
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 June 2020
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01249
This article is part of the Research Topic Interplay Between Immunity and Fibrosis View all 14 articles
 Aslıhan Avanoǧlu Güler1,2
Aslıhan Avanoǧlu Güler1,2 Francesca Wanda Rossi3
Francesca Wanda Rossi3 Silvia Bellando-Randone1*
Silvia Bellando-Randone1* Nella Prevete3
Nella Prevete3 Abdurrahman Tufan2
Abdurrahman Tufan2 Mirko Manetti1
Mirko Manetti1 Amato de Paulis3
Amato de Paulis3 Marco Matucci-Cerinic1
Marco Matucci-Cerinic1Resolvins, the member of specialized pro-resolving mediators, are produced from omega-3 polyunsaturated fatty acids as a response to an acute inflammatory process in that termination and resolution of inflammation. In the acute inflammation, these lipid mediators limit polymorphonuclear cells infiltration, proinflammatory cytokine production; promote efferocytosis, and regulate several cell types being important roles in innate and adaptive immunity. Any dysregulation or defect of the resolution phase result in prolonged, persistent inflammation and eventually fibrosis. Resolvins are implicated in the development of various chronic autoimmune diseases. Systemic sclerosis (SSc) is a very complicated, chronic autoimmune disorder proceeding with vasculopathy, inflammation, and fibrosis. Dysregulation of innate and adaptive immunity is another important contributing factor in the pathogenesis of SSc. In this review, we will focus on the different roles of this new family of lipid mediators, characterized by the ability to prevent the spread of inflammation and its chronicity in various ways and how they can control the development of fibrotic diseases like SSc.
Systemic sclerosis (SSc) is a complex immune-mediated connective tissue disorder characterized by microvascular damage, inflammatory cell infiltration, and excessive deposition of extracellular matrix proteins (ECMs) in the skin and various internal organs (1–3). Over the course of the disease, these pathologic alterations cause severe organ dysfunctions such as pulmonary fibrosis, pulmonary arterial hypertension, cardiac arrhythmias and heart failure, which are the major causes of mortality in SSc (4). Identification of the immune, vasculopathic, and fibrotic mechanisms involved in the pathogenesis of SSc is critical for the understanding of disease progression and developing new disease-modifying therapies (5). Despite the fact that innate and adaptive immunity components, including T cells, B cells, macrophages, dendritic cells (DCs), and multiple cytokines (e.g., interleukin (IL)-4, IL-6, transforming growth factor (TGF)-β) play roles in both the onset and the progression of SSc, the exact etiopathogenesis of the disease still remains elusive (1, 2).
It is well-known that acute inflammatory responses, triggered by a variety of noxious stimuli, including endogenous and exogenous signals, are protective, self-limited reactions that are essential for restoring homeostasis in the affected tissues. However, this benign process may not subside, leading to chronic systemic inflammatory disorders (6, 7). In fact, in a few autoimmune and chronic inflammatory diseases, including SSc, perturbation is observed in inflammation resolution (8). Until recently, termination of acute inflammation was considered as a passive process. However, the latest investigations have demonstrated that the resolution of inflammation is an active process controlled by the local release of various mediators called specialized pro-resolving mediators (SPMs). The biosynthesis of SPMs is induced by pro-inflammatory stimuli as a compensatory mechanism to downregulate the inflammatory response (7). In general, SPMs bind to G protein-coupled receptors (GPRs) to exert anti-inflammatory and pro-resolving biological actions; inhibition of polymorphonuclear leukocyte (PMNs) infiltration and pro-inflammatory cytokine/mediator secretion; promote bacterial removal; and evoke the efferocytosis of apoptotic PMNs through macrophages (9, 10). Thus, far, more than 20 different SPMs have been explored using lipid mediator metabolon lipidomics and proteomics, and these SPMs have been subdivided into four main classes based on distinct biosynthetic pathways and target receptors: lipoxins, resolvins (Rvs), protectins, and maresins (11, 12). The discovery of Rvs attracted significant interest because these lipid mediators play prominent roles in different pathological conditions by sustaining homeostasis owing to their anti-inflammatory properties (13). It is widely accepted that Rvs play significant roles in several chronic inflammatory diseases, such as rheumatoid arthritis, Sjogren's syndrome, and inflammatory bowel disease (14–17). Although many experimental studies have been conducted to define the preventive role of Rvs in pulmonary fibrosis and ischemia-reperfusion injury in animal models, their contribution to the pathogenesis of SSc is yet to be clarified (18, 19). In this review, we will focus on this new family of lipid mediators that can control the propagation and prolongation of inflammation, as well as their possible roles in the pathogenesis of fibrotic diseases such as SSc.
For the first time, Rvs were identified in inflammatory exudates triggered by tumor necrosis alpha (TNF-α) in mice exposed to omega (Ω)-3 polyunsaturated fatty acids (PUFAs) and aspirin. Usually, different immune cells, such as macrophages and PMNs generate Rvs from Ω-3 PUFAs, namely docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), originating from the dietary sources and cell membranes through phospholipase enzyme pathways (20). Two classes of Rvs have been identified: D-series resolvins (RvD1-6) derived from DHA through lipoxygenase (LO)-initiated pathways during the inflammation- resolution phase and the E-series family of Rvs (RvE1-4) produced from EPA via 5-LO and cytochrome P450 (12, 21–23). It has been shown that Rvs signal through specific GPRs (23–26).
RvD1 exerts anti-inflammatory and inflammation-resolution effects via A lipoxin and formyl peptide receptor 2 (ALX/FPR2), as well as via GPR32 (24, 25). RvD2 interacts with GPR18 expressed on PMNs, monocytes, and macrophages (26). In addition to RvD1, RvD3, and RvD5 activate GPR32 and enhance the phagocytosis and inhibition of neutrophil transmigration (27, 28). Furthermore, RvEs exert their function through GPCRs. Chemerin receptor 23 (ChemR23), which is selectively expressed on antigen-presenting cells (APCs), is a binding site for RvE1 (29). Additionally, RvE1 also interacts with leukotriene B4 receptor 1 (BLT1), as a partial agonist, which mediates the potent inflammatory effects of leukotriene B4 (LTB4) (30).
In SSc, endothelial cell activation is one of the earliest events, along with immune cell activation and inflammation (31). Unresolved or prolonged inflammation and immune cell activation could result in chronic inflammation and, subsequently, in fibrosis (7). The inflammatory process is characterized by the production of various mediators (e.g., cytokines, chemokines, vasoactive amines, and eicosanoids) by the innate immune cells, including PMNs, macrophages, dendritic cells, lymphocytes, endothelial cells, and fibroblasts, in the damaged tissue. This occurs concomitantly with the upregulation of cell-adhesion molecules on leukocytes and endothelial cells, and continue with the infiltration of neutrophils, monocytes, and phagocytes. It is now recognized that Rvs, which are produced by immune cells such as macrophages and PMNs, are pivotal in the resolution of acute inflammatory reactions. They significantly limit acute inflammation response and promote tissue repair (32) (Figure 1). Each type of Rvs is considered to have a unique role in the resolution phase of inflammation (Table 1). Therefore, the role of Rvs and their possible failure to resolve local inflammatory responses should be investigated in the pathogenesis of SSc.
