
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 23 June 2020
Sec. Immunological Tolerance and Regulation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01291
 Sohei Makita1†
Sohei Makita1† Hiroaki Takatori1,2*†
Hiroaki Takatori1,2*† Arifumi Iwata1
Arifumi Iwata1 Shigeru Tanaka1
Shigeru Tanaka1 Shunsuke Furuta1Kei Ikeda1Akira Suto1Kotaro Suzuki1
Shunsuke Furuta1Kei Ikeda1Akira Suto1Kotaro Suzuki1 Silvia B. V. Ramos3
Silvia B. V. Ramos3 Hiroshi Nakajima1*
Hiroshi Nakajima1*The zinc finger protein 36-like 2, ZFP36L2, is a member of a small family of RNA-binding proteins composed by ZFP36 (also known as tristetraprolin, TTP), ZFP36L1 and ZFP36L2 in humans, with corresponding murine orthologs. These proteins bind to adenine uridine-rich element (ARE) in the 3′untranslated region of target messenger RNA and stimulate target degradation. ZFP36 functions as an anti-inflammatory modulator in murine models of inflammatory diseases by down-regulating the production of inflammatory cytokines such as tumor necrosis factor-α. However, how ZFP36L1 and ZFP36L2 alter the function of CD4+ T cells is not completely understood. We addressed this issue by searching for the target genes of ZFP36L2 by comprehensive transcriptome analysis. We observed that ZFP36L2 is highly expressed in naïve CD4+ T cells; however, when CD4+ T cells are stimulated through their T cell receptors, ZFP36L2 expression is rapidly reduced in both humans and mice. Among CD4+ T cell populations, the expression levels of ZFP36L2 in regulatory T cells (Tregs) were significantly lower than those in naïve or effector CD4+ T cells. RNA-sequence analysis revealed that the forced expression of ZFP36L2 decreased Ikzf2 (encoding Helios) expression in Foxp3+ Tregs and inhibited the ability of induced Tregs (iTregs). ZFP36L2 directly bound to and destabilized the 3′untranslated region of Ikzf2 mRNA, which contains AU-rich elements. These results indicate that ZFP36L2 reduces the expression of Ikzf2 and suppresses iTreg function, raising the interesting possibility that the inhibition of ZFP36L2 in iTregs could be a therapeutic strategy for autoimmune diseases.
Zinc finger protein 36 (ZFP36) family proteins contain a similar tandem zinc finger domain, composed of three cysteines and one histidine. Because their tandem zinc finger domain can bind to adenine-uridine rich elements (AREs) present in the 3′untranslated region of some specific mRNAs, these proteins function as RNA-binding proteins. The family consists of three members in humans: ZFP36 (also known as tristetraprolin: TTP), ZFP36L1, and ZFP36L2 and one extra member in mice: ZFP36, ZFP36L1, ZFP36L2, and ZFP36L3 (1–4). TTP or ZFP36, the family prototype, physiologically binds to its target mRNAs and stimulates the subsequent target degradation (1, 2). Indeed, ZFP36-deficient mice develop a severe inflammatory disease due mainly to the over-production of tumor necrosis factor-α (TNF-α) by macrophages (5, 6). ZFP36 also directly downregulates the mRNA expression of other proinflammatory cytokines such as IL-2, IL-17, and IFN-γ, which can then modulate T-cell activation and anti-viral immunity (7–10). These mechanistic profiles of ZFP36 raise the possibility that the induction of ZFP36 expression could be an effective therapeutic strategy against inflammatory diseases, particularly in rheumatoid arthritis and other autoimmune diseases in which TNF-α secretion is increased (11, 12).
In contrast to these confirmed functions for ZFP36 as an anti-inflammatory protein, the role of the other ZFP36 family members in the immune system and its particular cells remains largely unknown. Interestingly, lipopolysaccharide (LPS) stimulation in macrophages induces ZFP36 expression, while downregulates the expression of ZFP36L1 and ZFP36L2 (13, 14), suggesting that the expression levels of these three molecules are differently regulated and thus, likely they play different roles. In an attempt to understand the physiological role of ZFP36L1 and ZFP36L2, these genes were knocked out in mice. Lack of ZFP36L1 leads to embryonic lethality at embryonic day 11 in mice due to placental defects (15). In contrast, homozygous Zfp36l2-deficient mice are viable; however, they die within ~2 weeks after birth as a consequence of pancytopenia due to hematopoietic stem cell failure (16). Intriguingly, mice carrying double-deficiency of Zfp36l1 and Zfp36l2 in T-cell lineage results in the arrest of thymopoiesis at the double-negative stage and develop T cell acute lymphoblastic leukemia (T-ALL) due to aberrant activation of Notch signaling (17). On the other hand, the single deletion of ZFP36L1 or ZFP36L2 in T-cell lineage does not lead to T-ALL (17). These findings suggest that ZFP36L2 plays a non-redundant role in hematopoietic stem cell differentiation. However, ZFP36L1 and ZFP36L2 have an interconnected role that seems to be redundant during T cell differentiation because the defect in thymopoiesis is observed only when both proteins are simultaneously absent.
Imbalance of T-cell response is present in various autoimmune diseases (18). Because ZFP36L2 is involved in hematopoietic stem cell differentiation and in thymopoiesis, it is not surprising that ZFP36L2 seems to be involved in the development of human autoimmune diseases, particularly in systemic lupus erythematosus (SLE) where ZFP36L2 was found to be significantly downregulated in peripheral blood mononuclear cells (PBMCs) of SLE patients in comparison to healthy individuals (19). Also, ZFP36L2 was found to be a disease-susceptibility gene in multiple sclerosis (MS), and decreased ZFP36L2 expression was observed in MS patients as compared with healthy controls (20). Collectively, these findings suggest that ZFP36L2 is involved in the physiopathology of autoimmune diseases in humans; however, the precise role of ZFP36L2 in a specific T cell population has not been elucidated. Thus, with the goal of better understanding the mechanistic role of ZFP36L2 in autoimmune diseases, we set up experiments to study the expression of ZFP36L2 in CD4+ T cells and find novel ZFP36L2-target mRNAs that could modulate regulatory T cells (Tregs). Our results suggest that ZFP36L2 is involved in the suppression function of induced Tregs (iTregs) by accelerating the degradation of Ikzf2 mRNA.
C57BL/6 mice and BALB/c mice were purchased from CLEA (Tokyo, Japan). RAG2−/− mice and Foxp3YFP−Cre mice on a C57BL/6 background were purchased from Jackson Laboratory (Bar Harbor, ME). Foxp3hCD2 mice on a BALB/c background were described previously (21). All mice were housed in microisolator cages under specific pathogen-free conditions, and all experiments were performed according to the guidelines of Chiba University established by Chiba University for experiments in animals, which conform to the “Guide for the Care and Use of Laboratory Animals” published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011).
Monoclonal antibodies to murine CD3ε (2C11), CD28 (37.51), CD4 (H129.19), CD44 (IM7), CD62L (MEL-14), CTLA-4 (UC10-4B9), IL-4 (11B11), IFN-γ (XMG1.2), and human NGFR (C40-1475) were purchased from BD Biosciences (San Jose, CA). Monoclonal antibodies to murine Glycoprotein A repetitions predominant (GARP) (F011-5) and Foxp3 (FJK-16s) and anti-mouse/human Helios antibody (22F6) were purchased from eBioscience (San Diego, CA). Anti-latency-associated peptide (LAP) antibody (TW7-16B4) was purchased from BioLegend (San Diego, USA). Human TGF-β was purchased from R&D Systems (Minneapolis, MN).
The human subject research component of this study was approved by the Ethics Committee of Chiba University, and written informed consent was obtained according to the Declaration of Helsinki. PBMCs from healthy donors were prepared by using Ficoll-Paque density gradient centrifugation (GE Healthcare, Piscataway, NJ). CD4+ T cells were purified from PBMCs with a CD4+ T Cell Isolation Kit II (Miltenyi Biotec, Sunnyvale, CA) according to the manufacturer's instructions. The purity of CD4+ T cells was routine >98% by FACS analysis. Isolated CD4+ T cells (1 × 106 cells/ml) were stimulated with Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific, Waltham, MA).
The bicistronic retrovirus vectors used in the experiments [pMX-IRES-hNGFR (pIN), MSCV-IRES-hNGFR (MIN), and MSCV-IRES-GFP (MIG)] have been described previously (22). Expression plasmids of murine ZFP36, ZFP36L1, and ZFP36L2 were kindly provided by Drs. Ching-Jin Chang (National Taiwan University, Taiwan) and Silvia B. V. Ramos. cDNA for Zfp36, Zfp36l1, or Zfp36l2 was subcloned into pIN, MIN, and MIG. cDNA for Zfp36l1 or Zfp36l2 was also subcloned into pcDNA3 (Invitrogen, Carlsbad, CA). pGL3-promoter vector (pGL3-pro) was purchased from Promega Biotech, Inc. (Madison, WI). 3′UTR of Ikzf2 (encoding Helios), which contains three AREs, was cloned into pGL3-pro to construct pGL3-pro-Ikzf2-3′UTR (pGL3-pro-Ikzf2) by PCR with KOD-Plus-Neo (Toyobo, Osaka, Japan). Primer sequences for cloning of 3′UTR of Ikzf2 mRNA are as follows: forward, 5′-GCCGTGTAATTCTAGGGCCTTTTCATTCCAAAGGGG-3′, reverse, 5′-GTCTGCTCGAAGCGGTGCTTGTTCCTGAGATTGGCTAAG-3′. Mutants of pGL3-pro-Ikzf2 lacking each ARE (Del ARE1, Del ARE2, and Del ARE3) were generated using pGL3-pro-Ikzf2 as a template by Gibson Assembly System (New England BioLabs, Ipswich, MA) according to the manufacturer's instructions. All constructs were verified by sequencing.
