 Sophie Jung
Sophie Jung Vincent Gies
Vincent Gies Anne-Sophie Korganow
Anne-Sophie Korganow Aurélien Guffroy
Aurélien Guffroy- 1Université de Strasbourg, Faculté de Chirurgie Dentaire, Strasbourg, France
- 2Hôpitaux Universitaires de Strasbourg, Centre de Référence Maladies Rares Orales et Dentaires (O-Rares), Pôle de Médecine et de Chirurgie Bucco-Dentaires, Strasbourg, France
- 3Université de Strasbourg, INSERM UMR_S 1109 “Molecular ImmunoRheumatology”, Strasbourg, France
- 4Université de Strasbourg, Faculté de Pharmacie, Illkirch-Graffenstaden, France
- 5Hôpitaux Universitaires de Strasbourg, Service d'Immunologie Clinique et de Médecine Interne, Centre de Référence des Maladies Auto-immunes Systémiques Rares (RESO), Centre de Compétences des Déficits Immunitaires Héréditaires, Strasbourg, France
- 6Université de Strasbourg, Faculté de Médecine, Strasbourg, France
The field of primary immunodeficiencies (PIDs) is rapidly evolving. Indeed, the number of described diseases is constantly increasing thanks to the rapid identification of novel genetic defects by next-generation sequencing. PIDs are now rather referred to as “inborn errors of immunity” due to the association between a wide range of immune dysregulation-related clinical features and the “prototypic” increased infection susceptibility. The phenotypic spectrum of PIDs is therefore very large and includes several orofacial features. However, the latter are often overshadowed by severe systemic manifestations and remain underdiagnosed. Patients with impaired innate immunity are predisposed to a variety of oral manifestations including oral infections (e.g., candidiasis, herpes gingivostomatitis), aphthous ulcers, and severe periodontal diseases. Although less frequently, they can also show orofacial developmental abnormalities. Oral lesions can even represent the main clinical manifestation of some PIDs or be inaugural, being therefore one of the first features indicating the existence of an underlying immune defect. The aim of this review is to describe the orofacial features associated with the different PIDs of innate immunity based on the new 2019 classification from the International Union of Immunological Societies (IUIS) expert committee. This review highlights the important role played by the dentist, in close collaboration with the multidisciplinary medical team, in the management and the diagnostic of these conditions.
Introduction
Primary Immunodeficiencies (PIDs)/Inborn Errors of Immunity
Primary immunodeficiencies (PIDs) constitute a large and heterogeneous group of inherited conditions caused by germline mutations impairing or not protein expression, but resulting in either loss-of-function (LOF; hypomorphic [partial LOF] or amorphic [complete LOF]), or gain-of-function (GOF) of the encoded protein (1–4). To date, 406 distinct disorders have been described with 430 identified gene defects that affect the immune system development and/or function (3, 4). Although PIDs are still considered as rare diseases, epidemiologic studies have suggested that they may be underdiagnosed (5, 6) and their collective prevalence is now estimated to range between 1/1,000 and 1/5,000 (3). The main clinical feature of PIDs is an increased susceptibility to severe, atypical, persistent, and/or recurrent infections. According to the gene defect, the susceptibility can vary from a strong predisposition to a large variety of microorganisms, to a selective susceptibility to a single type of infection (1). However, the phenotypic spectrum of PIDs is extremely large and extends beyond infections. It includes a wide range of clinical manifestations associated with immune dysregulation such as allergy, auto-immunity/inflammation, lymphoproliferation, and malignancies (7–9). Considering the growing recognition of immune dysregulation, the denomination “primary immune deficiencies” appears too restricted and one should now use the terminology “inborn errors of immunity” (2). For simplicity, we will however use the abbreviation “PID” in this review. According to the 2019 classification of the International Union of Immunological Societies (IUIS) expert committee of Inborn Errors of Immunity, PIDs are divided into 10 categories based on shared pathogenesis and/or clinical phenotypes (Table S1) (3, 4).
Oral Involvement in PIDs
PIDs can affect almost all organ systems and tissues including the orofacial region. Patients with compromised immunity, in particular when the innate immunity is affected, are therefore predisposed to a variety of oral manifestations including, among others, infections (e.g., candidiasis, herpes gingivostomatitis*, atypical dental infections), oral aphthous ulcers*, severe periodontal diseases*, and dental anomalies*. These lesions can in some cases be inaugural, preceding the other clinical manifestations and therefore being one of the first features of an underlying defect of immunity. In some PIDs of the innate immunity, they can even represent the main clinical manifestation, as in Papillon-Lefèvre syndrome (PLS). One of the most significant situation is the occurrence of aggressive periodontitis* with premature tooth loss in children/adolescents as it almost always indicates the existence of an underlying systemic or immunologic disorder, in particular neutrophil defects. The medical team, and especially the dentists, should be aware of the main oral features associated with the different PID disease categories and should be able to identify atypical oral lesions that may signal a previously undiagnosed PID. Although often underestimated, the role played by dentists in the detection of warning clinical signs is crucial as it can contribute to a timely diagnosis and an early implementation of adequate management. Indeed, the latter is essential to avoid the persistence of infectious and inflammatory foci that may have an impact on the course of the PID itself (i.e., development of autoimmune manifestations, malignant transformation).
The aim of this review is therefore to describe the spectrum of orofacial features associated with the different PIDs of innate immunity and to give clues for the management of these manifestations. We focused on defects of phagocyte number and function (Category 5; Table 1) as oral manifestations are very prevalent. However, several other PIDs of intrinsic and innate immunity (Category 6; Table 1) are also characterized by specific oral manifestations such as oral candidiasis in chronic mucocutaneous candidiasis (CMC) disease for example. We will not discuss in this review the deficiencies of the complement cascade (Category 8; Table S1).

Table 1. Disease categories described in this review.
Methods: Search Strategy
We performed a literature review using PubMed/MEDLINE database (up to April 2020). Relevant articles were selected, based on combinations of MeSH or other search terms, without language or time restriction.
The terms “primary immune deficiency” OR “primary immunodeficiency” OR “inborn errors of immunity” AND “innate immunity,” or the terms referring to the different diseases (e.g., “severe congenital neutropenia,” “Papillon-Lefevre syndrome,” “chronic granulomatous disease,” “mendelian susceptibility to mycobacterial disease”), were used in combination with the following terms: “oral,” “oral mucosa,” “mouth,” “orofacial,” “teeth,” “oral ulcer,” “periodontal disease,” “periodontitis,” “candidiasis,” “gingivostomatitis.”
Results
Congenital Defects of Phagocyte Number or Function [Category 5 (3)]
Multiple genetic defects associated with a reduction in the absolute neutrophil count (ANC) or with an aberrant function of these cells are clinically characterized by recurrent infections due to extracellular pathogens. In addition, they are associated with very aggressive forms of periodontitis, which already present in early childhood or adolescence. As “gatekeepers of oral immunity,” fully functional neutrophils are essential in the maintenance of periodontal homeostasis (10). Indeed, they represent the majority of immune cells (>95% of total leucocytes) recruited to the gingival crevice*, also called gingival sulcus*, and form a “defense wall” against the subgingival dental biofilm* (10–12) [for review see (11)].
Severe Congenital Neutropenias
Genetic, Pathophysiology, and Clinical Manifestations
Severe congenital neutropenias (SCNs) represent a group of PIDs characterized by an impaired differentiation of neutrophils and an accumulation of atypical promyelocytes in the bone marrow due to maturation arrest of myelopoiesis (13). Consequently, patients present a severe chronic neutropenia (i.e., blood ANC below 0.5 × 109 cells per liter or 500 cells per μl/mm3). The estimated prevalence of SNC ranges between 3 and 8.5 cases per million individuals (14). Already in the first months of life, SCN patients develop severe bacterial infections affecting the respiratory tract, the skin, and deep tissues and, to a lesser extent, fungal infections. One of the characteristic features is the absence of pus formation. In addition, SCN is considered as a pre-leukemic condition with a marked propensity for hematopoietic malignant transformation leading to acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) (13, 15).
Germline mutations have been identified in more than 20 genes (Table 2), but almost half of the patients carry heterozygous variants in ELANE gene (alternative name ELA2), which encodes neutrophil elastase (13).
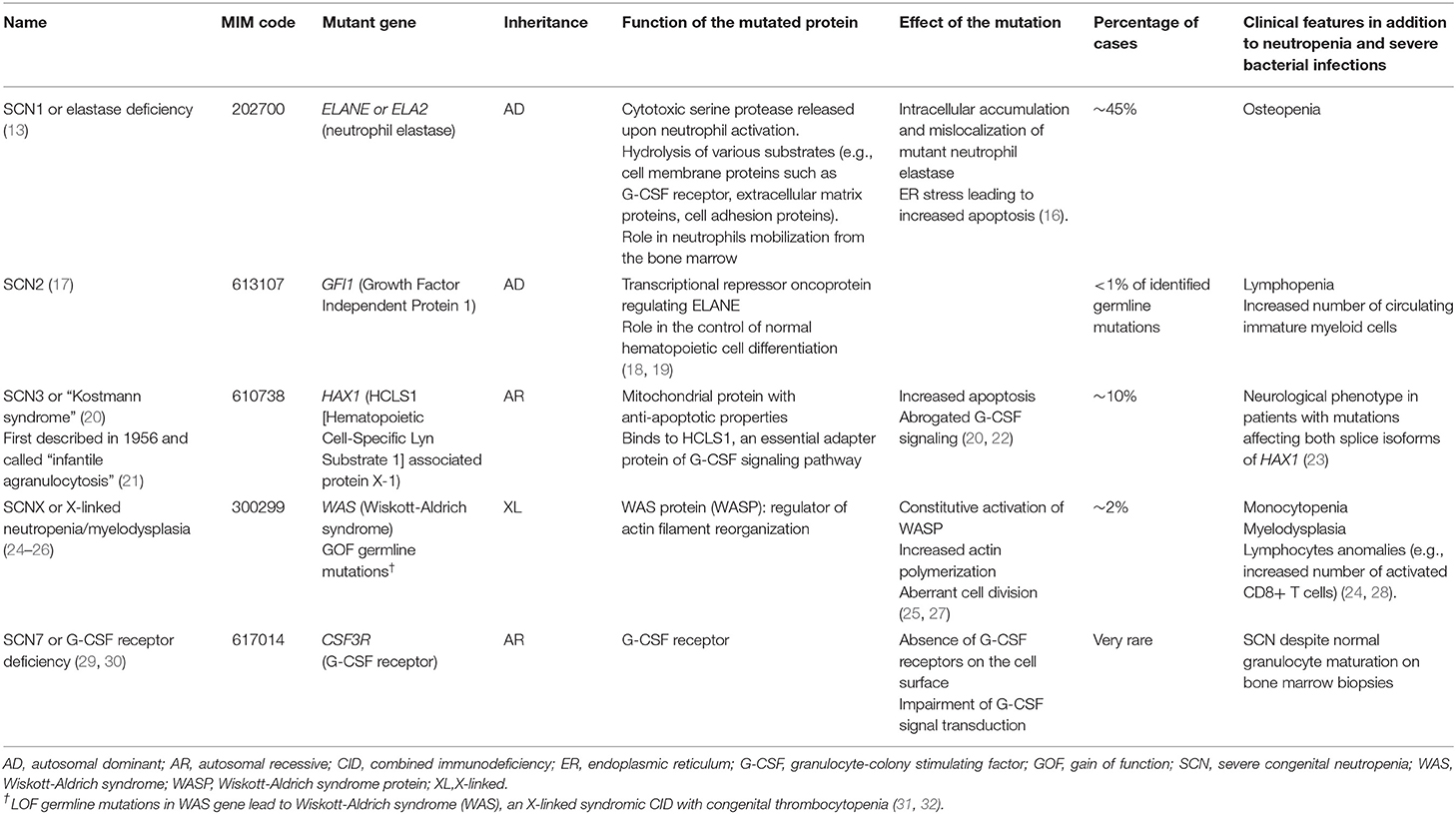
Table 2. Characteristics of the different forms of severe congenital neutropenias (SCNs).
Congenital neutropenia can also be found in association with additional immunologic and non-hematopoietic features in several syndromic disorders that are due to rare pathogenic variants in genes controlling ribosome maturation (e.g., SBDS, DNAJC21), lysosomal function (e.g., LAMTOR2, VPS13B), or glucose metabolism (e.g., G6PC3, SLC37A4), among others (13, 33) (Table 3).
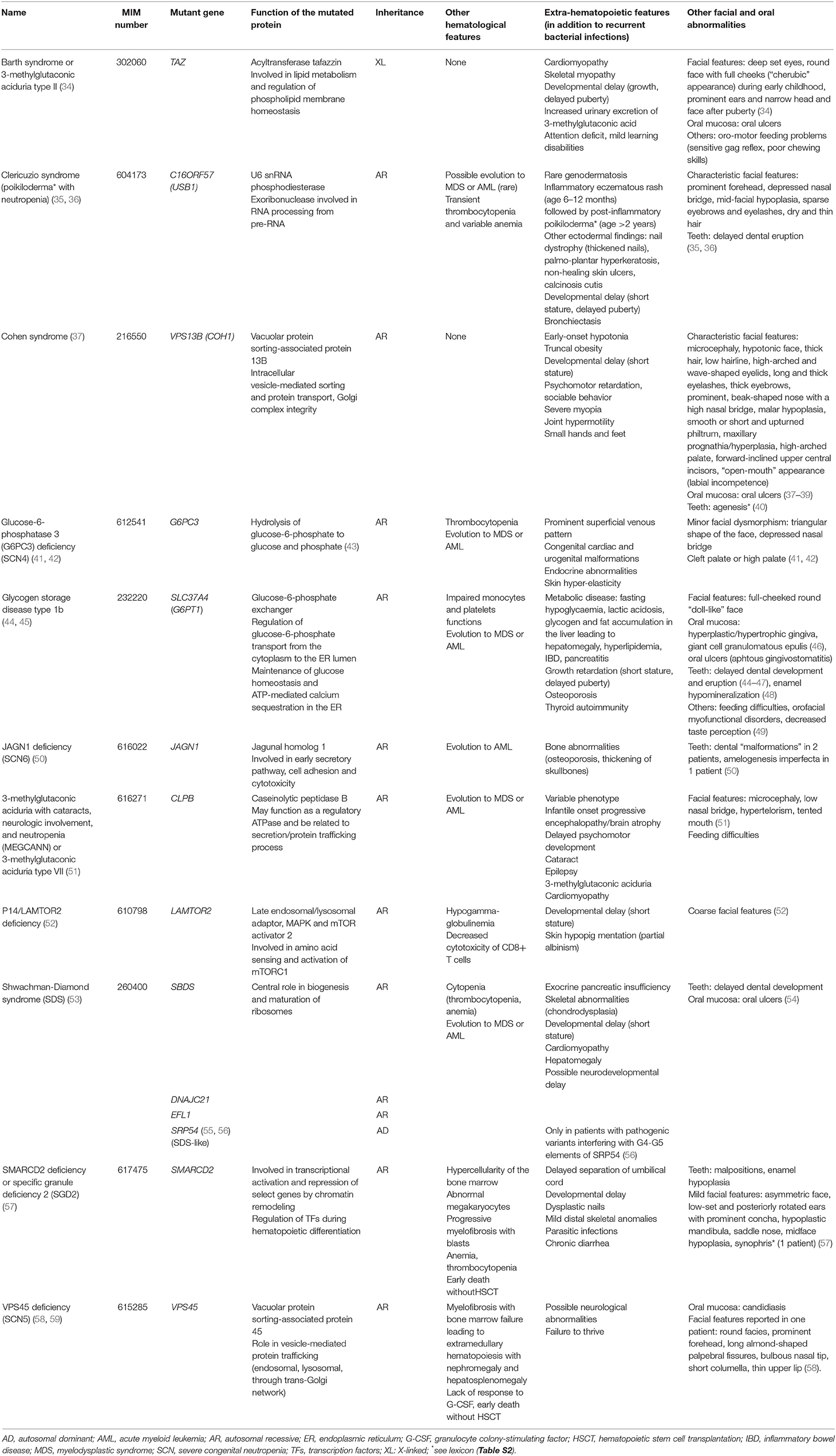
Table 3. Characteristics of syndromic forms of congenital neutropenias.
General Management
Since the availability of recombinant granulocyte colony-stimulating factor (G-CSF), which is currently the treatment of choice for SCN, the quality of life of patients has improved significantly and the overall survival exceeds 80% (13, 60, 61). Administration of G-CSF results in an increase in the neutrophil count that is associated with a significant reduction in the number and severity of infectious episodes (13). However, in patients who do not respond to G-CSF treatment and/or develop secondary malignancies, hematopoietic stem cell transplantation (HSCT) remains the only available treatment option (62). Prolonged exposure to high dosage of G-CSF can result in the acquisition of somatic CSF3R mutations that generate truncated G-CSF receptors. This is responsible for an hypersensitivity to G-CSF with clonal proliferation favoring leukemic transformation (63). Indeed, patients who require higher doses of G-CSF have a cumulative incidence of leukemia of 40% after 15 years compared to 11% in patients that are more responsive to G-CSF (61). In G-CSF receptor deficiency (SCN7; Table 2), neutropenia is unresponsive to G-CSF, but granulocyte-macrophage colony-stimulating factor (GM-CSF) treatment may be effective (29, 30).
Oral Manifestations and Management
The main oral manifestations associated with SCN include recurrent oral ulcers and periodontal diseases (64). Considering the key protective role of neutrophils in the periodontal tissues, a reduction of neutrophil numbers at the gingival sulcus leads to a marked increase in host susceptibility to periodontal diseases (65). Indeed, patients with SCN often suffer from early onset aggressive periodontitis, affecting both the primary and the permanent dentitions, with intense inflammation and severe bone loss leading to premature tooth loss (66). Periodontal involvement has been already reported in the SCN family originally described by Kostmann (67). A diagnostic score has been develop in order to differentiate congenital from non-congenital neutropenia using data collected during the first consultation (68). Apart from the medical history and previous severe infections, the key factors for congenital neutropenia prediction include the oral features classically associated to defects in neutrophils numbers (i.e., oral ulcers and/or gingivitis*) (68). This highlights the important role of the dentists in the diagnosis of this disease entity. In addition, the levels of plasmatic pro-LL-37, the precursor of the antimicrobial peptide cathelicidin LL-37 that is crucial to control periodontal normal flora, represents an early marker of severe disease. Therefore, pro-LL-37 may be used as a fast and simple tool to facilitate differential diagnosis of chronic neutropenia and to discriminate SCN from autoimmune and idiopathic neutropenia (69).
Ye et al. studied the genotype-phenotype correlation between the mutated gene and periodontal involvement in 14 SCN patients (70). They observed that patients with pathogenic variants in ELANE present with more severe periodontal disease than patients with HAX1 or unknown genetic defects. In addition, they found higher levels of the pro-inflammatory cytokine IL1β in the gingival crevicular fluid* as well as a skewed microflora in the periodontal pockets* of ELANE mutated patients (70). A correlation between the oral status and the treatment has also been studied. Several authors have observed that periodontal disease tends to persist in patients under G-CSF treatment, even after normalization of neutrophil counts (67, 70). In the family originally described by Kostmann, all surviving non-transplanted patients that have been treated by G-CSF presented periodontal disease (chronic gingival inflammation with or without bone loss) despite normal ANCs and prophylactic dental care (67). Pütsep et al. showed that G-CSF-treated SCN3 patients have proper numbers of circulating neutrophils but that these cells have an impaired production of the antimicrobial peptide cathelicidin LL-37 and its precursor (71). Professional periodontal maintenance, associated with strict oral hygiene, should therefore be continued even after normalization of ANCs in patients under G-CSF therapy (67). In some cases, the extraction of severely affected primary teeth could reduce the microbial load of periodontal pathogens and create a better environment for the eruption of permanent teeth. To date, HSCT remains the only available curative treatment of SCN and is associated with a stabilization of periodontal disease. Indeed, in the original Kostmann family, the patient who received HSCT had almost normal concentrations of LL-37 in neutrophils and saliva and no periodontal inflammation (67, 71).
