 Valentina Bianchi
Valentina Bianchi Alexandre Harari
Alexandre Harari George Coukos1†
George Coukos1†- 1Department of Oncology, Lausanne University Hospital, Ludwig Institute for Cancer Research, University of Lausanne, Lausanne, Switzerland
- 2Center of Experimental Therapeutics, Department of Oncology, University Hospital of Lausanne (CHUV), Lausanne, Switzerland
Mutation-derived neoantigens are taking central stage as a determinant in eliciting effective antitumor immune responses following adoptive T-cell therapies. These mutations are patient-specific, and their targeting calls for highly personalized pipelines. The promising clinical outcomes of tumor-infiltrating lymphocyte (TIL) therapy have spurred interest in generating T-cell infusion products that have been selectively enriched in neoantigen (or autologous tumor) reactivity. The implementation of an isolation step, prior to T-cell in vitro expansion and reinfusion, may provide a way to improve the overall response rates achieved to date by adoptive T-cell therapies in metastatic cancer patients. Here we provide an overview of the main technologies [i.e., peptide major histocompatibility complex (pMHC) multimers, cytokine capture, and activation markers] to enrich infiltrating or circulating T-cells in predefined neoantigen specificities (or tumor reactivity). The unique technical and regulatory challenges faced by such highly specialized and patient-specific manufacturing T-cell platforms are also discussed.
Introduction
In the new age of personalized immune-oncology, tumor-infiltrating lymphocytes (TILs) generated from surgical resections, expanded in vitro and adoptively transferred, provide a unique opportunity to harness the specificity and diversity of the patient's endogenous T-cell repertoire. Building on the promising clinical outcomes achieved by TIL therapy in melanoma and cervical cancer (1, 2), efforts are now made to generate even more tailored T-cell products with predefined antigen specificities and, potentially, with enhanced in vivo tumor reactivity. The success of personalized adoptive cell therapies (ACTs) is therefore tightly linked to the identification of tumor-associated antigens, which are essential for tumor control.
Against this background, cancer neoantigens deriving from private mutations represent an ideal class of cancer antigens to target in that they are highly tumor-specific by nature, therefore reducing the potential induction of central and peripheral tolerance (3, 4). Most studies predominantly focus on single-nucleotide variants (SNVs) when referring to immunogenic tumor-specific mutant peptides; however, small insertions and deletions (indels), gene fusions, and posttranslational modifications (such as phosphorylation or glycosylation, which often alter the protein structure and function) have also been recognized as important neoantigen sources, therefore expanding the plethora of potential targets for cancer immunotherapy (5–9). Furthermore, non-canonical major histocompatibility complex (MHC) peptides derived from annotated noncoding regions are emerging as critical immune regulators across cancer types and able to elicit tumor-specific T-cell responses (10, 11).
Neoantigen discovery is a multistep process performed on a patient-specific basis by cutting-edge preclinical pipelines integrating variant calling, in silico filtering, and immunogenicity evaluation, leading to private (and shared) neoantigen candidates (12–14). Briefly, mutations are called by whole-exome or whole-genome sequencing of tumor vs. germline DNA, are further filtered by in silico prediction algorithms and potentially tumor RNA sequencing immunopeptidomics, primarily taking into account peptide-MHC binding affinity and RNA expression as well as direct identification (15). Additional peptide features, such as stability, clonality, cleavage scores, variant allele frequency, dissimilarity to self, or mutation coverage, are now also taken into account as potential determinants of immunogenicity (16–18). The downstream number of short-listed neoepitopes varies among patients and tumor types and is further greatly reduced following cellular immunogenicity evaluation. Depending on the chosen experimental strategy, prioritized neoepitope candidates are synthesized in the form of short or long peptides, or mRNA encoding mutations, and screened for T-cell reactivities from patients' blood or tumor samples. In this context, functional assays [such as interferon (IFN)-γ ELISpot and CD137 assay] as well as peptide MHC (pMHC)-multimer direct stainings are typically used as sensitive readouts. Of note, cellular interrogation requires a significant number of patients' samples and often includes, prior to screening, a round of antigen-specific T-cell expansion with candidate neoepitope pools, which may alter the original clonotypes' composition.
Despite the variable mutational load across different human malignancies (19) and the technical challenges, tumor-infiltrating, as well as circulating, neoantigen-specific CD8+ and CD4+ T-cells have now been identified and characterized in several tumor types (20–25). Early clinical data also suggest that neoantigen load has a predictive role in patient response to checkpoint blockade and TIL ACT immunotherapy (26–29).
Bulk infiltrating T-cell populations can be very heterogeneous, and the frequency of private (and shared) tumor-associated antigen specificities is generally low (20, 21, 30). Dissection of melanoma and colorectal and lung cancers has highlighted that a significant fraction of TILs can contain antiviral CD8+ T cells [such as Epstein-Barr virus (EBV)- and cytomegalovirus (CMV)-specific], extending observations that many tumor infiltrates may be in fact not tumor-specific (30–32). A study by Scheper et al. (33) has assessed the intrinsic tumor reactivity of TILs in melanoma and ovarian and colorectal cancer, demonstrating how indeed only a small fraction of the intratumoral CD8+ T-cell receptor (TCR) repertoire is able to recognize autologous cancer cells. Yet, the frequency of CD8+ in TILs correlates with favorable prognosis, and increasing evidence has shown how a relatively limited set of neoantigen-specific T-cells from melanoma TILs can mediate tumor recognition, despite the tumor cells harboring hundreds of somatic mutations (34–37). Collectively, these data suggest that enriching TIL infusion products for a few T-cell clonotypes specific for key immunogenic neoantigens could guide more effective antitumor responses in vivo.
One might argue that the need for available resected tumor specimens, from which infiltrating T-cells are isolated ex vivo, limits a broader application of standard TIL therapies to other tumor types. In this regard, Cohen et al. (22) first provided a simplified and noninvasive blood-based strategy as an alternative to current TIL production by demonstrating that neoantigen and self-antigen reactive T-cells can be reliably isolated from the peripheral blood of melanoma patients. Detection of neoantigen-specific CD8+ and CD4+ lymphocytes from peripheral blood has been subsequently described in patients with relatively low tumor mutation burden, such as ovarian and gastrointesinal cancers (20, 38–40). However, circulating neoantigen-specific T-cells share with their infiltrating counterpart very low detection frequencies (ranging from 0.5 to 0.002%) (20, 22, 37, 41, 42), hence the need for specific enrichment strategies. Of note, novel evidence has shown that the patient neoantigen-reactive CD8+ TCR repertoire can be largely discordant (in terms of specificity and functional avidity) between circulating and infiltrating T-cells in ovarian cancer patients (20). In particular, neoantigen-specific TILs exhibited on average higher functional avidity than their peripheral blood lymphocyte (PBL) counterpart. Further studies are therefore required to assess whether PBL and TIL cultures can be an equally suitable source for successful personalized T-cell therapies. Of note, it has also been shown that non-tolerized CD8+ T-cell repertoires of healthy donors were able to specifically recognize neoantigens which were ignored by tumor-infiltrating T cells in melanoma patients (43, 44).
