 Lijie Zhai1
Lijie Zhai1 April Bell1
April Bell1 Erik Ladomersky1Kristen L. Lauing1Lakshmi Bollu1Jeffrey A. Sosman2,3
Erik Ladomersky1Kristen L. Lauing1Lakshmi Bollu1Jeffrey A. Sosman2,3 Bin Zhang2,3,4
Bin Zhang2,3,4 Jennifer D. Wu3,4,5
Jennifer D. Wu3,4,5 Stephen D. Miller4,6Joshua J. Meeks3,5,7
Stephen D. Miller4,6Joshua J. Meeks3,5,7 Rimas V. Lukas3,8
Rimas V. Lukas3,8 Eugene Wyatt9,10Lynn Doglio9,10
Eugene Wyatt9,10Lynn Doglio9,10 Gary E. Schiltz3,9,11
Gary E. Schiltz3,9,11 Robert H. McCusker12
Robert H. McCusker12 Derek A. Wainwright1,2,3,4*
Derek A. Wainwright1,2,3,4*- 1Department of Neurological Surgery, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 2Division of Hematology and Oncology, Department of Medicine, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 3Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Chicago, IL, United States
- 4Department of Microbiology-Immunology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 5Department of Urology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 6Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 7Department of Biochemistry and Molecular Genetics, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 8Division of Neuro-Oncology, Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 9Department of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 10Transgenic and Targeted Mutagenesis Laboratory, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 11Center for Molecular Innovation and Drug Discovery, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 12Department of Animal Sciences, University of Illinois at Urbana-Champaign, Urbana, IL, United States
Indoleamine 2, 3-dioxygenase 1 (IDO; IDO1; INDO) is a rate-limiting enzyme that metabolizes the essential amino acid, tryptophan, into downstream kynurenines. Canonically, the metabolic depletion of tryptophan and/or the accumulation of kynurenine is the mechanism that defines how immunosuppressive IDO inhibits immune cell effector functions and/or facilitates T cell death. Non-canonically, IDO also suppresses immunity through non-enzymic effects. Since IDO targeting compounds predominantly aim to inhibit metabolic activity as evidenced across the numerous clinical trials currently evaluating safety/efficacy in patients with cancer, in addition to the recent disappointment of IDO enzyme inhibitor therapy during the phase III ECHO-301 trial, the issue of IDO non-enzyme effects have come to the forefront of mechanistic and therapeutic consideration(s). Here, we review enzyme-dependent and -independent IDO-mediated immunosuppression as it primarily relates to glioblastoma (GBM); the most common and aggressive primary brain tumor in adults. Our group's recent discovery that IDO levels increase in the brain parenchyma during advanced age and regardless of whether GBM is present, highlights an immunosuppressive synergy between aging-increased IDO activity in cells of the central nervous system that reside outside of the brain tumor but collaborate with GBM cell IDO activity inside of the tumor. Because of their potential value for the in vivo study of IDO, we also review current transgenic animal modeling systems while highlighting three new constructs recently created by our group. This work converges on the central premise that maximal immunotherapeutic efficacy in subjects with advanced cancer requires both IDO enzyme- and non-enzyme-neutralization, which is not adequately addressed by available IDO-targeting pharmacologic approaches at this time.
Introduction
Tumors arise from cell-intrinsic pro-growth mutations, the catastrophic dysfunction of host immune defense, and/or the evolution of tumor cell-intrinsic mechanisms that facilitate host immune system evasion. Amino acid metabolism is a fundamental biological event intrinsic to all living organisms that has long been recognized to play a crucial regulatory checkpoint, resulting in the sculpting of cellular responses during pathophysiological conditions such as infectious disease, autoimmunity, neurodegeneration, psychiatric conditions, as well as cancer (1). Over the past two decades, the kynurenine (Kyn) pathway of tryptophan (Trp) metabolism (Figure 1) has been intensely studied throughout both preclinical and clinical therapeutic settings (2). The majority of this work has focused on the two primary rate-limiting enzymes that catalyze the first step of Trp metabolism: indoleamine 2, 3-dioxygenase 1 (IDO) and tryptophan dioxygenase (TDO). The central dogma underlying the immunosuppressive role for these enzymes is associated with their canonical Trp catabolic properties: IDO and/or TDO-mediated depletion of Trp and/or the accumulation of Kyn, which is associated with the suppression of immune effector cells and the upregulation, activation, and/or induction of tolerogenic immune cells (3, 4). Although a plethora of in vitro-motivated investigation has generated compelling evidence confirming IDO enzyme activity as a source of therapeutic value, data challenging the Trp depletion dogma as the only mechanism by which IDO suppresses the immune response began emerging almost a decade ago. Furthermore, the phase III Keynote-252/ECHO-301 clinical trial that evaluated combination treatment with the potent IDO enzyme inhibitor, epacadostat, and pembrolizumab [anti-programmed cell death 1 (PD-1)] in patients with unresectable or metastatic melanoma, failed to meet its primary end point as compared to individuals treated with pembrolizumab as a monotherapy (5). Despite certain variables that include objective in vivo efficacy and the underlying rationale for this combination of therapy which may have contributed to its clinical failure (6), a careful consideration for IDO-targeting approaches is warranted. Additional conflicting results that describe the role of IDO across different cancers and the cell types expressing the immunosuppressive mediator highlight the various underlying mechanisms that are context-dependent, multi-dimensional, and temporally-sensitive.
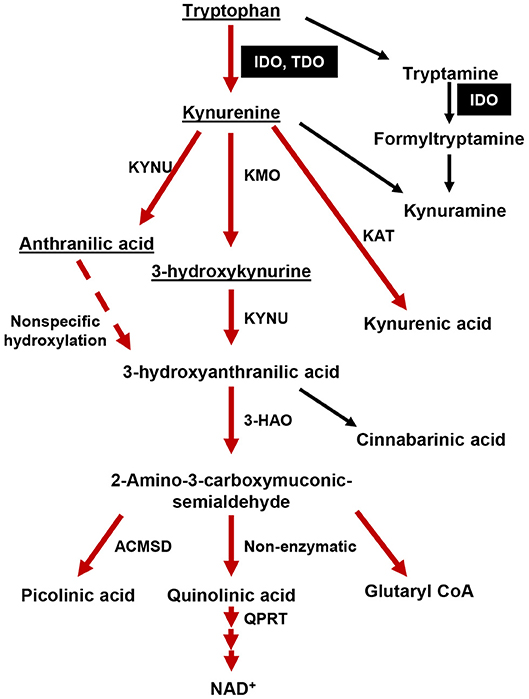
Figure 1. The tryptophan (Trp)→ kynurenine (Kyn) metabolic pathway. The majority of dietary tryptophan (95%) is metabolized via the TrpKyn pathway (red arrows). A minor pathway that converts tryptamine into kynuramine is also included. Metabolites capable of crossing the blood brain barrier (BBB) are underlined. IDO and TDO are highlighted in black boxes. KMO, kynurenine 3-monooxygenase; KYNU, kynureninase; KAT, kynurenine aminotransferase (I, II, III); 3-HAO, 3-hydroxyanthranilate 3,4-dioxygenase; ACMSD: 2-amino-3-carboxymuconate semialdehyde carboxylase; QPRT, quinolinic-acid phosphoribosyl transferase.
Here, we summarize current knowledge of IDO-mediated immunomodulation with a focus on how it affects the anti-cancer immune response. Potential mechanism(s) that reshape and/or revise current IDO dogma as it relates to the cancer immunity cycle are also explored. Recent advances in our understanding of IDO expression changes during aging and the potential contribution of these effects on suppressing immunosurveillance mechanisms during cancer cell initiation and/or tumor cell outgrowth are also discussed. Finally, the diametrically-opposed relationship between intratumoral IDO levels and overall survival among different types of cancer patients will provide a unique perspective on how cancer immunity dogma is not universally applicable.
IDO, Trp Metabolism and Its Association With Suppressing the Anti-cancer Immune Response
Less than 1% of dietary Trp is used for protein synthesis under physiological conditions while the remainder is degraded through decarboxylation, transamination, hydroxylation, or oxidation (7), which leads to the generation of physiologically active compounds including neuroactive tryptamine, neuroprotective melatonin, and/or immunomodulatory kynurenines. IDO and TDO catalyze the rate-limiting cleavage of the Trp indole ring 2, 3-double bond and incorporate molecular oxygen. The product of this reaction is N-formylkynurenine (NFK) that is rapidly converted into Kyn by formamidase. The latter catabolite is further transformed into downstream kynurenine-like intermediates, including 3-hydroxy-L-kynurenine (3-HK), 3-hydroxyanthranilate (3-HAA) and quinolinic acid (Quin), which also affect the immune response (8). The ultimate product of Kyn pathway metabolism is nicotinamide adenine dinucleotide (NAD+), a critical enzyme co-factor involved in a variety of fundamental biological events including acting as an electron transfer factor during glycolysis and oxidative phosphorylation, as well as being consumed by covalent modification of proteins and signaling enzymes. While IDO and TDO share similar functions as a mediator of Trp degradation, their biochemical properties are quite distinct. Monomeric IDO can act on a broad range of substrates that include L-Trp, D-Trp, tryptamine, 5-hydroxytryptophan and 5-hydroxytryptamine. In contrast, homotetrameric TDO is enantiomer specific and only metabolizes L-Trp (9). The affinity of IDO for Trp and Trp-derivative substrates is highly variable such that binding to D-Trp is low as compared to L-Trp, with a Km of human IDO for the D-isomer that is 200-fold higher than that for the L-isomer (10, 11). In contrast to IDO and TDO, the third Trp catabolic enzyme, IDO2, possesses a negligible level of intrinsic enzymic potential. The Km of human IDO and IDO2 for L-Trp is 20.90 ± 3.95 μmol/L and 6,809 ± 917 μmol/L, respectively (12) supporting the hypothesis that suggests IDO2 contributes a minimal role to overall Trp metabolism in humans. The lack of change in absolute Kyn among cell lines expressing high IDO2 cDNA levels, as well as the negligible change in systemic Kyn levels in mice with constitutional IDO2 deficiency (13–15), further support this hypothesis.
