
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 04 June 2020
Sec. Microbial Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01085
 Carl De Trez1*
Carl De Trez1* Benoit Stijlemans1,2
Benoit Stijlemans1,2 Viki Bockstal1†Jennifer Cnops1†
Viki Bockstal1†Jennifer Cnops1† Hannelie Korf3
Hannelie Korf3 Jacques Van Snick4,5
Jacques Van Snick4,5 Guy Caljon6
Guy Caljon6 Eric Muraille7,8
Eric Muraille7,8 Ian R. Humphreys9
Ian R. Humphreys9 Louis Boon10
Louis Boon10 Jo A. Van Ginderachter1,2
Jo A. Van Ginderachter1,2 Stefan Magez1,11
Stefan Magez1,11In many infectious diseases, the immune response operates as a double-edged sword. While required for protective immunity, infection-induced inflammation can be detrimental if it is not properly controlled, causing collateral body damage and potentially leading to death. It is in this context that the potent anti-inflammatory cytokine interleukin-10 (IL-10) is required to dampen the pro-inflammatory immune response that hallmarks trypanosomosis. Effective control of this infection requires not just the action of antibodies specific for the parasite's variable surface glycoprotein (VSG) coat antigens, but also a pro-inflammatory immune response mediated mainly by IFNγ, TNF, and NO. However, strict control of inflammation is mandatory, as IL-10-deficient mice succumb from an unrestrained cytokine storm within 10 days of a Trypanosome brucei infection. The relevant cellular source of IL-10 and the associated molecular mechanisms implicated in its trypanosomosis associated production are poorly understood. Using an IL-10 reporter mouse strain (Vert-X), we demonstrate here that NK cells, CD8+ T cells and CD4+ T cells as well as B cells and plasma cells constitute potential cellular sources of IL-10 within the spleen and liver during acute infection. The IL-10 wave follows peak pro-inflammatory cytokine production, which accompanied the control of peak parasitemia. Similar results were observed following conventional experimental needle infection and physiological infections via T. brucei-infected tsetse flies. Our results show that conditional T cell-specific ablation of the IL-10 regulating Prdm1 gene (encoding for the Blimp-1 transcription factor), leads to an uncontrolled trypanosome-induced pro-inflammatory syndrome like the one observed in infected IL-10-deficient mice. This result indicates that the biological role of IL-10-derived from non-T cells, including NK cells, is of minor importance when considering host survival. The cytokine IL-27 that is also considered to be an IL-10 regulator, did not affect IL-10 production during infection. Together, these data suggest that T. brucei activates a Blimp-1-dependent IL-10 regulatory pathway in T cells that acts as a critical anti-inflammatory rheostat, mandatory for host survival during the acute phase of parasitemia.
During inflammation, immune regulatory cytokines are mandatory to preserve host integrity by controlling pathogen-induced immune responses (1). IL-10 is a pleiotropic anti-inflammatory cytokine that dampens many inflammatory reactions (2). Hence, IL-10-deficient mice are prone to uncontrolled inflammation-mediated immunopathologies, such as spontaneous colitis potentially leading to tumorigenesis and chronic inflammation-driven auto-immunity (3). During infection, the genetic or pharmacological inhibition of IL-10 usually leads to a better control of infection, but this is often associated with enhanced immuno-pathology and, in some infectious models, death (4, 5). Macrophages and dendritic cells constitute targets of the suppressive function of IL-10, promoting their differentiation toward a suppressive and tolerizing phenotype (6, 7). Recently, many cell types have been shown to secrete IL-10 during infections, namely NK cells, dendritic cells, Th1 cells, CD8+ T, regulatory T and B cells, and even non-immune cells, such as hepatocytes and keratinocytes (8–15). Different transcription factors and cytokines have been implicated in the production of IL-10 depending on the cell type. For example, Foxp3, Aryl hydrocarbon Receptor, Blimp1 and IL-27 were shown to modulate IL-10 production by Tregs, NK, and Th1 cells, respectively (12, 16–22). African trypanosomes are extracellular protozoan hemoflagellated parasites transmitted by the bite of infected tsetse flies (genus Glossina). These parasites are endemic in Sub-Saharan Africa, causing African trypanosomosis in human (also called sleeping sickness) and Nagana disease in livestock. About 60 million people are at risk and Nagana causes three million cattle deaths every year due to weight loss and anemia. The associated economic loss in livestock production is estimated at 4 billion USD per year (23). Murine models are considered valuable tools to study the interactions between parasites and hosts that contribute to immunopathogenicity. Experimental T. brucei infections in mice have shown that clearance of the first parasitemia peak is dependent on an early strong type 1 inflammatory immune response, involving IFNγ, Nitric Oxide (NO) and Tumor Necrosis Factor (TNF) production, which correlates with an early activation of monocytes, the recruitment of splenic neutrophils and the development of anemia (24–28). Yet, the production of IL-10 is essential to dampen this type 1 immune response after parasitemia has been cleared to prevent the development of a hyper-inflammation syndrome and death (6, 29, 30). Despite the importance of IL-10 in T. brucei pathogenesis, the in vivo cellular source of IL-10 and the associated molecular mechanism(s) implicated in its production remain poorly understood. In this study, we report that increasing levels of IL-10 are being measured in both infected tissue and serum following clearance of the first parasitemia peak. Using IL-10 reporter [Vert-X (31)] mice, we show that NK cells, CD8+ T cells and CD4+ T cells are important cellular sources of IL-10 within infected liver and spleen tissues around day 6 post infection (p.i.), following the peak of pro-inflammatory cytokine production. Post-parasitemia peak (around day 8–9 p.i.), the cellular source of IL-10 is still similar in the liver, whereas, surprisingly, the main splenic IL-10-producing cells become plasma B cells. These results were first obtained in a conventional experimental infection model in which mice were challenged with T. brucei parasites via intraperitoneal needle injection. Subsequently, all results were confirmed following a natural infection via T. brucei-bearing tsetse flies. Using T cell conditional Blimp-1 knockout mice, we demonstrate the importance of this transcription factor in dampening trypanosome-mediated inflammation, mainly via the control of T cell activation and IL-10 production, and ultimately host survival.
All experiments complied with the ECPVA guidelines (CETS n° 123) and were approved by the VUB Ethical Committee (Permit Number: 14-220-10 and 14-220-05). Breeding and experimental work with tsetse flies was approved by the Scientific Institute Public Health Department Biosafety and Biotechnology (SBB 219.2007/1410). To minimize mouse suffering and distress during blood sampling, all animals were anesthetized with isoflurane using a UNO—Univentor Anesthesia Unit according to the manufacturer‘s protocol. Mice were monitored daily. Humane endpoints were used during the study, based on weight loss, animals with >25% weight loss were sacrificed using carbon dioxide treatment.
Eight to 12 weeks-old male and female wild type C57BL/6 (WT) mice were purchased from Janvier, France. IL-10−/− (B6.129P2-Il10tm1Cgn/J), Prdm1eYFP/+ (B6.Cg-Tg(Prdm1-EYFP)1Mnz/J), Vert-X (B6(Cg)-Il10tm1.1Karp/J), and B cell−/− (B6.129S2-Ighmtm1Cgn/J) mice were purchased from Jackson Laboratory, USA. CD4Cre/+ Prdm1fl/fl and littermate control Prdm1fl/fl were kindly provided by A. Scheffold at Charité - Universitätsmedizin Berlin, Berlin, Germany. CD4Cre/+ IL-10fl/fl and littermate control IL-10fl/fl mice were initially established in Cardiff University, Cardiff, United Kingdom (32). All mice were bred and maintained in the animal facility at the Vrije Universiteit Brussel.
Pleomorphic T. brucei AnTat 1.1E parasites were obtained from the Institute for Tropical Medicine, Belgium and stored at −80°C as blood aliquots containing 50% Alsever buffer (Sigma-Aldrich) and 10% glycerol (final V/V). Mice were infected with 5000 clonal AnTat1.1E trypanosomes via intraperitoneal (i.p.) injection in a volume of 200 μL PBS. Tsetse flies were infected at the Institute of Tropical Medicine with T. brucei AnTAR1 parasites and selected for mature salivary gland infections as described previously (33). For each mouse, one individual infected tsetse fly was used to initiate a natural infection by a fly bite.
Blood from non-infected control and infected mice at different time points of infection was harvested via tail-cut using heparinized capillaries and centrifuged at 8,000 g for 15 min. Serum was harvested and stored at −20°C.
Leukocyte liver cells were purified by perfusing the liver with 10 ml of cold PBS via the inferior vena cava, mechanical disruption of the liver, followed by passing cell suspensions over a 70 μm nylon mesh filter. The cells were washed twice with PBS and centrifuged at 582 g for 7 min at 4°C. After discarding the supernatant, the pellet was resuspended in a 33% Percoll solution and centrifuged at 394 g for 7 min at room temperature. After discarding the supernatant, the pellet was resuspended using ACK lysis buffer (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM Na2-EDTA) to lyse red blood cells (RBCs) and centrifuged at 394 g for 7 min at 4°C. The pellet was resuspended in complete medium buffer [RPMI supplemented with 10% FCS, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (all from Invitrogen Life Technologies)]. Spleen and lymph node cells were isolated as follows. The organs were mechanically disrupted and RBCs were lysed using ACK lysis buffer (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM Na2-EDTA). After washing twice with RPMI at 394 g for 7 min at 4°C, the cell suspensions were resuspended in complete medium and passed through a 70 μm nylon mesh filter.
Spleen, liver and lymph node cell populations from non-infected control and infected mice at different time points of infection were counted and cultured in vitro for cytokine ELISA or directly analyzed by flow cytometry.