CD4+ CD62L+ CD25− TCRαβ+ T cells (naïve CD4+ T cells) were isolated from spleen using an EasySep™ Mouse Naïve CD4+ T Cell Isolation Kit (STEMCELL Technologies, Vancouver, Canada) according to the manufacturer's instructions. The resultant cells were > 98% pure CD4+ CD62L+ T cells when analyzed by FACS. Cells (1 × 106 cells/ml) were then stimulated with plate-bound anti-CD3 mAb (1 μg/ml) in the presence of anti-CD28 mAb (1 μg/ml) in 48-well plates under neutral conditions [anti-IFN-γ mAb (5 μg/ml) and anti-IL-4 mAb (5 μg/ml)] or iTreg-inducing conditions [TGF-β (1 ng/ml), anti-IFN-γ mAb, and anti-IL-4 mAb].
Total RNA was isolated by using an RNeasy Mini Kit (Qiagen, Hilden, Germany). RNA-sequencing was performed as described previously (22). 3′UTR sequences were extracted from the table browser of the UCSC Genome Browser. Genes containing ARE “TATTTAT” in 3′UTR were identified using custom R script. RNA-seq data will be available from the National Center for Biotechnology Information's Gene Expression Omnibus upon publication of the manuscript.
Total RNA was extracted with Isogen II reagent (Nippon Gene, Tokyo, Japan), and reverse transcription was carried out using an iScript cDNA Synthesis Kit (BioRad, Hercules, CA). Quantitative real-time PCR analysis (qPCR) was performed as described previously (23, 24). The expression levels of each gene were normalized to the levels of β-actin. Sequences of primers are as follows:
Murine Zfp36 5′- GTCTCTTCACCAAGGCCATTC-3′,
reverse, 5′- CCGAGTTTATGTTCCAAAGTCC-3′.
Murine Zfp36l1 5′- CACGACACACCAGATCCTAGTC-3′,
reverse, 5′- CTGGGAGTGCTGTAGTTGAGC-3′.
Murine Zfp36l2, 5′- GGCCGCACAAGCACAAC−3′,
reverse, 5′-GAGACTCGAACCAAGATGAATAACG-3′,
Murine β -actin, 5′-CTGTCCCTGTATGCCTCTG-3′,
reverse, 5′-ATGTCACGCACGATTTCC-3′.
Human ZFP36L2, 5′-AAACATGTCGACCACACTTCTG-3′,
reverse, 5′-CATGTTGTTCAGGTTGAGGTTG-3′.
Human β-actin, 5′-AGGATGCAGAAGGAGATCACTG-3′,
reverse, 5′-GGGTGTAACGCAACTAAGTCATAG-3′.
The production of retroviruses and infection to murine CD4+ T cells was performed as described previously (22–25). In brief, naive CD4+ T cells that had been stimulated with plate-bound anti-CD3 mAb/anti-CD28 mAb under neutral conditions for 24 h were added to retrovirus-bound RetroNectin/anti-CD3 mAb-coated plates and were cultured for 24 h. Cells were then stimulated with anti-CD3 mAb/anti-CD28 mAb under neutral conditions or iTreg-inducing conditions for 3 days.
Intracellular staining of murine CD4+ T cells was performed as described previously (22–25) using the following antibodies: anti-Foxp3-FITC (259D/C7, BD Biosciences) and anti-mouse/human Helios-Alexa Fluor® 647 (22F6, BioLegend). Cells were analyzed on a FACS Canto II (Becton Dickinson, San Jose, CA) using FlowJo software (Tree Star, Ashland, OR).
Naïve CD4+ T cells of Foxp3hCD2 mice were infected with a retrovirus of MIN-Zfp36l2 or MIN (mock) in iTreg-inducing conditions as described above, and CD4+ hCD2+ hNGFR+ cells (ZFP36L2 iTregs or mock iTregs) were isolated by flow cytometry. These cells were treated with 10 μg/ml of actinomycin D, total RNA was harvested after indicated periods, and mRNA levels of Ikzf2 were evaluated by qPCR. GraphPad Prism software version 8.12 was used to calculate transcript half-lives based on a one-phase exponential decay model.
EL-4 cells were transfected with either pGL3-pro-Ikzf2 or ARE deletion mutants of pGL3-pro-Ikzf2 in the presence of pRL-TK with a Neon Transfection 100 μL kit with buffer R (Thermo Fisher Scientific, Waltham, MA). Where indicated, pcDNA3-Zfp36l1, pcDNA3-Zfp36l2, or pcDNA3 (as a control) was co-transfected. Four hours later, cells were treated with PMA/Ionomycin for 20 hours. Firefly luciferase activity of the reporters was normalized by Renilla luciferase activity of pRL-TK (Promega Biotech, Inc.).
The RNA probes were synthesized with the Riboprobe® System-T7 (Promega Biotech, Inc.) using synthetic DNA sequences immediately downstream a T7 promoter, and were body-labeled with [α-32P]UTP (3,000 Ci/mmol; Perkin Elmer) during the transcription process as previously described (26, 27). The RNA probes' sequences are Ikzf2 ARE1 (5′-UUUACUAGGGCUAUUUAUUCCACUAUUU-3′), Ikzf2 ARE2 (5′-AAGGAUAUUUAUUUCUGAAUGAGGUAAAUAAGUU-3′) and Ikzf2 ARE3 (5′-UUAUUCAUAUUUAUAUGUAGUGUGUUCU-3′). Protein extracts were prepared from HEK 293 cells transfected with a construct driven by the CMV promoter followed by murine ZFP36L2 or empty vector. The protein extracts were incubated for 15 min at room temperature with 1 x 105 cpm of 32P-labeled RNA probes as previously described (26, 27). The resultant reaction mixtures of protein-RNA complexes were then loaded onto 6% non-denaturing acrylamide (37.5:1) gels and subjected to electrophoresis at 150 V for 10 min, followed by electrophoresis at 200 V for 120 min in 0.4 x Tris-borate/EDTA running buffer. The gel was dried, exposed to film (Carestream BIOMAX MR Film), and developed after different times of exposure.
In vitro Treg suppression assays were performed as described previously (23, 24) with minor modifications. In brief, naïve CD4+ T cells of Foxp3YFP−Cre mice were prepared as described above. After naïve CD4+ T cells were infected with a retrovirus of MIN-Zfp36l2 or MIN (mock) and cultured for 4 days in iTreg-inducing conditions, CD4+ YFP+ hNGFR+ cells (ZFP36L2 iTregs or mock iTregs) were isolated by flow cytometry. Naïve CD4+ T cells from WT mice were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) and stimulated with soluble anti-CD3 mAb in the presence of irradiated splenocytes for 2 days. Where indicated, ZFP36L2 iTregs or mock iTregs were added at a 4:1 ratio. CFSE dilution profiles of naïve CD4+ T cells were assessed by flow cytometry, and data were transformed into Division Index using FlowJo software.
ZFP36L2 iTregs or mock iTregs were prepared as described above and intraperitoneally co-injected (2 × 105 cells/mouse) with naive CD4+ T cells (CD45Rbhigh YFP− CD4+ cells from Foxp3YFP−Cre mice, 4 × 105 cells/mouse) to 6–8-week RAG2−/− mice. These mice were weighed twice per week after cell transfer. Moribund mice were euthanized based on Institutional Animal Care and Use Committee protocol. Histopathological scoring was performed as described previously (28) in a blinded fashion.
Data are summarized as means ± SD. Multiple group comparison was performed by ANOVA followed by Tukey's test. A comparison between 2 groups was analyzed by Student's t-test or Chi-squared test. P < 0.05 were considered significant.
To determine how T cell receptor (TCR)-mediated stimulation differentially affects the expression of the ZFP36 family members in CD4+ T cells, we first isolated naïve CD4+ T cells (CD4+ CD62L+ CD25− TCRαβ+ T cells) from spleens of C57BL/6 wild-type (WT) mice and stimulated naïve CD4+ T cells with plate-coated anti-CD3 and soluble anti-CD28 antibodies for 100, 200, and 300 min. Then, total RNA was isolated at each time points of TCR-mediated stimulation, and the expression of Zfp36, Zfp36l1, and Zfp36l2 was quantified by qPCR. Consistent with previous studies (10, 29), TCR-mediated stimulation significantly induced Zfp36 (p < 0.005) and Zfp36l1 (p < 0.005) expression, resulting in a 3-fold increase at 100 min and then gradually decreasing thereafter at 200 and 300 min (Figure 1A). In contrast, TCR-mediated stimulation lead to a ~2.5 fold reduction (p < 0.005) of the mRNA levels of Zfp36l2 (Figure 1A). Interestingly, when analogous experiments were done using human CD4+ T cells, we also found that the expression of ZFP36L2 mRNA was reduced by TCR-mediated activation in cells isolated from healthy donors (Figure 1B). Thus, TCR-mediated stimulation resulted in a comparable downregulation of Zfp36l2 in the murine system as well as in humans, suggesting that the mechanism for this downregulation is conserved in both species. However, differentiation of naïve CD4+ T cells into effector T cells (Teffs) or Tregs in vivo requires other factors that could potentially affect the expression pattern of ZFP36 family members. So, to test if these genes behaved similarly when CD4+ T cells had been in vivo differentiated, we isolated three populations of CD4+ T cells from WT mice: (i) CD4+ CD62L+ CD25− TCRαβ+ T cells, namely naïve CD4+ T cells; (ii) CD4+ CD62L− CD25− T cells, referred as Teffs; and (iii) CD4+ CD25+ T cells, known as Tregs. We found that mRNA levels of Zfp36l2 were significantly lower in Tregs when compared to those in naïve CD4+ T cells or Teffs (Figure 1C). The levels of Zfp36 mRNA in naïve CD4+ T cells and Teffs were not significantly different; however, Tregs cells expressed significantly lower levels of Zfp36 mRNA than in naïve CD4+ T cells (Figure 1C). On the other hand, mRNA levels of Zfp36l1 were relatively similar among naïve CD4+ T cells, Teffs, and Tregs. These results indicate that the expression pattern of ZFP36L2 is regulated differently than ZFP36 or ZFP36L1, particularly in Tregs, suggesting that the lower expression of ZFP36L2 could be meaningful in Treg function.
