
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 May 2020
Sec. Microbial Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00983
This article is part of the Research Topic Advances in The Immunology of Host Defense Peptide: Mechanisms and Applications of Antimicrobial Functions and Beyond View all 29 articles
 Darren Shu Jeng Ting1,2,3
Darren Shu Jeng Ting1,2,3 Roger W. Beuerman3
Roger W. Beuerman3 Harminder S. Dua1,2
Harminder S. Dua1,2 Rajamani Lakshminarayanan3*†
Rajamani Lakshminarayanan3*† Imran Mohammed1*†
Imran Mohammed1*†The golden era of antibiotics, heralded by the discovery of penicillin, has long been challenged by the emergence of antimicrobial resistance (AMR). Host defense peptides (HDPs), previously known as antimicrobial peptides, are emerging as a group of promising antimicrobial candidates for combatting AMR due to their rapid and unique antimicrobial action. Decades of research have advanced our understanding of the relationship between the physicochemical properties of HDPs and their underlying antimicrobial and non-antimicrobial functions, including immunomodulatory, anti-biofilm, and wound healing properties. However, the mission of translating novel HDP-derived molecules from bench to bedside has yet to be fully accomplished, primarily attributed to their intricate structure-activity relationship, toxicity, instability in host and microbial environment, lack of correlation between in vitro and in vivo efficacies, and dwindling interest from large pharmaceutical companies. Based on our previous experience and the expanding knowledge gleaned from the literature, this review aims to summarize the novel strategies that have been employed to enhance the antimicrobial efficacy, proteolytic stability, and cell selectivity, which are all crucial factors for bench-to-bedside translation of HDP-based treatment. Strategies such as residues substitution with natural and/or unnatural amino acids, hybridization, L-to-D heterochiral isomerization, C- and N-terminal modification, cyclization, incorporation with nanoparticles, and “smart design” using artificial intelligence technology, will be discussed. We also provide an overview of HDP-based treatment that are currently in the development pipeline.
Antimicrobial resistance (AMR) currently represents a major global health threat of the twenty-first century and is estimated to cost 10 million deaths per year by 2050 (1, 2). Tackling AMR requires a multi-faceted and synergistic approach, including understanding of the main mechanisms and drivers of AMR at the microorganism, individual and population levels, antimicrobial stewardship within the healthcare and agriculture sectors, in tandem with discovery and development of new classes of antimicrobial therapy (3, 4).
Host defense peptides (HDPs), also known as antimicrobial peptides, are a group of evolutionary conserved molecules that play critical roles in the innate immune system (5–7). These molecules are usually small in size (around 12–50 amino acid residues), cationic (with a net charge of +2 to +13), and amphiphilic geometry (5, 6). So far, more than 3,000 naturally occurring and synthetic HDPs have been discovered across six life kingdoms (8, 9), underlining their essential roles during the adaptation and evolutionary process. HDPs have been shown to exhibit broad-spectrum antimicrobial activities against a plethora of microorganisms, including drug-resistant bacteria, fungi, parasites, and viruses (5, 10). In contrast to conventional antibiotics (which usually target a particular extracellular or intracellular binding site), cationic HDPs bind to the anionic surfaces of bacteria causing lytic cell death through varied mechanisms. Additionally, HDPs could also kill bacteria via inhibition of macromolecules that are involved in the biosynthesis of the surface membrane, and components of metabolic organelles (11, 12). Such unique mechanism of action accounts for the rapid and broad-spectrum antimicrobial action and the low risk of developing AMR as modification of the entire microorganisms' anionic membrane incurs a substantially higher fitness cost when compared to alteration of a particular binding site targeted by the conventional antibiotics (e.g., alteration in the 30S subunit inhibits the action of aminoglycosides) (13).
In addition to the antimicrobial activity, there has been emerging evidence highlighting the non-antimicrobial roles of HDPs, including antibiofilm (14), immunomodulatory (15), wound healing (16), anti-viral, and anti-cancer properties (17), rendering them an attractive and novel class of clinical therapeutics (18). For instance, corneal infection or infectious keratitis is a sight-threatening ocular disease that can be caused by a wide range of microorganisms, including bacteria, fungi, parasites, and viruses (19, 20). In addition, corneal infection may be caused by polymicrobial infection which can present significant therapeutic challenges to the clinicians (21). The dual broad-spectrum antimicrobial and wound healing properties of HDPs would not only provide a comprehensive antimicrobial coverage for the infection but also expedite the healing process of corneal ulceration, limiting the damage to the cornea and ultimately preserving the vision. The complete repertoire of antimicrobial and non-antimicrobial functions of HDPs has been recently summarized in a seminal review by Mookherjee et al. (22).
Despite decades of research efforts have been invested in the field of HDP, the mission of translating novel HDP-derived molecules from bench to beside has yet to be fully accomplished. The sluggishness in the HDP-derived drug development pipeline is primarily attributed to their complex structure-activity relationship (SAR), undesired toxicity to host tissues, instability in host and microbial environment, lack of correlation between in vitro and in vivo efficacies, and dwindling interest from large pharmaceutical companies (23–26). A list of synthetic derivatives of HDPs that are currently under clinical trials are summarized in Table 1 for readers' convenience. Detailed overview on the mechanisms of actions and clinical applications of HDPs have also been provided elsewhere in published review articles (27–29). Based on the literature and our experience, this review article aims to highlight the strategies that have been attempted to enhance the antimicrobial efficacy, proteolytic stability, and cell selectivity (for microbial cells instead of host cells; i.e., improved safety) of HDPs, which are all important factors to be considered during the translation of these molecules.
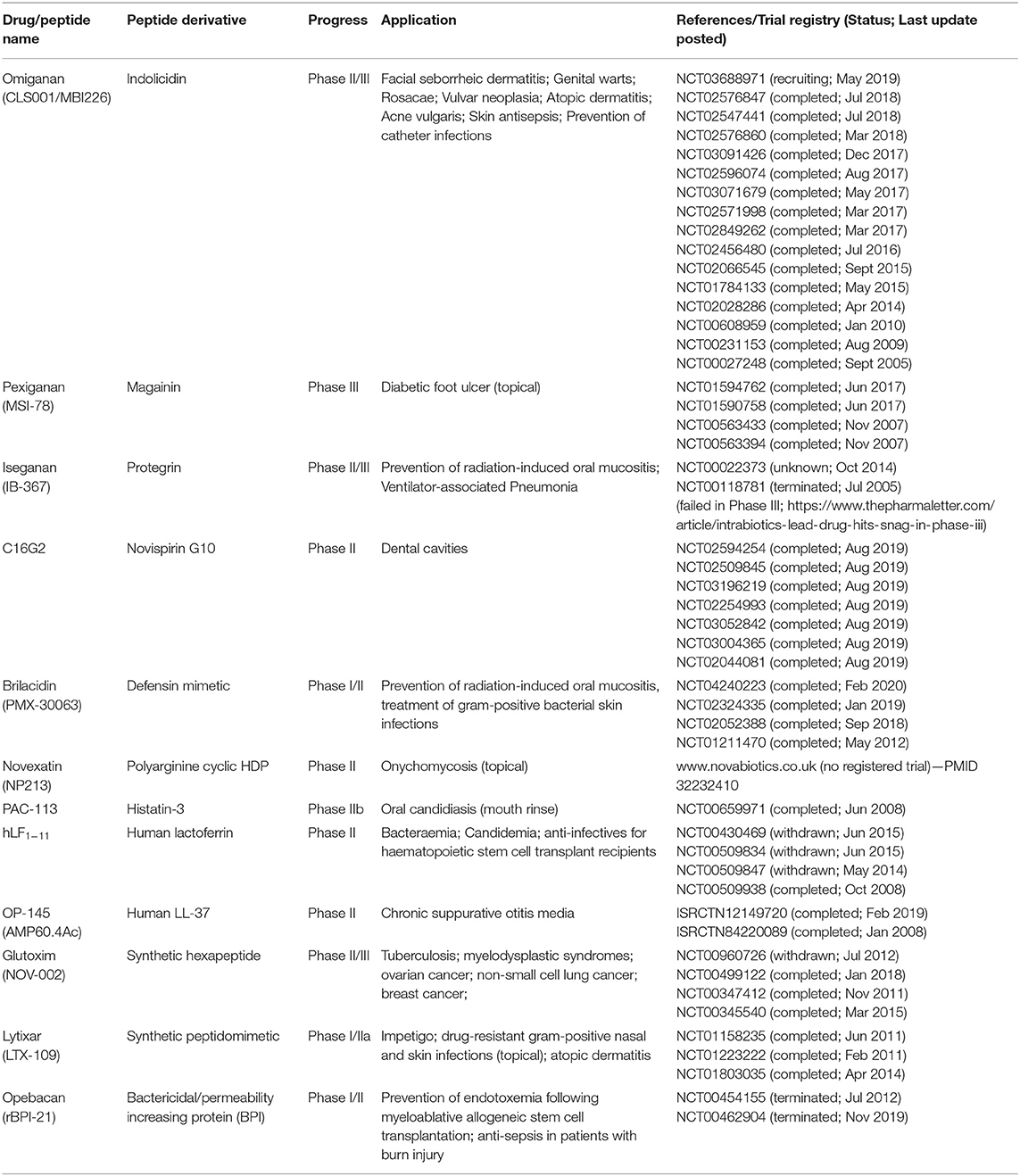
Table 1. Derivatives of host defense peptides in clinical trials.
Structurally, HDPs are mainly characterized by the presence of key charged residues (e.g., Arg and Lys), with a high proportion of hydrophobic residues (constituting 50% or more) and amphipathicity (7, 30). Optimization of HDP sequences to improve antimicrobial efficacy has been previously detailed (30, 31). Here, we aim to provide key strategies to improve the selectivity of HDPs toward microbial membranes through reported examples of model HDPs that were enhanced through rational design approaches (Table 1).
HDPs should fulfill key functional requirements to qualify for clinical use, including low toxicity, high antimicrobial activity, and good in vivo stability (24). These requirements are closely linked to their biochemical selectivity toward anionic and zwitterionic surfaces (24). The antimicrobial activity is attributed to a fine balance of hydrophobicity, cationic residues, amphipathicity, and structural conformation (e.g., α-helical, β-sheet, and cyclized) (32). On the other hand, the hydrophobic interaction between specific residues of HDPs (e.g., Leu, Ile, Val, Phe, Tyr, Trp) and zwitterionic phospholipids on host cell surfaces is responsible for its toxicity. For example, peptide derivatives of mastoparan (a key constituent of wasp venom) that were designed based on fixed five rules utilizing the quantitative structure-activity relationship (QSAR) approach showed potent antimicrobial efficacy against Bacillus subtilis (33). It was shown that the potency of these derivatives was mainly dependent on the presence of Trp, Lys, and His (33). Lata et al. (34) analyzed 486 HDPs from the antimicrobial peptide database (APD) for AA frequency in these sequences using the bioinformatic tools. Residues such as Gly, Arg, Lys, and Leu were shown to be commonly found in HDPs, whilst AAs such as Ser, Pro, Glu, and Asp were least common at both N- and C-terminus. A recent study from Hilpert group has demonstrated through in silico designed library of 3,000 de novo short peptides (9-mer in length) that the specific design characteristics of HDPs did not apply to short peptides (35). The peptide sequences that were grouped as “super active” based on their activity toward Pseudomonas aeruginosa were mainly composed of Lys, Arg, Trp, and Val/Ile/Leu (35). However, the activity of these super peptides toward host cells and in vivo stability was not yet reported.
Melittin, a key constituent of honey-bee venom, is a potent HDP with strong antimicrobial activity (36). However, its clinical use is largely limited by the high hemolytic activity (36). Blondelle et al. (37) studied the function of Trp in melittin activity through serial Trp substitution starting from N- to C-terminus. Substitution of Leu → Trp at 9th position was shown to decrease the hemolytic activity whereas substitution of Pro → Trp at 14th position improved the alpha-helical conformation and reduced the hemolytic effects compared to parent melittin peptide.
HDPs with Pro residues are widely known to display a disrupted helix conformation, which eventually affects their surface retention time and penetration into microbe cytoplasm (38, 39). A recent study using peptide analog (Anal 3, 19-AA long) from N-terminus of Helicobacter pylori ribosomal protein L1 has demonstrated that an insertion of Pro-hinge into Anal 3 (via Glu → Pro substitution at 9th position) significantly improves the peptide selectivity toward microbes with no effect on host cells (40, 41). This was attributed to the helix-hinge-helix conformation of Anal 3-Pro analog at the surface of bacteria allowing peptide penetration and DNA binding in the cytoplasm. This study suggested that rational insertion of Pro residue through SAR analysis could improve the biological membrane selectivity of microbicidal peptides. Proline-rich designed HDPs such as ARV-1502 (42), oncocin (43), and Bac-5 (44, 45) have shown significant efficacy against Gram-negative pathogens but not host cells membranes. Unlike cationic HDPs, proline-rich peptides kill bacteria through inhibition of protein synthesis (12, 45–47). Histidine-rich (48, 49), alanine-rich (50), and tryptophan-rich (51) short HDPs have also been developed. These were shown to be highly effective at acidic pH against a range of Gram-negative and Gram-positive bacteria (52).
