 Tomasz M. Grzywa1
Tomasz M. Grzywa1 Anna Sosnowska1,2
Anna Sosnowska1,2 Paweł Matryba1,3,4
Paweł Matryba1,3,4 Zuzanna Rydzynska1
Zuzanna Rydzynska1 Marcin Jasinski1
Marcin Jasinski1 Dominika Nowis1,5,6*
Dominika Nowis1,5,6* Jakub Golab1,7*
Jakub Golab1,7*- 1Department of Immunology, Medical University of Warsaw, Warsaw, Poland
- 2Postgraduate School of Molecular Medicine, Medical University of Warsaw, Warsaw, Poland
- 3Laboratory of Neurobiology BRAINCITY, Nencki Institute of Experimental Biology of Polish Academy of Sciences, Warsaw, Poland
- 4The Doctoral School of the Medical University of Warsaw, Medical University of Warsaw, Warsaw, Poland
- 5Laboratory of Experimental Medicine, Center of New Technologies, University of Warsaw, Warsaw, Poland
- 6Genomic Medicine, Medical University of Warsaw, Warsaw, Poland
- 7Centre of Preclinical Research, Medical University of Warsaw, Warsaw, Poland
Amino acid metabolism is a critical regulator of the immune response, and its modulating becomes a promising approach in various forms of immunotherapy. Insufficient concentrations of essential amino acids restrict T-cells activation and proliferation. However, only arginases, that degrade L-arginine, as well as enzymes that hydrolyze L-tryptophan are substantially increased in cancer. Two arginase isoforms, ARG1 and ARG2, have been found to be present in tumors and their increased activity usually correlates with more advanced disease and worse clinical prognosis. Nearly all types of myeloid cells were reported to produce arginases and the increased numbers of various populations of myeloid-derived suppressor cells and macrophages correlate with inferior clinical outcomes of cancer patients. Here, we describe the role of arginases produced by myeloid cells in regulating various populations of immune cells, discuss molecular mechanisms of immunoregulatory processes involving L-arginine metabolism and outline therapeutic approaches to mitigate the negative effects of arginases on antitumor immune response. Development of potent arginase inhibitors, with improved pharmacokinetic properties, may lead to the elaboration of novel therapeutic strategies based on targeting immunoregulatory pathways controlled by L-arginine degradation.
Introduction
The idea that the immune system can be harnessed to destroy tumors has been pursued for over a century (1). However, for decades the efforts have mainly focused on stimulating the immune system with recombinant cytokines, immune adjuvants, or co-stimulatory agonists that seemed critical for the induction of potent and sustained immune responses (1, 2). The rationale was that the immune system in cancer patients lacks sufficient power to mount anti-tumor response. It now seems however, that the interference with pathways dampening lymphocyte reactivity appears to be more effective in cancer patients than over-stimulation of effector mechanisms of immune system. The most successful approaches to impair tumor-elicited immunosuppressive mechanisms turned out to be monoclonal antibodies (referred to as immune checkpoint inhibitors) interfering with co-inhibitory molecules or their ligands, such as CTLA-4 (cytotoxic T-lymphocyte-associated protein 4), PD-1 (programmed cell death protein 1), or PD-L1 (programmed death-ligand 1). The spectacular therapeutic effects with unexpected ability to induce long-term tumor control led to clinical approval of checkpoint inhibitors (3–5).
Despite unprecedented antitumor efficacy, checkpoint inhibitors are effective in a minority of cancer patients, however. Thus, identification of response biomarkers as well as resistance mechanisms has become a priority for cancer researchers. A number of molecular mechanisms involved in the evasion of the anti-tumor immunity have been characterized in recent years (6). Central among them is the development of chronic inflammation (7, 8). Epidemiological data indicate that chronic inflammation is associated with poor prognosis (9). Mounting evidence indicates that the tumor microenvironment alters lymphoid and myeloid cells and converts them into potent immunosuppressive cells. It has become clear that tumor microenvironment, rich in inflammatory cells, is an indispensable component in the neoplastic process fostering proliferation, survival, and invasiveness of tumor cells (7). Chronic inflammation also triggers multiple regulatory pathways aimed at dampening immunity. The evolutionary rationale for this is to mitigate tissue damage and fibrosis. Coincidentally, the regulatory pathways impair development and/or activity of adaptive immune mechanisms that could be involved in eradication of tumor cells (8). Simultaneously, tumor cells frequently co-opt some of the signaling molecules participating in inflammation, such as adhesion molecules, cytokines, and growth factors for migration, invasion, and metastasis. Although there are many phenotypical and functional changes in different myeloid cell subpopulations, their precise role in the development of cancer resistance to immunotherapy is still not well-understood. This review will address the role of arginases (ARG), enzymes produced by tumor-infiltrating myeloid cells. The role of L-arginine (L-arg) metabolism in the regulation of immune response was of great interest in the 1980s and 1990s. However, further studies were focused mainly on L-arg-derived nitric oxide (NO) and its antimicrobial activity (10, 11), rather than immunosuppressive effects of L-arg deprivation. It is currently experiencing a renaissance due to increased awareness of the role of metabolic pathways in the regulation of immune cells function as well as due to the development of selective arginase inhibitors with improved pharmacokinetic properties. Novel tools and experimental models allowed to more precisely and comprehensively address the critical metabolic adaptations to microenvironmental changes experienced by immune cells. This is a clearly arginase-centered review, and it should be kept in mind that there are multiple other independent mechanisms of tumor immune evasion, including those affecting amino acids metabolism.
Arginine and Arginases—Basic Biochemistry
L-arginine is a dibasic cationic amino acid participating in a variety of metabolic pathways (Figure 1) (12). There are three major sources of L-arg in the body—dietary intake, endogenous de novo production from L-citrulline or recycling, i.e., retrieval from degraded proteins. Under pathological conditions (bleeding, sepsis, trauma, cancer, or chronic inflammation) endogenous sources of L-arg become insufficient (13). Thus, L-arg is considered to be a semi-essential or conditionally-essential amino acid that in stressful conditions must be supplied in diet. Most of the endogenous L-arg synthesis is carried out in the kidney proximal tubules from intestinal L-citrulline (14). L-Arg plasma concentrations range between 50 and 250 μM (15–18) and are much lower than those in subcellular compartments (up to 1 mM) (19). In mammalian cells, L-arg transport through the plasma membrane is mediated by at least eight transporters (20). The uptake of L-arg occurs mainly via cationic amino acid transporters (CAT-1, CAT-2A, CAT-2B, and CAT-3, SLC7A1-3) (21). In human T-cells L-arg transport is mediated mainly by CAT-1 (22), while in myeloid cells by CAT-2 (23). Moreover, L-arg is transported through the plasma membrane by b0, + AT (SLC7A9) and ATB0, + (SLC6A14) that also transport neutral amino acids (20, 24, 25). L-type amino acid transporters γ+LAT1 (SLC7A7) and γ+LAT2 (SLC7A6) mediate mostly arginine export from the cells (20, 24). L-arg is metabolized in animal cells by four groups of enzymes, some of which exist in various isoforms. These include arginases, nitric oxide synthases (NOS), arginine decarboxylase (ADC), and arginine:glycine amidinotransferase (AGAT). Moreover, arginine deiminase (ADI) that hydrolyzes L-arg to L-citrulline and ammonia is expressed by some bacteria (26, 27). It is the first enzyme of the arginine dihydrolase system (ADS) that generates alkali and ATP for growth (28). These enzymes are encoded by arginine catabolic mobile element (ACME) (29) that was detected in Staphylococcus aureus and Staphylococcus epidermidis (30). L-arg metabolism by ADS enables survival in acidic environments, including human skin, disrupts host arginine metabolism, and contributes to the success of community-associated methicillin-resistant S. aureus (CA-MRSA) (31).
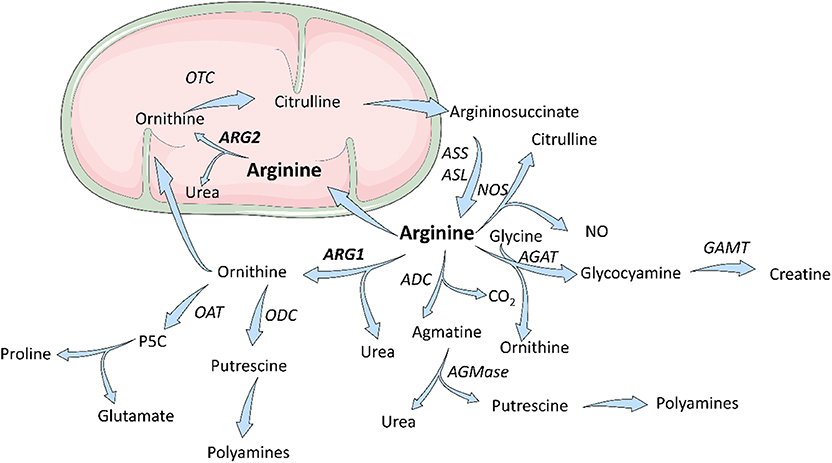
Figure 1. Scheme for arginine metabolism. In mammalian cells, L-Arginine is a substrate for four enzymes: ARG, NOS, ADC, AGAT. L-Arginine downstream metabolites are components of multiple metabolic pathways and are necessary for cells proliferation and collagen synthesis. ADC, arginine decarboxylase; AGAT, arginine:glycine amidinotransferase; AGMase, agmatinase; ARG, arginase; ASL, argininosuccinate lyase; ASS, argininosuccinate synthase; GAMT, guanidinoacetate N-methyltransferase; NOS, nitric oxide synthase; OAT; ornithine aminotransferase; OTC, ornithine transcarbamylase; P5C, pyrroline-5-carboxylic acid. Figure was modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. http://smart.servier.com/.
Arginases are manganese-containing enzymes that hydrolyze L-arg to L-ornithine and urea in the liver urea cycle (32). This is the most important pathway responsible for the conversion of highly toxic ammonia to excretable urea (33). L-Ornithine is a substrate for ornithine decarboxylase (ODC) that initiates polyamines synthesis, or it is metabolized by ornithine aminotransferase (OAT) to proline. Polyamines, such as putrescine, spermine, or spermidine are necessary for cell proliferation, while proline is necessary for collagen synthesis. Initially, it was thought that arginase is expressed only in the liver. However, further studies revealed that arginase is ubiquitously expressed in many types of cells (33), and that there are two different isoforms of this enzyme that catalyze the same biochemical reaction, but are expressed by different cells and are located in different cellular compartments. Human arginase 1 (ARG1) has 322 amino acids and is a cytosolic protein expressed primarily in the liver cells (34) as well as in the cells of the myeloid lineage (35). Human arginase 2 (ARG2) consists of 354 amino acids and can be found in mitochondria (36). It has ubiquitous expression, but usually at a lower level than ARG1. ARG2 has 58% sequence identity to ARG1 (37), but both enzymes are nearly identical within the catalytic region. There are also types of cells, such as endothelial cells, which have relatively high expression of both isoenzymes (38). The summary of the most important information on the two isoforms of arginase is presented in Table 1.