Taken together, the selective enrichment of bulk TIL or PBL cultures for private (and shared) tumor-antigen specificities may improve the response rates achieved to date by adoptive T-cell therapies. One can envision highly personalized and specialized platforms, which integrate tumor-antigen identification and the generation of T-cell infusion products with a predefined reactivity composition (Figure 1). Here, we provide an overview of the current toolbox of technologies for the tailored enrichment of T-cell products in tumor-specific reactivities, addressing main advantages and disadvantages of individual approaches. A first general distinction can be made between techniques which require T-cells to be reactivated with cognate antigen (or autologous tumor) prior to downstream readout and separation (i.e., cytokine production or surface activation marker expression), and methods (such as pMHC multimer-based labeling) in which unstimulated antigen-specific T-cells can be directly selected. Different ways to restimulate antigen-specific T-cells are beyond the scope of this review and have been extensively discussed elsewhere (4, 45). A second distinction can be made between antigen-specific purification pipelines based on predefined specificities of interest and requiring a thorough antigen discovery phase and tumor reactivity-based pipelines which aim for a more “agnostic” enrichment in that they do not require a priori target prediction and identification (Figure 2). The two main technologies for cell isolation are fluorescence-activated and magnetic bead-activated cell sorting (FACS and MACS, respectively), both of which are extensively employed in preclinical research environments. Finally, we will address some of the challenges and limitations that such individualized T-cell manufacturing platforms necessarily entail for clinical application from both a technical and regulatory point of view.

Figure 1. General workflow for personalized enrichment of antigen-specific T-cells from bulk tumor-infiltrating lymphocyte (TIL) [or peripheral blood lymphocyte (PBL)] cultures. T-cells can be isolated from the patient's infiltrating or circulating lymphocyte populations. Following neoantigen discovery and validation, antigen-specific T-cells are enriched by bulk cultures and expanded in vitro to meet the numbers required for reinfusion. FACS, fluorescence-activated cell sorting; MACS, magnetic bead-activated cell sorting; WES, whole-exome sequencing; MS, mass spectrometry.
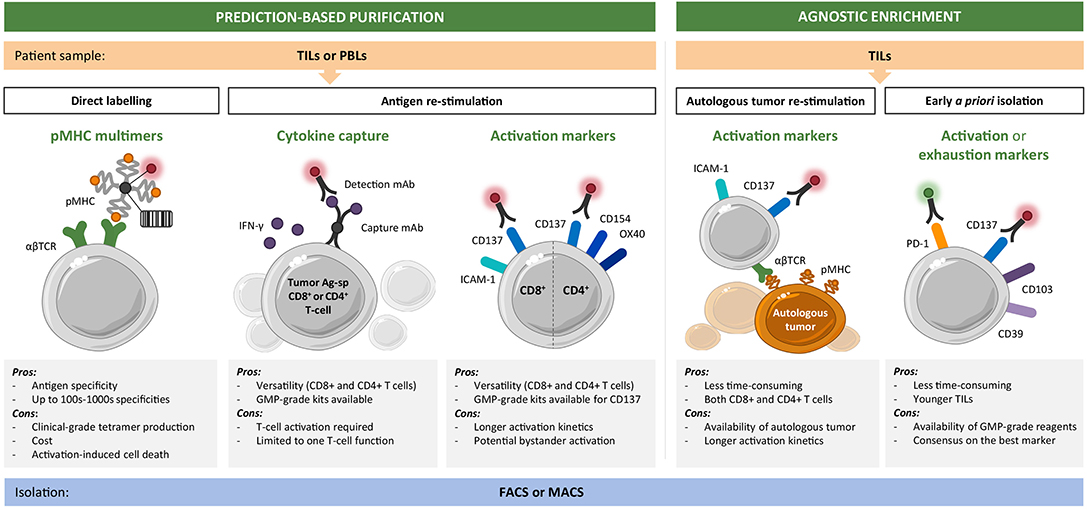
Figure 2. Toolset for personalized enrichment of T-cell infusion products. Current technologies can be grouped into neoantigen-specific purification strategies, which rely on predictions and predefined epitope selection, and agnostic enrichment strategies based on coculture with autologous tumor or a priori identification of tumor-reactive T-cells. The main advantages and disadvantages of each approach are listed. CD8+ (and CD4+) T-cells of interest can be isolated starting from tumor infiltrating lymphocyte (TIL) or peripheral blood lymphocyte (PBL) cultures by fluorescence-activated (FACS) or magnetic bead-activated (MACS) cell sorting.
Current Toolset for the Enrichment of Predefined Neoantigen Specificities
Peptide Major Histocompatibility Complex-Based Strategies
Labeling of a specific TCR by means of fluorochrome-conjugated pMHC multimers allows to directly identify CD8+ T-cell reactivities without restriction to functional parameters. MHC-based reagents have rapidly evolved from single fluorescent-labeled pMHC tetramers to increasingly advanced and optimized staining protocols with higher detection sensitivity (46, 47). For example, the screening of multiple T-cell reactivities can be achieved by combinatorial multimer staining either assigning a unique binary color code to each antigen specificity (48, 49) or using a high number of possible fluorochrome combinations (50). Several groups have speculated a possible clinical implementation of MHC multimer-based approaches in order to screen samples and generate neoantigen-enriched therapeutic cellular products (22, 51–53).
Alternative pMHC multimeric reagents, such as Streptamers and NTAmers, are built on reversible complexes and can therefore rapidly dissociate in the presence of biotin or imidazole, respectively (54–56). Antigen-specific T-cell staining with reversible multimers not only improves conventional pMHC reagents by reducing activation-induced T-cell death but also allows pMHC monomer dissociation kinetic measurements which have been shown to correlate with T-cell functionality (55, 57, 58). These technologies could therefore further aid in the precise selection of the “fittest” T-cell clonotypes within a single antigen specificity.
A more recent addition to the pMHC multimer portfolio is represented by a different labeling whereby DNA bar code tags are attached to the multimer scaffold. T-cells are collectively sorted based on one fluorescence, and distinct pMHCs are retrospectively revealed by means of large-scale sequencing of cognate bar codes (59). As a result, up to thousands of unique specificities can be potentially identified simultaneously, paving the way for high-throughput platforms and downstream applications such as TCR redirected T-cell therapy (60, 61). Of note, state-of-the-art microfluidic devices can help with potential sample size limitation by allowing on-chip detection and manipulation of multimer-sorted neoantigen-specific T cells for downstream analysis and applications (38).
If on one hand, pMHC-based strategies have facilitated the characterization of complex T-cell repertoires, on the other, they present some limitations for clinical application. First of all, the MHC restriction of the antigenic peptide has to be well-characterized; they do not provide information on T-cell function and are limited for CD4+ T-cell isolation (discussed below). Most importantly, given that pMHC multimer production is quite time-consuming and has to be manufactured under Good Manufacturing Practice (GMP) conditions, generation of a library of pMHC multimers on a patient-specific basis may be cost prohibitive for clinical implementation. In this regard, UV-based peptide exchange technologies (62) could aid the rapid engineering and manufacturing of multiple distinct pMHC reagents for individual patients.