Cells were centrifuged at 394 g for 7 min and resuspended in FACS medium (5% FCS in PBS) at a concentration of 2.106 cells/ml. Non-specific binding sites were blocked by incubating 20 min. at 4°C with an Fc-blocking antibody (anti-CD16/32, clone 2.4G2). Next, cell suspensions were stained with fluorescent conjugated antibodies for 30 min at 4°C. Fluorescent antibodies: CD11a Pe-Cy7 clone 2D7, CD11b PE-Cy7 clone M1/70, Ly6C APC clone AL-21, CD4 BV421 clone GK1.5, CD8 BV510 clone 53–67, NK11 PE clone PK136, TCRß APC clone H57-597, CD90.2 APC-Cy7 clone Thy1.2, CD138 APC clone 281-2 (BD Biosciences), B220 BV510 clone RA3-6B2, CD93 BV421 clone AA4.1, CD1d PE clone 1B1 and CD49d PE clone 9C10 (eBioscience). Following washing with FACS buffer the cell suspensions were analyzed on a FACS Canto II flow cytometer (BD Biosciences) and data was processed using FlowJo software (Tree Star Inc., Ashland, OR). The total number of live 7-AAD- cells in each population was determined by multiplying the percentages of subsets within a series of marker negative or positive gates by the total live cell number determined by microscopy counting with trypan blue for each tissue.
Spleen and liver cells were incubated in complete medium at 37°C for 4 h in the presence of eBioscience™ Cell Stimulation Cocktail (plus protein transport inhibitors) (500X) (ThermoFisher Scientific). Next, cells were washed in PBS at 394 g for 7 min at 4°C, stained for cell surface markers at 4°C, washed in PBS at 394 g for 7 min at 4°C and fixed in 1x BD Cytofix/Cytoperm solution (BD Pharmingen) for 15–20 min at 4°C. They were then washed using a saponin-based buffer (10 × Perm/Wash in FACS buffer; BD Pharmingen) at 394 g for 7 min at 4°C. After discarding the supernatant, cells were resuspended and stained with XMG1.2 (anti-IFNγ; BD Biosciences) in 1x BD Perm/wash solution for 30 min at 4°C. After washing the cells with 1x BD Perm/wash solution at 1,400 rpm for 7 min at 4°C, they were resuspended in PBS and analyzed on a FACS Canto II flow cytometer (BD Biosciences) and data was processed using FlowJo software (Tree Star Inc., Ashland, OR). No signal was detectable with the allophycocyanin-coupled anti-IgG1 isotype control (eBioscience).
Liver, splenic and lymph node cell populations isolated from infected and uninfected mice resuspended in complete medium buffer (cfr previous “Serum and cell isolation” section) were plated in 48-well plates at 2.106 cells per ml and incubated at 37°C in a 5% CO2 incubator for 48 h without any additional stimulation before supernatant was recovered for cytokine ELISA.
For depletion of CD8+ and CD4+ T cells, mice received the first i.p. injection of 500 μg anti-CD8 beta and anti-CD4 rat-anti-mouse monoclonal antibodies (clone YTS169 and GK1.5, Bioceros in the Netherlands, respectively) 24 h prior to infection. Subsequently, mice received a dose of 100 μg at 2 days interval post infection (34). NK and NKT cells were depleted with the anti-NK1.1 PK136 mouse-anti-mouse monoclonal antibody (PK136, BioXCell, USA and Bioceros, The Netherlands). Two hundred and fifty microgram was given 4 and 1 day prior to infection. A dose of 300 μg was given at 2–3 day intervals post infection (34). Control mice were treated with a respective control antibody isotype(all from BioXCell, USA), namely a monoclonal rat IgG2a isotype as control for the anti-NK1.1 depletion and a monoclonal mouse IgG2b as control for anti-CD8 and anti-CD4, and the same regimen. Depletion efficiency of NK and NKT cells as well as CD8+ and CD4+ T cells from both spleen and liver was assessed by flow cytometry and was confirmed to be above 90%.
For neutralization of IL-27, wild type mice were treated with 500 μg of an anti-IL-27 antibody (MM27.7B1) or a control IgG2a antibody once a week (32, 35–37).
Cytokines were quantified using a V-PLEX Custom Mouse Cytokine kit (catalog number K152A0H) from Meso Scale Discovery (Rockville, MD, USA) according to the manufacturer's protocol. Alternatively, culture medium and serum concentrations of IL-6, TNF, IFN-γ, and IL-10 (R&D Systems) were determined by ELISA as recommended by the suppliers.
Eight weeks-old C57BL/6 mice were irradiated at 1000 Rads using a Cesium source irradiator at IBMM, Gosselies, Belgium. The next day irradiated mice were transplanted intravenously with 107 bone marrow cells isolated from femurs and tibias of either IL-10-deficient mice or wild-type (WT) C57BL/6 mice. Briefly, bones were harvested and cells were flushed with PBS using a 27 gauge needle and syringe. Cell clumps were dissociated via up-and-down pipetting, filtered through a 70 μm nylon mesh filter and washed in PBS at 394 g for 7 min at 4°C. Red blood cells were lyzed using RBC lysis buffer, washed in PBS at 394 g for 7 min at 4°C and counted. Animals were kept under Sulfatrim antibiotic [sulfamethoxazole/trimethoprim, to be added to the drinking water (5/200 ml)] for 4 weekspost-irradiation.
The GraphPad Prism 7 software was used for statistical analyses (student t-test for paired analyses and Log-rank (Mantel-Cox) test for survival). Values are expressed as mean ± SEM, where *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
In Antat1.1E T. brucei-infected wild-type (WT) C57BL/6 mice, peak parasitemia is reached around day 6 post-infection (p.i.) (Supplementary Figure 1) and blood parasitemia control correlates with the development of a pro-inflammatory response characterized by increased levels of TNF, IFNγ, and IL-6, which peaks around day 7 and then decreases toward 10 p.i. (Figures 1A–C). Early after infection, IL-10 production is absolutely necessary to dampen the pathogenic effects of these pro-inflammatory cytokines, as mice deficient in IL-10 (IL-10 KO mice) exhibit increased levels of the pro-inflammatory cytokines IFNγ and TNF (Figures 1A–C), and die around 8–9 days p.i. (Figure 1D). Interestingly, the increased inflammation observed in IL-10 KO mice has no effect on parasitemia levels (6, 29).
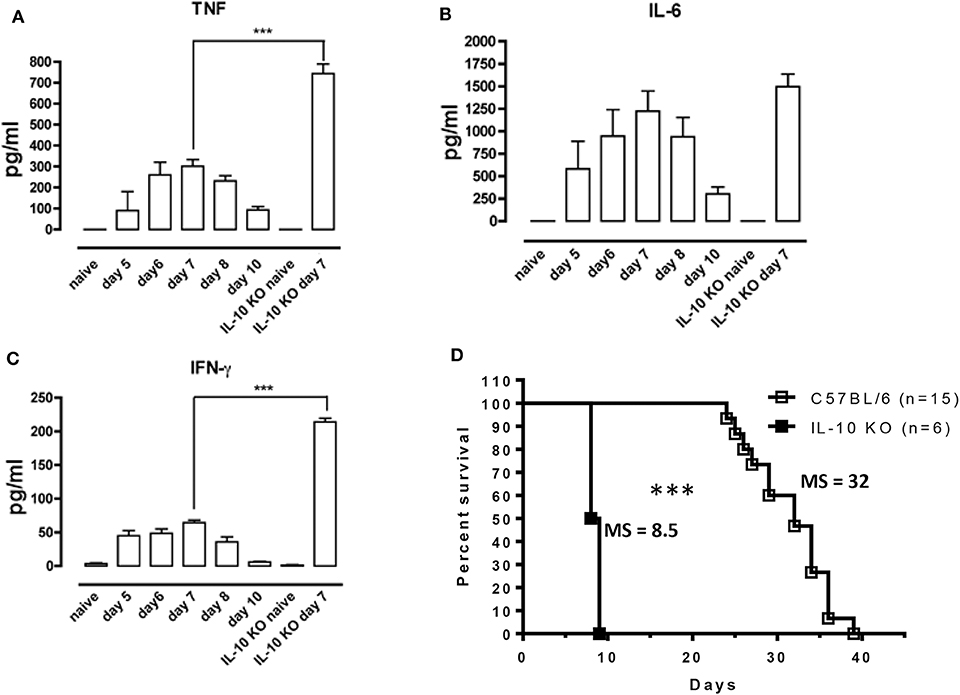
Figure 1. Uncontrolled inflammation in T. brucei-infected IL-10-deficient mice. (A–C) Cytokine levels were measured by ELISA in plasma of naive mice, infected WT mice on days 5, 6, 7, 8, and 10 p.i. and naive vs. infected IL-10-deficient mice on day 7 p.i. Data are represented as mean of 6 mice per group ± SEM and are representative of 2 independent experiments. (D) Survival of WT and IL-10-deficient mice following T. brucei infection i.p., where the median survival (MS) of each group is indicated in days. Data are presented as a pool of two representative independent experiments with a minimum of three mice per group, where ***p ≤ 0.001.
Given the essential role for IL-10 in controlling T. brucei pathogenesis, serum IL-10 levels and IL-10 secretion in ex vivo splenic, liver and pooled peripheral lymph node (axillary and inguinal) cell cultures were measured. IL-10 protein in the serum of T. brucei infected WT mice peaks around day 8 p.i., which corresponds to the time at which IL-10 KO mice start to succumb to the infection (Figure 2). In spleen and liver cell cultures, local production of IL-10 is detected at day 6 and 8 post-infection without the addition of ex vivo stimuli (Figure 2). This contrasts with the nearly complete absence of IL-10 secretion measured in pooled peripheral lymph nodes at the same time points (Figure 2). Together, these results suggest that IL-10 produced in liver and spleen is crucial in dampening pro-inflammatory cytokines induced during parasitemia.