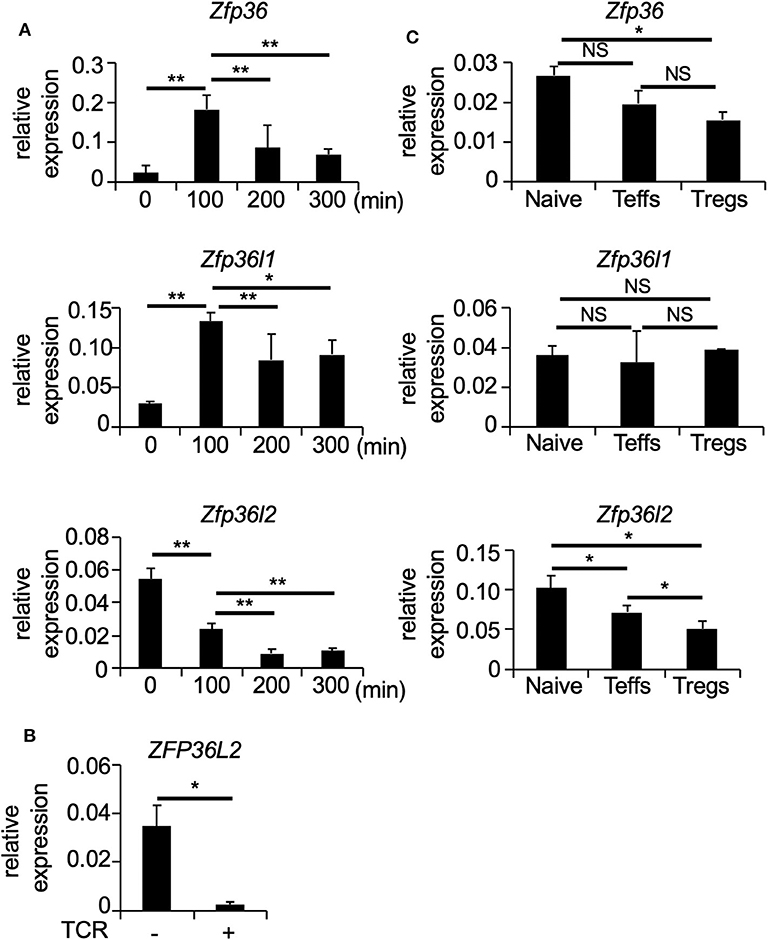
Figure 1. TCR-mediated stimulation downregulates Zfp36l2 expression. (A) Naïve CD4+ T cells were isolated from the spleen of C57BL/6 wild-type (WT) mice and stimulated with anti-CD3/anti-CD28 mAb under neutral conditions for indicated periods. Total RNAs were prepared from these cells, and mRNA levels of Zfp36, Zfp36l1, and Zfp36l2 were analyzed by quantitative PCR (qPCR) analysis. Data are means ± SD (n = 3, each), representative of three independent experiments. *p < 0.05, **p < 0.005. (B) CD4+ T cells isolated from PBMCs of healthy donors were stimulated with Dynabeads Human T-Activator CD3/CD28 (TCR+). Freshly isolated CD4+ T cells were used as controls (TCR–). Eight hours later, total RNAs were prepared from these cells, and mRNA levels of ZFP36L2 were analyzed by qPCR analysis. Data are means ± SD (n = 3, each). *p < 0.05. (C) CD25− CD62L+ CD4+ T cells (Naive), CD25− CD62L− CD4+ T cells (effector CD4+ T cells; Teffs), and CD25+ CD4+ T cells (regulatory CD4+ T cells; Tregs) were isolated from spleen of WT mice. Total RNAs were prepared from these cells, and mRNA levels of Zfp36, Zfp36l1, and Zfp36l2 were analyzed by qPCR analysis. Data are means ± SD (n = 3, each), representative of two independent experiments. *p < 0.05. NS, not significant.
Because ZFP36L2 can bind and stimulate the degradation of ARE-containing mRNAs (17, 26, 30), we raised the possibility that ZFP36L2 could alter the development and function of Tregs by modulating some specific mRNAs. To determine which target mRNAs were potentially subjected to ZFP36L2 modulation, we developed a system in which ZFP36L2 expression was comprehensively higher than the normal physiological levels. So, we could search for ARE-containing mRNAs that were downregulated when the expression of ZFP36L2 was artificially forced. Naïve CD4+ T cells were infected with a retrovirus of MIG-Zfp36l2 or with a control retrovirus MIG (mock). After infection, these cells were cultured under neutral conditions for three additional days, and then based on the simultaneous expression of CD4 and GFP by the retrovirus infection, the cells were sorted by flow cytometry. Two populations of cells were isolated: (i) one in which ZFP36L2 was highly expressed due to the retroviral infection and (ii) another “mock” retroviral infection, in which ZFP36L2 expression was derived only from the endogenous gene. Total RNA was extracted from both cell populations, and gene expression of these cells was evaluated by RNA-seq analysis.
Initially, we evaluated the presence or absence of AREs in the 3′UTR of all detected genes by the RNA-seq analysis and found that 4,368 over 24,941 (17.5%) contained an ARE (Figure 2A). Upregulated genes and downregulated genes were defined as: [1] Expression levels ≥2 FPKM (fragments per kilobase of exon per million mapped fragments) in mock-transfected CD4+ T cells or ZFP36L2-transfected CD4+ T cells, and [2] FPKM changed ≥1.5-fold by forced ZFP36L2 expression. As expected, genes containing AREs in 3′UTR were significantly enriched in the downregulated genes when ZFP36L2 was overexpressed; 233 over 620 downregulated genes contained an ARE sequence (37.6%) (Figure 2A). We next identified 33 candidate genes in CD4+ T cells that simultaneously fulfill three criteria (Figure 2B): [1] Expression level (FPKM ≥ 2) in mock-transfected CD4+ T cells or ZFP36L2-transfected CD4+ T cells; [2] FPKM was reduced ≥2-fold by forced ZFP36L2 expression; [3] Gene contains an ARE sequence in the 3′UTR. Among these 33 genes that satisfied the above three criteria (Figure S1), we then searched for transcription factors that could influence CD4+ T cell differentiation, and we found three transcription factors (Klf2, Irf8, and Ikzf2) (Figure 2C). We decided to focus on Ikzf2, which encodes Helios, because Ikzf2 has been described to be involved in Treg function and differentiation (24, 31).
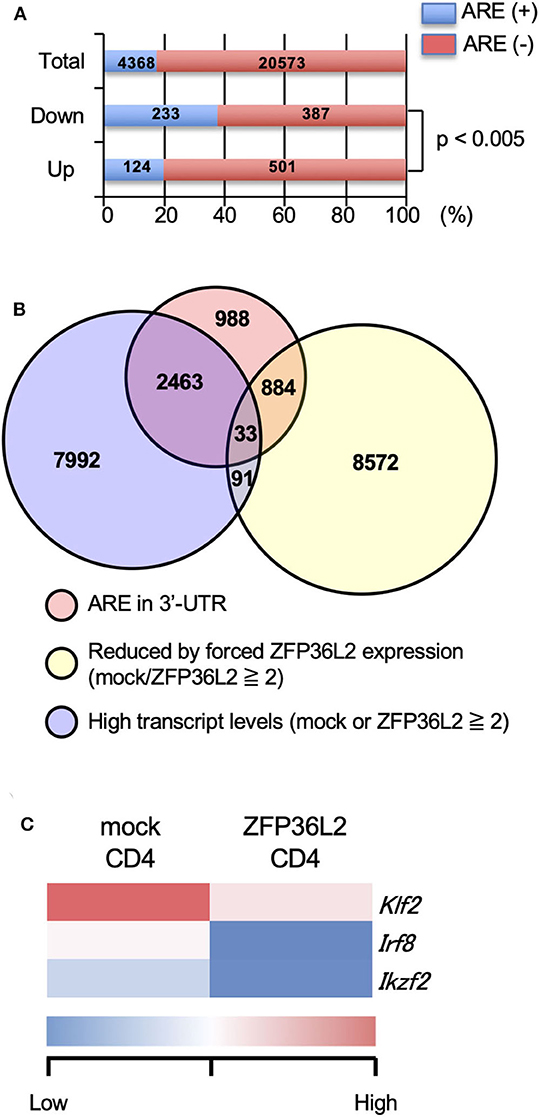
Figure 2. Ikzf2 (encoding Helios) is downregulated by forced ZFP36L2 expression. (A–C) After naïve CD4+ T cells were stimulated with anti-CD3/anti-CD28 mAb under neutral conditions for 24 h, cells were infected with retroviruses of MSCV-IRES-GFP-Zfp36l2 (MIG-Zfp36l2) or MIG, and cultured under neutral conditions. Three days later, GFP+ cells from MIG-Zfp36l2-infected CD4+ T cells and from MIG-infected CD4+ T cells were isolated by flow cytometry. Total RNAs were prepared from these cells and subjected to RNA-seq analysis. (A) Quantification of mRNAs with or without AREs in 3′UTR of mRNA downregulated (Down) or upregulated (Up) in CD4+ T cells by ZFP36L2 expression (transcript levels ≧2, fold change ≧1.5). (B) Venn diagram showing the overlap of indicated criteria in mRNAs expressed in CD4+ T cells. (C) Shown is a heat map of differentially expressed genes encoding transcriptional factors among 33 genes enriched in (B).
