 Cai Zhang
Cai Zhang Yuxia Liu
Yuxia Liu- Institute of Immunopharmaceutical Sciences, School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, China
Checkpoint blockade therapy, for example using antibodies against CTLA-4 and PD-1/PD-L1, relieves T cells from the suppression by inhibitory checkpoints in the tumor microenvironment; thereby achieving good outcomes in the treatment of different cancer types. Like T cells, natural killer (NK) cell inhibitory receptors function as checkpoints for NK cell activation. Upon interaction with their cognate ligands on infected cells, tumor cells, dendritic cells and regulatory T cells, signals from these receptors severely affect NK cells' activation and effector functions, resulting in NK cell exhaustion. Checkpoint inhibition with antagonistic antibodies (Abs) can rescue NK cell exhaustion and arouse their robust anti-tumor capacity. Most notably, the response to anti-PD-1 therapy can be enhanced by the increased frequency and activation of NK cells, thereby increasing the overall survival of patients with multiple types of cancer. In addition, rescue of NK cell activity could enhance adaptive T cells' anti-tumor activity. Some antagonistic Abs (e.g., anti-TIGIT and anti-NKG2A monoclonal Abs) have extraordinary potential in cancer therapy, as evidenced by their induction of potent anti-tumor immunity through recovering both NK and T cell function. In this review, we summarize the dysfunction of NK cells in the tumor microenvironment and the key NK cell checkpoint receptors or molecules that control NK cell function. We particularly focus on recent advances in the most promising strategies through blockade of NK cell checkpoints or their combination with other approaches to more effectively reject tumors.
Introduction
In the past few years, cancer immunotherapy has shown great promise. Among the most promising therapeutic modalities, chimeric antigen receptor T cell (CAR-T) and immune checkpoint blockade therapies have demonstrated unprecedented clinical success and represent a turning point in cancer treatment. In particular, checkpoint blockade, such as the use of monoclonal antibodies (mAbs) against cytotoxic T-lymphocyte associated protein 4 (CTLA-4), programmed death ligand 1 (PD-L1), and programmed cell death 1 (PD-1), has shown impressive potential to treat different cancer types by relieving the suppression from the inhibitory checkpoints in the tumor microenvironment (TME) (1, 2). Both CTLA-4 and PD-1 were originally identified as co-inhibitory receptors on T cells; therefore, checkpoint blockade immunotherapy has concentrated on recovering T cell-mediated anti-tumor capacity to regress tumors (2). Antibodies (Abs) against CTLA-4 (Ipilimumab), PD-1 (Pembrolizumab, Nivolumab, and Cemiplimab), and PD-L1 (Durvalumab, Atezolizumab, and Avelumab) have been approved by the Food and Drug Administration (FDA) to treat patients with advanced melanoma, lymphoma, non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC), bladder cancer, head and neck squamous cell cancer (HNSCC), liver cancer, and Merkel cell carcinoma. Currently, mAbs that target other checkpoint receptors, such as T cell immunoglobulin and mucin domain-3 (TIM-3) and lymphocyte-activation gene 3 (LAG-3), are being tested pre-clinically or are in clinical trials (3–5).
Natural killer (NK) cells are the most important effectors of innate immune surveillance because of their ability to kill virus-infected cells and tumor cells without the need for prior activation. NK cells not only recognize and exert direct cytotoxicity against target cells rapidly at the early stage of tumorigenesis, but also engage in cross-talk with immune cells, including dendritic cells (DCs), to alter adaptive immune responses, which enhances anti-tumor responses mediated by CD8+T cells. In multiple tumor settings, the antitumor abilities of CD8+ T cells require NK cell function (6). Intriguingly, NK cells have adaptive immunity features and can obtain memory-like responses similar to T and B cells (7–9). NK cells express multiple activating receptors, inhibitory receptors, and cytokine or chemokine receptors on their surfaces. NK cell activation and function rely on a balance between signaling from inhibitory receptors and activating receptors. Generally, host homeostasis is maintained by a preponderance of inhibitory signals derived from inhibitory receptors over activation signals (10). Similar to T cells, most of the inhibitory receptors on NK cells act as checkpoints to control NK cell activation (11–13). Increasing data indicate that chronic viral infection or the TME can upregulation killer immunoglobulin-like receptors (KIRs), NKG2A, PD-1, TIM-3, and T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibition motif domains (TIGIT) on the surfaces of NK cells. Signals from these inhibitory receptors, generated by interactions with their cognate ligands on tumor cells, infected cells, DCs, and regulatory T cells (Tregs), severely affect the activation and effector function of NK cells, even resulting in NK cell functional exhaustion (14). Checkpoint inhibition with antagonistic Abs can release the inhibitory signals that limit NK cell activity and restore NK cell function. Notably, some antagonistic Abs (e.g., anti-TIGIT and anti-NKG2A mAbs) can induce potent anti-tumor immunity via the recovery of the functions of both NK and T cells, thus revealing extraordinary potential in cancer therapy (6, 15). In this review, we summarize the dysfunction of NK cells in the TME and the key NK cell checkpoint receptors or molecules that control NK cell function. We particularly focus on recent advances in the most promising strategies for therapy, such as NK cell checkpoint blockade or its combination with other approaches to more effectively reject tumors.
NK Cell Dysfunction in the Tumor Microenvironment
Although NK cells play crucial roles in controlling tumor growth and clearance of viral infection, NK cell function is often impaired significantly during tumor progression and development, and in chronic infection. Numbers of NK cell infiltrating into tumor sites are usually decreased, and NK cell function and activation are severely inhibited. NK cell dysfunction promotes the progression and metastasis of tumors. Decreased NK cell cytolytic activity and cytokine secretion capacity are associated significantly with poor prognosis in various cancers (16, 17). Activating receptors of NK cells, such as NKG2D, CD226, and NKp30, are downregulated on NK cells and correlate inversely with the progression and metastasis of melanoma, colorectal carcinoma, hepatocellular carcinoma (HCC), breast cancer, prostate cancer, gastric cancer, and others (18). Pulmonary or hepatic metastases are accelerated if NK cells are depleted in multiple mouse models (19–21). High numbers of tumor-infiltrating NK cells, high levels of NK activating receptors, and a high capacity for NK lysis correlate positively with longer overall survival and better prognosis (18, 22).
Many factors in the TME prevent NK cells infiltrating inside the tumor sites and hamper their activation and anti-tumor activity (23–25). Extracellular matrix components, such as collagen and hyaluronic acid, are densely packed in pancreatic cancer tissues, which prevent NK cells migrating into the tumor bed (26, 27). Neuroblastoma cells prevent NK cell recruitment by modulating the expression of chemokine receptors on NK cells (25, 28). Stromal cells, tumor-associated macrophages (TAMs), myeloid-derived suppressive cells (MDSCs), and Tregs in the TME inhibit NK cell activation and function by the secretion of immunosuppressive cytokines or interfering with receptor expression of NK cells. Transforming growth factor-β (TGF-β), IL-10, prostaglandin E2 (PEG2), and indoleamine 2,3-dioxygenase (IDO) are the major soluble modulators secreted in the TME, which negatively regulate NK cell function (29, 30). TGF-β inhibits IFN-γ secretion and NK cell cytolytic activity by suppressing the SMAD3 signal pathway. It also silences the expression of NKG2D and NKp30 on NK cells, as well as decreases the expression of NKG2D ligands on tumor cells (31). TGF-β is reported to block IL-15-induced mTOR signaling, thus attenuating the metabolic activity, proliferation, receptor expression, and cytotoxicity of NK cells (32). The TME-derived TGF-β can induce NK cells to transdifferentiate into innate lymphoid cell (ILC) type 1 cells (ILC1), which have no cytotoxic activity, thus promoting tumor escape from immune attack (33). Tregs inhibit the effector function of NK cells by directly secreting TGF-β or downregulating the expression of NKp30 and NKG2D indirectly using membrane-bound TGF-β (34, 35). MDSCs drive the differentiation and expansion of Tregs, thus reducing the anti-tumor effect of both cytotoxic T lymphocytes (CTLs) and NK cells. Membrane-bound TGF-β on MDSCs can also downregulate NK cytokine secretion and cell-mediated cytotoxicity via producing nitric oxide (NO) or in a method dependent on NK-expressed NKp30 (36–38). Recently, NK cell metabolism has been highlighted in determining NK cell fate and facilitating NK cell survival, proliferation, and function. Many factors in the TME modulate NK cell metabolism and contribute to NK cell dysfunction. TGF-β can suppress NK cell metabolism either directly by impairing glycolysis-promoting mTOR signaling, or by inhibiting mitochondrial metabolism via canonical TGF-β signaling (32, 39). Tumor-infiltrating NK cells can upregulate the expression of fructose-1,6-bisphosphatase (FBP1), which impairs glycolysis, thus promoting NK cell dysfunction in the lung cancer microenvironment (40). Insufficient levels of glucose and amino acids (consumed by tumor cells) in the TME can inhibit NK cell metabolism, including oxidative phosphorylation (OXPHOS) and glycolysis. The high levels of metabolic end products in the TME, such as lactate, dampen NK cell metabolism by inducing mitochondrial dysfunction, leading to apoptosis of tumor-infiltrating liver-resident NK cells in patients with colorectal liver metastasis (41). Adenosine, a critical immunosuppressive purine metabolite in the TME, inhibits the activity of tumor-infiltrating immune cells, including T cells, DCs, macrophages and NK cells, and also enhances the immunosuppressive effect of Tregs and MDSCs (42, 43). By binding to purinergic adenosine receptors on surfaces of NK cells, adenosine inhibits the maturation, activation, proliferation, cytokine production and cytolytic capacity of NK cells (44–46).
Tumor cells, antigen-presenting cells (APCs), Tregs, and MDSCs in the TME express high levels of PD-L1 and other ligands of immune checkpoint molecules, which prevent NK cell activation, even resulting in NK cell dysfunction or exhaustion (23). Meanwhile, the immunosuppressive TME alters the balance between inhibitory or activating NK cell receptors by downregulating activating receptor expression and upregulating inhibitory receptor expression, including PD-1, TIM-3, LAG-3, NKG2A, and TIGIT, which are referred to as checkpoint receptors. The interaction between checkpoint receptors and their respective ligands is the major contributor to NK cell exhaustion. Checkpoint blockade can reverse NK cell function in multiple tumors (25). Therefore, NK cell-based immunotherapy targeting checkpoint molecules has shown marked promise to treat both solid tumors and hematological malignancies.