Cyclic Neutropenia
Genetic, Pathophysiology, and Clinical Manifestations
In addition to SCN1, heterozygous pathogenic variants in ELANE can cause cyclic neutropenia (CyN) (64, 72). CyN is a related disorder of granulopoiesis characterized by a regular oscillation in the number of circulating neutrophils (from normal levels to severe neutropenia) and other peripheral blood cells including monocytes, platelets, reticulocytes, and lymphocytes, usually with a 21-day periodicity. Fluctuations in blood cells numbers are due to an oscillatory production of cells by the bone marrow (64, 72). CyN is associated with a milder course of the disease and a lower risk of leukemic transformation. The main clinical manifestations that usually appear during early childhood include recurrent fever, painful oral mucosal ulcers as well as skin and oropharyngeal infections (64, 72). Between neutropenic intervals, affected individuals are usually healthy but life-threatening bacterial infections can occur during periods of severe neutropenia. Although neutrophil counts continue to cycle, clinical manifestations usually decrease in severity during adulthood (64, 72).
General Management
As for SCN, CyN management relies on G-CSF therapy.
Oral Manifestations and Management
The diagnosis of CyN is usually made based on a pattern of recurrent fever and oral ulcerations. Serial blood cell counts show regular oscillations with an average 3-week turnover frequency (64). Painful ulcerations that can affect any part of the oral mucosa during the neutropenic phases are often the initial manifestation of CyN, highlighting the crucial role of the dentist in the diagnosis of this condition. Severe periodontal diseases (i.e., gingivitis and periodontitis) also develop when ANCs are at their lowest point (73–77). In a recent systematic review, the authors reported oral ulcers in 18% of CyN patients but gingival inflammation was observed in almost all of the cases (65). Systemic symptoms such as fever usually decrease after puberty but adults with CyN continue to experience oral ulcers and periodontal disease (64, 70, 74). Although G-CSF therapy is associated with a reduction of oral ulcers (78), its combination with professional periodontal treatment and oral hygiene improvement is however required to control periodontal diseases.
Defects of Motility
Leukocyte Adhesion Deficiency
Genetic, pathophysiology, and clinical manifestations.
The interaction of leukocytes with vascular endothelial cells, which is mediated by several families of adhesion molecules, is crucial for their migration to the tissues (79, 80). Leukocyte adhesion deficiency (LAD) is a group of autosomal recessive (AR) PIDs due to defects in leukocyte adhesion cascade with altered extravasation into tissues. To date, three different forms have been described (79). LAD type I [>320 reported cases (81)] affects the firm adhesion of leukocytes to the endothelium (79, 82, 83). LAD type II (<10 patients reported worldwide) is characterized by a defect in the rolling adhesion phase that involves transient interactions between P- and E-selectins (expressed by endothelial cells) with their fucosylated ligands (expressed by neutrophils) (79, 82, 84, 85). Finally, LAD type III (described in about 20 patients) is due to abnormal integrin activation that is crucial to induce the immobilization of neutrophils (82, 86) (Table 4).
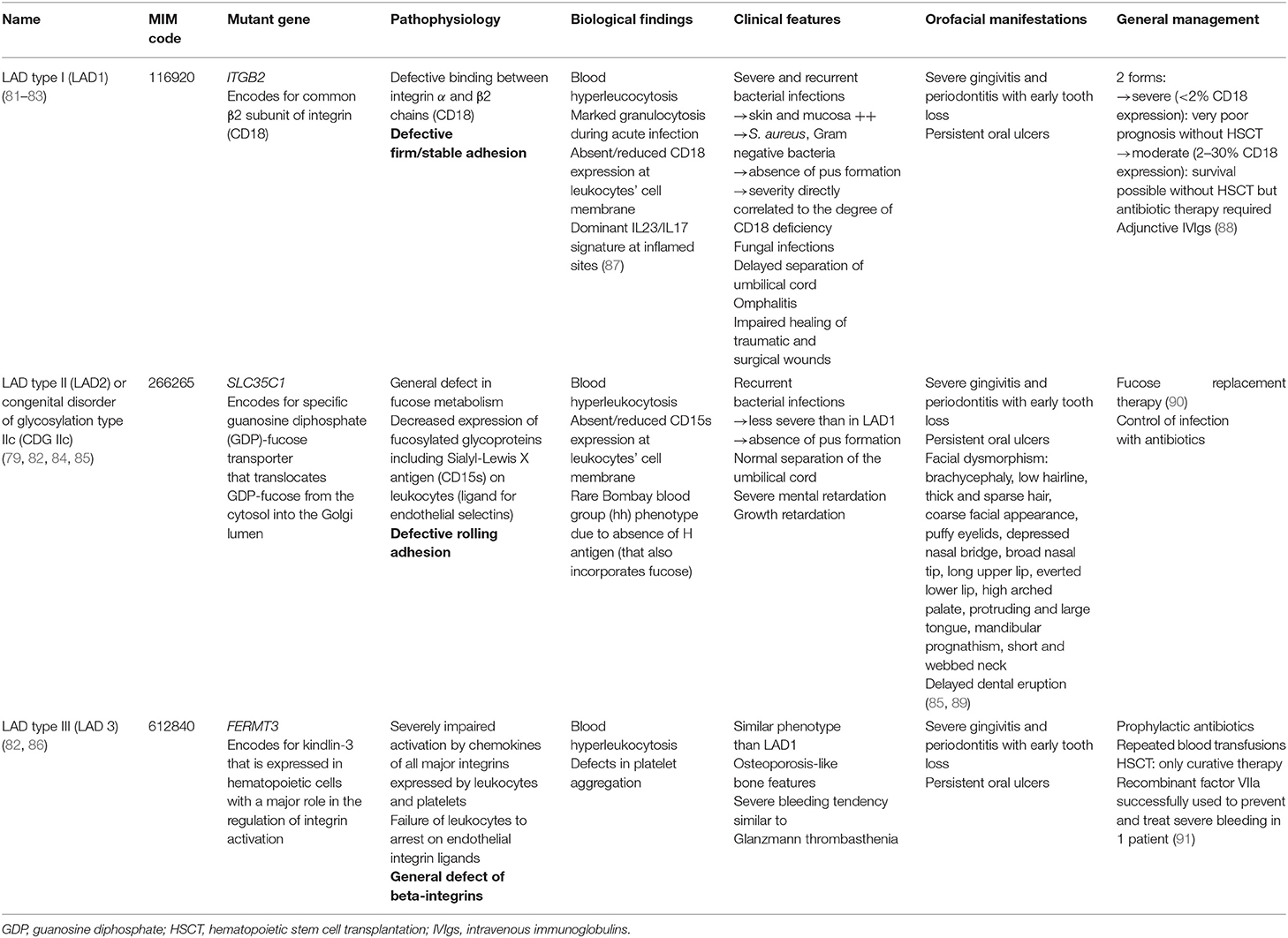
Table 4. Features of the different forms of leukocyte adhesion deficiency (LAD).
General management
The general management of each form of LAD is described in Table 4.
Oral manifestations and management
As LAD type II and type III have been reported in less than 50 cases worldwide, the management of oral manifestations has been mainly described for LAD type I (LAD1) patients, in particular those with moderate form that is characterized by residual CD18 expression (2–30%). These patients usually survive childhood without HSCT but they present recurrent infections and immune-related lesions of the skin and mucosal surfaces (81). Oral involvement is observed in more than 50% of the patients with moderate LAD1 and includes periodontal diseases as well as recurrent and painful oral ulcers (81). Palatal ulcer with perforation has been reported in one LAD1 patient (92). Periodontitis is extremely aggressive and has a very early onset, affecting already primary teeth. It is characterized by an intense inflammation and a rapid loss of periodontal tissues including alveolar bone* (66, 93). Periodontitis in LAD1 patients is mainly unresponsive to standard treatments (i.e., mechanical removal of tooth associated biofilm in combination with antibiotics), leading to premature tooth loss before young adulthood (66). Historically, LAD1-associated periodontitis has been attributed to defective neutrophil control of the periodontal infection. LAD subgingival microbiome is significantly different from the biofilm observed in healthy individuals and in individuals with localized aggressive periodontitis (94). Indeed, it is characterized by a reduced microbial diversity with a loss of health-associated microbial species and an over representation of periodontitis-associated species such as Parvimonas micra, Porphyromonas endodontalis, Eubacterium brachy, and Treponema species. Pseudomonas aeruginosa was also detected in LAD1 although it is a bacterial species that is not typically found in subgingival plaque (94). However, Moutsopoulos et al. have recently shown that LAD1 periodontitis does not represent a “raging infection” due to uncontrolled bacterial invasion of periodontal tissues but is rather caused by a dysregulated host inflammatory response, where the bacteria serve as initial triggers for local immunopathology (95–97). Indeed, the translocation of bacterial products such as lipopolysaccharide into the underlying tissues stimulates the local inflammatory response and the induction of IL23-mediated immunity. This leads to an excessive production of the proinflammatory and bone-resorptive cytokine IL17, implicating for the first time in humans the role of neutrophils in the regulation of IL17 responses (96). It has been shown that this cytokine is also overproduced in common forms of chronic periodontitis (98). This IL17 exaggerated response is mainly localized to the mucosal tissues. In the absence of tissue neutrophils, as in LAD1, the IL23 response fails to downregulate and continuously induces IL17 (66). Inhibition of the IL17 pathway in the murine model of LAD1 is associated with a reduction of both the inflammatory periodontal bone loss and the bacterial load. This suggests that dysregulated IL17-driven inflammation consecutive to impaired neutrophil recruitment fuels periodontal microbial overgrowth (95). Recently, the team of Moutsopoulos treated a patient with moderate form of LAD1, refractory periodontitis and non-healing cutaneous ulcers with anti-IL12/IL23 monoclonal antibody (ustekinumab) that inhibits IL23-dependent production of IL17 (87). The treatment allowed a significant reduction of oral inflammation and a complete resolution of the deep cutaneous wounds without significant adverse effect. Inhibition of IL23 and IL17 appears as a promising strategy in the management of moderate forms of LAD1, in particular for severe periodontal involvement (87), and an interventional protocol has been recently initiated (NCT03366142) (66). Indeed, despite strict oral hygiene regimen and regular periodontal treatment, most of the patients lose their teeth (99, 100). Their replacement relies on prosthetic rehabilitation with an age-specific approach. In growing patients, removable prostheses are favored and can be adapted depending on the teeth that are lost. To date, dental implants have been reported in only one patient with LAD (101).
Papillon-Lefèvre Syndrome (PLS)
Genetic, pathophysiology, and clinical manifestations
First described in a French family by the physicians Papillon and Lefèvre (102), Papillon-Lefevre syndrome (PLS; MIM 245000) is a rare AR condition characterized by the association of aggressive early-onset periodontitis and palmoplantar hyperkeratosis. The prevalence of PLS ranges between 1 to 4 cases per million individuals (103). PLS is caused by homozygous (2/3 of the cases) or compound heterozygous (1/3 of the cases) pathogenic variants in the gene CTSC that encodes a lysosomal cysteine protease called cathepsin C (CTSC) or dipeptidyl peptidase I (103, 104). CTSC is involved in posttranslational modification and activation of many serine proteases stored primarily in azurophilic granules from neutrophils (i.e., neutrophil elastase, cathepsin G, proteinase 3) (105). CTSC plays also a role in the activation of granzymes A and B in cytotoxic T lymphocytes (106). Whereas, mature neutrophils of PLS patients lack all serine proteases' activity, the latter is normal in immature neutrophils. PLS phenotype may therefore arise from functional defects affecting mature neutrophils within tissues. For example, they are incapable of producing neutrophil extracellular traps (NETs) in response to reactive oxygen species (ROS) (107). However, despite lack of active serine proteases in neutrophils and cytotoxic T lymphocytes from PLS patients, the associated immunodeficiency is remarkably mild as an only 15 to 20% of PLS patients are predisposed to recurrent bacterial infections (108). Most of these infections are mild skin pyodermas, but occasionally, severe and/or fatal pyogenic abscesses involving internal organs (i.e., liver abscesses) do occur (109).
In addition to activation of immune cells, the proteolytic activity of CTSC has also been proposed to play a role in epithelial differentiation and desquamation (110), likely explaining the skin phenotype that is dominated by palmoplantar hyperkeratosis. The latter can vary from mild psoriasiform scaly skin to overt hyperkeratosis. Keratosis can also affect other sites such as elbows and knees, and additional clinical findings may include intracranial calcifications, hyperkeratosis of the hair follicles, nail dystrophy, and hyperhidrosis (111).
Oral manifestations
Periodontitis in PLS patients is exceptionally severe with a very early-onset and a generalized pattern resulting in premature loss of both primary and permanent teeth (Figure 1). Although it has been associated with functional defects in neutrophils, the mechanisms by which CTSC deficiency leads to periodontitis have not been fully elucidated so far (66). Aggressive periodontitis in PLS patients could be attributed, at least in part, to a dysregulated inflammatory response rather than to an inefficient control of the periodontal bacteria. Indeed, CTSC deficiency results in failure to activate neutrophil-derived serine proteases, impairing proteolytic degradation of proinflammatory chemokines and cytokines, a mechanism important for periodontal tissue homeostasis (112). The subgingival microbiota in PLS patients is diverse with many periodontal pathogens commonly associated with chronic and aggressive periodontitis (e.g., Aggregatibacter actinomycetemcomitans, Fusobacterium nucleatum). Eruption of deciduous teeth occurs at expected ages with normal structure and form (113). Extremely intense gingival inflammation (e.g., erythema, edema, pain) develop shortly after eruption of primary teeth with a rapid periodontal destruction (e.g., deep periodontal pockets with pus exudate, extensive alveolar bone resorption, tooth mobility and migration) and premature tooth loss without signs of root resorption (Figures 1A,C) (111, 113, 114). Halitosis and lymphadenopathy are frequent. After the exfoliation or the extraction of the primary teeth, inflammation resolves rapidly. However, the cycle repeats itself with the eruption of permanent teeth (Figure 1B) (113). Radiological examination shows vertical alveolar bone loss around the teeth and in advanced stages, teeth can appear to be “floating” in the bone (Figure 1C) (113). Most PLS patients lose all of their primary and permanent teeth (66, 115) and only few patients without periodontal involvement have been reported (116, 117). Third molars remain however frequently unaffected (113). Gingival inflammation disappears in the totally edentulous patient. Although palmoplantar keratoderma and aggressive periodontitis are the cardinal clinical signs of PLS and usually manifest simultaneously between the age of 6 months and 4 years, no significant correlation has been found between the severity of these two conditions (118).
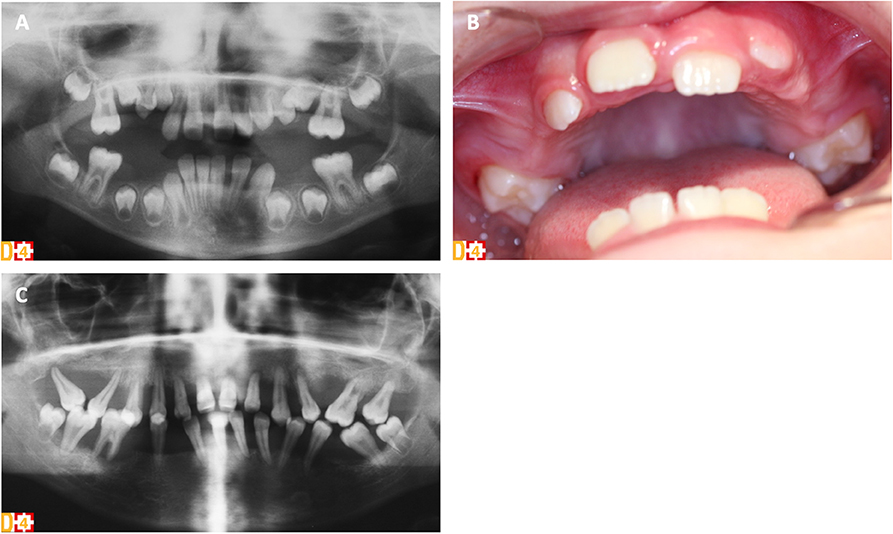
Figure 1. Clinical and radiological oral phenotype of two sisters with PLS syndrome (D[4]/Phenodent database (www.phenodent.org), Reference Center for Oral and Dental Rare Diseases, University Hospital, Strasbourg). (A,B) Premature tooth loss of the primary teeth in the 6-year-old patient. (A) Panoramic radiograph showing alveolar bone loss around the teeth, in particular at the mesial aspect of right permanent mandibular and maxillary first molars. (B) Intra-oral view showing the absence of all primary teeth. Absence of inflammation in the edentulous areas but presence of gingival inflammation around the erupting permanent maxillary first molars despite the perfect control of dental plaque. (C) Several periodontitis in the 16-year-old patient. Panoramic radiograph showing generalized severe vertical alveolar bone loss around all permanent teeth: typical radiological aspect of “floating” teeth.
Skin and oral management
To date, therapeutic strategies remain limited and the management of both skin and oral manifestations is known to be difficult.
Regarding periodontal treatment, the main goal is to eradicate the reservoirs of periodontopathogens and to limit the destruction of periodontal tissues. The management of periodontitis includes careful plaque control with professional and individual oral hygiene regimens, the possible use of antiseptic mouth rinses (e.g., 0.2% chlorhexidine), conventional mechanical periodontal treatment to remove tooth-associated biofilm (scaling and root planing) along with courses of systemic antibiotics, followed by regular supportive periodontal therapy. The treatment of teeth with deep periodontal pockets may require flap surgical procedures (113, 119, 120). The commonly used systemic antibiotics are tetracyclines (that should be avoided in pediatric patients under 8 years of age due to the risk of teeth discoloration and enamel hypoplasia), erythromycin, amoxicillin plus metronidazole, or clavulanic acid (113, 119, 120). Extraction of primary teeth with poor prognosis, in combination with the eradication of the periodontal pathogens, creates a safe environment for the eruption of permanent teeth (113). However, despite regular and appropriate periodontal and antibiotic treatment regimens, the majority of the PLS patients lose all of their teeth. Nickles et al. analyzed long-term results (≥10 years follow-up) of periodontal treatment in eight patients with PLS and the teeth were maintained in only two of them (120). The replacement of lost teeth relies on prosthetic rehabilitation with an age-specific approach in a similar way than for LAD patients. Implants have been used by several authors in adult patients to improve stability and support of the prostheses (120–124). However, in PLS patients, implant placement is often complicated by the severe alveolar bone resorption due to early tooth loss. Although bone augmentation techniques and the use of short implants can possibly be considered (125), PLS patients are at high risk of peri-implantitis and implants loss. In Nickles et al. study, implants have been placed in four patients but three of them showed peri-implantitis only 4 years after their insertion (120). A very regular maintenance is therefore required to avoid early implant loss (120).
Treatment of dermatological manifestations relies on topical applications of emollients, keratolytic agents containing salicylic or lactic acid, and topical steroids to reduce skin inflammation. Several authors have suggested that oral retinoids such as acitretin, etretinate, and isotretinoin (analogs of vitamin A), which are effectively used in the treatment of various types of keratinizing disorders (by decreasing the keratin content of keratinocytes), may be beneficial in the management of cutaneous lesions of PLS, but also useful to prevent the loss of permanent teeth in children with PLS (126–131). Whereas, palmoplantar keratoderma usually improves rapidly in patients receiving oral retinoids, periodontal disease requires longer periods of treatment (128). The safety of oral retinoids in children remains however controversial due to their side-effects, in particular on skeletal development (132). Recently, an enzyme replacement therapy with recombinant CTSC has been developed and allowed to correct pathophysiologic markers in fibroblasts from PLS patients. It can therefore be a promising therapeutic approach for the future treatment of CTSC deficiency (133). A multidisciplinary team including dermatologists, pediatricians and dentists (periodontology, pediatric dentistry, oral surgery, prosthodontics) is important for the overall care of PLS patients.
FPR1 Polymorphisms and Aggressive Periodontitis
Formyl peptide receptors (FPRs), which belong to a class of G-protein-coupled receptors, are highly expressed by neutrophils. FPR bind N-formylpeptides, produced by the degradation of bacterial cells, which are one of the major chemotactic stimuli guiding the migration of neutrophils to infection sites (134). This triggers an intracellular signaling cascade that coordinates cytoskeletal reorganization as well as the migration of neutrophils along the gradient of chemokines (134). Some studies highlight an association between FPR1 (the gene encoding FPR) single nucleotide polymorphisms (SNPs) and aggressive forms of periodontitis that are rapidly progressing (135) [grade C periodontitis in the new classification of periodontal diseases (136, 137)]. rs5030879 (c.348 C>T) SNP was particularly studied. Indeed, it has been shown that African Americans with a homozygous 348T/T genotype exhibit a significantly lower neutrophil chemotactic response to formylpeptides than that observed in subjects with the 348T/C or 348C/C genotypes, with an increased risk to develop aggressive periodontitis (135, 138, 139).