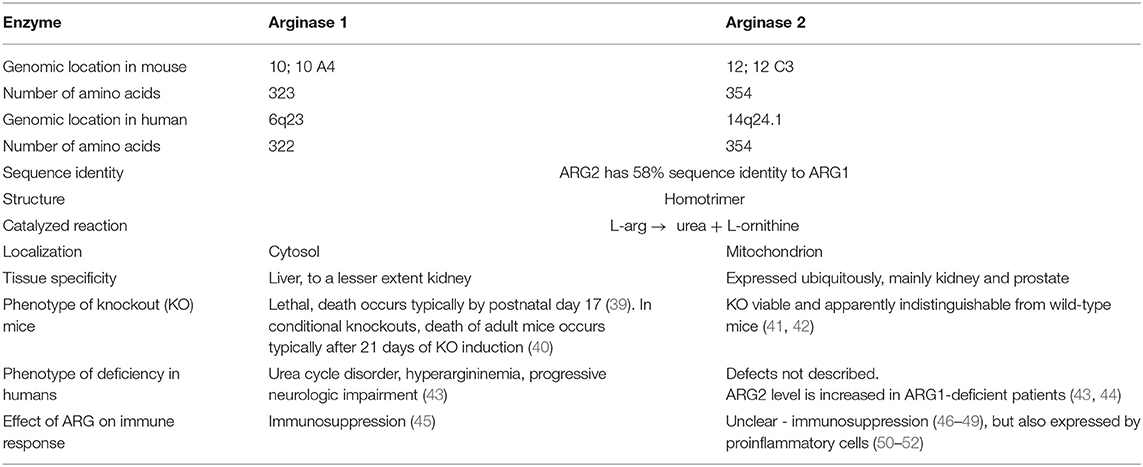
Table 1. Properties of the two arginase isoforms.
An important metabolic pathway of L-arg involves the activity of NOS. There are three isoforms of this enzyme—neuronal (nNOS or NOS1), inducible (iNOS or NOS2) and endothelial (eNOS or NOS3). NOS2 can be induced in many types of cells, but when present in activated myeloid cells it produces NO at a very high rate. There are multiple layers of competition between NOS2 and ARG1 in myeloid cells and both enzymes are induced by cytokines regulating different types of the immune response. NOS2 in myeloid cells is induced by type 1 cytokines (mainly IFN-γ), while ARG1 expression is regulated by IL-4 and IL-13. Considering that Km of ARG1 is ~1,000-fold higher than that of NOS2, the intracellular L-arg could be expected to be mainly metabolized to NO, rather than to L-ornithine and urea. However, Vmax of NOS is three orders of magnitude slower than that of ARG1 (53, 54). Thus, both enzymes compete for the same substrate. Intriguingly, insufficient L-arg concentrations lead to NOS uncoupling, whereby rather than NO these enzymes generate superoxide anions. Superoxide then rapidly reacts with any available NO molecules to form peroxinitrites that further decrease NOS activity by oxidizing tetrahydrobiopterin (BH4) (54). Moreover, induction of ARG1 that limits L-arg availability is involved in the regulation of NOS2 expression as L-arg is necessary for the translation of NOS2-encoding mRNA (55).
During acute wound healing resident myeloid cells express high levels of NADPH oxidase (NOX2) and NOS2, which participate in normal antimicrobial defense mechanisms by producing superoxide anion and NO, respectively. Then, after 3–5 days, a repair phase is initiated, which is associated with the appearance of ARG1+ macrophages. L-arg degradation produces L-ornithine that is converted by OAT to L-proline used as a substrate in collagen synthesis (56). ODC converts L-ornithine to polyamines that stimulate cell proliferation. This highly regulated process is perpetuated in tumors that are frequently described as wounds that never heal (57).
Arginine and Arginase in Tumors
Tumor progression is associated with alterations in metabolic pathways in tumor cells as well as in the cells forming the tumor microenvironment. Altered metabolic phenotype of tumors includes changes in L-arg concentrations. For example, the concentration of L-arg in the core regions of solid tumors is about 5 times lower as compared with tumor periphery and this difference turned out to be the highest among all of the measured amino acids (58). Quantification of interstitial fluid metabolites in murine tumors has also revealed that L-arg is the most strongly depleted amino acid in the tumor microenvironment (59). The mechanisms of L-arg depletion are incompletely elucidated. On the one hand, L-arg can be consumed by tumor cells that have increased metabolic demands and use it for protein synthesis, but it can also be used by enzymes such as arginases or NOS. Many studies reported that arginases can be produced by tumor cells (46, 60, 61), but even larger number of reports indicate that the major L-arg-metabolizing cells are found in the tumor stroma. It has not been studied in sufficient detail as to which cells in the tumor environment are mainly responsible for L-arg depletion. It is also entirely possible that this process is highly variable and changes in the course of tumor progression, with tumor cells or stromal cells predominating in L-arg metabolism at various stages of neoplastic disease.
Arginase and Tumor Prognosis
High ARG expression and activity have been reported in many types of human cancers, but its role as a prognosis factor remains vastly undetermined and usually studied on small populations of patients. Moreover, drawing conclusions from the limited number of studies is further complicated by a lack of standardized criteria for ARG measurements. For example, different cutoff criteria were applied to groups of patients with “low arginase” and “high arginase” expressing tumors, or studying either ARG1 or ARG2 expression profiles. Nonetheless, increasing evidence shows that overexpression of ARG1/2 (with or without subsequent decline in serum L-arg concentrations) should be perceived as a poor prognostic factor in a wide variety of cancer types including head and neck cancer (62), neuroblastoma (46), acute myeloid leukemia (AML) (61), pancreatic ductal carcinoma (63), ovarian carcinoma (64), or colorectal cancer (65). High expression of ARG1 in hepatocellular carcinoma also seems to play a role as a negative predictive factor that correlates with shorter median time to recurrence (66) and more aggressive tumors (67), but further evidence is required to support these observations as a contradictory report exists (68).
Although a number of studies provide strong evidence for increased ARG activity in both tissue (69) and blood (70–72) obtained from patients with breast cancer, so far no study was conducted to establish the role of ARG activity in determining the prognosis of breast cancer patients. Notably, contradictory reports exist that show a decrease in blood plasma ARG activity in breast cancer patients, however, these are based on very limited number of enrolled patients (73, 74). Similarly to breast cancer, increased ARG activity was found in skin (75), cervical (76), thyroid follicular (77), thyroid papillary and follicular variant of papillary (77), gastric, bile duct (78), and esophageal (79) cancers. However, again no study exists in these types of cancers that would demonstrate the impact of ARG activity/abundance on patients' prognosis.
Finally, there are tumors such as prostate (80–82) and lung cancer (83) as well as tumors that are auxotrophic for L-arg (these are not capable of re-synthesis of L-arg from citrulline due to the lack of expression of argininosuccinate synthetase-1, ASS-1), such as melanoma (84) and renal carcinoma (85, 86), where no correlation between ARG levels and survival has been found.
A critical question arises whether ARG in tumors is produced by tumor cells or by tumor-associated stromal cells that include mesenchymal as well as immune cells, among which myeloid cells seem to be the main source of the enzyme. Regrettably, no studies have been conducted that would directly address this issue and whether this is of any significance for cancer patients survival, whether ARG is expressed by tumor or tumor-infiltrating myeloid cells.
Arginase in Tumor-Infiltrating Myeloid Cells
Myeloid cells are major contributors to immune defense against pathogens and play an important role in tissue remodeling. During acute infections GM-CSF drives myelopoiesis in the bone marrow, and G-CSF as well as M-CSF induce further differentiation of granulocytes and macrophages, respectively (87). Some tissue macrophages develop from embryonic precursors that directly home to peripheral tissues and become a self-renewing population (88). Mature myeloid cells are specialized in killing infectious microorganisms and play an important role in promoting development of adaptive immunity. However, in cancer and other chronic inflammatory conditions constant production of low concentrations of myeloid growth factors and various inflammatory mediators dysregulate myeloid cells differentiation (Figure 2) (89–94). It is currently not well-understood what events trigger this disturbed myelopoiesis, but it must be emphasized that this process evolves over many years of tumor development and likely involves multiple independent mechanisms. Some of these might be completely stochastic, but in the course of tumor progression become promoted in a trial-and-error process that selects for mechanisms that best fit the demands of growing tumors.
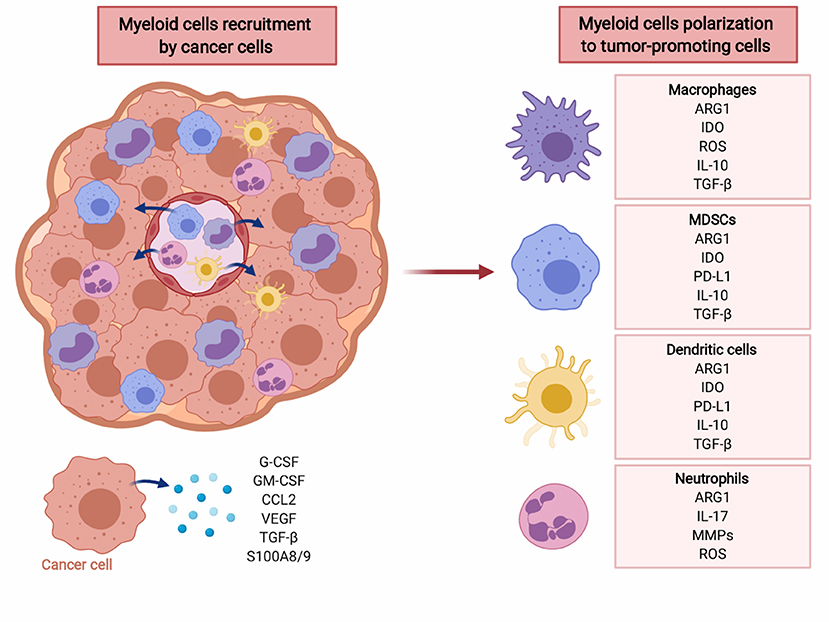
Figure 2. Cancer cells recruit myeloid cells to tumor microenvironment (TME) and induce their polarization to immunosuppressive phenotype. Myeloid cells, including macrophages, MDSC, dendritic cells, and neutrophils create tumor-promoting, immunosuppressive TME via multiple factors including reactive oxygen species (ROS), cytokines (IL-10, TGF-β), PD-L1, as well as ARG1. Created with BioRender.