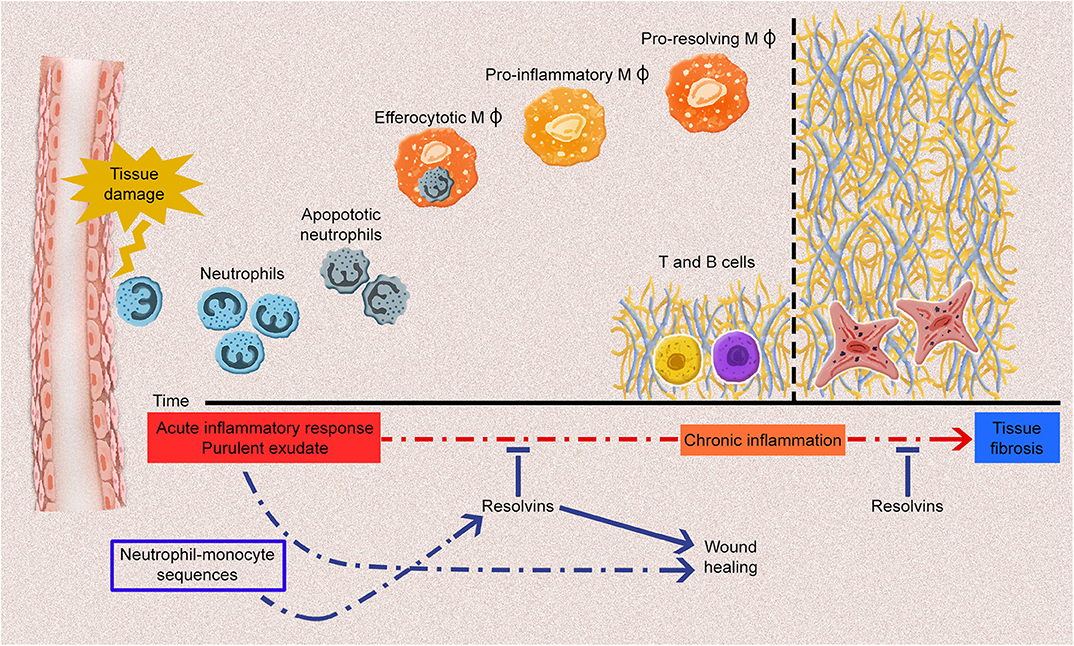
Figure 1. The acute inflammatory response and potential fates for the acute inflammatory process. Tissue damage induced by endogenous or exogenous stimuli leads to the generation of acute inflammatory responses, including various types of proinflammatory cell infiltrations and the production of plenty of proinflammatory mediators. Polymorphonuclear leukocytes infiltration especially neutrophils induce the influx of monocyte-derived macrophages to remove apoptotic cells and debris. Throughout the resolution phase of inflammation, resolvins (Rvs) promote the efferocytosis of macrophages and differentiation of proinflammatory macrophages (MΦ) into anti-inflammatory macrophages. At the post-resolution phase of inflammation, adaptive immunity response (B and T cells) establishes which contributes the wound healing. Any dysregulation of these processes may lead to chronic inflammation and fibrosis. Rvs limit the acute inflammatory process, thus, they prevent the development of chronic inflammation and fibrosis.
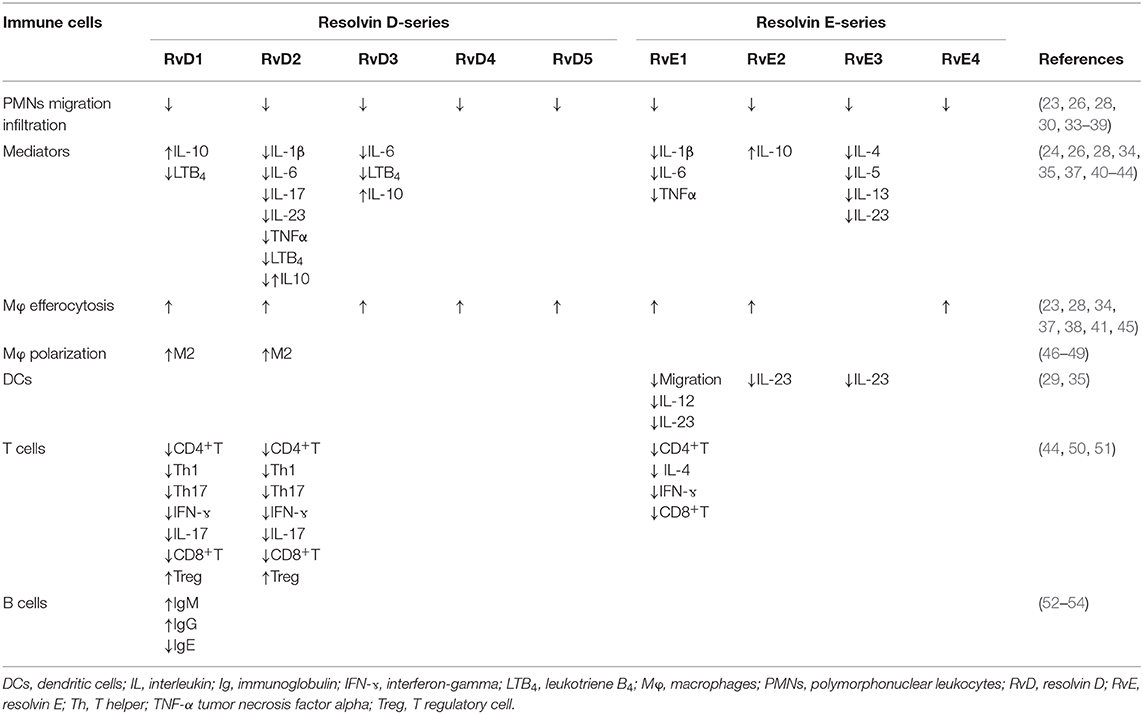
Table 1. Resolvins and their functions on immune cells.
RvE1 inhibits human neutrophil transendothelial migration and infiltration in vivo (20). RvE1 is characterized by the modulation of leukocytes adhesion molecules through the enhancement of L-selectin shedding, which inhibits the aggregation of leukocytes and reduces CD18 (LFA-1) expression, which is required for neutrophils adhesion and transmigration (30, 33). Animal studies have elucidated that RvE1 enhances efferocytosis through macrophages and reduces pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, in zymosan-induced peritonitis (40, 41). Similarly, the result of a mice animal model study indicated that the exogenous RvE1 induces the phagocytosis of neutrophil apoptosis via macrophages in pulmonary inflammation (45). Similar to RvE1, RvE2 actively participates in the resolution of inflammation by blocking neutrophil infiltration through chemotaxis modulation, reinforcement of phagocytosis, and macrophages-dependent production of IL-10 (34). Eosinophils mainly release RvE3, which limits the infiltration of PMNs in zymosan triggered peritonitis (55). In an allergic lung inflammation model, RvE3 significantly reduced the number of inflammatory cells and the secretion of pro-inflammatory cytokines in bronchoalveolar lavage (35). Recently, it has been demonstrated that the production of new RvE4 is accelerated by hypoxia, which induces the efferocytosis of neutrophils and erythrocytes through macrophages and inhibits the infiltration of neutrophil in hemorrhagic exudates in vivo (23).