In addition to the distinct metabolic properties associated with Trp metabolism, the factors regulating IDO gene transcription and translation are different as compared to those that regulate TDO and IDO2 expression. Pro-inflammatory mediators tend to be the strongest agonists of IDO transcription that include the prototypical antitumor-associated T cell effector cytokine, interferon-gamma (IFN-γ), as well as the toll-like receptor 9 (TLR9) and TLR4 agonists, CpG DNA and lipopolysaccharide (LPS), respectively (16–19). Tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6) and IL-1β also synergize with one another to facilitate the upregulation of IDO expression. Other modulators include soluble glucocorticoid-induced TNFR-related protein (GITR), prostaglandin E2, the oncogene c-Kit, as well as the tumor suppressor, Bin1 (20). The soluble fusion protein, cytotoxic T lymphocyte-associated protein 4-immunoglobulin (CTLA-4-Ig) induces IDO expression in dendritic cells (DCs) through the interaction of co-stimulatory receptors, CD80 (B7.1) and CD86 (B7.2) (21). Recent work further confirms that Wnt5α is another enhancer of IDO activity during β-catenin signaling in DCs (22).
Constitutive IDO expression can be found in healthy human subjects but is highly restricted to a select number of tissues and cells including mature DCs in secondary lymphoid organs, epithelial cells inside the female genital tract, as well as endothelial cells of term placenta and lung parenchyma (23). It should be noted that this superficial analysis does not capture the potential expression changes that occur across age groups as we previously found in the normal human brain parenchyma during advanced age (24). While some human tumor cell types constitutively express IDO, its presence is normal absent in human and mouse glioblastoma cells but is rapidly induced upon treatment with human and mouse IFNγ (25–29). Constitutive TDO expression is also expressed among different organs of healthy human subjects including bone marrow, muscle, gastrointestinal tract, kidney and urinary bladder, as well as inside the brain, with the highest levels in liver hepatocytes (30). TDO expression levels are regulated in-part by systemic levels of L-Trp and corticosteroids (31, 32). Notably, common corticosteroid-like treatments such as dexamethasone rapidly increases TDO levels across multiple cell types. Similar to IDO, TDO is expressed by a bevy of different tumor types including glioblastoma, melanoma, ovarian carcinoma, hepatic carcinoma, breast cancer, non-small-cell lung cancer, lymphoid and myeloid cancers, renal cell carcinoma, and bladder cancer, with high expression levels associated with a more rapid rate of tumor progression and/or decreased patient survival (33–36). IDO2 mRNA is constitutively expressed in the human liver, brain, thyroid, placenta, endometrium, and testis, and is induced in antigen-presenting cells (APCs) and B cells (37). However, the role of IDO2 in affecting the anti-cancer immune response is less clear with the analysis of 129 human tumor samples and 25 human tumor cell lines showing no detectable full-length IDO2 mRNA transcript (36).
The primary dogma establishing a relationship between IDO, TDO, IDO2 and immunosuppression is associated with their individual and/or collective contribution toward the metabolism of Trp. The premise for this dogma is based on the Trp starvation theory that postulates the near absolute depletion of Trp, at or below <1 μM, facilitates the accumulation of uncharged tRNAs which then activate the general control non de-repressible 2 (GCN2) kinase pathway and the dysfunction of T cells (38). In vitro studies support the hypothesis that Trp depletion inhibits the master metabolic regulator mammalian target of rapamycin (mTOR) and protein kinase C (PKC-Θ) in cancer cells, which consequently enhances autophagy and Treg development, respectively (39). Trp degradation may also suppress immune cell activities through the formation of Kyn and downstream derivative metabolites. In vitro and further requiring co-treatment with transforming growth factor-beta (TGF-β), Kyn facilitates the induction of FoxP3 in naïve CD4+ T cells by activating the aryl hydrocarbon receptor (AhR) (40), a ligand-activated transcription factor that exerts potent effects on immune cells (41) and is involved in the differentiation of inducible Tregs (42, 43). The downstream pathway Kyn metabolites including kynurenic acid (KA) (44), xanthurenic acid (XA) (35), and cinnabarinic acid (CA) (45) interact with AhR and may also play a role in modulating the immune response. In striking contrast, Trp catabolites have been demonstrated to also induce CD4+ T cell apoptosis. Terness et al. found that Kyn, 3-HK, and 3-HAA suppress T cell proliferation coincident with the induction of apoptosis (46). This finding was independently confirmed by Fallarino et al. (47) demonstrating in vitro that, Kyns induce the selective apoptosis of murine thymocytes and Th1-, but not Th2-cells. This immunoregulatory role of Kyns on different lymphocyte subsets may be important for maintaining peripheral lymphocyte homeostasis and for minimizing the accumulation of autoreactive and/or inflammatory lymphocytes. However, the exact molecular mechanism of Kyn-mediated selective apoptosis on T cell subsets remains unknown.
Despite a literature replete with previous work supporting the hypothesis that IDO-mediated Trp metabolism enhances suppression of the anti-cancer immune response, it's important to acknowledge that the predominant source for which these studies are based upon is derived from in vitro cell culture. Since Trp is an essential amino acid and not readily generated by mammalian cells, dietary consumption is critical for host survival and in vivo replenishment. It is therefore important to consider that the Trp concentrations required to inhibit T cell proliferation in vitro have been shown to be below 0.5–1 μM (38). Whether this cell culture condition can be transposed upon physiological conditions in vivo is controversial since human plasma Trp levels normally range between 50 and 100 μM. A number of conflicting results on this topic have recently arisen and was highlighted by Frumento et al. during their failure to observe an inhibition of T lymphocyte proliferation when the in vitro cell culture media was absent for Trp (48). Although it is arguable that high expression of IDO (or TDO) in tumor cells might lead to extremely low Trp levels within the tumor microenvironment, it's very difficult to fully validate this argument in vivo. Using the matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) imaging technique, Sonner et al. (49) recently reported on TDO-mediated immunosuppression in experimental murine melanoma and demonstrated that, even in B16 melanoma highly expressing TDO by virtue of a cDNA-expressing plasmid, the intratumoral Trp levels were maintained at a similar level as compared to those inside unmodified B16 melanoma; thus suggesting that in vivo Trp levels are very stable even when Trp metabolism is maintained at an abnormally high rate.
Another hypothetical explanation regarding Trp depletion relies on the alteration of Trp transporter activity. This theory posits that even while there is no dramatic decrease of Trp in the peripheral blood circulation, alteration of the Trp transporter activity may cause intracellular Trp depletion which further triggers downstream immunosuppressive signals. In addition to the well-known non-specific low affinity Trp transporter (system L) (50), two Trp specific transporters with high affinity have been reported to be expressed on monocyte-derived macrophages and tumor cell lines (51, 52). Unfortunately, neither of these novel Trp transporters has been successfully cloned. Comprehensive functional analyses of the Trp transporters expressed by different immune cells and/or tumor cells in vitro and in vivo remain largely unexplored.
Intriguingly, T cell specific GCN2 knockout mice have similar anti-tumor effect against syngeneic B16 melanoma as compared to their wild type counterparts (49). Moreover, despite the elegant in vitro work by Mezrich et al. reporting that the 50 μM Kyn treatment activates AhR in naïve T cells and subsequently induces FoxP3 expression (40), human serological Kyn levels are normally within the range of 2–3 μM in both healthy individuals and in patients with inflammatory conditions (53–57). Theoretically, local tissue Kyn levels might transiently increase when IDO expression is highly expressed (58), but more precise in vivo detection and quantification techniques remain critically unavailable. Moreover, whether Kyn possesses an inducible or inhibitory effect on DC-mediated Treg differentiation is also debatable. Somewhat conflicting with the report by Mezrich et al., Nguyen et al. reported that the addition of Kyn in a DC-T cell co-culture system led to the inhibition of naive T cell differentiation into FoxP3+ Tregs (59). This latter observation supports the hypothesis that the Kyn-AhR mechanism is likely not responsible for the direct generation of Tregs in solid tumors; especially since the infiltrating component of tumors tend to primarily consist of thymus-derived natural Treg (60, 61) that fail to express Ahr (62). It is also notable that other studies indicate the potential role of Kyn on Treg activation as evidenced by the Munn and Mellor group who showed IDO-expressing plasmacytoid DCs in tumor draining lymph nodes preferentially activate pre-existing Treg through the PD-1-PTEN pathway which was inhibited by the IDO pathway non-enzyme inhibitor, 1-methyl-D-tryptophan (1-MT) (63, 64). In addition to the sometimes conflicting outcomes discussed above, results from studies suggesting that Kyns directly suppress the actions of effector T cells have also been revisited due to the high doses evaluated during those studies, which ranged from 100 to 1,000 μM (46, 47). The panoply of different observations across various modeling systems highlight the complexity of IDO immunobiology that, may be conveying artifactual effects during in vitro study and thereby supports the re-evaluation of previous interpretations with the use of new physiologically-relevant in vivo modeling systems.
Pleiotropic IDO Effects Reflect Multiple Functions, Diverse Cellular Origins, and Varying Kinetics
Beyond the restricted tissues that constitutively express IDO is the induction of its expression in a variety of cell types under normal and pathological conditions. This includes the changes of IDO expression during progressive aging (24), as well as in the setting of malignancy whereby levels change in response to T cell infiltration (65). Further complicating IDO functionality has been highlighted by immunohistochemical IDO detection across 15 different human cancer types that found protein localization in myeloid cells, endothelial cells, tumor cells, or a combination of those origins (23). The select pattern of intratumoral IDO expression raises critical questions including: (i) does IDO perform the same role among different cell types; (ii) how do these cells coordinately contribute to tumor growth and/or suppression of anti-cancer immunity; (iii) what are the kinetics of endogenous IDO expression and its relationship to enzyme activity?