Cytokine Capture
Cytokines secreted by previously activated T-cells can be retained on the cell surface via a capture matrix, allowing the molecules detection and the isolation of viable antigen-specific T-cells via MACS (63, 64). In particular, IFN-γ secretion by activated CD8+ (and CD4+) T-cells has long been associated with effective tumor recognition and used as a functional readout to detect tumor-reactive T-cells (65, 66). However, there are only a couple of examples of preclinical isolation of tumor-specific T-cells by means of IFN-γ capture (65). Jedema et al. (67) describe a strategy to isolate leukemia-reactive CD8+ (and CD4+) T-cells upon specific IFN-γ secretion to be used for adoptive transfer. Another group has reported a GMP-grade isolation of protocol of polyclonal and polyfunctional antigen-specific T-cells from healthy donor PBLs, by IFN-γ labeling followed by FACS, using NY-ESO-1 as a model system (68). Taken together, fully automated IFN-γ-based T-cell enrichment procedures are commercially available and could be more easily implemented in a clinical pipeline. However, cytokine production is known to be restricted to certain T-cell subsets; therefore, the enrichment of antigen-specific T-cell frequencies uniquely based on cytokine secretion profile might be incomplete.
Activation Markers
An alternative approach to direct labeling and cytokine detection is the use of activation-induced surface markers, which are upregulated upon antigen-specific TCR engagement. Expression of some of these markers is independent of cytokine production or T-cell phenotype, therefore potentially allowing the capture of the total pool of functional and reactive T-cells. Several surface markers have been suggested over time; however, only a limited number has been selected and extensively characterized because of reduced bystander activation, high specificity, and upregulation kinetics (69, 70).
The tumor necrosis factor receptor (TNFR) family member CD137 (or 4-1BB) has been initially characterized as a specific marker of TCR-induced activation of viral-specific CD8+ T-cells (70–72). Following antigen-specific stimulation, CD137 is upregulated on CD8+ T-cells, allowing the detection of viable antigen-specific T-cells. CD137 surface expression is now being extensively used as a marker to detect shared as well as neoantigen-reactive circulating and infiltrating T-cells, in combination with standard IFN-gamma ELISPOT screening (34, 42, 73–75). For example, Parkhurst et al. (73) isolated CD137+ TILs by FACS following restimulation with dendritic cells (DCs) transfected with mutation-encoding RNA and showed that the expanded CD137+ fraction had indeed been enriched in neoantigen-specific T-cells. At this point in time, anti-CD137 and anti-IFN-γ are among the few clinical-grade commercially available antibodies for the selection of antigen-specific T-cells; several companies though now provide custom monoclonal antibody development and conjugation under GMP guidelines.
On a different note, structural changes of activated integrins upon early TCR engagement can be exploited as the inside-out signal to detect functional T-cells (76). Dimitrov et al. (76) have successfully applied this strategy in order to monitor viral-specific T-cell responses within minutes, thereby stressing the advantageous very short incubation time compared to other activation markers. A parallel assessment of pMHC multimer and activation-based sorting would be highly informative in highlighting whether distinct markers are able to isolate overlapping populations of heterogeneous antigen-specific T-cells.
CD4+ Neoantigen T-Cell Responses
Screening of naturally occurring or induced neoantigen T-cell responses in patients with solid tumors has provided evidence that both CD8+ and CD4+ T-cells recognize private mutated epitopes (24, 77–80). Furthermore, a number of single-patient case reports seem to indicate that neoantigen-specific CD4+ T-cells can mediate therapeutic immune responses to tumors (36, 81–83). A breakthrough paper by Tran et al. (36) provided the first demonstration of clinical activity of neoantigen-specific CD4+ T-cell infusion in a metastatic cancer patient.
Screening for MHC class-II-restricted T-cell has been long under-appreciated because of the limited accuracy of neoantigen prediction algorithms (84, 85). However, rapidly improving prediction tools for MHC class-II ligands (86–90) and the notion that TIL cultures can include a substantial fraction of functional CD4+ T-cells calls for flexible strategies to enable the enrichment of both CD4+ and CD8+ reactive compartments from bulk populations and downstream therapeutic infusion. In the framework of technologies validated for the CD8+ counterpart, MHC class-II multimers have so far progressed at a lower rate because of technical issues with recombinant pMHC class-II heterodimer production, and the assumption that CD4+ TCR binding affinity to cognate pMHC is significantly lower (91–93). On the other hand, activation marker upregulation following antigen restimulation offers the advantage of capturing cytokine-independent and heterogeneous CD4+ T-cell responses (71, 94). Indeed, CD137 may allow the capture of both CD4+ and CD8+ functional T-cells with high specificity (69, 95). However, a few publications have described the use of alternative TCR-dependent surface markers such as CD154 and CD134 (or OX-40) to detect neoantigen-specific CD4+ T-cells (34, 74, 75). A comprehensive comparison of activation-induced marker assays has yet not been investigated. In addition, care should be taken in discriminating regulatory T-cell from effector antigen-specific CD4+ T-cells when exploiting activation markers. For instance, the inverse expression of CD137 and CD154 has been described to discriminate between activated regulatory and effector CD4+ T-cells ex vivo (96).
Agnostic Enrichment of Tumor-Reactive Tumor-Infiltrating Lymphocytes
The identification and validation of patient-specific immunogenic neoantigen specificities require advanced technologies (such as high-throughput sequencing, mass spectrometry, and synthetic peptide production) and adds complexity and time (several weeks to months) to an already labor-intensive TIL production pipeline. Less time-consuming and unbiased methods are therefore being evaluated to generate patient-specific T-cell products, which are clinically feasible for adoptive transfer.
Coculture With Autologous Tumor
Using autologous tumor cells as targets circumvents the need for screening of immunogenic private or shared tumor antigens, while presenting to T-cells the complete range of naturally presented tumor antigens. In TIL production history, IFN-γ secretion has been exploited to prescreen which tumor fragments to expand: only TILs showing tumor reactivity above a predefined cutoff value were selected for downstream expansion and infused (97).
The role of activation marker CD137 was initially investigated by Ye et al. (98) for the quick and sensitive enrichment of tumor-reactive TILs from ovarian cancer and melanoma patients. The authors showed that CD137+-sorted TILs demonstrated increased reactivity against shared antigens following overnight incubation in the presence of MHC-matched tumor cell lines. Importantly, the CD137-enriched fraction resulted in enhanced in vitro and in vivo antitumor reactivity (98). Seliktar-Ofir et al. (99) presented proof of concept of a GMP-compatible CD137-based separation method for personalized adoptive cell therapy. Melanoma TILs were sorted by MACS based on CD137 surface upregulation following overnight coincubation with autologous tumor cultures (99). CD137+ TIL populations showed increased in vitro antitumor reactivity and contained a higher fraction of neoantigen and shared tumor antigen-specific T-cells when compared to the starting unseparated cultures. This approach might be limited by the establishment of autologous primary tumor cell lines, for which the success rate can be very low in tumors other than melanoma. In this regard, Dijkstra et al. (100) have presented a proof-of-concept study in which tumor-reactive T-cells from non-small-cell lung cancer and colon rectal cancer patients can be obtained by coculturing autologous PBLs with matched tumor epithelial organoids. Organoids are 3D cultures of primary solid tumors and can be established with a higher success rate from very limited amounts of tumor biopsies or surgical resections.