RvD1 modulates the regulatory action of PMNs by inhibiting rolling and adhesion to endothelium via GPR32, in addition to limiting the infiltration of leukocytes and neutrophils via FPR2/ALX and the production of pro-inflammatory mediators in zymosan-induced peritonitis (36). Through this binding with FPR2/ALX, RvD1 inhibits lipopolysaccharide (LPS)-induced acute lung inflammation. This is realized because of reduced neutrophil infiltration due to the suppression of macrophage inflammatory protein (MIP)2-α (CXCL2) expression on alveolar macrophages (56). Similarly, RvD2 plays an effective role in the resolution phase of inflammation by reducing neutrophil recruitment, increasing mononuclear and macrophage phagocytosis by binding with GPR18, and suppressing the pro-inflammatory mediators (26). Additionally, RvD2 suppresses pro-inflammatory mediators by decreasing the plasma levels of IL-1β, IL-6, IL-17, IL-23, and TNF-α, as well as the levels of prostaglandin (PG)E2 and LTB4 in peritoneal exudates, as demonstrated using an animal sepsis model Interestingly, RvD2 decreases the plasma levels of the potent anti-inflammatory cytokine IL-10, which is of interest because of its detrimental impact on survival in sepsis (42). By contrast, RvD2 increases the level of IL-10 mRNA in the porphyromonas gingivalis-induced periodontitis (43). RvD3, which appears later than RvD1 and RvD2 in the resolution phase of inflammation, has potent local and systemic anti-inflammatory activities, such as decreasing the recruitment of PMNs and reducing the levels of IL-6 and LTB4 and matrix-degrading enzymes (MMP-2 and MMP-9). This enhances the level of IL-10 and stimulates macrophage efferocytosis (28, 37). Moreover, RvD4 decreases PMNs infiltration and promotes macrophage efferocytosis in zymosan-induced peritonitis and Staphylococcus aureus-triggered skin infection, in addition to inducing the phagocytosis of dermal fibroblasts (38). Several studies have elucidated the dysregulation of neutrophils in SSc and the relationship between neutrophil infiltration in lung tissue and lung fibrosis or disease severity (57–61). Considering all of these results, it seems that blocking of neutrophil migration and infiltration from most of Rvs might be beneficial for SSc or dysregulation of these mediators might contribute to the pathogenesis of SSc. As mentioned above, most of Rvs stimulate macrophage efferocytosis, which has been found to be dysfunctional in autoimmune diseases (systemic lupus erythematosus, Sjogren's syndrome, and SSc) (62–64).
From the onset of inflammation to its resolution, macrophages, as a part of innate immunity, play a significant role in inflammatory responses because they have possessed a diversity of phenotypes and polarization abilities. Based on responses to various signals from the environment, macrophages convert into classically activated M1 or alternatively activated M2 phenotypes that are mainly stimulated by interferon (IFN)-ɤ/LPS and IL-4/IL-13, respectively (65, 66). M1 macrophages contribute to the initiation and progression of inflammation by secreting pro-inflammatory mediators (IL-12, IL-1β, IL-6, and TNF-α). M2 macrophages, by contrast, are implicated in the tissue repair, wound healing, and resolution phase of the inflammation through the production of cytokines (IL-4, IL-10, IL-13) and growth factors (TGF-β, vascular endothelial growth factor (VEGF), and endothelial growth factor (EGF) (67). Alternatively activated M2 macrophages may have four subtypes: M2a stimulated by IL-4 or IL-13, M2b stimulated by immune complex and LPS, M2c stimulated by IL-10 and TGF-β1, and M2d stimulated by IL-6 and adenosine (68). Activated macrophages may change their polarization in accordance with new environmental stimuli (69). In autoimmune diseases, an M1/M2 imbalance has been detected. In SSc, M2 macrophages produce profibrotic cytokines that promote ECM synthesis (31). The M2 polarization observed in SSc seems to be induced by increased IL-6 and IL-4 levels (2). Although previous results are mainly consistent with M2 activation, recent evidence has suggested that macrophages express mixed surface markers of the M1 and M2 phenotypes in SSc (70–72).
During the resolution phase of inflammation, M1 macrophages change into the M2 phenotype owing to the action of specific mediators, especially SPMs. It has been shown that RvD1 significantly reduces the expression of M1 phenotype markers (TNF-α, IL-6, monocyte chemoattractant protein (MCP)-1 expression) and increases the expression of M2 markers in peritoneal macrophages obtained from obese mice (46). In the mouse carotid ligation model, systemic RvD2 markedly enhanced the proportion of M2 macrophages among the monocytes/macrophages present in the injured arterial wall (47). Recently, an assessment of inflammation in an abdominal aortic aneurysm model animal study demonstrated that RvD2 improved M2 polarization and ameliorated pro-inflammatory markers (IL-1β, IL-6, MCP-1, and MIP-1α) (73). Moreover, RvD1 reinforced the activation of M2 macrophages in acute smoke-induced lung inflammation (48). The animal study has indicated that long-term treatment with aspirin-triggered (AT) RvD1 does not influence macrophage polarization in long-term smoke-induced lung inflammation. Additionally, tissue fibrosis is not observed with long-term AT-RvD1 treatment (49). These effects could suggest that the effects of RvD1 on macrophage polarization may be associated with the type of inflammation (acute or chronic) or the duration of Rvs exposure. At the moment, we don't have enough data to disclosure if M2 differentiation may be a negative event in the pathogenic cascade of SSc linked to the activity of Rvs. Therefore, it is still very difficult to regard M2 differentiation as in those by Rvs as beneficial or pathogenic.
Dendritic cells (DCs) are an important component of innate immunity. They recognize and present damage-associated molecular patterns and pathogens, as well as induce the adaptive immune response. Usually, DCs are composed of two main cell types: conventional (cDC) and plasmacytoid (pDC), which, especially, secretes interferon-alpha (IFN-α). Recent studies have revealed that pDCs infiltrate the skin and the lungs of SSc patients, and contribute to fibrosis and that the number of pDCs in the lungs of SSc patients correlates with the severity of the lung disease (74, 75). ChemR23, a receptor of RvEs, is highly expressed in pDCs, and it mediates the migration of pDCs to inflammatory sites (76, 77). In an animal study, ChemR23 deficiency in knockout mice reduced the migration of pDCs to atherosclerotic lesions (78). Only RvE1 restrained the migration of DCs and inhibited their production of IL-12 via ChemR23 (29, 79). The serum level of IL-12 is increased in patients with SSc (79). Although the role of cDCs in SSc is not as known as pDCs, the increase in the production of proinflammatory cytokines from cDCs is demonstrated in SSc (80).
Several reports have suggested that T cells, particularly CD4+ T helper 2 (Th2), play a significant role in both the inflammatory and fibrotic processes of SSc (81). Activated CD4+ Th2 cells produce the predominantly potent profibrotic cytokines IL-4 and IL-13, which induce fibroblast proliferation, their differentiation into myofibroblasts, and polarization of M2 macrophages. All of these features are implicated in the pathogenesis of SSc (2, 82, 83). Furthermore, IL-13 producing CD8+ T cells have been detected in the skin in the early phases of SSc (84). Studies have highlighted the importance of Rvs in T cell regulation. Exogenous RvD1 diminishes the infiltration of CD4+ and CD8+ T lymphocytes in endotoxin-induced uveitis (50). Similarly, in an animal study, it was found that exogenous RvE1 suppressed the infiltration of CD4+ and CD8+ T cells in atopic dermatitis in a dose-dependent manner. In addition, RvE1 treatment reduced the IL-4 and IFN-ɤ production of activated CD4+ T cells (51). Abnormal Th17 cell responses are encountered in many chronic inflammatory and autoimmune diseases (85). Th17 and IL-17 may play an important role in SSc due to proinflammatory and profibrotic effects. Some evidence has demonstrated that the level of Th17 and IL-17 increased in SSc (86–88). However, the results of several studies have not found an increase in the level of IL-17 in SSc. Therefore, the role of Th17 and IL-17 have not completely understood in the pathogenesis of SSc yet (89–91). RvD1 and RvD2 abate the inflammatory responses of activated CD8+ T, Th1, and Th17 cells by decreasing the production of TNF-α, IFN-ɤ, IL-2, and IL-17. RvD1 and RvD2 inhibit the differentiation of naïve CD4+ T cells into Th1 and Th17 cells while they improve the differentiation of CD4+ T cells into regulatory T (Treg) cells. However, they do not exert any effect on the apoptosis of both CD8+ and CD4+ cells (44). In contrast to RvD1-2, RvE1 amplifies the T cell apoptotic activity of DCs through indolamine 2,3 dioxygenase induction (92). Although the effect of RvE1 on Th17 is undefined, RvE1 diminishes the production of IL-23 and IL-6, which are crucial for the survival of Th17 cell, in allergic lung inflammation (93). Recently, it has been demonstrated that RvE1, RvE2, and RvE3 decrease the production of IL-23 from bone marrow DCs in vitro. In particular, the treatment of house dust-mite-sensitized mice with RvE3 promoted the reduction of inflammatory cells, including eosinophils, and decreased IL-23 and IL-17 levels in lavage fluid, thus supporting the role of RvE3 in the resolution of allergic airway inflammation (35). These effects of Rvs on T cell regulation might create a protective mechanism against the dysregulation of T cells in SSc.