WDR1 Deficiency
Genetic, pathophysiology, and clinical manifestations
Homozygous and heterozygous pathogenic variants in WDR1 gene that encodes an actin-interacting protein alter the regulation of neutrophil cytoskeleton, causing neutrophil dysfunction with abnormal morphology (herniation of nuclear lobes), chemotaxis and survival (140, 141). A more recent study also identified defects in the T- and B-cell compartments (aberrant assembly of immunological synapses) (141). Patients present recurrent infections and varying clinical manifestations including mild neutropenia, skin ulceration, impaired wound healing, and moderate intellectual disability (140, 141).
Oral manifestations
Patients with WDR1 deficiency can suffer from severe aphthous stomatitis* leading to oral stenosis, and from candidiasis. Facial dysmorphia has been observed in some patients (frontal bossing, hypertelorism, wide nasal nostrils) (141).
Defects of Respiratory Burst
Chronic Granulomatous Disease (CGD)
Genetic, pathophysiology, and clinical manifestations
Chronic granulomatous disease (CGD) is a PID [~1 case in 200 000 to 250 000 live births (142, 143)] due to functional impairment of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in all phagocytes (neutrophils, monocytes, macrophages and dendritic cells [DCs]) (144). NADPH oxidase (NOX) is a multiprotein enzyme complex comprising both membrane-bound (cytochrome b558: gp91phox and p22phox) and cytosolic (p40phox, p47phox, and p67phox) proteins that assemble upon phagocytes' activation. It catalyzes the transfer of electrons from NADPH to molecular oxygen to form superoxide ions that are used for the generation of ROS (e.g., hydrogen peroxide, hypochlorous acid). ROS production, called respiratory or oxidative burst, is a powerful antimicrobial mechanism essential for the destruction of phagocytosed bacteria and fungi (145). Pathogenic variants in the genes encoding any of the five structural subunits of NOX result in defective ROS production and in the development of CGD (144) (Table 5). More recently, it has been shown that homozygous germline mutations in CYBC1 abolish the expression of EROS, a chaperone protein required for stable expression of membrane-bound components of NOX, and represent therefore a novel cause of CGD (148–150). Two thirds of CGD patients have an X-linked form with various germline mutations in CYBB (Table 5) (144, 151) and the majority of the patients are diagnosed during early childhood (151). Survival is associated with residual ROS production independently of the gene that is mutated (152). CGD patients suffer from a variety of severe and recurrent bacterial and fungal infections, in particular due to Aspergillus species, Staphylococcus aureus, Burkholderia cepatia species, Serratia marcescens, and Nocardia species (153, 154). The lungs (pneumonia), skin (abscesses, granulomas…), lymph nodes (lymphadenitis), and the liver (abscesses) are the most common affected sites (144, 151). In developing countries, BCG (Bacille-Calmette-Guérin) and Mycobacterium tuberculosis are also important pathogens (155). CGD is associated with a very high prevalence of invasive fungal infections that affect up to 40% of the patients and can be life-threatening (156). “Mulch pneumonitis,” due to an intense inflammatory response to fungal elements in aerosolized decayed organic matter, is almost pathognomonic of CGD (144, 151). In addition to severe infections, CGD patients also suffer from dysregulated inflammation, in particular in the gastrointestinal [inflammatory bowel disease (IBD)-like] and genitourinary tracts, and can develop granulomatous obstructive disorders (157). Inflammatory and autoimmune manifestations (e.g., arthritis, discoid lupus, systemic lupus erythematosus, vasculitis, immune thrombocytopenia) are observed in respectively 70 and 10% of the cases, with the highest frequency in X-linked CGD patients (142, 143, 157). As NOX is active in other cell types than phagocytes, the clinical picture of CGD may even be more complex (144).
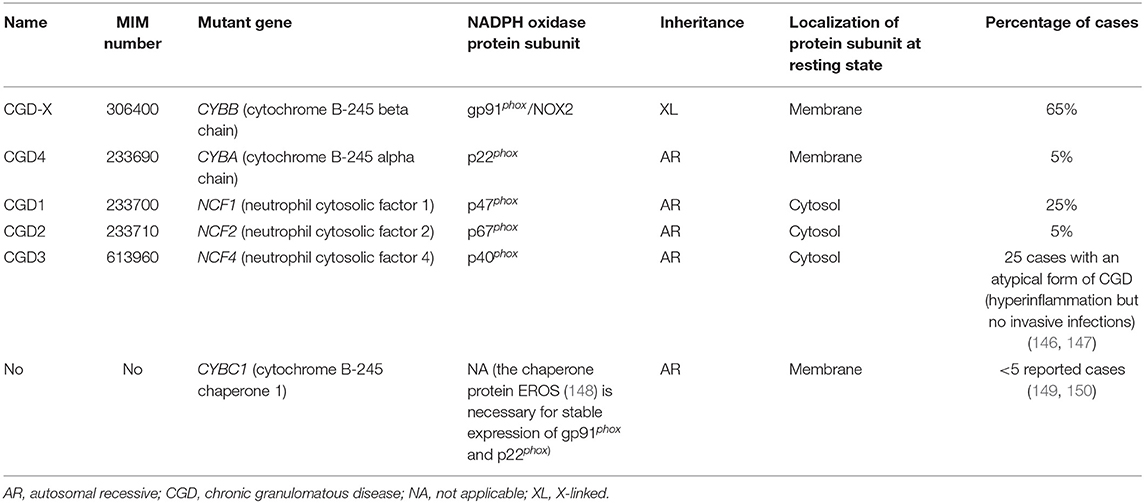
Table 5. Germline mutations in NADPH oxidase complex leading to different CGD subtypes.
General management
Diagnosis of CGD is made by functional evaluation of NADPH activity after phagocytes activation and by molecular confirmation. Conventional management predominantly relies on lifelong prophylactic antibiotics (trimethoprim-sulfamethoxazole) and antifungals (itraconazole), interferon (IFN)-γ therapy that can correct metabolic defects in phagocytes (158), along with the treatment of acute infections (144, 151). CGD-associated IBD is difficult to manage, in particular due to the combination with the inherent susceptibility to infections. The treatment of gastrointestinal (GI) tract inflammation frequently relies on corticosteroids although they remain controversial as they increase the growth retardation and the infectious risk (159). Allogeneic HSCT is currently the only curative treatment of CGD and may reverse both infectious and inflammatory manifestations (160).
Oral manifestations and gastrointestinal tract involvement
Several factors predispose CGD patients to oral manifestations. They include neutrophil dysfunction, but also the use of immunosuppressive therapies to manage inflammatory complications, as well as malnutrition due to GI complications. Indeed, GI tract inflammation is very frequent, with a reported incidence ranging from 30 to 60% (157, 159, 161, 162). Recent findings have demonstrated a crucial role of NOX complex in the regulation of gut immunity, regardless the susceptibility to infections (163). Although it remains a distinct entity, GI involvement in CGD mimics IBD with overlapping features of both ulcerative colitis and Crohn's disease (159). Every part of the GI tract can be affected from the oral cavity to the anus. In a series of 98 patients, the most frequent reported GI manifestation was non-infectious diarrhea, followed by oral ulcerations and anal fistulae (157). In an important cohort of 459 European CGD patients, oral ulcers have been observed in 11% of the cases (142) and in a long-term follow-up study on 39 patients, they have been reported in 26% of the cases, along with stomatitis (164). Oral ulcers are similar clinically to aphthae with frequent recurrences. Granulomatous inflammation of the oral mucosa with a nodular and cobblestoning aspect, which is typically observed in patients with Crohn's disease (orofacial granulomatosis), has however been rarely described in CGD patients (165). Most of CGD patients present very early-onset forms of IBD (159). Importantly, GI manifestations may precede the diagnosis of CGD as well as the development of infectious complications (166). CGD should be considered in all patients who present early-onset IBD. Recurrent oral ulceration can therefore represent one of the inaugural signs of the disease. To our knowledge, no cases of severe oral infection have been reported (167, 168). In the CGD cohort of Liese et al., only 5% of the patients had one episode of parotid glands infection during the 22-year follow-up (164). In addition, although some studies show that gingivitis is common in CGD patients, with a prevalence ranging from 11% (142) to 35% (169), severe periodontitis has been rarely observed (65, 165, 170). In a large survey on 368 CGD patients, severe gingival or periodontal inflammation has been found in only 2% of the cases (143). This is in contrast with other PIDs due to a defective function of neutrophils such as LAD1. One hypothesis of the reduced prevalence of periodontitis associated with this PID could be the absence of a respiratory burst in neutrophils, despite its importance in periodontal pathogens' destruction (65, 168). Indeed, enhanced ROS generation has been clearly involved in the pathophysiology of periodontal disease (168).
Other Non-lymphoid Defects
GATA2 Deficiency
Genetic, pathophysiology, and clinical manifestations
LOF heterozygous pathogenic variants in GATA2 (guanine-adenine-thymine-adenine 2), a zinc finger transcription factor regulating early hematopoietic differentiation as well as lymphatic and vascular development, cause GATA2 haploinsufficiency (GATA2 deficiency: MIM 137295). Germline mutations arise spontaneously (de novo) but are then transmitted with autosomal dominant (AD) inheritance. The age of clinical presentation ranges from early childhood to late adulthood, with most of the cases occurring during adolescence and early adulthood. GATA2 deficiency is a protean disorder with a broad phenotype encompassing (I) multi-lineage cytopenia [DCs, monocytes, NK (natural killer) cells, B cells], (II) immunodeficiency with increased susceptibility to human papillomavirus (HPV), invasive non-tuberculous mycobacterial (NTM) and fungal infections, (III) high risk of developing hematologic malignancies (MDS/AML), (IV) pulmonary alveolar proteinosis (pulmonary disease), and (V) congenital lymphedema (vascular/lymphatic dysfunction) (171, 172). However, GATA2 deficiency has a variety of presentations and offers a challenge in any classification system. Indeed, a small proportion of patients present with only asymptomatic mild neutropenia and no other discernible hematological abnormalities except monocytopenia or macrocytosis, but with high risk of hematologic transformation (173).
General management
Allogeneic HSCT remains currently the best therapeutic option to prevent or treat hematologic malignancies and life-threatening opportunistic infections (174, 175).
Oral manifestations
Oral manifestations associated with GATA2 deficiency have not been reviewed extensively yet. Authors report infectious oral lesions mainly caused by HPV (i.e., warts, condylomas) but also by herpes simplex virus 1 (HSV-1). A close monitoring of these lesions is strongly required, in particular due to their high risk of HPV-related malignant transformation (i.e., squamous intra-epithelial lesions, Bowenoid papulosis*, invasive squamous cell carcinoma) (176, 177). Other oral features have been described and include recurrent ulcerations and blistering, gingival hyperplasia and inflammation, as well as glossitis (177, 178).
Defects in Intrinsic and Innate Immunity [Category 6 (3)]
Defects in intrinsic and innate immunity are also characterized by several oral manifestations, in particular infections by various pathogens. However, in comparison with defects of phagocytes number and function, there are less data in the literature regarding the orofacial phenotypic spectrum as well as the oral management of these disorders.
Mendelian Susceptibility to Mycobacterial Disease (MSMD)
Genetic, Pathophysiology, and Clinical Manifestations
Mendelian susceptibility to mycobacterial disease (MSMD) is primarily characterized by a selective predisposition to infections caused by atypical and weakly virulent mycobacteria such as Mycobacterium bovis BCG vaccines and environmental NTM. MSMD patients may also suffer from bona fide tuberculosis caused by Mycobacterium tuberculosis. Otherwise, they don't show obvious immunological abnormalities (179). However, MSMD designation does not recapitulate the whole phenotype of the patients as they also show increased susceptibility to certain intracellular bacteria, in particular Listeria monocytogenes and Salmonella species, and to mucocutaneous fungal infections due to Candida species. More rarely, other severe infections have been reported, but mostly in single patients. They include infections caused by intramacrophagic bacteria (e.g., klebsiellosis, nocardiosis), fungi (e.g., histoplasmosis, coccidioido-, and paracoccidioidomycosis), parasites (e.g., leishmaniasis), and even viruses (e.g., CMV, varicella-zoster virus VZV, human herpes virus-8 HHV8). Although mycobacterial diseases are by far the most common infections in these patients, it is now clearly recognized that the clinical phenotype of MSMD extends beyond them (179).
To date, MSMD has been diagnosed in more than 500 individuals worldwide with a prevalence of almost 1:50,000 (180). MSMD is caused by germline mutations affecting 16 different genes i.e., 14 autosomal genes [AD or AR inheritance: IFNGR1 (181) and IRF8 (182); AR inheritance: IFNGR2 (183), IL12B (184), IL12RB1 (185), ISG15 (186), TYK2 (187), SPPL2A (188), IL12RB2 (189, 190), IL23R (189, 190), RORC (191), JAK1 (180), IFNG (192); AD inheritance STAT1 (193)] and two X-linked gene [NEMO (194) and CYBB (195)]. Recently, AR complete IFNγ deficiency (pathogenic variants in IFNG encoding the IFNγ cytokine itself) has been described in MSMD patients (192). In addition, allelic heterogeneity at the different loci has led to the definition of 31 different genetic disorders (190). All disorders affect the IFNγ-mediated immunity, in connection with IL12/IL23/ISG15 immunity (190). MSMD is therefore a good example of PID with a relatively narrow infectious phenotype that originates from germline mutations involving molecules belonging to the same functionally connected immunological pathway. Indeed, IL12/IL23 dependent IFNγ mediated immunity is crucial for the control of intracellular pathogens, in particular mycobacteria (179, 190).
General Management
Considering the impairment of IFNγ immunity, recombinant IFNγ therapy should be considered as the “natural” treatment of MSMD. However, in patients with defects in IFNγ receptors (IFNGR1 and IFNGR2 pathogenic variants) that have complete lack of cellular responses to this cytokine, treatment with IFN-γ is not indicated (179). All patients require prolonged antibiotic treatments against mycobacteria and the other involved pathogens. BCG vaccination should be avoided. Abdominal surgery may be needed to remove the splenic and/or mesenteric lesions in some cases (179). HSCT remains the only curative treatment, especially for patients with severe forms of MSMD (196).
Oral Manifestations
Susceptibility to mucocutaneous candidiasis
The most common genetic etiology of MSMD is AR complete IL12 receptor β1 (IL12Rβ1) deficiency. The latter is due to germline mutations in IL12RB1 gene that encodes one of the chains of IL12 and IL23 receptors (185). Mild forms of chronic mucocutaneous candidiasis (CMC) have been reported in about 25% of the patients (197–199). Whereas, IL12 is a key cytokine for IFNγ production, IL23 plays a role in the maintenance of IL17 producing T cells (Th17 cells) that are important effectors in host defense against fungi, in particular Candida albicans (200). Indeed, PID patients with impaired IL17 immunity [see “predisposition to mucocutaneous candidiasis (CMC)” section] are susceptible to Candida species and develop CMC (201). Ouederni et al. reported the clinical features of candidiasis in 35 patients with IL12Rβ1 deficiency and observed that recurrent oropharyngeal candidiasis was by far the most common presentation (in 34 patients). Although it was less severe than in patients with defects of IL17 axis, it tends to persist despite antifungal therapy (202). CMC is also observed in patients with pathogenic variants in IL12B gene, but not in other genetic etiologies of MSMD, as it is related to IL23-dependent impaired IL17 immunity. Indeed, IL12B encodes IL12p40, which is a common subunit of both IL12 and IL23 cytokines (179). However, patients with AR complete IL-12Rβ2 or IL23R deficiency that have been described more recently display mycobacteriosis without increased susceptibility to candidiasis (189). Bi-allelic germline mutations in RORC, which encodes RORγ and RORγT transcription factors, have been associated with impaired systemic IFNγ response to mycobacteria but also with defective IL-17 mucocutaneous immunity to Candida. RORγ- and RORγT-deficient individuals can therefore display both mycobacteriosis and mucocutaneous candidiasis (recurrent or persistent oral candidiasis in 70% of the cases) (191). MSMD must therefore been investigated in patients with CMC or persistent oropharyngeal candidiasis.
X-linked recessive MSMD type 1: NEMO pathogenic variants
NEMO (NF-κB essential modulator), also called IKBKG (inhibitor of NF-κB kinase regulatory subunit gamma) is an essential component of NF-κB (nuclear factor kappa B) signaling pathway. It is the regulatory subunit of the inhibitor of IκB kinase (IKK) complex, which activates NF-κB. Germline mutations in NEMO gene have long been known to cause different ectodermal dysplasia* (EDA) syndromes i.e., incontinentia pigmenti (IP) and anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) (Table 6). In its classical form, EDA is characterized by abnormalities of ectodermal structures including anodontia* or oligodontia* with cone shaped teeth, hypotrichosis, and hypohidrosis with heat intolerance (206, 207).
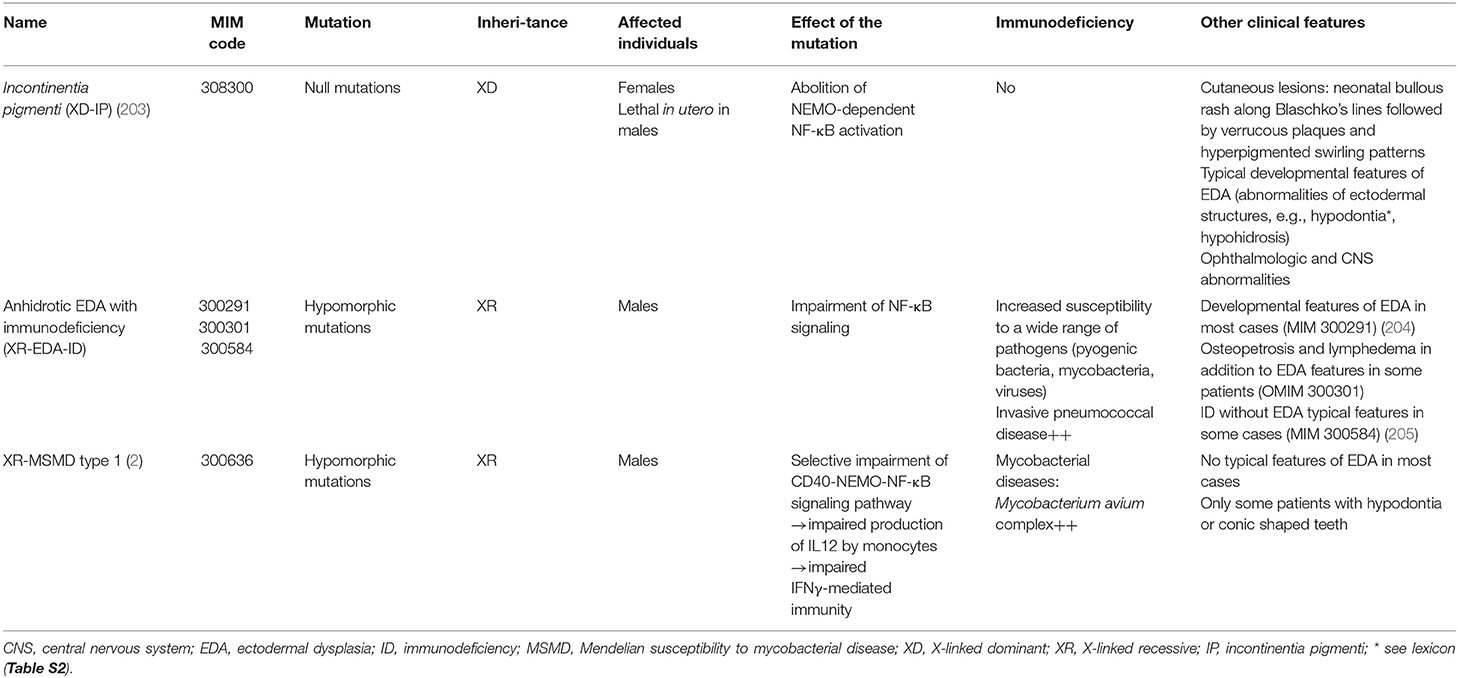
Table 6. Features of syndromes caused by germline mutations in NEMO other than XL-MSMD type 1.