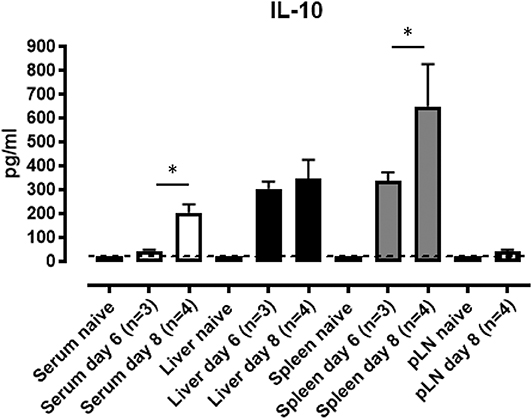
Figure 2. IL-10 production in mouse spleen and liver cell cultures following T. brucei infection. Serum and supernatants from ex vivo cultured total spleen cells, purified leukocyte liver cells from naïve, day 6 and day 8 p.i and pooled peripheral lymph node (pLN) cells from naïve and day 8 p.i. were analyzed by ELISA for IL-10 production. Data are represented as mean of at least three mice per group ± SEM, where *p ≤ 0.05, and are representative of three independent experiments. The dashed line represents the detection limit.
Numerous cell types, particularly hematopoietic cells, have the potential to produce IL-10. Hence, in order to understand the role of IL-10 in trypanosomosis, the main cellular origin needs to be known. Irradiated WT mice repopulated with bone marrow cells from IL-10 KO mice exhibit comparable susceptibility to T. brucei-induced death as compared to full IL-10 KO mice, while a transfer with WT-derived cells completely rescued the phenotype (Figure 3A). The analysis of cytokine levels present in serum at day 8 p.i. in these mice reveals that recipients of IL-10 KO bone marrow display significantly higher IFNγ (3,374 ± 66 pg/ml) and TNF (2,101 ± 83 pg/ml) as compared to WT mice that received WT bone marrow cells (Figure 3B). Coincidingly, a dramatic reduction in infection-associated IL-10 (105 ± 18 pg/ml) is observed (Figure 3B), phenocopying the results observed in IL-10 KO mice. Importantly, the small amount of IL-10 still present in irradiated WT mice receiving IL-10 KO bone marrow cells is insufficient to reduce the overall pro-inflammatory cytokine production in these mice.
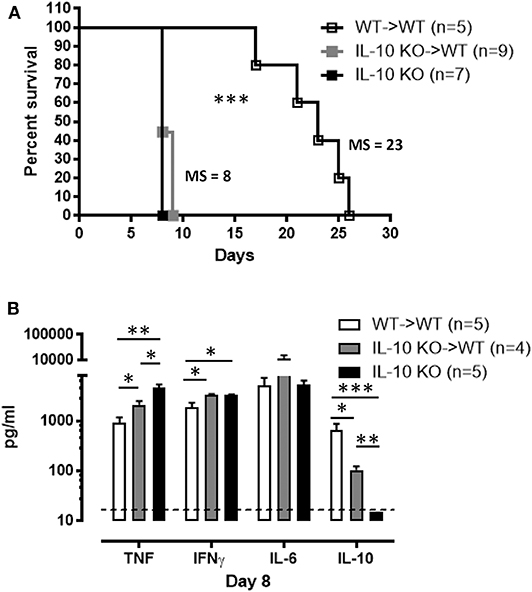
Figure 3. Hematopoietic cells constitute a major source of IL-10 production during T. brucei infection. (A) Survival of the chimeric mice 8 weeks post-transplantation as well as IL-10-deficient mice following T. brucei infection i.p., where the median survival (MS) of each group is indicated in days. (B) Cytokine levels were measured by ELISA in plasma of infected chimeric and IL-10-deficient mice 8 days p.i. Data are represented as mean of at least 4 mice per group ± SEM, where *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and are representative of 2 independent experiments. The dashed line represents the detection limit.
As IL-10 derived from the hematopoietic compartment is mandatory to counteract the excessive production of pro-inflammatory cytokines during early T. brucei infection and prevent early death, the radiosensitive source(s) of IL-10 was investigated using VERT-X IL-10eGFP reporter mice, encoding enhanced GFP (eGFP) in the 3' UTR of the Il10 gene (31). The specificity of the IL-10eGFP signal was tested in both liver and spleen, as infected non-reporter WT mice did not show any increase in eGFP signal compared to Vert-X IL-10eGFP reporter mice (Supplementary Figure 2). First, we follow the evolution of the IL-10eGFP signal expressed in percentage and total numbers within both the liver and spleen during the first 8 days p.i. (Figures 4A,B). In the liver a slight increase in the percentage of IL-10eGFP+ cells was observed toward day 3 p.i., peaking at day 6 p.i. and declining slightly around day 8 p.i. In the spleen, the percentage of IL-10eGFP+ cells only increase considerably at day 8 p.i. (Figure 4A). In absolute numbers of IL-10eGFP+ cells, there is a similar increase in both liver (4.8 106 ± 0.2) and spleen (5.9 106 ± 0.3) at day 6 p.i. compared to day 3 p.i. In the case of the spleen, but not the liver, we observed a substantial further increase in IL-10eGFP+ cell numbers by day 8 after infection (Figure 4B). These results mirror the IL-10 production data recorded in the corresponding ex vivo cultures of these organs (Figure 2).
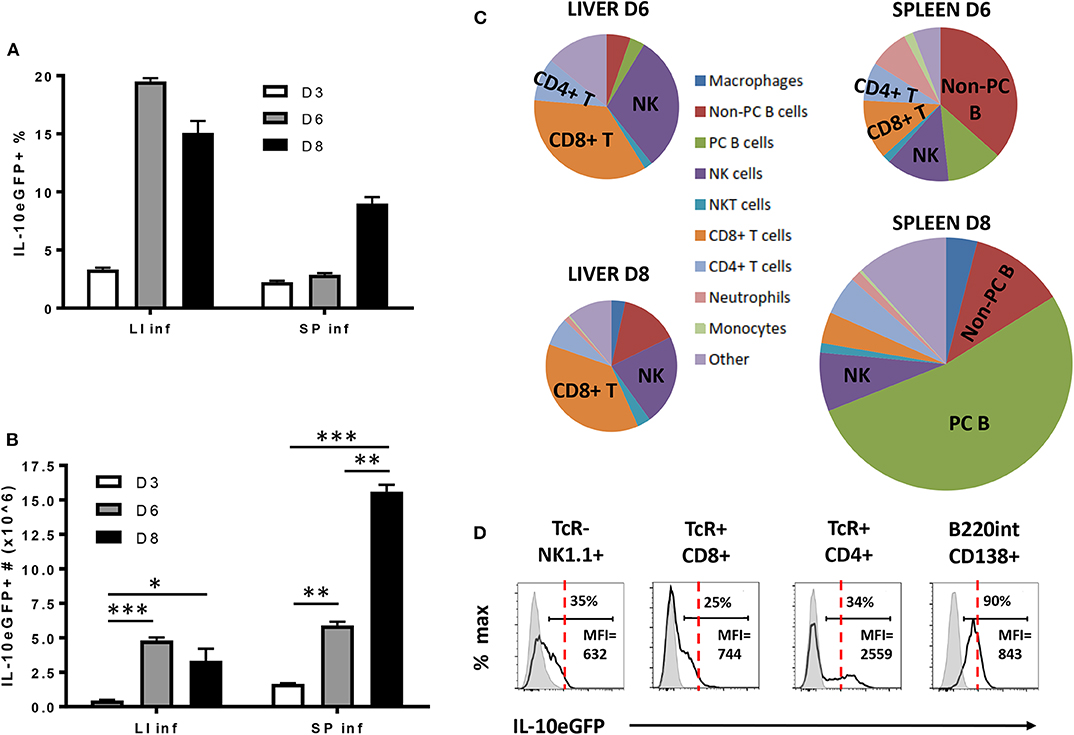
Figure 4. Cellular characterization of IL-10 producing cells in spleen and liver following T. brucei infection. (A) Percentages and (B) total cell number of IL-10 producing cells in infected liver (LI inf) and spleen (SP inf) at day 3, 6, and 8 p.i. (C) Relative contribution of the different cellular sources producing IL-10. The surface of the diagram is representative of the total number of IL-10+ cells present in liver and spleen at day 6 and day 8 p.i., which is described in (B). (D) Histogram showing the intensity of IL-10-eGFP signal as well as the percentage and MFI of IL-10+ cells in different cell subsets. The red dotted line is giving an indication of the relative IL-10-eGFP signal differences between the different cell subsets, whereas the gray histogram represent the relative IL-10-eGFP signal of these different cell subsets in infected non-reporter mice. Data are represented as mean of at least three mice per group ± SEM, where *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and are representative of 3 independent experiments.