In recent studies on SSc, the role of the B cells in the generation of fibrosis has been highlighted, especially in the lungs and the gastrointestinal tract (94, 95). In fact, an increase in the naive B cell count and a decrease in memory and regulatory B cell counts has been found in SSc. These impairments of B cell homeostasis result in the decline in the production of potent anti-inflammatory and anti-fibrotic cytokines (i.e., IL-10) and the enhancement of production of proinflammatory cytokines (i.e., IL-6) (96). However, information on the effects of Rvs on B cells is scarce. In the mouse spleen, 17-hydroxydosahexaenoic acid (17-HDHA) (a biomarker of Rvs), RvD1, and RvE1, but not RvD2 and RvD5, have been detected (52, 53). In activated B cells, RvD1 elevates antibody production, notably immunoglobulin (Ig)M, while 17-HDHA increases both IgM and IgG in proportion to the increasing differentiation of B cells into the antibody-secreting B cell phenotype (52). Interestingly, it has been found that RvD1 and 17-HDHA suppress the differentiation of naïve B cells into IgE-secreting cells, which induce a specific block of the epsilon germline transcription (54).
Raynaud's phenomenon (RP) is frequently encountered in SSc, and it influences the acral blood flow (97). In primary RP, impaired arterial inflow induced by sympathetic vasoconstriction causes mild reversible microvascular sufferance. In SSc, the impairment of arteriolar inflow is not compensated for by endothelial-dependent vasodilation. In fact, the disease affects the endothelial cells that are injured or dysfunctional (98, 99). Therefore, prolonged vasoconstriction leads to a loss of endothelial junctions, enhanced inflammatory immune cell migration and infiltration, and microvessel permeability (100). Moreover, repeated and sustained vasoconstriction attacks result in ischemia-reperfusion (IR) injury, which promotes the production of various proinflammatory mediators and reactive oxygen species, activation and migration of PMNs, and interaction with endothelial cells. This causes further microvascular damage (101, 102).
In this context, based on the data available about Rvs role in IR-induced injury models, they can be considered operative in a condition such as RP in SSc as well. In the IR-induced model, the levels of endogenous DHA and all types of RvDs, apart from RvD1 and RvD3, are known to increase in the plasma, while only DHA, RvD1, and RvD3 are detected in the affected kidney tissue. Exogenous Rvs (composed of RvD1-3) limit the infiltration of PMNs, and the deposition of interstitial collagen. RvD1 has a protective capacity for the kidney when administered after the development of IR (39). It has been shown that RvD1 treatment protects the lung tissue from IR-induced inflammation, thus limiting the homing of inflammatory cells, production of proinflammatory mediators, and apoptosis (19). IR injury elicits mitochondrial dysfunction and augments excessive ROS production (103). RvD1 limits IR-triggered liver damage by reducing mitochondrial oxidative stress and regulating mitochondrial homeostasis (104). Furthermore, RvD2 diminishes the infiltration of PMNs through GPR18 in IR-induced lung injury (26).
Fibrosis is the main eventual hallmark of SSc. Vasculopathy; immune dysregulation, including innate and adaptive immunity; and several cytokines contribute to the process leading to fibrosis. However, the exact mechanisms of fibrosis in SSc still remain undefined.
Rvs are mainly known for their anti-inflammatory and pro-resolutive effects. They prevent fibrosis by limiting inflammation, supporting efferocytosis, and suppressing proinflammatory and profibrotic cytokines. Furthermore, Rvs have direct anti-fibrotic effects: RvE1 prevents hepatic fibrosis induced by Schistosoma japonicum infection by decreasing the levels of fibrotic markers such as laminin, hyaluronic acid, procollagen type III, and type IV collagen (105). In the animal model study, the anti-fibrotic effects of RvE1 were evaluated by inducing unilateral ureteric obstruction. The interstitial fibrosis obtained using this model was driven not by an inflammatory process but by an irreversible surgical insult, and it was characterized by collagen deposition and the proliferation of α-smooth muscle actin (SMA)+ myofibroblasts. RvE1 treatment dramatically attenuated the accumulation of α-SMA+ myofibroblasts, deposition of type IV collagen, and production of platelet-derived growth factor (PDGF)-BB, which is a potent inducer of fibroblast proliferation through activation of the AKT and ERK pathways. Moreover, RvD1 markedly reduced myofibroblast accumulation, and mRNA levels of type I and III collagens in an injured kidney (106). In bleomycin-induced lung tissue, treatment with 17(R)-RvD1, an epimer of RvD1, diminished the mRNA-expression of IL-1β, TGF-β1, and connective tissue growth factor, in addition to sharply reducing the numbers of macrophages and neutrophils in the bronchoalveolar fluid. The anti-fibrotic capacity of 17(R)-RvD1 has been confirmed based on reductions in hydroxyproline content (marker of collagen deposition), type I collagen mRNA expression, and score of the fibrotic changes (via Ashcroft scale) in the lung tissue. Moreover, 17(R)-RvD1 treatment has anti-inflammatory and anti-fibrotic effects even when it is administered in the established fibrotic stage in lung tissue (18). Besides, RvD1 alleviates collagen deposition in heart tissue after a myocardial infarction (107).
Epithelial-mesenchymal transition (EMT) is thought to be a crucial mechanism in the development of fibrosis, particular in the lungs and kidneys (97, 98). In general, EMT is closely related to embryonic development, tissue repair, wound healing, and cell migration. During tissue repair or wound healing, epithelial cells lose their phenotype and gain mesenchymal phenotypes to produce fibroblasts and myofibroblasts (108). EMT may be a part of the cellular origins of fibrosis in SSc (109). The potent pro-resolving activity of RvD1 has been further investigated in a model of acute respiratory distress syndrome (ARDS) in which it was demonstrated that RvD1 can prevent EMT of lung epithelial cells with reversal of the TGF-β-smad2/3 signaling pathway and lung fibrosis via the FPR2/ALX receptor (110). Endothelial-to-mesenchymal (EndoMT) transition is also thought to play an important role in both SSc-related fibrosis and vasculopathy (101, 102). Of note, RvD1 has also been reported to significantly inhibit TGF-β1-induced EndoMT through increasing the expression of Smad7 (39).
The resolution of inflammation is vital for ensuring tissue homeostasis. Any defects in the resolution phase could lead to a prolonged inflammatory response, including increasing PMNs, exaggerated proinflammatory mediator production, increase in the number of apoptotic cells, and inappropriate activation of adaptive immune cells. This unresolved inflammation results in fibrosis of the affected tissue. After the identification of SPMs, many investigators have focused on the effects of SPM on the resolution of inflammation. Rvs are efficacious anti-inflammatory and pro-resolving mediators that play various roles in innate immunity cells. A myriad of studies has confirmed that they influence adaptive immune cells (Figure 2). This exciting anti-fibrotic effect has been supported by the direct effect of these mediators on the regulation of fibrotic cells and cytokines.