Considering the importance of costimulation in the context of successful tumor-specific T-cell activation, antigen presenting cells (APCs) should also be taken into account when establishing ex vivo cocultures of T cells and autologous tumor. The combination of natural or artificial APC with tumor lysate preparations can provide a wide array of tumor antigens in a more physiological costimulation context, therefore boosting the downstream antitumor activity of adoptively transferred T cells. Initial protocols used ex vivo-derived autologous DCs, stimulated with a defined maturation cocktail and pulsed with whole tumor lysate, to preferentially expand TILs to treat patients with melanoma (101–103). However, such strategies introduced additional time and numerous cytokines required for DC cell generation and maturation and prompted the quest for easily tailored and artificial APC (aAPC) platforms. Clinical-grade aAPCs have now limitless application potential: they can be coated with any number of costimulatory molecules (such as CD80, CD86, and CD137L) and membrane-bound cytokines to elicit rapid and improved TIL activation (104–106).
A Priori Enrichment of Tumor-Reactive Tumor-Infiltrating Lymphocytes
Efforts from several groups are focusing on improving the a priori identification of tumor-reactive TILs solely based on phenotypic profiling. The rationale behind this strategy lies in the fact that naturally occurring tumor-reactive TILs are chronically exposed to their cognate antigen in the tumor site, therefore expressing a defined set of surface activation- and/or exhaustion-associated markers, providing the opportunity for their direct isolation.
Initial evidence suggested that preselection of melanoma-infiltrating or peripheral blood T-cells by PD-1 expression prior to expansion could directly enrich tumor-reactive T-cells (39, 107). In another study, CD137 was identified as a better marker than PD-1 for the prospective selection of naturally occurring tumor-reactive fresh TILs in ovarian cancer (98). Building upon these previous works, a defined set of tissue residency markers (such as CD103, or integrin αE), necessary for recruitment and retention of TILs in the tumor site, has been suggested as a prospective marker of TIL tumor reactivity (108–110). Duhen et al. (111) have shown that co-expression of CD103 and CD39 further enriches the TIL population for tumor-reactive CD8+ T-cells. CD103+ CD39+ TILs were sorted from tumor digests and expanded in vitro, resulting in increased cytotoxicity toward autologous tumor cells when compared to the respective single positive populations (111).
Challenges and Limitations to Consider for Clinical Implementation
Compared to traditional biological molecules, personalized adoptive T-cell platforms are developed on a patient-specific basis, therefore presenting unique challenges not only for preclinical developers and manufacturers but also for regulatory authorities and healthcare providers. Starting from the initial step of private tumor antigen discovery and validation, throughout TIL (or PBL) enrichment and in vitro scale-up expansion, individual processes, facilities, and technologies must be carefully reviewed and adjusted according to clinical requirements. Indeed, specific regulations may differ slightly among countries and regions, but most challenges and limitations linked to clinical implementation are shared.
Isolation Phase
Starting from the isolation step itself, one has to consider not only the technical aspects of the sorting strategy but more importantly its compatibility with regulatory requirements. The choice of a FACS or MACS-based enrichment depends on several factors, including the number of cells in the source material (TILs or PBLs), the relative frequency of antigen-specific cells within, the level of purity needed for the final product. On one hand, magnetic beads are of lower technical complexity and clinical-grade isolation kits are already commercially available, on the other, FACS separation performs multiparameter analysis of single cells, achieving resolution and purity levels, which are not always possible by MACS. In addition, FACS analysis can characterize in real time the sorted bulk T-cell population (in terms of identity and purity), as a first in-process quality control. However, FACS is still not routinely applicable under GMP conditions, which require single-use and a closed fluidic system for clinical implementation.
Expansion Phase
As the absolute cell counts of neoantigen-specific T-cells after isolation are extremely low for direct reinfusion, a rapid expansion procedure (namely, REP) of sorted cells is typically performed with allogeneic irradiated feeder cells in the presence of high-dose interleukin (IL)-2 and anti-CD3 (97). Depending on the yield, the best scale-up closed-system expansion devices and culture conditions can be optimized to meet the numbers required for the adoptive transfer (typically in the order of 109 cells per patient). Several distributors supply culture bags or gas-permeable flasks, sterile tubing accessories, and welding to facilitate the conversion of research protocols to GMP closed manufacturing processes, where the risk of cross contamination has to be minimized.
Absolute numbers aside, critical parameters for a successful ACT is ensuring that the final TIL product has maintained purity, TCR clonal diversity, and tumor reactivity following in vitro expansion. Indeed, during the REP phase, there can be interclonal competition resulting in an increased or decreased frequency of given specificities of interest compared to the starting culture. In this sense, enriching for tumor-specific T-cells at appreciable frequencies prior to up-scale would higher the chances of obtaining a final TIL product with adequate tumor reactivity upon infusion. The extensive expansion can also drive progressive T-cell differentiation and phenotype changes which may affect TIL in vivo persistence, homing, and proliferative capacity shortly after transfer, as these cells reencounter cognate antigens within the tumor microenvironment (112–115). Increased TIL proliferation and reactivity toward autologous tumor have been recently reported in a study introducing CTLA-4 blockade in vitro during the initial TIL pre-REP from ovarian tumor fragments (26). As mentioned previously, cytokines used during the in vitro manufacturing of the product can also significantly affect TIL immune profiles. Alternative cytokines to standard IL-2 have been tested during TIL REP (114) or during initial priming period (e.g., IL-21) (116, 117).
An additional aspect to consider is that TILs may fail to perform their expected therapeutic effector functions upon infusion due to activation-induced cell death (AICD) and exhaustion; both are peripheral tolerance mechanisms restricting an escalating, therefore potentially damaging, immune response. Melanoma TILs undergoing intense polyclonal TCR stimulation during REP have been shown to be more sensitive to AICD when cocultured in vitro with autologous tumor, whereas “younger” and less differentiated TILs are less susceptible and have a better in vivo tumor control (118–121). In a similar fashion, alternative costimulatory pathways, such as through CD137 via the addition of agonist antibodies during or following REP, can increase the polyclonal expansion of infiltrating or circulating CD8+ TILs while preserving their responsiveness (122–124).
Overall, how infused antigen-specific TIL clonotypes persist in vivo and respond to tumor antigen restimulation upon transfer and how their gene signature correlates to clinical benefit should be studied systematically on a larger number of patients receiving the same infusion regimen.
General Improvements
Further pre-sensitization approaches could help increase neoantigen-specific T-cell frequency in starting TIL (or PBL) cultures and facilitate downstream sorting of the population of interest. Our group has reported that the addition of synthetic peptide pools of all predicted class-I neoantigens can improve conventional TIL generation in ovarian cancer (20). Primed TIL cultures were significantly enriched in neoepitope-specific CD8+ T-cells as compared with standard TILs generated from the same patients.
Finally, while it is tempting to focus on private neoantigens deriving from single point mutations, driver genes (such as RAS and BRAF) recurrently affected by mutation or fusion events across individuals and cancer types would be expected to yield semiprivate (or even “shared”) neoantigens. This seems to be especially the case for hematological malignancies, where immunogenic neoantigens have been reported to be mutated in up to 30% of patients (125, 126), along with case reports of mutation hot spots in solid tumors (127–130). In addition, potential semiprivate neoantigens derived from aberrant phosphorylation, resulting from dysregulated protein kinase activity during transformation, can be detected with mass spectrometry using relatively small amounts of patient samples (9, 131, 132). The targeting of shared or semiprivate neoantigens in solid tumors is a particularly desirable possibility which has to be further investigated, considering it would contribute to greatly reducing costs and production time of highly enriched T-cell infusion products. Given the multistep and laborious nature of these enriched T-cell therapies, which require coordination between highly specialized healthcare centers and manufacturing cell facilities, one has to ultimately consider if the added time frame is clinically reasonable. An additional month to the pipeline can result in significant patient dropout because of rapid disease progression (133).
