 Alba Rodriguez-Garcia
Alba Rodriguez-Garcia Asis Palazon
Asis Palazon Estela Noguera-Ortega
Estela Noguera-Ortega Daniel J. Powell Jr.
Daniel J. Powell Jr. Sonia Guedan
Sonia Guedan- 1Center for Cellular Immunotherapies, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 2Cancer Immunology and Immunotherapy Laboratory, Ikerbasque Basque Foundation for Science, CIC bioGUNE, Basque Research and Technology Alliance (BRTA), Derio, Spain
- 3Department of Hematology and Oncology, Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Hospital Clinic, Barcelona, Spain
Chimeric antigen receptor (CAR) T cell therapies have demonstrated remarkable efficacy for the treatment of hematological malignancies. However, in patients with solid tumors, objective responses to CAR-T cell therapy remain sporadic and transient. A major obstacle for CAR-T cells is the intrinsic ability of tumors to evade immune responses. Advanced solid tumors are largely composed of desmoplastic stroma and immunosuppressive modulators, and characterized by aberrant cell proliferation and vascularization, resulting in hypoxia and altered nutrient availability. To mount a curative response after infusion, CAR-T cells must infiltrate the tumor, recognize their cognate antigen and perform their effector function in this hostile tumor microenvironment, to then differentiate and persist as memory T cells that confer long-term protection. Fortunately, recent advances in synthetic biology provide a wide set of tools to genetically modify CAR-T cells to overcome some of these obstacles. In this review, we provide a comprehensive overview of the key tumor intrinsic mechanisms that prevent an effective CAR-T cell antitumor response and we discuss the most promising strategies to prevent tumor escape to CAR-T cell therapy.
Introduction
T cells that are genetically modified to express chimeric antigen receptors (CAR-T) constitute a potent new cancer therapy with curative potential (1, 2). CAR-T cell therapy has produced impressive response rates in patients with certain B-cell malignancies, resulting in the recent approval of two CAR-T cell products targeting CD19 (3, 4). Numerous CAR-T cell therapies targeting a variety of antigens are under clinical investigation, with anti-BCMA CAR-T cells showing very promising results for the treatment of multiple myeloma (5). Despite the impressive responses in patients with hematologic malignancies, early clinical trials using CAR-T cells in patients with solid tumors have reported limited antitumor activity, with objective responses observed only in a minority of patients (6–8).
The potential of T cells to induce complete responses in patients with solid tumors has been demonstrated by the success of immune checkpoint therapy (9). Also, objective responses to adoptive T cell therapy with tumor infiltrating lymphocytes (TILs) and T cells that are genetically engineered to express a transgenic T cell receptor (TCR) have been reported in patients with melanoma, sarcoma, cholangiocarcinoma, and breast cancer (10). While only a proportion of patients exhibit long term, durable responses, these results suggest that T cells have the potential to eliminate solid tumors under adequate conditions. However, to date only anecdotes of CAR-T cell mediated response have been reported (6, 8). Understanding the mechanisms that limit CAR-T cell efficacy in solid tumors is essential to design the next-generation of CAR-T cell therapies with increased therapeutic index.
Some of the key factors limiting the applicability of CAR-T cells for the treatment of solid tumors include: the lack of truly tumor-specific target antigens (11); tumor heterogeneity and plasticity that can lead to tumor escape due to loss of antigen expression (12); T cell dysfunction driven by CAR-mediated tonic signaling (13–15) or chronic antigen exposure (16); and the immunosuppressive tumor microenvironment (TME) (17). In this review, we summarize the key challenges that CAR-T cell encounter in the TME, with a particular emphasis on tumor intrinsic factors, such us hypoxia, extracellular matrix (ECM) and stromal and immune cells. We also discuss some of the efforts that are underway to overcome these challenges and expand the therapeutic window of CAR-T cells for the treatment of solid tumors (Table 1).

Table 1. Main challenges for CAR-T cell therapy in solid tumors and emerging strategies to address them.
Physical Barriers
Hypoxia
Defined as a shortage in oxygen availability, hypoxia is a prominent feature of solid tumors that results from an aberrant vascularization and rapidly proliferating tumor cells. Tumor hypoxia has been correlated with poor patient prognosis (101), resistance to neoadjuvant therapy (102, 103), and metastatic success (104). Importantly, reduced oxygenation can also influence antitumor immune responses (105).
Cellular adaptations to oxygen levels are governed by the hypoxia pathway and mediated by hypoxia-inducible factors (HIF). When oxygen is available, prolyl hydroxylase domain proteins (PHDs) are active and hydroxylate HIF, leading to HIF ubiquitination by Von-Hippel Lindau (VHL), and HIF degradation in the proteasome. When oxygen levels drop, hydroxylases become inactive leading to HIF stabilization and translocation to the nucleus, where it forms a transcriptional complex that directly binds to specific regions, termed hypoxia response elements (HREs). HREs are present in the promoters of several genes that encode for important proteins that mediate the cellular adaptation to hypoxia, such as glycolytic enzymes and the vascular endothelial growth factor-A (VEGF-A) (106). This family of transcription factors is mainly comprised of two isoforms: HIF-1α and HIF-2α (107), with HIF-1α being the main isoform expressed by activated T cells (108). HIF-1 accumulation in T cells promotes antitumor immunity in mouse models of solid tumors and metastases (109, 110).
After activation, T cells increase glucose uptake and glycolytic rate to support proliferation and the acquisition of effector functions (111). This process is supported by HIF stabilization after TCR engagement and augmented under hypoxia. A consequence of the T cell adaptation to hypoxia is metabolic rewiring, a process in which the reduced rate of oxidative phosphorylation (OXPHOS) is compensated by enhanced glycolysis. Competition for nutrients, persistent antigenic stimulation and immunosuppressive networks in the TME can lead to T cell exhaustion (112). Another consequence of metabolic adaptation in T cells is the accumulation of metabolites that impact epigenetic landscapes that influence the fate and function of T cells (113). One example is the increased production of the oncometabolite 2-hydroxyglutarate (2-HG) by hypoxic T cells. 2-HG inhibits 2-oxoglutarate-dependent epigenetic enzymes (114) resulting in the modulation of the T-cell terminal differentiation and favoring a central memory phenotype (115). Certain histone demethylases, such as KDM6A and KDM5A, can also be directly inhibited by a shortage of oxygen in a HIF- and 2-HG independent manner, leading to the control of gene expression and cell fate (116, 117).
The level of oxygenation impacts several aspects of CAR-T therapies (Figure 1). In vitro, hypoxia decreases the expansion capacity of CAR-T cells, blocking their differentiation into effector memory cells, and enriching the cultures with T cells with a central memory cell phenotype (118). Culturing and expanding CAR-T cells under controlled physiological oxygen concentrations might be an approach for enriching the cultures with memory-like T cells, which are known to have better persistence and efficacy than terminally differentiated effector T cells (119).
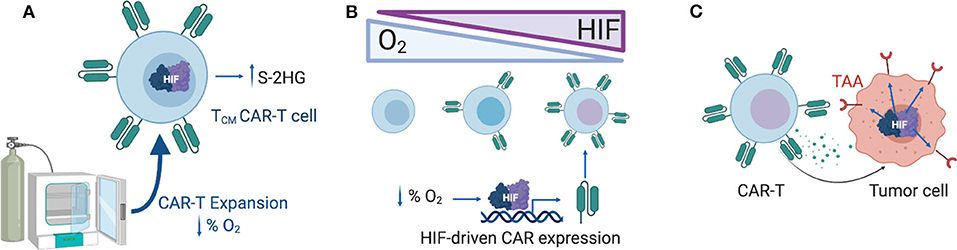
Figure 1. Exploiting the hypoxia response pathway for CAR-T therapy. (A) Expanding CAR-T cells ex vivo under reduced oxygen concentrations (1–5% O2) might support the enrichment of memory-like T cells, a process mediated by S-2HG. (B) CAR expression can be gradually modulated by increasing levels of HIF-1α in T cells, generating a hypoxia-responsive CAR-T with increased CAR expression in hypoxic tumors and reduced CAR expression in the periphery. (C) Selection of TAAs that are upregulated under hypoxic conditions in solid tumors might limit off-tumor CAR-T cell activity. HIF-1α, Hypoxia-inducible factor 1 alpha; S-2HG, S-2-hydroxyglutarate; TAA, tumor associated antigen.
After infusion, CAR-T cells must infiltrate solid tumors and carry out their cytotoxic activities. How hypoxia influences these processes remains largely unexplored. Recent development of in vitro tools will support the study of CAR-T function in relevant oxygenation conditions (120). In this context, the use of organoids and 3D tumor models (121–123) will support the preclinical development of CAR-T cells for the treatment of solid tumors.
The hypoxia pathway offers several opportunities for the design of CAR-T cells (Figure 1). The choice of the optimal costimulatory domains in the CAR might be influenced by oxygen availability in the TME, given that the metabolic consequences of signaling downstream of CD28 and 4-1BB are different (35–37). Another attractive approach is the design of CARs that are active in the TME, but inactive in better oxygenated environments in an attempt to reduce off-site toxicities. Novel strategies to confine CAR expression to the TME consists of introducing HRE regions on the promoter of the construct, or fusing HIF domains to the intracellular domain of the CAR to promote the hydroxylation and degradation of the CAR when oxygen is available (38). Both approaches rely on the endogenous T cell oxygen-sensing machinery to control the expression of the CAR. Alternatively, CAR-T cell activity can also be targeted to antigens that are known to be upregulated under hypoxic conditions in solid tumors, such as carbonic anhydrase IX (39).
Hypoxia also promotes immunosuppressive pathways in the TME that offer combinatorial therapeutic strategies with CAR-T cell approaches. Hypoxia and HIF promote the expression of program death ligand 1 (PD-L1) (86, 124) and adenosine levels (125, 126), as well as the recruitment of regulatory T (Treg) cells in the TME (127), all of which are known to inhibit T cell responses.
Extracellular Matrix
The ECM is an integral constituent of the tumor stroma composed of different macromolecules including fibrous proteins, glycosaminoglycans, and proteoglycans. The ECM is produced by tumor cells themselves as well as by cancer-associated fibroblasts (CAFs) and play an important role in cancer progression. Increased deposition of collagen or hyaluronan, constituents of the ECM, in tumors correlate with poor prognosis in different cancer types (128–131).
In addition, the ECM represents a physical barrier to various anticancer therapies, preventing their penetration and infiltration of tumors. Agents such as collagenase or hyaluronidase can degrade distinct components of the ECM and improve antitumor efficacy of diverse cancer therapies, including chemotherapy, oncolytic viruses, monoclonal antibodies, or checkpoint blockade (132–142).
While the role of ECM in resistance to adoptive T cell transfer therapies remains underexplored, some studies demonstrate that peritumoral ECM collagen fibers limit T cell access to tumors, and indeed, tumors with high-collagen density present lower levels of infiltrating T cells (142, 143). Here, the use of the matrix-degrading agents that facilitate T cell infiltration of tumors provides a rationale for matrix degradation as a means to improve efficacy of CAR-T cell therapy (140–142). In this regard, CAR-T cells engineered to express heparanase (HPSE), which degrades heparan sulfate proteoglycans, better infiltrated tumors and had increased antitumor activity in mouse models (23). Since matrix metalloproteinases (MMPs), mainly produced by macrophages, also regulate synthesis and degradation of most of the ECM components, an alternative strategy is to leverage the capacity of macrophages to secrete MMPs and remodel the ECM in order to clear the way for T cells to infiltrate tumors (24). This has been demonstrated in the context of endogenous T lymphocytes, but it could be hypothesized that the use of CAR-macrophages might benefit tumor infiltration of CAR-T cells, although it has not been experimentally tested yet.