If ZFP36L2 regulates the Helios expression in vivo, one would expect that increased expression levels of ZFP36L2 would result in lower Helios levels and vice-versa. Thus, we set up experiments to investigate the correlation between ZFP36L2 and Helios expression in murine Tregs in vivo. Foxp3 is a well-accepted marker of Tregs (32); thus, we used Foxp3YFP−Cre mice as a strategy to isolate Tregs by sorting YFP-expressing cells by flow cytometry. We isolated two distinct T cell populations from Foxp3YFP−Cre mice: (i) CD4+ YFP+ cells in the spleen [splenic Tregs], which mainly consist of thymus-derived Tregs and (ii) CD4+ YFP+ cells in the large intestine [colonic Tregs], which mainly consist of peripherally induced Tregs. We observed that the frequency of Helios-expressing cells was significantly higher in splenic Tregs (37.7%) than in colonic Tregs (7.67%) (p < 0.005) (Figure 3A), consistent with a previous report (33). Then, we used these two Treg populations to evaluate the expression of Zfp36l2 mRNA. Intriguingly, Zfp36l2 mRNA levels were significantly higher in colonic Tregs, which express low levels of Helios, than those in splenic Tregs, which express high levels of Helios (Figure 3B). These observations raise the possibility that Zfp36l2 would need to be downregulated for thymus-derived Treg differentiation, allowing high expression levels of Helios protein in this population. Conversely, to favor peripherally induced Treg differentiation, Zfp36l2 would need to be upregulated, consequently resulting in low levels of Helios protein.
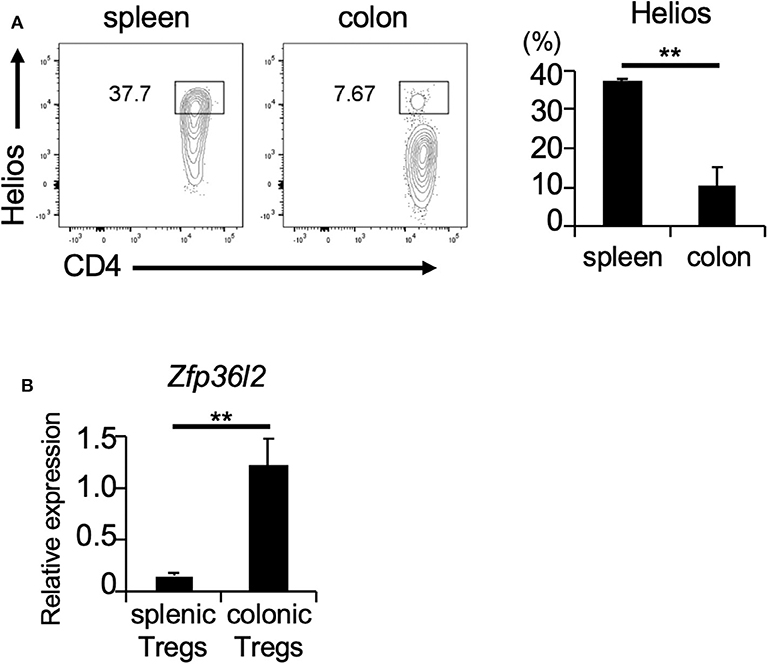
Figure 3. Zfp36l2 expression is inversely correlated with Helios expression in Tregs. (A) CD4+ YFP+ Tregs were isolated from the spleen and colon of Foxp3YFP−Cre mice. Representative CD4 vs. Helios staining of CD4+ YFP+ cells and the frequency of CD4+ Helios+ cells are shown. Data are means ± SD (n = 3, each), **p < 0.005, representative of two independent experiments. (B) Total RNAs were prepared from CD4+ YFP+ cells in spleen and colon of Foxp3YFP−Cre mice, and mRNA levels of Zfp36l2 were analyzed by qPCR analysis. Data are means ± SD (n = 3, each), **p < 0.005, representative of two independent experiments.
To test if any ZFP36 family members or only ZFP36L2 could modulate Helios proteins and iTreg differentiation, we used the retrovirus expression system to express each ZFP family member in naïve CD4+ T cells and examined the effect of each ZFP36 family member on Foxp3 and Helios expression in CD4+ T cells under iTreg-inducing conditions. Intracellular staining of Foxp3 and Helios in CD4+ T cells revealed that the percentage of Helios− iTregs (Helios− CD4+ Foxp3+ cells) among the total iTreg population was not significantly affected by the expression of ZFP36L2 in comparison to mock transfection, 61.4% vs. 63.3%, respectively (Figure 4A). In contrast, the percentage of Helios-expressing CD4+ Foxp3+ cells was markedly decreased when ZFP36L2 was expressed in CD4+ T cells compared to mock-transfected CD4+ T cells (Figure 4A). As suspected, forced expression of ZFP36 or ZFP36L1 did not significantly affect the frequency of Helios-expressing CD4+ Foxp3+ cells (Figure 4A). These findings suggest that ZFP36L2, but not ZFP or ZFP36L1, can modulate Ikzf2 mRNA and Helios protein expression in CD4+ T cells induced to differentiate into Helios-expressing iTregs.
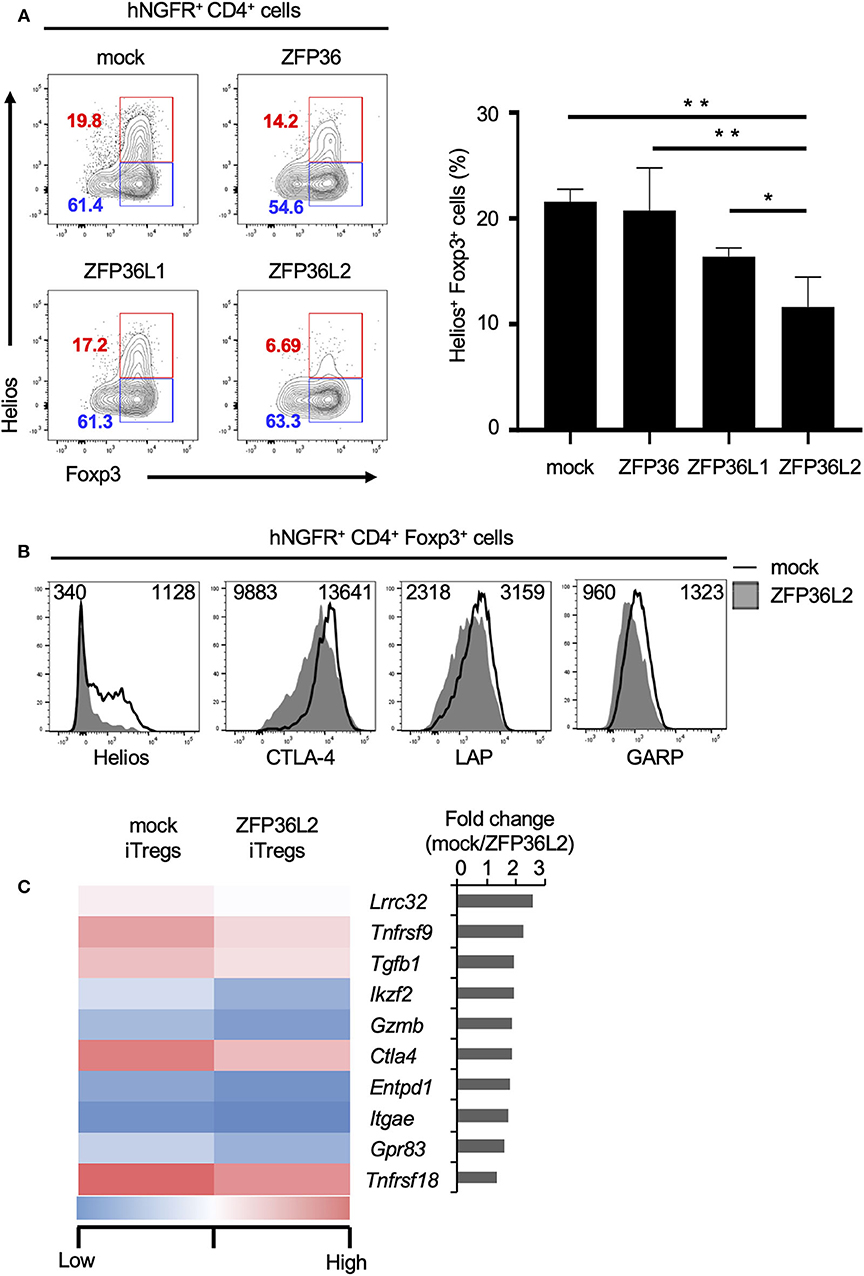
Figure 4. ZFP36L2 inhibits the development of Helios+ iTregs. (A,B) Naïve CD4+ T cells from BALB/c mice were stimulated with anti-CD3/anti-CD28 mAb under neutral conditions for 24 h, infected with retroviruses of MSCV-IRES-hNGFR (MIN)-Zfp36, MIN-Zfp36l1, MIN-Zfp36l2, or MIN (mock), and cultured under iTreg-inducing conditions. (A) The expression of Foxp3 and Helios in hNGFR+ CD4+ cells was analyzed. Shown are representative Foxp3 vs. Helios staining of hNGFR+ CD4+ cells and means ± SD of the percentages of Helios+ Foxp3+ cells n = 3. (B) The expression of indicated molecules in hNGFR+ CD4+ Foxp3+ cells were analyzed by flow cytometry. Shown are representative histograms with mean fluorescence intensity n = 3. (C) After naïve CD4+ T cells of Foxp3hCD2 mice were stimulated with anti-CD3/anti-CD28 mAb under neutral conditions for 24 h, cells were infected with retroviruses of MIG-Zfp36l2 or MIG, and cultured under iTreg-inducing conditions. Three days later, GFP+ hCD2+ cells from MIG-Zfp36l2-infected CD4+ T cells (ZFP36L2 iTregs) and from MIG-infected CD4+ T cells (mock iTregs) were isolated by flow cytometry. Total RNAs were prepared from these cells and subjected to RNA-seq analysis. Shown is a heat map of differentially expressed genes encoding Treg-associated molecules between Mock-iTregs and ZFP36L2-iTregs. *p < 0.05, **p < 0.005.