In the 2017 IUIS classification (2), hypomorphic mutations in NEMO gene were classified in MSMD disease category as they were previously shown to cause also X-linked recessive (XR; MIM 300636) MSMD (type 1) (208). These pathogenic variants interfere selectively with the CD40-NEMO-NF-κB signaling pathway, leading to an impaired T-cell dependent production of IL12 by monocytes and monocyte-derived DCs in response to CD40 (194). As a result of impaired IFNγ-mediated immunity, infections are mostly limited to mycobacterial diseases, in particular due to Mycobacterium avium complex. Unlike other patients with germline NEMO mutations, most of XR-MSMD type 1 patients lack the developmental features typical of EDA. Only some cases have been reported to have hypodontia* or conic shaped teeth, but to date, none of them has been described with oligo- or anodontia (180, 194, 208). In the most recent 2019 IUIS classification, NEMO mutations have however been removed from MSMD disease category and classified only as combined immunodeficiencies (CIDs) with syndromic features [disease category 2 (3)].
X-linked recessive MSMD type 2: CYBB pathogenic variants
As discussed before (see section on CGD), germline mutations in CYBB are responsible for the most common form of CGD (MIM 306400) (143, 144). More recently, specific pathogenic variants in CYBB have been associated with X-linked MSMD (type 2) in male subjects suffering from recurrent mycobacterial diseases. The MSMD-causing mutations in CYBB selectively affect the respiratory burst in macrophages that is crucial for protective immunity to mycobacteria. Unlike CGD patients, NADPH activity is normal in neutrophils and monocytes (195, 209). To our knowledge, no specific oral manifestations have been reported in these patients.
Epidermodysplasia Verruciformis
EVER1-EVER2-CIB1 Deficiencies
Genetic, pathophysiology, and clinical manifestations
Epidermodysplasia verruciformis (EV) is a rare genodermatosis characterized by a selective susceptibility to keratinocyte-tropic HPV infections (subgroup B1) and that typically presents in early childhood (210, 211). Disseminated, flat, wart-like hypo-, or hyper-pigmented papules develop on the trunk, the neck, the face, the head as well as the extremities and are mainly benign. Lesions with greater malignant potential present as verrucous or seborrheic keratosis-like lesions and occur more frequently on sun exposed surfaces. Indeed, patients with EV have a higher risk to develop actinic keratosis and non-melanoma skin cancers, in particular cutaneous squamous cell carcinomas (210, 211). Homozygous LOF pathogenic variants in EVER1 (MIM 226400) and EVER2 (MIM 618231) genes, also named TMC6 and TMC8, respectively, have been reported in ~75% of patients with EV (210, 211). More recently, biallelic germline mutations in CIB1 gene (calcium- and integrin-binding protein-1) have also been described (212). CIB protein forms a complex with EVER1 and 2 and it has been suggested that the disruption of the CIB1–EVER1–EVER2-dependent keratinocyte-intrinsic immunity may underly the selective susceptibility to beta-HPVs of EV patients (212).
General management
The development of EV lesions cannot be prevented, but regular monitoring and appropriate treatment of skin lesions (e.g., surgical excision, cryotherapy) that might transform into skin cancers are recommended (210).
Oral manifestations
To our knowledge, no HPV-related lesions or cancers have been described in the oral cavity, but they can develop on the facial skin.
WHIM Syndrome
Genetic, pathophysiology, and clinical manifestations
WHIM syndrome (MIM 193670) is a rare AD condition whose incidence is estimated to be about 1 in 4.3 millions live births (213, 214). The term “WHIM” is an acronym of the main clinical manifestations including warts, hypogammaglobulinemia, infections, and myelokathexis (i.e., bone marrow retention). WHIM is caused by dominant heterozygous GOF pathogenic variants in the gene encoding the CX chemokine receptor 4 (CXCR4). Since CXCR4 is involved in the retention of neutrophils in the bone marrow, GOF germline mutations will exaggerate this process, thereby retarding neutrophil egress, leading to neutropenia (214). WHIM is mainly characterized by susceptibility to extensive HPV infection, which causes multiple cutaneous, plantar, anogenital, and oral warts. Warts can also occur in atypical locations such as the limbs, chest and face, and are unusually resistant to destructive treatments such as cryotherapy or surgery. HPV-driven squamous cell carcinomas constitute a significant cause of morbidity. A regular monitoring of HPV lesions, especially in the mouth and anogenital regions, is therefore strongly required and must include frequent biopsies (211, 214). Patients present severe neutropenia, but also often lymphopenia and monocytopenia, as well as moderate hypogammaglobulinemia, and suffer from frequent oto-sinopulmonary infections. Recurrent lung infections can lead to bronchiectasis and be associated with colonization by Pseudomonas aeruginosa and Stenotrophomonas maltophilia. Nasal and skin infections due to Staphylococcus aureus or Streptococcus sp. are also reported and can lead to skin abscesses, cellulitis or even septicemia (arthritis, osteomyelitis) (214).
General management
The management of WHIM syndrome includes HPV vaccination (although WHIM patients respond less robustly than healthy individuals), prophylactic antibiotics, intravenous or subcutaneous immunoglobulins (IV Igs/SC Igs) substitution to prevent oto-sinopulmonary infections, as well as G-CSF injections to enhance the release of neutrophils from the bone marrow (214). More recently, some patients have been treated with low-dose plerixafor, a CXCR4 antagonist, with encouraging results (NCT02231879) (215).
Orofacial manifestations
In addition to oral warts and HPV-related oral squamous cell carcinomas described above (216), patients with WHIM syndrome can present severe pyogenic bacterial infections (e.g., cellulitis, recurrent acute and chronic sinusitis), or viral (e.g., HSV-1, VZV) infections (217, 218). The development of early-onset periodontitis has also been reported in several WHIM patients with rapid progression leading to early tooth-loss (219). Aphthous ulcers can be observed but they are less common than in individuals with SCN (214, 220). Rarely, odontogenic infections may disseminate and cause brain abscess or endocarditis, in particular in cases of associated congenital cardiopathies (e.g., tetralogy of Fallot) that are common in WHIM patients (214). The impact of G-CSF or plerixafor treatment on periodontitis evolution has however not been assessed yet (66).
Predisposition to Severe Viral Infection
Germline Mutations Affecting Interferon (IFN) Signaling Pathway
Genetic, pathophysiology, and clinical manifestations
IFN signaling is crucial for the defense against viral infections. Several gene defects affecting this pathway have been described (Table 7) and predispose to severe, even life-threatening, viral infections (e.g., encephalitis, pneumonitis), in particular due to herpes (e.g., HSV-1, VZV [varicella-zoster virus], CMV) and influenza viruses. As IFN signaling is also a central aspect of the response to other intracellular pathogens in macrophages and neutrophils, patients may in addition be susceptible to mycobacterial infections (239).
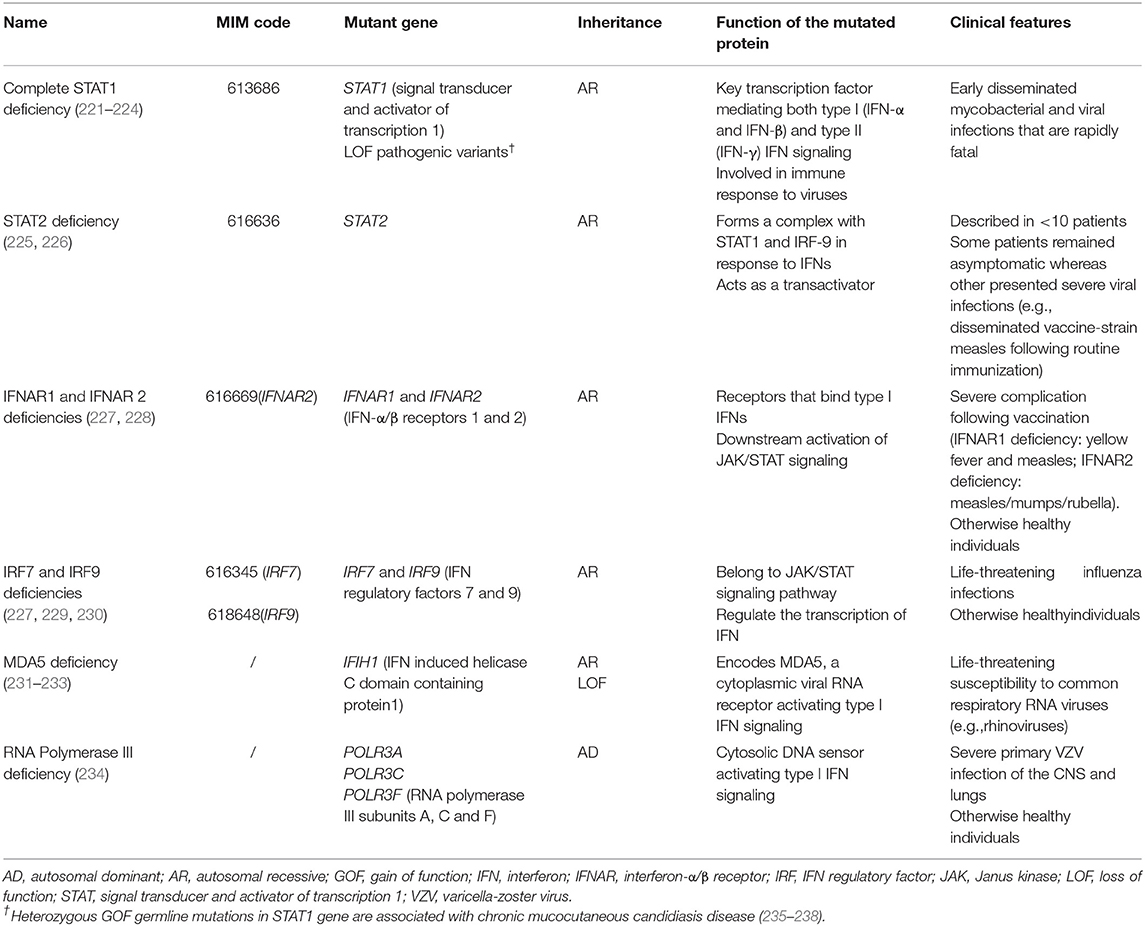
Table 7. Germline mutations affecting interferon signaling pathway and associated with a predisposition to severe viral infections.
Oral manifestations
Viral infections, in particular due to herpes viruses, can manifest in the oral cavity. However, considering the severity of the infections affecting the patients with pathogenic variants in IFN signaling pathway, their oral localization is rarely mentioned. HSV-1 gingivostomatitis has been described in STAT2 deficient patients (225, 226) but to our knowledge, specific oral features have been reported neither in IFNAR1/2, IRF7/9, MDA5, nor in RNA polymerase III-deficient patients.
CD16 Deficiency
Genetic, pathophysiology, and clinical manifestations
FcγRIIIA (CD16) is a low-affinity receptor for IgG Fc that is expressed by NK cells. Homozygous pathogenic variants in the FCGR3A gene lead to CD16 deficiency (MIM 615707) characterized by functional deficiency of NK cells with defective cytotoxic activity but retained antibody-dependent cellular cytotoxicity. Patients typically present early in childhood with severe herpes viral infections, in particular due to Epstein Barr Virus (EBV), VZV, and HPV (240–242).
Oral manifestations
Patients are prone to HSV-1 gingivostomatitis and require regular monitoring of the oral mucosa (240–242).
Herpes Simplex Encephalitis (HSE)
Genetic, Pathophysiology, and Clinical Manifestations
Pathogenic variants in genes that encode proteins belonging to the TLR3 signaling pathway (TRIF-dependent) result in early susceptibility to HSV-1 encephalitis (HSE) (243, 244). The spectrum of infections affecting the patients with TLR3 pathway defects is remarkably restricted to only one specific pathogen (HSV-1) and one specific type of infection (encephalitis).
Oral Manifestations
Surprisingly, children with HSE do not show an increased susceptibility to HSV-1-related diseases affecting other sites than the central nervous system (CNS) including herpes gingivostomatitis, which is the most common clinical symptom of HSV-1 infection in the general population (244, 245).
Predisposition to Invasive Fungal Diseases
CARD9 Deficiency
Genetic, pathophysiology, and clinical manifestations
CARD9 (caspase recruitment domain-containing protein 9) is an adaptor molecule expressed principally in myeloid cells downstream from C-type lectin receptors activation (e.g., Dectin-1) by fungal ligands. Activated CARD9 couples with BCL10 and MALT1, resulting in NF-κB and mitogen-activated protein kinases (MAPK) activation. This signaling pathway promotes the production of key cytokines (e.g., IL1β, IL6, IL23) for antifungal immune responses (246, 247). CARD9 deficiency (MIM 212050) is characterized by the spontaneous development of invasive fungal infections due to fungi belonging the phylum Ascomycota. They include CMC (see “predisposition to CMC” section), invasive Candida infections (in particular of the CNS but also of the eyes, the colon and the bones), extensive and/or deep dermatophytosis, subcutaneous and invasive phaeohyphomycosis, as well as extrapulmonary invasive aspergillosis (248–250). CARD9 deficiency is the consequence of homozygous or compound heterozygous LOF germline mutations in CARD9 that induce impaired cytokine production in response to fungal ligands, altered neutrophil killing and/or diapedesis, and defects of Th17 immunity. To date, more than 60 cases have been described with a very heterogeneous age of disease-onset ranging from childhood to adulthood (248–250).
General management
The treatment of patients with CARD9 deficiency is empirical, mainly based on antifungal therapies (e.g., azole agents, echinocandins) and on the surgical removal of fungal masses. In addition, CARD9-deficient patients should be given secondary prophylaxis with oral azole agents after the first episode of invasive fungal disease. In cases of persistent or relapsing Candida albicans infections of the CNS, adjuvant GM-CSF/G-CSF therapy can be considered. The potential value of HSCT still remains unclear due to the lack of available data (249).
Oral manifestations and management
Oral candidiasis (that is part of CMC due to Candida albicans) is very frequently associated to CARD9 deficiency and may reveal the disease (249, 251). It affects almost 30% of the patients as reported in a recent review (249). The use of oral fluconazole prophylaxis should also prevent the occurrence of CMC. In addition, frequent rigorous screening of the oral mucosa is required in order to diagnose CMC occurrence and/or relapse (249).
Predisposition to Mucocutaneous Candidiasis (CMC)
Genetic, Pathophysiology, and Clinical Manifestations
Chronic mucocutaneous candidiasis (CMC) is characterized by recurrent or persistent symptomatic mucocutaneous infections caused by fungi from the Candida genus, in particular the commensal Candida albicans. CMC affects the nails, the skin, but also the genital and the oral mucosae (oral candidiasis) (252). The term CMC disease (CMCD) is used to refer to patients presenting with CMC as the major clinical phenotype, with neither invasive fungal infections, nor other overt infectious or autoimmune manifestations (252, 253). Four of the five causative genes of CMCD are directly involved in IL17 signaling and encode the cytokine IL17F (IL17F), IL17 receptors (IL17RA and IL17RC) (201, 254), and ACT1 (TRAF3IP2), a membrane-proximal adaptor of IL17 receptor (255, 256) (Table 8). The discovery of these genetic defects highlighted the essential role of IL17 cytokines for mucocutaneous protection against Candida albicans. Heterozygous GOF germline mutations in STAT1 account for more than a half of CMCD cases (235–238). AD STAT1 GOF leads to defective Th1 and Th17 responses, with reduced production of IFN-γ, IL17, and IL22 (236). However, more recent studies revealed that STAT1 GOF pathogenic variants are associated with an unexpectedly wide range of clinical manifestations in addition to CMC. They include other infectious manifestations such as bacterial infections of the skin and the respiratory tract (Staphylococcus aureus), herpes virus infections (e.g., HSV-1, VZV), invasive fungal infections, mycobacterial disease, but also various autoimmune manifestations (>30% of the patients), cerebral aneurysms and malignancies, the latter two conferring a poor prognosis (237, 238). Very recently, AD germline mutations in the MAPK8 gene encoding the kinase JNK1 have been reported in a family with a combination of CMC and a previously undescribed form of connective tissue disorder resembling Ehlers-Danlos syndrome (257). JNK1 haploinsufficiency impairs both IL-17–dependent mucocutaneous immunity to Candida and TGFβ-dependent homeostasis of connective tissues (257).
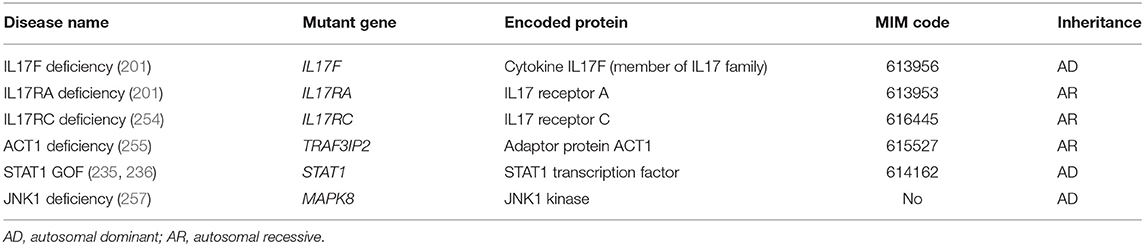
Table 8. Germline mutations leading to chronic mucocutaneous candidiasis disease (CMCD).
General Management
Most patients with CMCD are treated with a combination of topical and systemic antifungal agents, in particular azoles (fluconazole in first line, followed by itraconazole, posaconazole, and/or voriconazole) (253). They require long-lasting antifungal treatments and/or prophylaxis to manage persistent and prevent recurrences (253). Clinical resistance to at least one antifungal agent has however been observed in almost 40% of STAT1 GOF patients with long-term treatments (237). Topical therapy with polyenes (i.e., nystatin) has been proven to be a good alternative to triazoles (238). Oral ruxolitinib, a JAK1/2 kinase inhibitor that limits STAT1 mediated intracellular signaling, seems promising and allowed an improvement of CMC associated to STAT1 GOF. However, long-term administration seems necessary, as the effect is not sustained after treatment discontinuation (258, 259). HSCT might be considered in STAT1 GOF patients with progressive life-threatening disease unresponsive to conventional treatments (260). Finally, recombinant IL17 may also represent a promising therapeutic option.
Oral Manifestations
Considering the high susceptibility to Candida albicans mucosal infections associated with IL17 axis disruption, almost 100% of CMCD patients present recurrent/persistent and/or severe candidiasis of the oral mucosa (thrush, glossitis, and/or cheilitis) (253). Germline mutations affecting IL17 signaling pathway should therefore been investigated in patients with recurrent oral candidiasis. One third of patients with STAT1 GOF pathogenic variants also suffer from mucocutaneous viral infections including HSV-1 gingivostomatitis (237, 261). The treatment relies on appropriate systemic/topic antifungal and systemic antiviral agents.
In addition, CMCD patients, in particular those bearing AD STAT1 GOF pathogenic variants, present an increased risk of both oral and esophageal squamous cell carcinoma, in part due to chronic inflammation associated with persistent CMC (237, 262, 263). Regular examination of the oral mucosa is therefore required.
Oral anomalies have also been described and include progressive macroglossia, macrocheilitis, as well as dental abnormalities (peg-shaped incisors) in ACT1 deficiency (255, 264). Delayed exfoliation of primary molars and enamel erosions have been reported in only one patient with STAT1 GOF mutation (265). Although STAT1 has been involved in enamel formation in the rat (266), the link between STAT1 germline mutations and the observed tooth anomalies (that are frequently observed in the general population) needs to be further studied. Delayed exfoliation of primary teeth and dental crowding that have been reported in the family with JNK1 haploinsufficiency may be related to impaired TGFβ-dependent homeostasis of connective tissues (257).
TLR Signaling Pathway Deficiency With Bacterial Susceptibility
IRAK-4 and MyD88 Deficiencies
Genetic, pathophysiology, and clinical manifestations
AR IRAK-4 (IL-1 receptor-associated kinase-4) and MyD88 (myeloid differentiation factor 88) deficiencies selectively impair the signaling via the TLR and IL1 receptor pathway (267, 268). To date, more than 80 patients have been diagnosed worldwide [reviewed in (268)]. IRAK-4 (MIM 607676) and MyD88 (MIM 612260) deficiencies, which are phenocopies in term of clinical and immunological abnormalities, are characterized by a selective predisposition to pyogenic bacterial infections (267–270). Patients are highly susceptible to invasive bacterial infections caused by Streptococcus pneumoniae and, to a lesser extent, Staphylococcus aureus, as well as to non-invasive bacterial infections mainly restricted to the skin (Staphylococcus aureus) and the upper respiratory tract (Pseudomonas aeruginosa). Despite a large impact of these genetic defects on immune responses, affected individuals however present a normal resistance to common viruses, fungi, parasites and to many bacteria (270).