Myeloid cells, especially tumor-infiltrating myeloid cells (TIMs), are a highly heterogeneous population (95). TIMs include monocytes, macrophages, dendritic cells, granulocytes, mast cells, as well as their immature precursors that have not completed their differentiation processes. The latter cells are normally found in the bone marrow, but in the course of tumor development they frequently expand and relocate to the spleen, lymph nodes and the tumor itself, and can be found at increased numbers in the peripheral blood (96, 97). These cells express immune checkpoint molecules, deplete essential metabolites, release immunosuppressive adenosine and its metabolites, produce reactive oxygen species, secrete immunoregulatory cytokines, growth-promoting, and proangiogenic factors (Figure 2). Moreover, they induce various populations of regulatory T-cells that impair antitumor immune response (98). Due to their strong immunosuppressive functions these cells have been termed myeloid-derived suppressor cells (MDSCs). There are two major subsets of MDSCs—monocytic (M-MDSC) and granulocytic (polymorphonuclear, PMN-MDSC) (99). Both have been associated with dysregulation of immune response in murine cancer models and in cancer patients, although still the majority of studies report the suppressive potential of total MDSCs (100). In mouse tumor models that mostly involve transplantation of tumor cells, the expansion of MDSCs is very rapid. This is in contrast to slow-growing tumors, including diethylnitrosoamine (DEN)-induced or MYC-expressing hepatocellular carcinoma, that in terms of the rate of tumor progression more accurately reflect human cancer (101). In many types of humans tumors, including lung, colon, uterus, cervix, bladder, or thyroid gland cancers, the increased numbers of M-MDSCs in peripheral blood correlate with worse clinical outcomes (102). In melanoma or liver cancer, however the increased numbers of both PMN-MDSCs and M-MDSCs were associated with poorer outcomes (102), while in renal cell carcinoma PMN-MDSCs seem to predominate (103). Importantly, increased numbers of MDSCs are observed also in patients with pancreatic premalignancy—intraductal papillary mucinous neoplasm (IPMN), and in patients with colon adenomas, as compared with healthy controls (97).
Nearly all myeloid cells have been shown to produce ARG1 in mice (Figure 2). However, there are substantial differences in the expression of arginases by myeloid cells between mice and men (104). In humans, arginase is produced mainly by granulocytes and no arginase activity is detectable in monocytes, macrophages nor dendritic cells (105). The differences in expression of both isoforms of arginase by myeloid cells in mice and humans is summarized in Table 2.
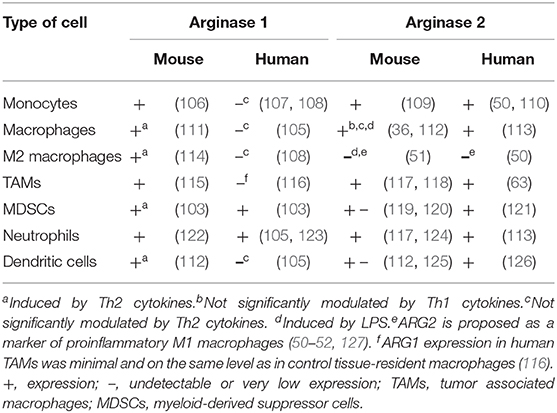
Table 2. Differences in arginases expression in myeloid cells between mouse and human.
The first report linking immunosuppression with arginase activity in macrophages was published over 40 years ago (128). However, the concept that L-arg metabolism is associated with regulation of the immune response did not gain much attention initially. It was suggested that suppressive effect of arginase may be just an interesting problem of in vitro culture (129). However, soon other studies described depletion of L-arg by macrophages expressing arginase both in vitro (130) and in vivo in tumor-bearing mice (131). The authors hypothesized that arginase may be an effector mechanism of macrophages against infectious microorganisms and tumor cells (131). After over 30 years we know that arginase plays an opposite role in immune response and is one of the main mechanism of immunosuppression.
L-arg depletion by suppressive myeloid cells in the tumor microenvironment can occur by increased L-arg uptake by CAT-2B transporters (132), which is followed by arginase-mediated hydrolysis (Figure 3). Myeloid cells also secrete arginase to the microenvironment (133), where it acts mainly locally due to short circulating half-life (134). Murine MDSCs deplete L-arg by increased uptake and intracellular degradation, in contrast to human MDSCs that mainly release arginase into the circulation (103). ARG1 may also be secreted in extracellular vesicles (EVs) by MDSCs (135). In EVs, arginase remains stable and may exert greater than local effects, for instance in draining lymph nodes (64).
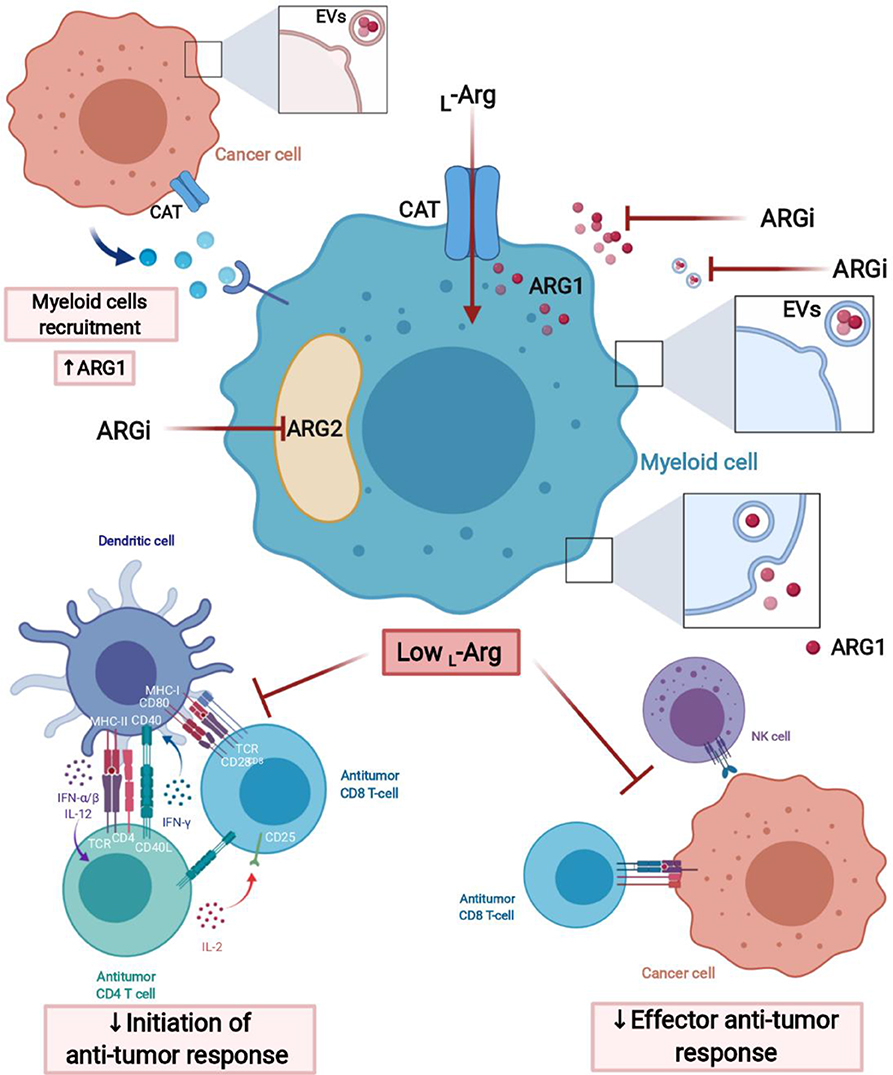
Figure 3. L-Arginine-depleting arginases lead to the impaired anti-tumor response. Arginases may act intracellularly (cytoplasmic ARG1 and mitochondrial ARG2) and extracellularly (secreted ARG1) leading to the local depletion of L-arginine in tumor microenvironment (TME). Moreover, ARG1 may have effects in sites distant from the TME, when packed into extracellular vesicles (EVs), transported over long distance and internalized by myeloid cells, for instance, in tumor-draining lymph node. Arginase inhibitors (ARGi) should target both isoforms (ARG1 and ARG2) and easily penetrate the cell membrane to block extracellular and intracellular arginases, as well as arginase in EVs. Created with BioRender.
Arginase in Myeloid-Derived Suppressor Cells
MDSCs have been the most intensively studied cells in terms of L-arg metabolism. Bronte et al. were the first to show that myeloid cells accumulating in the spleens of tumor-bearing mice express ARG1 and suppress the proliferation of allogeneic T-cells (141). Liu et al. showed that myeloid cells in the tumor microenvironment express arginase and suppress cytotoxic T lymphocyte (CTL) activity in NO-independent manner (142). Since then, many other studies confirmed that immature tumor myeloid cells express ARG1 in mice and humans with cancer and that the activity of this enzyme is involved in suppression of T-cell response (132, 143–146). The majority of studies indicate that arginase plays a more important role in PMN-MDSC rather than M-MDSC (103, 147–149). However, the role of this enzyme in the regulatory activities of the latter cells should not be completely dismissed. For example, iNOS inhibitor together with ARG inhibitor diminished the suppression driven by M-MDSC, with no effect on PMN-MDSC (150).
In humans, PMN-MDSCs store ARG1 in granules and release it to the extracellular milieu (103). It leads to the depletion of L-arg and suppression of anti-tumor response. In patients with pancreatic ductal adenocarcinoma CD13hi PMN-MDSCs were identified that produce ARG1 and suppress alloreactive T-cell responses in ARG1-dependent manner. Patients with more CD13hi PMN-MDSCs had significantly shorter survival than those with predominant CD13low PMN-MDSCs in the tumor infiltrates (149). Similarly, ARG1-producing MDSCs in patients with renal cell carcinoma turned out to be of granulocytic lineage (103). Interestingly, treatment of patients with IL-2 increased the number of these cells in peripheral blood, as well as in the plasma concentrations of ARG1 (103). Whole mount labeling and clearing followed by three-dimensional light sheet microscopy of head and neck carcinomas identified intratumoral hotspots of PMN-MDSCs that co-localized with T-cells. Those T-cells that were in close proximity to ARG1-positive PMN-MDSCs had strongly reduced expression of granzyme B (serpin participating in cytotoxic effects of T-cells) and Ki67 (a proliferation marker) (151). In multiple myeloma IL-18 was shown to induce ARG1+ PMN-MDSCs that suppress immune response (152). In KRASG12D genetically engineered mice that develop lung tumors resembling NSCLC, PMN-MDSCs were observed to cause T-cell suppression by L-arg depletion. Arginase inhibitor has not only restored T-cell function, but caused significant regressions of tumors in these mice (117). Arginase-expressing MDSCs were also shown to induce Tregs in murine tumor models (153) as well as in cancer patients (154). In some of these studies, this effect was abrogated by arginase inhibitor (153) indicating a specific role of this enzyme in Treg development (see below).