Tumor Vasculature
Aberrant tumor vasculature is required for tumor survival, progression, and metastasis, but also provides a physical barrier for T cell extravasation and infiltration into tumors (144). CAR-T cells capable of destroying tumor vasculature have been developed targeting molecules such as VEGFR-2 (18), VEGFR1 (19), PSMA (20), TEM8 (21), or the fibronectin splice variant EIIIB (22). All of these target antigens are also expressed by a range of tumor cell types, and some of them by immunosuppressive cell populations such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs, i.e., VEGFR2) (145, 146) or by the ECM (i.e., EIIIB), which may improve the outcome of the therapy in patients. Unfortunately, a clinical trial on metastatic cancer patient treated with VEGFR-2 CAR-T cells was terminated due to lack of objective responses (NCT01218867).
Fibroblasts
CAFs can contribute to up to 90% of the solid tumor mass in carcinomas (147) and represent a complex barrier to entry and activity of endogenous and adoptively transferred immune cells.
CAFs signal in a paracrine fashion with tumor cells and other components of the TME. Tumor promoting CAFs secrete factors, including VEGFs, that induce angiogenesis to improve oxygen and nutrient availability in the tumor. CAFs can also directly provide cancer cells with nutrients, growth factors and immunosuppressive cytokines such as transforming growth factor beta (TGF-β), epidermal growth factor (EGF), platelet-derived growth factor (PDGF), and fibroblast growth factor 2 (FGF2), and serve as a physical barrier to T cell infiltration (148, 149). CAFs heavily contribute to the survival, proliferation, metastasis initiation and, even, de-differentiation of tumor cells into more stem cell-like phenotype (150, 151).
Given their powerful and diverse protumoral effects, an attractive therapeutic approach could be generating CAR-T cells that target CAFs. In addition to eliminating their multiple negative effects, an advantage to targeting fibroblasts would be that they are more genetically stable than tumor cells, so they are less likely to lose antigen expression via immunoediting. Moreover, since mesenchymal tumoral stromal cells are present in almost all human adenocarcinomas, therapies against CAFs could potentially be used for multiple types of tumors (152).
In the setting of solid tumors, different subtypes of CAFs have been proposed to have disparate effects on tumor establishment, growth and progression, as well as in metastatic capacity (25, 153). Therefore, when choosing a CAR-targeted protein, it is important to consider which fibroblast cell subpopulation is going to be depleted (154). With this thought in mind, fibroblast activation protein (FAP) has been proposed as a potentially good target. FAP is a surface peptidase that also has gelatinase activity and is widely expressed in a subset of protumoral fibroblasts in many cancer types (155–157). FAP expression in pancreatic cancer (158, 159) and non-small cell lung cancer (160) is associated with worse clinical outcome. Depletion of FAP+ cells using genetic depletion strategies appeared to enhance T cell mediated antitumor activity in preclinical models of melanoma and pancreatic ductal adenocarcinoma (161–163). Antibodies against FAP have confirmed the suitability of FAP as a target by demonstrating efficient tumor stroma targeting capabilities in clinical trials (157). However, no therapeutic responses were observed, prompting the development of alternative strategies such as FAP antibody conjugates including immunostimulatory antibodies (164) and immunocytokines (165). One of those, an anti-FAP-IL-2v fusion protein, is currently being tested in clinical trials (NCT02627274, NCT03386721) (166). Alternatively, CAR-T cell therapy targeting FAP might be a more potent and efficacious strategy.
CAR-T Cells Targeting Fibroblasts: A Potential Double-Edged Sword
A number of groups have generated CAR-T cells targeted to mouse FAP and tested their ability to inhibit tumor growth. To date, eight studies have demonstrated antitumor activity of FAP-targeting CAR-T cells in several preclinical models including mesothelioma, lung, mammary, colon, pancreatic cancers (25–32), with a key measure of these studies being the potential for toxicity.
A key concern of targeting FAP is that, while it is highly expressed by CAFs and in wound healing, it is also expressed at low levels in healthy tissues including muscle, adipose tissue, bone marrow mesenchymal stem cells (BMMSCs), skin, and pancreas (167, 168). Complete ablation of FAP-expressing cells in mice using genetic approaches resulted in body weight loss, anemia, bone marrow hypoplasia and pancreatic toxicity (167). With these toxicities in mind, it is of interest to review the studies in which CAR-T cells targeting mouse FAP were tested, however, it is important to recognize that each study used a different single-chain fragment variable (scFv) antibody targeting FAP, different cytoplasmic domains, and different types of T cells (murine vs. human T cells).
Tran and colleagues observed minimal antitumor effect using a CAR with the FAP-5-scFv coupled with mouse CD28, 4-1BB, and CD3ζ intracellular signaling domains, but did observe severe toxicity indicated by significant cachexia and anemia (30). In contrast, Kakarla et al. showed that a FAP-CAR, using the MO35-scFv with human CD28 and CD3 derived domains, controlled tumor burden in a systemic lung carcinoma model without toxicity observed 2 days after T cell injection (27). However, this time point may be too early to see the negative effects exerted by the T cells.
The group at the University of Pennsylvania developed a FAP-CAR containing a scFv from the 73.3 anti-mouse FAP antibody and the human 41BB and CD3ζ intracellular domains (25, 28, 31, 32). These CAR-T cells slowed tumor growth in an immune-response dependent and independent manner in several tumor models in mice. Despite 73.3-FAP-CAR initial efficacy, CAR-T cells isolated from xenograft tumors became hypofunctional (28). Function was augmented by either using mouse T cells from mice lacking the inhibitory enzyme diacylgycerol kinase zeta (DGKZ) (32) or human T cells using the 73.3-CAR linked to the DAP12 signaling domain from natural killer (NK) cells (FAP-KIR CAR) (31). There was a link between enhanced CAR activity and toxicity: while no major toxicities were observed using the “basal” 73.3-FAP CAR-T cells, treatment with the more active DGKZ CAR-T cells resulted in a lymphocytic infiltrate observed in the pancreas (32). Likewise, treatment with the highly active FAP-KIR-CAR resulted in anemia, body weight loss and bone marrow hypoplasia (31). The “basal” 73.3-FAP-CAR targets cells with high FAP densities, like CAFs, while sparing low FAP expressing cells, which may provide a therapeutic window to obtain efficacy in the absence of toxicity. Unfortunately, the 73.3–FAP-CAR is mouse specific and cannot be used in the clinical setting.
There has been one reported clinical trial in which FAP CAR-T cells have been locally injected into the pleural effusion of mesothelioma patients (NCT01722149) (33). The authors reported the route of administration and the therapy to be safe in one patient (34) and, another patient showed stable disease for 1 year (26). Unfortunately, at the time of closure of the clinical trial in mid-2019 only 4 patients had been recruited.
In summary, FAP targeted CAR-T cells have clearly shown some antitumor activity in preclinical models, but they have also demonstrated the potential for toxicity. There does appear to be a viable therapeutic window, however. For this reason, it is likely that the role of FAP CAR-T cells will be in combination therapies. Combining FAP CAR-T cells with tumor-targeted CAR-T cells or with vaccines can result in additive or even synergistic effects (27, 32). Other target proteins like CD10 and GPR77 which identify a newly described CAF subpopulation with protumorigenic functions (169) provide alternative option for CAR development.
Tumor-Infiltrating Immune Cells as Barriers to Effective Car-T Cell Therapy
Solid tumors are highly infiltrated with immune cells such as Tregs, tumor-associated macrophages (TAMs) or MDSCs that contribute to the establishment of a hostile and immunosuppressive TME capable of limiting the efficacy of CAR-T cell therapy. In this section, we review the obstacles imposed by each of these cell populations and the different strategies that have been utilized in order for CAR-T cells to be efficacious in such context, as illustrated in Figure 2. These include strategies to directly target and deplete the immunosuppressive immune cell populations as well as indirect approaches consisting of genetically engineering the CAR-T cells to endow them with transgenes capable of modulating the TME or to confer them with resistance to immunosuppression.
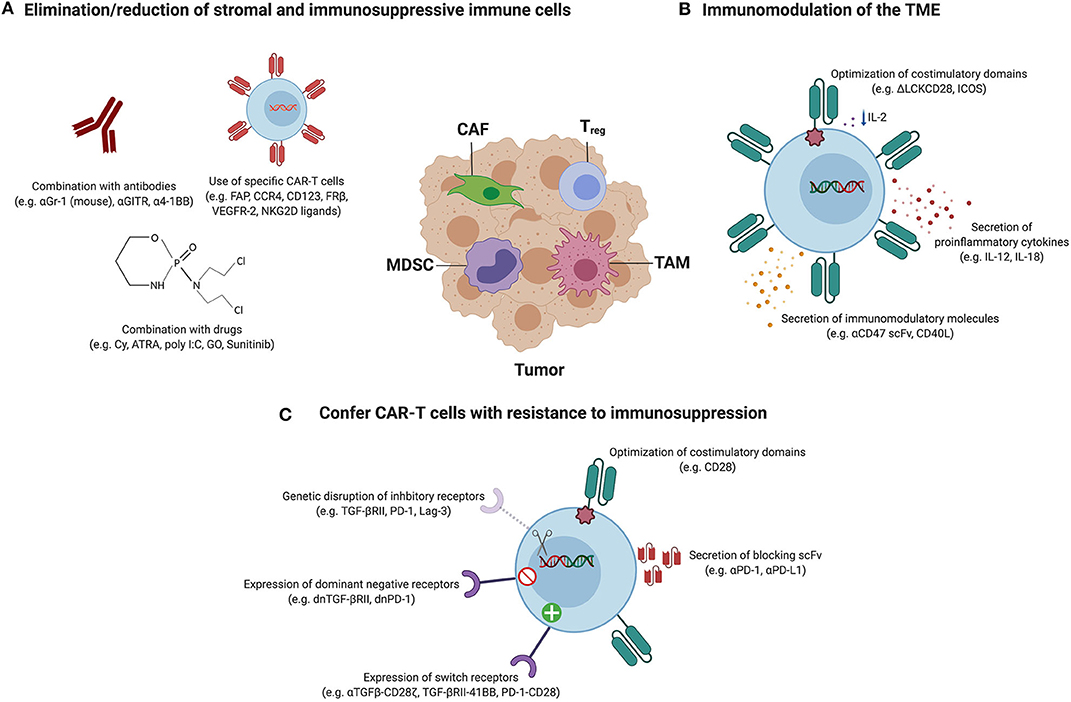
Figure 2. Therapeutic strategies to overcome the immunosuppressive TME. Tumors are infiltrated with stromal cells, such as cancer-associated fibroblasts (CAFs), and immune cells, including regulatory T cells (Tregs), tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) which support cancer progression and promote immunosuppression. Therapeutic strategies utilized to hit these components to enhance the efficacy of CAR-T cell therapy can be categorized in three classes, and the most relevant examples are represented in this figure. (A) Elimination or reduction of stromal and immunosuppressive immune cells: the combination of CAR-T cells with agents such as antibodies or drugs has resulted in decreased frequencies of Tregs and/or MDSCs. Alternatively, CARs have been designed to target antigens expressed on CAFs, Tregs, TAMs and MDSCs to directly deplete them. (B) Immunomodulation of the TME: this group of strategies aims at manipulating the TME to create a favorable environment that allows a better performance of the CAR-T cells. Some examples include the modulation of the tumor cytokine milieu by the expression of proinflammatory cytokines by CAR-T cells or by the optimization of costimulatory signaling domains in order to reduce IL-2 secretion and therefore impair Treg expansion and tumor infiltration. Immunomodulatory molecules that are able to polarize M2 TAMs into an antitumor M1 phenotype can also be expressed from CAR-T cells. (C) Confer CAR-T cells with intrinsic resistance to immunosuppression: CAR-T cells can be modified to be resistant to immunosuppression by endowing them with dominant-negative receptors (to disrupt signaling) or chimeric switch receptors (to convert negative signaling into positive), or by abrogating the expression of inhibitory receptors using genome-editing tools. Alternatively, antibodies blocking inhibitory receptors or ligands can be secreted by CAR-T cells. Also, it has been reported that the incorporation of particular costimulatory domains or the expression of some proinflammatory cytokines by CAR-T cells confer intrinsic resistance to Treg-mediated immunosuppression.