General management
In addition to vaccinations, in particular against Streptococcus pneumoniae, prophylactic antibiotic treatment (cotrimoxazole plus penicillin V) should be taken throughout the life. IV or SC Igs administrations during infancy seem to decrease the incidence of invasive bacterial infections (268). Empirical parenteral antibiotic treatment against Streptococcus pneumoniae, Staphylococcus aureus, and Pseudomonas aeruginosa must be initiated as soon as an infection is suspected, considering the high mortality risk due to invasive bacterial infections. Secondary adaptation of the treatment should then be done once the causal bacteria has been evidenced (270).
Oral manifestations
Bacterial infections involving the orofacial region such as maxillary sinusitis, necrotizing palate infection, cellulitis, and periodontal diseases have been reported in some patients with IRAK-4 and MyD88 deficiencies (268). Four patients also presented oral candidiasis (268). The treatment relies on appropriate systemic antibiotics and systemic/topic antifungal agents, respectively. Into et al. have shown in a murine model that MyD88 deficiency has an impact on the expression of several antimicrobial factors, which could also influence the susceptibility to oral infections (271).
Discussion
Patients with inborn errors of innate immunity are prone to several kind of fungal, viral and/or bacterial infections that frequently involve the oral cavity. Lesions affecting the oral mucosa and the periodontium* such as aphthous oral ulcers and early-onset aggressive periodontitis [grade C periodontitis in the new classification of periodontal diseases (136, 137)] respectively are also commonly observed, in particular in patients with defects of neutrophil number or function. Moreover, some PIDs of innate immunity, especially syndromic SCN (Table 3), are associated with developmental abnormalities including facial, oral, and even dental anomalies. Despite the large constellation of orofacial features, oral examination is sometimes overlooked from the global physical examination of the patients with a suspicion of PID, possibly because of a lack of habit or because the PID multidisciplinary team rarely involves dentists. In addition, dentists are not always familiar with these disorders. As a consequence, oral manifestations associated with inborn errors of immunity are frequently on the sideline and overshadowed by the other medical problems. However, oral features are of prime importance as they may reveal an underlying defect of immunity or further complicate the medical management of PID patients (see warning oral signs, Box 1). Indeed, they can promote a pro-inflammatory situation, lead to infectious complications, or even oncogenic transformation due to defective pathogen control (e.g., HPV). Both physicians and dentists should therefore be aware of the oral warning signs and the basic principles of oral management.
Box 1. Warning oral signs.
• Severe gingivitis in children that persists despite dental plaque/biofilm removal/control.
• Early-onset periodontitis with premature loss of primary teeth in children or loss of permanent teeth in adolescent/young adults.
• Aggressive forms of periodontitis that do not respond to conventional periodontal treatments.
• Recurrent and/or persistent oral ulcers.
• Severe and/or recurrent and/or persistent forms of herpetic gingivostomatitis or oral candidiasis that do not respond to treatment.
• Severe and/or atypical (involving uncommon pathogens) dental infections.
• In particular in children/adolescents.
Preventive measures including strict oral hygiene protocols, professional periodontal maintenance, nutritional advices as well as regular professional applications of topical fluorides (varnish) must be implemented in all patients with inborn errors of innate immunity (Box 2). A regular and rigorous screening of the oral cavity is also essential and persistent mucosal lesions should be biopsied considering the potential risk of malignant transformation (e.g., HPV-related oral squamous cell carcinomas in WHIM patients). The treatment of oral ulcers is mainly symptomatic and must include pain management. Indeed, mouth aphthous ulcers may be very painful and cause difficulties in eating, leading to nutritional problems with a potential impact on the patient's general condition. In addition to analgesics, oral film-forming agents containing hyaluronic acid can be used to form a protective barrier over the ulcerated oral mucosa. These protective agents are generally well-tolerated but provide only transient pain relief. In cancer patients with oral mucositis, the use of topical anesthetics such as lidocaine has been recommended for pain management although there are no studies available to assess their benefit and their potential toxicity (272). One should therefore be cautious when advising topical applications of anesthetics in PID patients as they may diminish the swallowing reflex (risk of food aspiration), alter taste sensation with a burning sensation, and be associated with possible cardiovascular effects. In addition, it mostly provides only short-term pain relief (272).
Box 2. Important Considerations for Oral Management of PID Patients (274, 275).
• Systematic discussion with the medical team.
• Immediately after the diagnosis of PID: complete oral assessment including clinical and radiographic (at least one panoramic radiography, complemented by appropriate radiographs such as bitewings or cone beam computed tomography) examinations.
• Implementation of intensive preventive measures: individual oral hygiene instructions, nutritional counseling, professional topical fluorides applications.
• Regular follow-up every 3–4 months including periodontal maintenance with professional plaque removal to prevent/limit periodontal inflammation.
• Before initiation of invasive dental procedures: discussion of antibiotic prophylaxis with the medical team (consider the high risk of antibiotic resistance development), complete blood count with a particular attention to neutrophil and platelets counts.
• In case of periodontitis: conventional mechanical periodontal treatment to remove tooth-associated biofilm +/– systemic antibiotics for severe forms, followed by regular supportive periodontal therapy.
• Immediate treatment of oral infections with appropriate antimicrobial agents considering the high risk of invasive infections.
Severe, recurrent and/or persistent oral infections may signal an underlying inborn error of immunity (after exclusion secondary immunodeficiencies). It is crucial to identify the causative pathogen as it gives clue on the signaling pathway that should be explored. For example, germline mutations affecting type I and II IFN signaling pathways should be investigated in patients with severe and relapsing herpetic gingivostomatitis. In a similar way to the management of systemic infections, the treatment of oral infections relies on the administration of antimicrobial agents (e.g., antibiotics, antifungal agents) depending on the type of pathogen that is involved. It should be initiated as soon as an oral infection is suspected considering the high risk of dissemination (Box 2). Since some patients are under continuous antibiotic and/or antifungal curative or prophylactic treatments, one should consider the use of another pharmacological class, after discussion with the medical team, in order to avoid the development of resistances. An antibiogram or even an antifungigram should be performed in order to determine the susceptibility of the causative microorganism and to adapt the treatment subsequently. One study suggested that human polyvalent IV IgGs, administered as a mouthwash, could constitute a novel adjuvant topical treatment of chronic oral candidiasis, in particular in cases of drug resistance, probably through their ability to opsonize Candida (273). Finally, antibiotic prophylaxis must also be discussed with the medical team before invasive dental treatment (i.e., all dental procedures that involve a manipulation of gingival tissue and the periapical region of teeth or induce a perforation of the oral mucosa) and oral surgery to prevent the onset of infections through the entrance way provided by the therapeutic action (Box 2) (274, 275). Pre-operative antibiotic prophylaxis must be systematically administrated in a single dose before invasive oral or dental procedures in patients with associated congenital cardiopathy, such as WHIM patients (214), that are at increased risk to develop infectious endocarditis (276).
Periodontal diseases (i.e., gingivitis and periodontitis) are particularly prevalent in congenital defects of neutrophil number and function. Indeed, neutrophils represent more than 95% of the total number of leukocytes found in the periodontium. Considered as “gatekeepers of oral immunity,” they form the first line of defense against the subgingival biofilm (11). In children and young adults, aggressive periodontitis with early tooth loss almost always indicates the existence of an underlying systemic or immunologic disorder. In some cases, it can even precede the other clinical manifestations. It is therefore crucial to refer the patient for appropriate medical investigation in order to allow a timely diagnosis and the implementation of adequate treatment. The management of the periodontal disease itself relies on careful plaque control with tailored oral hygiene regimens and on conventional mechanical periodontal treatment to remove or at least disrupt tooth-associated biofilm. The latter can possibly be complemented by systemic antibiotics in cases of severe forms of periodontitis, and by antiseptic mouth rinses (e.g., 0.2% chlorhexidine). Regular follow-up (every 3 months) is strongly recommended and must include periodontal maintenance with professional plaque removal (Box 2). The outcome of periodontal treatment in patients with defects of innate immunity seems however unpredictable and independent of the type of treatment provided (65). The highest rate of “stabilization” of the periodontal condition was reported for SCN patients but in only 61% of the cases (vs. less than 43% in other neutrophil-associated PIDs) (65). Unsuccessful outcome suggests that mechanisms other than a defective neutrophil defense against bacteria contribute to the development of periodontal disease (65, 277). Initially, increased host susceptibility to severe periodontitis was thought to be linked to the reduced number or the dysfunction of neutrophils at the gingival sulcus, leading to inefficient control of the periodontal pathogens. However, the implication of immunoregulatory defects responsible for a dysregulated inflammatory response has also been suggested, in particular in PLS syndrome and LAD1 (112). In PLS, despite CTSC deficiency, neutrophil remain capable to destroy pathogens. However, they fail to activate neutrophil proteases, which degrade certain chemokines and cytokines, a process that is crucial for periodontal homeostasis maintenance (112). In LAD1, aggressive periodontitis has been linked to the dysregulation of the IL23/Th17 axis, leading to an increased secretion of IL17 (95, 96). An overproduction of this pro-inflammatory cytokine has also been involved in the initiation and progression of chronic periodontitis, which is the most common form of periodontitis in the general population (98). The study of PID patients with pathogenic variants that alter key effectors of mucosal immunity provides therefore a better understanding of the immune pathways regulating oral mucosal homeostasis (112). The best example is the discovery of the role played by IL17 axis in the maintenance of oral equilibrium. High levels of IL17 lead to enhanced periodontal inflammation as stated above, whereas a decreased generation of Th17 cells and the impairment of IL17/IL23 signaling pathway are associated with a high susceptibility to oral candidiasis (CMC), highlighting the role of IL17 axis in antifungal immunity at barrier sites (250, 252). A better knowledge of the underlying mechanisms also gives clues for the management of these conditions. In moderate forms of LAD1, inhibition of IL23 and IL17 appears to be a promising therapeutic strategy, in particular in cases of severe periodontal involvement (87).
The treatment of the immunological defect itself may allow an improvement of the oral manifestations, especially mucosal lesions. One example is G-CSF therapy that is the “gold standard” treatment for SCN and CyN (13). Indeed, G-CSF administrations have been associated with a decrease of oral ulcers severity and recurrence (78). However, several authors have observed that periodontal disease tends to persist even after normalization of neutrophil counts (67, 70). HSCT provides a definitive correction for most PIDs and remains the only curative treatment for patients with severe forms (278, 279). This procedure therefore also allows an improvement of the lesions affecting the oral mucosa and the gingiva, in particular oral infections, aphthous ulcers and periodontal inflammation. For example, Carlsson et al. reported the case of one patient with SCN1 who did not experience any gingivitis since HSCT (67). However, if gingival inflammation is associated with alveolar bone loss (i.e,. periodontitis) before HSCT, the latter may not be reverted by the procedure. Similarly, preexisting dental developmental anomalies (i.e., alterations in the number, the shape, the size or the structure of the teeth) will not be corrected by HSCT. Despite the improved outcome observed after HSCT, patients still face severe short and long-term transplant-related complications (278, 279). HSCT can therefore also be responsible for several oral complications that include, among others, infections, mucositis, graft-vs.-host disease (GvHD) (280), secondary malignancies (281), and dental sequelae (e.g., agenesis, microdontia, enamel hypoplasia) (272).
Given the low prevalence of inborn errors of innate immunity, orofacial manifestations associated with these conditions have been rarely evaluated in dedicated clinical studies and most of the descriptions arise from case studies. Recently, Halai et al. published the first systematic review on the periodontal status of children with neutrophil associated PIDs. Although 118 studies were included, 98% of them were case reports or case series (65). In addition, these data are often difficult to interpret due to the lack of control groups in most clinical studies. This is particularly true for periodontal diseases that also have a high prevalence in the general population. Several questions regarding orofacial involvement in PIDs remain therefore open, such as the impact of the different oral manifestations on the course of the PID itself. Indeed, the existence of persistent oral infections or periodontal inflammation may contribute to the chronic stimulation of the immune system and also favor the development of secondary autoimmune and inflammatory complications. One should also take into consideration the influence of the treatments. For example, immunosuppressant drugs used in PIDs patients presenting autoimmune manifestations can predispose to the development of oral manifestations, worsen oral infections and affect the progression of periodontal inflammation. The impact of orofacial involvement on the quality of life has also only been poorly investigated, if ever, in PIDs patients and is certainly underestimated.
Further studies are therefore strongly required to better define the orofacial phenotypic spectrums associated with the different inborn errors of innate immunity as well as their impact on the disease course and on the quality of life of the patients. In addition, they will allow to get a better understanding of oral and mucosal immune mechanisms that is a prerequisite for the development of targeted therapeutic strategies.
Ethics Statement
Informed consents were obtained from the patients for photographs publication and are available at Reference Center for Oral Manifestations (O-Rares) from Strasbourg.
Author Contributions
SJ designed the study and performed literature search. SJ, VG, AG, and A-SK wrote the paper. SJ prepared the tables. All authors reviewed the manuscript and concur with the submission.
Funding
This work has been supported by the authors' institutions and by EU-funded (ERDF, European Regional Development Fund) project INTERREG V Upper Rhine program RARENET. Publication fees were supported by Hôpitaux Universitaires de Strasbourg.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Pr. Agnès Bloch-Zupan (Reference Center for Rare Diseases with Oral and Dental Manifestations, University Hospital, Strasbourg) for the photographs of PLS patients and Mrs. Marzena Kawczynski (Reference Center for Rare Diseases with Oral and Dental Manifestations, University Hospital, Strasbourg) for her help with the patients' data. We thank Pr. Olivier Huck (Department of Periodontology, University Hospital, Strasbourg) for careful proofreading of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01065/full#supplementary-material
Abbreviations
AD, autosomal dominant; AML, acute myeloid leukemia; ANC, absolute neutrophil count; AR, autosomal recessive; BCG, Bacille-Calmette-Guérin; CARD9, caspase recruitment domain–containing protein 9; CDG, congenital disorder of glycosylation; CGD, chronic granulomatous disease; CID, combined immunodeficiency; CMC, chronic mucocutaneous candidiasis; CMCD, chronic mucocutaneous candidiasis disease; CMV, cytomegalovirus; CN, congenital neutropenia; CNS, central nervous system; CTSC, cathepsin C; CXCR4, CX chemokine receptor 4; CyN, cyclic neutropenia; DC, dendritic cell; EBV, Epstein Barr Virus; EDA, ectodermal dysplasia; ER, endoplasmic reticulum; EV, epidermodysplasia verruciformis; FPR, formyl peptide receptor; GATA2, guanine-adenine-thymine-adenine 2; GDP, guanosine diphosphate; G-CSF, granulocyte colony-stimulating factor; GI, gastrointestinal; GM-CSF, granulocyte-macrophage colony-stimulating factor; GOF, gain-of-function; G6PC3, glucose-6-phosphatase 3; GvHD, graft-vs.-host disease; HPV, human papillomavirus; HSCT, hematopoietic stem cell transplantation; HSE, herpes simplex virus 1 encephalitis; HSV1, herpes simplex virus 1; IBD, inflammatory bowel disease; IFN, interferon; IFNAR, interferon-α/β receptor; IL, interleukin; IP, incontinentia pigmenti; IRAK-4, IL1 receptor-associated kinase-4; IRF, interferon regulatory factor; IUIS, International Union of Immunological Societies; IVIgs, intravenous immunoglobulins; JAK, Janus kinase; LAD, leukocyte adhesion deficiency; LOF, loss-of-function; MAPK, mitogen-activated protein kinase; MDS, myelodysplastic syndrome; MSMD, Mendelian susceptibility to mycobacterial disease; MyD88, myeloid differentiation factor 88; NADPH, nicotinamide adenine dinucleotide phosphate; NEMO, NF-κB essential modulator; NF-κB, nuclear factor kappa B; NET, neutrophil extracellular trap; NK, natural killer; NOX, NADPH oxidase; NTM, non-tuberculous mycobacteria; PID, primary immunodeficiency; PLS, Papillon-Lefèvre syndrome; ROS, reactive oxygen species; SC, subcutaneous; SCID, severe combined immunodeficiency; SCN, severe congenital neutropenia; SNP, single-nucleotide polymorphism; STAT, signal transducer and activator of transcription; VZV, varicella-zoster virus; WAS, Wiskott-Aldrich syndrome; WASP, Wiskott-Aldrich syndrome protein; XD, X-linked dominant; XR, X-linked recessive; WHIM, warts, hypogammaglobulinemia, infections, myelokathexis. *: refer to lexicon (Table S2).
References
1. Casanova J-L, Abel L. Primary immunodeficiencies: a field in its infancy. Science. (2007) 317:617–9. doi: 10.1126/science.1142963
2. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova J-L, Chatila T, et al. International Union of Immunological Societies: 2017 primary Immunodeficiency Diseases Committee Report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
3. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international Union of Immunological Societies Expert Committee. J Clin Immunol. (2020) 40:24–64. doi: 10.1007/s10875-020-00763-0
4. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol. (2020) 40:66–81. doi: 10.1007/s10875-020-00758-x
5. Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova J-L, et al. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol. (2013) 33:1–7. doi: 10.1007/s10875-012-9751-7
6. Mahlaoui N, Jais J-P, Brosselin P, Mignot C, Beaurain B, Brito C, et al. Prevalence of primary immunodeficiencies in France is underestimated. J Allergy Clin Immunol. (2017) 140:1731–33. doi: 10.1016/j.jaci.2017.06.020
7. Herber M, Mertz P, Dieudonné Y, Guffroy B, Jung S, Gies V, et al. Primary immunodeficiencies and lymphoma: a systematic review of literature. Leuk Lymphoma. (2019) 2019:1–11. doi: 10.1080/10428194.2019.1672056
8. Guffroy A, Gies V, Martin M, Korganow A-S. Déficit immunitaire primitif de l'adulte et auto-immunité. Rev Méd Intern. (2017) 38:383–92. doi: 10.1016/j.revmed.2016.10.388
9. Fischer A, Provot J, Jais J-P, Alcais A, Mahlaoui N, members of the CEREDIH French PID study group. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol. (2017) 140:1388–93.e8. doi: 10.1016/j.jaci.2016.12.978
10. Moutsopoulos NM, Konkel JE. Tissue-Specific immunity at the oral mucosal barrier. Trends Immunol. (2018) 39:276–87. doi: 10.1016/j.it.2017.08.005
11. Scott DA, Krauss J. Neutrophils in periodontal inflammation. Front Oral Biol. (2012) 15:56–83. doi: 10.1159/000329672
12. Hajishengallis E, Hajishengallis G. Neutrophil homeostasis and periodontal health in children and adults. J Dent Res. (2014) 93:231–7. doi: 10.1177/0022034513507956
13. Skokowa J, Dale DC, Touw IP, Zeidler C, Welte K. Severe congenital neutropenias. Nat Rev Dis Primer. (2017) 3:17032. doi: 10.1038/nrdp.2017.32
14. Donadieu J, Beaupain B, Mahlaoui N, Bellanné-Chantelot C. Epidemiology of congenital neutropenia. Hematol Oncol Clin North Am. (2013) 27:1–17, vii. doi: 10.1016/j.hoc.2012.11.003
15. Aprikyan AAG, Kutyavin T, Stein S, Aprikian P, Rodger E, Liles WC, et al. Cellular and molecular abnormalities in severe congenital neutropenia predisposing to leukemia. Exp Hematol. (2003) 31:372–81. doi: 10.1016/S0301-472X(03)00048-1
16. Grenda DS, Murakami M, Ghatak J, Xia J, Boxer LA, Dale D, et al. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. (2007) 110:4179–87. doi: 10.1182/blood-2006-11-057299
17. Person RE, Li F-Q, Duan Z, Benson KF, Wechsler J, Papadaki HA, et al. Mutations in proto-oncogene gFI1 cause human neutropenia and target eLA2. Nat Genet. (2003) 34:308–12. doi: 10.1038/ng1170
18. Hock H, Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S, et al. Intrinsic requirement for zinc finger transcription factor GFI-1 in neutrophil differentiation. Immunity. (2003) 18:109–20. doi: 10.1016/S1074-7613(02)00501-0
19. Zweidler-Mckay PA, Grimes HL, Flubacher MM, Tsichlis PN. Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol Cell Biol. (1996) 16:4024–34. doi: 10.1128/MCB.16.8.4024
20. Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schäffer AA, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet. (2007) 39:86–92. doi: 10.1038/ng1940
21. Kostmann R. Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria. Acta Paediatr Suppl. (1956) 45:1–78. doi: 10.1111/j.1651-2227.1956.tb06875.x
22. Skokowa J, Klimiankou M, Klimenkova O, Lan D, Gupta K, Hussein K, et al. Interactions among HCLS1, HAX1 and lEF-1 proteins are essential for G-CSF-triggered granulopoiesis. Nat Med. (2012) 18:1550–9. doi: 10.1038/nm.2958
23. Germeshausen M, Grudzien M, Zeidler C, Abdollahpour H, Yetgin S, Rezaei N, et al. Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood. (2008) 111:4954–7. doi: 10.1182/blood-2007-11-120667
24. Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Van Den Oord JJ, et al. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. (2001) 27:313–7. doi: 10.1038/85886
25. Westerberg LS, Meelu P, Baptista M, Eston MA, Adamovich DA, Cotta-de-Almeida V, et al. Activating WASP mutations associated with X-linked neutropenia result in enhanced actin polymerization, altered cytoskeletal responses, and genomic instability in lymphocytes. J Exp Med. (2010) 207:1145–52. doi: 10.1084/jem.20091245
26. Moulding DA, Record J, Malinova D, Thrasher AJ. Actin cytoskeletal defects in immunodeficiency. Immunol Rev. (2013) 256:282–99. doi: 10.1111/imr.12114
27. Moulding DA, Blundell MP, Spiller DG, White MRH, Cory GO, Calle Y, et al. Unregulated actin polymerization by WASP causes defects of mitosis and cytokinesis in X-linked neutropenia. J Exp Med. (2007) 204:2213–24. doi: 10.1084/jem.20062324
28. Boztug K, Klein C. Genetic etiologies of severe congenital neutropenia. Curr Opin Pediatr. (2011) 23:21–6. doi: 10.1097/MOP.0b013e32834262f8
29. Klimiankou M, Klimenkova O, Uenalan M, Zeidler A, Mellor-Heineke S, Kandabarau S, et al. GM-CSF stimulates granulopoiesis in a congenital neutropenia patient with loss-of-function biallelic heterozygous CSF3R mutations. Blood. (2015) 126:1865–7. doi: 10.1182/blood-2015-07-661264
30. Triot A, Järvinen PM, Arostegui JI, Murugan D, Kohistani N, Dapena Díaz JL, et al. Inherited biallelic CSF3R mutations in severe congenital neutropenia. Blood. (2014) 123:3811–7. doi: 10.1182/blood-2013-11-535419
31. Thrasher AJ, Burns SO. WASP: a key immunological multitasker. Nat Rev Immunol. (2010) 10:182–92. doi: 10.1038/nri2724
32. Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrich syndrome. Curr Opin Hematol. (2008) 15:30–6. doi: 10.1097/MOH.0b013e3282f30448
33. Klein C. Congenital neutropenia. Hematol Am Soc Hematol Educ Program. (2009) 2009:344–350. doi: 10.1182/asheducation-2009.1.344
34. Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, et al. Barth syndrome. Orphanet J Rare Dis. (2013) 8:1–7. doi: 10.1186/1750-1172-8-23
35. Colombo EA, Bazan JF, Negri G, Gervasini C, Elcioglu NH, Yucelten D, et al. Novel C16orf57 mutations in patients with poikiloderma with neutropenia: bioinformatic analysis of the protein and predicted effects of all reported mutations. Orphanet J Rare Dis. (2012) 7:7. doi: 10.1186/1750-1172-7-7
36. Wang L, Clericuzio C, Larizza L. Poikiloderma with Neutropenia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK459118/ (accessed October 22, 2019).