Also, we have previously shown that Helios effectively upregulates the expression of other molecules present in iTregs (24), such as CD103 and Glucocorticoid-induced TNFR-related protein (GITR). Particularly, Helios-expressing iTregs express higher mRNA levels of glycoprotein A repetitions predominant (GARP), folate receptor 4 (FR4), and G protein-coupled receptor 83 (GPR83) than in control iTregs (24). Because we observed that ZFP36L2 decreases the subpopulation of iTregs expressing Helios as well as the levels of Helios expression, we decided to investigate if the expression of these other molecules associated with Helios presence in iTregs were also affected by the forced expression of ZFP36L2. As expected, ZFP36L2-transfected iTregs expressed markedly reduced Helios levels when compared with mock-transfected control iTregs (Figure 4B). However, we observed only a modest decrease of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), LAP, and GARP expression levels. Subsequently, with the goal of broadly investigate the consequences of forced expression of Zfp36l2 in reducing gene expression of other Treg-associated molecules, we decided to use these two populations of cells, mock and ZFP36L2-transfected iTregs to isolate mRNAs and perform RNA-seq analysis. This allowed us to investigate other Treg-associated molecules, such as Lrrc32, Tnfrsf9, and Tgfb1, which encodes GARP, CD137/4-1BB, and TGF-β1, respectively. In Figure 4C, we summarized the top 10 Treg-associated genes that were found to be downregulated when ZFP36L2 was overexpressed. These results suggest that ZFP36L2 reduces the expression of Helios and successively some other Treg-associated molecules in iTregs.
We suspected that ZFP36L2 is downregulating the Helios expression in CD4+ T cells by binding to Ikzf2 mRNA, which triggers the destabilization of this mRNA, consequently leading to low expression levels of Helios protein. To investigate whether ZFP36L2 affects mRNA stability of Ikzf2 in iTregs, we measured the decay rate of Ikzf2 mRNA in sorted ZFP36L2- or mock-transfected iTregs treated with actinomycin D. We observed that Ikzf2 mRNA had a shorter half-life in ZFP36L2-transfected iTregs (t1/2 > 0.33 h) than that in mock-transfected iTregs (t1/2 > 3.01 h) (Figure 5A).
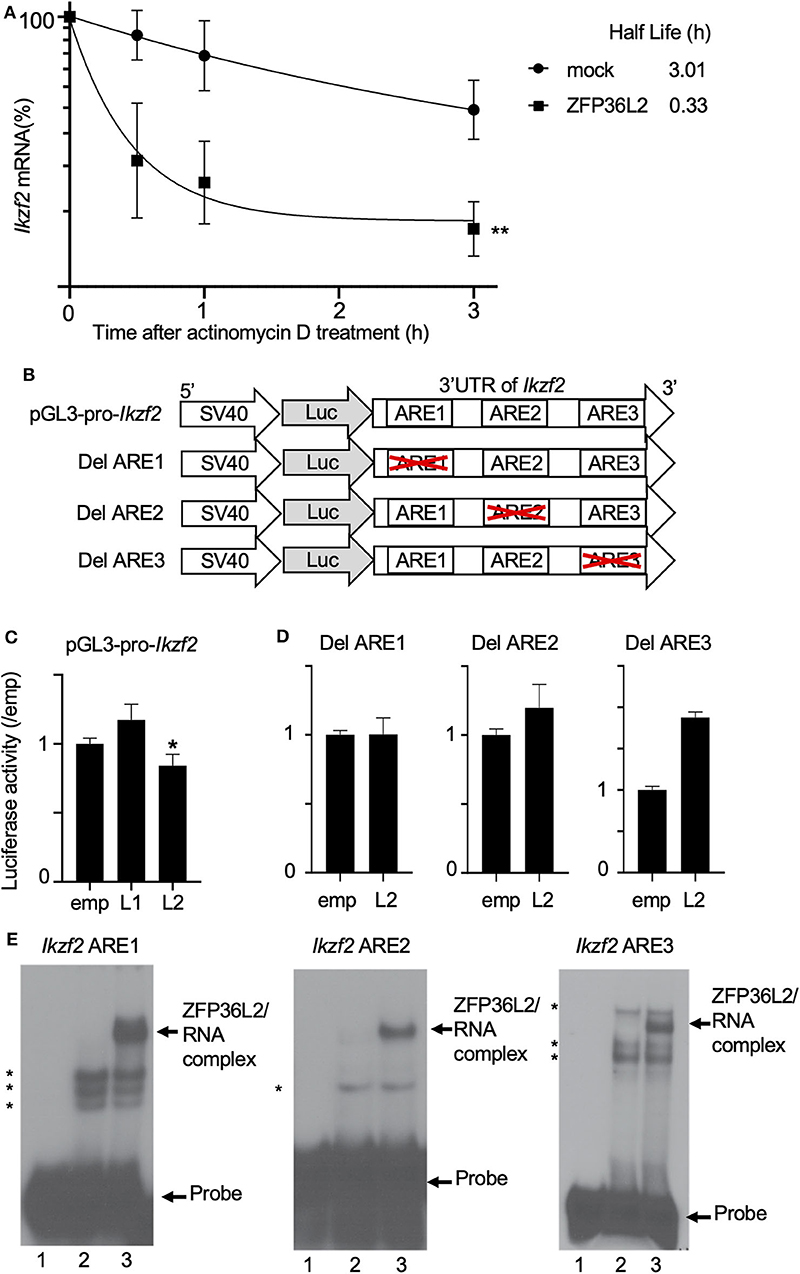
Figure 5. ZFP36L2 destabilizes 3′UTR of Ikzf2 mRNA. (A) Kinetics of Ikzf2 mRNA levels in sorted mock- or ZFP36L2-transfected iTregs after actinomycin D treatment. The amount of Ikzf2 mRNA at 0 h was assigned 100%. Trendlines (Graphpad Prism) were fitted to predict mRNA half-lives. Data are means ± SD (n = 3, each), **p < 0.005, representative of two independent experiments. (B) Schematic diagram of reporter constructs of pGL3-pro-Ikzf2-3′UTR (pGL3-pro-Ikzf2), and ARE deletion mutants. (C,D) EL-4 cells were transfected with pRL-TK and pGL3-pro-Ikzf2 in the presence of either pcDNA3-Zfp36l1 (L1), pcDNA3-Zfp36l2 (L2), or pcDNA3 (emp) (as a control) (C). EL-4 cells were transfected with pRL-TK and either Del ARE1, Del ARE2, or Del ARE3 in the presence of pcDNA3-Zfp36l2(L2) or pcDNA3 (emp) (D). Four hours later, cells were stimulated with PMA/Iono for 20 h. Luciferase activity was measured by the dual-luciferase reporter system. Data are means ± SD (n = 3, each), *p < 0.05, representative of two independent experiments. (E) RNA electrophoretic mobility shift assays were performed by incubating protein extracts from HEK 293 cells transfected with either an empty vector (10 μg, lanes 2) or a vector expressing ZFP36L2 (10 μg, lanes 3) with each Ikzf2 ARE probe, as indicated on top of the lanes. Lanes 1 in all three panels contain each probe without the addition of any protein extracts. ZFP36L2 caused a shift in the electrophoretic mobility of all three ARE probes (arrow). Note that when protein extracts lacking ZFP36L2 protein (lanes 2) were incubated with each probe, other bands appeared (indicated by asterisks). They correspond to complexes of proteins present in HEK 293 cells that interact with the probes.
Because the binding of ZFP36L2 to a particular target mRNA is dependent on the presence of ARE sequences in 3′UTR, we decided to investigate if the ZFP36L2 downregulation effect was dependent on the 3′UTR sequence of Ikzf2 mRNA. As illustrated in Figure 5B, we created a reporter construct in which the 3′UTR of Ikzf2 mRNA was inserted just after the coding sequence of the luciferase gene of pGL3-pro (pGL3-pro-Ikzf2). Three putative AREs were found to be present in the 3′UTR of the mouse Ikzf2 transcript. In order to investigate the contribution of each of these AREs on the destabilization of this transcript, we created mutant reporter constructs in which individual AREs were deleted (Del ARE1, Del ARE2, and Del ARE3, schematically represented in Figure 5B). As shown in Figure 5C, the luciferase activity of pGL3-pro-Ikzf2 was decreased by the expression of ZFP36L2 but not by ZFP36L1. Deletion of ARE1 or ARE2 did not significantly affect the luciferase activity in the presence of ZFP36L2 (Figure 5D), suggesting mild contribution of these AREs on the ZFP36L2-mediated downregulation of Ikzf2 in this experimental setting. Intriguingly, the deletion of ARE3 resulted in increased luciferase activity, although the reason remains unknown.