Regulatory T Cells
Tregs are a subset of T cells (phenotypically defined as CD4+CD25+FoxP3+) which play a crucial role in maintaining immune tolerance to self-antigens but can also suppress antitumor immunity (170). Cancer patients have increased numbers of Tregs in peripheral blood (171–173), and their presence in tumors is associated with poor prognosis in a variety of cancers (174–178).
Tregs suppress antigen-specific CD8+ T cell cytotoxicity by using different mechanisms: competitive consumption of IL-2; secretion of immunosuppressive cytokines such as IL-10 or TGF-β; CTLA-4-mediated suppression of antigen presenting cells (APCs); prevention of optimal T cell activation; or lysis of effector cells through the action of granzyme and/or perforin (170).
In the TME, Tregs play an inhibitory role in the antitumor efficacy of adoptively transferred tumor-targeted effector T cells (179). The frequency of Tregs in the blood of responder patients was lower than in samples obtained from non-responders in a combined analysis of multiple trials of adoptively transferred TILs (180). In a first-in-human study of an epidermal growth factor receptor variant III (EGFRvIII)-specific CAR in glioblastoma, the analysis of tumor specimens from patients who had post-treatment surgery revealed an increased influx of immunosuppressive Treg cells, which might have limited the antitumor effect of the CAR-T cell therapy (181). In addition, the importance of the effector to regulatory T cell balance in predicting responses to immunotherapy treatments has been highlighted (182).
A first and obvious way to address this limitation is to specifically eliminate Tregs. The combination of CAR-T cells with antibodies targeting GITR or 4-1BB (whose expression has been reported to be specific for tumor Tregs) has been explored in mice, resulting in decreased Treg frequencies and enhanced antitumor efficacy (40–43). The idea of directly depleting Tregs with a CAR has also been proposed by targeting the C-C chemokine receptor 4 (CCR4), which is expressed on T cell malignancies but also in Tregs (44).
Many clinical trials of CAR-T cells have failed to provide significant clinical benefit in the absence of prior lymphodepleting preconditioning (183–188). This lack of success may be explained, at least in part, by the fact that such preparative treatments are known to eradicate Tregs, which otherwise might suppress infused T cells (189). Illustrating this, the efficacy of CD19 CAR-T cells in a mouse model of lymphoma was completely abolished when Treg cells were previously injected and restored by preparative treatment with cyclophosphamide (45). Unfortunately, preconditioning regimens carry with them toxicities, which in some cases create a need for alternative strategies. In this line, several studies have demonstrated that IL-12 can help to overcome Treg-mediated immunosuppression and, therefore, the need for prior preconditioning. In mice, CAR-T cells engineered to constitutively produce IL-12 acquire intrinsic resistance to Tregs and are more efficacious in the absence of preconditioning (78, 79). Considering the potential clinical toxicity of constitutive IL-12 expression, safer approaches might involve the use of inducible systems to drive IL-12 production upon antigen recognition or the incorporation of elimination genes (80, 81). Constitutive IL-12-secreting mucin-16 ectodomain (MUC-16ecto)-specific CAR-T cells, which also express a truncated form of the human epidermal growth factor receptor (EGFRt) as a safety system, are currently being tested in an ovarian cancer phase I clinical trial (NCT02498912) (82, 83). CAR-T cells expressing alternative cytokines with safer clinical profiles such as IL-18 have also been tested in preclinical models with a similar impact on reducing tumor-infiltrating Treg numbers and improving antitumor activity (84).
A second, indirect approach to overcome CAR-T cell suppression by Tregs is to restrain their proliferation and survival by modulating the cytokines in the TME, specifically IL-2. IL-2 sustains the survival and function of both regulatory and effector T cells (190). In fact, IL-2 is often used to improve persistence of adoptively transferred T cells, albeit its administration leads to the expansion of Tregs in cancer patients (191). CAR-T cells release high levels of IL-2 upon antigen engagement, becoming a main source of this cytokine. It could be expected, then, that the use of CAR-T cells with reduced levels of secreted IL-2 would improve the antitumor efficacy of these engineered T cells as IL-2 would be no longer available to sustain Treg persistence. Cytokine levels can be modulated by selecting an appropriate co-stimulatory endodomain such as ICOS, which has been reported to generate CAR-T cells with increased IL-17 production and reduced secretion of IL-2 (46). Alternatively, more conventional co-stimulatory domains such as CD28 can be mutated for the same purpose. It is known that IL-2 secretion is initiated by CD28-mediated LCK recruitment and phosphorylation, therefore, the mutation of the LCK binding domain abolishes IL-2 secretion by CAR-T cells (52). This modification improved antitumor efficacy of CAR-T cells in the presence of previously inoculated Treg cells, which persisted less, compared to mice treated with CAR-T cells containing the wild type CD28 endodomain (52).
In apparent contradiction with this study, several groups report that the incorporation of a CD28 co-stimulatory domain in different CAR platforms provides increased resistance to Treg-mediated immunosuppression, and more specifically, to TGF-β-mediated suppression of T cell proliferation (47–50). Conversely, while sustaining Treg survival and function, IL-2 induced by CD28 activation of LCK and autocrine signaling through IL-2 receptor on tumor-specific effector T cells appears to be crucial to counteract the inhibitory effects of TGF-β (48, 49). In fact, the deletion of the LCK binding domain in CD28 reverted resistance to TGF-β-mediated suppression (49). One strategy proposed to compensate for the detrimental effect of LCK mutation while maintaining the benefits of abrogating IL-2 secretion is the addition of a 4-1BB co-stimulatory signaling domain (51). A different approach is the use of alternative cytokines to replace the CD28-induced IL-2 autocrine loop. For instance, CAR-T cells can be engineered to express IL-7Rα so that IL-7 can support their function (53). In a more sophisticated approach, CAR-T cells with a disrupted IL-2 axis can be engineered to release transgenic IL-7 and to co-express an IL-7Rα/IL-2β hybrid receptor to provide cell-intrinsic IL-2 signaling through IL-7 (49). Alternatively, cytokine stimulation can be provided by IL-15 through the expression of a tethered membrane-bound IL-15, which has been shown to favor the persistence and survival of CAR-T cells with a clinically desirable immature state of differentiation (54). This interesting approach avoids undesirable effects of soluble IL-15 coadministration or constitutive secretion by CAR-T cells such as o toxicity (192) or promotion of Tregs (193). In addition, there is great excitement on the use of engineered IL-2 mutants designed to preferentially signal into effector T cells but not Tregs, although this strategy has not yet been tested in the context of CAR-T cells (194–196).
Besides suppressing T cell proliferation, TGF-β induces a Treg-like phenotype on CAR-T cells (77). Therefore, conferring CAR-T cells with intrinsic resistance to TGF-β represents an opportunity for improvement. TGF-β signaling in CAR-T cells can be abrogated by knocking out TGF-βRII through CRISPR/Cas9 technology (77). In the same line, CAR-T cells can be endowed with a TGF-β dominant-negative receptor (dnTGF-βRII). A first-in-human trial in patients with refractory castration-resistant metastatic prostate cancer has been initiated with a prostate-specific membrane antigen (PSMA)-specific CAR incorporating this receptor (NCT04227275) (74). Alternatively, switch receptors can be created by fusing the extracellular part of the TGF-βRII to the endodomain of 4-1BB or by linking a TGF-β-specific scFv to the CD28-CD3ζ intracellular signaling domains, rendering CAR-T cells capable of converting the immunosuppressive signal from soluble TGF-β into an immunostimulatory one (75, 76).
Tumor-Associated Macrophages
TAMs are the most abundant immune cells infiltrating human cancers and their accumulation in tumors correlates with poor prognosis in a broad range of tumor types (197, 198). TAMs can sustain cancer progression by secreting growth factors which stimulate tumor cell proliferation, proteolytic enzymes that promote matrix remodeling and facilitate metastasis, proangiogenic factors which support angiogenesis, or reactive oxygen species (ROS) and nitric oxide (NO) that induce genetic instability on tumor cells (199). Furthermore, TAMs can suppress T cell-mediated antitumor immunity by releasing IL-10 and TGF-β, amino acid-depleting enzymes such as arginase 1 or indoleamine 2,3-dioxygenase (IDO) which cause metabolic starvation on T cells or prostaglandins with immunosuppressive effects, or by expressing immune checkpoint ligands like PD-L1, PD-L2, B7-H4, or VISTA. Moreover, TAMs can promote the recruitment and immunosuppressive activity of Tregs (199). TAMs can also prevent T cell-mediated antitumor immune responses by physically creating long-lasting interactions with CD8+ T cells, thus excluding them from tumors (200).
There is overt preclinical evidence of that TAMs can mediate resistance to immunotherapy, including adoptive cell transfer therapy. For instance, the depletion of TAMs through the administration of a CSF-1R inhibitor improved the efficacy of adoptively transferred tumor-specific T cells in syngeneic mouse models of melanoma (200). Superior antitumor activity of the combined treatment correlated with a decrease in the number of intratumoral macrophages, which subsequently facilitated an increase in expansion, intratumoral accumulation and functionality of the adoptively transferred T cells (201).
In the field of CAR-T cell therapy, the infusion of GD2-specific CAR-T cells in neuroblastoma patients provoked a striking expansion of circulating macrophages with immunosuppressive phenotype suggesting a role of macrophages limiting the antitumor efficacy (88).
Despite their overall tumor-promoting functions, certain subpopulations of TAMs can sustain antitumor activities including phagocytosis, antigen-presenting, or the release of proinflammatory cytokines such as TNF-α and IL-12. Indeed, in certain contexts, macrophages have been proven crucial for the development of effective immunotherapy (202–204). Several strategies have been proposed to either reprogram immunosuppressive “M2-like” TAMs into an antitumor “M1-like” phenotype which could cooperate with CAR-T cells to induce tumor regression, or to directly deplete TAMs to facilitate productive antitumor immunity.