Magainin-2 is 23 residues long antimicrobial peptide (AMP) isolated from frog skin, Xenopus laevis (53). Due to its non-hemolytic and broad-spectrum antimicrobial properties, magainin-2 was widely studied as a model peptide to understand the SAR of naturally occurring AMPs (54–58). Chen et al. (59) have demonstrated that the alpha-helical conformation of magainin-2 could be stabilized through Gly → Ala substitution (at both 13th and 18th position) and C-terminal amidation. This was shown to increase the antibacterial activity by 2-fold against a range of bacteria without modulating its safety against erythrocytes (59). Numerous groups have made attempts to improve the activity of magainin-2 against Gram-negative bacteria through residue substitution. It was demonstrated that the substitution of Phe → Trp in magainin-2 (F12W mutant) increased its activity against Gram-negative bacteria; however, this increased its selectivity toward erythrocytes, causing significant hemolysis (60). This could be attributed to the bulkiness of Trp compared to Phe and the presence of NH-group in Trp that is capable of forming hydrogen bonds with zwitterionic phospholipids (39, 61). Further modification through reduction of net charge of F12W mutant (Lys → Glu substitution at 10th position) was shown to reduce the hemolytic effect. However, this made the mutant magainin-2 less effective against Gram-negative bacteria (60). Extensive SAR studies from Zasloff's laboratory led to the development of MSI-78 (also known as pexiganan), a derivative of magainin-2, which showed improved safety/efficacy profile compared to parent magainin-2 sequence (62, 63). However, it failed in the phase III clinical trial for the treatment of infective diabetic foot ulcers. Further details on pexiganan clinical journey can be reviewed elsewhere (64–66).
Typically, natural HDPs display a net positive charge between +2 and +13. It has been widely demonstrated that modification of total net charge of synthetic HDPs through cationic residue substitution enhances electrostatic interaction between HDPs and lipopolysaccharide (LPS) (30, 60). However, this approach has shown to increase the toxicity of certain HDPs. For example, magainin-2 analogs with positive charge above +5 were shown to display hemolytic effects (rank order +6 > +5 > +4 > +3) (67). To overcome the inherent issues associated with peptide optimization, Mishra and Wang (68) adopted an ab initio design approach which involved utilization of novel database-filtering technology (DFT). This led to the development of a 13-AA long, leucine-rich, anti-MRSA peptide template—termed “DFTamP1” (68). A subsequent study demonstrated that DFT503, an optimized variant of DFTamP1, was shown to be safe and effective in in vivo killing of MRSA in a neutropenic mouse model. This anti-MRSA activity was attributed to its eight Leu residues and a single Lys at position 11 (net charge +1) (69). These studies suggested that lower cationic charge and high hydrophobicity is preferred for anti-MRSA synthetic peptides. This strategy could form the basis for the development of species-specific peptide-based therapy against multidrug resistant (MDR) pathogens.
LL-37 is a lone member of the cathelicidin family of HDPs reported in humans (70–72). It was widely studied due to its multi-functional abilities, including microbicidal (73), anti-cancer, immunomodulatory (74), chemotactic (75), and wound-healing properties (76). Numerous groups have exploited the structure of LL-37 to design a range of synthetic antimicrobial analogs through residue substitution (77, 78). FK-13 (residues 17-29 of LL-37) was identified as a core antimicrobial and anti-cancer domain using nuclear magnetic resonance (NMR) technique (79). Subsequently, the deletion of Phe at 17th position led to the development of KR-12, which showed potent antimicrobial efficacy equivalent to LL-37 and FK-13 against Escherichia coli, but devoid of toxic activity against host cells (80). KR-12 and KE-18 analogs were recently shown to possess anti-Candida and anti-Staphylococcal properties (81). Specifically, KE-18 showed anti-biofilm activity even at sub-killing concentration against yeast and bacteria (81). Further variants of KR-12 were also reported and the less cationic analogs, a5 and a6, were shown to possess potent immunomodulatory, antibiofilm, antimicrobial, and osteogenic properties (82–85). Variants of LL-23, corresponding to 23 N-terminal residues of LL-37, were generated through substitution of Ser → Ala and Ser → Val at the 9th position. LL-23V9 peptide was shown to display increased antimicrobial and immunosuppressive activities compared to LL-23 and parent LL-37 (86). Wang et al. (77), Mishra and Wang (87) have recently demonstrated that titanium surface immobilized FK-16 (a short variant of LL-37) is highly antimicrobial against ESKAPE pathogens. Our group has recently demonstrated that FK-16 could be used for repurposing conventional antibiotics such as vancomycin as a strategy to counter antimicrobial resistance (88). Further improvement of FK-16 by Narayana et al. (89) have also led to the development of GF-17, 17BIPHE2, and other related variants of superior efficacy and safety compared to LL-37. Nell et al. (90) designed a range of short peptides through residue substitution based on the LL-37 sequence for neutralization of lipopolysaccharide (LPS) and lipoteichoic acid (LTA). P60.4, a 24-AA derivative, was shown to possess similar LPS/LTA neutralization ability and antimicrobial effects compared to LL-37, but with negligible in vivo toxicity toward audible canal, skin, and eyes. This peptide was subsequently termed as OP-145 and was proven to be safe and efficacious in the treatment of chronic otitis media in phase I/II clinical trials (91). However, the activity of OP-145 was recently shown to be reduced in human plasma (92). Subsequent modification led to the development of synthetic antimicrobial and antibiofilm peptides (SAAPs) such as SAAP-145, −148, and −276, which showed potent anti-biofilm activity against a range of MDR pathogens (92).
HDPs are essentially a group of small bioactive molecules made of different combinations of 20 naturally occurring AAs. The nearly infinite chemical space (20n) and varying physicochemical properties account for the vast structural and functional diversities of naturally occurring HDPs (8, 9). However, susceptibility to host cell interaction (e.g., human erythrocytes, albumin, etc.) (93, 94) and proteolytic degradation from the host and bacterial proteases (e.g., human proteases in serum, staphylococcus aureolysin, pseudomonas elastase, etc.) (95–98) remains one of the key impediments in translating HDP-based treatment to clinical therapeutics. For instance, the anti-staphylococcal activity of cathelicidin (LL-37)—one of the most widely studied human HDPs—was shown to be inhibited by the proteases produced by Staphylococcus aureus, namely the aureolysin (a metalloproteinase) and V8 protease (glutamylendopeptidase), via cleavage and hydrolysis of the intramolecular peptide bonds (96).
To overcome this barrier, incorporation of unnatural or non-proteinogenic AAs has been employed to increase the proteolytic stability and/or antimicrobial efficacy of HDPs. It is known that the antimicrobial efficacy of HDPs is greatly influenced by the cationicity (99). To preserve the cationicity and thence efficacy, researchers have attempted to optimize the HDPs by replacing the cationic residues (e.g., lysine) with its analogs such as ornithine, 2,4-diamino-butyric acid (DAB), and 2,3-diamino-propionic acid (DAP), which have three, two, and one methylene (CH2) groups in the side chain, respectively (100, 101). Using Trp-rich peptides as the design template, Arias et al. (101) reported a 4-fold length-dependent increase in the antibacterial activity against E. coli when the side chain of lysine was shortened from 4-carbon (lysine) to 1-carbon (DAP). Such effect was likely attributed to an increase in membrane permeabilization based on calcein leakage study. In addition, a substantial improvement in the stability against trypsin was observed when the side chain of arginine or lysine was shortened (101). Oliva et al. (102) investigated the potential role of integrating unnatural AAs within the 9-residue synthetic HDPs and discovered that unnatural AAs such as 2-naphthyl-L-alanine (an aromatic residue) and S-tert-butylthio-L-cysteine residues enhanced the antimicrobial efficacy and proteolytic stability in 10% serum for 1 and 16 h (to a lesser extent). In addition, incorporation of unnatural AAs dipeptides (tetrahydroisoquionolinecarboxylic acid-octahydroindolecarboxylic acid; or Tic-Oic) within magainin analogs has been shown to induce an amphipathic and loose alpha-helical structure with enhanced antimicrobial potency and selectivity against Gram-positive, Gram-negative and mycobacterium (103).
Unnatural AAs have been successfully utilized for improving the efficacy and stability of various peptidomimetics (104). For example, Saralasin, a partial angiotensin II receptor agonist, was developed by incorporation of sarcosine (an unnatural AA) at a key position in angiotensin II molecule (105). This provided resistance against aminopeptidases and improved bioactivity. Carbetocin, a cyclic 8-AA derivative of oxytocin is currently used for the treatment of post-partum hemorrhage, was developed through incorporation of unnatural AAs such as methyltyrosine which improved its metabolic stability and overall therapeutic benefits (106).
It is widely known that a cocktail of HDPs are produced at the tissue sites in response to infection (107, 108). This natural synergism between HDPs was shown to be beneficial to the host, providing the first line of defense against pathogens. This was very well-exploited through in vitro and in vivo studies, which proved that the combination of two HDPs produces strong activity against bacteria (107, 108). However, this was not deemed as a cost-effective approach and the issue of host toxicity remains unresolved. Hybridization strategy was shown to circumvent these known issues, which involves the combination of key residues from two to three HDPs of different mechanisms of actions into a single sequence (109–111). In 1989, Boman et al. (112) elegantly showed that a hybrid of cecropin-A (1-13) and melittin (1-13) was highly bactericidal and less toxic toward host cells compared to parent cecropin-A and melittin. Subsequent modifications led to the development of numerous short hybrids of cecropin-A and melittin (15–18 residues in length), which showed similar activity as the first-generation hybrids (113, 114). Chimeras of cecropin-A (CA) and magainin-2 (MA) were also developed that exhibited potent antibacterial and antitumor activities. Insertion of hydrophobic residues through residue substitution in the hinge region (at 16th position) of CA(1-8)-MA(1-12) hybrid was shown to improve its antibacterial and antitumor activity with no hemolytic effects (115). A recent study has demonstrated that substitution of key residues in CA(1-8)-MA(1-12), specifically Phe5Lys, Lys7His, Phe13His, Leu14Phe, and His17Leu, could stabilize the alpha-helical conformation, resulting in improved LPS binding affinity, increased bactericidal activity against clinical Gram-negative isolates, and low cytotoxicity (116). Hybrids of human-derived and animal-derived HDPs were also developed to comprise the membrane-lytic and immunomodulatory properties of cationic HDPs. For example, hybrids of cecropin-A (1-8)-LL37(17-20) (117), melittin(1-13)-LL37(17-30) (118), and BMAP27(9-20)-LL37(17-29) (119) were shown to be highly bactericidal and improved the efficacy of conventional antibiotics against a variety of bacteria. Another study involving “triple hybrid” of cecropin-A, melittin, and LL-37 showed that this approach could significantly enhance the bactericidal against a range of Gram-negative and Gram-positive organisms (120). Similarly, Dutta et al. (121) reported good in vitro and in vivo efficacies of an antimicrobial contact lens coated with melimine (derived from melittin and protamine) for treating infectious keratitis in a rabbit model. These studies have clearly indicated that an optimized rational design approach could enable development of chimeras with improved biological selectivity.
In addition to overcoming the issue of host toxicity (as described above), the hybrid strategy has also led to the development of numerous species-specific and targeted bactericidal peptides to prevent damage to useful microbiome (122–124). Kim et al. (122) have developed a targeted chimeric peptide for the treatment of P. aeruginosa infection. Through phage-display library screening, they identified an outer-membrane porin F (OprF) binding peptide motif, termed PA2. Hybridization of this tag sequence to a membrane-lytic short peptide, GNU-7, was shown to improve the antimicrobial efficacy of parent GNU-7 by 16-fold toward P. aeruginosa both in in vitro and in vivo model systems. LPS-targeting GNU-7 variants were also developed through hybridization with lactoferrin (28-34), BPI (84-99), and de novo sequence (125). Chimeric bactericidal peptides targeted to E. faecalis (123) and S. mutans (124) were also developed based on the species-specific pheromones. This approach was proven to be highly targeted and would prevent the damage to commensal microbes.