Additionally, to test if ZFP36L2 could bind directly to these AREs located at 3′UTR of Ikzf2 transcript, we designed three different probes Ikzf2 ARE1, ARE2, and ARE3, each containing only one ARE. We then performed RNA gel shift assays with these probes using protein extracts overexpressing or lacking ZFP36L2. As illustrated in Figure 5E, ZFP36L2 prominently bound to each of these three probes (arrows in lane 3 of each panel). These results confirm a direct biochemical interaction between ZFP36L2 protein and each of these individual AREs of the Ikzf2 mRNA, confirming that ZFP36L2-mediated downregulation of Helios expression is dependent on specific ARE sequences present at the 3′UTR of Ikzf2 mRNA.
iTregs can inhibit the proliferation of naïve CD4+ T cells (33); however, it is not known if iTregs, which preferentially differentiate into Helios-low iTregs by forced expression of ZFP36L2, retain their ability to inhibit naïve CD4+ T cell proliferation. To address this question, naive CD4+ T cells isolated from Foxp3YFP−Cre mice were infected with a retrovirus of MIN-Zfp36l2 or MIN (as a control) and cultured for an additional 4 days in iTreg-inducing conditions. These cultured cells were subjected to flow cytometry to isolate only the retrovirus-infected iTregs (CD4+ YFP+ hNGFR+ cells), which were then co-cultured under neutral conditions with freshly isolated naïve CD4+ T cells (responder T cells:Tresps) to evaluate the ability of these iTregs to inhibit the proliferation of Tresps. iTregs overexpressing ZFP36L2 (ZFP36L2 iTregs) exhibit significantly weak ability to suppress Tresp proliferation than the control iTregs (mock iTregs), which suppress Tresp proliferation (Figures 6A,B).
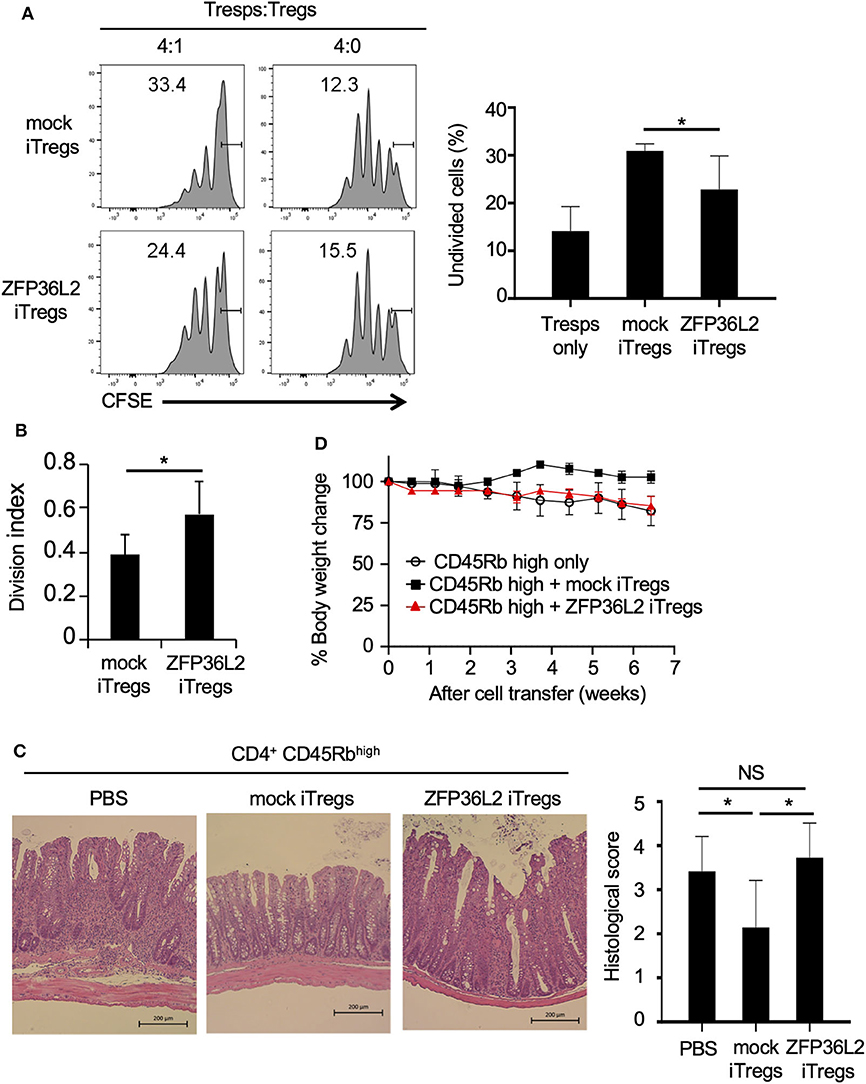
Figure 6. ZFP36L2 inhibits the suppression function of iTregs. (A,B) YFP+ hNGFR+ cells from MIN-Zfp36l2-infected CD4+ T cells (ZFP36L2 iTregs) and those from MIN-infected CD4+ T cells (mock iTregs) of Foxp3YFP−Cre mice were prepared as described in the Materials and Methods. Naïve CD4+ T cells were labeled with CFSE and stimulated with soluble anti-CD3 mAb in the presence of irradiated splenocytes. Where indicated, ZFP36L2 iTregs or mock iTregs were added at a 4:1 ratio. CFSE dilution profiles of naïve CD4+ T cells were assessed by flow cytometry. (A) Representative histograms of CFSE dilution and means ± SD of the percentages of undivided cells. (B) Division index using FlowJo software. Means ± SD (n = 6, each), *p < 0.05. Similar results were obtained in two independent experiments. (C,D) Naive CD4+ T cells (CD45Rbhigh YFP− CD4+ cells from Foxp3YFP−Cre mice) were injected into RAG2−/− mice to induce colitis. Where indicated, pMX-IN-Zfp36l2-transfected iTreg (ZFP36L2 iTregs) or pMX-IN-transfected iTregs (mock iTregs) were co-injected to the mice. (C) Shown are representative H&E stained sections of the colon and means ± SD of histopathological scores at 7 weeks after the cell transfer. *p < 0.05, bar: 200 μm. (D) Shown are changes in body weight after the cell transfer (three mice in each group).
We finally examined the function of ZFP36L2 iTregs in vivo by using a T cell transfer model of colitis (28). In this experiment, ZFP36L2 iTregs or mock iTregs were adoptively co-injected with naive CD4+ T cells into RAG2−/− mice, and the development of colitis was evaluated by histological analyses and by its effect on the mice weight. In the absence of iTreg injection, recipient RAG2−/− mice developed colitis (Figure 6C) and reduced body weight (Figure 6D, open circles). Notably, when the mice were co-injected with mock iTregs, the histological scores improved, and there was significant body weight gain (Figures 6C,D). However, when ZFP36L2 iTregs were co-injected, no significant improvement was observed on histological evaluation of the colitis nor the body weight, suggesting that ZFP36L2 iTregs have limited suppression function in vivo.
Imbalance of the immunological self-tolerance response is the basis of several autoimmune diseases (18). One of the critical cell populations in building up an effective immunological self-tolerant response is Tregs. These cells are particularly important in maintaining the induced tolerance to peripheral self-antigens as well as suppressing deleterious immune responses against the host itself. In this study, we focused on the role of the RNA-binding protein ZFP36L2 in modulating the differentiation and function of Tregs. Initially, we observed that TCR-mediated stimulation downregulates ZFP36L2 expression in human and murine CD4+ T cells (Figures 1A,B). Additionally, we found that ZFP36L2 expression levels were lower in Tregs than those in naïve CD4+ T cells (Figure 1C), suggesting that during the differentiation process of naïve CD4+ T cells into iTregs, TCR stimulation may negatively modulate the ZFP36L2 expression. In contrast, when we artificially forced ZFP36L2 expression, it inhibited the expression of several transcription factors, particularly Helios. The differentiation of iTregs in the presence of ZFP36L2 resulted in the differentiation of iTregs, preferentially not expressing Helios (Figure 4). The mechanism by which ZFP36L2 is downregulating Helios expression is likely to be the induction of Ikzf2 mRNA degradation since ZFP36L2 action is dependent on the endogenous 3′UTR sequence of Ikzf2 mRNA which contains ARE-binding elements (Figure 5). Interestingly, ZFP36L2-expressing iTregs have reduced suppression property on the proliferation of naïve CD4+ T cells in vitro as well as the development of the T cell transfer model of colitis (Figure 6). These results suggest that the reduction of ZFP36L2 expression during Treg differentiation boosts the preferential differentiation of Helios+ iTregs, which are known to efficiently suppress the proliferation of naïve CD4+ T cells, thus maintaining a more effective regulatory function over activated CD4+ T cells.