37. Wang H, Falk MJ, Wensel C, Traboulsi EI. Cohen Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1482/ (accessed October 22, 2019).
38. Hurmerinta K, Pirinen S, Kovero O, Kivitie-Kallio S. Craniofacial features in Cohen syndrome: an anthropometric and cephalometric analysis of 14 patients. Clin Genet. (2002) 62:157–64. doi: 10.1034/j.1399-0004.2002.620209.x
39. El Chehadeh-Djebbar S, Blair E, Holder-Espinasse M, Moncla A, Frances A-M, Rio M, et al. Changing facial phenotype in Cohen syndrome: towards clues for an earlier diagnosis. Eur J Hum Genet. (2013) 21:736–42. doi: 10.1038/ejhg.2012.251
40. García-Ballesta C, Pérez-Lajarín L, Lillo OC, Bravo-González LA. New oral findings in Cohen syndrome. Oral Radiol Endod. (2003) 95:681–7. doi: 10.1067/moe.2003.138
41. Boztug K, Appaswamy G, Ashikov A, Schäffer AA, Salzer U, Diestelhorst J, et al. A novel syndrome with congenital neutropenia caused by mutations in g6PC3. N Engl J Med. (2009) 360:32–43. doi: 10.1056/NEJMoa0805051
42. Banka S, Newman WG. A clinical and molecular review of ubiquitous glucose-6-phosphatase deficiency caused by G6PC3 mutations. Orphanet J Rare Dis. (2013) 8:84. doi: 10.1186/1750-1172-8-84
43. Martin CC, Oeser JK, Svitek CA, Hunter SI, Hutton JC, O'Brien RM. Identification and characterization of a human cDNA and gene encoding a ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein. J Mol Endocrinol. (2002) 29:205–22. doi: 10.1677/jme.0.0290205
44. Froissart R, Piraud M, Boudjemline AM, Vianey-Saban C, Petit F, Hubert-Buron A, et al. Glucose-6-phosphatase deficiency. Orphanet J Rare Dis. (2011) 6:1–12. doi: 10.1186/1750-1172-6-27
45. Bali DS, Chen Y-T, Austin S, Goldstein JL. Glycogen storage disease type I. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1312/ (accessed October 22, 2019).
46. Mortellaro C, Garagiola U, Carbone V, Cerutti F, Marci V, Bonda PLF. Unusual oral manifestations and evolution in glycogen storage disease type Ib. J Craniofac Surg. (2005) 16:45–52. doi: 10.1097/00001665-200501000-00010
47. Ma R, Moein Vaziri F, Sabino GJ, Sarmast ND, Zove SM, Iacono VJ, et al. Glycogen storage disease Ib and severe periodontal destruction: a case report. Dent J. (2018) 6:53. doi: 10.3390/dj6040053
48. Bartoli A, Bossù M, Sfasciotti G, Polimeni A. Glycogen storage disease type Ib: a paediatric case report. Eur J Paediatr Dent. (2006) 7:192–8.
49. Martinez CC, Tonon T, Nalin T, Refosco LF, de Souza CFM, Schwartz IVD. Feeding difficulties and orofacial myofunctional disorder in patients with hepatic glycogen storage diseases. JIMD Rep. (2019) 45:21–7. doi: 10.1007/8904_2018_131
50. Boztug K, Järvinen PM, Salzer E, Racek T, Mönch S, Garncarz W, et al. JAGN1 deficiency causes aberrant myeloid cell homeostasis and congenital neutropenia. Nat Genet. (2014) 46:1021–7. doi: 10.1038/ng.3069
51. Wortmann SB, Zietkiewicz S, Kousi M, Szklarczyk R, Haack TB, Gersting SW, et al. CLPB mutations cause 3-Methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am J Hum Genet. (2015) 96:245–57. doi: 10.1016/j.ajhg.2014.12.013
52. Bohn G, Allroth A, Brandes G, Thiel J, Glocker E, Schäffer AA, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein P14. Nat Med. (2007) 13:38–45. doi: 10.1038/nm1528
53. Nelson A, Myers K. Shwachman-Diamond Syndrome In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1756/ (accessed October 22, 2019).
54. Ho W, Cheretakis C, Durie P, Kulkarni G, Glogauer M. Prevalence of oral diseases in Shwachman-Diamond syndrome. Am Soc Geriatr Dent. (2007) 27:52–8. doi: 10.1111/j.1754-4505.2007.tb00328.x
55. Carapito R, Konantz M, Paillard C, Miao Z, Pichot A, Leduc MS, et al. Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond-like features. J Clin Invest. (2017) 127:4090–103. doi: 10.1172/JCI92876
56. Bellanné-Chantelot C, Schmaltz-Panneau B, Marty C, Fenneteau O, Callebaut I, Clauin S, et al. Mutations in the SRP54 gene cause severe congenital neutropenia as well as Shwachman-Diamond-like syndrome. Blood. (2018) 132:1318–31. doi: 10.1182/blood-2017-12-820308
57. Witzel M, Petersheim D, Fan Y, Bahrami E, Racek T, Rohlfs M, et al. Chromatin-remodeling factor SMARCD2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nat Genet. (2017) 49:742–52. doi: 10.1038/ng.3833
58. Shadur B, Asherie N, Newburger PE, Stepensky P. How we approach: severe congenital neutropenia and myelofibrosis due to mutations in VPS45. Pediatr Blood Cancer. (2019) 66:e27473. doi: 10.1002/pbc.27473
59. Vilboux T, Lev A, Malicdan MCV, Simon AJ, Järvinen P, Racek T, et al. A congenital neutrophil defect syndrome associated with mutations in VPS45. N Engl J Med. (2013) 369:54–65. doi: 10.1056/NEJMoa1301296
60. Souza LM, Boone TC, Gabrilove J, Lai PH, Zsebo KM, Murdock DC, et al. Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells. Science. (1986) 232:61–5. doi: 10.1126/science.2420009
61. Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, et al. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. (2006) 107:4628–35. doi: 10.1182/blood-2005-11-4370
62. Nickel A, Sykora K-W, Welte K. Improved outcome of stem cell transplantation for severe chronic neutropenia with or without secondary leukemia: a Long-Term analysis of european data for more than 25 years by the SCNIR. Blood. (2013) 122:3347. doi: 10.1182/blood.V122.21.3347.3347
63. Skokowa J, Steinemann D, Katsman-Kuipers JE, Zeidler C, Klimenkova O, Klimiankou M, et al. Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood. (2014) 123:2229–37. doi: 10.1182/blood-2013-11-538025
64. Dale DC, Makaryan V. ELANE-Related Neutropenia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1533/ (accessed December 13, 2019).
65. Halai H, Somani C, Donos N, Nibali L. Periodontal status of children with primary immunodeficiencies: a systematic review. Clin Oral Investig. (2019). doi: 10.1007/s00784-019-03055-z
66. Silva LM, Brenchley L, Moutsopoulos NM. Primary immunodeficiencies reveal the essential role of tissue neutrophils in periodontitis. Immunol Rev. (2019) 287:226–35. doi: 10.1111/imr.12724
67. Carlsson G, Wahlin Y-B, Johansson A, Olsson A, Eriksson T, Claesson R, et al. Periodontal disease in patients from the original Kostmann family with severe congenital neutropenia. J Periodontol. (2006) 77:744–51. doi: 10.1902/jop.2006.050191
68. Bejjani N, Beaupain B, Bertrand Y, Bellanne-Chantelot C, Donadieu J. How to differentiate congenital from noncongenital chronic neutropenia at the first medical examination? Proposal of score: a pilot study from the French Severe Chronic Neutropenia registry. Pediatr Blood Cancer. (2017) 64:e26722. doi: 10.1002/pbc.26722
69. Karlsson J, Carlsson G, Ramme KG, Hägglund H, Fadeel B, Nordenskjöld M, et al. Low plasma levels of the protein pro-LL-37 as an early indication of severe disease in patients with chronic neutropenia. Br J Haematol. (2007) 137:166–9. doi: 10.1111/j.1365-2141.2007.06530.x
70. Ye Y, Carlsson G, Wondimu B, Fahlén A, Karlsson-Sjöberg J, Andersson M, et al. Mutations in the ELANE gene are associated with development of periodontitis in patients with severe congenital neutropenia. J Clin Immunol. (2011) 31:936–45. doi: 10.1007/s10875-011-9572-0
71. Pütsep K, Carlsson G, Boman HG, Andersson M. Deficiency of antibacterial peptides in patients with morbus Kostmann: an observation study. Lancet Lond Engl. (2002) 360:1144–9. doi: 10.1016/S0140-6736(02)11201-3
72. Dale DC, Welte K. Cyclic and chronic neutropenia. Cancer Treat Res. (2011) 157:97–108. doi: 10.1007/978-1-4419-7073-2_6
73. Chen X, Peng W, Zhang Z, Wu Y, Xu J, Zhou Y, et al. ELANE gene mutation-induced cyclic neutropenia manifesting as recurrent fever with oral mucosal ulcer. Medicine. (2018) 97:10031. doi: 10.1097/MD.0000000000010031
74. Aota K, Kani K, Yamanoi T, Momota Y, Ninomiya M, Yumoto H, et al. Management of tooth extraction in a patient with ELANE gene mutation-induced cyclic neutropenia. Medicine. (2019) 98:17372. doi: 10.1097/MD.0000000000017372
75. Chen Y, Fang L, Yang X. Cyclic neutropenia presenting as recurrent oral ulcers and periodontitis. J Clin Pediatr Dent. (2013) 37:307–8. doi: 10.17796/jcpd.37.3.n8k0111177074828
76. Block MS, Brindis M, Block CA, Berron JM. Full-Arch rehabilitation of a patient with cyclic neutropenia. J Oral Maxillofac Surg. (2015) 73:1734.e1–e10. doi: 10.1016/j.joms.2015.03.073
77. Fonseca MA, Fontes F. Early tooth loss due to cyclic neutropenia: long-term follow-up of one patient. Spec Care Dent. (2000) 20:187–90. doi: 10.1111/j.1754-4505.2000.tb00017.x
78. Matarasso S, Daniele V, Iorio Siciliano V, Mignogna MD, Andreuccetti G, Cafiero C. The effect of recombinant granulocyte colony-stimulating factor on oral and periodontal manifestations in a patient with cyclic neutropenia: a case report. Int J Dent. (2009) 2009:654239. doi: 10.1155/2009/654239
79. Etzioni A. Leukocyte adhesion deficiencies: molecular basis, clinical findings, and therapeutic options. Adv Exp Med Biol. (2007) 601:51–60. doi: 10.1007/978-0-387-72005-0_5
80. Smith CW. 3. Adhesion molecules and receptors. J Allergy Clin Immunol. (2008) 121:S375–9. doi: 10.1016/j.jaci.2007.07.030
81. Almarza Novoa E, Kasbekar S, Thrasher AJ, Kohn DB, Sevilla J, Nguyen T, et al. Leukocyte adhesion deficiency-I: a comprehensive review of all published cases. J Allergy Clin Immunol Pract. (2018) 6:1418–20.e10. doi: 10.1016/j.jaip.2017.12.008
82. Hanna S, Etzioni A. Leukocyte adhesion deficiencies. Ann N Y Acad Sci. (2012) 1250:50–5. doi: 10.1111/j.1749-6632.2011.06389.x
83. Crowley CA, Curnutte JT, Rosin RE, André-Schwartz J, Gallin JI, Klempner M, et al. An inherited abnormality of neutrophil adhesion. Its genetic transmission and its association with a missing protein. N Engl J Med. (1980) 302:1163–8. doi: 10.1056/NEJM198005223022102
84. Etzioni A, Tonetti M. Leukocyte adhesion deficiency II - from A to almost Z. Immunol Rev. (2000) 178:138–47. doi: 10.1034/j.1600-065X.2000.17805.x
85. Etzioni A, Sturla L, Antonellis A, Green ED, Gershoni-Baruch R, Berninsone PM, et al. Leukocyte adhesion deficiency (LAD) type II/carbohydrate deficient glycoprotein (CDG) IIc founder effect and genotype/phenotype correlation. Am J Med Genet. (2002) 110:131–5. doi: 10.1002/ajmg.10423
86. Alon R, Etzioni A. LAD-III, a novel group of leukocyte integrin activation deficiencies. Trends Immunol. (2003) 24:561–6. doi: 10.1016/j.it.2003.08.001
87. Moutsopoulos NM, Zerbe CS, Wild T, Dutzan N, Brenchley L, DiPasquale G, et al. Interleukin-12 and interleukin-23 blockade in leukocyte adhesion deficiency type 1. N Engl J Med. (2017) 376:1141–6. doi: 10.1056/NEJMoa1612197
88. Yamazaki-Nakashimada M, Maravillas-Montero JL, Berrón-Ruiz L, López-Ortega O, Ramírez-Alejo N, Acevedo-Ochoa E, et al. Successful adjunctive immunoglobulin treatment in patients affected by leukocyte adhesion deficiency type 1 (LAD-1). Immunol Res. (2015) 61:260–8. doi: 10.1007/s12026-014-8619-8
89. Frydman M, Etzioni A, Eidlitz-Markus T, Avidor I, Varsano I, Shechter Y, et al. Rambam-Hasharon syndrome of psychomotor retardation, short stature, defective neutrophil motility, and Bombay phenotype. Am J Med Genet. (1992) 44:297–302. doi: 10.1002/ajmg.1320440307
90. Marquardt T, Lühn K, Srikrishna G, Freeze HH, Harms E, Vestweber D. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood. (1999) 94:3976–85. doi: 10.1182/blood.V94.12.3976.424k06_3976_3985
91. Saultier P, Szepetowski S, Canault M, Falaise C, Poggi M, Suchon P, et al. Long-term management of leukocyte adhesion deficiency type III without hematopoietic stem cell transplantation. Haematologica. (2018) 103:e264–e7. doi: 10.3324/haematol.2017.186304
92. Sharma A, Jindal AK, Pilania RK, Gautam P, Daroch S. Palatal ulcer in leukocyte adhesion deficiency: an unusual occurrence. J Clin Immunol. (2018) 38:736. doi: 10.1007/s10875-018-0545-4
93. Dababneh R, Al-Wahadneh AM, Hamadneh S, Khouri A, Bissada NF. Periodontal manifestation of leukocyte adhesion deficiency type I. J Periodontol. (2008) 79:764–8. doi: 10.1902/jop.2008.070323
94. Moutsopoulos NM, Chalmers NI, Barb JJ, Abusleme L, Greenwell-Wild T, Dutzan N, et al. Subgingival microbial communities in leukocyte adhesion deficiency and their relationship with local immunopathology. PLoS Pathog. (2015) 11:e1004698. doi: 10.1371/journal.ppat.1004698
95. Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. (2014) 6:229ra40. doi: 10.1126/scitranslmed.3007696
96. Hajishengallis G, Moutsopoulos NM. Etiology of leukocyte adhesion deficiency-associated periodontitis revisited: not a raging infection but a raging inflammatory response. Expert Rev Clin Immunol. (2014) 10:973–975. doi: 10.1586/1744666X.2014.929944
97. Hajishengallis G, Moutsopoulos NM. Role of bacteria in leukocyte adhesion deficiency-associated periodontitis. Microb Pathog. (2016) 94:21–6. doi: 10.1016/j.micpath.2015.09.003
98. Moutsopoulos NM, Kling HM, Angelov N, Jin W, Palmer RJ, Nares S, et al. Porphyromonas gingivalis promotes Th17 inducing pathways in chronic periodontitis. J Autoimmun. (2012) 39:294–303. doi: 10.1016/j.jaut.2012.03.003
99. Majorana A, Notarangelo LD, Savoldi E, Gastaldi G, Lozada-Nur F. Leukocyte adhesion deficiency in a child with severe oral involvement. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. (1999) 87:691–4. doi: 10.1016/S1079-2104(99)70162-9
100. Tewari N, Mathur VP, Yadav VS, Chaudhari P. Leukocyte adhesion defect-I: rare primary immune deficiency. Soc Geriatr Dent. (2017) 37:309–13. doi: 10.1111/scd.12249
101. Karaky AA, Sawair F, Tamimi Z, Hassona Y. Dental implants in a patient with suspected leucocyte adhesion deficiency. Case Rep. (2017) 2017:220975. doi: 10.1136/bcr-2017-220975
102. Papillon M, Lefèvre P. Deux cas de kératodermie palmaire et plantaire symétrique familale (maladie de Meleda) chez le frère et la soeur. Bull Soc Fr Dermatol Syph. (1924) 31:82–7.