Depending on the geometric arrangement, all naturally occurring AAs (except for glycine) can exist as stereoisomers, either in L- or D-form, albeit only the L-configuration can be utilized by cells (126). That said, there is emerging evidence showing that most organisms are able to produce D-AAs, primarily through spontaneous racemization of L-AAs or post-translational enzymatic modification (127). In addition, D-AAs such as D-alanine and D-glutamic acid are found in peptidoglycan, which is a key component of the cell wall of Gram-positive bacteria. These D-AAs have been shown to increase resistance to host proteases that usually cleave the peptide bonds between L-AAs, thereby maintaining their virulence (126).
Capitalizing on the evolutionarily advantageous strategy equipped by microbes, L-to-D isomerization has been utilized to enhance the proteolytic stability of HDPs against a range of host and microbes' proteases (128–133). L-to-D isomerization can be utilized to either modify specifically one or several L-AAs (131, 134), or the entire sequence of a L-form HDP (130–132). Carmona et al. (130) demonstrated that L-to-D isomerization of Panidin-2 (D-Pin2) improved the cell selectivity (i.e., reduced hemolysis) and proteolytic stability in human serum, elastase, and trypsin, while maintaining the antimicrobial activity against a range of Gram-positive and Gram-negative bacteria. In a similar vein, Jia et al. (131) reported an improved stability of D-AA derivative of polybia-CP (which was originally derived from the venom of social wasp Polybia paulista), in chymotrypsin and trypsin for 1 and 6 h and reduced hemolytic activity (D-lysine derivative). In addition to the beneficial effect of proteolytic stability and/or cell selectivity, the D-form AAs may enhance the antimicrobial efficacy of HDPs. For example, the D-form KLKLLLLLKLK-NH2 (derived from sapesin B) was shown to exhibit increased antimicrobial efficacy against S. aureus (due to increased interaction with the peptidoglycan), E. coli and Candida albicans when compared to the L-form (132). However, the enhanced antimicrobial efficacy was not observed in other tested D-forms of HDPs such as mastoparan M and temporin A, suggesting that the D-isomerization effect is sequence-dependent (132).
L-to-D isomerization has also been shown to confer unique changes to the peptide-folding and secondary structure of HDPs (128, 131, 133). Based on circular dichroism analysis, the D-form derivatives of naturally occurring alpha-helical HDPs typically exhibit a left-hand alpha-helical spectrum (instead of a right-hand spectrum) whereas partial D-isomerization of HDPs may result in some degree of loss of alpha-helicity, depending on the position and number of D-AAs being introduced (128, 131, 133). Such changes in the secondary structure are likely accountable for the reduced host toxicity and improved cell selectivity in some HDPs (131).
This strategy was found to be successful in the development of daptomycin, an antibacterial cyclic lipopeptide, which was approved by the US Food and Drug Administration (FDA) in 2003 for the treatment of skin and systemic Gram-positive infections (104, 135). Structurally, daptomycin is comprised of 13 residues including D-alanine and D-serine (134). In addition, it also contains non-canonical amino acids such as ornithine, L-kynurenine, and L-3-methylglutamic acid (134).
A range of N- and C-terminal modification strategies have been proposed to enhance the antimicrobial efficacy and/or cell selectivity of natural and synthetic HDPs (102, 136, 137). Amongst all, N-terminal acetylation (CH3CO-) and C-terminal amidation (-NH2) are the two most commonly attempted strategies (102). N-acetylation is a common protein modification observed in eukaryotic and prokaryotic cells (138). By neutralizing the positive charge (NH) at the N-terminal, N-acetylation can result in a range of irreversible changes to the protein properties, including the folding, stability, and protein-protein interactions (138). Saikia et al. (139) examined the antimicrobial efficacy and salt sensitivity of E. coli-derived MreB (a bacterial cytoskeleton protein found in non-spherical cells) and its N-acetylated analogs and found that N-acetylated W-MreB1−9 demonstrated a higher antimicrobial efficacy (in salt) compared to W-MreB1−9. However, N-acetylation may result in a decrease in antimicrobial efficacy of certain synthetic HDPs due to a reduction in the overall cationicity (139, 140), suggesting that the benefit of this modification strategy is only selective for certain HDPs.
On the other hand, C-amidation is a common post-translational modification that is widely observed in nature, including the natural synthesis of HDPs (137). C-amidation has been shown to improve the antimicrobial efficacy of certain HDPs, including aurein (141), melittin (142), modelin-5 (143), anoplin (144), and esculentin-1 (145), amongst others. The enhanced antimicrobial efficacy of these HDPs is likely ascribed to the increased alpha-helix stability at the peptide-membrane interfaces, enabling a greater membrane disruption and pore formation (141–144). In addition, Oliva et al. (102) demonstrated that simultaneous N-acetylation and C-amidation enhanced the proteolytic stability of HDP derived from human apolipoprotein B by more than 4-fold when exposed to fetal bovine serum 10% for 1 h. Similarly, the proteolytic stability of tachyplesin I (a beta-hairpin HDP from the horseshoe crab, Tachypleus tridentatus) in fresh human serum was significantly enhanced using the similar N-acetylation and C-amidation strategy (146).
Other N- or C-terminal modification strategies have also been described in the literature, including N-methylation of certain cyclic HDPs to enhance the antimicrobial efficacy (136), introduction of 6-aminocaproic acid at the N- and C-terminals to protect HDPs from the action of exopeptidases (102, 147), and pegylation of the C-terminus of M33, a branched peptide, to increase the resistance against P. aeruginosa elastase (148).
Cyclization is a common phenomenon observed in natural HDPs that can exist in three main forms: (a) sidechain to sidechain; (b) backbone to backbone; and (c) sidechain to backbone (137). It has been shown to demonstrate several favorable biological properties, including enhanced antimicrobial efficacy, stability against proteases (due to conformational rigidity), enhanced cell selectivity, and reduced host toxicity (137, 149), rendering it an attractive strategy for translating HDPs from bench to bedside. Some of the notable examples of cyclic glyco- or lipopeptides that are already in clinical use include vancomycin, daptomycin, and colistin/polymyxin, which are commonly used as last resorts for combatting MDR bacteria, albeit their widespread use are hindered by the inherent toxicity and emergence of AMR (149–151).
In view of the structural stability, Dathe et al. (152) were able to create a series of short cyclic hexapeptides (based on AcRRWWRF-NH2) with enhanced antimicrobial efficacy (up to >16-fold increase) against Bacillus subtilis and E. coli compared to the linear form, though the hemolytic activity was increased by 3-fold (152). It was also found that the antimicrobial activities of those small Arg/Trp-rich cyclic peptides were influenced by the self-assembling behavior of peptides at the bacterial membrane instead of their hydrophobic surface area, amphiphilicity, and ring size (153). In addition, a number of small cyclic D,L-alpha-peptides (with six or eight alternating D- and L-form residues) have also demonstrated strong antimicrobial efficacy against Gram-positive and/or Gram-negative bacteria via self-assembly on the bacterial membranes as organic nanotubules, which could increase membrane permeability and disrupt transmembrane ion potentials with resultant cell lysis (154, 155). Furthermore, molecular dynamic simulations and biophysical assays have provided further supportive evidence that cyclic peptides are able to bind to negatively charged membrane more strongly than the linear peptides and adopt a beta-sheet structure at the membrane surface (156).
Another form of cyclization that is found abundantly in natural HDPs, mainly in defensins, is the disulfide intramolecular cross-link between cysteine residues, which has been shown to enhance proteolytic stability (157–160). Inspired by the nature, Scudiero et al. (160) engineered a 17-residue cyclic synthetic hybrid HDP, based on the internal hydrophobic domain of human-beta defensin (HBD)-1 and positively charged C-terminal of HBD-3 (RRKK residues), and demonstrated good antimicrobial efficacy against Gram-positive and Gram-negative bacteria and herpes simplex virus, with low toxicity and good proteolytic stability. Similarly, Mwangi et al. (161) successfully developed a cyclic HDP-based molecule called ZY4 by introducing a disulfide bond to a derivative of cathelicidin-BF, which is an antimicrobial peptide derived from the snake venom of Bungarus fasciatus. This molecule was shown to exhibit significant in vivo antimicrobial efficacy against MDR P. aeruginosa and A. baumannii with high stability in mice lung infection and septicemia models (161).
Nanotechnology is a rapidly growing field in biotechnology that involves characterization, manipulation and synthesis of materials that are in nanoscales (or one billionth of meter; 10−9 m) (162). NPs, with sizes ranging from 1 to 100 nm, can exist in many forms, including lipid-based, metal-based, carbon-based, ceramics, semiconductor, and polymeric NPs (163). It has increasingly been applied in the field of antimicrobials, either employed as antimicrobial agents or nano-carriers for drug/peptide delivery in view of their enhanced protection against extracellular degradation, improved bioavailability, and cell selectivity (164–167). Recently, Biswaro et al. (166) have provided an excellent review on the role of nanotechnology in delivering HDPs. To avoid any significant overlap, this section aims to only recapitulate the fundamental principles of NPs and provide some notable examples regarding the potential values of incorporating NPs with HDPs.
In principle, there are two types of nano-delivery systems: (a) passive delivery where the intended drugs/peptides are encapsulated within the nanocarriers through hydrophobic interaction without any surface modification; and (b) active delivery where the drugs/peptides are directly conjugated with the nanocarriers with surface modification with ligands or other moieties to facilitate delivery to the targeted site (168). Among all, LL-37 and its mimics are some of the most commonly explored HDPs that have been incorporated with different types of NPs, including polymeric NPs [e.g., poly lactic-co-glycolic acid (PLGA)] (169), gold NP (170, 171), and magnetic NPs (172).
PLGA is a FDA-approved biodegradable and biocompatible polymer that has demonstrated promising potential as a drug delivery carrier (173). Chereddy et al. (169) described using PLGA as a nanoparticle carrier for delivering a sustained release of LL-37 treatment. Compared to PLGA or LL-37 alone, PLGA-LL37 nanoparticles were reported to expedite the wound healing process with significantly higher collagen deposition, re-epithelialization and neovascularization (169). It also demonstrated better antimicrobial activity against E. coli compared to PGLA alone, though the efficacy was lower than LL-37 alone (169). Cruz et al. (174) similarly reported that the encapsulated form of GIBIM-P5S9K peptide within PLGA or polylactic acid exhibited around 20 times stronger antimicrobial efficacy against methicillin-resistant S. aureus (MRSA), P. aeruginosa, and E. coli when compared to the free peptide.
In addition, gold NPs have been increasingly applied in the field of HDPs (175). Comune et al. (171) demonstrated that LL-37 conjugated with gold NPs (via an additional cysteine residue at the C-terminus of LL-37) demonstrated superior in vitro and in vivo wound healing properties compared to LL-37 alone. This was attributed to the prolonged phosphorylation of epidermal growth factor receptor (EGFR) and extracellular-signal-regulated kinase (ERK)1/2, which increased the migration of keratinocytes. Gold NPs have also been used as nanocarriers for other HDPs such as Esc(1-21), a derivative of a frog skin HDP called esculentin-1a (176). Compared to Esc(1-21), it was found that the conjugated form of Esc(1-21) with gold NPs via a poly-ethylene glycol linker improved the antimicrobial efficacy against P. aeruginosa by around 15-fold, with increased resistance to proteolytic degradation (176). Certain peptides have also demonstrated self-assembling ability as a nanocarrier for drug delivery (166), though this is beyond the scope of our review.
AI serves as one of the major breakthroughs in the mankind's history. Long been deployed in the automobile and technology industries, AI has only started gaining traction in the field of science and medicine, owing to the advancement in computer power, availability of big data, publicly available neural networks, and improvement in AI algorithms using machine learning and deep learning (177–180). In view of the infinite chemical space and complex SAR of natural and synthetic HDPs, AI serves as an attractive solution to identify and predict novel peptide sequences with potentially good antimicrobial efficacy (181–184).
To date, a number of machine learning algorithms such as artificial neural network (ANN) (34, 182), support vector machine (SVM)-based classifier (34, 183, 184), quantitative matrices (34), and fuzzy K-nearest neighbor (FKNN) (185) have been developed to search for the ideal synthetic HDPs. Cherkasov et al. (182) trained an atomic-based QSAR model using ANN and inductive chemical descriptors based on two large 9-mer peptide libraries. The model was then tested against 200 peptides that were chosen from a virtual library of 100,000 random 9-mer peptides. The model not only successfully predicted the antimicrobial efficacy of the synthetic peptides but also identified potent peptide candidates (HHC-10 and HHC-36) which were highly active against a range of Gram-positive and Gram-negative superbugs, with low risk of toxicity (182).