We found that in contrast to the other ZFP36 family members, ZFP36 and ZFP36L1, ZFP36L2 is expressed to a greater degree in naïve CD4+ T cells than in Teffs, whereas the expression of ZFP36 or ZFP36L1 in naïve CD4+ T cells was similar to that in Teffs (Figure 1C). We also found that TCR-mediated stimulation decreased ZFP36L2 expression but increased the expression of ZFP36 and ZFP36L1 in CD4+ T cells (Figures 1A,B). These findings were largely consistent with recent reports (10, 29). Moore et al. have shown that ZFP36 and ZFP36L1 were induced in CD4+ T cells upon activation (10), and Salerno et al. have demonstrated that the expression of ZFP36L2 mRNAs in CD4+ T cells was decreased by PMA/Ionomycin stimulation (29). We also found that genes that are known to be highly expressed in naïve CD4+ T cells and downregulated by TCR-mediated activation, including Stat5a and Tnfrsf10b, were upregulated by the forced expression of ZFP36L2 in CD4+ T cells (data not shown). These findings raise the possibility that the ZFP36L2 could be involved in maintaining CD4+ T cells in a naïve state.
Our findings suggest that maintaining a low ZFP36L2 expression level is important for maintaining Treg function in vivo. We found that the expression levels of ZFP36L2 in Tregs were significantly lower than those in Teffs or naïve CD4+ T cells (Figure 1C). We also found that the forced expression of ZFP36L2 decreased the suppression function of Tregs in the in vitro suppression assay as well as in the adoptive transfer model of colitis (Figure 6). Consistently, the forced expression of ZFP36L2 decreased the expression of Treg-associated molecules, including CTLA-4, LAP, and GARP, as well as Helios in iTregs (Figure 4B). In this regard, because 3′UTR of mRNAs of Ctla4, Tgfb1, or Lrrc32 does not contain AREs for binding of ZFP36L2 (data not shown), it is possible that ZFP36L2-mediated reduction of these Treg-associated molecules is achieved through indirect mechanisms rather than direct degradation of their mRNAs.
We showed that ZFP36L2 reduces Helios expression in CD4+ T cells under iTreg-inducing conditions. Regarding the roles of Helios in Treg function, it was initially reported that mice with a T cell-specific deletion of Helios appear to have normal Treg function (34). However, recent reports have shown that Foxp3-specific-Helios-deficient mice, as well as systemic Helios-deficient mice, spontaneously develop delayed-onset autoimmune-like conditions with multi-organ inflammation, systemic lymphocyte activation, and anti-nuclear and anti-DNA antibodies (35, 36). In addition, we have previously shown that the forced expression of Helios induces the expression of the Treg–related molecules GITR, CD103, GARP, and FR4 and enhances the suppression function in murine Tregs (24). Furthermore, it has been shown that Helios deficiency results in the unstable phenotypes in Tregs during inflammatory responses due to the production of proinflammatory cytokines, including IFN-γ, IL-17, and TNF-α (35–37). Moreover, mice with Helios-deficiency in Tregs exhibited enhanced antitumor immunity (36). Given that ZFP36L2 induction in CD4+ T cells profoundly reduced Helios expression with a modest reduction of other Treg-related molecules (Figure 4), we speculate that the fine-tuning of ZFP36L2 expression in Tregs is necessary for appropriate Helios expression and the function of Tregs.
Helios expression has been shown to be regulated in several ways. We have previously demonstrated that Helios is induced by TGF-β and suppressed by IL-6/STAT3 signaling in CD4+ T cells (24) and that cell-intrinsic Foxp3 expression is essential for Helios induction in murine iTregs (24). Yet numerous other explanations have been discovered for Helios induction and maintenance. Recent studies have shown that IL-2/STAT5 signaling pathway promotes Helios induction in Tregs in humans and mice (38, 39). Vitamin D receptor-mediated signaling has been demonstrated to activate Ikzf2 gene transcription in Tregs through vitamin D-responsive elements (40). On the other hand, Nakagawa et al. reported that activation of GITR using the agonistic anti-GITR antibody DTA-1 decreases Helios expression in Tregs and favors their conversion to Teffs in vivo (36). We demonstrated here that ZFP36L2 markedly inhibited Helios expression (Figure 4) and that 3′UTR of Ikzf2 mRNA seems to be a target of ZFP36L2-mediated destabilization (Figure 5). These findings suggest that Helios expression seems to be regulated by several distinct mechanisms in CD4+ T cells whose relative importance should be determined in future research.
Among ZFP36 family members, we found that ZFP36L2 but not ZFP36 or ZFP36L1 suppressed the expression of Helios in CD4+ T cells under iTreg-inducing conditions (Figure 4A). Also, the expression of these three members was differently regulated in CD4+ T cells (Figure 1A). These findings suggest that the roles of ZFP36 family members in Tregs differ from each other. On the other hand, Hodson et al. have shown that mice lacking both ZFP36L2 and ZFP36L1 but not either ZFP36L2 or ZFP36L1 in lymphoid lineages develop leukemia (17), suggesting some redundant roles.
Interestingly, Salerno et al. have recently reported that ZFP36L2 represses IFN-γ production in CD44hi CD4+ T cells at the translational level rather than effecting the mRNA stability (29). We showed here that ZFP36L2 suppresses iTreg function. Based on these findings, we suppose that ZFP36L2′s roles in CD4+ T cells seem to differ depending on the cell type. It is also possible that these different ZFP36L2 mechanisms may influence and complement each other in vivo. Indeed, the suppression of IFN-γ production by ZFP36L2 may partly influence the results of the colitis model because IFN-γ is well-known to be involved in the induction of colitis. Further studies, including specific deletion of ZFP36 family members in Tregs, are needed to elucidate the precise roles of ZFP36 family members in Helios expression and Treg suppression.
In conclusion, we show that ZFP36L2 suppresses iTreg function, possibly by destabilizing Ikzf2 mRNA. Although further studies are needed, selective fine-tuning of ZFP36L2 affecting Helios expression in iTregs could be an effective strategy for the treatment of autoimmune diseases.
The datasets generated for this study can be found in the National Center for Biotechnology Information's Gene Expression Omnibus (GSE150937) upon publication of the article.
The studies involving human participants were reviewed and approved by Ethics Committee of Chiba University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Chiba University for experiments in animals.
HT has full access to all data generated by this study and takes responsibility for the data and also designed and performed the experiments and wrote the manuscript. SM performed the experiments, prepared reagents, and wrote the manuscript. AI analyzed the data of RNA-seq analyses. ST, AS, and KS proposed the projects and designed the experiments. SF and KI proposed the projects. SR provided reagents, helped design constructs, performed and interpreted experiments, and wrote the manuscript. HN proposed the projects, designed the experiments, and wrote the manuscript. All authors were involved in drafting the manuscript and approved the final version to be published.
This work was supported in part by Grants-in-Aids for Scientific research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (17K09970), LGS (Leading Graduate School at Chiba University) Program, MEXT, Japan, and UNC TraCS grant no. 550KR11128 and CTSA—UL1TR002489 to SR.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Chang Ching-Jin (National Taiwan University, Taiwan) for ZFP36-, ZFP36L1-, and ZFP36L2-expressing vectors. We also thank Drs. Ken Nonaka and Osamu Ohara (Kazusa DNA Research Institute) for RNA-Seq analyses. We also thank Mss. Juri Iwata, Kazumi Nemoto, and Yumiko Hanabuchi (Chiba University) for technical support and Mr. Richard Youngblood for editing the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01291/full#supplementary-material
Figure S1. Genes whose expression is downregulated by the forced expression of ZFP36L2. Shown is a heat map of the 33 differentially expressed mRNAs described in Figure 2B.
ARE, AU-rich element; CTLA-4, cytotoxic T lymphocyte-associated protein 4; Foxp3, forkhead box p3; FPKM, fragments per kilobase of exon per million mapped fragments; GARP, glycoprotein-A repetitions predominant; Ikzf2, Ikaros family zinc finger 2; iTregs, induced Tregs; LAP, latency-associated peptide; MIG, MSCV-IRES-GFP; MIN, MSCV-IRES-hNGFR; PBMC, peripheral blood mononuclear cells; qPCR, quantitative real-time PCR; Teffs, effector T cells; Tregs, regulatory T cells; Zfp36l1, zinc finger protein 36, CCCH type-like 1; Zfp36l2, zinc finger protein 36, CCCH type-like 2.