Concluding Remarks
The identification of neoantigens as drivers of successful antitumor immunity is offering exciting new opportunities for cancer immunotherapies, including making T-cell infusion products highly individualized for more effective treatment. Moving forward, patient-specific T-cell enrichment technologies will need to be integrated into clinically compliant pipelines. In this respect, the first Food and Drug Administration (FDA) approval of the first adoptive cell therapy [i.e., chimeric antigen receptor (CAR) T-cell therapy] represents a huge achievement in the immune-oncology field and will hopefully pave the way for further approvals of personalized immunotherapies.
Author Contributions
VB wrote the manuscript and made the figures. VB, AH, and GC contributed to the concept, discussion of content, and editing of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. (2015) 348:62–8. doi: 10.1126/science.aaa4967
2. Stevanović S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. (2015) 33:1543–50. doi: 10.1200/JCO.2014.58.9093
3. Linette GP, Carreno BM. Neoantigen vaccines pass the immunogenicity test. Trends Mol Med. (2017) 23:869–71. doi: 10.1016/j.molmed.2017.08.007
4. Bobisse S, Foukas PG, Coukos G, Harari A. Neoantigen-based cancer immunotherapy. Ann Transl Med. (2016) 4:262. doi: 10.21037/atm.2016.06.17
5. Wei Z, Zhou C, Zhang Z, Guan M, Zhang C, Liu Z, et al. The landscape of tumor fusion neoantigens: a pan-cancer analysis. iScience. (2019) 21:249. doi: 10.1016/j.isci.2019.10.028
6. Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. (2017) 18:1009–21. doi: 10.1016/S1470-2045(17)30516-8
7. Raposo B, Merky P, Lundqvist C, Yamada H, Urbonaviciute V, Niaudet C, et al. T cells specific for post-translational modifications escape intrathymic tolerance induction. Scientific Reports. (2018) 9:353. doi: 10.1038/s41467-017-02763-y
8. Gao Q, Liang W-W, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. (2018) 23:227–38.e3. doi: 10.1016/j.celrep.2018.03.050
9. Solleder M, Guillaume P, Racle J, Michaux J, Pak H-S, Müller M, et al. Mass spectrometry based immunopeptidomics leads to robust predictions of phosphorylated HLA class I ligands. Mol Cell Proteomics. (2019) 19:390–404. doi: 10.1074/mcp.TIR119.001641
10. Müller M, Pak H, Harnett D, Huber F, Grun D, Leleu M, et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat Commun. (2020) 11:14968. doi: 10.1038/s41467-020-14968-9
11. Li Y, Jiang T, Zhou W, Li J, Li X, Wang Q, et al. Pan-cancer characterization of immune-related lncRNAs identifies potential oncogenic biomarkers. Nat Commun. (2020) 11:1000. doi: 10.1038/s41467-020-14802-2
12. Purcell AW, Ramarathinam SH, Ternette N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat Protoc. (2019) 14:1687–707. doi: 10.1038/s41596-019-0133-y
13. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. (2014) 515:572–6. doi: 10.1038/nature14001
14. Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. (2016) 7:13404–16. doi: 10.1038/ncomms13404
15. Garcia-Garijo A, Fajardo CA, Gros A. Determinants for neoantigen identification. Front Immunol. (2019) 10:1392. doi: 10.3389/fimmu.2019.01392
16. McGranahan N, Swanton C. Neoantigen quality, not quantity. Gene Ther. (2019) 11:eaax7918. doi: 10.1126/scitranslmed.aax7918
17. Hundal J, Kiwala S, Feng Y-Y, Liu CJ, Govindan R, Chapman WC, et al. Accounting for proximal variants improves neoantigen prediction. Nat Genet. (2018) 51:175–79. doi: 10.1038/s41588-018-0283-9
18. Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nat Cancer Rev. (2017) 551:512–6. doi: 10.1038/nature24462
19. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. (2013) 339:1546–58. doi: 10.1126/science.1235122
20. Bobisse S, Genolet R, Roberti A, Tanyi JL, Racle J, Stevenson BJ, et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8+ T cells in immunotherapy-naive ovarian cancer. Nat Commun. (2018) 9:1092. doi: 10.1038/s41467-018-03301-0
21. Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M, et al. TIL therapy broadens the tumor-reactive CD8+ T cell compartment in melanoma patients. OncoImmunology. (2012) 1:409–18. doi: 10.4161/onci.18851
22. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. (2015) 125:3981–91. doi: 10.1172/JCI82416
23. Zhang X, Kim S, Hundal J, Herndon JM, Li S, Petti AA, et al. Breast cancer neoantigens can induce CD8 T cell responses and antitumor immunity. Cancer Immunol Res. (2017) 5:516–23. doi: 10.1158/2326-6066.CIR-16-0264
24. Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Cancer Rev. (2013) 19:747–52. doi: 10.1038/nm.3161
25. Tran E, Ahmadzadeh M, Lu Y-C, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. (2015) 350:1387–90. doi: 10.1126/science.aad1253
26. Friese C, Harbst K, Borch TH, Westergaard MCW, Pedersen M, Kverneland A, et al. CTLA-4 blockade boosts the expansion of tumor-reactive CD8+ tumor-infiltrating lymphocytes in ovarian cancer. Sci Rep. (2020) 10:3914. doi: 10.1038/s41598-020-60738-4
27. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. (2017) 16:2598–608. doi: 10.1158/1535-7163.MCT-17-0386
28. Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell. (2018) 33:853–61.e4. doi: 10.1016/j.ccell.2018.04.001
29. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
30. Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebæk E, et al. Dissection of T-cell antigen specificity in human melanoma. Cancer Res. (2012) 72:1642–50. doi: 10.1158/0008-5472.CAN-11-2614
31. Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun. (2019) 10:567. doi: 10.1038/s41467-019-08534-1
32. Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
33. Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med. (2018) 25:89–94. doi: 10.1038/s41591-018-0266-5
34. Kalaora S, Wolf Y, Feferman T, Barnea E, Greenstein E, Reshef D, et al. Combined analysis of antigen presentation and T cell recognition reveals restricted immune responses in melanoma. Cancer Discov. (2018) 8:1366–75. doi: 10.1158/2159-8290.CD-17-1418
35. Lu YC, Yao X, Li YF, El-Gamil M, Dudley ME, Yang JC, et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J Immunol. (2013) 190:6034–42. doi: 10.4049/jimmunol.1202830
36. Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. (2014) 344:641–5. doi: 10.1126/science.1251102
37. Pritchard AL, Burel JG, Neller MA, Hayward NK, Lopez JA, Fatho M, et al. Exome sequencing to predict neoantigens in melanoma. Cancer Immunol Res. (2015) 3:992–8. doi: 10.1158/2326-6066.CIR-15-0088