On the other hand, Lee et al. (183) developed a SVM-based classifier coupled with Pareto-optimization (186) to deduce the functional and structural similarities of alpha-helical HDPs. By employing antimicrobial assays and small-angle X-ray scattering, it was found that the SVM distance to hyperplane σ correlated strongly with the ability of HDP in generating a negative Gaussian curvature (NGC), which is commonly responsible for the membrane disruption mechanism of HDP (183). Subsequently, Yount et al. (184) were able to identify a unifying physicochemical characteristic of alpha-helical HDPs in a 3-dimensional space, termed the alpha-core signature, using knowledge-based annotation and pattern recognition analysis of bioinformatics databases. The antimicrobial efficacy of this alpha-core signature (i.e., the ability to induce NGC) was further validated with the previously developed SVM-based classifier (184).
Since the serendipitous discovery of HDPs in nature during early 1980s, immense research effort have been invested in realizing the therapeutic potentials of HDPs in clinic (7). In this review, we provided a comprehensive overview on eight key strategies (with examples) in improving and translating the therapeutic potentials of HDP-based treatment from bench to bedside. Moreover, we summarized HDP derivatives that are currently in the development pipeline.
So far, a number of HDP-based treatment have entered advanced (phase II/III) clinical trials (Table 1) but none had reached the market due to regulatory hurdles (27, 187). Nevertheless, many lessons have been learnt from past experience. One of the notable examples (as described above) is MSI-78 or pexiganan, a magainin-derived HDP, which did not obtain the FDA approval after failing to demonstrate any superiority to the normal standard wound care with oral ofloxacin for infected diabetic foot ulcers in two phase 3 trials (188). Although the discouraging results have painted a gloomy outlook for HDP-based treatment at that time, a closer look at the development pathway of MSI-78 has shed light on the plausible reasons accounting for the failure. First, although the molecule demonstrated a broad-spectrum antimicrobial activity against 3,109 clinical isolates (with an average MIC90 of 32 μg/ml or less) (63), the activity remained considerably weaker than the conventional antibiotics (62). Second, peptide-based treatment including MSI-78 are more susceptible to proteolytic degradation when compared to the conventional small molecule antibiotics. That means HDP-based treatment need to be administered in a higher concentration to achieve the intended in vivo efficacy, which could inevitably lead to increased host toxicity. In addition, MSI-78 exhibits several favorable properties over conventional antibiotics, including low risk of developing AMR and good activity against MDR isolates (63), but the phase 3 clinical trials of MSI-78 were conducted for mild infective diabetic foot ulcers which did not fully capitalize on these strengths. This highlights the importance of setting the right research question during the development of HDP-based treatment.
Learning from the previous (unsuccessful) experience, a plethora of strategies have been proposed and attempted to overcome the inherent limitations of HDP-based treatment, with enhanced antimicrobial efficacy, proteolytic stability, and cell selectivity (for microbial cells). Although the design of ideal HDPs is not governed by a single overarching rule (7), it is apparent from the literature that peptide design guided by the fundamental principles and systematic SAR analysis is able to yield potential efficacious peptide candidates with desired properties (189). In fact, de novo designed synthetic peptides were successfully developed purely based on Arg and Trp with 50% hydrophobicity and demonstrated significantly antimicrobial and anti-biofilm efficacies against MDR staphylococci (190).
Based on the literature and our experience, we propose a potential strategy in streamlining the drug discovery and development pathway of HDP-based treatment, starting from designing new HDP treatment to conducting well-designed pre-clinical studies (Figure 1). So far, more than 3,000 naturally occurring and synthetic HDPs (with reported antimicrobial and/or non-antimicrobial functions) have been discovered (8, 9); therefore, it would be a good strategy to use an existing template with proven effect as a starting point for designing a new HDP-based treatment. Alternatively, employing artificial intelligence technology in predicting potentially efficacious molecules could be utilized. Once a starting template is identified (either a linear peptide or a cyclic peptide), systematic SAR analysis of the sequence via rational substitution of specific residues is required to optimize the antimicrobial efficacy and cell selectivity toward microbial cells. If hybridization strategy is used, functioning sequence of each single peptide should be first determined before being hybridized. This is then followed by further SAR analysis to determine the optimal sequence.
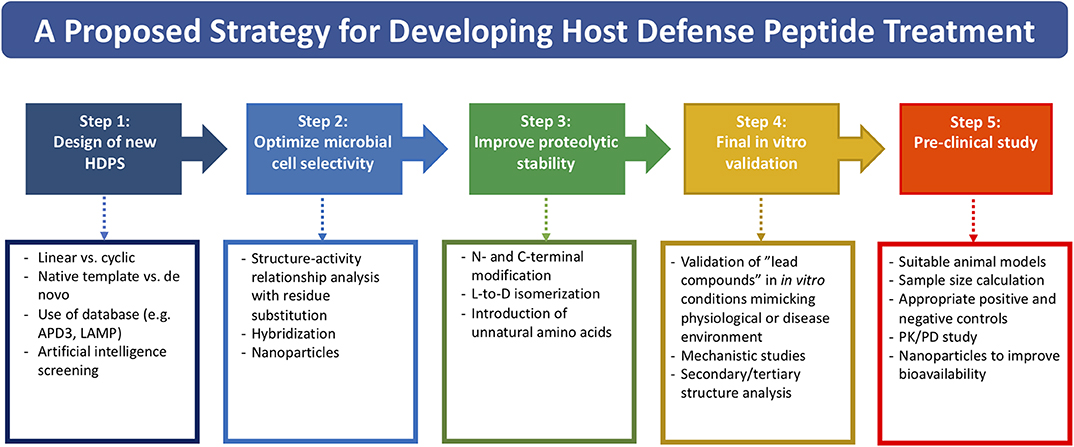
Figure 1. A potential strategy for streamlining the drug discovery and developmental pathway of HDP-based treatment, covering from designing of new HDP treatment to conducting well-designed pre-clinical studies.
Once the efficacy and safety are optimized, the next hurdle is to overcome the issue of proteolytic degradation, which could be achieved through the strategies (either singularly or in combination) mentioned in Table 2 and Figure 1. However, it is noteworthy to mention that the beneficial effects of these modifications may be unique to specific HDPs. In addition, antimicrobial efficacy and/or microbial cell selectivity of the HDPs may also be affected during the modification. Subsequently, the potential lead compound should be validated in in vitro conditions mimicking the physiological or host disease environment. For instance, when designing HDP-based treatment for corneal infection, the designed HDPs should be tested in tear fluid or in salt of physiological concentration, which are known to affect the efficacy and stability of HDPs. The findings enable a better prediction of the in vivo results and help minimize the unnecessary use of animals (191). Finally, well-designed pre-clinical studies need to be performed with appropriate sample size calculation and positive/negative controls, which will increase the success rate of clinical trials. For example, efficacy of the designed HDP needs to be compared with antibiotic treatment that reflects the current clinical practice for the disease of interest. Otherwise, subsequent clinical trials are likely to fail and necessary regulatory approval will not be obtained.
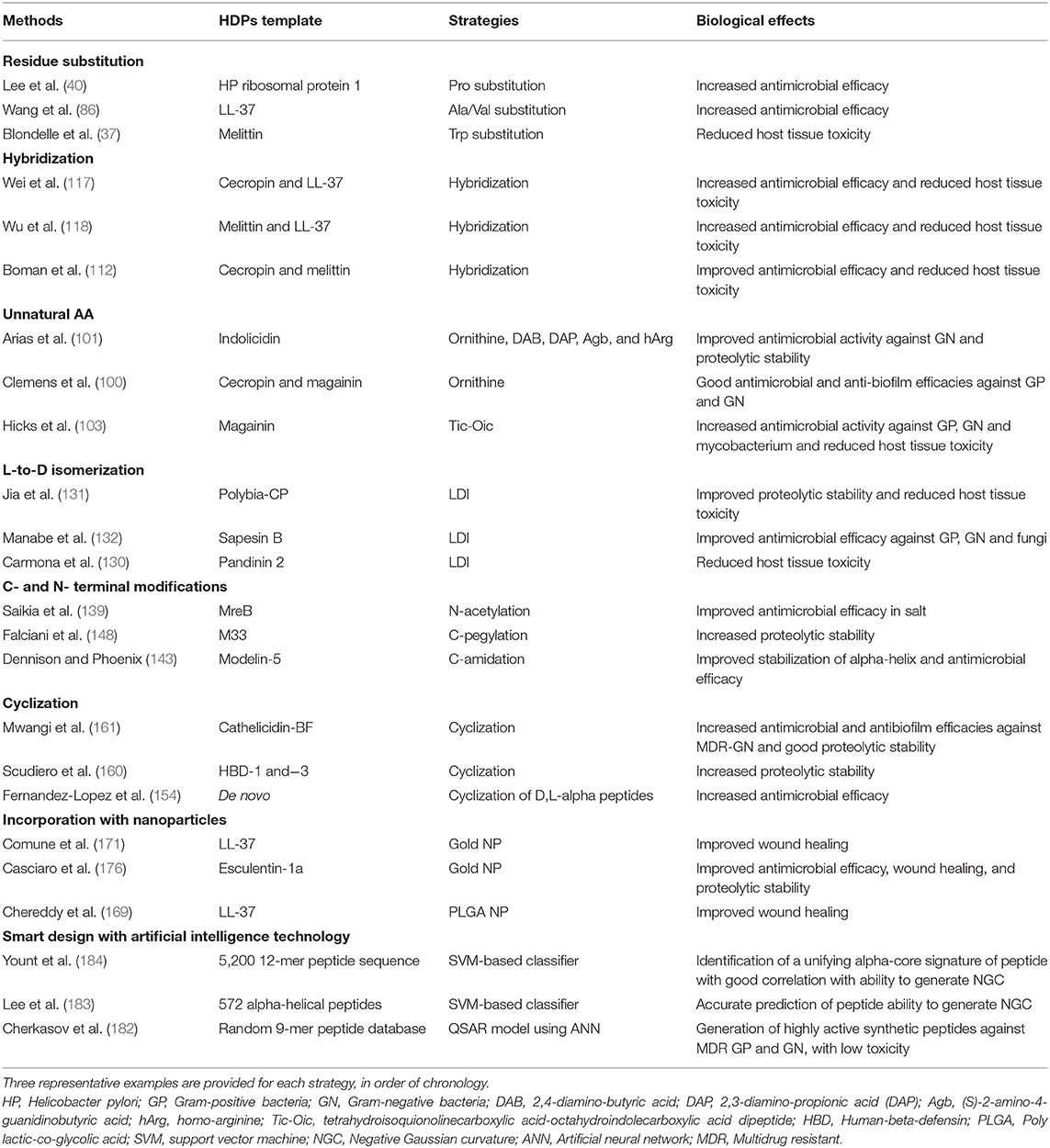
Table 2. Summary of different strategies in translating the therapeutic potentials of host defense peptides (HDPs).
Ideally, it is best to optimize the previous steps before progressing to the next step. For instance, validating the potential lead compound in in vitro conditions mimicking physiological environment is crucial before proceeding to pre-clinical studies. Modification strategies proposed in each step may also be applicable to other steps. For example, introduction of unnatural AAs primarily improves the proteolytic stability but may also enhance the antimicrobial efficacy (101), and incorporation of HDPs with nanoparticles may reduce host toxicity as well as improve bioavailability. In addition, several strategies may be employed in combination to achieve the intended therapeutic effect and stability.
There are also increasing reports examining and exploiting the strategy of using peptide-antibiotic combination to counter AMR, increase the lifespan of conventional antibiotics and HDPs, as well as to reduce the undesired toxicity to host tissues (88, 192–194). The synergistic effect of peptide-antibiotic combination treatment is likely attributed to the different underlying mechanism of action whereby the membrane perturbation effect of peptides facilitates the passive diffusion of conventional antibiotics into the cells for intracellular targeting action (192).
Although HDP-based treatment has long been envisioned as a novel solution to tackle AMR, emerging evidence are suggesting that HDPs could also develop AMR, albeit with a lower risk than the conventional antibiotics (7, 195). Spohn et al. (196) have highlighted the influence of physicochemical characteristics of HDPs, including the proportion of polar AAs, cationicity, and hydrophobicity, on the risk of developing HDP-related AMR, thereby providing invaluable insights into the design of future HDPs. Reassuringly, cross resistance between HDPs was found to be limited to those with similar modes of action, underscoring the importance and necessity of having HDPs with different antimicrobial mechanisms within the therapeutic armamentarium of antimicrobials (197).
With the advancement in peptide design strategy, synthesis techniques and AI technology, it is hopeful that clinical deployment of HDP-based treatment for a range of diseases will soon become a reality. However, further studies will need to be conducted to decipher the mechanism of HDP-related AMR in order to prepare for the potentially self-perpetuating vicious cycle of AMR in the future.