The Key NK Cell Checkpoint Receptors or Molecules That Control NK Cell Activation and Function
KIR
Human KIRs are encoded within the leukocyte receptor complex (LRC) on chromosome 19. They belong to the Ig superfamily, and interact with major histocompatibility complex (MHC) class I molecules on target cells. The KIR family comprises both inhibitory and activating receptors. In the inhibitory KIRs, inhibitory signaling is transduced via the immunoreceptor tyrosine-based inhibition motifs (ITIMs) located in the long cytoplasmic tails. The activating KIRs are short-tail and are associated with the adaptor molecule DAP12 or FcγR to provide activating signals to NK cells (47). Each inhibitory KIR contains two (KIR2DL) or three (KIR3DL) extracellular Ig domains which confer their specificity for human leukocyte antigen (HLA) molecules. Thus, in this way, they can sense the downregulation or alteration of HLA molecules during viral infection and malignancy (48). Generally, KIRs are constitutively expressed on NK cells, and the inhibitory signals from inhibitory KIRs typically predominate over activating signals from activating KIRs. Thus, the activation and function of NK cells are fine-tuned via inhibitory KIRs to prevent damage to normal healthy cells mediated by NK cells and the induction of virus infected cells or tumor cells.
However, during tumor malignancy, inhibitory KIRs are upregulated whereas activating KIRs are downregulated in various types of cancer, such as lymphoma, leukemia, breast cancer, NSCLC, skin cancer, and biliary cancer (14, 49). These alterations restrain the activation and anti-tumor capacity of NK cells, thus contributing to tumor escape from immunosurveillance. Inhibitory KIRs were the first identified NK cell checkpoint receptors (50). mAbs recognizing inhibitory KIRs might induce NK cells' anti-tumor activity by blockading the inhibitory signaling pathway, thus several anti-KIR mAbs, for example those targeting KIR2DL1-3, have been applied to treat patients with leukemia, lymphoma, multiple myeloma, and some solid tumors in multiple clinical trials, which have been demonstrated as safe with limited side effects (14, 51).
NKG2A
NKG2A, a C-type lectin family member, is a type II membrane receptor encoded by a gene on chromosome 12 within the NK gene complex. CD94, lacking an intracellular signal domain, is covalently associated with NKG2A, which has ITIM domains in its cytoplasmic tail, to form a CD94/NKG2A heterodimer. NKG2A is expressed on approximately 50% of NK cells and 5% of CD8+T cells in human peripheral blood. Cytokines, such as IL-15, and chronic inflammation, upregulate NKG2A expression. Through ligation with its ligand HLA-E in humans or Qa-1 in mice, NKG2A transduces negative signaling to NK cells to prevent NK cell activation and thus suppress cytokine secretion and cytotoxicity. Notably, the surface expression of HLA-E depends on peptides derived from the conserved region of the leader peptide sequences of HLA-A, HLA-B, HLA-C, and HLA-G. Therefore, the CD94-NKG2A axis can monitor the antigen presenting capacity and the expression of HLA class I molecules on target cells (48). Similar to KIRs, the ITIM-based inhibitory signal from CD94/NKG2A is dominant over the activation signaling from its counterpart CD94/NKG2C.
Tumor-infiltrating NK cells express high levels of NKG2A in comparison with peripheral or splenic NK cells, while HLA-E is upregulated on both hematopoietic and solid tumors (17, 52). The interaction of NKG2A and HLA-E inhibited the anti-tumor effector function of both NK cells and some CD8+T cell subsets that upregulate NKG2A in the TME. In patients with HCC, high levels of NKG2A on tumor-infiltrating NK cells and high levels of HLA-E in intratumor tissues were observed, and poor prognosis was associated with the functional exhaustion of high NKG2A-expressing CD56dim NK cells (17). Worse survival was associated with a high density of NKG2A+ tumor infiltrating NK cells and high levels of HLA-E in tumor tissues in patients with ovarian cancer, RCC, colorectal cancer, breast cancer, and lung cancer (51–53). NKG2A is regarded as the major immune checkpoint of NK cells because of the suppressive effect of NKG2A on NK cells. The use of anti-NKG2A mAbs for therapeutic blockade of NKG2A restored the cytolytic activity of NK cells against HLA-E+ chronic lymphocytic leukemia (CLL) targets, thus improving NK cell dysfunction (54). Increasing evidence indicates that interruption of the interaction between NKG2A and HLA-E can induce an effective anti-tumor immune response. NKG2A blockade enhanced both CD8+T cell and NK cell-mediated anti-tumor effects. In particular, combined co-blockade with an anti-PD-L1 mAb promoted the generation of protective anti-tumor memory in various tumors, including lymphoma in a mouse model, HNSCC patients, and CRC patients (15). mAbs targeting NKG2A, alone or combined with other therapeutic strategies, are being used currently to restore the effector function of NK and CD8+T cells in clinical trials (15).
TIGIT and CD96
TIGIT, a member of immunoglobulin (Ig) superfamily, consists of an extracellular Ig V domain, a transmembrane domain, and a cytoplasmic tail containing an ITIM motif and an Ig tail-tyrosine (ITT)-like motif, which transduces inhibitory signals. CD96 is also an Ig superfamily member and comprises three extracellular Ig-like domains and a short ITIM motif-containing cytoplasmic tail, mediating inhibitory functions. TIGIT and CD96 act as inhibitory receptors and compete with activating receptor DNAX accessory molecule-1 (DNAM-1) or CD226 to bind to CD155 and CD112. The affinity of TIGIT or CD96 binding to CD155 is higher than that to DNAM-1; therefore, similar to other NK cell inhibitory receptors, inhibitory signals from TIGIT or CD96 are usually dominant over the signals from DNAM-1-mediated co-stimulation. Human NK cells, Tregs, memory T cells, and some CD8+T cells, express TIGIT, while in the TME, tumor-infiltrating NK cells show upregulated TIGIT expression accompanied by decreased DNAM-1 levels. CD155 is barely expressed in healthy human tissues, but it is dramatically overexpressed in various cancers, being considered an independent prognostic marker for poor prognosis for patients with breast cancer (55). Binding of TIGIT with CD155 suppresses the NK cells' cytotoxicity and IFN-γ production, whereas blockade using an anti-TIGIT antibody could reverse this effect (56). TIGIT was reported to be the key checkpoint receptor associated with NK cell dysfunction and exhaustion in several tumor-bearing mouse models (including colon cancer, melanoma, breast cancer, and fibrosarcoma) and in patients with CRC. Blockade of TIGIT recovered the NK cell function and anti-tumor effector activity, improved memory responses after tumor rechallenge, and elicited tumor-specific T cell immune responses, which were dependent on the presence of NK cells (6). CD96's interaction with CD155 resulted in reduced IFN-γ production but not NK cell cytolysis. Blockade of CD96 or CD96-/- could enhance NK cell's capacity to produce IFN-γ and led to improved control of lung metastasis; these anti-metastatic effects depended on IFN-γ, CD226, and NK cells (57–59). Numbers of CD96+ NK cells were increased in tumor sites in HCC and exhibited functional exhaustion with decreased of IFN-γ and TNF-α production, low perforin and granzyme B levels, and high IL-10 and TGF-β expression. Moreover, the high CD96 levels in tumor tissues correlated with the shorter overall survival and disease-free survival (60). It was proposed that TIGIT and CD96 play complementary roles in the control of NK cell effector function (51). Therefore, TIGIT, CD96, and CD155 are deemed as key immune checkpoints for NK cells, and the blockade of these molecules has shown great promise in tumor immunotherapy (61–64). TIGIT is often coexpressed with PD-1; therefore, combination blockade of TIGIT and PD-1 could reverse functional exhaustion more effectively, as displayed in patients with advanced melanoma, who showed increased expansion and an elicited anti-tumor immune response (65).
PD-1
PD-1 is primarily induced in activated T cells, it is also expressed on NK cells, NKT cells, B cells, and myeloid cells. The PD-1+NK cell subset constitutes 25% of NK cells in healthy donors, it is confined to CD56dim mature NK cells (66). The proportions of PD-1+ NK subset and PD-1 expression on NK cells are increased in the TME of various cancers, including colon cancer, liver cancer, gastric cancer, esophageal cancer, ovarian cancer, head and neck cancer, and Hodgkin lymphoma, where its ligands, PD-L1 or PD-L2, are expressed at high level (67). The interaction between PD-1 and its ligands provides negative signals for NK cell activation, and inhibits the anti-tumor capacity of NK cells (67, 68). PD-1+NK cells exhibit impaired cytokine production, decreased cytolytic activity, and poor proliferation, even becoming functionally exhausted (69). There is an association between PD-1+NK cell levels and poor outcome. Blocking the PD-1/PD-L1 interaction with anti-PD-1 or PD-L1 antibodies could reverse the dysfunctional status of PD-1+NK cells, which manifests as significantly augmented production of cytokines, NK cell cytotoxicity, and marked suppression of tumor growth in vivo (67, 69–71). Therefore, clinically, PD-1 blockade not only unleashes T cells to attack tumor cells, but also restores the anti-tumor responses of NK cells. Notably, the enhancement of NK cell anti-tumor efficacy by blockade of PD-1/PD-L1 is more important for the treatment of patients with tumors that are defective in MHC class I expression or display low mutational loads, because T cells are often inactive in these settings. Indeed, most Hodgkin's lymphomas express decreased or negative MHC class I molecules but show upregulated PD-L1 expression, yet patients responded well to immunotherapy blockading PD-1/PD-L1, indicating the pivotal role of the anti-tumor efficacy of NK cells (70, 72).
TIM-3
TIM-3 is a type I transmembrane protein belonging to the Ig superfamily, expressed on CD4+T, CD8+T, Treg, NK, NKT and myeloid cells. TIM-3 ligands include phosphatidylserine (PtdSer), carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1), high mobility group protein B1 proteins (HMGB1), and galectin-9. The cytoplasmic tail of TIM-3 does not have an ITIM motif but comprises five conserved tyrosine residues that are important for TIM-3 signal transduction. Upon binding of TIM-3 with its ligands, the tyrosine residues recruit certain signaling components that transduce inhibitory signaling, thereby promoting the inhibition, anergy, or exhaustion of immune cells (51, 73). TIM-3 has been regarded as an activation or maturation marker on NK cells, because it induces IFN-γ production and promotes NK cell maturation at the early stage upon engagement with its ligand galectin-9 (74, 75). However, persistently high expression of TIM-3 contributes to NK cell dysfunction and exhaustion. TIM-3 is highly expressed on peripheral NK cells from patients with various types of solid tumors, such as lung cancer, gastric cancer, and advanced melanoma, and correlates with NK cell dysfunction and exhaustion (76–78). Tumor-infiltrating NK cells in particular show upregulated TIM-3 expression, which can predict poor prognosis in patients with liver cancer, NSCLC, endometrial cancer, and other types of tumors (79–81). Both conventional NK cells and liver-resident NK cells from patients with liver cancer express high levels of TIM-3, accompanied by decreased capacity of cytokine production and cytotoxicity (79). The percentages of tumor-infiltrating TIM-3+ NK cells correlated negatively with the survival of patients with HCC. TIM-3 blockade significantly restored IFN-γ production, cytotoxicity, and proliferation of both liver-resident NK and conventional NK cells. Mechanistically, the binding of the endogenous ligand PtdSer with TIM-3 induced the dysregulation of NK cells through interrupting the PI3K/mTORC1/p-S6 signaling pathway. Importantly, TIM-3 knockdown or antibody blockade reduced tumor growth and prolonged the overall survival of orthotopical liver tumor-bearing mice in an NK cell-dependent manner (79). TNF-α was reported to induce NK cell expression of TIM-3 and NK cell dysfunction via the NF-κB pathway. Tumor invasion, lymph node metastasis, and poor staging in patients with esophageal cancer was associated with high levels of TIM-3 on tumor-infiltrating NK cells (80). The high levels of TIM-3 on tumor-infiltrating NK cells hampered the functional potential of NK cells after stimulation with IL-2/IL-15/IL-21 (82). In addition, MHC class I-deficient tumor cells led to selective upregulation of TIM-3 and PD-1 expression on intratumoral NK cells, which showed an exhausted phenotype and dramatically reduced cytotoxicity and IFN-γ production. IL-21 could reverse the functions of exhausted TIM-3+PD-1+ NK cells by activating the STAT1 and PI3K-AKT-Foxo1 signaling pathways (83). Furthermore, TIM-3 and PD-1 blockade combined with IL-21 revived the anti-tumor effects of exhausted NK cells in patients with advanced MHC class I-deficient tumors (84).