A peculiar trait of tumor cells expressing high IDO expression is the resultant effect on an overall slower rate of growth as compared to cells with lower intrinsic IDO levels (66). A derivative and competing hypothesis arising from this observation suggests that IDO functions as a tumor suppressor. This was corroborated with findings of higher intratumoral IDO expression positively associated with longer survival in renal cell carcinoma-, hepatocellular carcinoma-, and melanoma-patients (3, 67–69). It should also be considered, however, that higher levels of T cell infiltration are often associated with higher intratumoral IDO levels and this relationship has an established survival improvement in human subjects diagnosed with select malignancies (3, 70). Therefore, the higher intratumoral IDO levels are likely associative and may not have any effect on tumor growth itself. Regardless and in contrast to the alleged beneficial effects of slower growth, higher IDO expression was associated with an improved motility of lung cancer cells that enhanced their metastatic formation in the brain, liver, and bone (71), whereas IDO deficiency decreased metastasic burden and improved the survival of subjects with breast carcinoma-derived pulmonary metastases (72, 73). Intratumoral IDO expression also correlated with the frequency of liver metastases in colorectal cancer (74), distant metastases in hepatocellular cancer (69), and nodal metastases in endometrial carcinoma (75). Among human cancer cells, IDO activity has been implicated in improving DNA repair and mediating the resistance to treatments such as the poly-ADP-ribosyltransferase (PARP) inhibitor, olaparib, γ-radiation, and the chemotherapeutic agent, cisplatin, through generation of NAD+ (76). It was reported that impaired Trp metabolism resulted in the inhibition of de novo NAD+ synthesis, which led to hepatic tumorigenesis through DNA damage (77), further supporting the previous finding that NAD+ serves as the only endogenous substrate of PARPs to facilitate the removal of oxidative DNA damage. However, it's important to note that IDO is not normally expressed in the liver, whereas, TDO is constitutively and highly expressed. Therefore, if either or both of these factors are associated with the impaired Trp metabolism, it may be associated with pathological injury and not necessarily the normal expression of those rate-limiting enzymes.
While IDO expression by tumor and immune cells receives the predominant attention, the immunosuppressive mediator is also expressed by endothelial cells (23). In healthy tissues, IDO is expressed by endothelial cells in a large proportion of placental and pulmonary blood vessels, as well as by a minority of blood vessels in select other organs. Intratumorally, vascular IDO expression is frequent among renal cell carcinoma, non-small lung carcinoma, endometrial carcinoma, and melanoma. Endothelial IDO appears to modulate vascular tone through several different signaling pathways including Kyn-mediated activation of soluble guanylate cyclase, adenylate cyclase, and voltage-dependent K+ channels (78, 79). The Kyn metabolite, xanthurenic acid, has been shown to possess greater potency as compared to L-Kyn in causing blood vessel relaxation that is dependent on nitric oxide (80). Stanley et al. found that in the presence of H2O2, IDO catalyzes Trp oxidation using singlet molecular oxygen (1O2), leading to the vasodilation product cis-WOOH (81). It's important to note that these experiments were conducted while utilizing high concentrations of the Trp catabolites between 300 μM to 1.5 mM in vitro. It's therefore unclear as to whether the vasodilating compounds attain a similar local level inside a solid tumor in vivo. However, other observations support the vaso-relaxing effects of endothelial IDO including a model of sepsis-induced hypotension (82). Intrinsic to the IDO expressed by endothelial cells there may also be a role for an effect on modulating tumor neovascularization (83), though the exact molecular mechanism explaining has yet to be elucidated (84).
Data suggesting that IDO possesses non-metabolic functions independent from those associated with its enzyme activity began emerging ~10 years ago. Pallotta et al. was the first to report that the in vitro treatment of TGF-β in cultures of mouse plasmacytoid DCs (pDCs) leads to the phosphorylation of IDO immunoreceptor tyrosine-based inhibitory motifs (ITIMs), which subsequently recruits and activates the tyrosine phosphatases, SHP-1 and SHP-2, as well as inositol polyphosphatase (SHIP) (85). Activation of the pDC TGF-β-IDO-SHP axis enabled an autocrine loop through the induction of non-canonical NF-κB signaling, which further enhanced intra-pDC IDO expression levels. Treatment of the pDCs with 1-methyl-D, L-tryptophan confirmed that the immunosuppressive IDO signaling mechanism was faithfully independent of its association with mediating Trp metabolism. However, it's notable that the 4 μM dose of 1-MT used in this study was lower than the EC50 of 1-MT (13). Future validation and replication of these results while utilizing a more potent IDO enzyme inhibitor will allow for confidence and verification of non-enzymic IDO functions. Our group found similar observations of IDO mediating both enzyme and non-enzyme effects that depend on context. Using a syngeneic brain tumor model, we previously demonstrated that the shRNA knockdown of GBM cell IDO leads to the suppression of intratumoral Treg accumulation and was associated with a significant improvement in long-term animal subject survival (86) independent of GBM cell IDO metabolism (29). We also showed that the forced expression of GBM cell IDO cDNA (IDO-O/E) enhances Treg recruitment even when animal subjects are treated with a potent blood brain barrier-penetrating pharmacologic IDO enzyme inhibitor (87). We further confirmed that while the IDO enzyme inhibitor significantly decreased intratumoral Kyn levels in GBM IDO-O/E in vivo, the reduction was not associated with decreased intratumoral Treg accumulation. Taken together, these observations support the hypothesis that new IDO-targeting approaches aimed at simultaneously reversing enzyme and non-enzymic activities will enhance the effectiveness of future cancer immunotherapy efforts.
IDO and Its Relationship to Host Age
Considering the suppressive role of the IDO/TDO axis in immunoregulation, as well as the changes in immunological status system during progressive aging, it's not surprising that several studies have investigated the relationship between the TrpKyn pathway as it relates to aging-dependent disease. The serological Kyn/Trp ratio is often used an indicator of IDO enzyme activity and progressively increases during normal aging in humans (88, 89). This increase has been associated with enhanced frailty in human subjects >65 years of age and predicts an increased mortality rate of individuals in their nineties (90). Additionally, meta-analysis of age-related gene expression changes in the peripheral blood of adult individuals identified the enzyme kynureninase (KYNU, Figure 1) as one of the most differentially expressed genes (91). In a large population screening analysis that included 7,074 human subjects that focused on studying bone mineralization, Apalset et al. reported that the serological Kyn/Trp ratio negatively correlated with bone mineral density in the 71–74 year old age group as compared to younger individuals that were 46–49 years of age (92). More recently, Ocampo et al. summarized the results from previous animal studies of brain diseases and the interaction with Kyns during aging that indicate a strong correlation between Kyn pathway metabolites across different rodent ages (93). They found an intriguing observation that while TDO and IDO expression decrease in the liver and kidney during progressive aging, TDO and IDO were dichotomized in the rat brain during advanced age such that TDO decreased and IDO increased in overall expression as compared to young animal subjects. Similarly, our group previously demonstrated that there is an ~400 fold increase in IDO mRNA expression in the normal naïve brain of 72–74 week old C57BL/6 WT mice as compared to 6–8 week old subjects (94). Our most recent study evaluating the interactions between normal human aging and its relationship to brain cancer incidence and mortality also found a significant increase in IDO mRNA expression in the normal human brain of individuals aged 60–69 years of age as compared to younger human subjects (24). The increased human brain IDO expression was associated with a maximal Treg/CD8+ cytolytic T cell ratio in the peripheral blood of the 60–69 year old age group confirming a simultaneous increase of systemic- and local-immune suppression (24). To address these observations, we are currently in the process of determining as to whether the advanced age-dependent increase of central nervous system IDO expression directly increases the peripheral Treg/CD8+ cytolytic T cell ratio and how this relationship affects the incidence and mortality rate, as well as responsiveness to immunotherapy of GBM.
Understanding how aging-dependent molecular mechanisms affect IDO enzyme and non-enzymic activity is in its infancy. Refaey et al. reported that 22-month old mice have significantly higher serum N-formylkynurenine as compared to younger counterparts at 4 and 13 months of age, suggesting a potential role for Kyn pathway metabolism in aging-dependent bone loss (95). Strikingly, either the in vitro addition of Kyn to bone marrow-derived mesenchymal stromal cells (BMSCs), dietary supplementation of Kyn, or a direct administration of Kyn into the peritoneum suppressed the formation of new bone; possibly by decreasing osteoblast formation, failed recruitment capability and/or loss of effector functions. It should be noted, however, that neither age-matched IDOKO nor TDOKO mice were used in the study and it's therefore inconclusive as to the primary source of aging-dependent changes in Kyn levels. Independently, Minhas et al. demonstrated that the catabolic enzyme converting quinolinic acid into NAD+, quinolate phosphoribosyltransferase (QPRT), significantly declines in aged human monocyte-derived macrophages (MDMs) from individuals ≥65 years old as compared to MDMs isolated from individuals ≤ 35 years of age (96). Loss of QPRT subsequently resulted in decreased de novo NAD+ synthesis and was associated with an enhanced pro-inflammatory status and lower phagocytic ability of MDMs. It's worth noting that in addition to the enzymes and metabolites of the Kyn pathway, changes in AhR activity and/or expression have also been implicated during aging (97). However, the direct relationship between IDO, TDO, Kyn, and AhR has not been comprehensively established during a combined investigation of aging and in a specific pathophysiological setting.
Based on the relationship between increased incidence of patients with cancer and advanced age (98), it may be surprising that few studies have investigated IDO and/or TDO changes in subjects with cancer and across the health-/life-span. Adult glioblastoma is an age-related disease with a median age of diagnosis at 65 years old (99). To explore the mechanistic underpinnings between advanced age and its effects on the GBM immune response, we previously compared the survival rate of young 6–8 week- and adult 72–74 week-old mice intracranially-engrafted with the syngeneic GL261 glioma cell line. The young mice survived slightly longer than the older subjects with a median overall survival (mOS) of 27.5 days and 21.5 days, respectively (p = 0.0292) (94). Intriguingly, advanced age was associated with a large increase of IDO expression in the contralateral brain without tumor and as compared to young mice, whereas there was no such difference within the tumor itself. These data suggest that the majority of brain parenchymal cells expressing increased IDO during advanced age do not migrate into the GBM. Intriguingly, serological, and intra-GBM Trp and Kyn levels showed no differences between young and adult subjects despite the increased IDO expression in the older brain (94). We also studied the effects of advanced age on immunotherapeutic efficacy in adult mice at 72 weeks of age, which is closer to the analogous time frame of a human GBM patient diagnosis as compared to the standard 6–12 week old mouse. When the young and adult mice underwent therapy (87) with whole brain radiotherapy, anti-PD-1 mAb and pharmacological IDO enzyme inhibitor, older mice showed higher IDO mRNA in the non-tumor brain tissues as compared to younger subjects. Functionally, the triple combination therapy resulted in a median survival of 31.5 days in the older subjects, which was significantly decreased as compared to the younger 8-week-old mice with a survival of 40.5 days (p < 0.001), suggesting that the increased brain IDO expression during advanced age has a directly negative effect on immunotherapeutic efficacy in subjects with GBM.