Electronic databases, including MEDLINE (January 1950–March 2020) and EMBASE (January 1980–March 2020), were searched for relevant articles related to host defense peptides. Keywords such as “host defense peptide,” “antimicrobial peptide,” “hybrid,” “cyclization,” “unnatural amino acid,” “D-amino acid,” “nanotechnology,” “nanoparticles,” “artificial intelligence,” and “machine learning” were used. Only articles published in English were included. Bibliographies of included articles were manually screened to identify further relevant studies. The final search was updated on 31 March 2020.
DT and IM: conception and design of work, data collection and interpretation, drafting the manuscript, and final approval of the work. RB and HD: data interpretation, critical revision, and final approval of the work. RL: conception and design of work, data interpretation, critical revision, and final approval of the work.
DT acknowledged funding support from the Medical Research Council/Fight for Sight (FFS) Clinical Research Fellowship (MR/T001674/1), the FFS/John Lee, Royal College of Ophthalmologists Primer Fellowship (24CO4), and the University of Nottingham International Research Collaboration Fund (A2RRG1). IM acknowledged funding support from the Medical Research Council—Confidence in Concept Scheme (MRC-CIC_2019-028).
HD receives travel honorarium from Thea, Dompe, and Santen and has shares in GlaxoSmithKline and NuVision Biotherapies.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. (2010) 74:417–33. doi: 10.1128/MMBR.00016-10
2. O'Neill J. Tackling drug-resistant infections globally: final report and recommendations. Rev Antimicrob Resist. (2016). Available online at: https://amr-review.org/sites/default/files/160525_Final%20paper_with%20cover.pdf
3. Holmes AH, Moore LS, Sundsfjord A, Steinbakk M, Regmi S, Karkey A, et al. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet. (2016) 387:176–87. doi: 10.1016/S0140-6736(15)00473-0
4. Johnson AP, Ashiru-Oredope D, Beech E. Antibiotic stewardship initiatives as part of the UK 5-year antimicrobial resistance strategy. Antibiotics. (2015) 4:467–79. doi: 10.3390/antibiotics4040467
5. Mahlapuu M, Hakansson J, Ringstad L, Bjorn C. Antimicrobial peptides: an emerging category of therapeutic agents. Front Cell Infect Microbiol. (2016) 6:194. doi: 10.3389/fcimb.2016.00194
6. Zasloff M. Antimicrobial peptides of multicellular organisms: my perspective. Adv Exp Med Biol. (2019) 1117:3–6. doi: 10.1007/978-981-13-3588-4_1
7. Haney EF, Straus SK, Hancock REW. Reassessing the host defense peptide landscape. Front Chem. (2019) 7:43. doi: 10.3389/fchem.2019.00043
8. Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucl Acids Res. (2016) 44(D1):D1087–93. doi: 10.1093/nar/gkv1278
9. Zhao X, Wu H, Lu H, Li G, Huang Q. LAMP: a database linking antimicrobial peptides. PLoS ONE. (2013) 8:e66557. doi: 10.1371/journal.pone.0066557
10. Mohammed I, Said DG, Dua HS. Human antimicrobial peptides in ocular surface defense. Prog Retin Eye Res. (2017) 61:1–22. doi: 10.1016/j.preteyeres.2017.03.004
11. Le CF, Fang CM, Sekaran SD. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob Agents Chemother. (2017) 61:AAC.02340-16. doi: 10.1128/AAC.02340-16
12. Cardoso MH, Meneguetti BT, Costa BO, Buccini DF, Oshiro KGN, Preza SLE, et al. Non-Lytic antibacterial peptides that translocate through bacterial membranes to act on intracellular targets. Int J Mol Sci. (2019) 20:4877. doi: 10.3390/ijms20194877
13. Kapoor G, Saigal S, Elongavan A. Action and resistance mechanisms of antibiotics: a guide for clinicians. J Anaesthesiol Clin Pharmacol. (2017) 33:300–5. doi: 10.4103/joacp.JOACP_349_15
14. Shahrour H, Ferrer-Espada R, Dandache I, Barcena-Varela S, Sanchez-Gomez S, Chokr A, et al. AMPs as anti-biofilm agents for human therapy and prophylaxis. Adv Exp Med Biol. (2019) 1117:257–79. doi: 10.1007/978-981-13-3588-4_14
15. Nijnik A, Hancock R. Host defence peptides: antimicrobial and immunomodulatory activity and potential applications for tackling antibiotic-resistant infections. Emerg Health Threats J. (2009) 2:e1. doi: 10.3402/ehtj.v2i0.7078
16. Mangoni ML, McDermott AM, Zasloff M. Antimicrobial peptides and wound healing: biological and therapeutic considerations. Exp Dermatol. (2016) 25:167–73. doi: 10.1111/exd.12929
17. Deslouches B, Di YP. Antimicrobial peptides with selective antitumor mechanisms: prospect for anticancer applications. Oncotarget. (2017) 8:46635–51. doi: 10.18632/oncotarget.16743
18. Brice DC, Diamond G. Antiviral activities of human host defense peptides. Curr Med Chem. (2020) 27:1420–43. doi: 10.2174/0929867326666190805151654
19. Ting DSJ, Settle C, Morgan SJ, Baylis O, Ghosh S. A 10-year analysis of microbiological profiles of microbial keratitis: the North East England study. Eye. (2018) 32:1416–7. doi: 10.1038/s41433-018-0085-4
20. Ung L, Bispo PJM, Shanbhag SS, Gilmore MS, Chodosh J. The persistent dilemma of microbial keratitis: global burden, diagnosis, and antimicrobial resistance. Surv Ophthalmol. (2019) 64:255–71. doi: 10.1016/j.survophthal.2018.12.003
21. Ting DSJ, Bignardi G, Koerner R, Irion LD, Johnson E, Morgan SJ, et al. Polymicrobial keratitis with cryptococcus curvatus, candida parapsilosis, and Stenotrophomonas maltophilia after penetrating keratoplasty: a rare case report with literature review. Eye Contact Lens. (2019) 45:e5–e10. doi: 10.1097/ICL.0000000000000517
22. Mookherjee N, Anderson MA, Haagsman HP, Davidson DJ. Antimicrobial host defence peptides: functions and clinical potential. Nat Rev Drug Discov. (2020) 19:311–32. doi: 10.1038/s41573-019-0058-8
23. Li J, Koh JJ, Liu S, Lakshminarayanan R, Verma CS, Beuerman RW. Membrane active antimicrobial peptides: translating mechanistic insights to design. Front Neurosci. (2017) 11:73. doi: 10.3389/fnins.2017.00073
24. Fjell CD, Hiss JA, Hancock RE, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov. (2011) 11:37–51. doi: 10.1038/nrd3591
25. Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. (2006) 24:1551–7. doi: 10.1038/nbt1267
26. Kmietowicz Z. Few novel antibiotics in the pipeline, WHO warns. BMJ. (2017) 358:j4339. doi: 10.1136/bmj.j4339
27. Boto A, Perez de la Lastra JM, Gonzalez CC. The road from host-defense peptides to a new generation of antimicrobial drugs. Molecules. (2018) 23:311. doi: 10.3390/molecules23020311
28. Nuti R, Goud NS, Saraswati AP, Alvala R, Alvala M. Antimicrobial peptides: a promising therapeutic strategy in tackling antimicrobial resistance. Curr Med Chem. (2017) 24:4303–14. doi: 10.2174/0929867324666170815102441
29. Afacan NJ, Yeung AT, Pena OM, Hancock RE. Therapeutic potential of host defense peptides in antibiotic-resistant infections. Curr Pharm Des. (2012) 18:807–19. doi: 10.2174/138161212799277617
30. Haney EF, Mansour SC, Hancock RE. Antimicrobial peptides: an introduction. Methods Mol Biol. (2017) 1548:3–22. doi: 10.1007/978-1-4939-6737-7_1
31. Haney EF, Hancock RE. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers. (2013) 100:572–83. doi: 10.1002/bip.22250
32. Raheem N, Straus SK. Mechanisms of action for antimicrobial peptides with antibacterial and antibiofilm functions. Front Microbiol. (2019) 10:2866. doi: 10.3389/fmicb.2019.02866
33. Avram S, Buiu C, Borcan F, Milac AL. More effective antimicrobial mastoparan derivatives, generated by 3D-QSAR-Almond and computational mutagenesis. Mol Biosyst. (2012) 8:587–94. doi: 10.1039/C1MB05297G
34. Lata S, Sharma BK, Raghava GP. Analysis and prediction of antibacterial peptides. BMC Bioinformatics. (2007) 8:263. doi: 10.1186/1471-2105-8-263
35. Mikut R, Ruden S, Reischl M, Breitling F, Volkmer R, Hilpert K. Improving short antimicrobial peptides despite elusive rules for activity. Biochim Biophys Acta. (2016) 1858:1024–33. doi: 10.1016/j.bbamem.2015.12.013
36. Memariani H, Memariani M, Shahidi-Dadras M, Nasiri S, Akhavan MM, Moravvej H. Melittin: from honeybees to superbugs. Appl Microbiol Biotechnol. (2019) 103:3265–76. doi: 10.1007/s00253-019-09698-y
37. Blondelle SE, Simpkins LR, Perez-Paya E, Houghten RA. Influence of tryptophan residues on melittin's hemolytic activity. Biochim Biophys Acta. (1993) 1202:331–6. doi: 10.1016/0167-4838(93)90024-L
38. O'Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. (1990) 250:646–51. doi: 10.1126/science.2237415
39. Creamer TP, Rose GD. Side-chain entropy opposes alpha-helix formation but rationalizes experimentally determined helix-forming propensities. Proc Natl Acad Sci USA. (1992) 89:5937–41. doi: 10.1073/pnas.89.13.5937
40. Lee JK, Gopal R, Park SC, Ko HS, Kim Y, Hahm KS, et al. A proline-hinge alters the characteristics of the amphipathic alpha-helical AMPs. PLoS ONE. (2013) 8:e67597. doi: 10.1371/journal.pone.0067597
41. Putsep K, Branden CI, Boman HG, Normark S. Antibacterial peptide from H. pylori. Nature. (1999) 398:671–2. doi: 10.1038/19439
42. Brakel A, Volke D, Kraus CN, Otvos L, Hoffmann R. Quantitation of a novel engineered anti-infective host defense peptide, ARV-1502: pharmacokinetic study of different doses in rats and dogs. Front Chem. (2019) 7:753. doi: 10.3389/fchem.2019.00753
43. Knappe D, Piantavigna S, Hansen A, Mechler A, Binas A, Nolte O, et al. Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): a novel antibacterial peptide optimized against gram-negative human pathogens. J Med Chem. (2010) 53:5240–7. doi: 10.1021/jm100378b
44. Mardirossian M, Sola R, Beckert B, Collis DWP, Di Stasi A, Armas F, et al. Proline-rich peptides with improved antimicrobial activity against E. coli, K. pneumoniae, and A. baumannii. ChemMedChem. (2019) 14:2025–33. doi: 10.1002/cmdc.201900465
45. Mardirossian M, Sola R, Degasperi M, Scocchi M. Search for shorter portions of the proline-rich antimicrobial peptide fragment Bac5(1-25) that retain antimicrobial activity by blocking protein synthesis. ChemMedChem. (2019) 14:343–8. doi: 10.1002/cmdc.201800734
46. Mardirossian M, Barriere Q, Timchenko T, Muller C, Pacor S, Mergaert P, et al. Fragments of the nonlytic proline-rich antimicrobial peptide Bac5 kill Escherichia coli cells by inhibiting protein synthesis. Antimicrob Agents Chemother. (2018) 62:e00534-18. doi: 10.1128/AAC.00534-18
47. Graf M, Mardirossian M, Nguyen F, Seefeldt AC, Guichard G, Scocchi M, et al. Proline-rich antimicrobial peptides targeting protein synthesis. Nat Prod Rep. (2017) 34:702–11. doi: 10.1039/C7NP00020K
48. Mason AJ, Gasnier C, Kichler A, Prevost G, Aunis D, Metz-Boutigue MH, et al. Enhanced membrane disruption and antibiotic action against pathogenic bacteria by designed histidine-rich peptides at acidic pH. Antimicrob Agents Chemother. (2006) 50:3305–11. doi: 10.1128/AAC.00490-06
49. Mason AJ, Moussaoui W, Abdelrahman T, Boukhari A, Bertani P, Marquette A, et al. Structural determinants of antimicrobial and antiplasmodial activity and selectivity in histidine-rich amphipathic cationic peptides. J Biol Chem. (2009) 284:119–33. doi: 10.1074/jbc.M806201200