LAG-3
LAG-3 is a member of the Ig superfamily of receptors and acts as an inhibitory receptor. LAG-3 expressed on plasmacytoid dendritic cells (pDCs), B cells, NK cells, and activated T cells. Its ligands include LSECtin, a member of the DC-SIGN family, and MHC class II molecules. LAG-3 is expressed in liver cancer and several other tumors (85). Recently, fibrinogen-like protein 1 (FGL1) was identified as another MHC II-independent functional LAG-3 ligand. FGL1 shows high expression on human cancer cells, and inhibits the antigen-specific T cell response, while blockade of the LAG-3–FGL1 interaction using antibodies enhanced its anti-tumor effects (86). LAG-3, PD-1 and other inhibitory receptors are often co-expressed on tumor-infiltrating T cells in various types of cancers, such as squamous cell carcinoma, NSCLC, ovarian cancer, melanoma, colorectal adenocarcinoma, and fibrosarcoma (87–90). Co-blockade of LAG-3 and PD-1 could reverse T cell exhaustion and disrupt tumor growth more effectively than single antibody treatment, particularly for tumors that are resistant to singe antibody therapy (91, 92). Although blockade of LAG-3 alone seemed to have no effect on NK cell lysis against various targets, several reports showed that chronic stimulation of NK cells could increase LAG-3 surface expression and that of other co-inhibitory receptors, which contributed to NK cell exhaustion and weakened the anti-tumor activity of NK cells. In this situation, blocking LAG-3 could restore NK cells' cytotoxic capacity (93, 94). In a 4T1 mouse lung metastatic breast cancer model, lung infiltrating NK cells manifested a dysfunctional state, with impaired lytic activity and low capacity to produce IFN-γ, concomitant with increased expression of inhibitory receptors and the loss of activating receptors. IL-12 treatment could rescue the NK cell cytotoxicity, but also upregulated the levels of coinhibitory checkpoints such as LAG-3, PD-1, and TIGIT, thereby restraining the NK cell-mediated anti-metastatic effect. Remarkably, a synergistic effect of IL-12 therapy combined with checkpoint blockade using anti-LAG-3 and PD-1 antibodies markedly reactivated and amplified the antitumor capacity on NK cells, leading to a forceful control of metastasis by NK cells (95).
Adenosine Pathway
Extracellular adenosine is a nucleoside and derivative of ATP. The levels of ATP and adenosine are highly elevated in the TME in response to hypoxia, apoptosis, and inflammation (42, 96). ATP catabolism in tumors is primarily mediated by ecto-nucleotidases CD39 and 5′-nucleotidase CD73, both of which are over-expressed on various types of cells, including tumor cells, stromal cells, Tregs, MDSCs, and T cells in the TME (42, 97). CD39 converts ATP or ADP to ADP or AMP, respectively; while CD73 dephosphorylates AMP to adenosine. The high levels of adenosine impair the function of T cells, DCs, macrophages and NK cells by binding to adenosine receptors, primarily A2A receptor (A2AR) (42). Tumor-infiltrating NK cells can obtain CD73 expression and play immunosuppressive role by producing adenosine (98). The accumulation of adenosine in TME strongly inhibits the anti-tumor efficacy of NK cells, including suppressing the maturation, activation, cytokine production and cytotoxicity by binding to A2ARs on the surface of NK cells. Accordingly, adenosine pathway is considered as a key checkpoint for both T cells and NK cells. Blockade of adenosine pathway with A2A receptor inhibitor or antagonist, or anti-CD73 mAb can reduce the tumor growth and metastasis, and prolong survival in several types of tumors (99–102).
IL-1R8
IL-1R8, also known as single immunoglobulin IL-1R-related receptor, SIGIRR, comprises a single extracellular domain, a transmembrane domain, a cytoplasmic TIR domain, and a long tail. It commonly functions to negatively regulate ILR and TLR signaling, therefore modulating innate and adaptive immune responses (103). Recently, IL-1R8 was identified as a checkpoint molecule for NK cells by virtue of its regulation of NK cell maturation and function (104). Although IL-1R8 is expressed in many cells (such as tumor cells, epithelial cells, and leukocytes), it is expressed at high level in NK cells, and is particularly upregulated in mature NK cells. IL-1R8 can directly regulate the IL-18 signaling pathway, thus impairing NK cell activation. IL-1R8 deficiency in NK cells enhanced the IL-18-mediated activation of JNK and mTOR pathways, which are important for the metabolism, differentiation, and activation of NK cells (104). Genetic blockade of IL-1R8 restored NK cell-mediated resistance to diethylnitrosamine (DEN)-induced liver carcinogenesis, colon cancer-derived liver metastasis and mucinous carcinoma associated antigen (MCA)-induced lung metastasis (104). Similarly, IL-1R8 deficiency unleased NK cell-mediated control of MCMV infection, which depends on increased NK cell cytotoxicity and production of IFN-γ (104). Thus, IL-1R8 blockade might be a novel therapeutic strategy to recover the anti-tumor and anti-viral efficacy of NK cells.
CIS
Cytokine-inducible Src-homology-2 containing protein (CIS), a member of the suppressor-of-cytokine-signaling (SOCS) family, is regarded as a potent intracellular NK cell checkpoint molecule in the anti-tumor immune response (105). In NK cells, CIS plays a vital role by negatively regulating IL-15 signaling. CIS depletion improved NK cells' response to IL-15, as evidenced by increased proliferation, survival, IFN-γ production, and cytolytic activity against tumors. The molecular mechanism revealed that CIS can directly target JAK1, a downstream protein of IL-15, by inhibiting the kinase activity of JAK and reducing the JAK protein level through ubiquitination and proteasomal degradation. Moreover, CIS-deficient NK cells protect mice by making them resistant to lung metastasis of melanoma, breast cancer, and prostate cancer (105). Further study showed that targeted deletion of CIS can lower the threshold of NK cell activation, thus decreasing the requirement for priming and enhancing NK cells' anti-tumor function (106). Accordingly, CIS is considered as a potent intracellular NK cell checkpoint. Ablation of CIS or blockade of the CIS-JAK pathway might unleash NK cell anti-tumor efficacy, and thus might become a novel therapeutic strategy for tumor immunotherapy, especially for the prevention of tumor metastasis.
E3 Ubiquitin Ligase CbL-b and TRIM29
E3 ubiquitin ligase Cbl-b (casitas B-lineage lymphoma-b) was reported to regulate cancer metastasis by suppressing NK cell function. Cbl-b depletion or inhibition of its ubiquitylation targets comprising TAM (Tyro3, Axl and Mer) tyrosine kinase receptors efficiently released the anti-metastatic activity of NK cells, as evidenced by reduced metastasis in various mouse tumor models, including mammary cancer and melanoma, dependent on NK cell activation (107). E3 ubiquitin ligase TRIM29 was recently identified as a checkpoint regulator of NK cell function. TRIM29 is upregulated in IL-12+IL-18− activated NK cells, and TRIM29 levels correlated negatively with IFN-γ production of NK cells. Conditional deletion of TRIM29 in NK cells increased the production of IFN-γ by NK cells and promoted host defense against viral infection (108). These findings suggest the importance of E3 ubiquitin ligase in the regulation of NK cell function, and some of the molecules, such as Cbl-b and TRIM29, might function as key checkpoint regulators of NK cell activity with important clinical implications.
Blockade of NK Cell Checkpoints to Rescue NK Cell Dysfunction in Cancer Immunotherapy
KIR Blockade-Based Cancer Immunotherapy
KIRs were the first discovered NK cell checkpoint receptor in the early 1990s. The first-in-class humanized IgG4 mAb IPH2101, targeting an epitope shared by KIR2D, was produced to block the binding of HLA-C to KIR2D to release NK cell inhibition and enhance NK cells' capacity to kill tumor targets. IPH2101, alone or in combination with Lenalidomide, has been used in phase I/II clinical trials to treat patients with multiple myeloma and acute myeloid leukemia, and have shown good safety and efficacy (NCT00552396, NCT00999830, NCT01222286, NCT01217203, NCT01248455, NCT01256073) (109). The second generation anti-KIR antibody IPH2102 (Lirilumab), a fully human IgG4, has been developed to treat patients with hematological malignancies and solid tumors. Clinical trials have shown evidence that IPH2102 is well-tolerated and had good efficacy (NCT01687387). Phase I/II clinical trials of the combination of PH2102 plus anti-CTLA4 mAb (Ipilimumab) or anti-PD-1 mAb (Nivolumab), or PH2102 plus Nivolumab and Ipilimumab are completed and displayed good safety, tolerability and efficacy in therapy of advanced solid tumors (NCT01750580, NCT01714739). The safety, tolerability, and efficacy assessment of IPH2102 combined with Nivolumab or IDO inhibitor (Epacadostat) is ongoing in the treatment of patients with advanced or metastatic malignancy (NCT03203876, NCT03341936, NCT03347123). IPH4102, a first-in-class humanized mAb selectively targeting KIR3DL2, is currently being applied in a phase I clinical trial to treat relapsed or refractory cutaneous T cell lymphoma (NCT02593045), which has shown good tolerance and encouraging clinical efficacy (110, 111). A multi-cohort phase II trial is ongoing to further confirm the clinical activity, safety, and effect of IPH4102 in other subtypes of KIR3DL2+T cell lymphoma (NCT03902184).