Mouse Models for Studying IDO
Among preclinical studies, transgenic mouse models have demonstrated their substantial importance to understanding the function, effects and mechanism of IDO in a physiologically-relevant environment. The first IDO knockout (IDOKO) mouse strain originated from the Munn and Mellor group whereby exons 3–5 of IDO were replaced with a β-gal and neomycin cassette (100, 101). Surprisingly, the homozygous IDOKO mice are viable, fertile and possess a phenotypically normal immune system without any obvious graft vs. host-like symptoms, suggesting that IDO-mediated immune suppression is dispensable for the normal maintenance of central and peripheral tolerance to self-antigens. Given the functional similarity between TDO and IDO for mediating Trp catabolism, it's possible that TDO plays a compensatory role in the absence of IDO. However, it's notable that Kyn levels are significantly decreased in naïve IDOKO mice, so if TDO compensates, there appears to be a limit to those potential effects (29). The expression and function of TDO has yet to be investigated in IDOKO mice with cancer. Though IDO expression is not required for immune tolerance under homeostatic conditions, it plays a critical role during acquisition of tumor-immune tolerance as indicated by studies demonstrating that IDOKO mice have increased tumor infiltration of effector T lymphocytes, decreased immunosuppressive immune cells, as well as increased survival (102–105), implicating non-redundant functions between IDO and TDO in the various cancer settings.
Because of their normal growth and immune system development, constitutional IDOKO mice are widely used preclinically. However, this transgenic model does not reveal the role of IDO in different tissues or cell types. Recently, Bishnupuri et al. generated an intestinal epithelium specific IDO knockout mouse strain by breeding Ido1tm1c(EUCOMM)Wtsi mice with the villin promoter Cre mice (105). The mouse colon cancer model utilizing this conditional IDO knockout mouse strain discovered neoplastic colon epithelium cell IDO to be a critical constituent of colon tumorigenesis and that Kyn metabolites rapidly activated PI3K-Akt signaling in the neoplastic epithelium to promote cellular proliferation and resistance to apoptosis. In addition to the constitutional and conditional IDO knockout mice that have previously been reported, our group has created new constructs that will allow for addressing novel questions including an IDO reporter mouse that expresses a IDO-GFP (Figure 2A), an IDO knock-in mouse under tissue-specific Cre using Rosa26Sor-loxp-IDO-FLAG-GFP (Figure 2B), and an IDO enzyme nullified mouse whereby the histidine of the 350th amino acid is substituted to an alanine (Figure 2C). We are currently characterizing each of these recently created models and will publish data supporting their phenotypes in the near future.
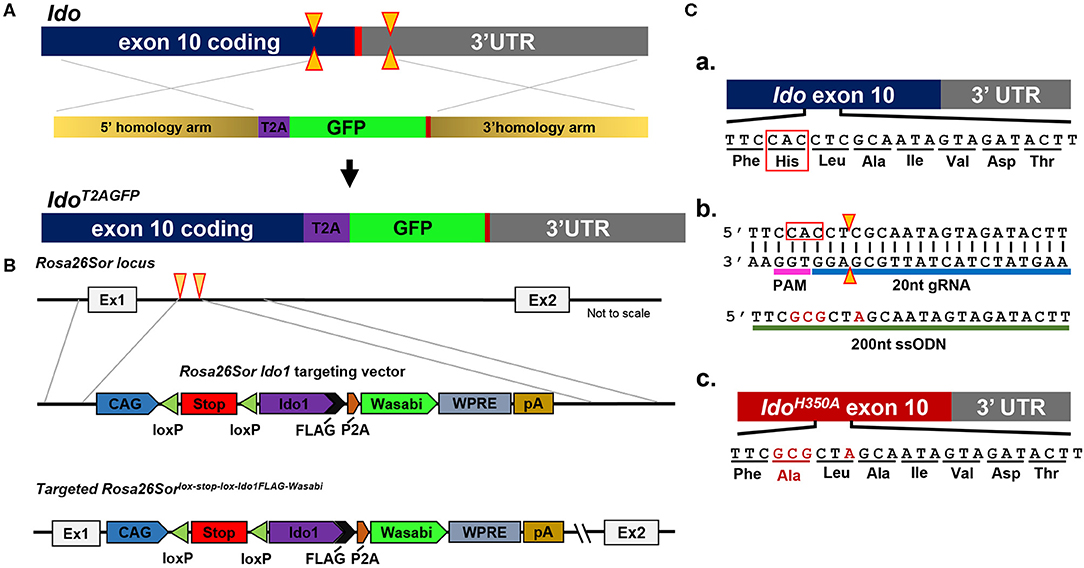
Figure 2. Schema utilized to generate IDO-targeted transgenic mouse strains. (A) Generation of the Ido1T2AGFP mouse line. CRISPR/Cas9 technology was employed in murine embryonic stem (ES) cells. Two separate guide RNAs directed Cas9 nuclease cleavage of the Ido1 locus, creating double strand DNA breaks (yellow arrow) before and after the stop codon (red bar). The CRISPR ES cell reaction included a plasmid repair template, which encoded the T2A and GFP elements flanked by 5′ and 3′ homology arms of 700nt (tan bars). The T2A and GFP elements were introduced into the endogenous Ido1 locus before the stop codon via homology directed repair (HDR). (B) Introduction of a Cre-dependent Ido1 expression construct into the Rosa26Sor locus by CRISPR/Cas9 technology. Two separate guide RNAs directed Cas9 nuclease cleavage at the Rosa26Sor locus, creating double strand DNA breaks (yellow arrows). A plasmid repair temple (Rosa26Sor Ido1 targeting vector) was included in the reaction and introduced via HDR. The edited locus encodes the CAG promoter driven, Cre-dependent conditional expression of a bi-cistronic Ido1 construct. The Ido1 transcript includes a C-terminal FLAG-tag, followed by a P2A element and expression of the Wasabi fluorophore. It also included a WPRE element and the bovine growth hormone polyadenylation (pA) signal. The construct is flanked by 800 bp 5′ and 750 bp 3′ homology arms (gray lines). (C) Generation of the Ido1H350A mouse line. (a) Schematic showing the last coding exon of the Ido1 gene, exon 10. The highlighted region encodes a histidine residue at position 350 (red box) that is essential for mouse IDO enzyme activity; (b) CRISPR/Cas9 technology generated a precise double strand DNA break in the Ido1 exon 10 locus in fertilized mouse embryos (yellow triangle). A 200nt single stranded oligodeoxynucleotide repair template was included in the reaction (green bar). The ssODN introduced point mutations (red letters) into the endogenous Ido1 locus via HDR. These point mutations altered the endogenous coding sequence from CAC→GCG, which changed the amino acid reside from histidine to alanine, H350A (red letters). An additional silent mutation (C→A) was included to add a restriction site for genotype analysis (red letter); (c) Schematic showing the mutated Ido1 exon 10 locus encoding H350A in red.
The Expression, Immunosuppression, and Targetability of IDO in Subjects With GBM
Tumors arising from glia within the central nervous system (CNS) are considered to be potently immunosuppressive due to their surrounding immunospecialized neuroanatomical landscape (106). GBM is as an immunologically “cold” malignancy and associated with an immune system microenvironment that has resulted in the failure of all phase III clinical trials evaluating immunotherapy in patients with GBM to-date. Considering the work by our group and others in repeatedly demonstrating the remarkable pathogenic influence of IDO in subjects with GBM, the elucidation of its role and multi-variate functions may provide a path for enhancing the effectiveness of cancer immunotherapy against malignant glioma in the future.
IDO mRNA is highly expressed in ≥90% of GBM patients presenting with wild-type isocitrate dehydrogenase (wtIDH) and while not normally expressed, is inducible among a majority of human GBM cell lines after exposure to proinflammatory cytokines (23, 66, 107–110). While TDO expression is expressed at an even higher mRNA level than IDO in patient-resected GBM (33, 109–111), IDO2 levels are negligible or undetectable at the mRNA level (65), despite an IHC-focused study suggesting high IDO2 protein levels in GBM (n = 52) (109); the latter of which likely reflects conclusions based on non-specific antibody immunostaining. The cognate receptor for Kyn is AhR and is present throughout all grades of glioma, with the highest level reported in GBM. A recent study screening 75 glioma tissue samples by immunohistochemistry also confirmed higher IDO and TDO expression while low/no expression of IDO2 at protein levels (112). Studies from our group and others' have independently demonstrated that both Trp and Kyn are significantly decreased in GBM patient plasma as compared to human subjects without a tumor of the CNS (56, 57). Strikingly, in contrast to the absolute levels of metabolites, the Kyn/Trp ratio is significantly increased in GBM patient plasma long after surgical resection of the tumor which may reflect a treatment-related effect rather than due to the malignancy itself. A recent study by Kesarwani et al. provided additional insights into metabolomic profiling of newly diagnosed GBM (n = 80) and lower grade gliomas (LGG, n = 28) demonstrating that, GBM patients have increased intratumoral Trp and Kyn levels, but decreased KA as compared to LGG patients (110). Interestingly, Trp and Kyn accumulation was specific to classical and mesenchymal GBM subtypes, whereas KA accumulation was only evident in the proneural subtype. It's not clear why the systemic Kyn decreases while intratumoral Kyn increases in human subjects with GBM, which requires further investigation for both prognostic and therapeutic purposes.