Arginase expression in tumor MDSCs is increased as compared with the cells of the same phenotype isolated from spleen (155). Both inflammatory and tumor-derived factors are involved in the regulation of ARG1 expression in MDSCs (Figure 4). For example, tumor-infiltrating MDSCs stimulated with TGF-β and IL-10 demonstrated high ARG1 activity (156). One mechanism involves stress sensor C/EBP-homologous protein (CHOP), which directly activates ARG1 gene through inhibition of LIP transcription suppressor. CHOP expression in MDSCs is induced by ROS and further by the activating-transcription factor-4 (ATF-4) (157). Intriguingly, diminished L-arg concentrations have been shown to induce accumulation of arginase-expressing MDSCs in the tumors after administration of pegylated recombinant ARG1 to tumor-bearing mice (158) indicating potential threats associated with L-arg-depleting therapeutic strategies for cancer. ARG1 levels in MDSCs from patients with head and neck cancer were regulated by STAT3 signaling (159). Accordingly, STAT3 silencing in MDSCs from prostate cancer patients abrogated their immunosuppressive activity (160). Chronic stress, which frequently accompanies cancer, was reported to increase the generation of ARG1+ MDSCs in mice and humans, through catecholamines stimulating β2 adrenergic receptors (β2AR). Induction of ARG1 by isoproterenol (a β2AR agonist) was associated with STAT3 phosphorylation in MDSCs (161). It was found that prostanoids produced by COX2 are responsible for mediating ARG1 overexpression in MDSCs by lung cancer cells in in vitro and in vivo models (162). The mechanism of ARG1 upregulation in MDSC is probably controlled by EP4 receptor for PGE2. Those findings were confirmed in other tumors (144, 163). MDSC not only infiltrate tumor and its environment, they were also found in peripheral blood. (103). MDSC abundance in blood correlated with staging in HNSCC patients. Moreover, MDSC in HNSCC have high level of pSTAT3 and ARG1 and potently inhibit T-cells proliferation (159).
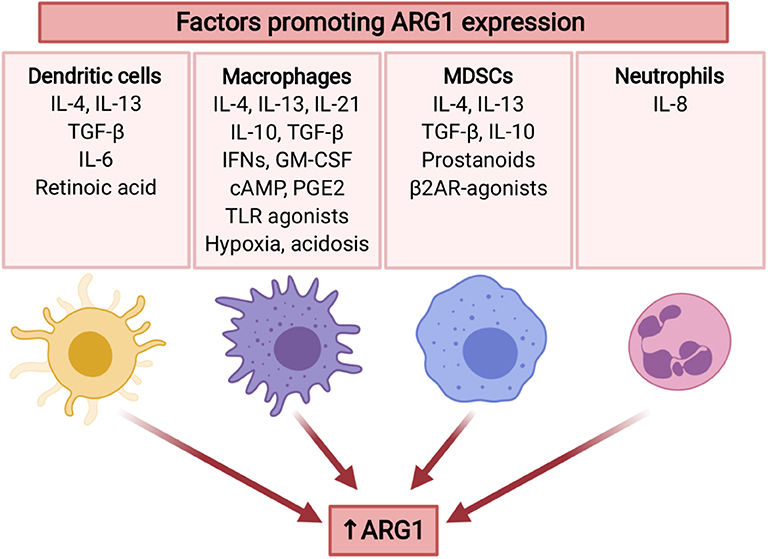
Figure 4. Several tumor-associated factors promote ARG1 expression in myeloid cells. ARG1 expression is mainly induced by type 2 cytokines (IL-4, IL-13), as well as immunosuppressive cytokines (TGF-β, IL-10). Moreover, it may be promoted by TME factors including hypoxia and acidosis, as well as stress mediators. ARG1 expression is also induced by GM-CSF (136), TLR agonists (137), and cAMP (138). IL-10 and IL-21 increase IL-4-induced ARG1 expression (139, 140). Created with BioRender.
Macrophages
Macrophages are the main phagocytic population of cells within tumors (47). However, contrary to their natural role in promoting immunity against infectious microorganisms, tumor-associated macrophages (TAMs) are involved in promoting tumor progression, partly through creating an immunosuppressive microenvironment (47). The majority of reports on macrophages in cancers describe their function in the context of in vitro polarization into M1 or M2 subsets (164). This classification is currently not recommended as TAMs are represented by a continuum of phenotypic variants (47), but will be incidentally used hereafter considering that the existing literature specifically refers to M1 and M2 macrophage subsets. It must be underscored however, that TAMs are highly diverse and form a wide range of populations with various functional roles (165). Additionally, these cells do not form a stable population, but are highly variable both in time and location within the tumor milieu (165, 166). So called M1 macrophages are induced by lipopolysaccharide (LPS) and type 1 cytokines (mainly IFN-γ), express high levels of tumor necrosis factor (TNF), IL-12, iNOS, and MHC class II molecules and are considered to participate in anti-tumor immunity (47). M2 macrophages are induced by type 2 cytokines and express ARG1, IL-4, IL-13, IL-10, and CD206 (47, 167). Cytokines, especially those associated with type 2 immune response (IL-4 and IL-13) that activate the transcription factors STAT6, PU.1, and CCAAT/enhancer binding protein β (C/EBPβ) were shown to directly induce signaling pathways leading to increased production of ARG1 in macrophages (168). IL-4- and IL-13-activated STAT6 with STAT3 and C/EBPβ bind to an enhancer in the ARG1 locus (169). Some cytokines, including IL-10 and IL-21, upregulate the expression of IL-4Rα and IL-13Rα1, leading to the increased IL-4-induced ARG1 expression (139, 140). M2 macrophages are the most abundant population of myeloid cells in tumors, and their presence is usually associated with poor prognosis, tumor cell invasion, metastasis, and neovascularization (170, 171). Importantly, TAMs are considered to be of either embryonic origin or to derive from hematopoietic stem cells (HSCs) (172–175). Both populations are found in the tumors in approximately similar ratio, but it seems that it is mainly the latter population that includes cells with immunosuppressive properties (47). HSC-derived macrophages in a tumor microenvironment sense local physicochemical conditions that are different than in many normal tissues and include hypoxia, acidosis, changes in the composition of extracellular matrix proteins (that affects rigidity of the tumor tissue), nutrient insufficiency, different cellular metabolites, various growth factors and inflammatory mediators (prostanoids, cytokines, etc.) (165). Necrosis and other forms of cell death lead to appearance of cell debris as well as cell death-associated molecular patterns [CDAMPs, also known as death-associated molecular patterns—DAMPs (176)] that additionally affect differentiation of macrophages. Many of these environmental conditions have been shown to induce ARG1 in TAMs including hypoxia via hypoxia-inducible factors (HIFs) (177), lactic acid (in a HIF-1α-dependent mechanism) (178), or COX2 via prostaglandin E2 (162) (Figure 4). Even local acidosis might be involved in ARG1 induction as resting macrophages at pH of 6.1 were observed to induce expression of VEGF, HIF-1α and ARG1 (179), and induction of ARG1 by IL-4 was stronger at pH of 6.8 (180). Cancer-associated fibroblasts (CAFs) have been shown to regulate macrophage differentiation and confer these immunosuppressive cells with the ability to secrete high levels of IL-6 and to produce collagen that leads to the development of tumor desmoplasia (181). Collagen forms a scaffold for many secreted mediators including TGF-β. The number of ARG1 positive macrophages was decreased in Mer tyrosine protein kinase (MERTK) knock-out mice (182). MERTK is involved in signaling triggered by recognition of apoptotic cells. Quite unexpectedly, a recent study revealed that type I interferons (IFNs) inhibit monocyte to macrophage differentiation within tumor and induce strong expression of ARG1 (183).
Macrophages are the main source of ARG1 within tumors in a murine model of colon adenocarcinoma (115). In vivo imaging of tumor macrophages revealed that in contrast to tumor periphery these cells are highly mobile within the tumor microenvironment, exhibit structural diversity and gene expression profile that includes increased ARG1. The number of these ARG+ macrophages significantly decreased after anti-PD-1 monoclonal antibody treatment (115). TAMs in lung cancer and melanoma also express more ARG1 than all other cells within tumor combined (178) and have over 20 times higher expression of ARG1 as compared with peritoneal macrophages (184).
Arginase production by macrophages not only leads to the inhibition of anti-tumor response via L-arg degradation, but also increases the proliferation of tumor cells, which is associated with the production of L-ornithine and then a polyamine—putrescine that promote tumor cells proliferation (185). Moreover, L-arg depletion in the tumor microenvironment attenuates NO production and reduces its cytotoxic effects on tumor cells (185). Several studies also indicate that arginase activity might be associated with delivery of additional metabolites with immunosuppressive properties. For example, inhibition of polyamines synthesis together with blocking of dietary polyamine transport was shown to exert antitumor effects that were associated with decreased numbers of intratumoral MDSCs and increased numbers of T-cells (186). Similar approach was shown to increase in granzyme B+IFN-γ+CD8+ T-cells and a decrease in immunosuppressive tumor-infiltrating cells including PMN-MDSCs, Tregs, and M2 macrophages (187).