50. Migliolo L, Silva ON, Silva PA, Costa MP, Costa CR, Nolasco DO, et al. Structural and functional characterization of a multifunctional alanine-rich peptide analogue from Pleuronectes americanus. PLoS ONE. (2012) 7:e47047. doi: 10.1371/journal.pone.0047047
51. Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Mietzner TA, Montelaro RC. Rational design of engineered cationic antimicrobial peptides consisting exclusively of arginine and tryptophan, and their activity against multidrug-resistant pathogens. Antimicrob Agents Chemother. (2013) 57:2511–21. doi: 10.1128/AAC.02218-12
52. Mishra AK, Choi J, Moon E, Baek KH. Tryptophan-rich and proline-rich antimicrobial peptides. Molecules. (2018) 23:815. doi: 10.3390/molecules23040815
53. Zasloff M. Magainins, a class of antimicrobial peptides from xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci USA. (1987) 84:5449–53. doi: 10.1073/pnas.84.15.5449
54. Zasloff M, Martin B, Chen HC. Antimicrobial activity of synthetic magainin peptides and several analogues. Proc Natl Acad Sci USA. (1988) 85:910–3. doi: 10.1073/pnas.85.3.910
55. Esmaili E, Shahlaei M. Analysis of the flexibility and stability of the structure of magainin in a bilayer, and in aqueous and nonaqueous solutions using molecular dynamics simulations. J Mol Model. (2015) 21:73. doi: 10.1007/s00894-015-2622-4
56. Nguyen KT, Le Clair SV, Ye S, Chen Z. Molecular interactions between magainin 2 and model membranes in situ. J Phys Chem B. (2009) 113:12358–63. doi: 10.1021/jp904154w
57. Westerhoff HV, Juretic D, Hendler RW, Zasloff M. Magainins and the disruption of membrane-linked free-energy transduction. Proc Natl Acad Sci USA. (1989) 86:6597–601. doi: 10.1073/pnas.86.17.6597
58. Maloy WL, Kari UP. Structure-activity studies on magainins and other host defense peptides. Biopolymers. (1995) 37:105–22. doi: 10.1002/bip.360370206
59. Chen HC, Brown JH, Morell JL, Huang CM. Synthetic magainin analogues with improved antimicrobial activity. FEBS Lett. (1988) 236:462–6. doi: 10.1016/0014-5793(88)80077-2
60. Matsuzaki K, Sugishita K, Harada M, Fujii N, Miyajima K. Interactions of an antimicrobial peptide, magainin 2, with outer and inner membranes of Gram-negative bacteria. Biochim Biophys Acta. (1997) 1327:119–30. doi: 10.1016/S0005-2736(97)00051-5
61. Manikandan K, Ramakumar S. The occurrence of C–H.O hydrogen bonds in alpha-helices and helix termini in globular proteins. Proteins. (2004) 56:768–81. doi: 10.1002/prot.20152
62. Gottler LM, Ramamoorthy A. Structure, membrane orientation, mechanism, and function of pexiganan–a highly potent antimicrobial peptide designed from magainin. Biochim Biophys Acta. (2009) 1788:1680–6. doi: 10.1016/j.bbamem.2008.10.009
63. Ge Y, MacDonald DL, Holroyd KJ, Thornsberry C, Wexler H, Zasloff M. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob Agents Chemother. (1999) 43:782–8. doi: 10.1128/AAC.43.4.782
64. Lamb HM, Wiseman LR. Pexiganan acetate. Drugs. (1998) 56:1047–54. doi: 10.2165/00003495-199856060-00011
65. Ramirez-Acuna JM, Cardenas-Cadena SA, Marquez-Salas PA, Garza-Veloz I, Perez-Favila A, Cid-Baez MA, et al. Diabetic foot ulcers: current advances in antimicrobial therapies and emerging treatments. Antibiotics. (2019) 8:193. doi: 10.3390/antibiotics8040193
66. Moore A. The big and small of drug discovery. Biotech versus pharma: advantages and drawbacks in drug development. EMBO Rep. (2003) 4:114–7. doi: 10.1038/sj.embor.embor748
67. Dathe M, Nikolenko H, Meyer J, Beyermann M, Bienert M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. (2001) 501:146–50. doi: 10.1016/S0014-5793(01)02648-5
68. Mishra B, Wang G. Ab initio design of potent anti-MRSA peptides based on database filtering technology. J Am Chem Soc. (2012) 134:12426–9. doi: 10.1021/ja305644e
69. Mishra B, Lakshmaiah Narayana J, Lushnikova T, Wang X, Wang G. Low cationicity is important for systemic in vivo efficacy of database-derived peptides against drug-resistant Gram-positive pathogens. Proc Natl Acad Sci USA. (2019) 116:13517–22. doi: 10.1073/pnas.1821410116
70. Scott MG, Davidson DJ, Gold MR, Bowdish D, Hancock RE. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J Immunol. (2002) 169:3883–91. doi: 10.4049/jimmunol.169.7.3883
71. Harder J, Bartels J, Christophers E, Schroder JM. A peptide antibiotic from human skin. Nature. (1997) 387:861. doi: 10.1038/43088
72. Cowland JB, Johnsen AH, Borregaard N. hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett. (1995) 368:173–6. doi: 10.1016/0014-5793(95)00634-L
73. Zanetti M, Gennaro R, Romeo D. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. (1995) 374:1–5. doi: 10.1016/0014-5793(95)01050-O
74. Bowdish DM, Davidson DJ, Scott MG, Hancock RE. Immunomodulatory activities of small host defense peptides. Antimicrob Agents Chemother. (2005) 49:1727–32. doi: 10.1128/AAC.49.5.1727-1732.2005
75. De Y, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, et al. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. (2000) 192:1069–74. doi: 10.1084/jem.192.7.1069
76. Ramos R, Silva JP, Rodrigues AC, Costa R, Guardao L, Schmitt F, et al. Wound healing activity of the human antimicrobial peptide LL37. Peptides. (2011) 32:1469–76. doi: 10.1016/j.peptides.2011.06.005
77. Wang G, Narayana JL, Mishra B, Zhang Y, Wang F, Wang C, et al. Design of antimicrobial peptides: progress made with human cathelicidin LL-37. Adv Exp Med Biol. (2019) 1117:215–40. doi: 10.1007/978-981-13-3588-4_12
78. Wang G, Mishra B, Epand RF, Epand RM. High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim Biophys Acta. (2014) 1838:2160–72. doi: 10.1016/j.bbamem.2014.01.016
79. Wang G. NMR of membrane-associated peptides and proteins. Curr Protein Pept Sci. (2008) 9:50–69. doi: 10.2174/138920308783565714
80. Wang G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J Biol Chem. (2008) 283:32637–43. doi: 10.1074/jbc.M805533200
81. Luo Y, McLean DT, Linden GJ, McAuley DF, McMullan R, Lundy FT. The naturally occurring host defense peptide, LL-37, and its truncated mimetics KE-18 and KR-12 have selected biocidal and antibiofilm activities against candida albicans, Staphylococcus aureus, and Escherichia coli in vitro. Front Microbiol. (2017) 8:544. doi: 10.3389/fmicb.2017.00544
82. Jacob B, Park IS, Bang JK, Shin SY. Short KR-12 analogs designed from human cathelicidin LL-37 possessing both antimicrobial and antiendotoxic activities without mammalian cell toxicity. J Pept Sci. (2013) 19:700–7. doi: 10.1002/psc.2552
83. Kim EY, Rajasekaran G, Shin SY. LL-37-derived short antimicrobial peptide KR-12-a5 and its d-amino acid substituted analogs with cell selectivity, anti-biofilm activity, synergistic effect with conventional antibiotics, and anti-inflammatory activity. Eur J Med Chem. (2017) 136:428–41. doi: 10.1016/j.ejmech.2017.05.028
84. Li H, Zhang S, Nie B, Long T, Qu X, Yue B. KR-12-a5 reverses adverse effects of lipopolysaccharides on HBMSC osteogenic differentiation by influencing BMP/Smad and P38 MAPK signaling pathways. Front Pharmacol. (2019) 10:639. doi: 10.3389/fphar.2019.00639
85. Fu L, Jin P, Hu Y, Lu H, Su L. KR12a6 promotes the osteogenic differentiation of human bone marrow mesenchymal stem cells via BMP/SMAD signaling. Mol Med Rep. (2020) 21:61–8. doi: 10.3892/mmr.2019.10843
86. Wang G, Elliott M, Cogen AL, Ezell EL, Gallo RL, Hancock RE. Structure, dynamics, and antimicrobial and immune modulatory activities of human LL-23 and its single-residue variants mutated on the basis of homologous primate cathelicidins. Biochemistry. (2012) 51:653–64. doi: 10.1021/bi2016266
87. Mishra B, Wang G. Titanium surfaces immobilized with the major antimicrobial fragment FK-16 of human cathelicidin LL-37 are potent against multiple antibiotic-resistant bacteria. Biofouling. (2017) 33:544–55. doi: 10.1080/08927014.2017.1332186
88. Mohammed I, Said DG, Nubile M, Mastropasqua L, Dua HS. Cathelicidin-derived synthetic peptide improves therapeutic potential of vancomycin against Pseudomonas aeruginosa. Front Microbiol. (2019) 10:2190. doi: 10.3389/fmicb.2019.02190
89. Narayana JL, Mishra B, Lushnikova T, Golla RM, Wang G. Modulation of antimicrobial potency of human cathelicidin peptides against the ESKAPE pathogens and in vivo efficacy in a murine catheter-associated biofilm model. Biochim Biophys Acta Biomembr. (2019) 1861:1592–602. doi: 10.1016/j.bbamem.2019.07.012
90. Nell MJ, Tjabringa GS, Wafelman AR, Verrijk R, Hiemstra PS, Drijfhout JW, et al. Development of novel LL-37 derived antimicrobial peptides with LPS and LTA neutralizing and antimicrobial activities for therapeutic application. Peptides. (2006) 27:649–60. doi: 10.1016/j.peptides.2005.09.016
91. Malanovic N, Leber R, Schmuck M, Kriechbaum M, Cordfunke RA, Drijfhout JW, et al. Phospholipid-driven differences determine the action of the synthetic antimicrobial peptide OP-145 on Gram-positive bacterial and mammalian membrane model systems. Biochim Biophys Acta. (2015) 1848(10 Pt A):2437–47. doi: 10.1016/j.bbamem.2015.07.010
92. Riool M, de Breij A, Drijfhout JW, Nibbering PH, Zaat SAJ. Antimicrobial peptides in biomedical device manufacturing. Front Chem. (2017) 5:63. doi: 10.3389/fchem.2017.00063
93. Starr CG, He J, Wimley WC. Host cell interactions are a significant barrier to the clinical utility of peptide antibiotics. ACS Chem Biol. (2016) 11:3391–9. doi: 10.1021/acschembio.6b00843
94. Svenson J, Brandsdal BO, Stensen W, Svendsen JS. Albumin binding of short cationic antimicrobial micropeptides and its influence on the in vitro bactericidal effect. J Med Chem. (2007) 50:3334–9. doi: 10.1021/jm0703542
95. Starr CG, Wimley WC. Antimicrobial peptides are degraded by the cytosolic proteases of human erythrocytes. Biochim Biophys Acta Biomembr. (2017) 1859:2319–26. doi: 10.1016/j.bbamem.2017.09.008
96. Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother. (2004) 48:4673–9. doi: 10.1128/AAC.48.12.4673-4679.2004
97. Moncla BJ, Pryke K, Rohan LC, Graebing PW. Degradation of naturally occurring and engineered antimicrobial peptides by proteases. Adv Biosci Biotechnol. (2011) 2:404–8. doi: 10.4236/abb.2011.26059
98. Bottger R, Hoffmann R, Knappe D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE. (2017) 12:e0178943. doi: 10.1371/journal.pone.0178943
99. Nguyen LT, Haney EF, Vogel HJ. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. (2011) 29:464–72. doi: 10.1016/j.tibtech.2011.05.001
100. Clemens LE, Jaynes J, Lim E, Kolar SS, Reins RY, Baidouri H, et al. Designed host defense peptides for the treatment of bacterial keratitis. Invest Ophthalmol Vis Sci. (2017) 58:6273–81. doi: 10.1167/iovs.17-22243
