prandi J, Song OR, Jang MS, Gicquel B, et al. Mycobacterium tuberculosis inhibits human innate immune responses via the production of TLR2 antagonist glycolipids. Proc Natl Acad Sci USA. (2017) 114:11205–10. doi: 10.1073/pnas.1707840114
57. Heitmann L, Schoenen H, Ehlers S, Lang R, Holscher C. Mincle is not essential for controlling Mycobacterium tuberculosis infection. Immunobiology. (2013) 218:506–16. doi: 10.1016/j.imbio.2012.06.005
58. Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. (2002) 168:554–61. doi: 10.4049/jimmunol.168.2.554
59. Marin ND, Dunlap MD, Kaushal D, Khader SA. Friend or foe: the protective and pathological roles of inducible bronchus-associated lymphoid tissue in pulmonary diseases. J Immunol. (2019) 202:2519–26. doi: 10.4049/jimmunol.1801135
60. Kaushal D, Foreman TW, Gautam US, Alvarez X, Adekambi T, Rangel-Moreno J, et al. Mucosal vaccination with attenuated Mycobacterium tuberculosis induces strong central memory responses and protects against tuberculosis. Nat Commun. (2015) 6:8533. doi: 10.1038/ncomms9533
61. Mehra S, Foreman TW, Didier PJ, Ahsan MH, Hudock TA, Kissee R, et al. The DosR regulon modulates adaptive immunity and is essential for Mycobacterium tuberculosis persistence. Am J Respir Crit Care Med. (2015) 191:1185–96. doi: 10.1164/rccm.201408-1502OC
62. Gautam US, Foreman TW, Bucsan AN, Veatch AV, Alvarez X, Adekambi T, et al. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. (2018) 115:E62–e71. doi: 10.1073/pnas.1711373114
63. Hudock TA, Lackner AA, Kaushal D. Microdissection approaches in tuberculosis research. J Med Primatol. (2014) 43:294–7. doi: 10.1111/jmp.12141
64. Hudock TA, Foreman TW, Bandyopadhyay N, Gautam US, Veatch AV, LoBato DN, et al. Hypoxia sensing and persistence genes are expressed during the intragranulomatous survival of Mycobacterium tuberculosis. Am J Respir Cell Mol Biol. (2017) 56:637–47. doi: 10.1165/rcmb.2016-0239OC
65. Dutta NK, Mehra S, Didier PJ, Roy CJ, Doyle LA, Alvarez X, et al. Genetic requirements for the survival of tubercle bacilli in primates. J Infect Dis. (2010) 201:1743–52. doi: 10.1086/652497
66. Dunlap MD, Howard N, Das S, Scott N, Ahmed M, Prince O, et al. A novel role for C-C motif chemokine receptor 2 during infection with hypervirulent Mycobacterium tuberculosis. Mucosal Immunol. (2018) 11:1727–42. doi: 10.1038/s41385-018-0071-y
67. Griffiths KL, Ahmed M, Das S, Gopal R, Horne W, Connell TD, et al. Targeting dendritic cells to accelerate T-cell activation overcomes a bottleneck in tuberculosis vaccine efficacy. Nat Commun. (2016) 7:13894. doi: 10.1038/ncomms13894
Keywords: Tuberculosis, lung, innate immunity, pulmonary, epithelial cells, iBALT
Citation: Dunlap MD, Prince OA, Rangel-Moreno J, Thomas KA, Scordo JM, Torrelles JB, Cox J, Steyn AJC, Zúñiga J, Kaushal D and Khader SA (2020) Formation of Lung Inducible Bronchus Associated Lymphoid Tissue Is Regulated by Mycobacterium tuberculosis Expressed Determinants. Front. Immunol. 11:1325. doi: 10.3389/fimmu.2020.01325
Received: 08 October 2019; Accepted: 26 May 2020;
Published: 30 June 2020.
Edited by:
Jérôme Nigou, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Marta Romano, Scientific Institute of Public Health (WIV-ISP), BelgiumMax Bastian, Friedrich Loeffler Institute, Germany
Copyright © 2020 Dunlap, Prince, Rangel-Moreno, Thomas, Scordo, Torrelles, Cox, Steyn, Zúñiga, Kaushal and Khader. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shabaana A. Khader, a2hhZGVyQHd1c3RsLmVkdQ==; Deepak Kaushal, ZGthdXNoYWxAdHhiaW9tZWQub3Jn
†These authors have contributed equally to this work and share first authorship