Neutrophils
Neutrophils are the most abundant leukocytes in peripheral blood and are produced in the bone marrow at a prodigious rate of 1 × 1011 cells per day (188). These cells constitute a rapidly reacting part of innate immune response, playing important role in defense against bacteria and fungi. Despite their important role in host defense, the increased numbers of neutrophils in blood of cancer patients correlate with poor prognosis (189). These cells can also be found in tumors, but their role in tumor has been largely neglected, mainly due to the belief that their life-span is one of the shortest among all leukocytes. However, tumor-associated neutrophils (TANs) persist in tumor microenvironment for extended time in response to GM-CSF and TGF-β (126). TANs are divided into two subtypes: N1 and N2, with anti-tumor and protumorigenic phenotype, respectively, but to date no specific molecular surface markers have been identified to distinguish them. Nonetheless, N2 neutrophils are characterized by high arginase expression (132, 190). ARG1 is in fact constitutively expressed in human neutrophils. However, these cells do not metabolize L-arg (123) possibly due to the confinement of ARG1 in gelatinase granules (191). Neutrophils can release ARG1 leading to the suppression of T-cells function (192). This process requires simultaneous exocytosis of ARG1-containg gelatinase granules and azurophil granules (192). It was assessed that 1 × 106 of neutrophils secrete ARG1 at amounts sufficient to catabolize all the L-arg contained in 5 ml of blood in 1 h (193). At least in some tumors ARG1+ neutrophils are quite abundant and the presence of ARG1+ neutrophils correlates with suppressed T-cell functions (193, 194). Intriguingly, in non-small cell lung cancers despite high arginase activity in tumor microenvironment, most of the TANs display low or no ARG1 expression, in contrast to neutrophils in peritumoral tissue that strongly stain for ARG1 (193). It turned out that tumor cells release IL-8 that induces ARG1 exocytosis from neutrophils into extracellular milieu (193) (Figure 4). Degranulated neutrophils are also expanded in peripheral circulation of cancer patients, and ARG1 released from these cells strongly contributes to general suppression of T-cell functions (195). ARG1 released from neutrophils has also been shown to inhibit the proliferation of NK cells and IL-12/IL-18-induced production of IFN-γ (196). Zoledronic acid, a bisphosphonate used in the treatment of osteoporosis has been shown to induce ARG1 in neutrophils that suppress the activity of γδ T-cells (197). All these observations indicate that ARG1+ neutrophils seem to play a detrimental role in tumor progression, mainly due to immunosuppressive effects. Notably however, a recent study indicated that high intratumoral neutrophil numbers expressing ARG1 correlate with better survival of patients with colorectal cancer (198).
Dendritic Cells
Dendritic cells (DCs) are classically described as professional antigen-presenting cells that produce cytokines and provide co-stimulatory molecules, leading to naïve T-cells activation and differentiation into effector cells (199). There are conventional DCs (cDCs), plasmacytoid DCs (pDCs), and monocyte-derived DCs (MoDCs) that have different origin and differ in function. Within cDCs there are additional subsets both in mice and in humans that are referred to as cDC1 and cDC2. cDCs1 are presumed to be primarily involved in cross-presentation of antigens to CD8+ T-cells, while cDCs2 seem to be largely associated with stimulating CD4+ T-cells (200). Another layer of subdivision into migratory and lymph node (LN)-resident DCs reflects location and the mechanisms of antigen acquisition by these cells. Migratory CD103+ DCs take up antigens in non-lymphoid tissues (including tumors) and traffic through lymphatic vessels into LNs. LN-resident CD8αα+ DCs enter the LNs from the blood and acquire antigens draining through the lymphatics or transported to LNs by other cells (200).
Tumors are frequently infiltrated by various populations of DCs. During infections DCs acquire, process and present antigen in association with MHC molecules, deliver co-stimulatory signals and release cytokines that shape T-cell responses. The same role is expected to be played by DCs in tumors. However, the stimulatory activity of these cells is often compromised and tumor DCs often drive tolerance rather than immunity in cancer patients (201). The mechanisms of tumor-infiltrating DCs that hamper development of antitumor immune response include decrease in MHC class I and II levels as well as in co-stimulatory molecules (CD40, CD80, CD86), rise in co-inhibitory molecules (such as PD-L1, PD-L2, VISTA), increased tryptophan degradation by indoleamine 2,3-dioxygenase (IDO1), decreased release of IL-12, but increased secretion of IL-10 and TGF-β, among others (201). Arginases can be added to this expanding list, based on numerous reports.
Lung cancer cells isolated from murine tumors induced DCs to differentiate into regulatory cells that suppressed T-cell response through ARG1 (202). In another study tumor-infiltrating DCs were observed to decrease the expression of CD3ζ in T-cells in ARG1-dependent manner and induced anergy in naïve CD8+ T-cells (203). ARG1 produced by DCs promotes the generation of FoxP3+ Tregs (204, 205). Not only ARG1 was shown to be expressed by DCs. Human fetal cDC2 cells uniquely express constitutively high levels of ARG2, through which these cells inhibit T-cell activation and TNF-α release (206).
The expression of ARG1 in DCs is regulated by a number of cytokines and tissue factors (Figure 4). As in other myeloid cells, ARG1 is induced by type 2 cytokines, including IL-4 and IL-13. Tregs were reported to induce ARG1 in DCs in a TGF-β-dependent mechanism (207). Supernatants from tumor cells experiencing endoplasmic reticulum (ER) stress and unfolded protein response (UPR) was shown to induce ARG1 in DCs (208). Retinoic acid was also shown to be a key mediator regulating expression of ARG1 in DCs, mediated by retinoic acid-responsive elements in the 5′ non-coding region of the ARG1 gene. Blockade of retinoic acid receptors makes DCs less responsive to IL-4 and GM-CSF (205).
Mechanisms of Immunoregulatory Function of Arginase
An obvious question in understanding the role of amino acid-degrading enzymes in the regulation of the immune response is why do myeloid cells degrade L-arg Perhaps the best answers come from studies in mice with targeted deficiency of ARG1 in myeloid cells and the regulation of immune response and inflammation triggered by infectious microorganisms. ARG1 induced in macrophages during Schistosoma mansoni infection prevented cachexia, neutrophilia, and endotoxemia during acute schistosomiasis. Moreover, ARG1+ macrophages promoted TGF-β production and Foxp3 expression, suppressed antigen-specific T-cell proliferation, and limited Th17 differentiation. In mice with deficiency of ARG1 in myeloid cells infection with Schistosoma mansoni triggered a lethal T-cell-dependent immunopathology with non-resolving inflammation (209). On the other side, ARG deficiency in myeloid cells results in substantially decreased tumor growth (210) and increased CD8+ T-cells numbers and activity as compared with wild-type mice (211).
Effects on Effector Functions in T-Cells
Lack of any single essential amino acids restricts T-cells activation and proliferation and this phenomenon is not specific to L-arg. Depletion of L-histidine, L-leucine, L-lysine, L-phenylalanine, L-threonine, and L-valine inhibited the proliferation of T-cells to a similar extent as L-arg depletion (207). Of importance, however, only arginases as well as IDO that hydrolyzes L-tryptophan (212, 213) are substantially increased in cancer.
Role of L-arg in T-Cell Proliferation
One of the hallmarks of ARG activity in the immune system is impaired T-cell proliferation (Figure 5). Proliferation of both human and murine T-cells is completely inhibited in L-arg-free medium after stimulation with anti-CD3- and anti-CD28-coupled beads or different types of mitogens. A similar inhibition of the T-cells proliferation is also triggered by ARG-producing cells, and this effect is restored by L-arg supplementation or arginase inhibitors (123, 132, 203, 214, 215). It is of note that T-cells remain viable in L-arg-depleted medium (123) and resume proliferation as soon as L-arg is added to the culture medium. The minimum L-arg concentration in cell culture medium necessary for one division of murine T-cell was determined to be 23 μM (216). Upon activation, when large amounts of L-arg are needed, T-cells rely mainly on the extracellular L-arg transport. A potent increase in the expression of cationic amino acid transporter-1 (CAT-1) is observed in both naïve and memory CD4+ and CD8+ T-cells after activation (22). Silencing of CAT-1 expression leads to the inhibition of T-cell proliferation, but not impaired TNF-α, IFN-γ, IL-2, IL-6 production (22).
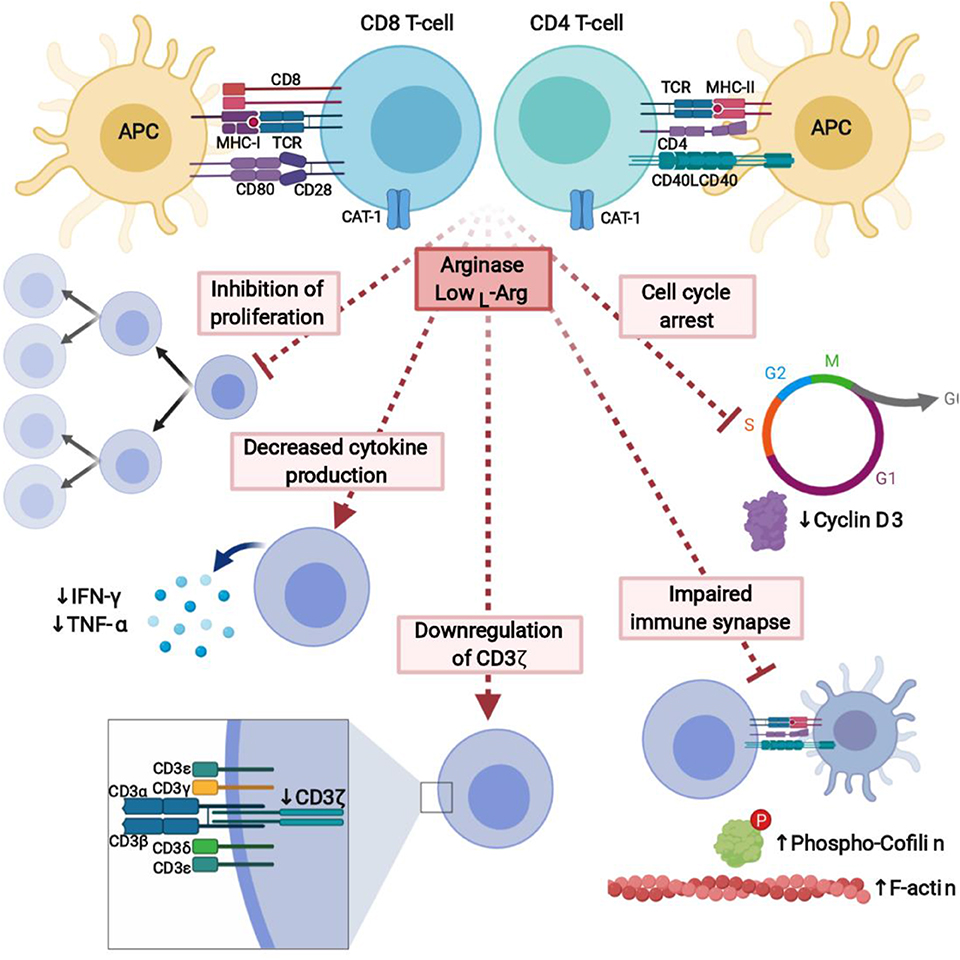
Figure 5. L-Arginine depletion by arginase potently inhibits immune response. Lack of L-arg completely inhibits proliferation of T-cells and leads to the decreased cytokine production. It is caused by downregulation of signal-transducing CD3ζ chain, cell cycle arrest, and affected formation of the immune synapse between T-cells and APC. Created with BioRender.