38. Peng S, Zaretsky JM, Chour W, Bethune MT, Choi J, Hsu A, et al. Sensitive detection and analysis of neoantigen-specific T cell populations from tumors and blood. Cell Rep. (2019) 28:2728. doi: 10.1016/j.celrep.2019.07.106
39. Gros A, Tran E, Parkhurst MR, Ilyas S, Pasetto A, Groh EM, et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. Nat Med. (2019) 129:4992. doi: 10.1172/JCI127967
40. Cafri G, Yossef R, Pasetto A, Deniger DC, Lu Y-C, Parkhurst M, et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat Commun. (2019) 10:449. doi: 10.1038/s41467-019-08304-z
41. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Cancer Rev. (2016) 22:433–8. doi: 10.1038/nm.4051
42. Martin SD, Wick DA, Nielsen JS, Little N, Holt RA, Nelson BH. A library-based screening method identifies neoantigen-reactive T cells in peripheral blood prior to relapse of ovarian cancer. Oncoimmunology. (2017) 7:e1371895. doi: 10.1080/2162402X.2017.1371895
43. Strønen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. (2016) 352:1337–41. doi: 10.1126/science.aaf2288
44. Ali M, Foldvari Z, Giannakopoulou E, Böschen M-L, Strønen E, Yang W, et al. Induction of neoantigen-reactive T cells from healthy donors. Nat Protoc. (2019) 14:1926–43. doi: 10.1038/s41596-019-0170-6
45. Arnaud M, Duchamp M, Bobisse S, Renaud P, Coukos G, Harari A. Biotechnologies to tackle the challenge of neoantigen identification. Curr Opin Biotechnol. (2020) 65:52. doi: 10.1016/j.copbio.2019.12.014
46. Dolton G, Tungatt K, Lloyd A, Bianchi V, Theaker SM, Trimby A, et al. More tricks with tetramers: a practical guide to staining T cells with peptide-MHC multimers. Immunology. (2015) 146:11–22. doi: 10.1111/imm.12499
47. Bentzen AK, Hadrup SR. Evolution of MHC-based technologies used for detection of antigen-responsive T cells. Cancer Immunol Immunother. (2017) 66:657–66. doi: 10.1007/s00262-017-1971-5
48. Hadrup SR, Bakker AH, Shu CJ, Andersen RS, Veluw J, Hombrink P, et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods. (2009) 6:520–6. doi: 10.1038/nmeth.1345
49. Hadrup SR, Schumacher TN. MHC-based detection of antigen-specific CD8+ T cell responses. Cancer Immunol Immunother. (2010) 59:1425–33. doi: 10.1007/s00262-010-0824-2
50. Newell E, Klein L, Yu W, Davis M. Simultaneous detection of many T-cell specificities using combinatorial tetramer staining. Nat methods. (2009) 6:497–9. doi: 10.1038/nmeth.1344
51. Kelderman S, Heemskerk B, Fanchi L, Philips D, Toebes M, Kvistborg P, et al. Antigen-specific TIL therapy for melanoma: a flexible platform for personalized cancer immunotherapy. Eur J Immunol. (2016) 46:1351–60. doi: 10.1002/eji.201545849
52. Tubb VM, Schrikkema DS, Croft NP, Purcell AW, Linnemann C, Freriks MR, et al. Isolation of T cell receptors targeting recurrent neoantigens in hematological malignancies. J Immuno Therapy Cancer. (2018) 6:70. doi: 10.1186/s40425-018-0386-y
53. Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. (2015) 125:3413–21. doi: 10.1172/JCI80008
54. Neudorfer J, Schmidt B, Huster KM, Anderl F, Schiemann M, Holzapfel G, et al. Reversible HLA multimers (Streptamers) for the isolation of human cytotoxic T lymphocytes functionally active against tumor- and virus-derived antigens. J Immunol Methods. (2007) 320:119–31. doi: 10.1016/j.jim.2007.01.001
55. Schmidt J, Guillaume P, Irving M, Baumgaertner P, Speiser D, Luescher IF. Reversible major histocompatibility complex I-peptide multimers containing Ni2+-nitrilotriacetic acid peptides and histidine tags improve analysis and sorting of CD8+ T cells. J Biol Chem. (2011) 286:41723–35. doi: 10.1074/jbc.M111.283127
56. Hebeisen M, Schmidt J, Guillaume P, Baumgaertner P, Speiser DE, Luescher I, et al. Identification of rare high-avidity, tumor-reactive CD8+ T cells by monomeric TCR-ligand off-rates measurements on living cells. Cancer Res. (2015) 75:1983–91. doi: 10.1158/0008-5472.CAN-14-3516
57. Allard M, Couturaud B, Carretero-Iglesia L, Duong MN, Schmidt J, Monnot GC, et al. TCR-ligand dissociation rate is a robust and stable biomarker of CD8+ T cell potency. JCI Insight. (2017) 2:e92570. doi: 10.1172/jci.insight.92570
58. Gannon PO, Wieckowski S, Baumgaertner P, Hebeisen M, Allard M, Speiser DE, et al. Quantitative TCR:pMHC dissociation rate assessment by NTAmers reveals antimelanoma T cell repertoires enriched for high functional competence. J Immunol. (2015) 195:356–66. doi: 10.4049/jimmunol.1403145
59. Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat Biotechnol. (2016) 34:1037–45. doi: 10.1038/nbt.3662
60. Bentzen AK, Such L, Jensen KK, Marquard AM, Jessen LE, Miller NJ, et al. T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide-MHC complexes. Nat Biotechnol. (2018) 36:1191–6. doi: 10.1038/nbt.4303
61. Zhang S-Q, Ma K-Y, Schonnesen AA, Zhang M, He C, Sun E, et al. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat Biotechnol. (2018) 36:1156–9. doi: 10.1101/457069
62. Toebes M, Coccoris M, Bins A, Rodenko B, Gomez R, Nieuwkoop NJ, et al. Design and use of conditional MHC class I ligands. Nat Med. (2006) 12:246–51. doi: 10.1038/nm1360
63. Brosterhus H, Brings S, Leyendeckers H, Manz RA, Miltenyi S, Radbruch A, et al. Enrichment and detection of live antigen-specific CD4+ and CD8+ T cells based on cytokine secretion. Eur J Immunol. (1999) 29:4053–9. doi: 10.1002/(SICI)1521-4141(199912)29:12<4053::AID-IMMU4053>3.0.CO;2-C
64. Manz R, Assenmacher M, Pflüger E, Miltenyi S, Radbruch A. Analysis and sorting of live cells according to secreted molecules, relocated to a cell-surface affinity matrix. Proc Natl Acad Sci U S A. (1995) 92:1921–5. doi: 10.1073/pnas.92.6.1921
65. Becker C, Pohla H, Frankenberger B, Schüler T, Assenmacher M, Schendel DJ, et al. Adoptive tumor therapy with T lymphocytes enriched through an IFN-γ capture assay. Nat Med. (2001) 7:1159–62. doi: 10.1038/nm1001-1159
66. Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. (1998) 95:7556–61. doi: 10.1073/pnas.95.13.7556
67. Jedema I, Meij P, Steeneveld E, Hoogendoorn M, Nijmeijer BA, van de Meent M, et al. Early detection and rapid isolation of leukemia-reactive donor T cells for adoptive transfer using the IFN-γ secretion assay. Clin Cancer Res. (2007) 13:636–43. doi: 10.1158/1078-0432.CCR-06-2093
68. Kayser S, Boβ C, Feucht J, Witte K-E, Scheu A, Bülow H-J, et al. Rapid generation of NY-ESO-1-specific CD4+ T HELPER1 cells for adoptive T-cell therapy. Oncoimmunology. (2015) 4:e1002723. doi: 10.1080/2162402X.2014.1002723