1. Lai WS, Carballo E, Thorn JM, Kennington EA, Blackshear PJ. Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin-related zinc finger proteins to Au-rich elements destabilization of mRNA. J Biol Chem. (2000) 275:17827–37. doi: 10.1074/jbc.M001696200
2. Lai WS, Blackshear PJ. Interactions of CCCH zinc finger proteins with mRNA: tristetraprolin-mediated AU-rich element-dependent mRNA degradation can occur in the absence of a poly(A) tail. J Biol Chem. (2001) 276:23144–54. doi: 10.1074/jbc.M100680200
3. Fu M, Blackshear PJ. RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat Rev Immunol. (2017) 17:130–43. doi: 10.1038/nri.2016.129
4. Blackshear PJ, Phillips RS, Ghosh S, Ramos SB, Richfield EK, et al. Zfp36l3, a rodent X chromosome gene encoding a placenta-specific member of the tristetraprolin family of CCCH tandem zinc finger proteins. Biol Reprod. (2005) 73:297–307. doi: 10.1095/biolreprod.105.040527
5. Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. (1996) 4:445–54. doi: 10.1016/S1074-7613(00)80411-2
6. Lai WS, Kennington EA, Blackshear PJ. Tristetraprolin and its family members can promote the cell-free deadenylation of AU-rich element-containing mRNAs by poly(A) ribonuclease. Mol Cell Biol. (2003) 23:3798–812. doi: 10.1128/MCB.23.11.3798-3812.2003
7. Ogilvie RL, Abelson M, Hau HH, Vlasova I, Blackshear PJ, Bohjanen PR. Tristetraprolin down-regulates IL-2 gene expression through AU-rich element-mediated mRNA decay. J Immunol. (2005) 174:953–61. doi: 10.4049/jimmunol.174.2.953
8. Ogilvie RL, Sternjohn JR, Rattenbacher B, Vlasova IA, Williams DA, Hau HH, et al. Tristetraprolin mediates interferon-gamma mRNA decay. J Biol Chem. (2009) 284:11216–23. doi: 10.1074/jbc.M901229200
9. Lee HH, Yoon NA, Vo MT, Kim CW, Woo JM, Cha HJ, et al. Tristetraprolin down-regulates IL-17 through mRNA destabilization. FEBS Lett. (2012) 586:41–6. doi: 10.1016/j.febslet.2011.11.021
10. Moore MJ, Blachere NE, Fak JJ, Park CY, Sawicka K, Parveen S, et al. ZFP36 RNA-binding proteins restrain T cell activation and anti-viral immunity. Elife. (2018) 7:e33057. doi: 10.7554/eLife.33057.035
11. Patial S, Blackshear PJ. Tristetraprolin as a therapeutic target in inflammatory disease. Trends Pharmacol Sci. (2016) 37:811–21. doi: 10.1016/j.tips.2016.07.002
12. Patial S, Curtis AD, Lai WS, Stumpo DJ, Hill GD, Flake GP, et al. Enhanced stability of tristetraprolin mRNA protects mice against immune-mediated inflammatory pathologies. Proc Natl Acad Sci USA. (2016) 113:1865–70. doi: 10.1073/pnas.1519906113
13. Wang KT, Wang HH, Wu YY, Su YL, Chiang PY, Lin NY, et al. Functional regulation of Zfp36l1 and Zfp36l2 in response to lipopolysaccharide in mouse RAW264.7 macrophages. J Inflamm (Lond). (2015) 12:42. doi: 10.1186/s12950-015-0088-x
14. Ramos SB. Characterization of DeltaN-Zfp36l2 mutant associated with arrest of early embryonic development and female infertility. J Biol Chem. (2012) 287:13116–27. doi: 10.1074/jbc.M111.330837
15. Stumpo DJ, Byrd NA, Phillips RS, Ghosh S, Maronpot RR, Castranio T, et al. Chorioallantoic fusion defects and embryonic lethality resulting from disruption of Zfp36L1, a gene encoding a CCCH tandem zinc finger protein of the Tristetraprolin family. Mol Cell Biol. (2004) 24:6445–55. doi: 10.1128/MCB.24.14.6445-6455.2004
16. Stumpo DJ, Broxmeyer HE, Ward T, Cooper S, Hangoc G, Chung YJ, et al. Targeted disruption of Zfp36l2, encoding a CCCH tandem zinc finger RNA-binding protein, results in defective hematopoiesis. Blood. (2009) 114:2401–10. doi: 10.1182/blood-2009-04-214619
17. Hodson DJ, Janas ML, Galloway A, Bell SE, Andrews S, Li CM, et al. Deletion of the RNA-binding proteins ZFP36L1 and ZFP36L2 leads to perturbed thymic development and T lymphoblastic leukemia. Nat Immunol. (2010) 11:717–24. doi: 10.1038/ni.1901
18. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. (2018) 19:665–73. doi: 10.1038/s41590-018-0120-4
19. Mandel M, Gurevich M, Pauzner R, Kaminski N, Achiron A. Autoimmunity gene expression portrait: specific signature that intersects or differentiates between multiple sclerosis and systemic lupus erythematosus. Clin Exp Immunol. (2004) 138:164–70. doi: 10.1111/j.1365-2249.2004.02587.x
20. Parnell GP, Gatt PN, Krupa M, Nickles D, McKay FC, Schibeci SD, et al. The autoimmune disease-associated transcription factors EOMES and TBX21 are dysregulated in multiple sclerosis and define a molecular subtype of disease. Clin Immunol. (2014) 151:16–24. doi: 10.1016/j.clim.2014.01.003
21. Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. (2012) 36:262–75. doi: 10.1016/j.immuni.2011.12.012
22. Tanaka S, Suto A, Iwamoto T, Kashiwakuma D, Kagami S, Suzuki K, et al. Sox5 and c-Maf cooperatively induce Th17 cell differentiation via RORγt induction as downstream targets of Stat3. J Exp Med. (2014) 211:1857–74. doi: 10.1084/jem.20130791
23. Kawashima H, Takatori H, Suzuki K, Iwata A, Yokota M, Suto A, et al. Tumor suppressor p53 inhibits systemic autoimmune diseases by inducing regulatory T cells. J Immunol. (2013) 191:3614–23. doi: 10.4049/jimmunol.1300509
24. Takatori H, Kawashima H, Matsuki A, Meguro K, Tanaka S, Iwamoto T, et al. Helios enhances Treg cell function in cooperation with FoxP3. Arthritis Rheumatol. (2015) 67:1491–502. doi: 10.1002/art.39091
25. Hiramatsu Y, Suto A, Kashiwakuma D, Kanari H, Kagami S, Ikeda K, et al. c-Maf activates the promoter and enhancer of the IL-21 gene, and TGF-beta inhibits c-Maf-induced IL-21 production in CD4+ T cells. J Leukoc Biol. (2010) 87:703–12. doi: 10.1189/jlb.0909639
26. Ball CB, Rodriguez KF, Stumpo DJ, Ribeiro-Neto F, Korach KS, Blackshear PJ, et al. The RNA-binding protein, ZFP36L2, influences ovulation and oocyte maturation. PLoS ONE. (2014) 9:e97324. doi: 10.1371/journal.pone.0097324
27. Ball CB, Solem AC, Meganck RM, Laederach A, Ramos SB. Impact of RNA structure on ZFP36L2 interaction with luteinizing hormone receptor mRNA. RNA. (2017) 23:1209–23. doi: 10.1261/rna.060467.116
28. Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. (1999) 190:995–1004. doi: 10.1084/jem.190.7.995
29. Salerno F, Engels S, van den Biggelaar M, van Alphen FPJ, Guislain A, Zhao W, et al. Translational repression of pre-formed cytokine-encoding mRNA prevents chronic activation of memory T cells. Nat Immunol. (2018) 19:828–37. doi: 10.1038/s41590-018-0155-6
30. Cargnin F, Nechiporuk T, Müllendorff K, Stumpo DJ, Blackshear PJ, Ballas N, et al. An RNA binding protein promotes axonal integrity in peripheral neurons by destabilizing REST. J Neurosci. (2014) 34:16650–61. doi: 10.1523/JNEUROSCI.1650-14.2014
31. Thornton AM, Shevach EM. Helios: still behind the clouds. Immunology. (2019) 158:161–70. doi: 10.1111/imm.13115
32. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
33. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. (2014) 259:88–102. doi: 10.1111/imr.12160
34. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. (2010) 184:3433–41. doi: 10.4049/jimmunol.0904028
35. Kim HJ, Barnitz RA, Kreslavsky T, Brown FD, Moffett H, Lemieux ME, et al. Stable inhibitory activity of regulatory T cells requires the transcription factor helios. Science. (2015) 350:334–9. doi: 10.1126/science.aad0616
36. Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, Kim HJ. Instability of helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc Natl Acad Sci USA. (2016) 113:6248–53. doi: 10.1073/pnas.1604765113
37. Chougnet C, Hildeman D. Helios-controller of Treg stability and function. Transl Cancer Res. (2016) 5:S338–41. doi: 10.21037/tcr.2016.07.37
38. Hirakawa M, Matos TR, Liu H, Koreth J, Kim HT, Paul NE, et al. Low-dose IL-2 selectively activates subsets of CD4. JCI Insight. (2016) 1:e89278. doi: 10.1172/jci.insight.89278
39. Luu M, Jenike E, Vachharajani N, Visekruna A. Transcription factor c-Rel is indispensable for generation of thymic but not of peripheral Foxp3. Oncotarget. (2017) 8:52678–89. doi: 10.18632/oncotarget.17079
40. Moore JR, Hubler SL, Nelson CD, Nashold FE, Spanier JA, Hayes CE. 1,25-dihydroxyvitamin D3 increases the methionine cycle, CD4+ T cell DNA methylation and Helios+Foxp3+ T regulatory cells to reverse autoimmune neurodegenerative disease. J Neuroimmunol. (2018) 324:100–14. doi: 10.1016/j.jneuroim.2018.09.008
Keywords: ZFP36L2, Helios, regulatory T cells, AU-rich element, 3′untranslated region
Citation: Makita S, Takatori H, Iwata A, Tanaka S, Furuta S, Ikeda K, Suto A, Suzuki K, Ramos SBV and Nakajima H (2020) RNA-Binding Protein ZFP36L2 Downregulates Helios Expression and Suppresses the Function of Regulatory T Cells. Front. Immunol. 11:1291. doi: 10.3389/fimmu.2020.01291
Received: 08 December 2019; Accepted: 21 May 2020;
Published: 23 June 2020.
Edited by:
Shohei Hori, The University of Tokyo, JapanReviewed by:
Osamu Takeuchi, Kyoto University, JapanCopyright © 2020 Makita, Takatori, Iwata, Tanaka, Furuta, Ikeda, Suto, Suzuki, Ramos and Nakajima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroaki Takatori, dGFrYXRvcmloQGZhY3VsdHkuY2hpYmEtdS5qcA==; Hiroshi Nakajima, bmFrYWppbWhAZmFjdWx0eS5jaGliYS11Lmpw
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.