Role of L-arg in T-Cell Cytokine Production
Secretion of several cytokines that play a critical role in T-cell differentiation and effector functions is also diminished in L-arg-starved cells (Figure 5). Conspicuously, this especially refers to the secretion of Th1 cytokines, including IFN-γ and tumor necrosis factor β (TNF-β) (123, 214, 217), although T-cells cultured in L-arg-free medium also secrete lower amounts of IL-5, and IL-10 as compared with T-cells cultured in complete medium (218). The decrease in IFN-γ secretion is also induced by ARG+ tumor-infiltrating DCs (203) and ARG inhibitors administered in vivo increase IFN-γ secretion (219). On the contrary, the synthesis of IL-2, IL-6, and IL-8 seems to be unaffected by the absence of L-arg (217), although in another study PMN-MDSCs were shown to suppress IL-2 production from T-cells and this effect was restored by ARG inhibitor (220).
Role of L-arg in T-Cell Differentiation
Upon antigen recognition naïve T-cells proliferate and acquire effector functions that are dependent on multiple additional signals delivered in the microenvironment of secondary lymphoid organs. The signals include various cytokines, growth factors, and surface-associated molecules (including co-stimulatory and adhesion molecules) (221, 222). Accumulating evidence indicates that L-arg metabolism plays an important role in regulating T-cell differentiation. For example, oral administration of L-arg in a mouse model of breast cancer increased the levels of T-bet, a transcription factor associated with Th1 cells (223). Moreover, it increased the frequency of CD8+ T-cells, and mRNA levels of granzyme B and IFN-γ in the tumor (223). High extracellular L-arg increased the survival of T-cells stimulated with IL-2 after cytokine withdrawal and favored the formation of central memory T-cells (224). Inhibition of L-arg transport into the cell decreased T-cell longevity further confirming the role of L-arg in human T-cells survival (224).
Somewhat enigmatic and to some extent contradictory reports refer to L-arg metabolism and Treg cells development. In an interesting study FoxP3+ Tregs were shown to induce ARG1 (as well as other enzymes involved in amino acid metabolism) in DCs, thereby increasing amino acid consumption in local microenvironment. This reduced mTOR signaling and favored development of additional Tregs (207). Inhibition of mTOR signaling by rapamycin or amino acid depletion was shown to induce FoxP3, but L-arg deficiency was effective only when TGF-β was added (207). Moreover, ARG2 was found in Tregs from normal skin and its expression increased in metastatic melanoma (225). ARG2 in Tregs was demonstrated to attenuate mTOR activity and conferred Tregs with enhanced suppressive activity (225) suggesting that low intracellular L-arg concentrations may facilitate Tregs development. Consistently, with these findings, T-cells from mTOR-deficient mice preferentially become regulatory, but not effector T-cells (226).
However, another study showed that mice fed with L-arg-deficient diet had modestly reduced number of peripheral effector Tregs and these cells had reduced expression of ICOS and CTLA4. L-arg turned out to be essential for sustaining mTORC1 activity, functional programming, and Treg cell-mediated immunosuppression (227). Moreover, disruption of mTORC1 in FoxP3+ T-cells caused a loss of Treg suppressive activity in vivo and led to development of systemic immunopathology in mice indicating that Treg cell responses are critically dependent on mTORC1 signaling (228). Clearly, the effects of L-arg metabolism on T-cell differentiation are very complex and require further studies, especially that still another report indicated that ARG1 in MDSCs is participating in promotion of Th17 differentiation (229).
Molecular Mechanisms of Immunoregulatory Effects Associated With L-arg Metabolism
The exact molecular mechanisms of L-arg starvation-mediated immunosuppression still remain to a large extent enigmatic. Up to now, L-arg starvation was shown to affect T-cell antigen receptor ζ chain (CD3ζ) expression (230) and phosphorylation of other signal-transducing proteins (231), and therefore to impair transduction of activation signal, cell cycle progression (232), as well as formation of the immune synapse (231) (Figure 5).
Downregulation of the CD3ζ and Impaired Signal Transduction
The main mechanism by which L-arg starvation inhibits T-cells proliferation is through downregulation of the CD3ζ chain (230, 233). CD3ζ is a critical component of the TCR complex that couples antigen recognition to the intracellular signaling pathways (234). After T-cells stimulation, TCR proteins including CD3ζ undergo internalization followed by re-expression, externalization, or sorting to lysosomes for degradation (235, 236). A common finding in cancer patients is a marked decrease in the expression of CD3ζ in T-cells (143, 237).
Many studies reported that L-arg depletion in culture medium leads to a rapid decrease of CD3ζ levels (132, 230, 238). Of note, the changes in TCR receptor subunits expression during L-arg starvation are observed only in stimulated T-cells (218). This decrease is specific to L-arg-starvation, since lack of glutamine or leucine (233) as well as glycine or lysine (218) did not change the levels of CD3ζ. However, a decrease in CD3ζ was also reported to be caused by hydrogen peroxide secreted from tumor macrophages (239). The decrease in CD3ζ is completely reversed by L-arg supplementation in cell medium (230) or ARG inhibition when co-culture with ARG-producing cells is used (132). A similar downregulation of CD3ζ and CD3ε levels is induced by tumor-associated myeloid cells, which express ARG1 (132). This effect is prevented by the addition of ARG inhibitor (N-hydroxy-nor-L-arg) or L-arg supplementation, but not by the catalase, a hydrogen peroxide scavenger (132), as suggested before (239).
How L-arg starvation selectively impairs CD3ζ expression still remains unclear. L-arg starvation of human T-cells did not affect the degradation of CD3ζ in proteasome or lysosomes (218). Therefore, it was suggested that L-arg depletion may impair CD3ζ synthesis (218) or the stability of mRNA for CD3ζ (230).
Cell Cycle Arrest
Another defined mechanisms by which L-arg starvation restricts T-cells activation and proliferation is the regulation of cell cycle progression (232) via modulation of cyclin D3 mRNA stability (240). Cyclins, including cyclin D3 (241), are critical regulators of the cell cycle, immune cells development and proliferation (242). L-arg starvation arrests human T-cells in G0-G1 phase (232). The levels of cyclin D3 as well as CDK4 significantly increase after T-cells activation, however, not in absence of L-arg. Moreover, silencing of cyclin D3 in Jurkat cells reproduces effects induced by L-arg starvation (232). Cyclin D3 was shown to be regulated by L-arg through transcriptional, posttranscriptional, and translational mechanisms (232). In the absence of L-arg human T-cells have decreased phosphorylation of the retinoblastoma protein (Rb), which is the major substrate for the cyclin D/cyclin-dependent kinase complex, as well as decreased levels of E2F-1, which is crucial for the initiation of the transcription of genes involved in the G2/S transition (232). In the absence of L-arg there is a global arrest in de novo protein synthesis. L-arg starvation also affects the expression of HuR, RNA binding protein, that stabilizes mRNA of cyclin D3 by the binding to the 3'-untranslated region (UTR) and shuttles its transport to the cytoplasm. Silencing of HuR exerts similar effect on T-cells proliferation as L-arg starvation (240).
Changes in the Immune Synapse Between APC and T-Cells
Proliferation of T-cells after antigen presenting cells (APC)-based cellular activation is also completely inhibited in the absence of L-arg (231). The formation of immune synapse between T-cell and APC is critical for the activation of effector cell (243). In L-arg-depleted medium, the formation of the immune synapse is impaired. T-cells activated in the absence of L-arg have increased F-actin concentration, which may be caused by impaired cofilin dephosphorylation (231). Cofilin is a small actin-remodeling protein that couples T-cell activation via the TCR and co-stimulatory receptors in the immune synapse (231, 244). Phosphorylation of ERK1/2 is significantly reduced in L-arg absence, however, the phosphorylation of AKT is increased to the higher level compared to the cells activated in L-arg-containing medium (231). It leads to the impaired dephosphorylation of cofilin that results in impair immune synapse formation. Impaired dephosphorylation of cofilin in human T-cells was also induced by cell-free human pus supernatant, which is known to contain high arginase activity (123). This effect may be prevented by arginase inhibitor (231).
L-arg in Metabolic Regulation of T-Cells
Proliferation and differentiation of T-cells can occur only if sufficient access to metabolites and nutrients is ensured (245). A recent metabolomic analysis of activated T-cells revealed that out of 429 measured metabolites only 14 were less abundant in activated T-cells, and L-arg was the only protein amino acid among them (224). A drop in intracellular L-arg levels was observed despite induction of CAT-1 transporters. Interestingly, the intracellular levels of L-glutamine, which is also intensively metabolized in activated cells, remained high. Along with CAT-1 induction, T-cell activation was associated with increased expression of L-arg metabolism-related enzymes including ARG2, OAT, and spermidine synthase (SRM). Once entering the cell, L-arg turned out to be rapidly converted into L-ornithine, agmatine, and putrescine. Importantly, increasing L-arg concentration in the culture medium upregulated gluconeogenesis-related genes, serine biosynthesis pathway, and mitochondrial tricarboxylic acid cycle, while downregulating glucose transporter and glycolytic enzymes. These changes promoted mitochondrial OXPHOS in activated T-cells, while downregulating glycolysis (224). Global analysis of T-cells proteome changes in response to high L-arg concentration revealed several proteins that are responsible for increased T-cells survival. These can be assigned into four functional groups, including mRNA splicing, DNA repair mechanisms, regulation of the cytoskeleton and the ribosome (224).
Oral supplementation of L-arg that increased its serum concentration over 4-fold allowed more robust induction of antigen-specific T-cell proliferation in mice. Moreover, T-cells from ARG2−/− mice, incubated with supplemental L-arg or treated with ARG inhibitor reveled much better survival after cytokine withdrawal (224). In a complementary study, CD8+ T-cells from ARG2-deficient mice showed markedly superior antitumor activity in mice and turned out to respond stronger to PD-1 blockade as compared with ARG2+ T-cells (246). Moreover, ARG2-deficient T-cells were characterized by faster acquisition of effector functions, increased persistence and enhanced differentiation into memory cells.