69. Wehler TC, Karg M, Distler E, Konur A, Nonn M, Meyer RG, et al. Rapid identification and sorting of viable virus-reactive CD4+ and CD8+ T cells based on antigen-triggered CD137 expression. J Immunol Methods. (2008) 339:23–37. doi: 10.1016/j.jim.2008.07.017
70. Wölfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakley M, et al. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. (2007) 110:201–10. doi: 10.1182/blood-2006-11-056168
71. Wölfl M, Kuball J, Eyrich M, Schlegel PG, Greenberg PD. Use of CD137 to study the full repertoire of CD8+ T cells without the need to know epitope specificities. Cytometry. (2008) 73A:1043–9. doi: 10.1002/cyto.a.20594
72. Zandvliet ML, van Liempt E, Jedema I, Kruithof S, Kester MGD, Guchelaar H-J, et al. Simultaneous isolation of CD8+ and CD4+ T cells specific for multiple viruses for broad antiviral immune reconstitution after allogeneic stem cell transplantation. J Immunother. (2011) 34:307–19. doi: 10.1097/CJI.0b013e318213cb90
73. Parkhurst MR, Gros A, Pasetto A, Prickett TD, Crystal JS, Robbins PF, et al. Isolation of T cell receptors specifically reactive with mutated tumor associated antigens from tumor infiltrating lymphocytes based on CD137 expression. Clin Cancer Res. (2016) 23:2491–505. doi: 10.1186/2051-1426-3-S2-P40
74. Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight. (2018) 3:4579. doi: 10.1172/jci.insight.122467
75. Deniger DC, Pasetto A, Robbins PF, Gartner JJ, Prickett TD, Paria BC, et al. T-cell responses to TP53 “Hotspot” mutations and unique neoantigens expressed by human ovarian cancers. Clin Cancer Res. (2018) 24:5562–73. doi: 10.1158/1078-0432.CCR-18-0573
76. Dimitrov S, Gouttefangeas C, Besedovsky L, Jensen ATR, Chandran PA, Rusch E, et al. Activated integrins identify functional antigen-specific CD8+ T cells within minutes after antigen stimulation. Proc Natl Acad Sci U S A. (2018) 115:E5536–E45. doi: 10.1073/pnas.1720714115
77. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nat Cancer Rev. (2017) 547:222–6. doi: 10.1038/nature23003
78. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217–21. doi: 10.1038/nature22991
79. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. (2013) 31:e439–e42. doi: 10.1200/JCO.2012.47.7521
80. Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. (2015) 21:81–5. doi: 10.1038/nm.3773
81. Friedman KM, Prieto PA, Devillier LE, Gross CA, Yang JC, Wunderlich JR, et al. Tumor-specific CD4+ melanoma tumor-infiltrating lymphocytes. J Immunother. (2012) 35:400–8. doi: 10.1097/CJI.0b013e31825898c5
82. Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol. (2008) 180:6116. doi: 10.4049/jimmunol.180.9.6116
83. Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. (2015) 520:692–96. doi: 10.1038/nature14426
84. Lundegaard C, Lund O, Nielsen M. Prediction of epitopes using neural network based methods. J Immunol Methods. (2011) 374:26–34. doi: 10.1016/j.jim.2010.10.011
85. Nielsen M, Lund O, Buus S, Lundegaard C. MHC class II epitope predictive algorithms. Immunology. (2010) 130:319–28. doi: 10.1111/j.1365-2567.2010.03268.x
86. Chen B, Khodadoust MS, Olsson N, Wagar LE, Fast E, Liu CL, et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat Biotechnol. (2019) 37:1332. doi: 10.1038/s41587-019-0280-2
87. Racle J, Michaux J, Rockinger GA, Arnaud M, Bobisse S, Chong C, et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat Biotechnol. (2019) 37:1283–6. doi: 10.1038/s41587-019-0289-6
88. Gfeller D, Bassani-Sternberg M. Predicting antigen presentation-what could we learn from a million peptides? Front Immunol. (2018) 9:1716. doi: 10.3389/fimmu.2018.01716
89. Schneidman-Duhovny D, Khuri N, Dong GQ, Winter MB, Shifrut E, Friedman N, et al. Predicting CD4 T-cell epitopes based on antigen cleavage, MHCII presentation, and TCR recognition. PLoS One. (2018) 13:e0206654. doi: 10.1371/journal.pone.0206654
90. Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, Nielsen M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics. (2015) 67:641–50. doi: 10.1007/s00251-015-0873-y
91. Holland CJ, Dolton G, Scurr M, Ladell K, Schauenburg AJ, Miners K, et al. Enhanced detection of antigen-specific CD4+ T cells using altered peptide flanking residue peptide-MHC class II multimers. J Immunol. (2015) 195:5827–36. doi: 10.4049/jimmunol.1402787
92. Ayyoub M, Dojcinovic D, Pignon P, Raimbaud I, Schmidt J, Luescher I, et al. Monitoring of NY-ESO-1 specific CD4+ T cells using molecularly defined MHC class II/His-tag-peptide tetramers. Proc Natl Acad Sci U S A. (2010) 107:7437–42. doi: 10.1073/pnas.1001322107
93. Cole DK, Pumphrey NJ, Boulter JM, Sami M, Bell JI, Gostick E, et al. Human TCR-binding affinity is governed by MHC class restriction. J Immunol. (2007) 178:5727–34. doi: 10.4049/jimmunol.178.9.5727
94. Chattopadhyay PK, Yu J, Roederer M. A live-cell assay to detect antigen-specific CD4+ T cells with diverse cytokine profiles. Nat Med. (2005) 11:1113–7. doi: 10.1038/nm1293
95. Danilova L, Anagnostou V, Caushi JX, Sidhom J-W, Guo H, Chan HY, et al. The mutation-associated neoantigen functional expansion of specific T cells (MANAFEST) assay: a sensitive platform for monitoring antitumor immunity. Cancer Immunol Res. (2018) 6:888–99. doi: 10.1158/2326-6066.CIR-18-0129
96. Nowak A, Lock D, Bacher P, Hohnstein T, Vogt K, Gottfreund J, et al. CD137+CD154– expression as a regulatory T cell (Treg)-specific activation signature for identification and sorting of stable human Tregs from in vitro expansion cultures. Front Immunol. (2018) 9:3921–15. doi: 10.3389/fimmu.2018.00199
97. Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. (2003) 3:666–75. doi: 10.1038/nrc1167
98. Ye Q, Song D-G, Poussin M, Yamamoto T, Best A, Li C, et al. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res. (2014) 20:44–55. doi: 10.1158/1078-0432.CCR-13-0945
99. Seliktar-Ofir S, Merhavi-Shoham E, Itzhaki O, Yunger S, Markel G, Schachter J, et al. Selection of shared and neoantigen-reactive T cells for adoptive cell therapy based on CD137 separation. Front Immunol. (2017) 8:4550–14. doi: 10.3389/fimmu.2017.01211
100. Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell. (2018) 174:1586–98.e12. doi: 10.1016/j.cell.2018.07.009
101. Lou Y, Wang G, Lizée G, Kim GJ, Finkelstein SE, Feng C, et al. Dendritic cells strongly boost the antitumor activity of adoptively transferred T cells in vivo. Cancer Res. (2004) 64:6783–90. doi: 10.1158/0008-5472.CAN-04-1621
102. Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res. (2014) 20:2457–65. doi: 10.1158/1078-0432.CCR-13-3017