One strategy of TAM reeducation consists in making them more phagocytic. CD47 is expressed on tumor cells and interacts with SIRPα expressed on macrophages to deliver a “don't eat me” signal. CAR-T cells can be engineered to express CD47-blocking antibodies in order to prevent that interaction, thus stimulating phagocytosis of tumor cells and improving engagement of the innate immune system (58, 59). A clever approach to hijack the phagocytic capacities of TAMs and redirect them toward tumor-associated antigens is to engineer macrophages themselves to express a CAR. Interestingly, macrophage transduction with chimeric adenoviral vectors promoted a gene expression change toward a proinflammatory M1 phenotype, which subsequently converted bystander M2 TAMs into an M1 phenotype and boosted endogenous antitumor T cell responses (60).
TAMs can also be manipulated to become more functionally activated. CD40 is expressed in antigen presenting cells (APCs) including dendritic cells (DCs), B cells, monocytes and macrophages. Interaction of CD40 with its ligand, CD40L, is known to induce activation and IL-12 secretion by APCs. Preliminary studies using a bispecific antibody to mediate the interaction between a c-myc tag on CAR-T cells and CD40 on APCs demonstrated enhanced CAR-T cell function (61). Constitutive expression of CD40L by CAR-T cells improved their therapeutic efficacy in part through the induction of maturation and IL-12 secretion by monocyte-derived DCs and macrophages (62, 63). By means of a different pathway, the administration of the multikinase inhibitor sorafenib in combination with CAR-T cells also induced an increase in IL-12 production by TAMs which contributed to antitumor activity (64).
Not surprisingly, cytokines secreted by CAR-T cells upon antigen encounter can alter the TME and convert TAMs from immunosuppressive to immunostimulatory. For instance, secretion of GM-CSF and IFN-γ by CAR-T cells upon antigen engagement has been shown to elicit a recruitment of myeloid cells to the TME and to activate newly recruited as well as re-educate resident suppressive TAMs thus potentiating their IL-12 production, capacity of antigen presentation, and tumoricidal activity (205). Armoring CAR-T cells with additional cytokines can improve their capacity to modulate the TME. In mice, inducible IL-12 secretion by CAR-T cells resulted in the recruitment of activated TNF-α-producing macrophages which directly contributed to tumor elimination in a TNF-α-dependent manner (206). In addition, IL-12 secretion by CAR-T cells indirectly mediated the depletion of TAMs as a result of Fas engagement on TAMs by FasL on CAR-T cells and altered the phenotype of remaining TAMs toward a proinflammatory one (83). IL-18-secreting CAR-T cells also led to a reduction in “M2-like” macrophages in tumors as well as Tregs (84).
A different strategy to overcome immunosuppression in the TME is to develop CARs that target antigens expressed by TAMs to directly eliminate them. CAR-T cells targeting the antigen CD123, with shared expression in malignant cells and TAMs, have been proposed for the treatment of Hodgkin lymphoma which contains a highly immunosuppressive TME (55). Alternatively, rather than hitting all macrophages by using a pan-macrophage target, it would be desirable to design CARs that are able selectively deplete TAMs with protumor “M2-like” properties while sparing other TAM populations with antitumor “M1-like” functions. CAR-T cells targeting folate receptor β (FRβ), which is expressed only in the immunosuppressive TAM population, have been developed for that aim (56). Similarly, CAR-T cell targeting B7-H4, a molecule expressed by cancer cells and TAMs, mediated antitumor responses in a preclinical ovarian cancer model, but was also toxic due to possible targeting of tissue resident macrophages (57).
Finally, CAR-T cells can also be combined with agents that protect them from TAM-related immunosuppressive pathways, such as that mediated by IDO. IDO is produced by tumor cells and TAMs and mediates the metabolism of tryptophan into immunosuppressive metabolites that can suppress CAR-T cell function. The use of IDO inhibitors or preconditioning with fludarabine, which can inhibit IDO expression, are strategies that can be used to improve the activity of CAR-T cells in immunosuppressive microenvironments (85).
Myeloid-Derived Suppressor Cells
MDSCs are a highly diverse population of immature myeloid cells which include two major subsets: the mononuclear MDSCs (M-MDSCs), which are morphologically and phenotypically similar to monocytes and can differentiate into TAMs, and the polymorphonuclear MDSC (PMN-MDSCs), which resemble neutrophils and are precursors of tumor-associated neutrophils (TANs), as well as a small group of myeloid progenitors (207). MDSCs play a role in supporting tumor progression, and according to a meta-analysis of the literature, their accumulation is associated with poor clinical outcome in cancer patients (208). The hallmark feature of MDSCs is their strong capacity to inhibit immune responses, with T cells being the main targets of these effects. Mechanisms implicated in MDSC-induced immunosuppression are common to those reported for TAMs, including production of NO and ROS, elimination of key nutrition factors needed for T cell proliferation such as arginine, cysteine, or tryptophan, production of IL-10 and TGF-β, and induction of Tregs (209). MDSCs have also been implicated in limiting the effects of CAR-T cell therapy. In a clinical trial of third generation CD19 CAR-T cell therapy, low levels of M-MDSCs was associated with response in patients with lymphoma and leukemia (210).
The detrimental effect of MDSCs on CAR-T cell proliferation and cytolytic function has been demonstrated by using CARs targeting a number of different antigens (65–67). As a proof of concept, depletion of MDSCs with anti-Gr-1 antibody resulted in improved antitumor efficacy of CAR-T cells in mouse models (40, 65, 66). Unfortunately, the lack of a suitable marker for human MDSCs prevents their targeting by using a single antibody. It has been demonstrated that GM-CSF and STAT3 signaling through GM-CSF and/or IL-6 can drive the expansion of MDSCs and support PD-L1 expression by these cells, promoting suppression of CAR-T cells through the PD-1/PD-L1 axis. Therefore, GM-CSF neutralization, STAT3 inhibition or PD-L1 blockade might represent alternative targets to limit the impact of MDSCs in humans (65, 68). The combination of CAR-T cells with compounds such as polyinosinic-polycytidylic acid (poly I:C), all-trans retinoic acid (ATRA), gemtuzumab ozogamicin (GO) or sunitinib also resulted in improved antitumor efficacy attributed to a reduction in the content and suppressive function of MDSCs (66, 67, 69, 70).
Interestingly, some studies combining CAR-T cell therapy with anti-PD-1 or anti-4-1BB antibodies have reported a decrease in the percentage of MDSCs in the TME, correlating with improved antitumor effects (43, 71). However, mechanisms underlying MDSC depletion mediated by immune checkpoint blockade are not fully understood.
A more direct approach of depleting MDSCs by using CAR-T cell therapy is to target antigens expressed on their surface. For instance, CAR-T cells targeting tumor vasculature through VEGFR-2 were able to reduce the frequency of MDSCs in the TME, which also expressed VEGFR-2 (72). Parihar and colleagues engineered NK cells to express a chimeric activating receptor comprised of the extracellular domain of NKG2D receptor fused to the T cell signaling domain CD3ζ (73). Engineered NK cells achieved efficient depletion of MDSCs, which express NKG2D ligands, and increased the recruitment and tumor infiltration of tumor-specific CAR-T cells when given in combination.
Neutrophils can also be immunosuppressive in the context of cancer, and their presence in tumors has been associated with poor outcome (211). In a CAR-T cell therapy trial targeting CEA, increased neutrophil to lymphocyte ratios correlated with poor responses in colon cancer patients with liver metastasis (212). Like TAMs, tumor-associated neutrophils (TANs) can be generally classified into antitumorigenic “N1” or protumorigenic “N2” phenotypes (213). Although strategies to target “N2” TANs have not been reported yet in the context of CAR-T cell therapy, some of the above-mentioned strategies could be used to counteract immunosuppressive pathways common with Tregs, TAMs, or MDSCs.
Inhibitory Receptors and Their Ligands
Tumor cells, tumor-infiltrating immune cells and tumor-derived exosomes frequently express an array of ligands that bind to inhibitory receptors on T cells to suppress antitumor immunity. Blocking these interactions with therapeutic antibodies, known as immune checkpoint inhibitors, releases the brakes from suppressed T cells, allowing them to recover their antitumor activity. This therapeutic approach can mediate long-term responses, especially in a subset of tumors that are infiltrated with neoantigen-specific T cells. Therapeutic antibodies targeting the inhibitory receptors CTLA-4 and PD-1 or the PD-1 ligand PD-L1 have been approved for clinical use in patients with different solid cancer types (9). Checkpoint blockade has revolutionized cancer treatment, highlighting the tremendous power of T cells in controlling solid tumors.
Among the different immune checkpoints, the PD-1/PD-L1 axis has gained increasing attention. PD-1 is expressed in the surface of activated or dysfunctional T cells, while PD-L1 is frequently expressed in the surface of tumor cells and immune cells, and can also be found in extracellular forms (214, 215). PD-L1 upregulation is mainly associated with IFN-γ release in response to T cell activation (216); however more recent findings suggest that multiple cytokines found in the TME (including IL-10, IL-1α, IL-27, and IL-32γ) can induce PD-L1 expression (217). Of note, some cancer cells can constitutively express the PD-L1 gene due to hypomethylation of its promoter, while TAMs have been reported to also express PD-L1 naturally or via trogocytosis from tumor cells (218). Expression of PD-L1 in the tumor restrain tumor infiltrating lymphocytes from full and persistent activation. Moreover, PD-L1 expression in the stroma can prevent T cells from infiltrating the tumor, excluding them to the margin of the tumor (219). Blocking the PD-1-PD-L1 interaction can promote T cell proliferation and infiltration into the tumor, and results in durable antitumor responses (219).
The success of checkpoint immune therapies targeting CTLA-4 or the PD-1/PD-L1 axis has prompted intense investigation into new inhibitory receptors, including TIM-3, LAG-3, and TIGIT. A new wave of therapeutic agents targeting these receptors are being investigated in clinical trials, with encouraging initial results (220). However, little is known about the biology of these receptors and the interactions with their ligands. TIM-3 ligands include the cell surface ligands Ceacam-1 and Phosphatidyl serine-PTdSer (221) and the soluble factors, Galectin-9 (222) and HMGB1, that are released to the TME. LAG-3 also interacts with various ligands in the TME, including MHC class II expressed in APC and tumor cells; Galectin-3 (223) and LSECtin, expressed on tumor-associated stromal cells and tumor cells; and FGL-1, a soluble factor produced in some tumors (224). TIGIT interacts with the ligands CD112 and CD155, which are expressed on APCs and tumor cells. Expression of these ligands in tumors is associated with tumor progression and inhibition of antitumor T cell responses (224–227).