NKG2A Blockade-Based Cancer Immunotherapy
Convincing evidence indicates that blockade of NKG2A can recover the anti-tumor function of tumor-infiltrating NK and CD8+T cells. A first-in-class humanized IgG4 mAb against CD94/NKG2A, IPH2201 (Monalizumab), has been employed in phase I trials for therapy of gynecological cancer (NCT02459301) and hematological malignancy after allogenic stem cell transplantation (NCT02921685), which is suggested to be safe, tolerable, and effective. Monalizumab in combination with other Abs or therapeutic agents has shown more promising results. Monalizumab treatment can restore CD107 and IFN-γ production in NK cells against various tumor cells, whereas the combination of monalizumab and anti-PD-L1 mAb (Durvalumab) showed additive effects, as demonstrated by rescue of the anti-tumor activity of both CD8+T and NK cells. Monalizumab combined with the anti-EGFR Ab cetuximab enhanced NK cell-mediated antibody-dependent cell-mediated cytotoxicity (ADCC), thus amplifying anti-cancer capacity against Ab-coated tumor cells (15). The phase II clinical trial NCT02643550 is ongoing to assess the safety and validity of monalizumab combined with cetuximab to treat patients who have received prior systemic regimens for recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN). The combined therapy was well-tolerated, and yielded an encouraging outcome comprising an overall response rate (ORR) of 27.5% as compared to the efficacy of ORR of 13% when given cetuximab alone (15, 112). Another phase II clinical trial applying a combination of monolizumab and Durvalumab to treat patients with recurrent or metastatic CRC has just been completed (NCT02671435). The preliminary results indicate encouraging activity with well-tolerated toxicity in patients with microsatellite-stable CRC (MSS-CRC) who were non-responsive to checkpoint blockade for PD-1/PD-L1 (113). Clinical trials assessing the safety and efficacy of anti-NKG2A mAb combined with other agents, such as the BTK inhibitor ibrutinib, are currently ongoing (NCT02557516). A recent interesting study developed NKG2A protein expression blockers (PEBLs) containing an anti-NKG2A ScFv antibody fragment linked to endoplasmic reticulum (ER)-retention domains that set traps to prevent NKG2A transport to the NK cell-surface. Notably, the PEBL-transduced NKG2Anull NK cells displayed a more effective antitumor capacity against HLA-E+ tumor cells in vivo than blockade with anti-NKG2A mAb (114, 115). These impressive results suggested that the adoptive transfer of NKG2Anull NK cells could represent a promising therapeutic strategy to treat HLA-E-expression tumors.
TIGIT Blockade-Based Cancer Immunotherapy
Increasing evidence has shown that the TIGIT/CD96/DNAM-1/CD155 axis controls T cell and NK cell activation and effector function. CD96 and TIGIT-derived inhibitory signals counteract the activating signals from DNAM-1 to form key checkpoints for both CD8+T and NK cells. CD155, CD96, or TIGIT blockade is able to reverse the dysfunction of T cells and NK cells in the TME in many types of cancers.
TIGIT is preferentially expressed on CD16+NK cells, and blockade of TIGIT significantly augmented trastuzumab-triggered NK cell-mediated anti-tumor immune responses against breast cancer (116). In vivo blockade of TIGIT or CD96 using Abs inhibited tumor growth and metastasis in an NK cell-dependent manner and thus improved the overall survival in melanoma and experimental or spontaneous lung metastasis models (6, 58). TIGIT knockout or Ab blockade led to increased IFN-production and enhanced CD8+T cells' and NK cells' anti-tumor effects, resulting in colon cancer growth inhibition (117). Combined blockade of PD-1/PD-L1 and TIGIT exhibited a more powerful anti-tumor effect in colon cancer, melanoma, and glioblastoma (GBM) compared with that of monotherapy (6, 65, 118, 119). Currently, phase I and phase II clinical trials using anti-TIGIT mAbs, alone or combined with anti-PD-1/PD-L1 mAbs, are ongoing to evaluate the safety and efficacy in patients with metastatic or locally advanced solid tumors, such as RCC, NSCLC, SCCHN, breast cancer, melanoma, and CRC (NCT02794571, NCT03119428, NCT03563716, NCT03628677, NCT04047862). A phase I/II randomized clinical trial with anti-TIGIT and anti-LAG-3 mAbs, alone or combined with pomalidomide and dexamethasone is recruiting patients with relapsed refractory multiple myeloma who have relapsed after treatment with prior therapies (NCT04150965). A phase III clinical trial is ongoing to evaluate the efficacy of anti-TIGIT mAb (tiragolumab) plus anti-PD-L1 mAb (atezolizumab) and carboplatin and etoposide in patients with chemotherapy-naive extensive-stage small cell lung cancer (NCT04256421). Another phase III clinical trial is about to begin to assess the safety and scientific validity of anti-TIGIT antibody (tiragolumab) combined with atezolizumab in patients with previously untreated locally advanced unresectable or metastatic PD-L1-selected NSCLC (NCT04294810).
TIM-3 Blockade-Based Cancer Immunotherapy
TIM-3 has gained prominence as a checkpoint receptor and as a potential candidate for cancer immunotherapy. TIM-3 and PD-1 are often co-expressed on tumor-infiltrating T and NK cells, and synergistically mediate immune cell exhaustion (83, 120). Upregulation of TIM-3 correlated with resistance to PD-1 blockade therapy (121–123). TIM-3 blockade in vivo, particularly combined with PD-1 blockade, has shown encouraging results, as evidenced by restoration of T and NK cell effector function, and a significant reduction of tumor growth in various preclinical cancer models and clinical trials (78, 121). In some preclinical models, such as sarcoma, prostate cancer, and colon carcinoma, TIM-3 blockade alone appeared to be effective. The co-blockade of TIM-3 and PD-1 displayed increased anti-tumor efficacy, with more complete tumor regression and longer survival, in models of melanoma, fibrosarcoma, colon cancer and leukemia (124–126). NK cells from patients with advanced melanoma expressed high levels of TIM-3 and displayed an exhausted phenotype and function. The high levels of TIM-3 on NK cells correlated with the advance stage and poor prognosis of patients with melanoma. Moreover, TIM-3 blockade could induce the internalization of cell surface TIM-3 and upregulated the expression of IL-2R, thereby reversing the dysfunction and exhaustion of NK cells from patients with metastatic melanoma (78).
Currently, clinical trials (phase I and II) to evaluate the safety and efficacy of anti-TIM-3 antibodies alone or in combination with anti-PD-1/PD-L1 antibodies in patients with advanced solid tumors, lymphoma and leukemia are ongoing. A multicenter, open-label, first-in-human phase I study using anti-TIM-3 antibody (TSR-022) as a monotherapy and in combination with an anti-PD-1 antibodies (TSR-042) or anti-LAG-3 antibodies (TSR-033) is recruiting patients with advanced solid tumors (NCT02817633). A phase II clinical trial (NCT03680508) to study the safety and validity of TSR-042 and TSR-022 during the therapy of patients with primary, locally advanced or metastatic liver cancer has begun to recruit patients. Another phase I clinical trial (NCT03099109) is studying the safety and efficacy of anti-TIM-3 antibody (LY3321367) alone, or combined with anti-PD-L1 antibodies (LY3300054), to treat patients with advanced relapsed/refractory solid tumors. An open-label, multicenter, non-randomized phase I and phase II clinical trial using a humanized mAb against TIM-3 (BGB-A425) combined with humanized mAb against PD-1 (tislelizumab) is recruiting to test the safety and anti-tumor effect in patients with advanced solid tumors (NCT03744468). Novartis Pharmaceuticals are recruiting patients for phase I/II clinical trials to characterize the efficacy, tolerability, and safety of the anti-TIM-3 antibody MGB453 as a single agent or combined with anti-PD-1 antibody (PDR001) and/or chemotherapy drugs to treat patients with acute myeloid leukemia (AML), high risk myelodysplastic syndrome (MDS), or advanced solid tumors (NCT03066648, NCT02608268, NCT03946670). A phase II clinical trial has been initiated to evaluate the safety and efficacy of MBG453 in combination with azacitidine and the BCL2 inhibitor venetoclax to treat patients with AML (NCT04150029). A phase III multi-center, randomized study of MBG453 or placebo added to azacitidine will be recruiting to treat patients with intermediate, high, or very high risk MDS or chronic myelomonocytic leukemia (NCT04266301).
LAG-3 Blockade-Based Cancer Immunotherapy
As one of the key checkpoint receptors for both T and NK cells, LAG-3 is becoming a major target for blockade to restore the anti-tumor capacity in the therapy of various types of cancer. To date, multiple clinical trials are evaluating several anti-LAG-3 mAbs for both solid and hematological cancers, both alone and in combination with anti-PD-1 mAbs (NCT01968109, NCT02061761, NCT02658981, NCT02966548, NCT03005782, NCT03250832, NCT03311412, NCT03365791, NCT03459222, NCT03489369, NCT03623854, NCT03743766, and NCT04080804). Clinical trials applying anti-LAG-3 mAbs in combination with other checkpoint inhibitors, such as anti-TIM-3 mAbs or anti-CTLA-4 mAbs are also recruiting patients to evaluate the safety and scientific validity of their use to treat patients with advanced solid tumors (NCT02817633, NCT03459222). Some of these clinical trials have indicated promising results, in which the combination of anti-PD-1 and anti-LAG-3 blockade has a synergistic anti-tumor effect compared with monotherapy, and could improve the efficacy in patients who are resistant to anti-PD-1 therapy (3, 127).
PD-1 Blockade-Based Cancer Immunotherapy
Antibodies targeting PD-L1 and PD-1 have been approved to treat solid and hematological malignancies, including melanoma, bladder cancer, head and neck cancer, RCC, NSCLC, HCC, and other tumors. Although it is assumed that these checkpoint blockade therapies primarily rescue T cell's anti-tumor efficacy, increasing evidence suggests that NK cells potentially respond to PD-1/PD-L1 blockade, particularly for MHC I-deficient tumors, as evidenced by attenuated anti-tumor efficacy after depletion of NK cells (70). A phase II clinical trial has enrolled patients to assess the efficacy of the anti-PD-1 mAb, Pembrolizumab, in terms of the function and exhaustion of NK cells in patients with unresectable stage III or IV melanoma (NCT03241927). Accordingly, blockade of PD-L1/PD-1 could rescue NK and T cell-mediated anti-tumor activity. Notably, the mobilization and re-activation of NK cells using PD-1 blockade is critically important for therapy of patients with HLAneg tumors (e.g., Hodgkin lymphoma) that are resistant to T cell-mediated therapy. Innate and adaptive immune responses may be activated simultaneously by combined blockade of PD-1 and other checkpoints. Currently, multiple clinical trials are ongoing utilizing anti-PD1/PD-L1 antibodies, alone or in combination with other checkpoint blockade antibodies, antiangiogenic bevacizumab, or chemotherapy to treat solid and hematological tumors.
Blockade of Adenosine Pathway in Cancer Immunotherapy
Targeting adenosine pathway, either blockade CD39 and CD73 with respective mAb to prevent the adenosine production or blockade the A2AR with inhibitors to interrupt adenosine-induced signal pathway, has been evaluated for cancer immunotherapy in pre-clinical studies and clinical trials. Both CD73-deficient mice and administration of anti-CD73 mAbs have displayed significant reduction of tumor growth and metastasis in several types of tumor models (99, 101, 102, 128). Anti-CD73 mAbs (Oleculumab, NZV930) are currently evaluated alone or in combination with other therapies (such as anti-PD-1 mAb) for the treatment of a variety of solid tumors (Table 1). Combination therapy with oleculumab and PD-L1 blockade in patients with pancreatic cancer and MSS-CRC revealed partial responses in 2 out of 20 and 1 out of 21 patients, respectively. Similarly, genetic deletion of A2AR or given A2AR antagonists or inhibitors have shown reduced tumor growth and metastasis, leading to prolonged survival (129–131). Moreover, treatment with A2AR antagonists can enhance the infiltration of CD8+T cells and NK cells into tumor sites, while simultaneously reduce the frequency of Tregs (132, 133). Currently, several A2AR antagonists (such as CPI-444, AZD4635, NIR178, and PBF-509) are being evaluated as single agent or in combination with anti-PD-1 mAb in clinical trials for treatment of cancer patients bearing solid tumors (Table 1). Combinations of anti-CD73 mAb with A2AR antagonist for therapy of patients with solid tumors are also ongoing in several phase I/II studies (NCT03274479, NCT03454451, NCT03549000, and NCT03381274).