103. Nagy N, Vályi P, Csoma Z, Sulák A, Tripolszki K, Farkas K, et al. CTSC and Papillon-Lefèvre syndrome: detection of recurrent mutations in Hungarian patients, a review of published variants and database update. Mol Genet Genomic Med. (2014) 2:217–28. doi: 10.1002/mgg3.61
104. Machado RA, Cuadra-Zelaya FJM, Martelli-Júnior H, Miranda RT, Casarin RCV, Corrêa MG, et al. Clinical and molecular analysis in Papillon-Lefèvre syndrome. Am J Med Genet A. (2019) 179:2124–31. doi: 10.1002/ajmg.a.61285
105. Rao NV, Rao GV, Hoidal JR. Human dipeptidyl-peptidase I gene characterization, localization, and expression. J Biol Chem. (1997) 272:10260–5. doi: 10.1074/jbc.272.15.10260
106. Pham CT, Ley TJ. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc Natl Acad Sci USA. (1999) 96:8627–32. doi: 10.1073/pnas.96.15.8627
107. Sørensen OE, Clemmensen SN, Dahl SL, Østergaard O, Heegaard NH, Glenthøj A, Nielsen FC, et al. Papillon-Lefèvre syndrome patient reveals species-dependent requirements for neutrophil defenses. J Clin Invest. (2014) 124:4539–48. doi: 10.1172/JCI76009
108. Pham CTN, Ivanovich JL, Raptis SZ, Zehnbauer B, Ley TJ. Papillon-Lefèvre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/Dipeptidyl peptidase I Deficiency in humans. J Immunol. (2004) 173:7277–81. doi: 10.4049/jimmunol.173.12.7277
109. Almuneef M, Khenaizan SA, Ajaji SA, Al-Anazi A. Pyogenic liver abscess and Papillon-Lefèvre syndrome: not a rare association. Pediatrics. (2003) 111:e85–8. doi: 10.1542/peds.111.1.e85
110. Toomes C, James J, Wood AJ, Wu CL, McCormick D, Lench N, et al. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat Genet. (1999) 23:421–4. doi: 10.1038/70525
111. Hattab FN, Amin WM. Papillon-Lefèvre syndrome with albinism: a review of the literature and report of 2 brothers. Oral Radiol Endod. (2005) 100:709–16. doi: 10.1016/j.tripleo.2004.08.030
112. Moutsopoulos NM, Lionakis MS, Hajishengallis G. Inborn errors in immunity. J Dent Res. (2015) 94:753–8. doi: 10.1177/0022034515583533
113. Sreeramulu B, Shyam ND, Ajay P, Suman P. Papillon-Lefèvre syndrome: clinical presentation and management options. Clin Cosmet Investig Dent. (2015) 7:75–81. doi: 10.2147/CCIDE.S76080
114. Dhanrajani PJ. Papillon-Lefevre syndrome: clinical presentation and a brief review. Oral Radiol Endod. (2009) 108:e1–7. doi: 10.1016/j.tripleo.2009.03.016
115. Ishikawa I, Umeda M, Laosrisin N. Clinical, bacteriological, and immunological examinations and the treatment process of two Papillon-Lefèvre syndrome patients. J Periodontol. (1994) 65:364–71. doi: 10.1902/jop.1994.65.4.364
116. Fardal O, Drangsholt E, Olsen I. Palmar plantar keratosis and unusual periodontal findings. Observations from a family of 4 members. J Clin Periodontol. (1998) 25:181–4. doi: 10.1111/j.1600-051X.1998.tb02425.x
117. Soskolne WA, Stabholz A, van Dyke TE, Hart TC, Meyle J. Partial expression of the Papillon-Lefèvre syndrome in 2 unrelated families. J Clin Periodontol. (1996) 23:764–9. doi: 10.1111/j.1600-051X.1996.tb00607.x
118. Ullbro C, Crossner C-G, Nederfors T, Alfadley A, Thestrup-Pedersen K. Dermatologic and oral findings in a cohort of 47 patients with Papillon-Lefèvre syndrome. J Am Acad Dermatol. (2003) 48:345–51. doi: 10.1067/mjd.2003.197
119. Pacheco JJ, Coelho C, Salazar F, Contreras A, Slots J, Velazco CH. Treatment of Papillon-Lefèvre syndrome periodontitis. J Clin Periodontol. (2002) 29:370–4. doi: 10.1034/j.1600-051X.2002.290414.x
120. Nickles K, Schacher B, Ratka-Krüger P, Krebs M, Eickholz P. Long-term results after treatment of periodontitis in patients with Papillon-Lefèvre syndrome: success and failure. J Clin Periodontol. (2013) 40:789–98. doi: 10.1111/jcpe.12120
121. Ullbro C, Crossner CG, Lundgren T, Stålblad PA, Renvert S. Osseointegrated implants in a patient with Papillon-Lefèvre syndrome. A 4 1/2-year follow up. J Clin Periodontol. (2000) 27:951–4. doi: 10.1034/j.1600-051x.2000.027012951.x
122. Etöz OA, Ulu M, Kesim B. Treatment of patient with Papillon-Lefevre syndrome with short dental implants: a case report. Implant Dent. (2010) 19:394–9. doi: 10.1097/ID.0b013e3181ed0798
123. Ahmadian L, Monzavi A, Arbabi R, Hashemi HM. Full-mouth rehabilitation of an edentulous patient with Papillon-Lefèvre syndrome using dental implants: a clinical report. J Prosthodont. (2011) 20:643–8. doi: 10.1111/j.1532-849X.2011.00768.x
124. Senel FC, Altintas NY, Bagis B, Cankaya M, Pampu AA, Satiroglu I, et al. A 3-year follow-up of the rehabilitation of Papillon-Lefèvre syndrome by dental implants. Oral Maxillofac Surg. (2012) 70:163–7. doi: 10.1016/j.joms.2011.03.058
125. Kinaia B, Hope K, Zuhaili A, Tulasne J. Full-Mouth rehabilitation with calvarium bone grafts and dental implants for a Papillon-Lefèvre syndrome patient: case report. Int J Oral Maxillofac Implants. (2017) 32:e259–4. doi: 10.11607/jomi.6282
126. Al-Khenaizan S. Papillon-Lefévre syndrome: the response to acitretin. Int J Dermatol. (2002) 41:938–41. doi: 10.1046/j.1365-4362.2002.01664_3.x
127. Lee MR, Wong L-CF, Fischer GO. Papillon-Lefèvre syndrome treated with acitretin. Australas J Dermatol. (2005) 46:199–201. doi: 10.1111/j.1440-0960.2005.00180.x
128. Balci DD, Serarslan G, Sangun O, Homan S. Acitretin for Papillon-Lefevre syndrome in a five-year-old girl. Indian J Dermatol Venereol Leprol. (2008) 74:71–3. doi: 10.4103/0378-6323.38423
129. Sarma N, Ghosh C, Kar S, Bazmi BA. Low-dose acitretin in Papillon-Lefèvre syndrome: treatment and 1-year follow-up. Dermatol Ther. (2015) 28:28–31. doi: 10.1111/dth.12177
130. Kellum RE. Papillon-Lefèvre syndrome in four siblings treated with etretinate a Nine-Year evaluation. Int J Dermatol. (1989) 28:605–8. doi: 10.1111/j.1365-4362.1989.tb02539.x
131. Gelmetti C, Nazzaro V, Cerri D, Fracasso L. Long-Term preservation of permanent teeth in a patient with Papillon-Lefevre syndrome treated with etretinate. Pediatr Dermatol. (1989) 6:222–5. doi: 10.1111/j.1525-1470.1989.tb00822.x
132. Bronckers IMGJ, Seyger MMB, West DP, Lara-Corrales I, Tollefson M, Tom WL, et al. Safety of systemic agents for the treatment of pediatric psoriasis. JAMA Dermatol. (2017) 153:1147–57. doi: 10.1001/jamadermatol.2017.3029
133. Bullón P, Castejón-Vega B, Román-Malo L, Jimenez-Guerrero MP, Cotán D, Forbes-Hernandez TY, et al. Autophagic dysfunction in patients with Papillon-Lefèvre syndrome is restored by recombinant cathepsin C treatment. J Allergy Clin Immunol. (2018) 142:1131–43.e7. doi: 10.1016/j.jaci.2018.01.018
134. Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. (2002) 23:541–8. doi: 10.1016/S1471-4906(02)02316-5
135. Maney P, Walters JD. Formylpeptide receptor single nucleotide polymorphism 348T>C and its relationship to polymorphonuclear leukocyte chemotaxis in aggressive periodontitis. J Periodontol. (2009) 80:1498–505. doi: 10.1902/jop.2009.090103
136. Caton JG, Armitage G, Berglundh T, Chapple ILC, Jepsen S, Kornman KS, et al. A new classification scheme for periodontal and peri-implant diseases and conditions - Introduction and key changes from the 1999 classification. J Clin Periodontol. (2018) 45:S1–8. doi: 10.1111/jcpe.12935
137. Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, et al. Periodontitis: consensus report of workgroup 2 of the 2017 world workshop on the classification of periodontal and peri-implant diseases and conditions. J Clin Periodontol. (2018) 45:S162–70. doi: 10.1111/jcpe.12946
138. Gonçalves PF, Harris TH, Elmariah T, Aukhil I, Wallace MR, Shaddox LM. Genetic polymorphisms and periodontal disease in populations of African descent: a review. J Periodontal Res. (2018) 53:164–73. doi: 10.1111/jre.12505
139. Maney P, Emecen P, Mills JS, Walters JD. Neutrophil formylpeptide receptor single nucleotide polymorphism 348T>C in aggressive periodontitis. J Periodontol. (2009) 80:492–8. doi: 10.1902/jop.2009.080225
140. Kuhns DB, Fink DL, Choi U, Sweeney C, Lau K, Priel DL, et al. Cytoskeletal abnormalities and neutrophil dysfunction in WDR1 deficiency. Blood. (2016) 128:2135–43. doi: 10.1182/blood-2016-03-706028
141. Pfajfer L, Mair NK, Jiménez-Heredia R, Genel F, Gulez N, Ardeniz Ö, et al. Mutations affecting the actin regulator WD repeat-containing protein 1 lead to aberrant lymphoid immunity. J Allergy Clin Immunol. (2018) 142:1589–604.e11. doi: 10.1016/j.jaci.2018.04.023
142. van den Berg JM, van Koppen E, Åhlin A, Belohradsky BH, Bernatowska E, Corbeel L, Español T, et al. Chronic granulomatous disease: the European experience. PLoS ONE. (2009) 4:e5234. doi: 10.1371/journal.pone.0005234
143. Winkelstein JA, Marino MC, Johnston RBJ, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease: report on a National Registry of 368 patients. Medicine. (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
144. Holland SM. Chronic granulomatous disease. Hematol Oncol Clin North Am. (2013) 27:89–99. doi: 10.1016/j.hoc.2012.11.002
145. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: mechanisms of nADPH oxidase activation and bacterial resistance. Front Cell Infect Microbiol. (2017) 7:373. doi: 10.3389/fcimb.2017.00373
146. van de Geer A, Nieto-Patlán A, Kuhns DB, Tool AT, Arias AA, Bouaziz M, et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest. (2018) 128:3957–75. doi: 10.1172/JCI97116
147. Matute JD, Arias AA, Wright NAM, Wrobel I, Waterhouse CCM, Li XJ, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40phox and selective defects in neutrophil NADPH oxidase activity. Blood. (2009) 114:3309–15. doi: 10.1182/blood-2009-07-231498
148. Thomas DC, Clare S, Sowerby JM, Pardo M, Juss JK, Goulding DA, et al. Eros is a novel transmembrane protein that controls the phagocyte respiratory burst and is essential for innate immunity. J Exp Med. (2017) 214:1111–28. doi: 10.1084/jem.20161382
149. Arnadottir GA, Norddahl GL, Gudmundsdottir S, Agustsdottir AB, Sigurdsson S, Jensson BO, et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat Commun. (2018) 9:1–9. doi: 10.1038/s41467-018-06964-x
150. Thomas DC, Charbonnier L-M, Schejtman A, Aldhekri H, Coomber EL, Dufficy ER, et al. EROS mutations: decreased NADPH oxidase function and chronic granulomatous disease. J Allergy Clin Immunol. (2019) 143:782–5.e1. doi: 10.1016/j.jaci.2018.09.019
151. Arnold DE, Heimall JR. A review of chronic granulomatous disease. Adv Ther. (2017) 34:2543–57. doi: 10.1007/s12325-017-0636-2
152. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. (2010) 363:2600–10. doi: 10.1056/NEJMoa1007097
153. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine. (2000) 79:170. doi: 10.1097/00005792-200005000-00004
154. Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, et al. Common severe infections in chronic granulomatous disease. Infect Dis Soc Am. (2015) 60:1176–183. doi: 10.1093/cid/ciu1154
155. Conti F, Lugo-Reyes SO, Blancas Galicia L, He J, Aksu G, Borges de Oliveira E, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. (2016) 138:241–8.e3. doi: 10.1016/j.jaci.2015.11.041
156. Beauté J, Obenga G, Le Mignot L, Mahlaoui N, Bougnoux M-E, Mouy R, et al. Epidemiology and outcome of invasive fungal diseases in patients with chronic granulomatous disease: a multicenter study in france. Pediatr Infect Dis J. (2011) 30:57–62. doi: 10.1097/INF.0b013e3181f13b23
157. Magnani A, Brosselin P, Beauté J, de Vergnes N, Mouy R, Debré M, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol. (2014) 134:655–62.e8. doi: 10.1016/j.jaci.2014.04.014
158. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. The International Chronic Granulomatous Disease Cooperative Study Group. N Engl J Med. (1991) 324:509–16. doi: 10.1056/NEJM199102213240801
159. Angelino G, Angelis PD, Faraci S, Rea F, Romeo EF, Torroni F, et al. Inflammatory bowel disease in chronic granulomatous disease: an emerging problem over a twenty years' experience. Pediatr Allergy Immunol. (2017) 28:801–9. doi: 10.1111/pai.12814
160. Connelly JA, Marsh R, Parikh S, Talano J-A. Allogeneic hematopoietic cell transplantation for chronic granulomatous disease: controversies and state of the art. J Pediatr Infect Dis Soc. (2018) 7:S31–9. doi: 10.1093/jpids/piy015
161. Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O'Brien S, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. (2004) 114:462–8. doi: 10.1542/peds.114.2.462
162. Dunogué B, Pilmis B, Mahlaoui N, Elie C, Coignard-Biehler H, Amazzough K, et al. Chronic granulomatous disease in patients reaching adulthood: a Nationwide study in France. Infect Dis Soc Am. (2017) 64:767–75. doi: 10.1093/cid/ciw837
163. Dhillon SS, Fattouh R, Elkadri A, Xu W, Murchie R, Walters T, et al. Variants in nicotinamide adenine dinucleotide phosphate oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology. (2014) 147:680–9.e2. doi: 10.1053/j.gastro.2014.06.005
164. Liese J, Kloos S, Jendrossek V, Petropoulou T, Wintergerst U, Notheis G, et al. Long-term follow-up and outcome of 39 patients with chronic granulomatous disease. J Pediatr. (2000) 137:687–93. doi: 10.1067/mpd.2000.109112
165. Dar-Odeh NS, Hayajneh WA, Abu-Hammad OA, Hammad HM, Al-Wahadneh AM, Bulos NK, et al. Orofacial findings in chronic granulomatous disease: report of twelve patients and review of the literature. BMC Res Notes. (2010) 3:37. doi: 10.1186/1756-0500-3-37
166. Uzzan M, Ko HM, Mehandru S, Cunningham-Rundles C. Gastrointestinal disorders associated with common variable immune deficiency (CVID) and chronic granulomatous disease (CGD). Curr Gastroenterol Rep. (2016) 18:17. doi: 10.1007/s11894-016-0491-3
167. Hasui M. Chronic granulomatous disease in Japan: incidence and natural history. The study group of phagocyte disorders of Japan. Pediatr Int. (1999) 41:589–593. doi: 10.1046/j.1442-200x.1999.01129.x
168. Giannopoulou C, Krause K-H, Müller F. The NADPH oxidase NOX2 plays a role in periodontal pathologies. Semin Immunopathol. (2008) 30:273–8. doi: 10.1007/s00281-008-0128-1
169. Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, et al. Clinical features, long-term follow-up and outcome of a large cohort of patients with chronic granulomatous disease: an Italian multicenter study. Clin Immunol. (2008) 126:155–64. doi: 10.1016/j.clim.2007.09.008
170. Buduneli N, Baylas H, Aksu G, Kütükçüler N. Prepubertal periodontitis associated with chronic granulomatous disease. J Clin Periodontol. (2001) 28:589–93. doi: 10.1034/j.1600-051x.2001.028006589.x
171. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. (2014) 123:809–21. doi: 10.1182/blood-2013-07-515528
172. Hsu AP, McReynolds LJ, Holland SM. GATA2 deficiency. Curr Opin Allergy Clin Immunol. (2015) 15:104–9. doi: 10.1097/ACI.0000000000000126
173. Pasquet M, Bellanné-Chantelot C, Tavitian S, Prade N, Beaupain B, Larochelle O, et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood. (2013) 121:822–9. doi: 10.1182/blood-2012-08-447367
174. Simonis A, Fux M, Nair G, Mueller NJ, Haralambieva E, Pabst T, et al. Allogeneic hematopoietic cell transplantation in patients with GATA2 deficiency-a case report and comprehensive review of the literature. Ann Hematol. (2018) 97:1961–73. doi: 10.1007/s00277-018-3388-4
175. Donadieu J, Lamant M, Fieschi C, de Fontbrune FS, Caye A, Ouachee M, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica. (2018) 103:1278–87. doi: 10.3324/haematol.2017.181909
176. Spinner MA, Ker JP, Stoudenmire CJ, Fadare O, Mace EM, Orange JS, et al. GATA2 deficiency underlying severe blastomycosis and fatal herpes simplex virus-associated hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. (2016) 137:638–40. doi: 10.1016/j.jaci.2015.07.043
177. Polat A, Dinulescu M, Fraitag S, Nimubona S, Toutain F, Jouneau S, et al. Skin manifestations among GATA2-deficient patients. Br J Dermatol. (2018) 178:781–5. doi: 10.1111/bjd.15548
178. Sanyi A, Jaye DL, Rosand CB, Box A, Shanmuganathan C, Waller EK. Diagnosis of GATA2 haplo-insufficiency in a young woman prompted by pancytopenia with deficiencies of B-cell and dendritic cell development. Biomark Res. (2018) 6:13. doi: 10.1186/s40364-018-0127-x
179. Bustamante J, Boisson-Dupuis S, Abel L, Casanova J-L. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. (2014) 26:454–470. doi: 10.1016/j.smim.2014.09.008
180. Rosain J, Kong X-F, Martinez-Barricarte R, Oleaga-Quintas C, Ramirez-Alejo N, Markle J, et al. Mendelian susceptibility to mycobacterial disease: 2014-2018 update. Immunol Cell Biol. (2019) 97:360–7. doi: 10.1111/imcb.12210
181. Newport MJ, Huxley CM, Huston S, Hawrylowicz CM, Oostra BA, Williamson R, et al. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med. (1996) 335:1941–9. doi: 10.1056/NEJM199612263352602
182. Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. (2011) 365:127–38. doi: 10.1056/NEJMoa1100066
183. Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. (1998) 101:2364–9. doi: 10.1172/JCI2901
184. Picard C, Fieschi C, Altare F, Al-Jumaah S, Al-Hajjar S, Feinberg J, et al. Inherited interleukin-12 deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds. Am J Hum Genet. (2002) 70:336–48. doi: 10.1086/338625
185. Fieschi C, Bosticardo M, de Beaucoudrey L, Boisson-Dupuis S, Feinberg J, Santos OF, et al. A novel form of complete IL-12/IL-23 receptor beta1 deficiency with cell surface-expressed nonfunctional receptors. Blood. (2004) 104:2095–101. doi: 10.1182/blood-2004-02-0584
186. Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, et al. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science. (2012) 337:1684–8. doi: 10.1126/science.1224026
187. Kreins AY, Ciancanelli MJ, Okada S, Kong X-F, Ramírez-Alejo N, Kilic SS, et al. Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med. (2015) 212:1641–62. doi: 10.1084/jem.20140280
188. Kong X-F, Martinez-Barricarte R, Kennedy J, Mele F, Lazarov T, Deenick EK, et al. Disruption of an antimycobacterial circuit between dendritic and helper t cells in human SPPL2a deficiency. Nat Immunol. (2018) 19:973–85. doi: 10.1038/s41590-018-0178-z
189. Martínez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramírez-Alejo N, Mele F, et al. Human IFN-γ immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol. (2018) 3:6759. doi: 10.1126/sciimmunol.aau6759
190. Bustamante J. Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum Genet. (2020). doi: 10.1007/s00439-020-02120-y
191. Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, et al. Impairment of immunity to candida and mycobacterium in humans with bi-allelic RORC mutations. Science. (2015) 349:606–13. doi: 10.1126/science.aaa4282
192. Kerner G, Rosain J, Guérin A, AlKhabaz A, Oleaga-Quintas C, Rapaport F, et al. Inherited human IFNγ deficiency underlies mycobacterial disease. J Clin Invest. (2020). doi: 10.1172/JCI135460
193. Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. (2001) 293:300–3. doi: 10.1126/science.1061154
194. Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku C-L, Puel A, et al. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. (2006) 203:1745–59. doi: 10.1084/jem.20060085
195. Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. (2011) 12:213–21. doi: 10.1038/ni.1992
196. Patel S, Uppuluri R, Vellaichamy Swaminathan V, Ravichandran N, Melarcode Ramanan K, Raj R. Mendelian susceptibility to mycobacterial disease-Challenges in hematopoietic stem cell transplantation. Pediatr Blood Cancer. (2020) 67:e28187. doi: 10.1002/pbc.28187
197. de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rβ1 deficiency. Medicine. (2010) 89:381–402. doi: 10.1097/MD.0b013e3181fdd832
198. Aytekin C, Dogu F, Tuygun N, Tanir G, Guloglu D, Boisson-Dupuis S, et al. BCG lymphadenitis and recurrent oral candidiasis in an infant with a new mutation leading to interleukin-12 receptor beta-1 deficiency. J Investig Allergol. (2011) 21:401–4.