Investigating the cellular source of the IL-10eGFP signal at day 6 and day 8 p.i. in both liver and spleen shows that NK cells (NK1.1+ TcRαβ−) and CD8+ T (CD8α+ TcRαβ+) cells are the main source of IL-10 at day 6 p.i. (30.7% ± 0.3% and 35.2% ± 9.1% of all eGFP+ cells, respectively), as well as at day 8 p.i. (22.4% ± 2.3% and 37.0% ± 1.8%, respectively) in livers of infected mice (Figure 4C). Interestingly, NK cells (NK1.1+ TcRαβ−) and CD8+ T (CD8α+ TcRαβ+) cells were also both recently identified as the main producers of IFNγ in response to T. brucei infection (34), In the spleen, the main IL-10eGFP+ cell populations are non-plasma B cells (B220hi CD138−) at day 6 p.i. (36.9% ± 4.7%), whereas IL-10+ plasma cells (B220int CD138+) predominant (52.9% ± 4.5%) at day 8 p.i. with nearly all plasma B cells showing positive IL-10eGFP expression at the later timepoint (Figure 4D). Importantly, CD4+ T cells show a double expression profile, with the IL-10 positive population expressing a 3- to-4-fold increased mean fluorescence intensity (MFI) compared to the other subsets (red dashed line) (Figure 4D), suggesting a higher IL-10 expression by this cell subset.
These data reveal that different cell types, such as NK cells, CD8+ T cells, and CD4+ T cells, and B cells, have the potential to produce IL-10 during early experimental trypanosomosis.
In most experimental trypanosomosis studies, mice are infected by intraperitoneal (i.p.) injections of parasites. To evaluate the relevance of our results under a more biologically relevant condition, the survival of IL-10-deficient mice following T. brucei-infected tsetse fly-mediated infection was monitored. IL-10 KO mice infected via the natural route all succumb within 10 days p.i., similar to intraperitoneally infected IL-10 KO mice (Figure 5A). In agreement with the results obtained following i.p. infection (Figure 1), tsetse fly-mediated infection induces a significant increase in IFNγ and TNF levels, which is further aggravated in IL-10 KO mice, at day 6 p.i. (Figure 5B).
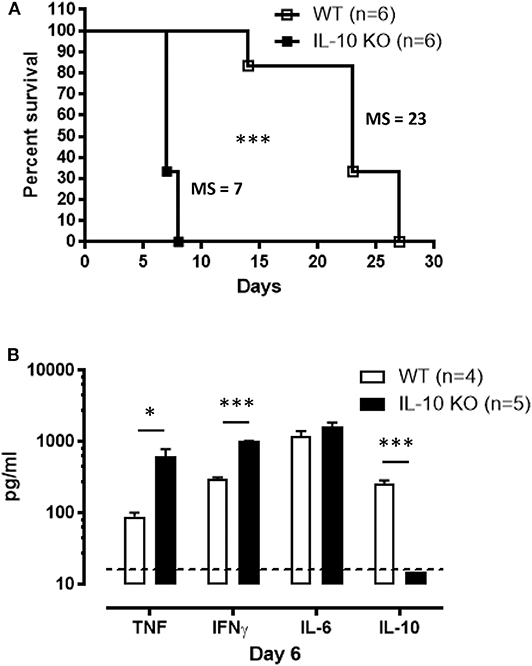
Figure 5. Survival and inflammation profile of IL-10-deficient mice inoculated with T. brucei infected tsetse fly. (A) Survival of WT and IL-10-deficient mice after T. brucei infection via tsetse fly, where the median survival (MS) of each group is indicated in days. Data are represented as mean of at least 6 mice per group ± SEM and are representative of 2 independent experiments. (B) Cytokine levels were measured by ELISA in plasma of infected WT and IL-10-deficient mice at 6 days p.i. Data are represented as mean of at least four mice per group ± SEM, where *p ≤ 0.05, ***p ≤ 0.001, and are representative of three independent experiments. The dashed line represents the detection limit.
Following the natural route of a T. brucei infection, the liver exhibits the highest frequency of IL-10eGFP+ cells, as compared to the spleen, both at day 4 and day 7 p.i. (Figure 6A). However, due to the occurrence of splenomegaly, the spleen contains a higher actual number of IL-10eGFP+ cells at day 7 p.i. as compared to the liver (12.6 106 ± 2.5 vs. 3.8 106 ± 0.6, respectively) (Figure 6B), which mirrors the results obtained following i.p. infection. Also here, NK cells, CD8+ T cells, CD4+ T cells subsequently followed by non-plasma and plasma B cells remain the most abundant cell types producing IL-10 at day 7 p.i., the time point when IL-10 KO mice infected via the natural route start to succumb from the pro-inflammatory cytokine storm (Figure 6C).
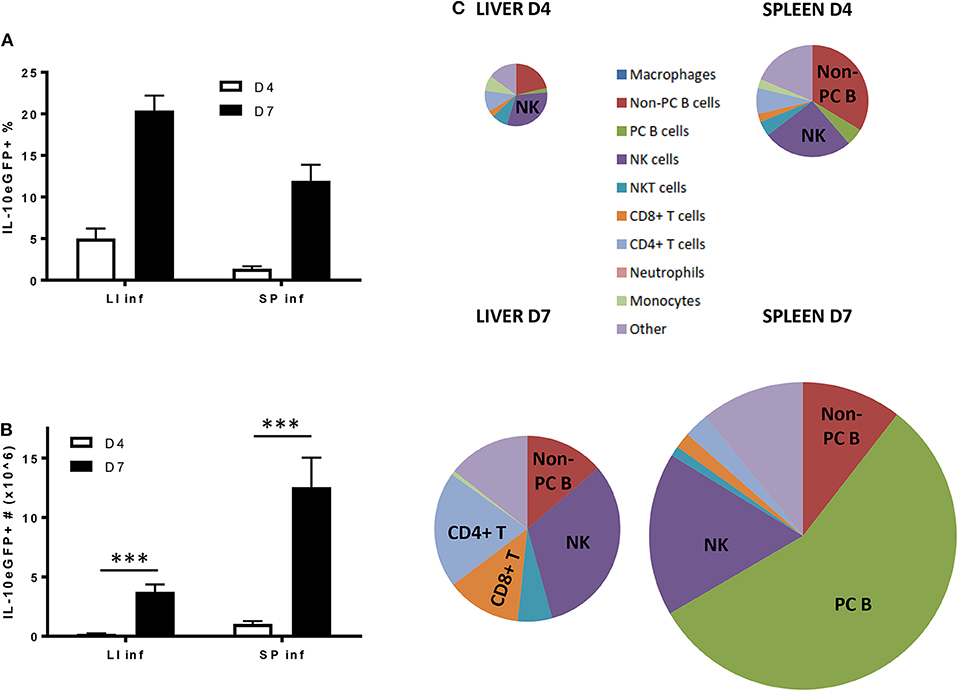
Figure 6. Cellular characterization of IL-10 producing cells in spleen and liver following T. brucei infection by tsetse fly. (A) Percentages and (B) total cell number of IL-10 producing cells in infected liver (LI inf) and spleen (SP inf) at day 4 and 7 p.i. (C) Relative contribution of the different cellular sources producing IL-10. The surface of the diagram is representative of the total number of IL-10+ cells present in liver and spleen at day 4 and day 7 p.i., which is described in (B). Data are represented as mean of at least three mice per group ± SEM, where ***p ≤ 0.001, and are representative of three independent experiments.
As shown by the data compiled above, NK and T cells constitute a potential early source of IL-10 during T. brucei infection (Figure 4). Therefore, these cells were pharmacologically depleted and subsequently the levels of IL-10 in the circulation at day 6 and day 9 post-infection were measured (Figure 7A). The results confirm the contribution of NK and T cells on systemic IL-10 levels at both day 6 (184 ± 34 pg/ml vs. 76 ± 7 pg/ml) and day 9 (502 ± 68 pg/ml vs. 152 ± 15 pg/ml) post-infection. As we confirmed that the peak of IL-10 in the serum occurs post peak parasitemia, round day 8–9 post infection, the role of these individual cell subsets, i.e., NK cells as well as CD8 and CD4 T cells, on the circulating levels of IL-10 was evaluated at day 9 post-infection (new Figure 7B). The results demonstrate that both CD8+ and CD4+ T cells, in contrast to NK cells, play a role in the systemic production of IL-10 at day 9 post-infection.
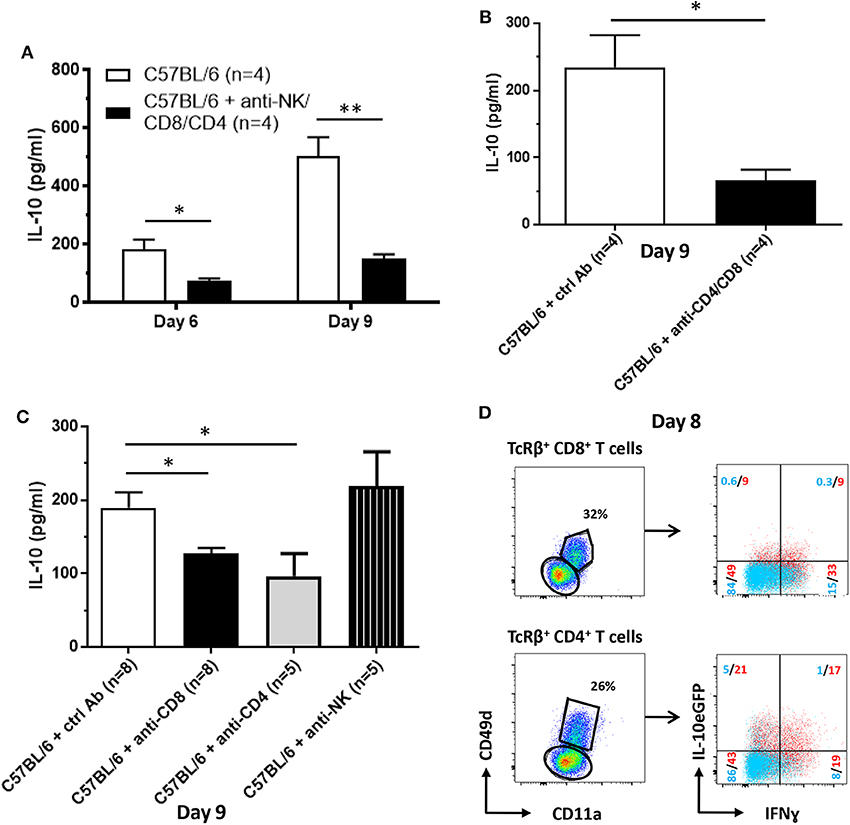
Figure 7. Cellular source of IL-10 production following T. brucei infection. IL-10 levels were measured by ELISA in plasma of infected WT and anti-NK-, anti-CD8- and CD4-treated WT at day 6 and 9 p.i. (A) as well as in WT and anti-CD8- or CD4-treated or anti-NK1.1-treated WT mice (B) and in WT and anti-CD8- and CD4-treated 9 days p.i. (C). (D) At day 8 p.i., CD11a and CD49d expression on splenic CD8+ and CD4+ T cells was analyzed. Both CD11a+ CD49d+ and CD11a− CD49d− CD8+ and CD4+ T cell subsets were analyzed for the expression of IL-10eGFP and intracellular expression of IFNg. Data are represented as mean of at least four mice per group ± SEM, where *p ≤ 0.05, **p ≤ 0.01, and are representative of two independent experiments.