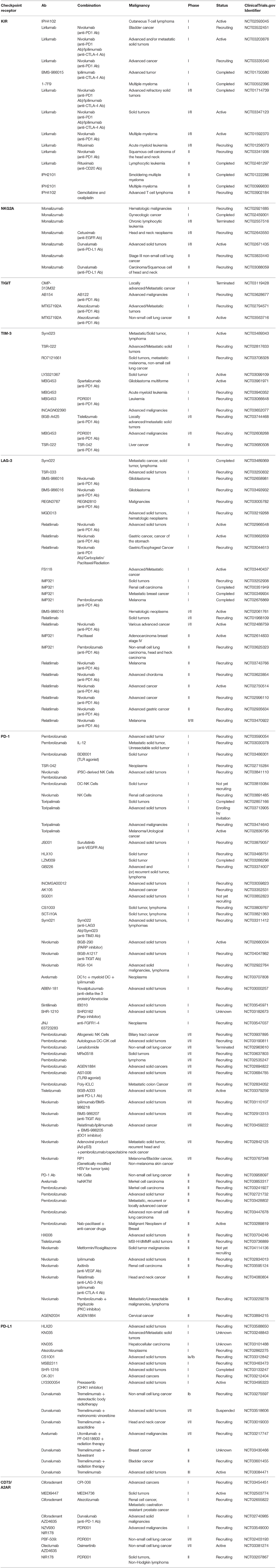
Table 1. Current clinical trials based on NK cell checkpoint receptors or molecules.
Combination Therapies Comprising Different Checkpoint Receptor Blockades or Combined With Other Therapeutic Strategies
Although therapy with checkpoint inhibitors, particularly anti-PD-1/PD-L1 and anti-CTLA-4 mAbs, has demonstrated marked success in recovering anti-tumor immune responses and improving the survival of patients with advanced malignancy, the response rates are only approximately 20 to 40% using given a checkpoint inhibitor alone (122). Many patients do not respond or develop resistance to these therapeutic approaches (134). It is likely that interrupting a single checkpoint is not enough to release the dysfunction of NK or T cells, and some cancers develop primary resistance or may adapt to acquire resistance to further therapies (134). Combination therapies with other immune checkpoint inhibitors are currently being investigated to overcome this resistance. Increasing evidence indicates the promise of this approach by enhancing anti-tumor efficacy and increasing the response rates (135). Exploiting the different mechanisms that impair T cell response, co-blockade of CTLA-4 and PD-1 (Ipilimumab + Nivolumab) revealed additional anti-tumor efficacy but also high levels of side effects in treatment of melanoma (136). A newly report of the 5-year outcome of combination therapy with Nivolumab and Ipilimumab in advanced melanoma has shown encouraging results (137). At a minimum follow-up of 60 months, the median overall survival is more than 60 months in Ipilimumab + Nivolumab group, 36.9 months in the nivolumab group, and 19.9 months in the ipilimumab group. The 5-year overall survival rate is 52% in the Ipilimumab + Nivolumab group, as compared with 44% in the nivolumab group and 26% in the ipilimumab group. These results demonstrate that the combination of anti-PD-1 mAb and anti-CTLA-4 mAb can prolong the response rate and overall survival of patients with advanced melanoma, supporting the promise of combination approach. Other combinations of checkpoint blockades, such as anti-PD-1 or PD-L1 mAbs in combination with Abs targeting TIM-3, LAG-3, or TIGIT, are also being assessed in ongoing clinical trials (Table 1) or are under investigation.
Notably, the common reasons leading to tumor resistance to checkpoint inhibitors include low antigenicity and the defects in the presentation of tumor-cell antigens (e.g., mutation or deficiency in MHC molecules), which are necessary to prime anti-tumor T cell immunity (134, 138). In these circumstances, NK cells can play critical anti-tumor role because the activity of NK cells is not limited by antigenicity and lacks MHC restriction. Consequently, it is critical to recover the NK cell's anti-tumor function by blockade of NK cell-specific checkpoint receptors or using combination blockade with NK cell checkpoint receptor and monoclonal antibodies recognizing CTLA-4 or PD-1/PD-L1. More importantly, activated NK cells can further promote T cell priming and activation, thus leading to enhanced innate and adaptive anti-tumor activity. Recent research reported that inhibitory KIRs and PD-1 correlated positively in NSCLC, suggesting that a combination of anti-KIR mAbs and anti-PD-1 mAbs could be a critical therapeutic strategy for patients with NSCLC (139). Currently, phase I/II clinical trials of a combination of anti-KIR mAbs and anti-PD-1 or anti-CTLA-4 mAbs have finished and indicated that the therapy was safe and effective for patients with advanced solid tumors (NCT01750580, NCT01714739). Indeed, targeting NK cell checkpoint receptors using anti-TIGIT and anti-NKG2A mAbs could restore the anti-tumor efficacy of both NK and T cells, and more significantly, that anti-tumor capacity could be amplified using a combination with anti-PD-1/PD-L1 mAbs in various tumor models (6, 15). A phase II clinical trial (NCT02671435) of the combination of anti-NKG2A mAbs and anti-PD-1 mAbs showed encouraging results for patients with CRC, particularly those resistance to therapy with anti-PD-1/PD-L1 mAbs (113). Recently, combinations of tumor antigen-targeted IgG Abs with checkpoint inhibitors have displayed promising results. One of the most critical effects of IgG Abs is to induce NK cell-mediated ADCC. Accordingly, the combination of checkpoint inhibitors with IgG mAbs was observed to enhance innate and adaptive anti-tumor capacity, thereby increasing the efficacy of tumor elimination. The combination of monalizumab and cetuximab to treat patients with SCCHN has yielded an encouraging outcome with an improved response rate (NCT02643550) (15). Currently, checkpoint inhibitors in combination with mAbs targeting HER-2, EGFR, and CD20 have been evaluated in pre-clinical and clinical trials for the treatment of various types of tumors, such as CRC, HNSCC, non-Hodgkin's lymphoma, and CLL (140).
Combining checkpoint inhibitors with conventional therapy (e.g., radiation therapy, targeted therapy, and chemotherapy) or other immunotherapies are also being investigated in clinical trials and/or animal models. Low-dosage chemotherapies plus checkpoint blockers appeared promising in experimental animal models and clinical studies. A phase III clinical trial applying anti-PD-1 mAbs (Pembrolizumab) in combination with chemotherapy to treat metastatic NSCLC has demonstrated markedly increased overall survival compared with that of chemotherapy alone (NCT02578680) (141, 142). Low-dose decitabine is reported to increase the expression of MHC and cytokines, and improve lymphocyte infiltration into tumor sites, thereby enhancing the efficiency of PD-1 blockade in CRC with microsatellite stability by re-modulating the TME (143). Radiation therapy can upregulate PD-1/PD-L1 expression on tumor cells and immune cells, thus increasing sensitivity to therapy with PD-1/PD-L1 inhibitors. In particular, radiation therapy can promote NK cells long-term migration and infiltration into tumor sites, and the long-term survival of NK cells (144). Certain ongoing and completed clinical trials of the combination of checkpoint blockade with chemotherapy or radiation therapy to treat different types of tumors have shown beneficial effect (145). A number of studies or clinical trials investigating checkpoint inhibitors combined with targeted therapies (e.g., anti-angiogenic therapy, B-RAF and MEK inhibitors, and CDK4/6 inhibitors), epigenetic modulators, and TLR agonists are currently being carried out in various types of cancers. For instance, a large phase III clinical trial with anti-PD-L1 mAb plus anti-VEGF mAb (bevacizumab) has recently confirmed that the combination therapy can extend the progression-free survival of patients with previously untreated metastatic RCC when compared to therapy with sunitinib (146).
It is noteworthy that the NK cell frequency appeared to correlate with responsiveness to checkpoint blockade therapies and increased survival in various types of cancers (147). In patients with melanoma at different stages of treatment, a high frequency of tumor-infiltrating NK cells predicted their responsiveness to anti-PD-1 mAb therapy (148). Not only checkpoint inhibitor therapies promote the direct anti-tumor efficacy of NK cells, but also, NK cells can promote stimulatory DCs recruitment to tumor sites by secreting cytokine Fms-related tyrosine kinase 3 ligand (FLT3LG), which subsequently enhances the responses of patients to anti-PD-1 therapy and promotes increased overall survival (148, 149). Accordingly, intervention strategies combining the recruitment and activation of NK cells (such as cytokines, TLR agonists, STING agonists) suggest great promise to improve the responsiveness and outcomes of checkpoint blockade therapy. For example, the combination of anti-PD-1 and/or anti-LAG-3 mAbs with IL-12, which promotes NK cell recruitment and activation, significantly enhanced the anti-metastatic effect that entirely depended on NK cells in breast cancer (95). Pre-treatment with IFN-γ, the TLR3 ligand poly (I:C), and anti-IL-10 Ab, which attract IFN-γ-producing NK cell infiltration into tumor sites, rendered tumors sensitive to checkpoint blockade therapy (e.g., mAbs recognizing CTLA-4 and PD-1), leading to increased cure rates. Depletion of NK cells abrogated this sensitizing effect (150). Currently, there are several ongoing clinical trials investigating polyICLC, a stable derivative of poly (I:C), combined with checkpoint inhibitors (NCT02643303, NCT02834052, NCT03721679). In addition, combining checkpoint blockade with the transfer of activated NK cells or CAR-engineered NK cells is also a beneficial approach to improve the response rates and therapeutic efficacy of immune checkpoint therapy.