Altogether, these studies indicate that ARG2 might be a metabolic gatekeeper in T-cells. In activated T-cells ARG2 degrades L-arg and generates agmatine and polyamines. In case of accessible L-arg in the extracellular environment the intracellular pool of this amino acid can be replenished. However, at sites, where extracellular L-arg is depleted (by tumor cells or tumor-infiltrating myeloid cells) the intracellular pool cannot be restored leading to T-cell suppression (224).
Mechanisms of arginine-starvation sensing in immune cells are still unclear. It is suggested that mTOR together with GCN2 kinase regulate amino acid metabolism and response to arginine starvation (207, 227, 232, 247, 248), however, the exact mechanism is unknown and requires further investigation.
B Cells and L-arg
The role of L-arg in B-cells functions was much less investigated and is poorly understood. It was shown that L-arg deficiency due to high ARG1 activity in F/A-2+/+ transgenic mice, that overexpress arginase in enterocytes, potently impairs early B cell maturation with no major impact on T-cells (249). F/A-2+/+ mice have reduced number of B cells, decreased serum IgM concentration and hampered B cell maturation in the early pre-B cell stage (249). L-Arg-free diet fed mice which have significantly lower concentration of plasma L-arg compared to L-arg-supplemented diet had also impaired antigen-specific mucosal immune response against tetanus toxoid (TT). After oral administration, no TT-specific fecal IgA antibodies were detected in L-arg-free diet fed mice (250). Both PMN-MDSCs and M-MDSCs were shown to regulate key B-cell functions, particularly B-cell proliferation and antibody production. PMN-MDSC-mediated B-cell suppression turned out to be cell contact dependent and involved ARG1 (251). A recent study from the same group indicated that M-MDSCs suppress B-cell proliferation, and downregulate IgM, HLA-DR, CD80, CD86, TACI, and CD95 in contact independent, but ARG1 and iNOS-dependent mechanism (252).
Myeloid Cells and L-arg
The role of L-arg in differentiation of myeloid cells is poorly investigated. Most of the studies focused on the role of ARG1 produced by myeloid cells rather than the dependence of these cells on L-arg. Individual results in vitro show no influence of L-arg on macrophages differentiation, maturation, and effector functions. In the absence of L-arg, maturation of macrophages into classically activated macrophages (M1) and alternatively activated macrophages (M2) was unaffected (253). Moreover, the production of cytokines by both macrophage subtypes was unimpaired under L-arg-starvation (253). Likewise, the expression of iNOS by M1 cells as well as the expression of ARG by M2 cells turned out to be independent from the L-arg concentration (253). However, ARG1 expression was essential for monocytic DC differentiation (254). ARG1 was also recently shown to be crucial in efferocytic clearance of apoptotic cells by macrophages (255).
In vivo however, L-arg supplementation was shown to promote Gr-1+CD11b−F4/80+, but suppressed Gr-1+CD11b+F4/80+ macrophages in a murine model of breast cancer (223). However, these effects might not be caused directly, but rather result from the effects on T-cells activation. Another study showed that L-arg starvation promotes tumor G-MDSC accumulation, which further suppress T-cells anti-tumor response (158). Similar results were obtained with PEG-asparaginase administration, suggesting that generally amino acid starvation results in MDSC accumulation. PEG-ARG1-induced MDSC accumulation was found to be regulated by GCN2, since the accumulation of MDSCs in GCN2-deficient mice treated with PEG-ARG1 was negligible (158). Importantly, MDSCs isolated from GCN2-deficient mice had similar immunosuppressive properties as compared with MDSCs isolated from wild-type mice, which suggests that GCN2 is involved in the accumulation of MDSC, but not in their effector functions. Moreover, it was observed that ARG2-releasing AML blasts as well as ARG2-rich plasma of patients with AML promotes the differentiation of monocytes toward M2 macrophages. These effects were diminished by L-arg supplementation or arginase inhibitors (61).
NK Cells and L-arg
NK cells are less sensitive to low L-arg concentrations as compared with T-cells however, L-arg starvation affects the main effector functions of NK cells (196). L-arg starvation decreases NK cells proliferation and viability, as well as cytotoxic activity (210, 256). Depletion of L-arg leads to the reduction in the expression of NKp46 and NKp30 activating receptors, as well as the NK cell ζ chain expression in the FcγRIIIA, similar to the CD3ζ chain in T-cells. Moreover, in the absence of L-arg the production of IFN-γ by NK cells is significantly decreased (256). Similar effect is exert by arginase from human neutrophils (196). However, NK cell degranulation and cytotoxicity seems to be unaffected by L-arg depletion (196).
L-arg Metabolites and Immune Response
L-arg is in the center of many metabolic pathways. Arginase not only depletes L-arg, but also creates multiple downstream metabolites including L-ornithine and urea, as well as L-proline, glutamate, agmatine, putrescine, L-citrulline, and polyamines.
Ornithine
L-arg is degraded by arginase to L-ornithine and urea. While the concentration of L-arg substantially decreases in cancer, the concentration of L-ornithine increases (59, 75, 257). High concentration of L-ornithine in tumor interstitial fluid may inhibit anti-tumor cytotoxic response of CD8+ T-cells (258, 259), and together with L-arg depletion, that affects T-cells properties but not cytotoxicity (214), provide effective tumor evasion of the immune system. Reversible inhibition of cytotoxicity of T-cells in the presence of L-ornithine is independent from the type of stimulation and it seems that it affects early stages of CTL activation (258). However, L-ornithine did not impair mitogenic response to the stimulation (258, 259), as well as IL-2 and IL-3 production (258). ODC catalyzes the conversion of L-ornithine to polyamines.
Polyamines
A diamine putrescine, triamine spermidine, and tetraamine spermine are ubiquitous L-ornithine metabolites associated with important cellular processes. Polyamines are essential for cell growth and proliferation during development, wound healing, and tissue regeneration. ODC catalyzes the conversion of L-ornithine into putrescine, which is then metabolized to spermidine by spermidine synthase and spermine by spermine synthase (260). At physiological pH polyamines are positively charged and bind to acidic sites in DNA and RNA, controlling gene expression (261). Moreover, polyamines have antioxidative properties, bind to K+ channels, NMDA receptors, and modulate the activity of various enzymes (261).
Growth promoting functions of polyamines are best described in tumors. However, it seems that polyamines are also important in T-cell clonal expansion. It has been suggested that the synthesis of polyamines in T-cells is under the direction of Myc, as Myc-deficient T-cells fail to induce ODC and other genes involved in polyamine synthesis, leading to decreased polyamine production (262). Spermidine is also a precursor of hypusine, which post-translationally binds to eukaryotic initiation factor 5a (eIF5a). Intriguingly, eIF5a, which prevents ribosomal stalling during translation of certain mRNAs, is one of the most strongly expressed proteins in activated T-cells (263).
Polyamines were reported to exert anti-inflammatory effects in macrophages by restraining activation of M1 while promoting differentiation of M2 subtype. For example, LPS-induced expression of TNF, IL-1, IL-6, IL-12, iNOS, and CD80 was suppressed by polyamines (264–266). Polyamines also modulate immunoregulatory activities of DCs. IDO1 activity in TGF-β-treated DCs requires ARG-1-dependent spermidine synthesis that activates Src tyrosine kinase, which participates in IDO1 phosphorylation (267).
Arginase Inhibitors
Expanding knowledge on the biological role of arginases prompts the idea of therapeutic inhibition of these enzymes. The interplay between ARG and NOS resulting mainly from the competition for the common substrate L-arg makes ARG inhibition an attractive approach in the treatment of cardiovascular and inflammatory conditions (such as asthma, diabetes, hypertension, atherosclerosis, coronary artery disease, heart failure or erectile dysfunctions). Furthermore, inhibition of immunosuppressive functions of arginases is being explored in the treatment of cancer. Modulation of L-arg metabolism is also being explored as a therapeutic strategy in Alzheimer's disease (268).
As many pathogenic bacteria (such as Helicobacter, Mycobacterium, Salmonella), fungi (Candida) and parasites (Trypanosoma, Leishmania, Schistosoma) express species-specific isoforms of ARG to facilitate their survival in the host, finding pathogen-ARG-specific inhibitors emerges as a timely approach in the antibiotic-resistance era. Interestingly, Leishmania parasites induce ARG1 expression in infected macrophages to decrease L-arg availability for iNOS and thus to avoid NO toxicity (269). The latter observation further supports the potential use of ARG inhibitors in the treatment of infectious diseases.
Currently, almost all ARG inhibitors being developed as drug candidates are competitive inhibitors of both isoenzymes (ARG1 and ARG2) and in vast majority are L-arg analogs (270). Finding an isoform-specific ARG inhibitor is challenging as 100% homology exists in the active site between human ARG1 and ARG2. As the results of the preclinical, mainly in vitro, testing of ARG inhibitors have been extensively reviewed elsewhere (270, 271), here we just briefly summarize the data on in vivo and clinical activity of selected ARG inhibitors.
So called first generation of ARG inhibitors such as N-hydroxy-nor-L-arginine (nor-NOHA) (272), (S)-2-amino-6-boronohexanoic acid (ABH) (273) and (S)-(2-boronoethyl)-L-cysteine (BEC) (274) are reversible, modest inhibitors of ARG1 and ARG2 enzymatic activity with either poor pharmacokinetic properties or insufficient penetration through the plasma membrane. In mouse models nor-NOHA has been shown to inhibit local tumor growth in B- and T-cells-dependent manner as well as to reduce metastatic burden (132, 178, 275). Second generation compounds are characterized by better pharmacokinetic and pharmacodynamic properties. As an example, so called compound 9 [(R)-2-amino-6-borono-2-(2-(piperidin-1-yl)ethyl)hexanoic acid] has been recently showed to decrease the growth of KRAS mutated murine lung tumors via inhibition of ARG activity in tumor-infiltrating myeloid cells (117).
Up to date, there are only two ARG inhibitors being tested in clinical trials. Both drug candidates have been developed by Calithera Biosciences and are orally available small-molecule compounds. INCB001158 (CB-1158) is being evaluated in Phase 2 as a single agent and in combination with immune checkpoint inhibitors in cancer (both solid tumors and multiple myeloma), while CB-280 in Phase 1 in cystic fibrosis, exploiting the novel idea of increasing NO production to improve lung function. CB-1158 has been shown ex vivo to reverse human T-cell immunosuppression mediated by ARG1 produced by neutrophils as well as MDSCs (210). It also exerts immune-based antitumor effects in syngeneic mouse tumor models in vivo as a single agent as well as in combination with the immune checkpoint inhibitors (210). An interesting ARG inhibitor to watch is OATD-02 (276), a compound being developed by Oncoarendi Therapeutics. In preclinical models it has been shown to delay ovarian cancer progression and to revert ARG1-mediated inhibition of antigen-specific T-cells proliferation and to restore their CD3ζ levels (64). Moreover, in syngeneic mouse tumors it potentiated the antitumor efficacy of immune checkpoint inhibitors (277). The company claims OATD-02 Phase 1 trial in cancer patients to begin in 2020-2021.