Taken the importance of both T cell subsets and the very high level of IL-10eGFP expression in the cytokine positive CD4+ T cell population, the role of these cells was assessed in more detail using T. brucei infected mice, which are pharmacologically depleted in both CD8+ and CD4+ T cells. In these mice, serum IL-10 levels at day 9 p.i.were 70–75% lower compared to WT mice (234 ± 48 pg/ml vs. 66 ± 16 pg/ml) (Figure 7C). Together, these results confirm that T cells constitute an important source of systemic IL-10 as well as the data obtained at day 9 p.i. in Figure 7A.
To further characterize these IL-10-producing T cells, they were assessed for their levels of CD11a and CD49d expression. These are typical markers for activated and antigen-experienced effector type 1 regulatory T (Tr1) cells that are known for their combined capacity to produce both IFNγ and IL-10 (38–40). Figure 7D shows that ~18 and 38% of the antigen-experienced CD11a+ CD49d+ CD8+ and CD4+ T cells (red), respectively, are IL-10eGFP+, whereas <1 and 6% of the non-antigen experienced CD8+ and CD4+ T cells (blue) are IL-10eGFP+ at day 8 p.i. (Figure 7D). Moreover, within the IL-10eGFP+ T cell populations, 50% of the CD8+ T cells and 45% of the CD4+ T cells co-express IFNγ, implying the presence of Tr1 cells in T. brucei-infected WT mice.
To gain further insight into the mechanism of IL-10 upregulation following T. brucei infection the role of IL-27 was assessed. This member of the IL-12 cytokine family has been shown to promote IL-10 production by various T cell subsets via IL-27 receptor engagement and subsequent STAT3-dependent signaling (19, 41, 42). However, blocking IL-27, using an anti-IL-27 neutralizing antibody, in T. brucei-infected Vert-X mice did not alter frequencies of splenic IL-10eGFP+ cells, nor did it change the IL-10 levels in ex vivo cultured spleen cells or in serum at day 9 p.i. (Figures 8A,B). Hence measurement of systemic IL-10 production either before (day 6) or after (day 12) the day 9 peak shows a gradual increase of IL-10 during the course of the anti-IL-27 treatment and T. brucei infection (Figure 8C), which is in line with previous results (43). Also, similar to published data obtained in the T. congolense model using IL-27R-deficient mice (43), blocking of IL-27 during T. brucei infection results in an increased pro-inflammatory cytokine storm characterized by increased serum levels of IFNγ (886 ± 52 pg/ml vs. 156 ± 17 pg/ml) and TNF (353 ± 18 pg/ml vs. 202 ± 5 pg/ml), which is associated with a shortened survival (15 days vs. 55 days in control antibody-treated mice) (Supplementary Figures 3A,B). Together, these data does confirm the importance of IL-27 with respect to its anti-inflammatory role during T. brucei infection. However, in contrast to the expectation, the data show that IL-27 does not provide a co-stimulatory signal for the actual infection-associated IL-10 production by T cells or the induction of IL-10eGFP+ cells.
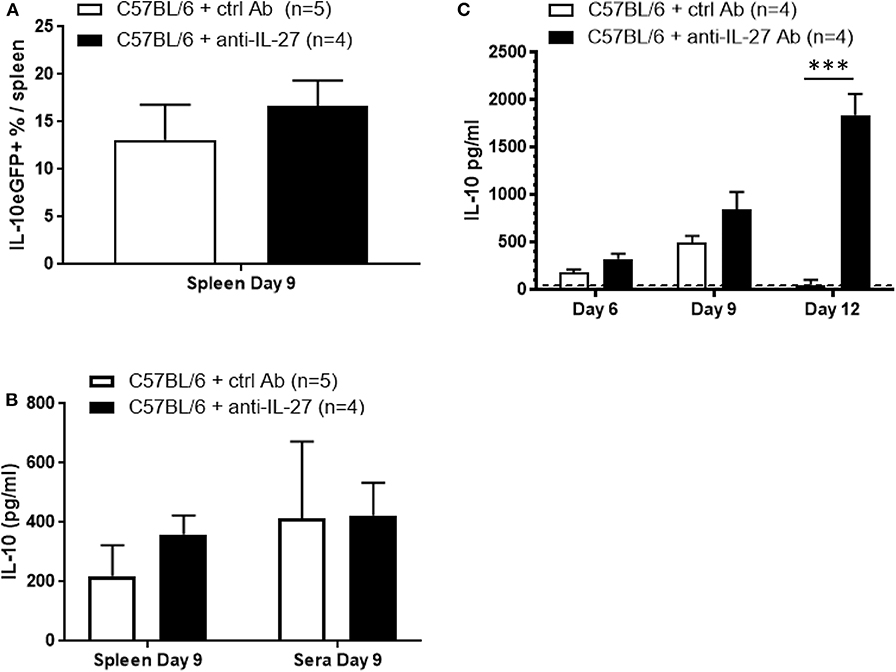
Figure 8. IL-27-independent IL-10 production following T. brucei infection. (A) Splenic percentage of IL-10eGFP+ as well as (B) IL-10 levels in supernatant of ex vivo spleen cell cultures and in serum were assessed in untreated and anti-IL-27-treated infected Vert-X mice. IL-10 levels were measured by ELISA in plasma of infected WT and anti-IL-27-treated WT at day 6, 9, and 12 p.i. (C). Data are represented as mean of minimum 4 mice per group ± SEM, where ***p ≤ 0.001, and are representative of at least TWO independent experiments.
Recently, numerous reports have demonstrated a role for the transcriptional regulator B lymphocyte-induced maturation protein-1 (Blimp-1), encoded by the Prdm1 gene, in T cell homeostasis and function, but also in the control of systemic inflammation predominantly via the regulation of IL-10 production (17, 18, 44, 45). However, Blimp-1 has also been described to directly dampen T cell and NK cell activation and proliferation as well as the production of pro-inflammatory cytokines such TNF and IFNγ (45–47). In addition, plasma cells are known to express high levels of Blimp-1, as this factor is mandatory for the differentiation of B cells into antibody secreting cells (48, 49). Using Blimp-1eYFP reporter mice (Blimp-1eYFP/+), we found that the major cellular sources of Blimp-1 are similar to those producing IL-10eGFP during the first 9 days of infection, namely NK cells and T cells in the liver at day 6 p.i. as well as mainly T cells in the liver and plasma B cells in the spleen, at day 9 p.i (Supplementary Figure 4).
To evaluate a possible correlation between IL-10 production and the expression of the Prdm1 gene in T cells, the production of IL-10 by both CD8+ and CD4+ T cells was analyzed in mice with a conditional deletion of Prdm1 within the T cell lineage (CD4Cre/+ Prdm1fl/fl, Prdm1 CKO) and littermate CD4+/+ Prdm1fl/fl controls (Prdm1 WT) following T. brucei infection. Interestingly, the loss of Prdm1 expression in T cells leads to an almost complete absence of IL-10 production by splenic CD8+ and CD4+ T cell subsets compared to WT mice at day 9 p.i. (Figure 9A). As mentioned previously, Prmd1 is also implicated in dampening T cell activation and pro-inflammatory cytokine production. This is confirmed by the finding that during T. brucei infection of Prdm1 CKO mice, an increased frequency of activated antigen-experienced splenic CD8+ and CD4+ T cells is observed by day 12 p.i. (based on CD11a and CD49d expression) (Figure 9B). Consequently, these mice also display increased serum level of pro-inflammatory cytokines, e.g., IFNγ (205 ± 26 pg/ml vs. 79 ± 19 pg/ml) and TNF (3518 ± 625 pg/ml vs. 1,040 ± 188 pg/ml), at day 12 p.i. (Figure 9C). This sustained systemic increase in pro-inflammatory cytokines is associated with a premature death of infected conditional KO mice (Figure 9D).