199. Hatipoglu N, Güvenç BH, Deswarte C, Koksalan K, Boisson-Dupuis S, Casanova J-L, et al. Inherited IL-12Rβ1 deficiency in a child with bCG adenitis and oral candidiasis: a Case report. Pediatrics. (2017) 140:e20161668. doi: 10.1542/peds.2016-1668
200. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
201. Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity*. Science. (2011) 332:65–8. doi: 10.1126/science.1200439
202. Ouederni M, Sanal O, Ikinciogullari A, Tezcan I, Dogu F, Sologuren I, et al. Clinical features of candidiasis in patients with inherited interleukin 12 receptor β1 deficiency. Infect Dis Soc Am. (2014) 58:204–13. doi: 10.1093/cid/cit722
203. Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature. (2000) 405:466–72. doi: 10.1038/35013114
204. Döffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. (2001) 27:277–85. doi: 10.1038/85837
205. Niehues T, Reichenbach J, Neubert J, Gudowius S, Puel A, Horneff G, et al. Nuclear factor kappaB essential modulator-deficient child with immunodeficiency yet without anhidrotic ectodermal dysplasia. J Allergy Clin Immunol. (2004) 114:1456–62. doi: 10.1016/j.jaci.2004.08.047
206. Clauss F, Manière M-C, Obry F, Waltmann E, Hadj-Rabia S, Bodemer C, et al. Dento-craniofacial phenotypes and underlying molecular mechanisms in hypohidrotic ectodermal dysplasia (HED): a review. J Dent Res. (2008) 87:1089–99. doi: 10.1177/154405910808701205
207. de La Dure-Molla M, Fournier BP, Manzanares MC, Acevedo AC, Hennekam RC, Friedlander L, et al. Elements of morphology: standard terminology for the teeth and classifying genetic dental disorders. Am J Med Genet A. (2019) 179:1913–81. doi: 10.1002/ajmg.a.61316
208. Bustamante J, Picard C, Boisson-Dupuis S, Abel L, Casanova J-L. Genetic lessons learnt from X-linked Mendelian susceptibility to mycobacterial diseases. Ann N Y Acad Sci. (2011) 1246:92–101. doi: 10.1111/j.1749-6632.2011.06273.x
209. Bustamante J, Picard C, Fieschi C, Filipe-Santos O, Feinberg J, Perronne C, et al. A novel X-linked recessive form of Mendelian susceptibility to mycobaterial disease. J Med Genet. (2007) 44:e65. doi: 10.1136/jmg.2006.043406
210. de Jong SJ, Imahorn E, Itin P, Uitto J, Orth G, Jouanguy E, et al. Epidermodysplasia verruciformis: inborn errors of immunity to human beta-Papillomaviruses. Front Microbiol. (2018) 9:1222. doi: 10.3389/fmicb.2018.01222
211. Leiding JW, Holland SM. Warts and all: HPV in primary immunodeficiencies. J Allergy Clin Immunol. (2012) 130:1030–48. doi: 10.1016/j.jaci.2012.07.049
212. de Jong SJ, Créquer A, Matos I, Hum D, Gunasekharan V, Lorenzo L, et al. The human CIB1-EVER1-EVER2 complex governs keratinocyte-intrinsic immunity to β-papillomaviruses. J Exp Med. (2018) 215:2289–310. doi: 10.1084/jem.20170308
213. Zuelzer WW. “Myelokathexis”–A new form of chronic granulocytopenia. Report of a case. N Engl J Med. (1964) 270:699–704. doi: 10.1056/NEJM196404022701402
214. McDermott DH, Murphy PM. WHIM syndrome: immunopathogenesis, treatment and cure strategies. Immunol Rev. (2019) 287:91–102. doi: 10.1111/imr.12719
215. McDermott DH, Pastrana DV, Calvo KR, Pittaluga S, Velez D, Cho E, et al. Plerixafor for the treatment of wHIM syndrome. N Engl J Med. (2019) 380:163–70. doi: 10.1056/NEJMoa1808575
216. Cipriani NA, Blair E, Taxy JB. WHIM syndrome and oral squamous cell carcinoma. Oral Radiol Endodontology. (2010) 109:105–8. doi: 10.1016/j.tripleo.2009.08.011
217. Tarzi MD, Jenner M, Hattotuwa K, Faruqi AZ, Diaz GA, Longhurst HJ. Sporadic case of warts, hypogammaglobulinemia, immunodeficiency, and myelokathexis syndrome. J Allergy Clin Immunol. (2005) 116:1101–5. doi: 10.1016/j.jaci.2005.08.040
218. Mc Guire PJ, Cunningham-Rundles C, Ochs H, Diaz GA. Oligoclonality, impaired class switch and B cell memory responses in WHIM syndrome. Clin Immunol Orlando Fla. (2010) 135:412–21. doi: 10.1016/j.clim.2010.02.006
219. Bock I, Dugué F, Loppinet E, Bellanné-Chantelot C, Bénet B. [WHIM syndrome: presumptive diagnosis based on myelokathexis on bone marrow smear]. Ann Biol Clin. (2014) 72:111–9. doi: 10.1684/abc.2013.0928
220. Dale DC, Bonilla MA, Davis MW, Nakanishi AM, Hammond WP, Kurtzberg J, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. (1993) 81:2496–502. doi: 10.1182/blood.V81.10.2496.bloodjournal81102496
221. Naviglio S, Soncini E, Vairo D, Lanfranchi A, Badolato R, Porta F. Long-Term survival after hematopoietic stem cell transplantation for complete sTAT1 deficiency. J Clin Immunol. (2017) 37:701–6. doi: 10.1007/s10875-017-0430-6
222. Boisson-Dupuis S, Kong X-F, Okada S, Cypowyj S, Puel A, Abel L, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol. (2012) 24:364–78. doi: 10.1016/j.coi.2012.04.011
223. Chapgier A, Wynn RF, Jouanguy E, Filipe-Santos O, Zhang S, Feinberg J, et al. Human complete STAT-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol Baltim Md. (2006) 176:5078–83. doi: 10.4049/jimmunol.176.8.5078
224. Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. (2003) 33:388–91. doi: 10.1038/ng1097
225. Moens L, Van Eyck L, Jochmans D, Mitera T, Frans G, Bossuyt X, et al. A novel kindred with inherited STAT2 deficiency and severe viral illness. J Allergy Clin Immunol. (2017) 139:1995–7.e9. doi: 10.1016/j.jaci.2016.10.033
226. Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SMB, Valappil M, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci USA. (2013) 110:3053–8. doi: 10.1073/pnas.1220098110
227. Hernandez N, Melki I, Jing H, Habib T, Huang SSY, Danielson J, et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J Exp Med. (2018) 215:2567–85. doi: 10.1084/jem.20180628
228. Duncan CJA, Mohamad SMB, Young DF, Skelton AJ, Leahy TR, Munday DC, et al. Human IFNAR2 deficiency: lessons for antiviral immunity. Sci Transl Med. (2015) 7:307ra154. doi: 10.1126/scitranslmed.aac4227
229. Ciancanelli MJ, Huang SXL, Luthra P, Garner H, Itan Y, Volpi S, et al. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. (2015). doi: 10.1126/science.aaa1578
230. Bravo García-Morato M, Calvo Apalategi A, Bravo-Gallego LY, Blázquez Moreno A, Simón-Fuentes M, Garmendia JV, et al. Impaired control of multiple viral infections in a family with complete IRF9 deficiency. J Allergy Clin Immunol. (2019) 144:309–12.e10. doi: 10.1016/j.jaci.2019.02.019
231. Asgari S, Schlapbach LJ, Anchisi S, Hammer C, Bartha I, Junier T, et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc Natl Acad Sci USA. (2017) 114:8342–7. doi: 10.1073/pnas.1704259114
232. Zaki M, Thoenes M, Kawalia A, Nürnberg P, Kaiser R, Heller R, et al. Recurrent and prolonged infections in a child with a homozygous IFIH1 nonsense mutation. Front Genet. (2017) 8:130. doi: 10.3389/fgene.2017.00130
233. Lamborn IT, Jing H, Zhang Y, Drutman SB, Abbott JK, Munir S, et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med. (2017) 214:1949–72. doi: 10.1084/jem.20161759
234. Ogunjimi B, Zhang S-Y, Sørensen KB, Skipper KA, Carter-Timofte M, Kerner G, et al. Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J Clin Invest. (2017) 127:3543–56. doi: 10.1172/JCI92280
235. Liu L, Okada S, Kong X-F, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. (2011) 208:1635–48. doi: 10.1084/jem.20110958
236. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LAB, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. (2011) 365:54–61. doi: 10.1056/NEJMoa1100102
237. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. (2016) 127:3154–64. doi: 10.1182/blood-2015-11-679902
238. Depner M, Fuchs S, Raabe J, Frede N, Glocker C, Doffinger R, et al. The extended clinical phenotype of 26 patients with chronic mucocutaneous candidiasis due to gain-of-function mutations in STAT1. J Clin Immunol. (2016) 36:73–84. doi: 10.1007/s10875-015-0214-9
239. Jing H, Su HC. New immunodeficiency syndromes that help us understand the IFN-mediated antiviral immune response. Curr Opin Pediatr. (2019) 31:815–20. doi: 10.1097/MOP.0000000000000827
240. Grier JT, Forbes LR, Monaco-Shawver L, Oshinsky J, Atkinson TP, Moody C, et al. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest. (2012) 122:3769–80. doi: 10.1172/JCI64837
241. de Vries E, Koene HR, Vossen JM, Gratama JW, von dem Borne AE, Waaijer JL, et al. Identification of an unusual Fc gamma receptor IIIa (CD16) on natural killer cells in a patient with recurrent infections. Blood. (1996) 88:3022–7. doi: 10.1182/blood.V88.8.3022.bloodjournal8883022
242. Jawahar S, Moody C, Chan M, Finberg R, Geha R, Chatila T. Natural killer (NK) cell deficiency associated with an epitope-deficient fc receptor type iIIA (CD16-II). Clin Exp Immunol. (1996) 103:408–13. doi: 10.1111/j.1365-2249.1996.tb08295.x
243. Zhang S-Y, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. (2007) 317:1522–7. doi: 10.1126/science.1139522
244. Maglione PJ, Simchoni N, Cunningham-Rundles C. Toll-like receptor signaling in primary immune deficiencies. Ann N Y Acad Sci. (2015) 1356:1–21. doi: 10.1111/nyas.12763
245. Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. (2011) 208:2083–98. doi: 10.1084/jem.20101568
246. Drummond RA, Franco LM, Lionakis MS. Human CARD9: a Critical molecule of fungal immune surveillance. Front Immunol. (2018) 9:1836. doi: 10.3389/fimmu.2018.01836
247. Gross O, Gewies A, Finger K, Schäfer M, Sparwasser T, Peschel C, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. (2006) 442:651–6. doi: 10.1038/nature04926
248. Glocker E-O, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. (2009) 361:1727–35. doi: 10.1056/NEJMoa0810719
249. Corvilain E, Casanova J-L, Puel A. Inherited CARD9 deficiency: invasive disease caused by ascomycete fungi in previously healthy children and adults. J Clin Immunol. (2018) 38:656–93. doi: 10.1007/s10875-018-0539-2
250. Lionakis MS, Netea MG, Holland SM. Mendelian genetics of human susceptibility to fungal infection. Cold Spring Harb Perspect Med. (2014) 4:19638. doi: 10.1101/cshperspect.a019638
251. Grumach AS, de Queiroz-Telles F, Migaud M, Lanternier F, Filho NR, Palma SMU, et al. A homozygous CARD9 mutation in a Brazilian patient with deep dermatophytosis. J Clin Immunol. (2015) 35:486–90. doi: 10.1007/s10875-015-0170-4
252. Puel A, Cypowyj S, Maródi L, Abel L, Picard C, Casanova J-L. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol. (2012) 12:616–22. doi: 10.1097/ACI.0b013e328358cc0b
253. Okada S, Puel A, Casanova J-L, Kobayashi M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin Transl Immunol. (2016) 5:e114. doi: 10.1038/cti.2016.71
254. Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med. (2015) 212:619–631. doi: 10.1084/jem.20141065
255. Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. (2013) 39:676–86. doi: 10.1016/j.immuni.2013.09.002
256. Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. (2006) 281:35603–7. doi: 10.1074/jbc.C600256200
257. Li J, Ritelli M, Ma CS, Rao G, Habib T, Corvilain E, et al. Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-β. Sci Immunol. (2019) 4:7965. doi: 10.1126/sciimmunol.aax7965
258. Bloomfield M, Kanderová V, Paračková Z, Vrabcová P, Svaton M, Fronková E, et al. Utility of ruxolitinib in a child with chronic mucocutaneous candidiasis caused by a novel Stat1 gain-of-function mutation. J Clin Immunol. (2018) 38:589–601. doi: 10.1007/s10875-018-0519-6
259. Higgins E, Al Shehri T, McAleer MA, Conlon N, Feighery C, Lilic D, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol. (2015) 135:551–3. doi: 10.1016/j.jaci.2014.12.1867
260. Kiykim A, Charbonnier LM, Akcay A, Karakoc-Aydiner E, Ozen A, Ozturk G, et al. Hematopoietic stem cell transplantation in patients with heterozygous STAT1 gain-of-Function mutation. J Clin Immunol. (2019) 39:37–44. doi: 10.1007/s10875-018-0575-y
261. Baghad B, Benhsaien I, El Fatoiki FZ, Migaud M, Puel A, Chiheb S, et al. [Chronic mucocutaneous candidiasis with STAT1 gain-of-function mutation associated with herpes virus and mycobacterial infections]. Ann Dermatol Venereol. (2019) 147:41–5. doi: 10.1016/j.annder.2019.09.597
262. Mohd Bakri M, Mohd Hussaini H, Rachel Holmes A, David Cannon R, Mary Rich A. Revisiting the association between candidal infection and carcinoma, particularly oral squamous cell carcinoma. J Oral Microbiol. (2010) 2:5780. doi: 10.3402/jom.v2i0.5780
263. Carey B, Lambourne J, Porter S, Hodgson T. Chronic mucocutaneous candidiasis due to gain-of-function mutation in STAT1. Oral Dis. (2019) 25:684–92. doi: 10.1111/odi.12881
264. Bhattad S, Dinakar C, Pinnamaraju H, Ganapathy A, Mannan A. Chronic mucocutaneous candidiasis in an adolescent boy due to a novel mutation in TRAF3IP2. J Clin Immunol. (2019) 39:596–9. doi: 10.1007/s10875-019-00664-x
265. Frans G, Moens L, Schaballie H, Van Eyck L, Borgers H, Wuyts M, et al. Gain-of-function mutations in signal transducer and activator of transcription 1 (STAT1): chronic mucocutaneous candidiasis accompanied by enamel defects and delayed dental shedding. J Allergy Clin Immunol. (2014) 134:1209–13.e6. doi: 10.1016/j.jaci.2014.05.044
266. Tanase S, Bawden JW. The immunohistochemical localization of signal-transduction pathway components Jak1, Jak2, Jak3, Tyk2 and STAT-1 during early enamel and dentine formation in rat molars. Arch Oral Biol. (1996) 41:925–40. doi: 10.1016/S0003-9969(96)00048-9
267. von Bernuth H, Picard C, Puel A, Casanova J-L. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur J Immunol. (2012) 42:3126–35. doi: 10.1002/eji.201242683
268. Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore). (2010) 89:403–25. doi: 10.1097/MD.0b013e3181fd8ec3
269. Ku C-L, von Bernuth H, Picard C, Zhang S-Y, Chang H-H, Yang K, et al. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med. (2007) 204:2407–22. doi: 10.1084/jem.20070628
270. Picard C, Casanova J-L, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IκBα deficiency. Clin Microbiol Rev. (2011) 24:490–7. doi: 10.1128/CMR.00001-11
271. Into T, Takigawa T, Niida S, Shibata K. MyD88 deficiency alters expression of antimicrobial factors in mouse salivary glands. PLoS ONE. (2014) 9:e113333. doi: 10.1371/journal.pone.0113333
272. Guideline on dental management of pediatric patients receiving chemotherapy hematopoietic cell transplantation and/or radiation therapy. Pediatr Dent. (2016) 38:334–2.
273. Pedraza-Sánchez S, Méndez-León JI, Gonzalez Y, Ventura-Ayala ML, Herrera MT, Lezana-Fernández JL, et al. Oral administration of human polyvalent IgG by mouthwash as an adjunctive treatment of chronic oral candidiasis. Front Immunol. (2018) 9:2956. doi: 10.3389/fimmu.2018.02956
274. Atkinson JC, O'Connell A, Aframian D. Oral manifestations of primary immunological diseases. J Am Dent Assoc 1939. (2000) 131:345–56. doi: 10.14219/jada.archive.2000.0178
275. Szczawinska-Poplonyk A, Gerreth K, Breborowicz A, Borysewicz-Lewicka M. Oral manifestations of primary immune deficiencies in children. Oral Radiol Endod. (2009) 108:e9–20. doi: 10.1016/j.tripleo.2009.03.049
276. Wilson W, Taubert KA, Gewitz M, Lockhart PB, Baddour LM, Levison M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association rheumatic fever, endocarditis and Kawasaki disease committee, council on cardiovascular disease in the young, and the council on clinical cardiology, council on cardiovascular surgery and anesthesia, and the quality of care and outcomes research interdisciplinary working group. J Am Dent Assoc. (2008) 139 Suppl:3S–24S. doi: 10.1161/CIRCULATIONAHA.106.183095
277. Peiseler M, Kubes P. More friend than foe: the emerging role of neutrophils in tissue repair. J Clin Invest. (2019) 129:2629–39. doi: 10.1172/JCI124616
278. Castagnoli R, Delmonte OM, Calzoni E, Notarangelo LD. Hematopoietic stem cell transplantation in primary immunodeficiency diseases: current status and future perspectives. Front Pediatr. (2019) 7:295. doi: 10.3389/fped.2019.00295
279. Gennery AR, Albert MH, Slatter MA, Lankester A. Hematopoietic stem cell transplantation for primary immunodeficiencies. Front Pediatr. (2019) 7:445. doi: 10.3389/fped.2019.00445
280. Mawardi H, Hashmi SK, Elad S, Aljurf M, Treister N. Chronic graft-vs. -host disease: Current management paradigm and future perspectives. Oral Dis. (2019) 25:931–48. doi: 10.1111/odi.12936
Keywords: inborn errors of immunity, innate immunity, orofacial manifestations, oral management, primary immunodeficiencies
Citation: Jung S, Gies V, Korganow A-S and Guffroy A (2020) Primary Immunodeficiencies With Defects in Innate Immunity: Focus on Orofacial Manifestations. Front. Immunol. 11:1065. doi: 10.3389/fimmu.2020.01065
Received: 04 March 2020; Accepted: 04 May 2020;
Published: 18 June 2020.
Edited by:
Guzide Aksu, Ege University, TurkeyReviewed by:
Jacinta Bustamante, Université Paris Descartes, FranceSaul Oswaldo Lugo Reyes, National Institute of Pediatrics, Mexico
Copyright © 2020 Jung, Gies, Korganow and Guffroy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Jung, cy5qdW5nQHVuaXN0cmEuZnI=
†These authors have contributed equally to this work