Mechanistic studies investigating the full range of IDO-mediated immune modulation have primarily focused on the Kyn-AhR-Treg-MDSC (myeloid-derived suppressor cell) axis with data from several studies implying that alternative mechanisms of IDO behavior can occur. The inhibition of IDO enzyme activity in cultured human astroglioma cell lines decreases de novo synthesis of NAD+, which is associated with decreased tumor growth (113). The triple combination of IDO pathway inhibitor with chemo-radiation achieved better overall survival as compared to the dual combination of chemo-radiation (55). Interestingly, the survival benefit was abrogated in mice deficient for complement C3, potentially suggesting that IDO activity conveys immune evasion properties to tumor cells by suppressing complement activation (55). Finally, human GBM cell lines overexpress a translesion DNA polymerase, hpol κ, which helps to restore genome stability. Inhibition of AhR or the blockade of TDO enzyme activity decreases hpol κ expression, indicating that TDO contributes to tumor cell survival by supporting genome stability (114).
Different therapeutic interventions have profoundly different effects on both the systemic and local tumor-immune environment which include the alteration of IDO levels. In two recent glioma studies, both PCC0208009 (PCC), a potent IDO enzyme inhibitor with an IC50 of 4.52 nM, and RY103, an IDO-TDO dual inhibitor, have demonstrated suppression of tumor cell line- and intra-tumoral-IDO expression, respectively (112, 115). In contrary to the downregulation of IDO, a recent phase I clinical trial evaluating epidermal growth factor receptor variant III (EGFRvIII)-targeted chimeric antigen receptor (CAR) T cell therapy in GBM patients demonstrated a dramatic induction of intratumoral IDO expression after the adoptive transfer of CAR T cells (116). At almost the same time as when this clinical observation was reported, our group published a similar T cell-inducing IDO effect in humanized mice with intracranial human GBM and reconstituted with human immune cells (65). While the IDO-promoting effects of intratumoral T cell-infiltration appears to extend across human cancer types (3, 4), the association between IDO expression and overall survival depends on the type of tumor under investigation. This likely reflects the different composition of IDO expressing cells within tumors, the different function of intratumoral IDO that depends on the cell of origin, as well as for IDO expressing cells outside of the tumor microenvironment that also possess anti-cancer mechanisms depending on anatomical context. It remains notable that, to effectively target IDO with an enzyme inhibitor, the treatment must include an approach that yields robust inflammation such as irradiation. Accordingly, while studying the syngeneic GL261 in young C57BL/6 mice, we previously found that while neither radiation, anti-PD-1 mAb, nor IDO enzyme inhibitor treatment improved long-term survival as single- or dual-agent approaches, the simultaneous combination of all three modalities led to a remarkable synergistic durable survival improvement (87). Presumably, this was due to the induction of IDO by radiation, the neutralization of IDO enzyme activity by pharmacologic neutralization, and the enhancement of the anti-tumor immune response with PD-1 blockade. It's notable, however, that this triple combination was significantly less effective in older animal subjects when IDO expression is increased in the brain independent of tumor burden. As a means to explain this observation, we are currently investigating the working hypothesis that dendritic cells accumulate in the brain during advanced age (117, 118), express IDO and suppress immunotherapeutic efficacy (Figures 3A,B).
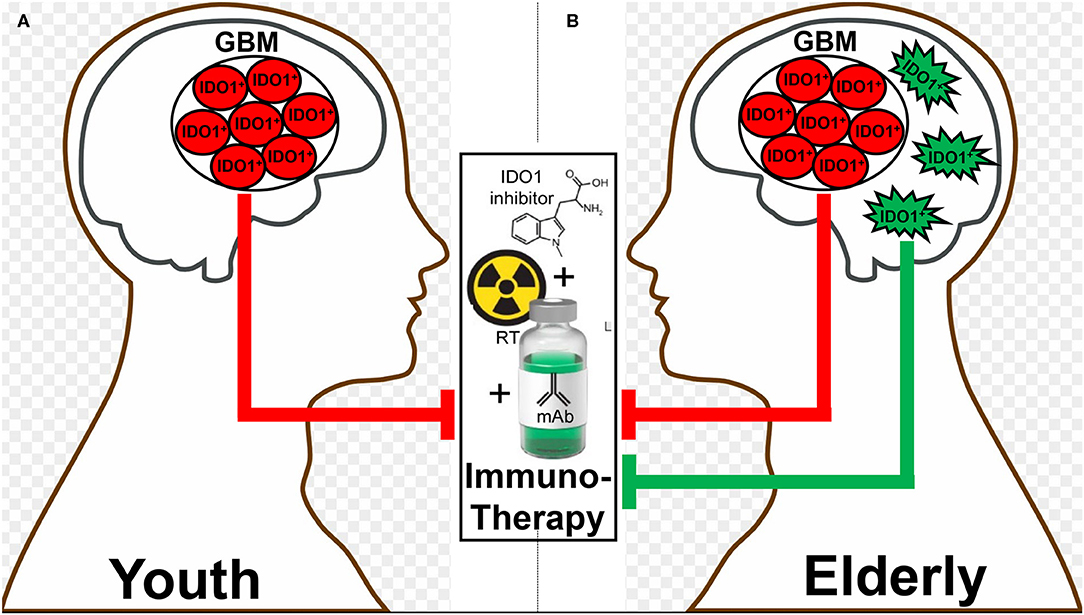
Figure 3. A hypothetic schematic for how advanced aging decreases immunotherapeutic efficacy in subjects with glioblastoma (GBM). There is a basal level of IDO-mediated immunosuppression by GBM cells (red) that does not change between (A) young and (B) elderly (as defined by ≥65 years of age) individuals. In contrast, there is additional immunosuppression in the elderly brain due to the accumulation of brain-resident IDO-expressing dendritic cells (green), which synergize with the GBM cell IDO to suppress the anti-GBM immune response facilitated by combination radiation, anti-PD-1 mAb, and IDO enzyme inhibitor treatment (middle box).
Concluding Remarks
It has been >20 years since the initial study by Munn et al. that uncovered the immunosuppressive role of IDO (119). Although extensive studies have been conducted to elucidate the underlying mechanisms of IDO-mediated immunosuppression, our knowledge remains incomplete. Notwithstanding, a growing list of clinical trials aimed at inhibiting the immunosuppressive effects of IDO have been undertaken as single agent and combinatorial regimens (Table 1)—without any remarkable success stories to-date. To this end, there are questions in the field including those that we previously addressed (3), as well as new considerations such as: (i) what are the immunosuppressive contributions of enzyme- and non-enzyme-IDO activity; (ii) will an IDO neutralizing pharmacologic that degrades protein, rather than only inhibiting enzyme activity, provide a superior therapeutic effect; (iii) given that IDO is expressed in tumor-draining lymph nodes (29), within the tumor itself, and inside the brain parenchyma during advanced age, are all anatomical sites required for mitigation to achieve optimal immunotherapeutic efficacy; (iv) among the growing compendium of possible immunological modifiers (i.e., radiation, anti-CTLA-4 mAb, anti-PD-(L)1 mAb, etc.), what is the optimal immunotherapeutic cocktail for combining with an IDO pharmacologic inhibitor? Also, the functional similarity between IDO and TDO requires further study and the use of an IDO-TDO double knockout mouse model might be helpful in validating their potency and half-life. Future investigations should also include the rigorous assessment of expression levels combined with an in vivo analysis of Trp and Kyn for better understanding how this immunosuppressive mediator functions among tissues, treatments, and across the lifespan. Understanding the full immunobiology of IDO and the generation of better neutralizing agents will allow for the potential future achievement of applying optimal therapeutic effects in human subjects with malignant cancer(s).
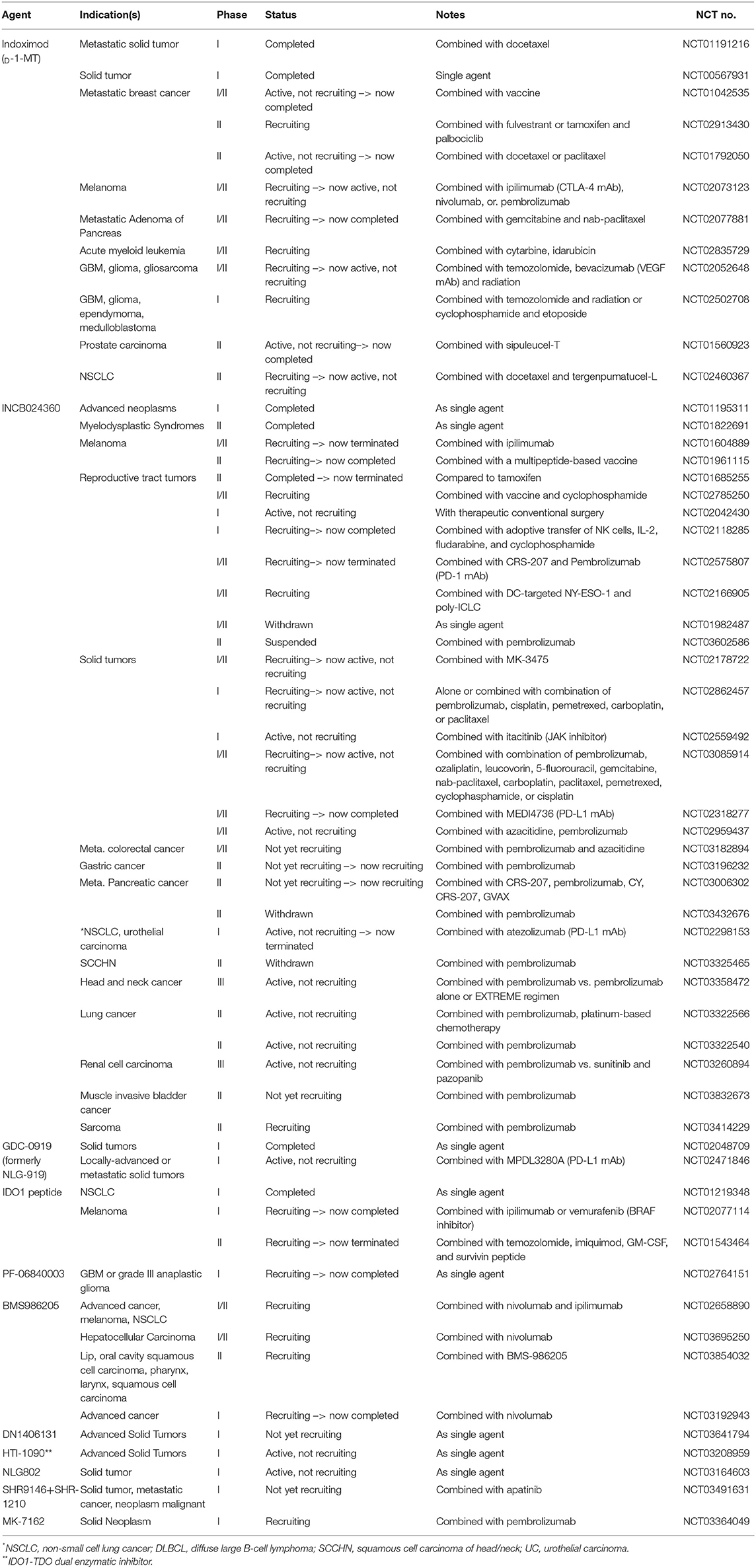
Table 1. Ongoing and historical clinical trials that target IDO in cancer.