Perspectives
Recently, immune checkpoint blockade, particularly using mAbs targeting PD-L1, PD-1, or CTLA-4 has achieved outstanding therapeutic efficacy for patients with metastatic melanoma, NSCLC, microsatellite stable colorectal cancer, and other tumor types. These therapies rely mainly on generating robust T cell anti-tumor immune responses. Increasing evidence shows that NK cell-based therapy is an effective supplement to T-cell therapy in that they have unique advantages over conventional T cells in certain respects. Blockade of NK cell-specific checkpoint receptors to rescue TME-induced dysfunction of NK cells (Figure 1) can reinvigorate NK cells' direct cytotoxic activity against tumors and further prime and enhance the T cell-mediated adaptive anti-tumor immunity, especially for those low antigenicity and MHCneg tumors that are resistant to T cell recognition and cytolysis. Although the preliminary data from clinical trials indicate that blockade of NK cell-specific checkpoints seems not as efficient as anti-PD-1 mAbs, combination with other checkpoint inhibitors or other therapeutics has shown great promise in clinical trials and pre-clinical studies. In particular, the combination of anti-NKG2A mAbs or anti-TIGIT mAbs plus anti-PD-1 mAbs or cetuximab have demonstrated encouraging outcome in the treatment of patients with advanced solid tumors (6, 15). Other combination strategies are under investigation or in clinical trials, such as combining checkpoint inhibitors with agents that promote NK cell infiltration into tumors, promote the persistence and activation of NK cells (such as cytokines and TLR or STING agonists), and resist the inhibitory TME (e.g., anti-TGF-β mAbs), to further increase anti-tumor capacity of NK cells and improve the response rates to immunotherapies. A combination of NK checkpoint inhibition with adoptive transfer of CAR-engineered NK cells or allogeneic activated NK cells would seem a reasonable approach. Specific stimulation or induction of the anti-tumor capacity of certain NK cell subpopulations, such as memory-like or adaptive NK cells, might achieve more powerful efficacy and better clinical outcomes. Recently, increasing evidence has highlighted the crucial role of the cellular metabolic program in directing the development, survival, proliferation, and function of immune cells, and tumors and metabolic products in the TME induce the dysfunction and exhaustion of T and NK cells by modulating cell metabolism (40, 151–154). Novel strategies might emerge from increased knowledge of the metabolic requirements of activated NK cells through targeting metabolic reprogramming and related pathways or factors to improve NK cell function more effectively within the TME (155). TLR signaling is involved in the metabolic reprogramming of the TME, thus regulating T cell function. Therefore, TLR agonists combined with checkpoint inhibitors and/or adoptive T cell therapy is proposed as a novel strategy for cancer immunotherapy (156). Similar strategies are also appropriate for NK cell-based immunotherapy. The tumor heterogeneity and the complex TME across different patients and tumor types form major barriers to anti-tumor responses and impair the responsiveness of patients to specific checkpoint blockade therapy. A deeper understanding of the immune phenotypes of tumors, lymphocyte infiltration, and the TME composition, are important to identify appropriate patient populations to receive specific therapies and could help to prioritize the most effective therapies or therapeutic combinations. It is crucial to identify suitable biomarkers to predict and monitor the response to specific therapy, appropriate therapeutic combinations, and therapeutic efficacy (157–159). Personalized therapy based on particular patients is a future goal.
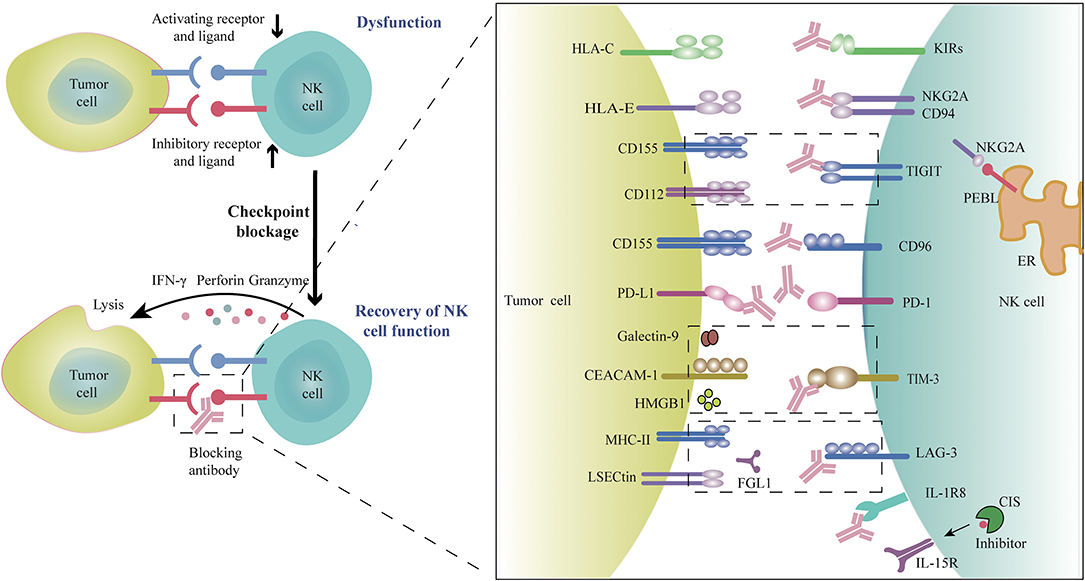
Figure 1. Blockade of checkpoint inhibitors or molecules promotes NK cell anti-tumor efficacy. Many factors in the tumor microenvironment induce NK cell dysfunction, as evidenced by the upregulation of inhibitory checkpoint receptors and the downregulation of activating receptors on tumor-infiltrating NK cells, which can result in NK cell exhaustion. Blocking these immune checkpoint receptors or molecules using monoclonal antibodies would restore the anti-tumor activity of NK cells. Rationally, co-combination with different checkpoint inhibitors or combinations of checkpoint inhibitors with other therapeutic agents could further amplify anti-tumor efficacy.
Author Contributions
CZ designed and wrote the manuscript. YL searched the literature and drafted the figures. All authors revised and approved the final version of the manuscript.
Funding
The present study was supported financially by the National Natural Science Foundation of China (91842305, 81771686), the Shandong Provincial Key Research and Development Program (Major Scientific and Technological Innovation Project) (2019JZZY021013), and the National Major Science & Technology Project for Control and Prevention of Major Infectious Diseases in China (2018ZX10301401). The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Galluzzi L, Chan TA, Kroemer G, Wolchok JD, López-Soto A. The hallmarks of successful anticancer immunotherapy. Sci Transl Med. (2018) 10:eaat7807. doi: 10.1126/scitranslmed.aat7807
2. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol. (2016) 34:539–73. doi: 10.1146/annurev-immunol-032414-112049
3. Egen JG, Ouyang W, Wu LC. Human anti-tumor immunity: insights from immunotherapy clinical trials. Immunity. (2020) 52:36–54. doi: 10.1016/j.immuni.2019.12.010
4. Topalian SL, Taube JM, Pardoll DM. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science. (2020) 367:eaax0182. doi: 10.1126/science.aax0182
5. Xu W, Hiêu T, Malarkannan S, Wang L. The structure, expression, and multifaceted role of immune-checkpoint protein VISTA as a critical regulator of anti-tumor immunity, autoimmunity, and inflammation. Cell Mol Immunol. (2018) 15:438–46. doi: 10.1038/cmi.2017.148
6. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
7. O'sullivan TE, Sun JC, Lanier LL. Natural killer cell memory. Immunity. (2015) 43:634–45. doi: 10.1016/j.immuni.2015.09.013
8. Chen Y, Tian Z, Peng H. Immunological memory: ILC1s come into view. Cell Mol Immunol. (2019) 16:895–6. doi: 10.1038/s41423-019-0311-4
9. Wu LSH, Wang JY. Warm up, cool down, and tearing apart in NK cell memory. Cell Mol Immunol. (2018) 15:1095–7. doi: 10.1038/s41423-018-0188-7
10. Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev. Immunol. (2015) 15:243–54. doi: 10.1038/nri3799
12. Bi J, Tian Z. NK Cell dysfunction and checkpoint immunotherapy. Front Immunol. (2019) 10:1999. doi: 10.3389/fimmu.2019.01999
13. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. (2019) 16:430–41. doi: 10.1038/s41423-019-0206-4
14. Sun H, Sun C. The rise of NK cell checkpoints as promising therapeutic targets in cancer immunotherapy. Front Immunol. (2019) 10:2354. doi: 10.3389/fimmu.2019.02354
15. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. (2018) 175:1731–43.e13. doi: 10.1016/j.cell.2018.10.014
16. Sun C, Sun H, Zhang C, Tian Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol. (2015) 12:292–302. doi: 10.1038/cmi.2014.91
17. Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology. (2017) 6:e1264562. doi: 10.1080/2162402X.2016.1264562
18. Denkert C, Von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. (2018) 19:40–50. doi: 10.1016/S1470-2045(17)30904-X
19. Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils suppress intraluminal NK cell-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. (2016) 6:630–49. doi: 10.1158/2159-8290.CD-15-1157
20. Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. (2001) 413:165–71. doi: 10.1038/35093109
21. Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. (2016) 165:45–60. doi: 10.1016/j.cell.2016.02.025
22. Muntasell A, Rojo F, Servitja S, Rubio-Perez C, Cabo M, Tamborero D, et al. NK cell infiltrates and HLA class I expression in primary HER2 breast cancer predict and uncouple pathological response and disease-free survival. Clin Cancer Res. (2019) 25:1535–45. doi: 10.1158/1078-0432.CCR-18-2365
23. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front Immunol. (2019) 10:3038. doi: 10.3389/fimmu.2019.03038
24. Rossi GR, Trindade ES, Souza-Fonseca-Guimaraes F. Tumor microenvironment-associated extracellular matrix components regulate NK cell function. Front Immunol. (2020) 11:73. doi: 10.3389/fimmu.2020.00073
25. Habif G, Crinier A, André P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. (2019) 16:415–22. doi: 10.1038/s41423-019-0224-2
26. Neesse A, Bauer CA, Öhlund D, Lauth M, Buchholz M, Michl P, et al. Stromal biology and therapy in pancreatic cancer: ready for clinical translation? Gut. (2019) 68:159–71. doi: 10.1136/gutjnl-2018-316451
27. Huang Q, Huang M, Meng F, Sun R. Activated pancreatic stellate cells inhibit NK cell function in the human pancreatic cancer microenvironment. Cell Mol Immunol. (2019) 16:87–9. doi: 10.1038/s41423-018-0014-2
28. Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, et al. Neuroblastoma-derived TGF-β1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol. (2013) 190:5321–8. doi: 10.4049/jimmunol.1202693
29. Zou W. Mechanistic insights into cancer immunity and immunotherapy. Cell Mol Immunol. (2018) 15:419–20. doi: 10.1038/s41423-018-0011-5
30. Zhai L, Ladomersky E, Lenzen A, Nguyen B, Patel R, Lauing KL, et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol. (2018) 15:447–57. doi: 10.1038/cmi.2017.143
31. Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
32. Viel S, Marçais A, Guimaraes FSF, Loftus R, Rabilloud J, Grau M, et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. (2016) 9:ra19. doi: 10.1126/scisignal.aad1884
33. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
34. Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris JH, Noman MZ, et al. Critical role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol. (2015) 6:482. doi: 10.3389/fimmu.2015.00482
35. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
36. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol. (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
37. Stiff A, Trikha P, Mundy-Bosse B, Mcmichael E, Mace TA, Benner B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor-mediated natural killer cell function. Clin Cancer Res. (2018) 24:1891–904. doi: 10.1158/1078-0432.CCR-17-0691
38. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: roles in tumor progression. Front Immunol. (2019) 10:771. doi: 10.3389/fimmu.2019.00771
39. Zaiatz-Bittencourt V, Finlay DK, Gardiner CM. Canonical TGF-β Signaling Pathway Represses Human NK Cell Metabolism. J Immunol. (2018) 200:3934–41. doi: 10.4049/jimmunol.1701461
40. Cong J, Wang X, Zheng X, Wang D, Fu B, Sun R, et al. Dysfunction of natural killer cells by FBP1-induced inhibition of glycolysis during lung cancer progression. Cell Metab. (2018) 28:243–55.e5. doi: 10.1016/j.cmet.2018.06.021
41. Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD, et al. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liver-resident NK cells in colorectal liver metastasis. Cancer Immunol Res. (2019) 7:335–46. doi: 10.1158/2326-6066.CIR-18-0481
42. Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol. (2019) 10:925. doi: 10.3389/fimmu.2019.00925
43. Mastelic-Gavillet B, Navarro Rodrigo B, Décombaz L, Wang H, Ercolano G, Ahmed R, et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8 T cells. J Immunother Cancer. (2019) 7:257. doi: 10.1186/s40425-019-0719-5
44. Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. (2018) 78:1003–16. doi: 10.1158/0008-5472.CAN-17-2826
45. Lokshin A, Raskovalova T, Huang X, Zacharia LC, Jackson EK, Gorelik E. Adenosine-mediated inhibition of the cytotoxic activity and cytokine production by activated natural killer cells. Cancer Res. (2006) 66:7758–65. doi: 10.1158/0008-5472.CAN-06-0478
46. Zhang H, Vijayan D, Li XY, Robson SC, Geetha N, Teng MWL, et al. The role of NK cells and CD39 in the immunological control of tumor metastases. Oncoimmunology. (2019) 8:e1593809. doi: 10.1080/2162402X.2019.1593809
47. Thielens A, Vivier E, Romagné F. NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. Curr Opin Immunol. (2012) 24:239–45. doi: 10.1016/j.coi.2012.01.001
48. Pende D, Falco M, Vitale M, Cantoni C, Vitale C, Munari E, et al. Killer Ig-like receptors (KIRs): their role in NK cell modulation and developments leading to their clinical exploitation. Front Immunol. (2019) 10:1179. doi: 10.3389/fimmu.2019.01179
49. Cornillet M, Jansson H, Schaffer M, Hertwig L, Berglin L, Zimmer CL, et al. Imbalance of Genes encoding natural killer immunoglobulin-like receptors and human leukocyte antigen in patients with biliary cancer. Gastroenterology. (2019) 157:1067–80.e9. doi: 10.1053/j.gastro.2019.06.023
50. Pesce S, Greppi M, Grossi F, Del Zotto G, Moretta L, Sivori S, et al. PD/1-PD-Ls checkpoint: insight on the potential role of NK cells. Front Immunol. (2019) 10:1242. doi: 10.3389/fimmu.2019.01242
51. Kim N, Kim HS. Targeting checkpoint receptors and molecules for therapeutic modulation of natural killer cells. Front Immunol. (2018) 9:2041. doi: 10.3389/fimmu.2018.02041
52. Bossard C, Bézieau S, Matysiak-Budnik T, Volteau C, Laboisse CL, Jotereau F, et al. HLA-E/β2 microglobulin overexpression in colorectal cancer is associated with recruitment of inhibitory immune cells and tumor progression. Int J Cancer. (2012) 131:855–63. doi: 10.1002/ijc.26453
53. Andersson E, Poschke I, Villabona L, Carlson JW, Lundqvist A, Kiessling R, et al. Non-classical HLA-class I expression in serous ovarian carcinoma: correlation with the HLA-genotype, tumor infiltrating immune cells and prognosis. Oncoimmunology. (2016) 5:e1052213. doi: 10.1080/2162402X.2015.1052213
54. Mcwilliams EM, Mele JM, Cheney C, Timmerman EA, Fiazuddin F, Strattan EJ, et al. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology. (2016) 5:e1226720. doi: 10.1080/2162402X.2016.1226720
55. Stamm H, Oliveira-Ferrer L, Grossjohann EM, Muschhammer J, Thaden V, Brauneck F, et al. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. Oncoimmunology. (2019) 8:e1674605. doi: 10.1080/2162402X.2019.1674605
56. Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, et al. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur J Immunol. (2013) 43:2138–50. doi: 10.1002/eji.201243072
57. Blake SJ, Dougall WC, Miles JJ, Teng MWL, Smyth MJ. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. (2016) 22:5183–8. doi: 10.1158/1078-0432.CCR-16-0933
58. Blake SJ, Stannard K, Liu J, Allen S, Yong MCR, Mittal D, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. (2016) 6:446–59. doi: 10.1158/2159-8290.CD-15-0944
59. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. (2014) 15:431–8. doi: 10.1038/ni.2850
60. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 Correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. (2019) 70:168–83. doi: 10.1002/hep.30347
61. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. (2017) 276:112–20. doi: 10.1111/imr.12518
62. Solomon BL, Garrido-Laguna I. TIGIT: a novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother. (2018) 67:1659–67. doi: 10.1007/s00262-018-2246-5
63. Georgiev H, Ravens I, Papadogianni G, Bernhardt G. Coming of age: CD96 emerges as modulator of immune responses. Front Immunol. (2018) 9:1072. doi: 10.3389/fimmu.2018.01072
64. Kučan Brlić P, Lenac Roviš T, Cinamon G, Tsukerman P, Mandelboim O, Jonjić S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell Mol Immunol. (2019) 16:40–52. doi: 10.1038/s41423-018-0168-y
65. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8? T cells in melanoma patients. J Clin Invest. (2015) 125:2046–58. doi: 10.1172/JCI80445
66. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J Allergy Clin Immunol. (2017) 139:335–46.e3. doi: 10.1016/j.jaci.2016.04.025
67. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene. (2017) 36:6143–53. doi: 10.1038/onc.2017.209
68. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vély F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget. (2016) 7:72961–77. doi: 10.18632/oncotarget.12150
69. Concha-Benavente F, Kansy B, Moskovitz J, Moy J, Chandran U, Ferris RL. PD-L1 mediates dysfunction in activated PD-1 NK cells in head and neck cancer patients. Cancer Immunol Res. (2018) 6:1548–60. doi: 10.1158/2326-6066.CIR-18-0062
70. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. (2018) 128:4654–68. doi: 10.1172/JCI99317
71. Oyer JL, Gitto SB, Altomare DA, Copik AJ. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology. (2018) 7:e1509819. doi: 10.1080/2162402X.2018.1509819
72. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. (2015) 372:311–9. doi: 10.1056/NEJMoa1411087
73. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. (2015) 517:386–90. doi: 10.1038/nature13848
74. Gleason MK, Lenvik TR, Mccullar V, Felices M, O'brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood. (2012) 119:3064–72. doi: 10.1182/blood-2011-06-360321
75. Ndhlovu LC, Lopez-Vergès S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. (2012) 119:3734–43. doi: 10.1182/blood-2011-11-392951
76. Wang Z, Zhu J, Gu H, Yuan Y, Zhang B, Zhu D, et al. The clinical significance of abnormal tim-3 expression on NK cells from patients with gastric cancer. Immunol Invest. (2015) 44:578–89. doi: 10.3109/08820139.2015.1052145
77. Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol. (2015) 29:635–41. doi: 10.1016/j.intimp.2015.09.017
78. Da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. (2014) 2:410–22. doi: 10.1158/2326-6066.CIR-13-0171
79. Tan S, Xu Y, Wang Z, Wang T, Du X, Song X, et al. Tim-3 hampers tumor surveillance of liver-resident and conventional NK cells by disrupting PI3K signaling. Cancer Res. (2020) 80:1130–42. doi: 10.1158/0008-5472.CAN-19-2332
80. Zheng Y, Li Y, Lian J, Yang H, Li F, Zhao S, et al. TNF-α-induced Tim-3 expression marks the dysfunction of infiltrating natural killer cells in human esophageal cancer. J Transl Med. (2019) 17:165. doi: 10.1186/s12967-019-1917-0
81. Degos C, Heinemann M, Barrou J, Boucherit N, Lambaudie E, Savina A, et al. Endometrial tumor microenvironment alters human NK cell recruitment, and resident NK cell phenotype and function. Front Immunol. (2019) 10:877. doi: 10.3389/fimmu.2019.00877
82. Zhang W, Feng H, Chen Q, Lu X, Ge J. The functional potency of natural killer cells in response to IL-2/IL-15/IL-21 stimulation is limited by a concurrent upregulation of Tim-3 in bladder cancer. Exp Cell Res. (2018) 372:92–8. doi: 10.1016/j.yexcr.2018.09.013
83. Seo H, Jeon I, Kim BS, Park M, Bae EA, Song B, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun. (2017) 8:15776. doi: 10.1038/ncomms15776
84. Seo H, Kim BS, Bae EA, Min BS, Han YD, Shin SJ, et al. IL21 Therapy combined with PD-1 and Tim-3 blockade provides enhanced NK cell antitumor activity against MHC class I-deficient tumors. Cancer Immunol Res. (2018) 6:685–95. doi: 10.1158/2326-6066.CIR-17-0708
85. Goldberg MV, Drake CG. LAG-3 in Cancer Immunotherapy. Curr Top Microbiol Immunol. (2011) 344:269–78. doi: 10.1007/82_2010_114
86. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell. (2019) 176:334–47.e12. doi: 10.1016/j.cell.2018.11.010
87. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
88. Mishra AK, Kadoishi T, Wang X, Driver E, Chen Z, Wang XJ, et al. Squamous cell carcinomas escape immune surveillance via inducing chronic activation and exhaustion of CD8+ T Cells co-expressing PD-1 and LAG-3 inhibitory receptors. Oncotarget. (2016) 7:81341–56. doi: 10.18632/oncotarget.13228
89. Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res. (2019) 25:4663–73. doi: 10.1158/1078-0432.CCR-18-4142
90. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
91. Lichtenegger FS, Rothe M, Schnorfeil FM, Deiser K, Krupka C, Augsberger C, et al. Targeting LAG-3 and PD-1 to Enhance T cell activation by antigen-presenting cells. Front Immunol. (2018) 9:385. doi: 10.3389/fimmu.2018.00385
92. Khair DO, Bax HJ, Mele S, Crescioli S, Pellizzari G, Khiabany A, et al. Combining immune checkpoint inhibitors: established and emerging targets and strategies to improve outcomes in melanoma. Front Immunol. (2019) 10:453. doi: 10.3389/fimmu.2019.00453
93. Ben-Shmuel A, Biber G, Barda-Saad M. Unleashing natural killer cells in the tumor microenvironment-the next generation of immunotherapy? Front Immunol. (2020) 11:275. doi: 10.3389/fimmu.2020.00275
94. Merino A, Zhang B, Dougherty P, Luo X, Wang J, Blazar BR, et al. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J Clin Invest. (2019) 130:3770–85. doi: 10.1172/JCI125916
95. Ohs I, Ducimetière L, Marinho J, Kulig P, Becher B, Tugues S. Restoration of natural killer cell antimetastatic activity by IL12 and checkpoint blockade. Cancer Res. (2017) 77:7059–71. doi: 10.1158/0008-5472.CAN-17-1032
96. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
97. Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. (2010) 29:5346–58. doi: 10.1038/onc.2010.292
98. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
99. Stagg J, Divisekera U, Mclaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci USA. (2010) 107:1547–52. doi: 10.1073/pnas.0908801107
100. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-tumor Immune Responses. Cancer Cell. (2016) 30:391–403. doi: 10.1016/j.ccell.2016.06.025
101. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, Mcglinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology. (2016) 5:e1208875. doi: 10.1080/2162402X.2016.1208875
102. Terp MG, Olesen KA, Arnspang EC, Lund RR, Lagerholm BC, Ditzel HJ, et al. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J Immunol. (2013) 191:4165–73. doi: 10.4049/jimmunol.1301274
103. Molgora M, Supino D, Mantovani A, Garlanda C. Tuning inflammation and immunity by the negative regulators IL-1R2 and IL-1R8. Immunol Rev. (2018) 281:233–47. doi: 10.1111/imr.12609
104. Molgora M, Bonavita E, Ponzetta A, Riva F, Barbagallo M, Jaillon S, et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature. (2017) 551:110–4. doi: 10.1038/nature24293
105. Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. (2016) 17:816–24. doi: 10.1038/ni.3470
106. Delconte RB, Guittard G, Goh W, Hediyeh-Zadeh S, Hennessy RJ, Rautela J, et al. NK cell priming from endogenous homeostatic signals is modulated by CIS. Front Immunol. (2020) 11:75. doi: 10.3389/fimmu.2020.00075
107. Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. (2014) 507:508–12. doi: 10.1038/nature12998
108. Dou Y, Xing J, Kong G, Wang G, Lou X, Xiao X, et al. Identification of the E3 Ligase TRIM29 as a critical checkpoint regulator of NK cell functions. J Immunol. (2019) 203:873–80. doi: 10.4049/jimmunol.1900171
109. Benson DM, Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A Phase I trial of the Anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res. (2015) 21:4055–61. doi: 10.1158/1078-0432.CCR-15-0304
110. Van Der Weyden C, Bagot M, Neeson P, Darcy PK, Prince HM. IPH4102, a monoclonal antibody directed against the immune receptor molecule KIR3DL2, for the treatment of cutaneous T-cell lymphoma. Expert Opin Investig Drugs. (2018) 27:691–7. doi: 10.1080/13543784.2018.1498081
111. Bagot M, Porcu P, Marie-Cardine A, Battistella M, William BM, Vermeer M, et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: an international, first-in-human, open-label, phase 1 trial. Lancet Oncol. (2019) 20:1160–70. doi: 10.1016/S1470-2045(19)30320-1
112. Creelan BC, Antonia SJ. The NKG2A immune checkpoint - a new direction in cancer immunotherapy. Nat Rev Clin Oncol. (2019) 16:277–8. doi: 10.1038/s41571-019-0182-8
113. Van Hall T, André P, Horowitz A, Ruan DF, Borst L, Zerbib R, et al. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J Immunother Cancer. (2019) 7:263. doi: 10.1186/s40425-019-0761-3
114. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Invest. (2019) 129:2094–106. doi: 10.1172/JCI123955
115. Cichocki F, Miller JS. Setting traps for NKG2A gives NK cell immunotherapy a fighting chance. J Clin Invest. (2019) 129:1839–41. doi: 10.1172/JCI128480
116. Xu F, Sunderland A, Zhou Y, Schulick RD, Edil BH, Zhu Y. Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol Immunother. (2017) 66:1367–75. doi: 10.1007/s00262-017-2031-x
117. Zhou XM, Li WQ, Wu YH, Han L, Cao XG, Yang XM, et al. Intrinsic expression of immune checkpoint molecule TIGIT could help tumor growth by suppressing the function of NK and CD8 T cells. Front Immunol. (2018) 9:2821. doi: 10.3389/fimmu.2018.02821
118. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
119. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. (2018) 7:e1466769. doi: 10.1080/2162402X.2018.1466769
120. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. (2010) 207:2175–86. doi: 10.1084/jem.20100637
121. Zhang X, Chen H, Li G, Zhou X, Shi Y, Zou F, et al. Increased Tim-3 expression on TILs during treatment with the Anchored GM-CSF vaccine and anti-PD-1 antibodies is inversely correlated with response in prostate cancer. J Cancer. (2020) 11:648–56. doi: 10.7150/jca.29705
122. Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl Oncol. (2020) 13:100738. doi: 10.1016/j.tranon.2019.12.010
123. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
124. Ngiow SF, Von Scheidt B, Akiba H, Yagita H, Teng MWL, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. (2011) 71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096
125. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
126. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. (2011) 117:4501–10. doi: 10.1182/blood-2010-10-310425
127. Ruffo E, Wu RC, Bruno TC, Workman CJ, Vignali DaA. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin Immunol. (2019) 42:101305. doi: 10.1016/j.smim.2019.101305
128. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MWL, Darcy PK, et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. (2011) 71:2892–900. doi: 10.1158/0008-5472.CAN-10-4246
129. Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother. (2012) 61:917–26. doi: 10.1007/s00262-011-1155-7
130. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci USA. (2013) 110:14711–6. doi: 10.1073/pnas.1308209110
131. Kjaergaard J, Hatfield S, Jones G, Ohta A, Sitkovsky M. A Adenosine receptor gene deletion or synthetic A antagonist liberate tumor-reactive CD8 T cells from tumor-induced immunosuppression. J Immunol. (2018) 201:782–91. doi: 10.4049/jimmunol.1700850
132. Ma SR, Deng WW, Liu JF, Mao L, Yu GT, Bu LL, et al. Blockade of adenosine A2A receptor enhances CD8 T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol Cancer. (2017) 16:99. doi: 10.1186/s12943-017-0665-0
133. Young A, Ngiow SF, Madore J, Reinhardt J, Landsberg J, Chitsazan A, et al. Targeting adenosine in BRAF-mutant melanoma reduces tumor growth and metastasis. Cancer Res. (2017) 77:4684–96. doi: 10.1158/0008-5472.CAN-17-0393
134. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
135. Seliger B. Combinatorial approaches with checkpoint inhibitors to enhance anti-tumor immunity. Front Immunol. (2019) 10:999. doi: 10.3389/fimmu.2019.00999
136. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369
137. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2019) 381:1535–46. doi: 10.1056/NEJMoa1910836
138. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. (2017) 7:1420–35. doi: 10.1158/2159-8290.CD-17-0593
139. He Y, Liu S, Mattei J, Bunn PA, Zhou C, Chan D. The combination of anti-KIR monoclonal antibodies with anti-PD-1/PD-L1 monoclonal antibodies could be a critical breakthrough in overcoming tumor immune escape in NSCLC. Drug Des Devel Ther. (2018) 12:981–6. doi: 10.2147/DDDT.S163304
140. Ferris RL, Lenz HJ, Trotta AM, García-Foncillas J, Schulten J, Audhuy F, et al. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat Rev. (2018) 63:48–60. doi: 10.1016/j.ctrv.2017.11.008
141. Xu X, Huang Z, Zheng L, Fan Y. The efficacy and safety of anti-PD-1/PD-L1 antibodies combined with chemotherapy or CTLA4 antibody as a first-line treatment for advanced lung cancer. Int J Cancer. (2018) 142:2344–54. doi: 10.1002/ijc.31252
142. Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
143. Yu G, Wu Y, Wang W, Xu J, Lv X, Cao X, et al. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell Mol Immunol. (2019) 16:401–9. doi: 10.1038/s41423-018-0026-y
144. Kim KW, Jeong JU, Lee KH, Uong TNT, Rhee JH, Ahn SJ, et al. Combined NK cell therapy and radiation therapy exhibit long-term therapeutic and antimetastatic effects in a human triple negative breast cancer model. Int J Radiat Oncol Biol Phys. (2019) 33868–4. doi: 10.1016/j.ijrobp.2019.09.041
145. Kang J, Demaria S, Formenti S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J Immunother Cancer. (2016) 4:51. doi: 10.1186/s40425-016-0156-7
146. Rini BI, Powles T, Atkins MB, Escudier B, Mcdermott DF, Suarez C, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet. (2019) 393:2404–15. doi: 10.1016/S0140-6736(19)30723-8
147. Zemek RM, Chin WL, Nowak AK, Millward MJ, Lake RA, Lesterhuis WJ. Sensitizing the tumor microenvironment to immune checkpoint therapy. Front Immunol. (2020) 11:223. doi: 10.3389/fimmu.2020.00223
148. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
149. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
150. Zemek RM, De Jong E, Chin WL, Schuster IS, Fear VS, Casey TH, et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci Transl Med. (2019) 11:eaav7816. doi: 10.1126/scitranslmed.aav7816
151. Mohamed E, Al-Khami AA, Rodriguez PC. The cellular metabolic landscape in the tumor milieu regulates the activity of myeloid infiltrates. Cell Mol Immunol. (2018) 15:421–7. doi: 10.1038/s41423-018-0001-7
152. Poznanski SM, Ashkar AA. What defines NK cell functional fate: phenotype or metabolism? Front Immunol. (2019) 10:1414. doi: 10.3389/fimmu.2019.01414
153. Terrén I, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. NK cell metabolism and tumor microenvironment. Front Immunol. (2019) 10:2278. doi: 10.3389/fimmu.2019.02278
154. Chambers AM, Lupo KB, Matosevic S. Tumor microenvironment-induced immunometabolic reprogramming of natural killer cells. Front Immunol. (2018) 9:2517. doi: 10.3389/fimmu.2018.02517
155. O'brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol. (2019) 19:282–90. doi: 10.1038/s41577-019-0139-2
156. Huang L, Xu H, Peng G. TLR-mediated metabolic reprogramming in the tumor microenvironment: potential novel strategies for cancer immunotherapy. Cell Mol Immunol. (2018) 15:428–37. doi: 10.1038/cmi.2018.4
157. Trotta AM, Pacelli R, Scala S. Predictive immune biomarkers: an unattainable chimera? Cell Mol Immunol. (2018) 15:740–2. doi: 10.1038/cmi.2017.162
158. Jacquelot N, Roberti MP, Enot DP, Rusakiewicz S, Ternès N, Jegou S, et al. Predictors of responses to immune checkpoint blockade in advanced melanoma. Nat Commun. (2017) 8:592. doi: 10.1038/s41467-017-00608-2
Keywords: checkpoint receptors, natural killer cells, cancer immunotherapy, checkpoint blockade, tumor microenvironment
Citation: Zhang C and Liu Y (2020) Targeting NK Cell Checkpoint Receptors or Molecules for Cancer Immunotherapy. Front. Immunol. 11:1295. doi: 10.3389/fimmu.2020.01295
Received: 20 April 2020; Accepted: 22 May 2020;
Published: 23 June 2020.
Edited by:
Zhigang Tian, University of Science and Technology of China, ChinaReviewed by:
Jiacheng Bi, Chinese Academy of Sciences (CAS), ChinaXiaodong Zheng, University of Science and Technology of China, China
Copyright © 2020 Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cai Zhang, Y2FpemhhbmdzZEBzZHUuZWR1LmNu