103. Mackensen A, Meidenbauer N S, Vogl S, Vogl R, Laumer M, et al. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J Clin Oncol. (2006) 24:5060–9. doi: 10.1200/JCO.2006.07.1100
104. Forget M-A, Malu S, Liu H, Toth C, Maiti S, Kale C, et al. Activation and propagation of tumor-infiltrating lymphocytes on clinical-grade designer artificial antigen-presenting cells for adoptive immunotherapy of melanoma. J Immunother. (2014) 37:448–60. doi: 10.1097/CJI.0000000000000056
105. Eggermont LJ, Paulis LE, Tel J, Figdor CG. Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends Biotechnol. (2014) 32:456–65. doi: 10.1016/j.tibtech.2014.06.007
106. Su Q, Igyártó BZ. One-step artificial antigen presenting cell-based vaccines induce potent effector CD8 T cell responses. Sci Rep. (2019) 9:18949. doi: 10.1038/s41598-019-55286-5
107. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. (2014) 124:2246–59. doi: 10.1172/JCI73639
108. Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpréville V, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol. (2015) 194:3475–86. doi: 10.4049/jimmunol.1402711
109. Ganesan A-P, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. (2017) 18:940–50. doi: 10.1038/ni.3775
110. Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res. (2014) 20:434–44. doi: 10.1158/1078-0432.CCR-13-1877
111. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. (2018) 9:56–13. doi: 10.1038/s41467-018-05072-0
112. Lu Y-C, Jia L, Zheng Z, Tran E, Robbins PF. Single-cell transcriptome analysis reveals gene signatures associated with T-cell persistence following adoptive cell therapy. Cancer Immunol Res. (2019) 7:1824–36. doi: 10.1158/2326-6066.CIR-19-0299
113. Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. (2012) 18:6758–70. doi: 10.1158/1078-0432.CCR-12-1177
114. Li Y, Liu S, Hernandez J, Vence L, Hwu P, Radvanyi L. MART-1-specific melanoma tumor-infiltrating lymphocytes maintaining CD28 expression have improved survival and expansion capability following antigenic restimulation in vitro. J Immunol. (2010) 184:452–65. doi: 10.4049/jimmunol.0901101
115. Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. (2004) 173:7125–30. doi: 10.4049/jimmunol.173.12.7125
116. Chapuis AG, Lee SM, Thompson JA, Roberts IM, Margolin KA, Bhatia S, et al. Combined IL-21-primed polyclonal CTL plus CTLA4 blockade controls refractory metastatic melanoma in a patient. J Exp Med. (2016) 213:1133–9. doi: 10.1084/jem.20152021
117. Chapuis AG, Desmarais C, Emerson R, Schmitt TM, Shibuya KC, Lai IP, et al. Tracking the fate and origin of clinically relevant adoptively transferred CD8 +T cells in vivo. Sci Immunol. (2017) 2:eaal2568. doi: 10.1126/sciimmunol.aal2568
118. Scheffel MJ, Scurti G, Simms P, Garrett-Mayer E, Mehrotra S, Nishimura MI, et al. Efficacy of adoptive T-cell therapy is improved by treatment with the antioxidant N-acetyl cysteine, which limits activation-induced T-cell death. Cancer Res. (2016) 76:6006–16. doi: 10.1158/0008-5472.CAN-16-0587
119. Gattinoni L. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. (2005) 115:1616–26. doi: 10.1172/JCI24480
120. Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR, et al. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother. (2008) 31:742–51. doi: 10.1097/CJI.0b013e31818403d5
121. Hernandez-Chacon JA, Li Y, Wu RC, Bernatchez C, Wang Y, Weber JS, et al. Costimulation through the CD137/4-1BB pathway protects human melanoma tumor-infiltrating lymphocytes from activation-induced cell death and enhances antitumor effector function. J Immunother. (2011) 34:236–50. doi: 10.1097/CJI.0b013e318209e7ec
122. Chacon JA, Wu RC, Sukhumalchandra P, Molldrem JJ, Sarnaik A, Pilon-Thomas S, et al. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8+ melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One. (2013) 8:e60031. doi: 10.1371/journal.pone.0060031
123. Choi BK, Lee SC, Lee MJ, Kim YH, Kim Y-W, Ryu K-W, et al. 4-1BB-based isolation and expansion of CD8+ T cells specific for self-tumor and non-self-tumor antigens for adoptive T-cell therapy. J Immunother. (2014) 37:225–36. doi: 10.1097/CJI.0000000000000027
124. Sakellariou-Thompson D, Forget M-A, Creasy C, Bernard V, Zhao L, Kim YU, et al. 4-1BB agonist focuses CD8+ tumor-infiltrating T-cell growth into a distinct repertoire capable of tumor recognition in pancreatic cancer. Clin Cancer Res. (2017) 23:7263–75. doi: 10.1158/1078-0432.CCR-17-0831
125. Yang W, Lee K-W, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. (2019) 25:767–75. doi: 10.1038/s41591-019-0434-2
126. van der Lee DI, Reijmers RM, Honders MW, Hagedoorn RS, de Jong RC, Kester MG, et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. Nat Med. (2019) 129:774–85. doi: 10.1172/JCI97482
127. Inderberg EM, Wälchli S, Myhre MR, Trachsel S, Almåsbak H, Kvalheim G, et al. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology. (2017) 6:e1302631. doi: 10.1080/2162402X.2017.1302631
128. Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. (2018) 215:141–57. doi: 10.1084/jem.20171046
129. Wang QJ, Yu Z, Griffith K, Hanada K-I, Restifo NP, Yang JC. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol Res. (2016) 4:204–14. doi: 10.1158/2326-6066.CIR-15-0188
130. Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
131. Cobbold M, La Peña De H, Norris A, Polefrone JM, Qian J, English AM, et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci Transl Med. (2013) 5:203ra125. doi: 10.1126/scitranslmed.3006061
132. Abelin JG, Trantham PD, Penny SA, Patterson AM, Ward ST, Hildebrand WH, et al. Complementary IMAC enrichment methods for HLA-associated phosphopeptide identification by mass spectrometry. Nat Protoc. (2015) 10:1308–18. doi: 10.1038/nprot.2015.086
Keywords: cancer immunotherapy, adoptive cell therapy (ACT), tumor-infiltrating lymphocyte (TIL), neoantigens, enrichment
Citation: Bianchi V, Harari A and Coukos G (2020) Neoantigen-Specific Adoptive Cell Therapies for Cancer: Making T-Cell Products More Personal. Front. Immunol. 11:1215. doi: 10.3389/fimmu.2020.01215
Received: 31 March 2020; Accepted: 15 May 2020;
Published: 26 June 2020.
Edited by:
Nikolaos G. Sgourakis, University of California, Santa Cruz, United StatesReviewed by:
Maria Parkhurst, National Institutes of Health (NIH), United StatesGilles Marodon, INSERM U1135 Centre d'Immunologie et de Maladies Infectieuses, France
Copyright © 2020 Bianchi, Harari and Coukos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Bianchi, dmFsZW50aW5hLmJpYW5jaGlAY2h1di5jaA==
†These authors have contributed equally to this work