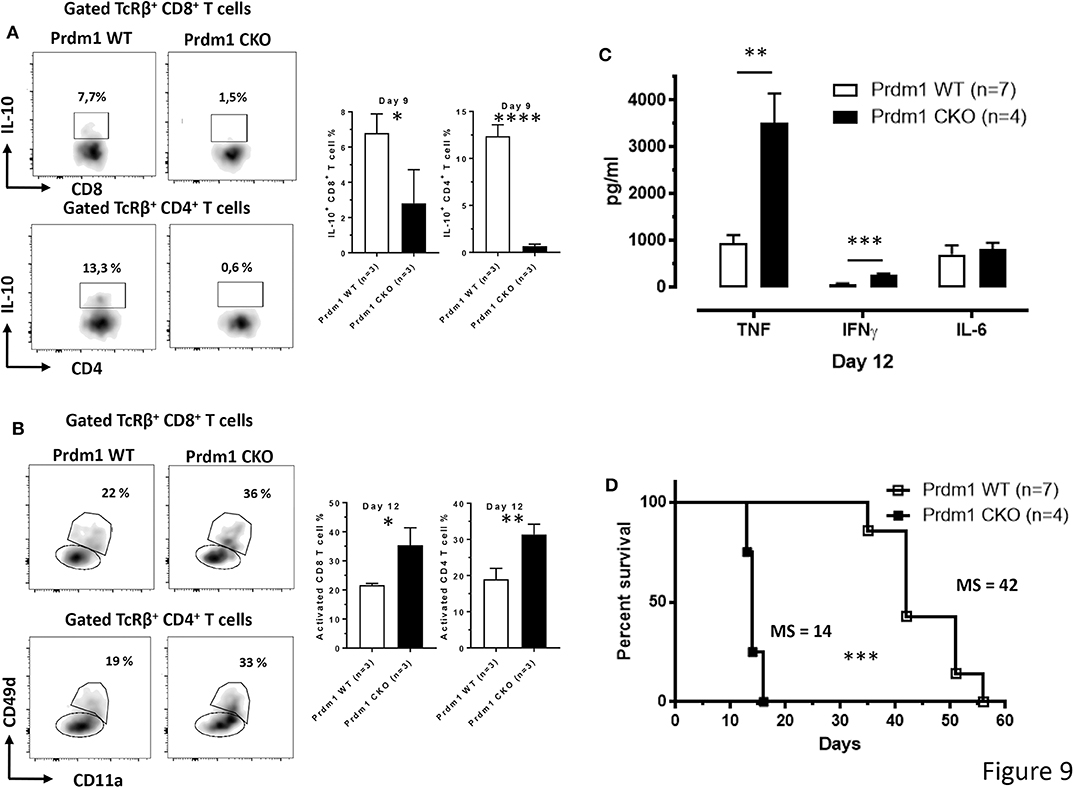
Figure 9. T cell-derived Blimp-1 signaling pathway is required to dampen inflammation following T. brucei infection. (A) Percentage of splenic IL-10+ CD8+ and CD4+ T cell subsets was analyzed at day 9 p.i. both in WT and T cell-specific Prdm1-deficient (Prdm1 CKO) mice following T. brucei infection. (B) The CD11a and CD49d expression on splenic CD8+ and CD4+ T cells were analyzed at day 12 p.i. in WT and T cell-specific Prdm1-deficient (Prdm1 CKO) mice. Data are represented as mean of at least 3 mice per group ± SEM and are representative of 3 independent experiments. (C) Cytokine levels in serum at day 12 p.i. and (D) survival of WT and Prdm1 CKO mice were monitored following T. brucei infection, where the median survival (MS) of each group is indicated in days. Data are represented as mean of at least four mice per group ± SEM, where *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, and are representative of two independent experiments.
The crucial role of IL-10 in dampening pro-inflammatory responses in order to avoid immunopathology is well-established (4, 50). In the case of T. brucei infection, IL-10 is necessary to down-modulate the inflammatory response and to prevent death of the host from a hyper-inflammation syndrome. Indeed, IL-10 KO mice started to succumb on day 8 p.i. due to the uncontrolled presence of high pro-inflammatory cytokine levels, such as IFNγ and TNF. In contrast, the IL-6 cytokine does not seem to play a major role in this phenomenon as we did not detect any significant difference in IL-6 levels between IL-10 KO and WT mice in nearly all experimental settings. In some infectious settings, the absence of IL-10 can be associated to the development of immunopathology mediated by the pro-inflammatory IL-17 cytokine, which is mainly driven by the IL-6 production. For example, the absence of IL-10 leads to IL-17-mediated immunopathology during infection with the intracellular Leishmania major parasite (51). However, our unpublished observation shows that the lack of IL-10 does not induce any systemic IL-17 levels above detection limit, which correlates with similar IL-6 levels observed between IL-10 KO and WT mice during T. brucei infection.
Until now, the cell types responsible for IL-10 secretion in the T. brucei trypanosome infection model have never been well-characterized. Since many immune hematopoietic-derived cells have the capacity to produce IL-10 (8–10, 14, 52), we first confirmed that the radiosensitive bone marrow compartment is the main source of this cytokine. Also, day 8 p.i. coincides with the highest level of IL-10 in the serum and ex vivo liver and spleen cell cultures, whereas no IL-10 could be detected in cell cultures from peripheral and mesenteric lymph nodes. Moreover, the data obtained on IL-10 production from ex vivo cell cultures correlates with the total number of IL-10eGFP+ cells in liver and spleen at day 6 and 8 p.i. using the Vert-X mice. For example, no difference in the IL-10 level and IL-10eGFP+ cell number were observed at day 6 p.i. in liver and spleen as well as day 8 p.i. in liver.
In order to characterize the different immune cell types producing IL-10 in these tissues at different time points after infection, Vert-X IL-10eGFP reporter mice were used. The study identified NK, T and B cells as producers of IL-10 during the acute phase of T. brucei infection. For example, in the liver, the main sources of IL-10 are NK and CD8+ T cells, both at early and later time points. The early production of IL-10 by NK cells also plays a crucial role during the infection with rapidly disseminating parasites, e.g., Toxoplasma gondii, Listeria monocytogenes, and Yersinia pestis (8) as well as during the late stage of murine visceral leishmaniasis (53), by counteracting the production of inflammatory cytokines such as IL-12. Another study also reported the anti-inflammatory IL-10 property of antiviral effector CD8+ T cells in the lung during acute influenza virus infection (13). In the spleen, non-plasma and plasma B cells predominate as a source of IL-10 at day 6 and 8 p.i., respectively. Recently, many B cell types, such as T2-MZP cells (CD19+CD21hiCD23hiCD1dhi), B10 cells (CD5+CD1dhi), and MZB cells (CD23−CD21hiCD1dhi), were shown to possess intrinsic regulatory properties mainly via the production of IL-10 (54–56). Madan et al. already described that CD19+ B cells represent a dominant population of IL-10-expressing cells in the spleen after diverse in vivo stimuli, such as lipopolysaccharide, CpG, goat anti-IgD and mouse cytomegalovirus infection (31). IL-10-producing plasmablasts can also exert a regulatory function in autoimmune inflammation (57). However, the protective role of B cell-derived IL-10 was negligible in our experimental model, at least during the acute phase of infection, as the genetic deletion of B cells does not significantly impact the levels of IL-10 in the serum at day 8 p.i., a time point when IL-10 KO mice starts to succumb from an uncontrolled inflammatory response.
Our results also demonstrated that IL-10 KO mice infected with T. brucei via the natural route, using tsetse flies, succumbed with a similar kinetic from an uncontrolled pro-inflammatory cytokine storm, mainly consisting of IFNγ and TNF. In addition, the different IL-10-producing cell types in this model are comparable to the ones following i.p. administration of T. brucei. Together, these results fit with the hypothesis that systemically disseminating parasites induce a chronological cell programming, leading to IL-10 production in order to counterbalance the production of pro-inflammatory cytokines and to avoid premature host death.
The resistance to T. brucei infection relies on the development of a typical Type 1 immune response characterized by the production of pro-inflammatory cytokines such as IFNγ and TNF (24, 58). However, this inflammatory response, which is mainly driven by effector T cells, must be tightly regulated in order to avoid immunopathology and potentially an early death. Our study demonstrates that NK and T cells constitute sequential sources of IL-10. These are the exact same cell types that have recently been shown to produce IFNγ following T. brucei infection (34). Effector NK and T cells possess the capacity to produce both inflammatory and anti-inflammatory cytokines in various pathogenic models. For example, Tr1 cells can produce IL-10 and IFNγ at the same time (10, 18, 20, 59). In our murine model, we have shown that antigen-experienced IFNγ+ CD11a+ CD49d+ T cells are the main producers of IL-10, suggesting that this activated CD4+ T cell subset corresponds to Tr1 cells. The genetic and pharmacological depletion of T cells drastically reduced the levels of IL-10 in the serum of T. brucei-infected mice.
As IL-27 is known to play an important role in the regulation of IFNγ during trypanosome infection as well as to modulate IL-10 production in other models (19, 32, 41, 42), we investigated whether IL-27 was affecting IL-10 levels during T brucei infection. As previously shown by the group of Shi in the T. congolense model (43), we did not find any clear role for IL-27 in IL-10 production during the early stages of T. brucei infection. For example, within the first 12 days p.i. during African trypanosomosis, the levels of IL-10 in serum and ex vivo spleen culture supernatant from infected anti-IL-27-treated mice were similar, or even higher at day 12 p.i., to the ones monitored in untreated WT mice. In addition, the percentage of IL-10+ cells is also similar between both groups. Therefore, the exact immunoregulatory role of IL-27 in the context of African trypanosomosis remains ambiguous. However, different studies focusing on the anti-inflammatory role of IL-27 in the context of parasitic infections, such as malaria, Trypanosoma cruzi and African Trypanosomosis, but also in viral infection, assign an important role of IL-27 in the regulation of CD4+ T cell activation and recruitment, to avoid IFNγ-mediated immunopathology (43, 60–63). For example, during cytomegalovirus infection, IL-27 signaling restricts the development of virus-specific CD4+ T cells displaying a cytotoxic phenotype via the inhibition of the transcription factor T-bet expression (63).