Author Contributions
LZ and DW generated a draft of the manuscript. AB, EL, KL, LB, JS, BZ, JW, SM, JM, RL, EW, LD, GS, and RM provided feedback on the draft and/or data for inclusion. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants R01 NS097851-01 (DW), P50 CA221747 Project 2 (DW and RL), T32 CA070085 (EL), BrainUp grant 2136 (DW and RL), the Gail Boyter Magness (GBM) Foundation (DW), and the Grace Giving Foundation (DW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. McGaha TL, Huang L, Lemos H, Metz R, Mautino M, Prendergast GC, et al. Amino acid catabolism: a pivotal regulator of innate and adaptive immunity. Immunol Rev. (2012) 249:135–57. doi: 10.1111/j.1600-065X.2012.01149.x
2. Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. (2019) 18:379–401. doi: 10.1038/s41573-019-0016-5
3. Zhai L, Ladomersky E, Lenzen A, Nguyen B, Patel R, Lauing KL, et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol. (2018) 15:447–57. doi: 10.1038/cmi.2017.143
4. Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, et al. Molecular pathways: targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res. (2015) 21:5427–33. doi: 10.1158/1078-0432.CCR-15-0420
5. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
6. Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol. (2019) 41:41–8. doi: 10.1007/s00281-018-0702-0
7. Badawy AA-B, Guillemin G. The plasma [kynurenine]/[tryptophan] ratio and indoleamine 2,3-dioxygenase: time for appraisal. Int J Tryptophan Res. (2019) 12:1178646919868978. doi: 10.1177/1178646919868978
8. Grohmann U, Fallarino F, Puccetti Tolerance P. DCs tryptophan: much ado about IDO. Trends Immunol. (2003) 24:242–8. doi: 10.1016/S1471-4906(03)00072-3
9. Shimizu T, Nomiyama S, Hirata F, Hayaishi O. Indoleamine 2,3-dioxygenase. Purification and some properties. J Biol Chem. (1978) 253:4700–6.
10. Batabyal D, Yeh SR. Human tryptophan dioxygenase: a comparison to indoleamine 2,3-dioxygenase. J Am Chem Soc. (2007) 129:15690–701. doi: 10.1021/ja076186k
11. Basran J, Rafice SA, Chauhan N, Efimov I, Cheesman MR, Ghamsari L, et al. A kinetic, spectroscopic, and redox study of human tryptophan 2,3-dioxygenase. Biochemistry. (2008) 47:4752–60. doi: 10.1021/bi702393b
12. Pantouris G, Serys M, Yuasa HJ, Ball HJ, Mowat CG. Human indoleamine 2,3-dioxygenase-2 has substrate specificity and inhibition characteristics distinct from those of indoleamine 2,3-dioxygenase-1. Amino Acids. (2014) 46:2155–63. doi: 10.1007/s00726-014-1766-3
13. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. (2010) 115:3520–30. doi: 10.1182/blood-2009-09-246124
14. Yuasa HJ, Ball HJ, Austin CJ, Hunt NH. 1-L-methyltryptophan is a more effective inhibitor of vertebrate IDO2 enzymes than 1-D-methyltryptophan. Comp Biochem Physiol B Biochem Mol Biol. (2010) 157:10–5. doi: 10.1016/j.cbpb.2010.04.006
15. Metz R, Smith C, DuHadaway JB, Chandler P, Baban B, Merlo LMF, et al. IDO2 is critical for IDO1-mediated T-cell regulation and exerts a non-redundant function in inflammation. Int Immunol. (2014) 26:357–67. doi: 10.1093/intimm/dxt073
16. Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, tryptophan catabolism. FASEB J. (1991) 5:2516–22. doi: 10.1096/fasebj.5.11.1907934
17. Mellor AL, Baban B, Chandler PR, Manlapat A, Kahler DJ, Munn DH. Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J Immunol. (2005) 175:5601–5. doi: 10.4049/jimmunol.175.9.5601
18. Fallarino F, Volpi C, Zelante T, Vacca C, Calvitti M, Fioretti MC, et al. IDO mediates TLR9-driven protection from experimental autoimmune diabetes. J Immunol. (2009) 183:6303–12. doi: 10.4049/jimmunol.0901577
19. Fujigaki H, Saito K, Fujigaki S, Takemura M, Sudo K, Ishiguro H, et al. The signal transducer and activator of transcription 1α and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-κB pathways, and synergistic effect of several proinflammatory cytokines. J Biochem. (2006) 139:655–62. doi: 10.1093/jb/mvj072
20. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. (2011) 17:1094–100. doi: 10.1038/nm.2438
21. Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. (2002) 3:1097–101. doi: 10.1038/ni846
22. Holtzhausen Zhao F, Evans KS, Tsutsui M, Orabona C, Tyler DS, et al. Melanoma-derived Wnt5a promotes local dendritic-cell expression of IDO and immunotolerance: opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol Res. (2015) 3:1082–95. doi: 10.1158/2326-6066.CIR-14-0167
23. Theate A, van Baren N, Pilotte L, Moulin P, Larrieu P, Sempoux C, et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res. (2015) 3:161–72. doi: 10.1158/2326-6066.CIR-14-0137
24. Ladomersky E, Scholtens DM, Kocherginsky M, Hibler EA, Bartom ET, Otto-Meyer S, et al. The coincidence between increasing age, immunosuppression, and the incidence of patients with glioblastoma. Front Pharmacol. (2019) 10:200. doi: 10.3389/fphar.2019.00200
25. Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget. (2014) 5:1038–51. doi: 10.18632/oncotarget.1637
26. Li Q, Harden JL, Anderson CD, Egilmez NK. Tolerogenic phenotype of IFN-gamma-induced IDO+ dendritic cells is maintained via an autocrine IDO-kynurenine/AhR-IDO loop. J Immunol. (2016) 197:962–70. doi: 10.4049/jimmunol.1502615
27. Hennequart M, Pilotte L, Cane S, Hoffmann D, Stroobant V, Plaen E, et al. Constitutive IDO1 expression in human tumors is driven by cyclooxygenase-2 and mediates intrinsic immune resistance. Cancer Immunol Res. (2017) 5:695–709. doi: 10.1158/2326-6066.CIR-16-0400
28. Miyazaki T, Moritake K, Yamada K, Hara N, Osago H, Shibata T, et al. Indoleamine 2,3-dioxygenase as a new target for malignant glioma therapy. Laboratory investigation. J Neurosurg. (2009) 111:230–7. doi: 10.3171/2008.10.JNS081141
29. Zhai L, Ladomersky E, Dostal CR, Lauing KL, Swoap K, Billingham LK, et al. Non-tumor cell IDO1 predominantly contributes to enzyme activity and response to CTLA-4/PD-L1 inhibition in mouse glioblastoma. Brain Behav Immun. (2017) 62:24–9. doi: 10.1016/j.bbi.2017.01.022
30. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. (2015) 347:1260419. doi: 10.1126/science.1260419
31. Comings DE, Muhleman D, Dietz G, Sherman M, Forest GL. Sequence of human tryptophan 2,3-dioxygenase (TDO2): presence of a glucocorticoid response-like element composed of a GTT repeat and an intronic CCCCT repeat. Genomics. (1995) 29:390–6. doi: 10.1006/geno.1995.9990
32. Li DD, Gao YJ, Tian XC, Yang ZQ, Cao H, Zhang QL, et al. Differential expression and regulation of Tdo2 during mouse decidualization. J Endocrinol. (2014) 220:73–83. doi: 10.1530/JOE-13-0429
33. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. (2011) 478:197–203. doi: 10.1038/nature10491
34. Pilotte L, Larrieu P, Stroobant V, Colau D, Dolušić E, Frédérick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci USA. (2012) 109:2497–502. doi: 10.1073/pnas.1113873109
35. Novikov O, Wang Z, Stanford EA, Parks AJ, Ramirez-Cardenas A, Landesman E, et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in ER-/PR-/Her2-human breast cancer cells. Mol Pharmacol. (2016) 90:674–88. doi: 10.1124/mol.116.105361
36. van Baren N, Van den Eynde BJ. Tryptophan-degrading enzymes in tumoral immune resistance. Front Immunol. (2015) 6:34. doi: 10.3389/fimmu.2015.00034
37. Prendergast GC, Metz R, Muller AJ, Merlo LM, Mandik-Nayak L. IDO2 in immunomodulation and autoimmune disease. Front Immunol. (2014) 5:585. doi: 10.3389/fimmu.2014.00585
38. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. (2005) 22:633–42. doi: 10.1016/j.immuni.2005.03.013
39. Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. (2012) 1:1460–8. doi: 10.4161/onci.21716
40. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
41. Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol. (2014) 32:403–32. doi: 10.1146/annurev-immunol-032713-120245
42. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature. (2008) 453:65–71. doi: 10.1038/nature06880
43. Marshall NB, Kerkvliet NI. Dioxin and immune regulation: emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann N Y Acad Sci. (2010) 1183:25–37. doi: 10.1111/j.1749-6632.2009.05125.x
44. DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci. (2010) 115:89–97. doi: 10.1093/toxsci/kfq024
45. Lowe MM, Mold JE, Kanwar B, Huang Y, Louie A, Pollastri MP, et al. Identification of cinnabarinic acid as a novel endogenous aryl hydrocarbon receptor ligand that drives IL-22 production. PLoS ONE. (2014) 9:e87877. doi: 10.1371/journal.pone.0087877