101. Arias M, Piga KB, Hyndman ME, Vogel HJ. Improving the activity of Trp-rich antimicrobial peptides by Arg/Lys substitutions and changing the length of cationic residues. Biomolecules. (2018) 8:19. doi: 10.3390/biom8020019
102. Oliva R, Chino M, Pane K, Pistorio V, De Santis A, Pizzo E, et al. Exploring the role of unnatural amino acids in antimicrobial peptides. Sci Rep. (2018) 8:8888. doi: 10.1038/s41598-018-27231-5
103. Hicks RP, Bhonsle JB, Venugopal D, Koser BW, Magill AJ. De novo design of selective antibiotic peptides by incorporation of unnatural amino acids. J Med Chem. (2007) 50:3026–36. doi: 10.1021/jm061489v
104. Qvit N, Rubin SJS, Urban TJ, Mochly-Rosen D, Gross ER. Peptidomimetic therapeutics: scientific approaches and opportunities. Drug Discov Today. (2017) 22:454–62. doi: 10.1016/j.drudis.2016.11.003
105. Gavras H, Brunner HR. Role of angiotensin and its inhibition in hypertension, ischemic heart disease, and heart failure. Hypertension. (2001) 37(2 Pt 2):342–5. doi: 10.1161/01.HYP.37.2.342
106. Gruber CW, Koehbach J, Muttenthaler M. Exploring bioactive peptides from natural sources for oxytocin and vasopressin drug discovery. Future Med Chem. (2012) 4:1791–8. doi: 10.4155/fmc.12.108
107. Grassi L, Maisetta G, Esin S, Batoni G. Combination strategies to enhance the efficacy of antimicrobial peptides against bacterial biofilms. Front Microbiol. (2017) 8:2409. doi: 10.3389/fmicb.2017.02409
108. Yan H, Hancock RE. Synergistic interactions between mammalian antimicrobial defense peptides. Antimicrob Agents Chemother. (2001) 45:1558–60. doi: 10.1128/AAC.45.5.1558-1560.2001
109. Almaaytah A, Qaoud MT, Abualhaijaa A, Al-Balas Q, Alzoubi KH. Hybridization and antibiotic synergism as a tool for reducing the cytotoxicity of antimicrobial peptides. Infect Drug Resist. (2018) 11:835–47. doi: 10.2147/IDR.S166236
110. Wade HM, Darling LEO, Elmore DE. Hybrids made from antimicrobial peptides with different mechanisms of action show enhanced membrane permeabilization. Biochim Biophys Acta Biomembr. (2019) 1861:182980. doi: 10.1016/j.bbamem.2019.05.002
111. Wang C, Yang C, Chen YC, Ma L, Huang K. Rational design of hybrid peptides: a novel drug design approach. Curr Med Sci. (2019) 39:349–55. doi: 10.1007/s11596-019-2042-2
112. Boman HG, Wade D, Boman IA, Wahlin B, Merrifield RB. Antibacterial and antimalarial properties of peptides that are cecropin-melittin hybrids. FEBS Lett. (1989) 259:103–6. doi: 10.1016/0014-5793(89)81505-4
113. Andreu D, Ubach J, Boman A, Wahlin B, Wade D, Merrifield RB, et al. Shortened cecropin A-melittin hybrids. Significant size reduction retains potent antibiotic activity. FEBS Lett. (1992) 296:190–4. doi: 10.1016/0014-5793(92)80377-S
114. Wade D, Andreu D, Mitchell SA, Silveira AM, Boman A, Boman HG, et al. Antibacterial peptides designed as analogs or hybrids of cecropins and melittin. Int J Pept Protein Res. (1992) 40:429–36. doi: 10.1111/j.1399-3011.1992.tb00321.x
115. Shin SY, Kang JH, Hahm KS. Structure-antibacterial, antitumor and hemolytic activity relationships of cecropin A-magainin 2 and cecropin A-melittin hybrid peptides. J Pept Res. (1999) 53:82–90. doi: 10.1111/j.1399-3011.1999.tb01620.x
116. Lee JK, Seo CH, Luchian T, Park Y. Antimicrobial peptide CMA3 derived from the CA-MA hybrid peptide: antibacterial and anti-inflammatory activities with low cytotoxicity and mechanism of action in Escherichia coli. Antimicrob Agents Chemother. (2016) 60:495–506. doi: 10.1128/AAC.01998-15
117. Wei XB, Wu RJ, Si DY, Liao XD, Zhang LL, Zhang RJ. Novel hybrid peptide cecropin A (1-8)-LL37 (17-30) with potential antibacterial activity. Int J Mol Sci. (2016) 17:983. doi: 10.3390/ijms17070983
118. Wu R, Wang Q, Zheng Z, Zhao L, Shang Y, Wei X, et al. Design, characterization and expression of a novel hybrid peptides melittin (1-13)-LL37 (17-30). Mol Biol Rep. (2014) 41:4163–9. doi: 10.1007/s11033-013-2900-0
119. Tall YA, Al-Rawashdeh B, Abualhaijaa A, Almaaytah A, Masadeh M, Alzoubi KH. Functional characterization of a novel hybrid peptide with high potency against gram-negative bacteria. Curr Pharm Des. (2020) 26:376–85. doi: 10.2174/1381612826666200128090700
120. Fox MA, Thwaite JE, Ulaeto DO, Atkins TP, Atkins HS. Design and characterization of novel hybrid antimicrobial peptides based on cecropin A, LL-37 and magainin II. Peptides. (2012) 33:197–205. doi: 10.1016/j.peptides.2012.01.013
121. Dutta D, Vijay AK, Kumar N, Willcox MD. Melimine-coated antimicrobial contact lenses reduce microbial keratitis in an animal model. Invest Ophthalmol Vis Sci. (2016) 57:5616–24. doi: 10.1167/iovs.16-19882
122. Kim H, Jang JH, Kim SC, Cho JH. Development of a novel hybrid antimicrobial peptide for targeted killing of Pseudomonas aeruginosa. Eur J Med Chem. (2020) 185:111814. doi: 10.1016/j.ejmech.2019.111814
123. Xu L, Shao C, Li G, Shan A, Chou S, Wang J, et al. Conversion of broad-spectrum antimicrobial peptides into species-specific antimicrobials capable of precisely targeting pathogenic bacteria. Sci Rep. (2020) 10:944. doi: 10.1038/s41598-020-58014-6
124. Eckert R, He J, Yarbrough DK, Qi F, Anderson MH, Shi W. Targeted killing of Streptococcus mutans by a pheromone-guided “smart” antimicrobial peptide. Antimicrob Agents Chemother. (2006) 50:3651–7. doi: 10.1128/AAC.00622-06
125. Kim H, Jang JH, Kim SC, Cho JH. Enhancement of the antimicrobial activity and selectivity of GNU7 against Gram-negative bacteria by fusion with LPS-targeting peptide. Peptides. (2016) 82:60–6. doi: 10.1016/j.peptides.2016.05.010
126. Aliashkevich A, Alvarez L, Cava F. New insights into the mechanisms and biological roles of D-Amino acids in complex eco-systems. Front Microbiol. (2018) 9:683. doi: 10.3389/fmicb.2018.00683
127. Li H, Anuwongcharoen N, Malik AA, Prachayasittikul V, Wikberg JE, Nantasenamat C. Roles of d-amino acids on the bioactivity of host defense peptides. Int J Mol Sci. (2016) 17:1023. doi: 10.3390/ijms17071023
128. Wade D, Boman A, Wahlin B, Drain CM, Andreu D, Boman HG, et al. All-D amino acid-containing channel-forming antibiotic peptides. Proc Natl Acad Sci USA. (1990) 87:4761–5. doi: 10.1073/pnas.87.12.4761
129. Cardoso MH, Candido ES, Oshiro KGN, Rezende SB, Franco OL. Peptides containing d-amino acids and retro-inverso peptides: general applications and special focus on antimicrobial peptides. In: Koutsopoulos S, editor. Peptide Applications in Biomedicine, Biotechnology and Bioengineering. Duxford: Woodhead Publishing (2018). p. 1–81.
130. Carmona G, Rodriguez A, Juarez D, Corzo G, Villegas E. Improved protease stability of the antimicrobial peptide Pin2 substituted with D-amino acids. Protein J. (2013) 32:456–66. doi: 10.1007/s10930-013-9505-2
131. Jia F, Wang J, Peng J, Zhao P, Kong Z, Wang K, et al. D-amino acid substitution enhances the stability of antimicrobial peptide polybia-CP. Acta Biochim Biophys Sin. (2017) 49:916–25. doi: 10.1093/abbs/gmx091
132. Manabe T, Kawasaki K. D-form KLKLLLLLKLK-NH2 peptide exerts higher antimicrobial properties than its L-form counterpart via an association with bacterial cell wall components. Sci Rep. (2017) 7:43384. doi: 10.1038/srep43384
133. Mangoni ML, Papo N, Saugar JM, Barra D, Shai Y, Simmaco M, et al. Effect of natural L- to D-amino acid conversion on the organization, membrane binding, and biological function of the antimicrobial peptides bombinins H. Biochemistry. (2006) 45:4266–76. doi: 10.1021/bi052150y
134. Heidary M, Khosravi AD, Khoshnood S, Nasiri MJ, Soleimani S, Goudarzi M. Daptomycin. J Antimicrob Chemother. (2018) 73:1–11. doi: 10.1093/jac/dkx349
135. Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. (2011) 52:285–92. doi: 10.1093/cid/cir034
136. Dahiya R, Kumar S, Khokra SL, Gupta SV, Sutariya VB, Bhatia D, et al. Toward the synthesis and improved biopotential of an N-methylated analog of a proline-rich cyclic tetrapeptide from marine bacteria. Mar Drugs. (2018) 16:305. doi: 10.3390/md16090305
137. Wang G. Post-translational modifications of natural antimicrobial peptides and strategies for peptide engineering. Curr Biotechnol. (2012) 1:72–9. doi: 10.2174/2211550111201010072
138. Ree R, Varland S, Arnesen T. Spotlight on protein N-terminal acetylation. Exp Mol Med. (2018) 50:1–13. doi: 10.1038/s12276-018-0116-z
139. Saikia K, Sravani YD, Ramakrishnan V, Chaudhary N. Highly potent antimicrobial peptides from N-terminal membrane-binding region of E. coli MreB. Sci Rep. (2017) 7:42994. doi: 10.1038/srep42994
140. Crusca E Jr, Rezende AA, Marchetto R, Mendes-Giannini MJ, Fontes W, Castro MS, et al. Influence of N-terminus modifications on the biological activity, membrane interaction, and secondary structure of the antimicrobial peptide hylin-a1. Biopolymers. (2011) 96:41–8. doi: 10.1002/bip.21454
141. Mura M, Wang J, Zhou Y, Pinna M, Zvelindovsky AV, Dennison SR, et al. The effect of amidation on the behaviour of antimicrobial peptides. Eur Biophys J. (2016) 45:195–207. doi: 10.1007/s00249-015-1094-x
142. Irudayam SJ, Berkowitz ML. Binding and reorientation of melittin in a POPC bilayer: computer simulations. Biochim Biophys Acta. (2012) 1818:2975–81. doi: 10.1016/j.bbamem.2012.07.026
143. Dennison SR, Phoenix DA. Influence of C-terminal amidation on the efficacy of modelin-5. Biochemistry. (2011) 50:1514–23. doi: 10.1021/bi101687t
144. Dos Santos Cabrera MP, Arcisio-Miranda M, Broggio Costa ST, Konno K, Ruggiero JR, Procopio J, et al. Study of the mechanism of action of anoplin, a helical antimicrobial decapeptide with ion channel-like activity, and the role of the amidated C-terminus. J Pept Sci. (2008) 14:661–9. doi: 10.1002/psc.960
145. Islas-Rodriguez AE, Marcellini L, Orioni B, Barra D, Stella L, Mangoni ML. Esculentin 1-21: a linear antimicrobial peptide from frog skin with inhibitory effect on bovine mastitis-causing bacteria. J Pept Sci. (2009) 15:607–14. doi: 10.1002/psc.1148
146. Kuzmin DV, Emelianova AA, Kalashnikova MB, Panteleev PV, Ovchinnikova TV. Effect of N- and C-terminal modifications on cytotoxic properties of antimicrobial peptide tachyplesin I. Bull Exp Biol Med. (2017) 162:754–7. doi: 10.1007/s10517-017-3705-2
147. Purwin M, Markowska A, Bruzgo I, Rusak T, Surazynski A, Jaworowska U, et al. Peptides with 6-aminohexanoic acid: synthesis and evaluation as plasmin inhibitors. Int J Pept Res Ther. (2017) 23:235–45. doi: 10.1007/s10989-016-9555-3
148. Falciani C, Lozzi L, Scali S, Brunetti J, Bracci L, Pini A. Site-specific pegylation of an antimicrobial peptide increases resistance to Pseudomonas aeruginosa elastase. Amino Acids. (2014) 46:1403–7. doi: 10.1007/s00726-014-1686-2
149. Zorzi A, Deyle K, Heinis C. Cyclic peptide therapeutics: past, present and future. Curr Opin Chem Biol. (2017) 38:24–9. doi: 10.1016/j.cbpa.2017.02.006
150. Falagas ME, Kasiakou SK. Colistin: the revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin Infect Dis. (2005) 40:1333–41. doi: 10.1086/429323