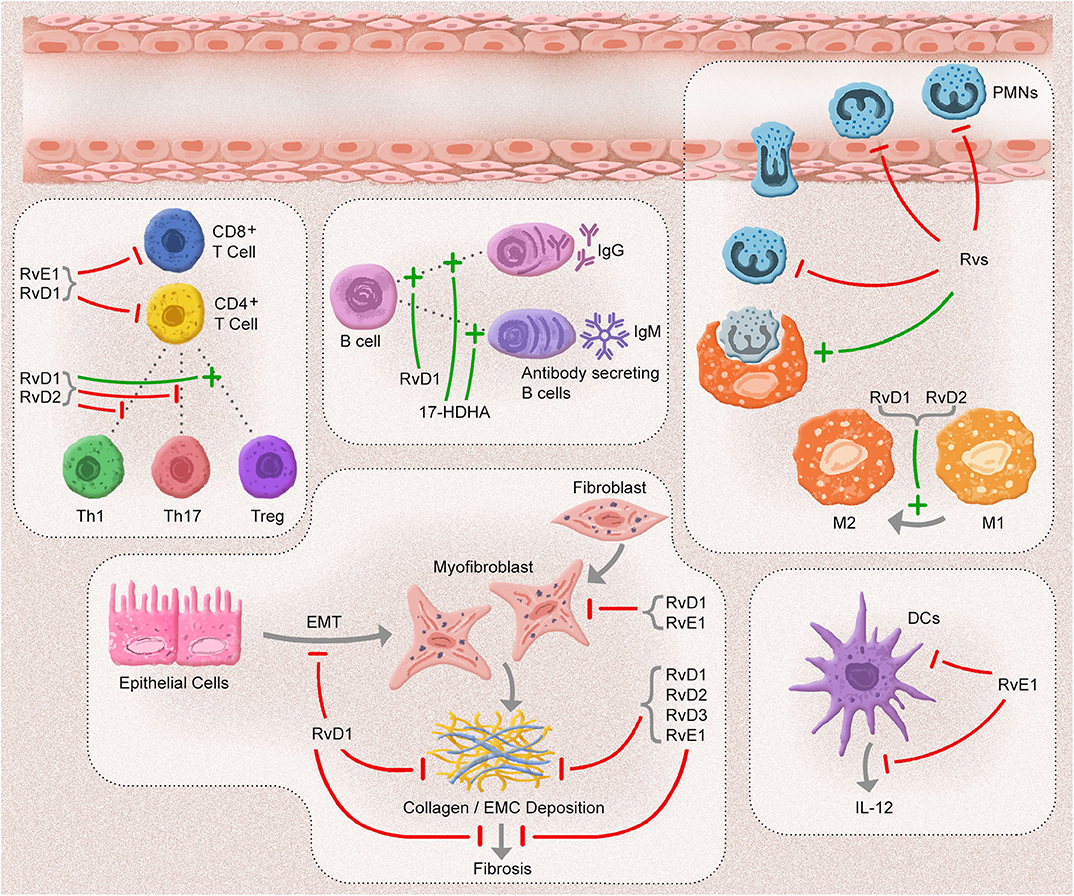
Figure 2. The anti-inflammatory, pro-resolution, and anti-fibrotic effects of Resolvins. In an acute inflammatory response, resolvins (Rvs) inhibit the adhesion, migration, and infiltration of polymorphonuclear leukocytes (PMNs) and enhance the efferocytosis capacity of macrophages. D-series Rvs (RvD1 and RvD2) induce the polarization of macrophages toward to phenotype M2. One of the E-series Rvs, RvE1 blocks the migration and production of interleukin (IL)-12 in dendritic cells (DCs) and the infiltration of CD8+ and CD4+ cells. RvD1 and RvD2 suppress the inflammatory responses of CD8+ T, T helper (Th)1, and Th17 cells, in addition to limiting the differentiation of CD4+ T cells into T helper (Th)1 and Th17 cells and promoting the conversion of T regulatory (Treg) cells. 17-hydroxydosahexaenoic acid (17-HDHA) and RvD1 enhance the antibody secretion of B cells. After an inflammatory response, most of Rvs block the development of fibrosis by decreasing collagen deposition and myofibroblast infiltration, as well as by inhibiting epithelial-mesenchymal cell transition (EMT).
The pathogenesis of SSc is associated with vasculopathy, immune dysregulation, and fibrosis (1). It is well-known that progressive chronic inflammation is a part of the disease, while the connection between the resolution of inflammation and SSc still remains unclear. From this viewpoint, any dysfunction of the well-defined anti-fibrotic, anti-inflammatory, and pro-resolving abilities of Rvs may contribute to the progression of SSc. In the future, an accurate understanding of Rvs in SSc may foster the development of novel treatment strategies (4, 111).
AA: substantial contributions to the conception of the work, acquisition, and interpretation of data, drafting the article, revising the manuscript critically, providing approval for publication of the content. FR: acquisition of data, drafting the article, revising the manuscript critically, providing approval for publication of the content. SB-R and NP: revising the manuscript critically, providing approval for publication of the content. AT: drafting the article, revising the manuscript critically, providing approval for publication of the content. MM: drafting the article, revising the manuscript critically, providing approval for publication of the content. AP: substantial contributions to the conception of the work, revising the manuscript critically, providing approval for publication of the content. MM-C: substantial contributions to the conception of the work, revising the manuscript critically, providing approval for publication of the content.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Varga J, Trojanowska M, Kuwana M. Pathogenesis of systemic sclerosis: recent insights of molecular and cellular mechanisms and therapeutic opportunities. J Scleroderma Relat Disord. (2017) 2:137–52. doi: 10.5301/jsrd.5000249
2. Brown M, O'Reilly S. The immunopathogenesis of fibrosis in systemic sclerosis. Clin Exp Immunol. (2019) 195:310–21. doi: 10.1111/cei.13238
3. Hamaguchi Y, Takehara K. Anti-nuclear autoantibodies in systemic sclerosis: news and perspectives. J Scleroderma Relat Disord. (2018) 3:201–13. doi: 10.1177/2397198318783930
4. Tyndall AJ, Bannert B, Vonk M, Airo P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR scleroderma trials and research (EUSTAR) database. Ann Rheum Dis. (2010) 69:1809–15. doi: 10.1136/ard.2009.114264
5. Volkmann ER, Varga J. Emerging targets of disease-modifying therapy for systemic sclerosis. Nat Rev Rheumatol. (2019) 15:208–24. doi: 10.1038/s41584-019-0184-z
6. Medzhitov R. Origin and physiological roles of inflammation. Nature. (2008) 454:428–35. doi: 10.1038/nature07201
7. Ariel A, Timor O. Hanging in the balance: endogenous anti-inflammatory mechanisms in tissue repair and fibrosis. J Pathol. (2013) 229:250–63. doi: 10.1002/path.4108
8. Abdolmaleki F, Kovanen PT, Mardani R, Gheibi-Hayat SM, Bo S, Sahebkar A. Resolvins: emerging players in autoimmune and inflammatory diseases. Clin Rev Allergy Immunol. (2019) 58:82–91. doi: 10.1007/s12016-019-08754-9
9. Serhan CN. A search for endogenous mechanisms of anti-inflammation uncovers novel chemical mediators: missing links to resolution. Histochem Cell Biol. (2004) 122:305–21. doi: 10.1007/s00418-004-0695-8
10. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. (2008) 8:349–61. doi: 10.1038/nri2294
11. Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. (2005) 174:4345–55. doi: 10.4049/jimmunol.174.7.4345
12. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. (2014) 510:92–101. doi: 10.1038/nature13479
13. Fetterman JW Jr, Zdanowicz MM. Therapeutic potential of n-3 polyunsaturated fatty acids in disease. Am J Health Syst Pharm. (2009) 66:1169–79. doi: 10.2146/ajhp080411
14. Arnardottir HH, Dalli J, Norling LV, Colas RA, Perretti M, Serhan CN. Resolvin D3 is dysregulated in arthritis and reduces arthritic inflammation. J Immunol. (2016) 197:2362–8. doi: 10.4049/jimmunol.1502268
15. Dean S, Wang CS, Nam K, Maruyama CL, Trump BG, Baker OJ. Aspirin triggered resolvin D1 reduces inflammation and restores saliva secretion in a sjogren's syndrome mouse model. Rheumatology (Oxford). (2019) 58:1285–92. doi: 10.1093/rheumatology/kez072
16. Schwanke RC, Marcon R, Bento AF, Calixto JB. EPA- and DHA-derived resolvins' actions in inflammatory bowel disease. Eur J Pharmacol. (2016) 785:156–64. doi: 10.1016/j.ejphar.2015.08.050
17. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. (2018) 128:2657–69. doi: 10.1172/JCI97943
18. Yatomi M, Hisada T, Ishizuka T, Koga Y, Ono A, Kamide Y, et al. 17(R)-resolvin D1 ameliorates bleomycin-induced pulmonary fibrosis in mice. Physiol Rep. (2015) 3:e12628. doi: 10.14814/phy2.12628
19. Xia J, Xue JY, Du J, Wu GW, Hu XT, Zhao QF. [Role and related mechanism of resolvin D1 in lung ischemia reperfusion injury in rats]. Zhonghua yi xue za zhi. (2019) 99:1111–5. doi: 10.3760/cma.j.issn.0376-2491.2019.14.015
20. Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. (2000) 192:1197–204. doi: 10.1084/jem.192.8.1197
21. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. (2002) 196:1025–37. doi: 10.1084/jem.20020760
22. Norris PC, Libreros S, Chiang N, Serhan CN. A cluster of immunoresolvents links coagulation to innate host defense in human blood. Sci Signal. (2017) 10:eaan1471. doi: 10.1126/scisignal.aan1471
23. Norris PC, Libreros S, Serhan CN. Resolution metabolomes activated by hypoxic environment. Sci Adv. (2019) 5:eaax4895. doi: 10.1126/sciadv.aax4895
24. Krishnamoorthy S, Recchiuti A, Chiang N, Fredman G, Serhan CN. Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am J Pathol. (2012) 180:2018–27. doi: 10.1016/j.ajpath.2012.01.028
25. Prevete N, Liotti F, Amoresano A, Pucci P, de Paulis A, Melillo RM. New perspectives in cancer: modulation of lipid metabolism and inflammation resolution. Pharmacol Res. (2018) 128:80–7. doi: 10.1016/j.phrs.2017.09.024
26. Chiang N, Dalli J, Colas RA, Serhan CN. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J Exp Med. (2015) 212:1203–17. doi: 10.1084/jem.20150225
27. Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. (2012) 484:524–8. doi: 10.1038/nature11042
28. Dalli J, Winkler JW, Colas RA, Arnardottir H, Cheng CY, Chiang N, et al. Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem Biol. (2013) 20:188–201. doi: 10.1016/j.chembiol.2012.11.010
29. Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. (2005) 201:713–22. doi: 10.1084/jem.20042031
30. Arita M, Ohira T, Sun YP, Elangovan S, Chiang N, Serhan CN. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J Immunol. (2007) 178:3912–7. doi: 10.4049/jimmunol.178.6.3912
31. Dowson C, Simpson N, Duffy L, O'Reilly S. Innate immunity in systemic sclerosis. Curr Rheumatol Rep. (2017) 19:2. doi: 10.1007/s11926-017-0630-3
32. Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. (2016) 15:551–67. doi: 10.1038/nrd.2016.39
33. Dona M, Fredman G, Schwab JM, Chiang N, Arita M, Goodarzi A, et al. Resolvin E1, an EPA-derived mediator in whole blood, selectively counterregulates leukocytes and platelets. Blood. (2008) 112:848–55. doi: 10.1182/blood-2007-11-122598
34. Oh SF, Dona M, Fredman G, Krishnamoorthy S, Irimia D, Serhan CN. Resolvin E2 formation and impact in inflammation resolution. J Immunol. (2012) 188:4527–34. doi: 10.4049/jimmunol.1103652
35. Sato M, Aoki-Saito H, Fukuda H, Ikeda H, Koga Y, Yatomi M, et al. Resolvin E3 attenuates allergic airway inflammation via the interleukin-23-interleukin-17A pathway. FASEB J. (2019) 33:12750–9. doi: 10.1096/fj.201900283R
36. Norling LV, Dalli J, Flower RJ, Serhan CN, Perretti M. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: receptor-dependent actions. Arterioscler Thromb Vasc Biol. (2012) 32:1970–8. doi: 10.1161/ATVBAHA.112.249508
37. Norris PC, Arnardottir H, Sanger JM, Fichtner D, Keyes GS, Serhan CN. Resolvin D3 multi-level proresolving actions are host protective during infection. Prostag Leukot Essent Fatty Acids. (2018) 138:81–9. doi: 10.1016/j.plefa.2016.01.001
38. Winkler JW, Orr SK, Dalli J, Cheng CY, Sanger JM, Chiang N, et al. Resolvin D4 stereoassignment and its novel actions in host protection and bacterial clearance. Sci Rep. (2016) 6:18972. doi: 10.1038/srep18972
39. Duffield JS, Hong S, Vaidya VS, Lu Y, Fredman G, Serhan CN, et al. Resolvin D series and protectin D1 mitigate acute kidney injury. J Immunol. (2006) 177:5902–11. doi: 10.4049/jimmunol.177.9.5902
40. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. (2007) 447:869–74. doi: 10.1038/nature05877
41. Oh SF, Pillai PS, Recchiuti A, Yang R, Serhan CN. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J Clin Invest. (2011) 121:569–81. doi: 10.1172/JCI42545
42. Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. (2009) 461:1287–91. doi: 10.1038/nature08541
43. Mizraji G, Heyman O, Van Dyke TE, Wilensky A. Resolvin D2 restrains Th1 immunity and prevents alveolar bone loss in murine periodontitis. Front Immunol. (2018) 9:785. doi: 10.3389/fimmu.2018.00785
44. Chiurchiu V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, et al. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med. (2016) 8:353ra111. doi: 10.1126/scitranslmed.aaf7483
45. El Kebir D, Gjorstrup P, Filep JG. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci U S A. (2012) 109:14983–8. doi: 10.1073/pnas.1206641109
46. Titos E, Rius B, Gonzalez-Periz A, Lopez-Vicario C, Moran-Salvador E, Martinez-Clemente M, et al. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol. (2011) 187:5408–18. doi: 10.4049/jimmunol.1100225
47. Akagi D, Chen M, Toy R, Chatterjee A, Conte MS. Systemic delivery of proresolving lipid mediators resolvin D2 and maresin 1 attenuates intimal hyperplasia in mice. FASEB J. (2015) 29:2504–13. doi: 10.1096/fj.14-265363
48. Hsiao HM, Sapinoro RE, Thatcher TH, Croasdell A, Levy EP, Fulton RA, et al. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS ONE. (2013) 8:e58258. doi: 10.1371/journal.pone.0058258
49. Hsiao HM, Thatcher TH, Colas RA, Serhan CN, Phipps RP, Sime PJ. Resolvin D1 reduces emphysema and chronic inflammation. Am J Pathol. (2015) 185:3189–201. doi: 10.1016/j.ajpath.2015.08.008
50. Settimio R, Clara DF, Franca F, Francesca S, Michele D. Resolvin D1 reduces the immunoinflammatory response of the rat eye following uveitis. Mediat Inflamm. (2012) 2012:318621. doi: 10.1155/2012/318621
51. Kim TH, Kim GD, Jin YH, Park YS, Park CS. Omega-3 fatty acid-derived mediator, Resolvin E1, ameliorates 2,4-dinitrofluorobenzene-induced atopic dermatitis in NC/Nga mice. Int Immunopharmacol. (2012) 14:384–91. doi: 10.1016/j.intimp.2012.08.005
52. Ramon S, Gao F, Serhan CN, Phipps RP. Specialized proresolving mediators enhance human B cell differentiation to antibody-secreting cells. J Immunol. (2012) 189:1036–42. doi: 10.4049/jimmunol.1103483