46. Terness P, Bauer TM, Röse L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase–expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. (2002) 196:447–57. doi: 10.1084/jem.20020052
47. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. (2002) 9:1069–77. doi: 10.1038/sj.cdd.4401073
48. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. (2002) 196:459–68. doi: 10.1084/jem.20020121
49. Sonner JK, Deumelandt K, Ott M, Thome CM, Rauschenbach KJ, Schulz S, et al. The stress kinase GCN2 does not mediate suppression of antitumor T cell responses by tryptophan catabolism in experimental melanomas. Oncoimmunology. (2016) 5:e1240858. doi: 10.1080/2162402X.2016.1240858
50. Segawa H, Fukasawa Y, Miyamoto K, Takeda E, Endou H, Kanai Y. Identification and functional characterization of a Na+-independent neutral amino acid transporter with broad substrate selectivity. J Biol Chem. (1999) 274:19745–51. doi: 10.1074/jbc.274.28.19745
51. Seymour RL, Ganapathy V, Mellor AL, Munn DH. A high-affinity, tryptophan-selective amino acid transport system in human macrophages. J Leuk Biol. (2006) 80:1320–7. doi: 10.1189/jlb.1205727
52. Silk JD, Lakhal S, Laynes R, Vallius L, Karydis I, Marcea C, et al. IDO induces expression of a novel tryptophan transporter in mouse and human tumor cells. J Immunol. (2011) 187:1617–25. doi: 10.4049/jimmunol.1000815
53. Schrocksnadel K, Wirleitner B, Winkler C, Fuchs D. Monitoring tryptophan metabolism in chronic immune activation. Clin Chim Acta. (2006) 364:82–90. doi: 10.1016/j.cca.2005.06.013
54. Huang Fuchs D, Widner B, Glover C, Henderson DC, Allen-Mersh TG. Serum tryptophan decrease correlates with immune activation and impaired quality of life in colorectal cancer. Br J Cancer. (2002) 86:1691–6. doi: 10.1038/sj.bjc.6600336
55. Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J Immunother Cancer. (2014) 2:21. doi: 10.1186/2051-1426-2-21
56. Zhai L, Dey M, Lauing KL, Gritsina G, Kaur R, Lukas RV, et al. The kynurenine to tryptophan ratio as a prognostic tool for glioblastoma patients enrolling in immunotherapy. J Clin Neurosci. (2015) 22:5. doi: 10.1016/j.jocn.2015.06.018
57. Adams S, Teo C, McDonald KL, Zinger A, Bustamante S, Lim CK, et al. Involvement of the kynurenine pathway in human glioma pathophysiology. PLoS ONE. (2014) 9:e112945. doi: 10.1371/journal.pone.0112945
58. Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci. (2019) 22:729–40. doi: 10.1038/s41593-019-0370-y
59. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci USA. (2010) 107:19961–6. doi: 10.1073/pnas.1014465107
60. Wainwright DA, Sengupta S, Han Y, Lesniak MS. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro Oncol. (2011) 13:1308–23. doi: 10.1093/neuonc/nor134
61. Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science. (2013) 339:1219–24. doi: 10.1126/science.1233913
62. Ye J, Qiu J, Bostick JW, Ueda A, Schjerven H, Li S, et al. The aryl hydrocarbon receptor preferentially marks and promotes gut regulatory T cells. Cell Rep. (2017) 21:2277–90. doi: 10.1016/j.celrep.2017.10.114
63. Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. (2007) 117:2570–82. doi: 10.1172/JCI31911
64. Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD, et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv. (2015) 1:e1500845. doi: 10.1126/sciadv.1500845
65. Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infiltrating T cells increase IDO1 expression in glioblastoma and contribute to decreased patient survival. Clin Cancer Res. (2017) 23:6650–60. doi: 10.1158/1078-0432.CCR-17-0120
66. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. (2003) 9:1269–74. doi: 10.1038/nm934
67. Riesenberg R, Weiler C, Spring O, Eder M, Buchner A, Popp T, et al. Expression of indoleamine 2,3-dioxygenase in tumor endothelial cells correlates with long-term survival of patients with renal cell carcinoma. Clin Cancer Res. (2007) 13:6993–7002. doi: 10.1158/1078-0432.CCR-07-0942
68. Ishio T, Goto S, Tahara K, Tone S, Kawano K, Kitano S. Immunoactivative role of indoleamine 2,3-dioxygenase in human hepatocellular carcinoma. J Gastroenterol Hepatol. (2004) 19:319–26. doi: 10.1111/j.1440-1746.2003.03259.x
69. Pan K, Wang H, Chen MS, Zhang HK, Weng DS, Zhou J, et al. Expression and prognosis role of indoleamine 2,3-dioxygenase in hepatocellular carcinoma. J Cancer Res Clin Oncol. (2008) 134:1247–53. doi: 10.1007/s00432-008-0395-1
70. Piras F, Colombari R, Minerba L, Murtas D, Floris C, Maxia C, et al. The predictive value of CD8, CD4, CD68, and human leukocyte antigen-D-related cells in the prognosis of cutaneous malignant melanoma with vertical growth phase. Cancer. (2005) 104:1246–54. doi: 10.1002/cncr.21283
71. Tang D, Yue L, Yao R, Zhou L, Yang Y, Lu L, et al. P53 prevent tumor invasion and metastasis by down-regulating IDO in lung cancer. Oncotarget. (2017) 8:54548–57. doi: 10.18632/oncotarget.17408
72. Levina V, Su Y, Gorelik E. Immunological and nonimmunological effects of indoleamine 2,3-dioxygenase on breast tumor growth and spontaneous metastasis formation. Clin Dev Immunol. (2012) 2012:173029. doi: 10.1155/2012/173029
73. Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Flick HE, et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. (2012) 2:722–35. doi: 10.1158/2159-8290.CD-12-0014
74. Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res. (2006) 12:1144–51. doi: 10.1158/1078-0432.CCR-05-1966
75. Ino K, Yamamoto E, Shibata K, Kajiyama H, Yoshida N, Terauchi M, et al. Inverse correlation between tumoral indoleamine 2,3-dioxygenase expression and tumor-infiltrating lymphocytes in endometrial cancer: its association with disease progression and survival. Clin Cancer Res. (2008) 14:2310–7. doi: 10.1158/1078-0432.CCR-07-4144
76. Vareki SM, Rytelewski M, Figueredo R, Chen D, Ferguson PJ, Vincent M, et al. Indoleamine 2,3-dioxygenase mediates immune-independent human tumor cell resistance to olaparib, gamma radiation, and cisplatin. Oncotarget. (2014) 5:2778–91. doi: 10.18632/oncotarget.1916
77. Tummala KS, Gomes AL, Yilmaz M, Grana O, Bakiri L, Ruppen I, et al. Inhibition of de novo NAD(+) synthesis by oncogenic URI causes liver tumorigenesis through DNA damage. Cancer Cell. (2014) 26:826–39. doi: 10.1016/j.ccell.2014.10.002
78. Wang Y, Liu H, McKenzie G, Witting PK, Stasch JP, Hahn M, et al. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat Med. (2010) 16:279–85. doi: 10.1038/nm.2092
79. Sakakibara K, Feng GG, Li J, Akahori T, Yasuda Y, Nakamura E, et al. Kynurenine causes vasodilation and hypotension induced by activation of KCNQ-encoded voltage-dependent K(+) channels. J Pharmacol Sci. (2015) 129:31–7. doi: 10.1016/j.jphs.2015.07.042
80. Fazio F, Carrizzo A, Lionetto L, Damato A, Capocci L, Ambrosio M, et al. Vasorelaxing action of the kynurenine metabolite, xanthurenic acid: the missing link in endotoxin-induced hypotension? Front Pharmacol. (2017) 8:214. doi: 10.3389/fphar.2017.00214
81. Stanley CP, Maghzal GJ, Ayer A, Talib J, Giltrap AM, Shengule S, et al. Singlet molecular oxygen regulates vascular tone and blood pressure in inflammation. Nature. (2019) 566:548–52. doi: 10.1038/s41586-019-0947-3
82. Changsirivathanathamrong D, Wang Y, Rajbhandari D, Maghzal GJ, Mak WM, Woolfe C, et al. Tryptophan metabolism to kynurenine is a potential novel contributor to hypotension in human sepsis. Crit Care Med. (2011) 39:2678–83. doi: 10.1097/CCM.0b013e31822827f2
83. Mondal Smith C, DuHadaway JB, Sutanto-Ward E, Prendergast GC, Bravo-Nuevo A, et al. IDO1 is an integral mediator of inflammatory neovascularization. EBioMedicine. (2016) 14:74–82. doi: 10.1016/j.ebiom.2016.11.013
84. Prendergast GC, Mondal A, Dey S, Laury-Kleintop LD, Muller AJ. Inflammatory Reprogramming with IDO1 inhibitors: turning immunologically unresponsive ‘cold’ tumors ‘hot’. Trends Cancer. (2018) 4:38–58. doi: 10.1016/j.trecan.2017.11.005
85. Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. (2011) 12:870–8. doi: 10.1038/ni.2077
86. Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. (2012) 18:6110–21. doi: 10.1158/1078-0432.CCR-12-2130