151. Johnson AP, Uttley AH, Woodford N, George RC. Resistance to vancomycin and teicoplanin: an emerging clinical problem. Clin Microbiol Rev. (1990) 3:280–91. doi: 10.1128/CMR.3.3.280
152. Dathe M, Nikolenko H, Klose J, Bienert M. Cyclization increases the antimicrobial activity and selectivity of arginine- and tryptophan-containing hexapeptides. Biochemistry. (2004) 43:9140–50. doi: 10.1021/bi035948v
153. Bagheri M, Amininasab M, Dathe M. Arginine/tryptophan-rich cyclic α/β-antimicrobial peptides: the roles of hydrogen bonding and hydrophobic/hydrophilic solvent-accessible surface areas upon activity and membrane selectivity. Chemistry. (2018) 24:14242–53. doi: 10.1002/chem.201802881
154. Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, et al. Antibacterial agents based on the cyclic D,L-alpha-peptide architecture. Nature. (2001) 412:452–5. doi: 10.1038/35086601
155. Dartois V, Sanchez-Quesada J, Cabezas E, Chi E, Dubbelde C, Dunn C, et al. Systemic antibacterial activity of novel synthetic cyclic peptides. Antimicrob Agents Chemother. (2005) 49:3302–10. doi: 10.1128/AAC.49.8.3302-3310.2005
156. Mika JT, Moiset G, Cirac AD, Feliu L, Bardaji E, Planas M, et al. Structural basis for the enhanced activity of cyclic antimicrobial peptides: the case of BPC194. Biochim Biophys Acta. (2011) 1808:2197–205. doi: 10.1016/j.bbamem.2011.05.001
157. Falanga A, Nigro E, De Biasi MG, Daniele A, Morelli G, Galdiero S, et al. Cyclic peptides as novel therapeutic microbicides: engineering of human defensin mimetics. Molecules. (2017) 22:1217. doi: 10.3390/molecules22071217
158. Menendez A, Brett Finlay B. Defensins in the immunology of bacterial infections. Curr Opin Immunol. (2007) 19:385–91. doi: 10.1016/j.coi.2007.06.008
159. Sher Khan R, Iqbal A, Malak R, Shehryar K, Attia S, Ahmed T, et al. Plant defensins: types, mechanism of action and prospects of genetic engineering for enhanced disease resistance in plants. 3 Biotech. (2019). 9:192. doi: 10.1007/s13205-019-1725-5
160. Scudiero O, Nigro E, Cantisani M, Colavita I, Leone M, Mercurio FA, et al. Design and activity of a cyclic mini-beta-defensin analog: a novel antimicrobial tool. Int J Nanomedicine. (2015) 10:6523–39. doi: 10.2147/IJN.S89610
161. Mwangi J, Yin Y, Wang G, Yang M, Li Y, Zhang Z, et al. The antimicrobial peptide ZY4 combats multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii infection. Proc Natl Acad Sci USA. (2019) 116:26516–22. doi: 10.1073/pnas.1909585117
162. Bayda S, Adeel M, Tuccinardi T, Cordani M, Rizzolio F. The history of nanoscience and nanotechnology: from chemical-physical applications to nanomedicine. Molecules. (2019) 25:112. doi: 10.3390/molecules25010112
163. Khan I, Saeed K, Khan I. Nanoparticles: properties, applications and toxicities. Arab J Chemistry. (2019) 12:908–31. doi: 10.1016/j.arabjc.2017.05.011
164. Reshma VG, Syama S, Sruthi S, Reshma SC, Remya NS, Mohanan PV. Engineered nanoparticles with antimicrobial property. Curr Drug Metab. (2017) 18:1040–54. doi: 10.2174/1389200218666170925122201
165. Natan M, Banin E. From nano to micro: using nanotechnology to combat microorganisms and their multidrug resistance. FEMS Microbiol Rev. (2017) 41:302–22. doi: 10.1093/femsre/fux003
166. Biswaro LS, da Costa Sousa MG, Rezende TMB, Dias SC, Franco OL. Antimicrobial peptides and nanotechnology, recent advances and challenges. Front Microbiol. (2018) 9:855. doi: 10.3389/fmicb.2018.00855
167. Lakshminarayanan R, Ye E, Young DJ, Li Z, Loh XJ. Recent advances in the development of antimicrobial nanoparticles for combating resistant pathogens. Adv Healthc Mater. (2018) 7:e1701400. doi: 10.1002/adhm.201701400
168. Patra JK, Das G, Fraceto LF, Campos EVR, Rodriguez-Torres MDP, Acosta-Torres LS, et al. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnology. (2018) 16:71. doi: 10.1186/s12951-018-0392-8
169. Chereddy KK, Her CH, Comune M, Moia C, Lopes A, Porporato PE, et al. PLGA nanoparticles loaded with host defense peptide LL37 promote wound healing. J Control Release. (2014) 194:138–47. doi: 10.1016/j.jconrel.2014.08.016
170. Ferreira AF, Comune M, Rai A, Ferreira L, Simoes PN. Atomistic-level investigation of a LL37-conjugated gold nanoparticle by well-tempered metadynamics. J Phys Chem B. (2018) 122:8359–66. doi: 10.1021/acs.jpcb.8b05717
171. Comune M, Rai A, Chereddy KK, Pinto S, Aday S, Ferreira AF, et al. Antimicrobial peptide-gold nanoscale therapeutic formulation with high skin regenerative potential. J Control Release. (2017) 262:58–71. doi: 10.1016/j.jconrel.2017.07.007
172. Wnorowska U, Fiedoruk K, Piktel E, Prasad SV, Sulik M, Janion M, et al. Nanoantibiotics containing membrane-active human cathelicidin LL-37 or synthetic ceragenins attached to the surface of magnetic nanoparticles as novel and innovative therapeutic tools: current status and potential future applications. J Nanobiotechnol. (2020) 18:3. doi: 10.1186/s12951-019-0566-z
173. Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. (2011) 3:1377–97. doi: 10.3390/polym3031377
174. Cruz J, Florez J, Torres R, Urquiza M, Gutierrez JA, Guzman F, et al. Antimicrobial activity of a new synthetic peptide loaded in polylactic acid or poly(lactic-co-glycolic) acid nanoparticles against Pseudomonas aeruginosa, Escherichia coli O157:H7 and methicillin resistant Staphylococcus aureus (MRSA). Nanotechnology. (2017) 28:135102. doi: 10.1088/1361-6528/aa5f63
175. Yeh YC, Creran B, Rotello VM. Gold nanoparticles: preparation, properties, and applications in bionanotechnology. Nanoscale. (2012) 4:1871–80. doi: 10.1039/C1NR11188D
176. Casciaro B, Moros M, Rivera-Fernandez S, Bellelli A, de la Fuente JM, Mangoni ML. Gold-nanoparticles coated with the antimicrobial peptide esculentin-1a(1-21)NH2 as a reliable strategy for antipseudomonal drugs. Acta Biomater. (2017) 47:170–81. doi: 10.1016/j.actbio.2016.09.041
177. Hamet P, Tremblay J. Artificial intelligence in medicine. Metabolism. (2017) 69s:S36–40. doi: 10.1016/j.metabol.2017.01.011
178. Ting DSJ, Ang M, Mehta JS, Ting DSW. Artificial intelligence-assisted telemedicine platform for cataract screening and management: a potential model of care for global eye health. Br J Ophthalmol. (2019) 103:1537–8. doi: 10.1136/bjophthalmol-2019-315025
179. Schneider P, Walters WP, Plowright AT, Sieroka N, Listgarten J, Goodnow RA, et al. Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov. (2019) 19:353–64. doi: 10.1038/s41573-019-0050-3
180. LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. (2015) 521:436–44. doi: 10.1038/nature14539
181. Cardoso MH, Orozco RQ, Rezende SB, Rodrigues G, Oshiro KGN, Candido ES, et al. Computer-aided design of antimicrobial peptides: are we generating effective drug candidates? Front Microbiol. (2019) 10:3097. doi: 10.3389/fmicb.2019.03097
182. Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, et al. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem Biol. (2009) 4:65–74. doi: 10.1021/cb800240j
183. Lee EY, Fulan BM, Wong GC, Ferguson AL. Mapping membrane activity in undiscovered peptide sequence space using machine learning. Proc Natl Acad Sci USA. (2016) 113:13588–93. doi: 10.1073/pnas.1609893113
184. Yount NY, Weaver DC, Lee EY, Lee MW, Wang H, Chan LC, et al. Unifying structural signature of eukaryotic alpha-helical host defense peptides. Proc Natl Acad Sci USA. (2019) 116:6944–53. doi: 10.1073/pnas.1819250116
185. Xiao X, Wang P, Lin WZ, Jia JH, Chou KC. iAMP-2L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal Biochem. (2013) 436:168–77. doi: 10.1016/j.ab.2013.01.019
186. Shoval O, Sheftel H, Shinar G, Hart Y, Ramote O, Mayo A, et al. Evolutionary trade-offs, pareto optimality, and the geometry of phenotype space. Science. (2012) 336:1157–60. doi: 10.1126/science.1217405
187. Fox JL. Antimicrobial peptides stage a comeback. Nat Biotechnol. (2013) 31:379–82. doi: 10.1038/nbt.2572
188. Lipsky BA, Holroyd KJ, Zasloff M. Topical versus systemic antimicrobial therapy for treating mildly infected diabetic foot ulcers: a randomized, controlled, double-blinded, multicenter trial of pexiganan cream. Clin Infect Dis. (2008) 47:1537–45. doi: 10.1086/593185
189. Hilpert K, Volkmer-Engert R, Walter T, Hancock RE. High-throughput generation of small antibacterial peptides with improved activity. Nat Biotechnol. (2005) 23:1008–12. doi: 10.1038/nbt1113
190. Mohamed MF, Abdelkhalek A, Seleem MN. Evaluation of short synthetic antimicrobial peptides for treatment of drug-resistant and intracellular Staphylococcus aureus. Sci Rep. (2016) 6:29707. doi: 10.1038/srep29707
191. Mannis MJ. The use of antimicrobial peptides in ophthalmology: an experimental study in corneal preservation and the management of bacterial keratitis. Trans Am Ophthalmol Soc. (2002) 100:243–71.
192. Pizzolato-Cezar LR, Okuda-Shinagawa NM, Machini MT. Combinatory therapy antimicrobial peptide-antibiotic to minimize the ongoing rise of resistance. Front Microbiol. (2019) 10:1703. doi: 10.3389/fmicb.2019.01703
193. Lakshminarayanan R, Tan WX, Aung TT, Goh ET, Muruganantham N, Li J, et al. Branched peptide, B2088, disrupts the supramolecular organization of lipopolysaccharides and sensitizes the gram-negative bacteria. Sci Rep. (2016) 6:25905. doi: 10.1038/srep25905
194. Lewies A, Du Plessis LH, Wentzel JF. Antimicrobial peptides: the achilles' heel of antibiotic resistance? Probiotics Antimicrob Proteins. (2019) 11:370–81. doi: 10.1007/s12602-018-9465-0
195. Moravej H, Moravej Z, Yazdanparast M, Heiat M, Mirhosseini A, Moosazadeh Moghaddam M, et al. Antimicrobial peptides: features, action, and their resistance mechanisms in bacteria. Microb Drug Resist. (2018) 24:747–67. doi: 10.1089/mdr.2017.0392
196. Spohn R, Daruka L, Lazar V, Martins A, Vidovics F, Grezal G, et al. Integrated evolutionary analysis reveals antimicrobial peptides with limited resistance. Nat Commun. (2019) 10:4538. doi: 10.1038/s41467-019-12364-6
Keywords: antibiotic, antimicrobial peptide, antimicrobial resistance, artificial intelligence, host defense peptide, nanoparticle, peptide
Citation: Ting DSJ, Beuerman RW, Dua HS, Lakshminarayanan R and Mohammed I (2020) Strategies in Translating the Therapeutic Potentials of Host Defense Peptides. Front. Immunol. 11:983. doi: 10.3389/fimmu.2020.00983
Received: 28 February 2020; Accepted: 27 April 2020;
Published: 22 May 2020.
Edited by:
Charles Lee Bevins, University of California, Davis, United StatesReviewed by:
Henk Peter Haagsman, Utrecht University, NetherlandsCopyright © 2020 Ting, Beuerman, Dua, Lakshminarayanan and Mohammed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajamani Lakshminarayanan, bGFrc2htaW5hcmF5YW5hbi5yYWphbWFuaUBzZXJpLmNvbS5zZw==; Imran Mohammed, SW1yYW4ubW9oYW1tZWRAbm90dGluZ2hhbS5hYy51aw==
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.