Releasing the Breaks on CAR-T Cells
A promising strategy to increase the antitumor efficacy of CAR-T cells is to prevent or revert T cell dysfunction driven by engagement of inhibitory receptors with their ligands in the tumor. Upon antigen recognition, CAR-T cells up-regulate different inhibitory receptors, similarly to endogenous tumor-specific T cells. CAR-T cells isolated from xenograft tumors typically express high PD-1 levels, with a fraction of these cells co-expressing TIM-3 and LAG-3 (86, 228). Overexpression of PD-L1 by tumor cells has been shown to inhibit CAR-T cell function, while combining CAR-T cell therapy with antibodies that block the PD1/PD-L1 interaction has proved to increase the antitumor effects of each therapy alone (71, 86, 87). One study using syngeneic mouse models showed therapeutic responses when combining CAR-T cells with PD1-blocking antibodies, which was correlated with a decrease in MDSCs (71). Several ongoing clinical trials are testing the combination of CAR-T cells with anti-PD-1/PD-L1 blocking antibodies in patients with hematologic malignancies or solid tumor (NCT02414269, NCT01822652, NCT03980288, NCT03726515), with some preliminary results with small groups of patients showing safety and encouraging efficacy results (88–90).
Novel alternative approaches to target the PD-1/PD-L1 axis include the genetic modification of CAR-T cells to release a PD-1- or PD-L1-blocking scFv in the tumor (92, 93), to express PD-1 dominant negative receptors (86), or chimeric switch receptors (94). These strategies may avoid the toxicities associated with systemic delivery of checkpoint inhibitors and bypass the requirement for repeated antibody administration. Expression of chimeric switch receptors has the advantage of converting an inhibitory signal (PD-1) into a costimulatory signal (i.e., CD28) (94). Compared to PD-1 chimeric receptors, the delivery of PD-1 or PD-L1 blocking antibodies (by combination therapy or genetic modification) offers the possibility to re-invigorate endogenous tumor-specific T cells (92), which may be required to achieve complete responses in solid tumors. In this line, combination of CAR-T cells with oncolytic viruses releasing an anti-PD-L1 mini-body locally in the tumor resulted in enhanced therapeutic effects (91). Oncolytic viruses provide a danger signal able to diminish tumor immunosuppression while inducing tumor debulking, and may be ideal partners to combine with CAR-T cells and immune checkpoint inhibitors (229).
Another strategy to counteract tumor-induced T cell inhibition is to disrupt T cell inhibitory receptors by genome editing. Several studies have demonstrated that PD-1 gene editing, using TALEN or the CRISPR/Cas9 system, can augment T cell-mediated killing in vitro and enhance clearance of PD-L1+ tumors in vivo (95–97). However, reported in vivo results testing this strategy seem to be contradictory and conflicting. Recent studies suggest that PD-1 ablation or knockdown can accelerate T cell exhaustion, prevent memory formation and reduce long-term antitumor efficacy (230, 231). Enhanced antitumor effects with PD-1 knockout (KO) CAR-T cells are usually observed in animal experiments using tumor cell lines genetically modified to express constitutive and uniform levels of PD-L1. So, it is possible that PD-1 disruption is only beneficial in tumors with high PD-L1 tumor densities. Different clinical trials are actively testing PD-1 KO engineered T cells for the treatment of solid tumors (NCT03747965, NCT03525782, NCT03706326, NCT03399448). A first-in-human phase 1 clinical trial has recently published the safety and feasibility of deleting three genes (TRAC, TRBC, and PDCD1, the gene encoding PD-1) using CRISPR-Cas9 in cancer-specific T cells for the treatment of patients with refractory cancer (98). Initial results in three patients demonstrated engraftment of PD-1–deficient T cells with no evidence of autoimmunity or T cell genotoxicity. Surprisingly, it was found that, in one patient, the percentage of tumor-specific T cells with mutations in the PD-1 locus decreased from 25% in the infusion product to 5% 4 months post-infusion. While further investigations are required to interpret these results, loss of PD-1 edited T cells would be consistent with mouse studies highlighting the role of PD-1 in preserving T cells from overstimulation and terminal differentiation. In this same line, initial reports have established the feasibly of knocking out other inhibitory receptors, such as CTLA-4 or LAG-3, but it remains unclear as to whether these modifications result in enhanced CAR-T cell activity (99, 100). A better understanding on the mechanisms by which inhibitory receptor negatively regulate T cell function together with preclinical models that better recapitulate the TME are required to design the next-generation CAR-T cell therapies.
Conclusions and Future Directions
Unprecedented durable responses in cancer patients treated with checkpoint blockade antibodies or CAR-T cell therapy is generating considerable optimism. Augmenting the therapeutic outcome of CAR-T cell therapy in the context of solid tumors represents the next big challenge and opportunity for the field. Clearly, a major obstacle for CAR-T cells in solid tumors is the immunosuppressive TME. There is now an understanding that physical barriers and stromal and immune cells that express and release an array of immunosuppressive molecules limit CAR-T cell persistence and efficacy. In these hostile circumstances, strategies aimed at remodeling the tumor microenvironment or conferring intrinsic CAR-T cell resistance to immunosuppression may be more promising than targeting only one specific pathway. The cellular component of TME is characterized by considerable diversity and a high degree of plasticity (232, 233). Several strategies directed to regulating this plasticity and reversing immunosuppression are being explored. Armored CAR-T cells expressing proinflammatory cytokines or combination of CAR-T cells with oncolytic viruses could serve this purpose (234). Gene ablation technology will allow CAR-T cells to avoid immunosuppresssive signals in the TME. By a different approach, direct elimination of stroma or immune suppressive cells could revert immunosuppression, tackling different pathways simultaneously. Ongoing efforts seek to develop a new generation of CAR-T cell therapies targeting fibroblasts, Tregs, M2 macrophages or MDSCs.
Other factors such as the effect of gut microbiota on response to immune therapies might be also considered. It has been recently reported by many groups that microbiome composition modulates the antitumor response to immune checkpoint inhibitors. This effect is described to be mediated by IL-12 and to correlate with a decrease of Tregs and MDSCs in the TME (235). Similar observations have been made in preclinical mouse studies in the context of adoptive cell transfer therapy (236). In the field of CAR-T cell therapy, a preliminary study of microbiota composition in cancer patients prior to CAR-T cells infusion found a correlation between the presence of certain bacterial families and efficacy and toxicity of the therapy (237). This observation warrants future consideration of strategies such as the use of specific antibiotics or fecal microbial transplantation in combination with CAR-T cell therapy (238).
One of the greatest challenges in developing effective and safe CAR-T cells that tackle the TME is the lack of clinically relevant models that reflect the challenges of solid tumors. Currently available preclinical models have been unable to predict the toxicities observed in clinical trials and the lack of antitumor activity, especially in patients with solid tumors. Advanced preclinical models relevant to study the impact of tumor heterogeneity and the role of the TME in CAR-T cell efficacy are required to test the next-generation of CAR-T cells as monotherapy or in combination with other agents. The testing of such CAR-T cell approaches in canines with spontaneous solid cancer represents a promising avenue of investigation (239). Current clinical studies will hopefully reveal information on the safety and efficacy of novel CAR-T cell approaches, including those addressing barriers of the TME. Lessons learned from these early-phase clinical trials will be important to continue to develop novel CAR-T cell therapies for the treatment of solid tumors.
Author Contributions
SG and AR-G conceptualized, wrote, and edited the manuscript. AP and EN-O wrote and edited the manuscript. AR-G and AP designed the figures. DP edited the manuscript.
Funding
AR-G has received funding from the Ovarian Cancer Research Alliance (OCRA) grant agreement number 599349. AP has received funding from the European Research Council (ERC), grant agreement number 804236 (Horizon 2020). SG has received funding from the European Union's Horizon 2020 research and innovation program (Marie Sklodowska-Curie, 839566) and from the Spanish Ministry of Science and Innovation under a Ramon y Cajal grant (RYC2018-024442-I).
Conflict of Interest
DP and SG are inventors on patents related to CAR-T cell therapy, filed by the University of Pennsylvania and licensed to Novartis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Steven M. Albelda for helpful comments and feedback on this manuscript. Figures were created with biorender.com.
References
1. Guedan S, Calderon H, Posey AD, Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Develop. (2019) 12:145–56. doi: 10.1016/j.omtm.2018.12.009
2. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
3. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
4. Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. (2019) 25:1341–55. doi: 10.1038/s41591-019-0564-6
5. D'Agostino M, Raje N. Anti-BCMA CAR T-cell therapy in multiple myeloma: can we do better? Leukemia. (2019) 34:21–34. doi: 10.1038/s41375-019-0669-4
6. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
7. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-Specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
8. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
9. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. (2018) 359:1350–5. doi: 10.1126/science.aar4060
10. Guedan S, Ruella M, June CH. Emerging cellular therapies for cancer. Annu Rev Immunol. (2019) 37:145–71. doi: 10.1146/annurev-immunol-042718-041407
11. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Dis. (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
12. Watanabe K, Kuramitsu S, Posey AD Jr, June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol. (2018) 9:2486. doi: 10.3389/fimmu.2018.02486
13. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. C-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. (2019) 576:293–300. doi: 10.1038/s41586-019-1805-z
14. Calderon H, Mamonkin M, Guedan S. (2020) Analysis of CAR-Mediated Tonic Signaling. In: Swiech K, Malmegrim KCR, Picanço-Castro V, et al. editors. Chimeric Antigen Receptor T Cells: Development Production(New York, NY: Springer). 223–36. doi: 10.1007/978-1-0716-0146-4_17
15. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
16. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
17. Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. (2013) 25:268–76. doi: 10.1016/j.coi.2013.02.009
18. Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest. (2010) 120:3953–68. doi: 10.1172/JCI43490
19. Wang W, Ma Y, Li J, Shi HS, Wang LQ, Guo FC, et al. Specificity redirection by CAR with human VEGFR-1 affinity endows T lymphocytes with tumor-killing ability and anti-angiogenic potency. Gene Ther. (2013) 20:970–8. doi: 10.1038/gt.2013.19
20. Santoro SP, Kim S, Motz GT, Alatzoglou D, Li C, Irving M, et al. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res. (2015) 3:68. doi: 10.1158/2326-6066.CIR-14-0192
21. Byrd TT, Fousek K, Pignata A, Szot C, Samaha H, Seaman S, et al. Croix B, Ahmed N. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res. (2018) 78:489. doi: 10.1158/0008-5472.CAN-16-1911
22. Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci USA. (2019) 116:7624–31. doi: 10.1073/pnas.1817147116
23. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. (2015) 21:524–9. doi: 10.1038/nm.3833
24. Zhang W, Liu L, Su H, Liu Q, Shen J, Dai H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br J Cancer. (2019) 121:837–45. doi: 10.1038/s41416-019-0578-3
25. Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. (2015) 75:2800–10. doi: 10.1158/0008-5472.CAN-14-3041
26. Gulati P, Ruhl J, Kannan A, Pircher M, Schuberth P, Nytko KJ, et al. Aberrant Lck signal via CD28 costimulation augments antigen-specific functionality and tumor control by redirected T cells with PD-1 blockade in humanized mice. Clin Cancer Res. (2018) 24:3981–93. doi: 10.1158/1078-0432.CCR-17-1788
27. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. (2013) 21:1611–20. doi: 10.1038/mt.2013.110
28. Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. (2014) 20:4262–73. doi: 10.1158/1078-0432.CCR-13-2627
29. Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med. (2013) 11:187. doi: 10.1186/1479-5876-11-187
30. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. (2013) 210:1125–35. doi: 10.1084/jem.20130110
31. Wang E, Wang LC, Tsai CY, Bhoj V, Gershenson Z, Moon E, et al. Generation of Potent T-cell immunotherapy for cancer using DAP12-based, multichain, chimeric immunoreceptors. Cancer Immunol Res. (2015) 3:815–26. doi: 10.1158/2326-6066.CIR-15-0054
32. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
33. Petrausch U, Schuberth PC, Hagedorn C, Soltermann A, Tomaszek S, Stahel R, et al. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC Cancer. (2012) 12:615. doi: 10.1186/1471-2407-12-615
34. Pircher MSP, Gulati P, Sulser S, Weder W, Curioni A, Renner, Petrausch U. FAP-specific re-directed T cells first in-man study in malignant pleural mesothelioma: experience of the first patient treated. J ImmunoTher Cancer. (2015) 3 (Suppl. 2):120. doi: 10.1186/2051-1426-3-S2-P120
35. Teijeira A, Labiano S, Garasa S, Etxeberria I, Santamaría E, Rouzaut A, et al. Mitochondrial morphological and functional reprogramming following CD137 (4-1BB) costimulation. Cancer Immunol Res. (2018) 6:798–811. doi: 10.1158/2326-6066.CIR-17-0767
36. Zhang H, Snyder KM, Suhoski MM, Maus MV, Kapoor V, June CH, et al. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol. (2007) 179:4910–8. doi: 10.4049/jimmunol.179.7.4910
37. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
38. Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. (2017) 7:39833. doi: 10.1038/srep39833
39. Cui J, Zhang Q, Song Q, Wang H, Dmitriev P, Sun MY, et al. Targeting hypoxia downstream signaling protein, CAIX, for CAR T-cell therapy against glioblastoma. Neuro Oncol. (2019) 21:1436–46. doi: 10.1093/neuonc/noz117
40. Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, et al. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. (2016) 23:142–8. doi: 10.1038/cgt.2016.14
41. Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, et al. Antibodies to Costimulatory Receptor 4-1BB Enhance Anti-tumor Immunity via T Regulatory Cell Depletion and Promotion of CD8 T Cell Effector Function. Immunity. (2018) 49:958–70 e7. doi: 10.1016/j.immuni.2018.09.014
42. Freeman ZT, Nirschl TR, Hovelson DH, Johnston RJ, Engelhardt JJ, Selby MJ, et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J Clin Invest. (2020) 3:1405–16. doi: 10.1172/JCI128672
43. Mardiana S, John LB, Henderson MA, Slaney CY, von Scheidt B, Giuffrida L, et al. A multifunctional role for adjuvant anti-4-1BB therapy in augmenting antitumor response by chimeric antigen receptor T cells. Cancer Res. (2017) 77:1296–309. doi: 10.1158/0008-5472.CAN-16-1831
44. Perera LP, Zhang M, Nakagawa M, Petrus MN, Maeda M, Kadin ME, et al. Chimeric antigen receptor modified T cells that target chemokine receptor CCR4 as a therapeutic modality for T-cell malignancies. Am J Hematol. (2017) 92:892–901. doi: 10.1002/ajh.24794
45. Lee JC, Hayman E, Pegram HJ, Santos E, Heller G, Sadelain M, et al. In vivo inhibition of human CD19-targeted effector T cells by natural T regulatory cells in a xenotransplant murine model of B cell malignancy. Cancer Res. (2011) 71:2871–81. doi: 10.1158/0008-5472.CAN-10-0552
46. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. (2014) 124:1070–80. doi: 10.1182/blood-2013-10-535245
47. Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner MK. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. (2006) 20:1819–28. doi: 10.1038/sj.leu.2404366
48. Koehler H, Kofler D, Hombach A, Abken H. CD28 costimulation overcomes transforming growth factor-beta-mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res. (2007) 67:2265–73. doi: 10.1158/0008-5472.CAN-06-2098
49. Golumba-Nagy V, Kuehle J, Hombach AA, Abken H. CD28-zeta CAR T Cells Resist TGF-beta repression through il-2 signaling, which can be mimicked by an engineered IL-7 autocrine loop. Mol Ther. (2018) 26:2218–30. doi: 10.1016/j.ymthe.2018.07.005
50. Kegler A, Koristka S, Bergmann R, Berndt N, Arndt C, Feldmann A, et al. T cells engrafted with a UniCAR 28/z outperform UniCAR BB/z-transduced T cells in the face of regulatory T cell-mediated immunosuppression. Oncoimmunology. (2019) 8:e1621676. doi: 10.1080/2162402X.2019.1621676
51. Suryadevara CM, Desai R, Farber SH, Choi BD, Swartz AM, Shen SH, et al. Preventing Lck Activation in CAR T Cells Confers Treg Resistance but Requires 4-1BB Signaling for Them to Persist and Treat Solid Tumors in Nonlymphodepleted Hosts. Clin Cancer Res. (2019) 25:358–68. doi: 10.1158/1078-0432.CCR-18-1211
52. Kofler DM, Chmielewski M, Rappl G, Hombach A, Riet T, Schmidt A, et al. CD28 costimulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory t cells which can be overcome by preventing Lck activation. Mol Ther. (2011) 19:760–7. doi: 10.1038/mt.2011.9
53. Perna SK, Pagliara D, Mahendravada A, Liu H, Brenner MK, Savoldo B, et al. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res. (2014) 20:131–9. doi: 10.1158/1078-0432.CCR-13-1016
54. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proceedings of the National Academy of Sciences. (2016) 113:E7788. doi: 10.1073/pnas.1610544113
55. Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO, et al. Overcoming the immunosuppressive tumor microenvironment of hodgkin lymphoma using chimeric antigen Receptor T cells. Cancer Discov. (2017) 7:1154–67. doi: 10.1158/2159-8290.CD-16-0850
56. Rodriguez-Garcia A, Lynn RC, Matsuyama T, Powell DJ. ASGCT 21st Annual Meeting Abstracts. Mol Ther. (2018) 26:1–459. doi: 10.1016/j.ymthe.2018.05.001
57. Smith JB, Lanitis E, Dangaj D, Buza E, Poussin M, Stashwick C, et al. Tumor regression and delayed onset toxicity following B7-H4 CAR T cell therapy. Mol Ther. (2016) 24:1987–99. doi: 10.1038/mt.2016.149
59. Xie YJ, Dougan M, Ingram JR, Pishesha N, Fang T, Momin N, et al. Improved anti-tumor efficacy of chimeric antigen receptor T cells that secrete single-domain antibody fragments. Cancer Immunol Res. (2020) 4:518–29. doi: 10.1158/2326-6066.CIR-19-0734
60. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. (2020) doi: 10.1038/s41587-020-0462-y. [Epub ahead of print].
61. von Scheidt B, Wang M, Oliver AJ, Chan JD, Jana MK, Ali AI, et al. Enterotoxins can support CAR T cells against solid tumors. Proc Natl Acad Sci U S A. (2019) 116:25229–35. doi: 10.1073/pnas.1904618116
62. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. (2015) 23:769–78. doi: 10.1038/mt.2015.4
63. Kuhn NF, Purdon TJ, van Leeuwen DG, Lopez AV, Curran KJ, Daniyan AF, et al. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell. (2019) 35:473–88 e6. doi: 10.1016/j.ccell.2019.02.006
64. Wu X, Luo H, Shi B, Di S, Sun R, Su J, et al. Combined antitumor effects of sorafenib and gpc3-car t cells in mouse models of hepatocellular carcinoma. Mol Ther. (2019) 27:1483–94. doi: 10.1016/j.ymthe.2019.04.020
65. Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. (2015) 64:817–29. doi: 10.1007/s00262-015-1692-6
66. Di S, Zhou M, Pan Z, Sun R, Chen M, Jiang H, et al. Combined adjuvant of poly i:c improves antitumor effects of CAR-T cells. Front Oncol. (2019) 9:241. doi: 10.3389/fonc.2019.00241
67. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res. (2016) 4:869–80. doi: 10.1158/2326-6066.CIR-15-0230
68. Guha P, Gardell J, Darpolor J, Cunetta M, Lima M, Miller G, et al. STAT3 inhibition induces Bax-dependent apoptosis in liver tumor myeloid-derived suppressor cells. Oncogene. (2019) 38:533–48. doi: 10.1038/s41388-018-0449-z
69. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine. (2019) 47:235–46. doi: 10.1016/j.ebiom.2019.08.025
70. Li H, Ding J, Lu M, Liu H, Miao Y, Li L, et al. CAIX-specific CAR-T cells and sunitinib show synergistic effects against metastatic renal cancer models. J Immunother. (2020) 43:16–28. doi: 10.1097/CJI.0000000000000301
71. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
72. Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. (2012) 18:1672–83. doi: 10.1158/1078-0432.CCR-11-3050
73. Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol Res. (2019) 7:363–75. doi: 10.1158/2326-6066.CIR-18-0572
74. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
75. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. (2018) 14:317–24. doi: 10.1038/nchembio.2565
76. Sukumaran S, Watanabe N, Bajgain P, Raja K, Mohammed S, Fisher WE, et al. Enhancing the potency and specificity of engineered T cells for cancer treatment. Cancer Discov. (2018) 8:972–87. doi: 10.1158/2159-8290.CD-17-1298
77. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGFbeta inhibition via CRISPR promotes the long-term efficacy of CAR-T cells against solid tumors. JCI Insight. (2020) 5:e133977. doi: 10.1172/jci.insight.133977
78. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
79. Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T cells expressing il-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics. (2018) 8:41–51. doi: 10.1016/j.omto.2017.12.003
80. Liu Y, Di S, Shi B, Zhang H, Wang Y, Wu X, et al. armored inducible expression of il-12 enhances antitumor activity of glypican-3-targeted chimeric antigen receptor-engineered t cells in hepatocellular carcinoma. J Immunol. (2019) 203:198–207. doi: 10.4049/jimmunol.1800033
81. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. (2015) 4:e994446. doi: 10.4161/2162402X.2014.994446
82. Koneru M, O'Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med. (2015) 13:102. doi: 10.1186/s12967-015-0460-x
83. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep. (2017) 7:10541. doi: 10.1038/s41598-017-10940-8
84. Chmielewski M, Abken H. CAR T cells releasing il-18 convert to T-Bet(high) FoxO1(low) effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. (2017) 21:3205–19. doi: 10.1016/j.celrep.2017.11.063
85. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. (2015) 125:3905–16. doi: 10.1182/blood-2015-01-621474
86. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
87. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with car t cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. (2019) 36:471–82. doi: 10.1016/j.ccell.2019.09.006
88. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. (2017) 25:2214–24. doi: 10.1016/j.ymthe.2017.05.012
89. Adusumilli PS, Zauderer MG, Rusch VW, O'Cearbhaill R, Zhu A, Ngai D, et al. Regional delivery of mesothelin-targeted CAR T cells for pleural cancers: Safety and preliminary efficacy in combination with anti-PD-1 agent. J Clin Oncol. (2019) 37:2511. doi: 10.1200/JCO.2019.37.15_suppl.2511
90. Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. (2017) 129:1039–41. doi: 10.1182/blood-2016-09-738245
91. Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, Gottschalk S, et al. Armed oncolytic adenovirus-expressing pd-l1 mini-body enhances antitumor effects of chimeric antigen receptor t cells in solid tumors. Cancer Res. (2017) 77:2040–51. doi: 10.1158/0008-5472.CAN-16-1577
92. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–56. doi: 10.1038/nbt.4195
93. Suarez ER, Chang D-K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. (2016) 7:23. doi: 10.18632/oncotarget.9114
94. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting pd1 augments the efficacy of second-generation car t cells in advanced solid tumors. Cancer Res. (2016) 76:1578–90. doi: 10.1158/0008-5472.CAN-15-2524
95. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. (2017) 7:737. doi: 10.1038/s41598-017-00462-8
96. Menger L, Sledzinska A, Bergerhoff K, Vargas FA, Smith J, Poirot L, et al. TALEN-Mediated Inactivation of PD-1 in tumor-reactive lymphocytes promotes intratumoral t-cell persistence and rejection of established tumors. Cancer Res. (2016) 76:2087–93. doi: 10.1158/0008-5472.CAN-15-3352
97. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. (2017) 23:2255–66. doi: 10.1158/1078-0432.CCR-16-1300
98. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. (2020) 367:eaba7365. doi: 10.1126/science.aba7365
99. Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. (2017) 11:554–62. doi: 10.1007/s11684-017-0543-6
100. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. (2017) 8:17002–11. doi: 10.18632/oncotarget.15218
101. Vaupel P. Hypoxia and aggressive tumor phenotype: implications for therapy and prognosis. The Oncologist. (2008) 13:21–6. doi: 10.1634/theoncologist.13-S3-21
102. Jean-Philippe C, Carine M. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anti Cancer Agents Med Chem. (2008) 8:790–7. doi: 10.2174/187152008785914798
103. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA. (2014) 111:E5429. doi: 10.1073/pnas.1421438111
104. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. (2016) 352:175. doi: 10.1126/science.aaf4405
105. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. (2014) 41:518–28. doi: 10.1016/j.immuni.2014.09.008
106. Schito L, Semenza GL. Hypoxia-Inducible factors: master regulators of cancer progression. Trends in Cancer. (2016) 2:758–70. doi: 10.1016/j.trecan.2016.10.016
107. Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. (2012) 12:9–22. doi: 10.1038/nrc3183
108. Palazon A, Tyrakis PA, Macias D, Velica P, Rundqvist H, Fitzpatrick S, et al. An HIF-1alpha/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell. (2017) 32:669–83.e5. doi: 10.1016/j.ccell.2017.10.003
109. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat Immunol. (2013) 14:1173–82. doi: 10.1038/ni.2714
110. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, et al. Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell. (2016) 166:1117–31.e14. doi: 10.1016/j.cell.2016.07.032
111. Pearce EL, Poffenberger MC, Chang C-H, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. (2013) 342:1242454. doi: 10.1126/science.1242454
112. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
113. Phan AT, Goldrath AW, Glass CK. Metabolic and epigenetic coordination of T cell and macrophage immunity. Immunity. (2017) 46:714–29. doi: 10.1016/j.immuni.2017.04.016
114. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H, et al. Oncometabolite 2-Hydroxyglutarate Is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
115. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Veliça P, et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature. (2016) 540:236–41. doi: 10.1038/nature20165
116. Chakraborty AA, Laukka T, Myllykoski M, Ringel AE, Booker MA, Tolstorukov MY, et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science. (2019) 363:1217. doi: 10.1126/science.aaw1026
117. Batie M, Frost J, Frost M, Wilson JW, Schofield P, Rocha S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science. (2019) 363:1222. doi: 10.1126/science.aau5870
118. Berahovich R, Liu X, Zhou H, Tsadik E, Xu S, Golubovskaya V, et al. Hypoxia selectively impairs CAR-T cells in vitro. Cancers. (2019)11:602. doi: 10.3390/cancers11050602
119. McLellan AD, Ali Hosseini Rad SM. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol. (2019) 97:664–74. doi: 10.1111/imcb.12254
120. Ando Y, Siegler EL, Ta HP, Cinay GE, Zhou H, Gorrell KA, et al. Evaluating CAR-T cell therapy in a hypoxic 3d tumor model. Adv Healthcare Mater. (2019) 8:1900001. doi: 10.1002/adhm.201900001
121. Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, et al. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. (2016) 76:2465. doi: 10.1158/0008-5472.CAN-15-2402
122. Schnalzger TE, de Groot MHP, Zhang C, Mosa MH, Michels BE, Röder J, et al. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. (2019) 38:e100928. doi: 10.15252/embj.2018100928
123. Bar-Ephraim YE, Kretzschmar K, Clevers H. Organoids in immunological research. Nat Rev Immunol. (2019) 20:279–93. doi: 10.1038/s41577-019-0248-y
124. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
125. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. (2017) 127:929–41. doi: 10.1172/JCI89455
126. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. (2018) 6:57. doi: 10.1186/s40425-018-0360-8
127. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. (2011) 475:226–30. doi: 10.1038/nature10169
128. Auvinen P, Tammi R, Parkkinen J, Tammi M, Agren U, Johansson R, et al. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am J Pathol. (2000) 156:529–36. doi: 10.1016/S0002-9440(10)64757-8
129. Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. (2011) 178:1221–32. doi: 10.1016/j.ajpath.2010.11.076
130. Li HX, Zheng JH, Fan HX, Li HP, Gao ZX, Chen D. Expression of alphavbeta6 integrin and collagen fibre in oral squamous cell carcinoma: association with clinical outcomes and prognostic implications. J Oral Pathol Med. (2013) 42:547–56. doi: 10.1111/jop.12044
131. Ohno S, Tachibana M, Fujii T, Ueda S, Kubota H, Nagasue N. Role of stromal collagen in immunomodulation and prognosis of advanced gastric carcinoma. Int J Cancer. (2002) 97:770–4. doi: 10.1002/ijc.10144
132. Eikenes L, Bruland OS, Brekken C, Davies Cde L. Collagenase increases the transcapillary pressure gradient and improves the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts. Cancer Res. (2004) 64:4768–73. doi: 10.1158/0008-5472.CAN-03-1472
133. Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, et al. Phase Ib Study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clin Cancer Res. (2016) 22:2848–54. doi: 10.1158/1078-0432.CCR-15-2010
134. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, et al. HALO 202: Randomized Phase II Study of PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol. (2018) 36:359–66. doi: 10.1200/JCO.2017.74.9564
135. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. (2013) 62:112–20. doi: 10.1136/gutjnl-2012-302529
136. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. (2012) 21:418–29. doi: 10.1016/j.ccr.2012.01.007
137. Rodriguez-Garcia A, Gimenez-Alejandre M, Rojas JJ, Moreno R, Bazan-Peregrino M, Cascallo M, et al. Safety and efficacy of VCN-01, an oncolytic adenovirus combining fiber HSG-binding domain replacement with RGD and hyaluronidase expression. Clin Cancer Res. (2015) 21:1406–18. doi: 10.1158/1078-0432.CCR-14-2213
138. Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther. (2010) 18:1275–83. doi: 10.1038/mt.2010.79
139. Singha NC, Nekoroski T, Zhao C, Symons R, Jiang P, Frost GI, et al. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol Cancer Ther. (2015) 14:523–32. doi: 10.1158/1535-7163.MCT-14-0580
140. Clift R, Souratha J, Garrovillo SA, Zimmerman S, Blouw B. Remodeling the tumor microenvironment sensitizes breast tumors to anti-programmed death-ligand 1 immunotherapy. Cancer Res. (2019) 79:4149–59. doi: 10.1158/0008-5472.CAN-18-3060
141. Blair AB, Kim VM, Muth ST, Saung MT, Lokker N, Blouw B, et al. Dissecting the stromal signaling and regulation of myeloid cells and memory effector t cells in pancreatic cancer. Clin Cancer Res. (2019) 25:5351–63. doi: 10.1158/1078-0432.CCR-18-4192
142. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. (2012) 122:899–910. doi: 10.1172/JCI45817
143. Kuczek DE, Larsen AMH, Thorseth ML, Carretta M, Kalvisa A, Siersbaek MS, et al. Collagen density regulates the activity of tumor-infiltrating T cells. J Immunother Cancer. (2019) 7:68. doi: 10.1186/s40425-019-0556-6
144. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opinion Immunol. (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
145. Suzuki H, Onishi H, Wada J, Yamasaki A, Tanaka H, Nakano K, et al. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. Eur J Immunol. (2010) 40:197–203. doi: 10.1002/eji.200939887
146. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. (2004) 6:409–21. doi: 10.1016/j.ccr.2004.08.031
147. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
148. Ahirwar DK, Nasser MW, Ouseph MM, Elbaz M, Cuitino MC, Kladney RD, et al. Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene. (2018) 37:4428–42. doi: 10.1038/s41388-018-0263-7
149. Poggi A, Varesano S, Zocchi MR. How to hit mesenchymal stromal cells and make the tumor microenvironment immunostimulant rather than immunosuppressive. Front Immunol. (2018) 9:262. doi: 10.3389/fimmu.2018.00262
150. LeBleu VS, Kalluri R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Model Mech. (2018) 11:dmm029447. doi: 10.1242/dmm.029447
151. Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. (2019) 10:128. doi: 10.3389/fimmu.2019.00128
152. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. (2019) 18:99–115. doi: 10.1038/s41573-018-0004-1
153. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
154. Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. (2016) 30:1002–19. doi: 10.1101/gad.279737.116
155. Loktev A, Lindner T, Mier W, Debus J, Altmann A, Jager D, et al. A tumor-imaging method targeting cancer-associated fibroblasts. J Nucl Med. (2018) 59:1423–9. doi: 10.2967/jnumed.118.210435
156. Park JE, Lenter MC, Zimmermann RN, Garin-Chesa P, Old LJ, Rettig WJ. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J Biol Chem. (1999) 274:36505–12. doi: 10.1074/jbc.274.51.36505
157. Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. (2003) 9:1639–47.
158. Cohen SJ, Alpaugh RK, Palazzo I, Meropol NJ, Rogatko A, Xu Z, et al. Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas. (2008) 37:154–8. doi: 10.1097/MPA.0b013e31816618ce
159. Lo A, Li CP, Buza EL, Blomberg R, Govindaraju P, Avery D, et al. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight. (2017) 2:92232. doi: 10.1172/jci.insight.92232
160. Liao Y, Ni Y, He R, Liu W, Du J. Clinical implications of fibroblast activation protein-alpha in non-small cell lung cancer after curative resection: a new predictor for prognosis. J Cancer Res Clin Oncol. (2013) 139:1523–8. doi: 10.1007/s00432-013-1471-8
161. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
162. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. (2010) 330:827–30. doi: 10.1126/science.1195300
163. Zhang Y, Ertl HC. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget. (2016) 7:23282–99. doi: 10.18632/oncotarget.7818
164. Hornig N, Kermer V, Frey K, Diebolder P, Kontermann RE, Müller D. Combination of a bispecific antibody and costimulatory antibody-ligand fusion proteins for targeted cancer immunotherapy. J Immunother. (2012) 35:418–29. doi: 10.1097/CJI.0b013e3182594387
165. Bauer S, Adrian N, Williamson B, Panousis C, Fadle N, Smerd J, et al. Targeted bioactivity of membrane-anchored TNF by an antibody-derived TNF fusion protein. J Immunol. (2004) 172:3930. doi: 10.4049/jimmunol.172.6.3930
166. Herter S, Morra L, Schlenker R, Sulcova J, Fahrni L, Waldhauer I, et al. A novel three-dimensional heterotypic spheroid model for the assessment of the activity of cancer immunotherapy agents. Cancer Immunology, Immunotherapy. (2017) 66:129–40. doi: 10.1007/s00262-016-1927-1
167. Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. (2013) 210:1137–51. doi: 10.1084/jem.20122344
168. Pure E, Blomberg R. Pro-tumorigenic roles of fibroblast activation protein in cancer: back to the basics. Oncogene. (2018) 37:4343–57. doi: 10.1038/s41388-018-0275-3
169. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. (2018) 172:841–56 e16. doi: 10.1016/j.cell.2018.01.009
170. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
171. Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. (2002) 169:2756–61. doi: 10.4049/jimmunol.169.5.2756
172. Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. (2003) 9:606–12.
173. Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. (2001) 61:4766–72.
174. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol. (2006) 24:5373–80. doi: 10.1200/JCO.2006.05.9584
175. Beyer M, Schultze JL. Regulatory T cells in cancer. Blood. (2006) 108:804–11. doi: 10.1182/blood-2006-02-002774
176. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. (2004) 10:942–9. doi: 10.1038/nm1093
177. Flammiger A, Weisbach L, Huland H, Tennstedt P, Simon R, Minner S, et al. High tissue density of FOXP3+ T cells is associated with clinical outcome in prostate cancer. Eur J Cancer. (2013) 49:1273–9. doi: 10.1016/j.ejca.2012.11.035
178. Sayour EJ, McLendon P, McLendon R, De Leon G, Reynolds R, Kresak J, et al. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol Immunother. (2015) 64:419–27. doi: 10.1007/s00262-014-1651-7
179. June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. (2007) 117:1466–76. doi: 10.1172/JCI32446
180. Yao X, Ahmadzadeh M, Lu YC, Liewehr DJ, Dudley ME, Liu F, et al. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood. (2012) 119:5688–96. doi: 10.1182/blood-2011-10-386482
181. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
182. Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. (2006) 116:1935–45. doi: 10.1172/JCI27745
183. Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. (2011) 118:4817–28. doi: 10.1182/blood-2011-04-348540
184. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183
185. Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood. (2011) 117:72–82. doi: 10.1182/blood-2010-07-294520
186. Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. (2007) 15:825–33. doi: 10.1038/sj.mt.6300104
187. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. (2011) 121:1822–6. doi: 10.1172/JCI46110
188. Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. (2008) 112:2261–71. doi: 10.1182/blood-2007-12-128843
189. Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. (2004) 34:336–44. doi: 10.1002/eji.200324181
190. Shameli A, Yamanouchi J, Tsai S, Yang Y, Clemente-Casares X, Moore A, et al. IL-2 promotes the function of memory-like autoregulatory CD8+ T cells but suppresses their development via FoxP3+ Treg cells. Eur J Immunol. (2013) 43:394–403. doi: 10.1002/eji.201242845
191. Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood. (2006) 107:2409–14. doi: 10.1182/blood-2005-06-2399
192. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and cd8 t cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. (2015) 33:74–82. doi: 10.1200/JCO.2014.57.3329
193. Xu S, Sun Z, Sun Y, Zhu J, Li X, Zhang X, et al. IL-15 and dendritic cells induce proliferation of CD4+CD25+ regulatory T cells from peripheral blood. Immunol Lett. (2011) 140:59–67. doi: 10.1016/j.imlet.2011.06.005
194. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. (2018) 359:1037. doi: 10.1126/science.aar3246
195. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 'superkine'. Nature. (2012) 484:529–33. doi: 10.1038/nature10975
196. Parisi G, Saco JD, Salazar FB, Tsoi J, Krystofinski P, Puig-Saus C, et al. Persistence of adoptively transferred T cells with a kinetically engineered IL-2 receptor agonist. Nature Commun. (2020) 11:660. doi: 10.1038/s41467-019-12901-3
197. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. (2018) 17:887–904. doi: 10.1038/nrd.2018.169
198. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One. (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
199. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
200. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A. (2018) 115:E4041–E50. doi: 10.1073/pnas.1720948115
201. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. (2014) 74:153–61. doi: 10.1158/0008-5472.CAN-13-1816
202. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. (2011) 331:1612–6. doi: 10.1126/science.1198443
203. Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. (2017) 32:253–67 e5. doi: 10.1016/j.ccell.2017.07.006
204. Thoreau M, Penny HL, Tan K, Regnier F, Weiss JM, Lee B, et al. Vaccine-induced tumor regression requires a dynamic cooperation between T cells and myeloid cells at the tumor site. Oncotarget. (2015) 6:27832–46. doi: 10.18632/oncotarget.4940
205. Spear P, Barber A, Rynda-Apple A, Sentman CL. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-gamma and GM-CSF. J Immunol. (2012) 188:6389–98. doi: 10.4049/jimmunol.1103019
206. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. (2011) 71:5697–706. doi: 10.1158/0008-5472.CAN-11-0103
207. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. (2015) 125:3356–64. doi: 10.1172/JCI80005
208. Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The Role of Myeloid-Derived Suppressor Cells in Patients with Solid Tumors: A Meta-Analysis. PLoS ONE. (2016) 11:e0164514. doi: 10.1371/journal.pone.0164514
209. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
210. Enblad G, Karlsson H, Gammelgard G, Wenthe J, Lovgren T, Amini RM, et al. A phase I/IIa trial using CD19-targeted third-generation CAR T cells for lymphoma and leukemia. Clin Cancer Res. (2018) 24:6185–94. doi: 10.1158/1078-0432.CCR-18-0426
211. Donskov F. Immunomonitoring and prognostic relevance of neutrophils in clinical trials. Sem Cancer Biol. (2013) 23:200–7. doi: 10.1016/j.semcancer.2013.02.001
212. Saied A, Licata L, Burga RA, Thorn M, McCormack E, Stainken BF, et al. Neutrophil:lymphocyte ratios and serum cytokine changes after hepatic artery chimeric antigen receptor-modified T-cell infusions for liver metastases. Cancer Gene Ther. (2014) 21:457–62. doi: 10.1038/cgt.2014.50
213. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
214. Daassi D, Mahoney KM, Freeman GJ. (2020) The importance of exosomal PDL1 in tumour immune evasion. Nat Rev Immunol. doi: 10.1038/s41577-019-0264-y
215. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. (2018) 560:382–6. doi: 10.1038/s41586-018-0392-8
216. Sun C, Mezzadra R, Schumacher TN. Regulation and Function of the PD-L1 Checkpoint. Immunity. (2018) 48:434–52. doi: 10.1016/j.immuni.2018.03.014
217. Chen S, Crabill GA, Pritchard TS, McMiller TL, Wei P, Pardoll DM, et al. Mechanisms regulating PD-L1 expression on tumor and immune cells. J Immunother Cancer. (2019) 7:305. doi: 10.1186/s40425-019-0770-2
218. Kawashima M, Carreras J, Higuchi H, Kotaki R, Hoshina T, Okuyama K, et al. PD-L1/L2 protein levels rapidly increase on monocytes via trogocytosis from tumor cells in classical Hodgkin lymphoma. Leukemia. (2020) doi: 10.1038/s41375-020-0737-9. [Epub ahead of print].
219. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
220. Andrews LP, Yano H, Vignali DAA. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat Immunol. (2019) 20:1425–34. doi: 10.1038/s41590-019-0512-0
221. DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim Y-LE, Lee H-H, et al. T Cell/transmembrane, ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. (2010) 184:1918. doi: 10.4049/jimmunol.0903059
222. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
223. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol Res. (2015) 3:412–23. doi: 10.1158/2326-6066.CIR-14-0150
224. Kang R, Zhang Q, Zeh HJ, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res. (2013) 19:4046. doi: 10.1158/1078-0432.CCR-13-0495
225. Dankner M, Gray-Owen SD, Huang Y-H, Blumberg RS, Beauchemin N. CEACAM1 as a multi-purpose target for cancer immunotherapy. Oncoimmunology. (2017) 6:e1328336–e. doi: 10.1080/2162402X.2017.1328336
226. Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Different. (2016) 23:962–78. doi: 10.1038/cdd.2016.11
227. Ruvolo PP. Galectin 3 as a guardian of the tumor microenvironment. Biochimica et Biophysica Acta (BBA) – Mol Cell Res. (2016) 1863:427–37. doi: 10.1016/j.bbamcr.2015.08.008
228. Wing A, Fajardo CA, Posey AD Jr, Shaw C, Da T. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol Res. (2018) doi: 10.1158/2326-6066.CIR-17-0314
229. Guedan S, Alemany R. CAR-T cells and oncolytic viruses: joining forces to overcome the solid tumor challenge. Front Immunol. (2018) 9:2460. doi: 10.3389/fimmu.2018.02460
230. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. (2015) 212:1125–37. doi: 10.1084/jem.20142237
231. Wei J, Luo C, Wang Y, Guo Y, Dai H, Tong C, et al. PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J Immunother Cancer. (2019) 7:209. doi: 10.1186/s40425-019-0685-y
232. Poltavets V, Kochetkova M, Pitson SM, Samuel MS. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front Oncol. (2018) 8:431. doi: 10.3389/fonc.2018.00431
233. Granot Z, Fridlender ZG. Plasticity beyond cancer cells and the “immunosuppressive switch”. Cancer Res. (2015) 75:4441. doi: 10.1158/0008-5472.CAN-15-1502
234. Russell SJ, Barber GN. Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell. (2018) 33:599–605. doi: 10.1016/j.ccell.2018.03.011
235. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. (2018) 359:91. doi: 10.1126/science.aan3706
236. Uribe-Herranz M, Bittinger K, Rafail S, Guedan S, Pierini S, Tanes C, et al. Gut microbiota modulates adoptive cell therapy via CD8alpha dendritic cells and IL-12. JCI Insight. (2018) 3:e94952. doi: 10.1172/jci.insight.94952
237. Smith M, Littmann ER, Slingerland JB, Clurman A, Slingerland AE, Taur Y, et al. Intestinal microbiota composition prior to CAR T cell infusion correlates with efficacy and toxicity. Blood. (2018) 132:3492. doi: 10.1182/blood-2018-99-118628
238. Routy B, Gopalakrishnan V, Daillère R, Zitvogel L, Wargo JA, Kroemer G. The gut microbiota influences anticancer immunosurveillance and general health. Nat Rev Clin Oncol. (2018) 15:382–96. doi: 10.1038/s41571-018-0006-2
Keywords: chimeric antigen receptors (CAR), solid tumors, immunotherapy, immunosuppressive tumor microenvironment, adoptive cell transfer (ACT), inhibitory receptors
Citation: Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ Jr and Guedan S (2020) CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 11:1109. doi: 10.3389/fimmu.2020.01109
Received: 01 April 2020; Accepted: 07 May 2020;
Published: 17 June 2020.
Edited by:
Francisco Martin, Andalusian Autonomous Government of Genomics and Oncological Research (GENYO), SpainReviewed by:
Fernando Aranda, University of Navarra, SpainPedro Berraondo, University of Navarra, Spain
Copyright © 2020 Rodriguez-Garcia, Palazon, Noguera-Ortega, Powell and Guedan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sonia Guedan, c2d1ZWRhbkBjbGluaWMuY2F0