Overall, these data suggest that T cells play an important role in the generation of an early IL-10-mediated anti-inflammatory response, which occurs independently of IL-27.
Prdm1, the gene coding for Blimp-1, is a master regulator of plasma cell differentiation (49, 64) and, more recently, the expression of Blimp-1 in CD8+ and CD4+ T cells has been demonstrated to regulate homeostasis and activation via induction of IL-10 and dampening of IL-2 and IFNγ expression (17, 45). Using Blimp-1eYFP reporter mice, we demonstrated that Blimp-1-expressing cells largely overlap with IL-10 expressing cells, namely NK cells and T cells in liver and plasma B cells in spleen. Importantly, T brucei-infected mice harboring a conditional deletion of Prdm1 in T cells succumbed from uncontrolled inflammation within 2 weeks p.i. Numerous recent publications have identified Blimp-1 as a major regulator of T cell activation, mainly by controlling IL-10 and pro-inflammatory cytokine production by both CD8+ and Tr1 CD4+ T cells (18, 20, 44, 46, 47). In our T. brucei infection model, we demonstrated a similar role for Prdm1 in positively regulating IL-10 expression in T cells as well as dampening their activation and subsequent pro-inflammatory status.
Together, these results suggest that the production of IL-10 during acute T. brucei infection constitutes a tightly regulated process both at the cellular and molecular level. Our data demonstrates that the production of IL-10 by CD8+ and CD4+ T cells is required to dampen the production of pro-inflammatory cytokines by these same cell types early after infection. This study also highlights the importance of the Prdm1 transcription factor within the T cell compartment, which controls their IL-10 production, their activation status as well as the production of inflammatory cytokines. In conclusion, Prdm1 acts independently of IL-27 as a master regulator of inflammation during T. brucei infection.
All datasets generated for this study are included in the article/Supplementary Material.
All experiments complied with the ECPVA guidelines (CETS n° 123) and were approved by the VUB Ethical Committee (Permit Number: 14-220-10 and 14-220-05).
CD, BS, VB, JC, HK, and GC performed the research work. CD, VB, and JC analyzed the data. JVS, IH, LB, and EM provided materials. CD, BS, JVS, EM, IH, LB, JVG, and SM participated in writing of the manuscript.
This work was performed in frame of an Interuniversity Attraction Pole Program (PAI-IAP N. P7/41, http://www.belspo.be/belspo/iap/index_en.stm) as well as supported by the Fonds Wetenschappelijke Onderzoek (FWO, Research Project #G015016N) and by the Strategic Research Program (SRP#3, SRP#47, and SRP#63, VUB).
LB was employed by company Bioceros.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Christopher Karp and Christopher Hunter for kindly providing the IL-10 reporter mice.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01085/full#supplementary-material
1. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
2. O'Garra A, Saraiva M. The regulation of IL-10 production by immune cells. Nat Rev Immunol. (2010) 10:170. doi: 10.1038/nri2711
3. Sturlan S, Oberhuber G, Beinhauer BG, Tichy B, Kappel S, Wang J, et al. Interleukin-10-deficient mice and inflammatory bowel disease associated cancer development. Carcinogenesis. (2001) 22:665–71. doi: 10.1093/carcin/22.4.665
4. Gazzinelli R. In the absence of endogenous il-10, mice acutely infected with toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma, and TNF-alpha. J Immunol. (1996) 157:798–805.
5. Loebbermann J, Schnoeller C, Thornton H, Durant L, Sweeney NP, Schuijs M, et al. IL-10 regulates viral lung immunopathology during acute respiratory syncytial virus infection in mice. PLoS ONE. (2012) 7:e0032371. doi: 10.1371/journal.pone.0032371
6. Guilliams M, Movahedi K, Bosschaerts T, VandenDriessche T, Chuah MK, Herin M, et al. IL-10 dampens TNF/Inducible nitric oxide synthase-producing dendritic cell-mediated pathogenicity during parasitic infection. J Immunol. (2009) 182:1107–18. doi: 10.4049/jimmunol.182.2.1107
7. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, et al. Macrophage IL-10 blocks CD8+T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
8. Perona-Wright G, Mohrs K, Szaba FM, Kummer LW, Madan R, Karp CL, et al. Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe. (2009) 6:503–12. doi: 10.1016/j.chom.2009.11.003
9. Corinti S, Albanesi C, la Sala A, Pastore S, Girolomoni G. Regulatory activity of autocrine IL-10 on dendritic cell functions. J Immunol. (2001) 166:4312–8. doi: 10.4049/jimmunol.166.7.4312
10. Villegas-Mendez A, Shaw TN, Inkson CA, Strangward P, Brian de Souza J, Couper KN. Parasite-specific CD4+ IFN-γ+ IL-10+ T cells distribute within both lymphoid and nonlymphoid compartments and are controlled systemically by interleukin-27 and ICOS during blood-stage malaria infection. Infect Immun. (2015) 84:34–46. doi: 10.1128/IAI.01100-15
11. Trandem K, Zhao J, Fleming E, Perlman S. Highly activated cytotoxic CD8 T cells express protective IL-10 at the peak of coronavirus-induced encephalitis. J Immunol. (2011) 186:3642–52. doi: 10.4049/jimmunol.1003292
12. Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, et al. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3- precursor cells in the absence of interleukin 10. Nat Immunol. (2007) 8:931–41. doi: 10.1038/ni1504
13. Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med. (2009) 15:277–84. doi: 10.1038/nm.1929
14. Hilgenberg E, Shen P, Dang VD, Ries S, Sakwa I, Fillatreau S. Interleukin-10-producing B cells and the regulation of immunity. Curr Top Microbiol Immunol. (2014) 380:69–92. doi: 10.1007/978-3-662-43492-5_4
15. Alfrey EJ, Most D, Wang X, Lee LK, Holm B, Krieger NR, et al. Interferon-gamma and interleukin-10 messenger RNA are up-regulated after orthotopic liver transplantation in tolerant rats: evidence for cytokine-mediated immune dysregulation. Surgery. (1995) 118:399–405. doi: 10.1016/S0039-6060(05)80351-4
16. Wagage S, John B, Krock BL, Hall AO, Randall LM, Karp CL, et al. The aryl hydrocarbon receptor promotes IL-10 production by NK cells. J Immunol. (2014) 192:1661–70. doi: 10.4049/jimmunol.1300497
17. Kallies A, Hawkins ED, Belz GT, Metcalf D, Hommel M, Corcoran LM, et al. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat Immunol. (2006) 7:466–74. doi: 10.1038/ni1321
18. Neumann C, Heinrich F, Neumann K, Junghans V, Mashreghi M-F, Ahlers J, et al. Role of blimp-1 in programing Th effector cells into IL-10 producers. J Exp Med. (2014) 211:1807–19. doi: 10.1084/jem.20131548
19. Zhu C, Sakuishi K, Xiao S, Sun Z, Zaghouani S, Gu G, et al. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat Commun. (2015) 6:7072. doi: 10.1038/ncomms7072
20. Montes de Oca M, Kumar R, de Labastida Rivera F, Amante FH, Sheel M, Faleiro RJ, et al. Blimp-1-dependent IL-10 production by Tr1 cells regulates TNF-mediated tissue pathology. PLoS Pathog. (2016) 12:e1005398. doi: 10.1371/journal.ppat.1005398
21. Bankoti R, Ogawa C, Nguyen T, Emadi L, Couse M, Salehi S, et al. Differential regulation of effector and regulatory T cell function by blimp1. Sci Rep. (2017) 7:3. doi: 10.1038/s41598-017-12171-3
22. Sukhbaatar O, Kimura D, Miyakoda M, Nakamae S, Kimura K, Hara H, et al. Activation and IL-10 production of specific CD4+ T cells are regulated by IL-27 during chronic infection with Plasmodium chabaudi. Parasitol Int. (2020) 74:101994. doi: 10.1016/j.parint.2019.101994
24. Hertz C, Filutowicz H, Mansfield JM. Resistance to the African trypanosomes is IFN-γ dependent. J Immunol. (1998) 161:6775–783.