87. Ladomersky E, Zhai L, Lenzen A, Lauing KL, Qian J, Scholtens DM, et al. IDO1 Inhibition synergizes with radiation and PD-1 blockade to durably increase survival against advanced glioblastoma. Clin Cancer Res. (2018) 24:2559–73. doi: 10.1158/1078-0432.CCR-17-3573
88. Frick B, Schroecksnadel K, Neurauter G, Leblhuber F, Fuchs D. Increasing production of homocysteine and neopterin and degradation of tryptophan with older age. Clin Biochem. (2004) 37:684–7. doi: 10.1016/j.clinbiochem.2004.02.007
89. Pertovaara M, Raitala A, Lehtimaki T, Karhunen PJ, Oja SS, Jylha M, et al. Indoleamine 2,3-dioxygenase activity in nonagenarians is markedly increased and predicts mortality. Mech Ageing Dev. (2006) 127:497–9. doi: 10.1016/j.mad.2006.01.020
90. Marcos-Perez D, Sanchez-Flores M, Maseda A, Lorenzo-Lopez L, Millan-Calenti JC, Strasser B, et al. Frailty status in older adults is related to alterations in indoleamine 2,3-dioxygenase 1 and guanosine triphosphate cyclohydrolase I enzymatic pathways. J Am Med Direc Assoc. (2017) 18:1049–57. doi: 10.1016/j.jamda.2017.06.021
91. Sutphin GL, Backer G, Sheehan S, Bean S, Corban C, Liu T, et al. Caenorhabditis elegans orthologs of human genes differentially expressed with age are enriched for determinants of longevity. Aging Cell. (2017) 16:672–82. doi: 10.1111/acel.12595
92. Apalset EM, Gjesdal CG, Ueland PM, Midttun O, Ulvik A, Eide GE, et al. Interferon (IFN)-gamma-mediated inflammation and the kynurenine pathway in relation to bone mineral density: the Hordaland Health Study. Clin Exp Immunol. (2014) 176:452–60. doi: 10.1111/cei.12288
93. Reyes Ocampo J, Lugo Huitron R, Gonzalez-Esquivel D, Ugalde-Muniz P, Jimenez-Anguiano A, Pineda B, et al. Kynurenines with neuroactive and redox properties: relevance to aging and brain diseases. Oxidat Med Cell Longev. (2014) 2014:646909. doi: 10.1155/2014/646909
94. Ladomersky E, Zhai L, Gritsina G, Genet M, Lauing KL, Wu M, et al. Advanced age negatively impacts survival in an experimental brain tumor model. Neurosci Lett. (2016) 630:203–8. doi: 10.1016/j.neulet.2016.08.002
95. Refaey ME, McGee-Lawrence ME, Fulzele S, Kennedy EJ, Bollag WB, Elsalanty M, et al. Kynurenine, a tryptophan metabolite that accumulates with age, induces bone loss. J Bone Miner Res. (2017) 32:2182–93. doi: 10.1002/jbmr.3224
96. Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat Immunol. (2019) 20:50–63. doi: 10.1038/s41590-018-0255-3
97. Eckers Jakob S, Heiss C, Haarmann-Stemmann T, Goy C, Brinkmann V, et al. The aryl hydrocarbon receptor promotes aging phenotypes across species. Sci Rep. (2016) 6:19618. doi: 10.1038/srep19618
98. Sharpless N. The Challenging Landscape of Cancer and Aging: Charting a Way Forward. National Cancer Institute (2018).
99. Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol. (2017) 19:v1–88. doi: 10.1093/neuonc/nox158
100. Mellor AL, Baban B, Chandler P, Marshall B, Jhaver K, Hansen A, et al. Cutting Edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol. (2003) 171:1652–5. doi: 10.4049/jimmunol.171.4.1652
101. Baban B, Chandler P, McCool D, Marshall B, Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J Reprod Immunol. (2004) 61:67–77. doi: 10.1016/j.jri.2003.11.003
102. Moreno ACR, Porchia B, Pagni RL, Souza PDC, Pegoraro R, Rodrigues KB, et al. The combined use of melatonin and an indoleamine 2,3-dioxygenase-1 inhibitor enhances vaccine-induced protective cellular immunity to HPV16-associated tumors. Front Immunol. (2018) 9:1914. doi: 10.3389/fimmu.2018.01914
103. Schafer CC, Wang Y, Hough KP, Sawant A, Grant SC, Thannickal VJ, et al. Indoleamine 2,3-dioxygenase regulates anti-tumor immunity in lung cancer by metabolic reprogramming of immune cells in the tumor microenvironment. Oncotarget. (2016) 7:75407–24. doi: 10.18632/oncotarget.12249
104. Takamatsu M, Hirata A, Ohtaki H, Hoshi M, Ando T, Ito H, et al. Inhibition of indoleamine 2,3-dioxygenase 1 expression alters immune response in colon tumor microenvironment in mice. Cancer Sci. (2015) 106:1008–15. doi: 10.1111/cas.12705
105. Bishnupuri KS, Alvarado DM, Khouri AN, Shabsovich M, Chen B, Dieckgraefe BK, et al. IDO1 and kynurenine pathway metabolites activate PI3K-Akt signaling in the neoplastic colon epithelium to promote cancer cell proliferation and inhibit apoptosis. Cancer Res. (2019) 79:1138–50. doi: 10.1158/0008-5472.CAN-18-0668
106. Reardon DA, Freeman G, Wu C, Chiocca EA, Wucherpfennig KW, Wen PY, et al. Immunotherapy advances for glioblastoma. Neuro Oncol. (2014) 16:1441–58. doi: 10.1093/neuonc/nou212
107. Avril T, Saikali S, Vauleon E, Jary A, Hamlat A, De Tayrac M, et al. Distinct effects of human glioblastoma immunoregulatory molecules programmed cell death ligand-1 (PDL-1) and indoleamine 2,3-dioxygenase (IDO) on tumour-specific T cell functions. J Neuroimmunol. (2010) 225:22–33. doi: 10.1016/j.jneuroim.2010.04.003
108. Mitsuka K, Kawataki T, Satoh E, Asahara T, Horikoshi T, Kinouchi H. Expression of indoleamine 2,3-dioxygenase and correlation with pathological malignancy in gliomas. Neurosurgery. (2013) 72:1031–9. doi: 10.1227/NEU.0b013e31828cf945
109. Guastella AR, Michelhaugh SK, Klinger NV, Fadel HA, Kiousis S, Ali-Fehmi R, et al. Investigation of the aryl hydrocarbon receptor and the intrinsic tumoral component of the kynurenine pathway of tryptophan metabolism in primary brain tumors. J Neurooncol. (2018) 139:239–9. doi: 10.1007/s11060-018-2869-6
110. Kesarwani P, Prabhu A, Kant S, Kumar P, Graham SF, Buelow KL, et al. Tryptophan metabolism contributes to radiation-induced immune checkpoint reactivation in glioblastoma. Clin Cancer Res. (2018) 24:3632–43. doi: 10.1158/1078-0432.CCR-18-0041
111. Ott M, Litzenburger UM, Rauschenbach KJ, Bunse L, Ochs K, Sahm F, et al. Suppression of TDO-mediated tryptophan catabolism in glioblastoma cells by a steroid-responsive FKBP52-dependent pathway. Glia. (2015) 63:78–90. doi: 10.1002/glia.22734
112. Du L, Xing Z, Tao B, Li T, Yang D, Li W, et al. Both IDO1 and TDO contribute to the malignancy of gliomas via the Kyn-AhR-AQP4 signaling pathway. Signal Transduc Target Ther. (2020) 5:10. doi: 10.1038/s41392-019-0103-4
113. Grant R, Kapoor V. Inhibition of indoleamine 2,3-dioxygenase activity in IFN-gamma stimulated astroglioma cells decreases intracellular NAD levels. Biochem Pharmacol. (2003) 66:1033–6. doi: 10.1016/S0006-2952(03)00464-7
114. Bostian AC, Maddukuri L, Reed MR, Savenka T, Hartman JH, Davis L, et al. Kynurenine signaling increases DNA polymerase kappa expression and promotes genomic instability in glioblastoma cells. Chem Res Toxicol. (2016) 29:101–8. doi: 10.1021/acs.chemrestox.5b00452
115. Sun S, Du G, Xue J, Ma J, Ge M, Wang H, et al. PCC0208009 enhances the anti-tumor effects of temozolomide through direct inhibition and transcriptional regulation of indoleamine 2,3-dioxygenase in glioma models. Int J Immunopathol Pharmacol. (2018) 32:2058738418787991. doi: 10.1177/2058738418787991
116. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
117. Bulloch K, Miller MM, Gal-Toth J, Milner TA, Gottfried-Blackmore A, Waters EM, et al. CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J Comp Neurol. (2008) 508:687–710. doi: 10.1002/cne.21668
118. Kaunzner UW, Miller MM, Gottfried-Blackmore A, Gal-Toth J, Felger JC, McEwen BS, et al. Accumulation of resident and peripheral dendritic cells in the aging CNS. Neurobiol Aging. (2012) 33:681–93 e1. doi: 10.1016/j.neurobiolaging.2010.06.007
Keywords: aging, immunotherapy, glioblastoma, tryptophan, immunosuppression, Treg, IDO1, kynurenine
Citation: Zhai L, Bell A, Ladomersky E, Lauing KL, Bollu L, Sosman JA, Zhang B, Wu JD, Miller SD, Meeks JJ, Lukas RV, Wyatt E, Doglio L, Schiltz GE, McCusker RH and Wainwright DA (2020) Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front. Immunol. 11:1185. doi: 10.3389/fimmu.2020.01185
Received: 01 February 2020; Accepted: 13 May 2020;
Published: 16 June 2020.
Edited by:
Alexander Muller, Lankenau Institute for Medical Research, United StatesReviewed by:
Graham Robert Leggatt, The University of Queensland, AustraliaKawaljit Kaur, University of California, Los Angeles, United States
Copyright © 2020 Zhai, Bell, Ladomersky, Lauing, Bollu, Sosman, Zhang, Wu, Miller, Meeks, Lukas, Wyatt, Doglio, Schiltz, McCusker and Wainwright. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Derek A. Wainwright, ZGVyZWt3YWlud3JpZ2h0QG5vcnRod2VzdGVybi5lZHU=; ZGVyZWt3YWlud3JpZ2h0QGhvdG1haWwuY29t