53. Hong S, Porter TF, Lu Y, Oh SF, Pillai PS, Serhan CN. Resolvin E1 metabolome in local inactivation during inflammation-resolution. J Immunol. (2008) 180:3512–9. doi: 10.4049/jimmunol.180.5.3512
54. Kim N, Ramon S, Thatcher TH, Woeller CF, Sime PJ, Phipps RP. Specialized proresolving mediators (SPMs) inhibit human B-cell IgE production. Eur J Immunol. (2016) 46:81–91. doi: 10.1002/eji.201545673
55. Isobe Y, Arita M, Matsueda S, Iwamoto R, Fujihara T, Nakanishi H, et al. Identification and structure determination of novel anti-inflammatory mediator resolvin E3, 17,18-dihydroxyeicosapentaenoic acid. J Biol Chem. (2012) 287:10525–34. doi: 10.1074/jbc.M112.340612
56. Zhang HW, Wang Q, Mei HX, Zheng SX, Ali AM, Wu QX, et al. RvD1 ameliorates LPS-induced acute lung injury via the suppression of neutrophil infiltration by reducing CXCL2 expression and release from resident alveolar macrophages. Int Immunopharmacol. (2019) 76:105877. doi: 10.1016/j.intimp.2019.105877
57. Maugeri N, Capobianco A, Rovere-Querini P, Ramirez GA, Tombetti E, Valle PD, et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci Transl Med. (2018) 10:eaao3089. doi: 10.1126/scitranslmed.aao3089
58. Barnes TC, Anderson ME, Edwards SW, Moots RJ. Neutrophil-derived reactive oxygen species in SSc. Rheumatology (Oxford, England). (2012) 51:1166–9. doi: 10.1093/rheumatology/ker520
59. Kowal-Bielecka O, Kowal K, Highland KB, Silver RM. Bronchoalveolar lavage fluid in scleroderma interstitial lung disease: technical aspects and clinical correlations: review of the literature. Semin Arthritis Rheum. (2010) 40:73–88. doi: 10.1016/j.semarthrit.2008.10.009
60. Cakmak G, Selcuk Can T, Gundogdu S, Akman C, Ikitimur H, Musellim B, et al. Relationship between abnormalities on high-resolution computerized tomography, pulmonary function, and bronchoalveolar lavage in progressive systemic sclerosis. Sarcoidosis Vasc Diffuse Lung Dis. (2016) 33:349–54.
61. Antoniou KM, Wells AU. Scleroderma lung disease: evolving understanding in light of newer studies. Curr Opin Rheumatol. (2008) 20:686–91. doi: 10.1097/BOR.0b013e3283126985
62. Janko C, Schorn C, Grossmayer GE, Frey B, Herrmann M, Gaipl US, et al. Inflammatory clearance of apoptotic remnants in systemic lupus erythematosus (SLE). Autoimmun Rev. (2008) 8:9–12. doi: 10.1016/j.autrev.2008.07.015
63. Manoussakis MN, Fragoulis GE, Vakrakou AG, Moutsopoulos HM. Impaired clearance of early apoptotic cells mediated by inhibitory IgG antibodies in patients with primary sjogren's syndrome. PLoS ONE. (2014) 9:e112100. doi: 10.1371/journal.pone.0112100
64. Ballerie A, Lescoat A, Augagneur Y, Lelong M, Morzadec C, Cazalets C, et al. Efferocytosis capacities of blood monocyte-derived macrophages in systemic sclerosis. Immunol Cell Biol. (2019) 97:340–7. doi: 10.1111/imcb.12217
65. Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. (2007) 117:1155–66. doi: 10.1172/JCI31422
66. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
67. Lech M, Anders HJ. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta. (2013) 1832:989–97. doi: 10.1016/j.bbadis.2012.12.001
68. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. (2018) 154:186–95. doi: 10.1111/imm.12910
69. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. (2005) 175:342–9. doi: 10.4049/jimmunol.175.1.342
70. Higashi-Kuwata N, Jinnin M, Makino T, Fukushima S, Inoue Y, Muchemwa FC, et al. Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res Ther. (2010) 12:R128. doi: 10.1186/ar3066
71. Trombetta AC, Soldano S, Contini P, Tomatis V, Ruaro B, Paolino S, et al. A circulating cell population showing both M1 and M2 monocyte/macrophage surface markers characterizes systemic sclerosis patients with lung involvement. Respir Res. (2018) 19:186. doi: 10.1186/s12931-018-0891-z
72. Soldano S, Trombetta AC, Contini P, Tomatis V, Ruaro B, Brizzolara R, et al. Increase in circulating cells coexpressing M1 and M2 macrophage surface markers in patients with systemic sclerosis. Ann Rheum Dis. (2018) 77:1842–5. doi: 10.1136/annrheumdis-2018-213648
73. Pope NH, Salmon M, Davis JP, Chatterjee A, Su G, Conte MS, et al. D-series resolvins inhibit murine abdominal aortic aneurysm formation and increase M2 macrophage polarization. FASEB J. (2016) 30:4192–201. doi: 10.1096/fj.201600144RR
74. Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med. (2018) 10:eaam8458. 10:eaam8458. doi: 10.1126/scitranslmed.aam8458
75. Kafaja S, Valera I, Divekar AA, Saggar R, Abtin F, Furst DE, et al. pDCs in lung and skin fibrosis in a bleomycin-induced model and patients with systemic sclerosis. JCI Insight. (2018) 3:e98380. doi: 10.1172/jci.insight.98380
76. Zabel BA, Silverio AM, Butcher EC. Chemokine-like receptor 1 expression and chemerin-directed chemotaxis distinguish plasmacytoid from myeloid dendritic cells in human blood. J Immunol. (2005) 174:244–51. doi: 10.4049/jimmunol.174.1.244
77. Vermi W, Riboldi E, Wittamer V, Gentili F, Luini W, Marrelli S, et al. Role of chemr23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J Exp Med. (2005) 201:509–15. doi: 10.1084/jem.20041310
78. van der Vorst EPC, Mandl M, Muller M, Neideck C, Jansen Y, Hristov M, et al. Hematopoietic chemr23 (chemerin receptor 23) fuels atherosclerosis by sustaining an M1 macrophage-phenotype and guidance of plasmacytoid dendritic cells to murine lesions-brief report. Arterioscler Thromb Vasc Biol. (2019) 39:685–93. doi: 10.1161/ATVBAHA.119.312386
79. Sato S, Hanakawa H, Hasegawa M, Nagaoka T, Hamaguchi Y, Nishijima C, et al. Levels of interleukin 12, a cytokine of type 1 helper T cells, are elevated in sera from patients with systemic sclerosis. J Rheumatol. (2000) 27:2838–42.
80. Carvalheiro T, Zimmermann M, Radstake T, Marut W. Novel insights into dendritic cells in the pathogenesis of systemic sclerosis. Clin Exp Immunol. (2020). doi: 10.1111/cei.13417. [Epub ahead of print].