25. Drennan MB, Stijlemans B, Den J Van, Quesniaux VJ, Barkhuizen M, Baetselier P De, et al. The induction of a type 1 immune response following a Trypanosoma brucei infection is MyD88 dependent. J Immunol. (2005) 175:2501–9. doi: 10.4049/jimmunol.175.4.2501
26. Bosschaerts T, Guilliams M, Stijlemans B, Morias Y, Engel D, Tacke F, et al. Tip-DC development during parasitic infection is regulated by IL-10 and requires CCL2/CCR2, IFN-γ and MyD88 signaling. PLoS Pathog. (2010) 6:e1001045. doi: 10.1371/journal.ppat.1001045
27. Deleeuw V, Phạm HTT, De Poorter I, Janssens I, De Trez C, Radwanska M, et al. Trypanosoma brucei brucei causes a rapid and persistent influx of neutrophils in the spleen of infected mice. Parasite Immunol. (2019) 41:e12664. doi: 10.1111/pim.12664
28. Stijlemans B, De Baetselier P, Magez S, Van Ginderachter JA, De Trez C. African trypanosomiasis-associated anemia: the contribution of the interplay between parasites and the mononuclear phagocyte system. Front Immunol. (2018) 9:218. doi: 10.3389/fimmu.2018.00218
29. Namangala B, Noël W, De Baetselier P, Brys L, Beschin A. Relative contribution of interferon-γ and interleukin-10 to resistance to murine African trypanosomosis. J Infect Dis. (2001) 183:1794–800. doi: 10.1086/320731
30. Shi M, Pan W, Tabel H. Experimental African trypanosomiasis: IFN-gamma mediates early mortality. Eur J Immunol. (2003) 33:108–18. doi: 10.1002/immu.200390013
31. Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J Immunol. (2009) 183:2312–20. doi: 10.4049/jimmunol.0900185
32. Clement M, Marsden M, Stacey MA, Abdul-Karim J, Gimeno Brias S, Costa Bento D, et al. Cytomegalovirus-specific IL-10-producing CD4+T cells are governed by type-I IFN-induced IL-27 and promote virus persistence. PLoS Pathog. (2016) 12:e1006050. doi: 10.1371/journal.ppat.1006050
33. Caljon G, Van Reet N, De Trez C, Vermeersch M, Pérez-Morga D, Van Den Abbeele J. The dermis as a delivery site of Trypanosoma brucei for tsetse flies. PLoS Pathog. (2016) 12:e1005744. doi: 10.1371/journal.ppat.1005744
34. Cnops J, De Trez C, Stijlemans B, Keirsse J, Kauffmann F, Barkhuizen M, et al. NK-, NKT- and CD8-derived IFNγ drives myeloid cell activation and erythrophagocytosis, resulting in trypanosomosis-associated acute anemia. PLoS Pathog. (2015) 11:e1004964. doi: 10.1371/journal.ppat.1004964
35. Uyttenhove C, Marillier RG, Tacchini-Cottier F, Charmoy M, Caspi RR, Damsker JM, et al. Amine-reactive OVA multimers for auto-vaccination against cytokines and other mediators: perspectives illustrated for GCP-2 in L. major infection. J Leukoc Biol. (2011) 89:1001–7. doi: 10.1189/jlb.1210699
36. Marillier RG, Uyttenhove C, Goriely S, Marbaix E, Van Snick J. IL-27p28 is essential for parent-to-F1 acute graft-versus-host disease. Eur J Immunol. (2014) 44:2064–73. doi: 10.1002/eji.201444491
37. Belle L, Agle K, Zhou V, Yin-Yuan C, Komorowski R, Eastwood D, et al. Blockade of interleukin-27 signaling reduces GVHD in mice by augmenting treg reconstitution and stabilizing Foxp3 expression. Blood. (2016) 128:2068–82. doi: 10.1182/blood-2016-02-698241
38. Christiaansen AF, Dixit UG, Coler RN, Marie Beckmann A, Reed SG, Winokur PL, et al. CD11a and CD49d enhance the detection of antigen-specific T cells following human vaccination. Vaccine. (2017) 35:4255–61. doi: 10.1016/j.vaccine.2017.06.013
39. McDermott DS, Varga SM. Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T cell responses are larger than we think. J Immunol. (2011) 187:5568–76. doi: 10.4049/jimmunol.1102104
40. Jankovic D, Kugler DG, Sher A. IL-10 production by CD4+ effector T cells: a mechanism for self-regulation. Mucosal Immunol. (2010) 3:239–46. doi: 10.1038/mi.2010.8
41. Batten M, Kljavin NM, Li J, Walter MJ, de Sauvage FJ, Ghilardi N. Cutting edge: IL-27 Is a potent inducer of IL-10 but not FoxP3 in murine T cells. J Immunol. (2008) 180:2752–6. doi: 10.4049/jimmunol.180.5.2752
42. Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, et al. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol. (2007) 8:1363–71. doi: 10.1038/ni1537
43. Liu G, Xu J, Wu H, Sun D, Zhang X, Zhu X, et al. IL-27 signaling is crucial for survival of mice infected with African trypanosomes via preventing lethal effects of CD4+ T cells and IFN-gamma. PLoS Pathog. (2015) 11:e1005065. doi: 10.1371/journal.ppat.1005065
44. Lu P, Youngblood BA, Austin JW, Rasheed Mohammed AU, Butler R, Ahmed R, et al. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J Exp Med. (2014) 211:515–27. doi: 10.1084/jem.20130208
45. Martins GA. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat Immunol. (2006) 7:457–65. doi: 10.1038/ni1320
46. Cimmino L, Martins GA, Liao J, Magnusdottir E, Grunig G, Perez RK, et al. Blimp-1 attenuates Th1 differentiation by repression of ifng, tbx21, and bcl6 gene expression. J Immunol. (2008) 181:2338–47. doi: 10.4049/jimmunol.181.4.2338
47. Smith MA, Maurin M, Cho HI, Becknell B, Freud AG, Yu J, et al. PRDM1/Blimp-1 controls effector cytokine production in human NK cells. J Immunol. (2010) 185:6058–67. doi: 10.4049/jimmunol.1001682
48. Shaffer a L. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. (2002) 17:51–62. doi: 10.1016/S1074-7613(02)00335-7
49. Angelin-Duclos C, Cattoretti G, Lin K-I, Calame K. Commitment of B lymphocytes to a plasma cell fate is associated with blimp-1 expression in vivo. J Immunol. (2000) 165:5462–71. doi: 10.4049/jimmunol.165.10.5462
50. Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci USA. (2005) 102:8686–91. doi: 10.1073/pnas.0500419102
51. Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and eomes control key checkpoints of natural killer cell maturation. Immunity. (2012) 36:55–67. doi: 10.1016/j.immuni.2011.11.016
52. Yao Y, Simard AR, Shi FD, Hao J. IL-10-Producing lymphocytes in inflammatory disease. Int Rev Immunol. (2013) 32:324–36. doi: 10.3109/08830185.2012.762361
53. Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, Kaye PM. Post-transcriptional regulation of IL-10 gene expression allows NK cells to express immunoregulatory function. Immunity. (2009) 29:295–305. doi: 10.1016/j.immuni.2008.06.012
54. Evans JG, Chavez-Rueda K a, Eddaoudi a, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR, et al. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol. (2007) 178:7868–78. doi: 10.4049/jimmunol.178.12.7868
55. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. (2008) 118:3420–30. doi: 10.1172/JCI36030
56. Lee C-C, Kung JT. Marginal zone b cell is a major source of Il-10 in listeria monocytogenes susceptibility. J Immunol. (2012) 189:3319–27. doi: 10.4049/jimmunol.1201247
57. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity. (2014) 41:1040–51. doi: 10.1016/j.immuni.2014.10.016
58. Magez S, Radwanska M, Beschin A, Magez S, Radwanska M, Beschin A, et al. Tumor necrosis factor alpha is a key mediator in the regulation of experimental Trypanosoma brucei infections tumor necrosis factor alpha is a key mediator in the regulation of experimental Trypanosoma brucei infections. (1999) 67:3128–32. doi: 10.1128/IAI.67.6.3128-3132.1999
59. Jankovic D, Kullberg MC, Feng CG, Goldszmid RS, Collazo CM, Wilson M, et al., Conventional T-bet + Foxp3 – Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. (2007) 204:273–83. doi: 10.1084/jem.20062175
60. Findlay EG, Greig R, Stumhofer JS, Hafalla JCR, de Souza JB, Saris CJ, et al. Essential role for IL-27 receptor signaling in prevention of th1-mediated immunopathology during malaria infection. J Immunol. (2010) 185:2482–92. doi: 10.4049/jimmunol.0904019
61. Villarino A, Hibbert L, Lieberman L, Wilson E, Mak T, Yoshida H, et al. The IL-27R (WSX-1) is required to suppress T cell hyperactivity during infection. Immunity. (2003) 19:645–55. doi: 10.1016/S1074-7613(03)00300-5
62. Hamano S, Himeno K, Miyazaki Y, Ishii K, Yamanaka A, Takeda A, et al. WSX-1 is required for resistance to Trypanosoma cruzi infection by regulation of proinflammatory cytokine production. Immunity. (2003) 19:657–67. doi: 10.1016/S1074-7613(03)00298-X
63. Wehrens EJ, Wong KA, Gupta A, Khan A, Benedict CA, Zuniga EI. IL-27 regulates the number, function and cytotoxic program of antiviral CD4 T cells and promotes cytomegalovirus persistence. PLoS ONE. (2018) 13:e0201249. doi: 10.1371/journal.pone.0201249
Keywords: T. brucei, IL-10, inflammation, T cells, IL-27, Blimp-1
Citation: De Trez C, Stijlemans B, Bockstal V, Cnops J, Korf H, Van Snick J, Caljon G, Muraille E, Humphreys IR, Boon L, Van Ginderachter JA and Magez S (2020) A Critical Blimp-1-Dependent IL-10 Regulatory Pathway in T Cells Protects From a Lethal Pro-inflammatory Cytokine Storm During Acute Experimental Trypanosoma brucei Infection. Front. Immunol. 11:1085. doi: 10.3389/fimmu.2020.01085
Received: 30 December 2019; Accepted: 05 May 2020;
Published: 04 June 2020.
Edited by:
Gregoire S. Lauvau, Albert Einstein College of Medicine, United StatesReviewed by:
Manuel Antonio Franco, Pontifical Javeriana University, ColombiaCopyright © 2020 De Trez, Stijlemans, Bockstal, Cnops, Korf, Van Snick, Caljon, Muraille, Humphreys, Boon, Van Ginderachter and Magez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carl De Trez, Y2FybC5kZS50cmV6QHZ1Yi5iZQ==; Y2FybGRldHJlekB5YWhvby5mcg==
†Present address: Viki Bockstal, The Janssen Pharmaceutical Companies of Johnson & Johnson, The Hague, Netherlands
Jennifer Cnops, Glaxo-Smith Kline, Rixensart, Belgium
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.