81. Gasparini G, Cozzani E, Parodi A. Interleukin-4 and interleukin-13 as possible therapeutic targets in systemic sclerosis. Cytokine. (2020) 125:154799. doi: 10.1016/j.cyto.2019.154799
82. Salmon-Ehr V, Serpier H, Nawrocki B, Gillery P, Clavel C, Kalis B, et al. Expression of interleukin-4 in scleroderma skin specimens and scleroderma fibroblast cultures. Potential role in fibrosis. Arch Dermatol. (1996) 132:802–6. doi: 10.1001/archderm.132.7.802
83. Huang XL, Wang YJ, Yan JW, Wan YN, Chen B, Li BZ, et al. Role of anti-inflammatory cytokines IL-4 and IL-13 in systemic sclerosis. Inflamm Res. (2015) 64:151–9. doi: 10.1007/s00011-015-0806-0
84. Fuschiotti P, Larregina AT, Ho J, Feghali-Bostwick C, Medsger TA Jr. Interleukin-13-producing CD8+ T cells mediate dermal fibrosis in patients with systemic sclerosis. Arthritis Rheum. (2013) 65:236–46. doi: 10.1002/art.37706
85. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. (2009) 27:485–517. 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
86. Kurasawa K, Hirose K, Sano H, Endo H, Shinkai H, Nawata Y, et al. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum. (2000) 43:2455–63. 43:2455–63. doi: 10.1002/1529-0131(200011)43:11<2455::AID-ANR12>3.0.CO;2-K
87. Rolla G, Fusaro E, Nicola S, Bucca C, Peroni C, Parisi S, et al. Th-17 cytokines and interstitial lung involvement in systemic sclerosis. J Breath Res. (2016) 10:046013. 10:046013. doi: 10.1088/1752-7155/10/4/046013
88. Yang X, Yang J, Xing X, Wan L, Li M. Increased frequency of Th17 cells in systemic sclerosis is related to disease activity and collagen overproduction. Arthritis Res Ther. (2014) 16:R4. doi: 10.1186/ar4430
89. Chizzolini C, Dufour AM, Brembilla NC. Is there a role for IL-17 in the pathogenesis of systemic sclerosis? Immunol Lett. (2018) 195:61–7. doi: 10.1016/j.imlet.2017.09.007
90. Gourh P, Arnett FC, Assassi S, Tan FK, Huang M, Diekman L, et al. Plasma cytokine profiles in systemic sclerosis: associations with autoantibody subsets and clinical manifestations. Arthritis Res Ther. (2009) 11:R147. doi: 10.1186/ar2821
91. Olewicz-Gawlik A, Danczak-Pazdrowska A, Kuznar-Kaminska B, Gornowicz-Porowska J, Katulska K, Trzybulska D, et al. Interleukin-17 and interleukin-23: importance in the pathogenesis of lung impairment in patients with systemic sclerosis. Int J Rheum Dis. (2014) 17:664–70. doi: 10.1111/1756-185X.12290
92. Vassiliou EK, Kesler OM, Tadros JH, Ganea D. Bone marrow-derived dendritic cells generated in the presence of resolvin E1 induce apoptosis of activated CD4+ T cells. J Immunol. (2008) 181:4534–44. doi: 10.4049/jimmunol.181.7.4534
93. Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-γ and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. (2008) 9:873–9. doi: 10.1038/ni.1627
94. Manetti M, Neumann E, Muller A, Schmeiser T, Saar P, Milia AF, et al. Endothelial/lymphocyte activation leads to prominent CD4+ T cell infiltration in the gastric mucosa of patients with systemic sclerosis. Arthritis Rheum. (2008) 58:2866–73. doi: 10.1002/art.23806
95. Lafyatis R, O'Hara C, Feghali-Bostwick CA, Matteson E. B cell infiltration in systemic sclerosis-associated interstitial lung disease. Arthritis Rheum. (2007) 56:3167–8. doi: 10.1002/art.22847
96. Sanges S, Guerrier T, Launay D, Lefevre G, Labalette M, Forestier A, et al. Role of B cells in the pathogenesis of systemic sclerosis. Rev Med Interne. (2017) 38:113–24. doi: 10.1016/j.revmed.2016.02.016
97. Wigley FM, Flavahan NA. Raynaud's phenomenon. N Engl J Med. (2016) 375:556–65. doi: 10.1056/NEJMra1507638
98. Cutolo M, Smith V, Furst DE, Khanna D, Herrick AL. Points to consider-Raynaud's phenomenon in systemic sclerosis. Rheumatology (Oxford). (2017) 56(suppl. 5):v45–v8. doi: 10.1093/rheumatology/kex199
99. Cutolo M, Soldano S, Smith V. Pathophysiology of systemic sclerosis: current understanding and new insights. Expert Rev Clin Immunol. (2019) 15:753–64. doi: 10.1080/1744666X.2019.1614915
100. Bruni C, Frech T, Manetti M, Rossi FW, Furst DE, De Paulis A, et al. Vascular leaking, a pivotal and early pathogenetic event in systemic sclerosis: should the door be closed? Front Immunol. (2018) 9:2045. doi: 10.3389/fimmu.2018.02045
101. Flavahan NA. A vascular mechanistic approach to understanding raynaud phenomenon. Nat Rev Rheumatol. (2015) 11:146–58. doi: 10.1038/nrrheum.2014.195
102. Collard CD, Gelman S. Pathophysiology, clinical manifestations, and prevention of ischemia-reperfusion injury. Anesthesiology. (2001) 94:1133–8. doi: 10.1097/00000542-200106000-00030
103. Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. (2018) 46:1650–67. doi: 10.1159/000489241
104. Kang JW, Choi HS, Lee SM. Resolvin D1 attenuates liver ischaemia/reperfusion injury through modulating thioredoxin 2-mediated mitochondrial quality control. Br J Pharmacol. (2018) 175:2441–53. doi: 10.1111/bph.14212
105. Qiu W, Guo K, Yi L, Gong Y, Huang L, Zhong W. Resolvin E1 reduces hepatic fibrosis in mice with schistosoma japonicum infection. Exp Ther Med. (2014) 7:1481–5. doi: 10.3892/etm.2014.1641
106. Qu X, Zhang X, Yao J, Song J, Nikolic-Paterson DJ, Li J. Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. J Pathol. (2012) 228:506–19. doi: 10.1002/path.4050
107. Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, et al. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol. (2015) 84:24–35. doi: 10.1016/j.yjmcc.2015.04.003
108. Rout-Pitt N, Farrow N, Parsons D, Donnelley M. Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res. (2018) 19:136. doi: 10.1186/s12931-018-0834-8
109. Postlethwaite AE, Shigemitsu H, Kanangat S. Cellular origins of fibroblasts: possible implications for organ fibrosis in systemic sclerosis. Curr Opin Rheumatol. (2004) 16:733–8. doi: 10.1097/01.bor.0000139310.77347.9c
110. Yang Y, Hu L, Xia H, Chen L, Cui S, Wang Y, et al. Resolvin D1 attenuates mechanical stretch-induced pulmonary fibrosis via epithelial-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L1013–l24. doi: 10.1152/ajplung.00415.2018
Keywords: resolvins, resolution of inflammation, systemic sclerosis, innate immunity, adaptive immunity, fibrosis
Citation: Avanoǧlu Güler A, Rossi FW, Bellando-Randone S, Prevete N, Tufan A, Manetti M, de Paulis A and Matucci-Cerinic M (2020) The Role of Endogenous Eicosapentaenoic Acid and Docosahexaenoic Acid-Derived Resolvins in Systemic Sclerosis. Front. Immunol. 11:1249. doi: 10.3389/fimmu.2020.01249
Received: 12 February 2020; Accepted: 18 May 2020;
Published: 19 June 2020.
Edited by:
Dimitrios Petrou Bogdanos, University of Thessaly, GreeceReviewed by:
Dimitrios Daoussis, University of Patras Medical School, GreeceCopyright © 2020 Avanoǧlu Güler, Rossi, Bellando-Randone, Prevete, Tufan, Manetti, de Paulis and Matucci-Cerinic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Bellando-Randone, cy5iZWxsYW5kb3JhbmRvbmVAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.