Arginase inhibition cannot be replaced, however, by chronic L-arg supplementation. Dietary intake of L-arg results only in a transient increase of L-arg plasma concentration (278). Moreover, if arginases are active in blood or body tissues, it is very likely that they easily degrade the excessive amounts of this amino acid.
Global ARG1 inhibition rises significant safety concerns. ARG1 gene knockout mice die 10–14 days post-birth (39). Similarly, induction of whole body Arg1 KO in adult “floxed” Arg1 CreETT2 transgenic mice leads to the animals death in up to 2 weeks post-tamoxifen administration (279). The major cause of death in Arg1 KO mice is hyperammonemia resulting from the defect of the liver urea cycle. It is the lack of Arg1 expression in the liver that is fatal, as hepatocyte-specific knockout of Arg1 mimics the whole body deficiency of this enzyme (280). Lack of Arg1 expression leads to altered hepatocytes morphology, significantly increased plasma L-arg and L-citrulline concentrations accompanied by decreased plasma L-ornithine and L-proline concentrations (39). Interestingly, Arg2 knockout mice are viable and do not have a disabling phenotype apart from high plasma L-arg concentrations and decreased male fertility. Moreover, Arg2 KO mice have significantly extended lifespan, indicating some role of this enzyme in aging (41). Double Arg1 and Arg2 KO mice show the same phenotype as Arg1-lacking animals. Unexpectedly, in Arg1 KO mouse embryo no compensatory Arg2 expression was observed (281), suggesting non-overlapping role of both arginase isoenzymes in murine embryonal development. In humans, ARG1 deficiency is a rare autosomal recessive disorder, resulting from over 40 reported mutations in ARG1. In the most severe form ARG1 deficiency results in hyperargininemia, neurological impairment and eventually fatal episodes of hyperammonemia (282). ARG1 deficiency is frequently accompanied by a compensatory increase in ARG2 activity in the kidney, ameliorating metabolic disturbances (44). The latter observation encourages a still very challenging attempt to develop ARG1-specific inhibitors.
Animal studies confirmed that there is a safe therapeutic window for tested ARG inhibitors. In both mice and rats, over 2-months long daily systemic administration of nor-NOHA did not result in detectable toxicity. It is likely, that due to the quantitative differences in ARG1 expression between the liver and other tissues way lower ARG inhibitors concentrations are needed to exert immunomodulatory and/or vascular effects than to block the Krebs cycle in hepatocytes (270).
Initial results of the investigational trial of the oral ARG inhibitor INCB001158 in colon cancer patients proved acceptable safety profile of this drug candidate. A maximum tolerated dose was not reached even for the twice daily total dose of 150 mg. Moreover, clinically significant urea cycle inhibition was not observed. In microsatellite stable (MSS) colorectal cancer patients involved in this study, 7 and 6% of partial responses to the INCB001158 and pembrolizumab (anti-PD1 monoclonal antibody) combination or INCB001158 monotherapy, respectively, were reported. Importantly, objective pharmacodynamic parameters such as an increase in the intratumoral CD8+ T-cells as well as dose-related increase in plasma L-arg were achieved in the treated individuals (283).
To evaluate the clinical efficacy of ARG inhibition in a comprehensive way we need much more data. Nonetheless, existing preclinical and initial clinical evidence seems to support the idea that therapeutic targeting of the immunomodulatory ARG might serve as a potent addition to the other immunotherapeutic strategies rather than as an effective single agent treatment. Moreover, it would be crucial not only to evaluate proper dosing, timing and treatment duration but also to find reliable biomarkers predicting desirable clinical effects.
Although recent data support the idea that ARG overexpression correlates with poor prognosis, a number of studies indicate that arginine depletion may also be beneficial for subgroups of patients, especially those with inactivation of ASS1 in cancer cells that leads to the dependence on exogenous L-arg (284). L-Arg deprivation by ADI conjugated with polyethylene glycol (ADI-PEG) (84, 285) as well as pegylated recombinant human ARG (rhARG-PEG) (286, 287) were applied to the treatment of arginine-auxotrophic tumors and showed potent anticancer effects [reviewed in (288)].
Noteworthy, L-arg-restriction as the regulation of immune response is not specific to the cancer. It was shown that Helicobacter pylori by arginase not only produces urea which can be used to CO2 and NH3 production by urease to support acid tolerance (289). H. pylori using ARG also depletes L-arg which leads to the downregulation of CD3ζ and inhibition of T-cells proliferation during infection (290). T-cells response is also suppressed via ARG by human embryonic stem cells (291). ARG also mediates T-cells hyporesponsiveness in human pregnancy (292), post-stroke immunosuppression (293), as well as in the control of autoimmunity (294). Moreover, H. pylori induces ARG2 expression in macrophages contributing to the immune evasion by limiting production of antimicrobial NO (48). Crucial role of ARG in the regulation of immune response by impairing NO production was also described in the model of cutaneous contact hypersensitivity (295) as well as in immune response to Leishmania major infections (296). Importantly, some intracellular pathogens induce expression of ARG1 in macrophages that hampers effective immune response (137). A recent study revealed that increased ARG levels may play a role in fatigue intensification in cancer patients undergoing external-beam radiation therapy (297)
Final Remarks
ARG expression is substantially elevated in myeloid cells in cancer and mitigate antitumor response via multiple mechanisms. Intriguingly, cytotoxic effects of T-cells are unaffected by a lack of L-arg, despite the fact that CD3ζ and CD3ε are downregulated and thus TCR signal transduction should be inhibited. In contrast, T-cell proliferation is strongly suppressed, but it must be emphasized that T-cells proliferate extensively in tumor-draining lymph nodes, and not in the tumor. L-arg concentrations in tumor-draining LN have not been measured so far. It would also be interesting to see whether increased ARG activity contributes to fibrotic processes leading to desmoplastic changes in some types of tumors, such as pancreatic cancer. Increased activity of arginases could limit L-arg availability to NOS—could it be responsible for vascular abnormalities frequently described in tumorsxx Altogether, increasing evidence indicates that arginases become potentially important targets for therapeutic interventions that might improve the efficacy of immunotherapy, decrease infectious complications and improve quality of life of cancer patients.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants iONKO (Regionalna Inicjatywa Doskonałości) from the Polish Ministry of Science and Higher Education and 2017/25/B/NZ6/01139 from the National Science Center in Poland.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Galon J, Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity. (2020) 52:55–81. doi: 10.1016/j.immuni.2019.12.018
2. Decker WK, da Silva RF, Sanabria MH, Angelo LS, Guimaraes F, Burt BM, et al. Cancer immunotherapy: historical perspective of a clinical revolution and emerging preclinical animal models. Front Immunol. (2017) 8:829. doi: 10.3389/fimmu.2017.00829
3. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
4. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. (2013) 369:134–44. doi: 10.1056/NEJMoa1305133
5. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
6. O'Donnell JS, M.Teng WL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
7. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
8. Shalapour S, Karin M. Control of anti-tumor immunity by cancer-associated inflammation. Immunity. (2019) 51:15–26. doi: 10.1016/j.immuni.2019.06.021
9. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. (2013) 339:286–91. doi: 10.1126/science.1232227
10. Hibbs J, Taintor R, Vavrin Z. Macrophage cytotoxicity: role for L-arginine deiminase and imino nitrogen oxidation to nitrite. Science. (1987) 235:473–76. doi: 10.1126/science.2432665
11. Stuehr DJ, Marletta MA. Mammalian nitrate biosynthesis: mouse macrophages produce nitrite and nitrate in response to Escherichia coli lipopolysaccharide. Proc Natl Acad Sci USA. (1985) 82:7738–42. doi: 10.1073/pnas.82.22.7738
12. Tapiero H, Mathé G, Couvreur P, Tew KD. I. Arginine. Biomed Pharmacother. (2002) 56:439–45. doi: 10.1016/S0753-3322(02)00284-6
13. Morris SM Jr. Arginine metabolism revisited. J Nutr. (2016) 146:2579S−86S. doi: 10.3945/jn.115.226621
14. Brosnan ME, Brosnan JT. Renal arginine metabolism. J Nutr. (2004) 134:2791S−5S. doi: 10.1093/jn/134.10.2791S
15. Lüneburg N, Xanthakis V, Schwedhelm E, Sullivan LM, Maas R, Anderssohn M, et al. Reference intervals for plasma L-arginine and the L-arginine:asymmetric dimethylarginine ratio in the framingham offspring cohort. J Nutr. (2011) 141:2186–90. doi: 10.3945/jn.111.148197
16. Böger RH. The Pharmacodynamics of L-Arginine. J Nutr. (2007) 137:1650S−5S. doi: 10.1093/jn/137.6.1650S
17. Castillo L, Chapman TE, Sanchez M, Yu YM, Burke JF, Ajami AM, et al. Plasma arginine and citrulline kinetics in adults given adequate and arginine-free diets. Proc Natl Acad Sci USA. (1993) 90:7749–53. doi: 10.1073/pnas.90.16.7749
18. Wu G, Morris SM. Jr. Arginine metabolism: nitric oxide and beyond. Biochem J. (1998) 336 (Pt 1):1–17. doi: 10.1042/bj3360001
19. Baydoun AR, Emery PW, Pearson JD, Mann GE. Substrate-dependent regulation of intracellular amino acid concentrations in cultured bovine aortic endothelial cells. Biochem Biophys Res Commun. (1990) 173:940–8. doi: 10.1016/S0006-291X(05)80876-9
20. Werner A, Pieh D, Echchannaoui H, Rupp J, Rajalingam K, Theobald M, et al. Cationic amino acid transporter-1-mediated arginine uptake is essential for chronic lymphocytic leukemia cell proliferation and viability. Front Oncol. (2019) 9:1268. doi: 10.3389/fonc.2019.01268
21. Closs EI, Boissel JP, Habermeier A, Rotmann A. Structure and function of Cationic Amino Acid Transporters (CATs). J Membr Biol. (2006) 213:67–77. doi: 10.1007/s00232-006-0875-7
22. Werner A, Amann E, Schnitzius V, Habermeier A, Luckner-Minden C, Leuchtner N, et al. Induced arginine transport via cationic amino acid transporter-1 is necessary for human T-cell proliferation. Eur J Immunol. (2016) 46:92–103. doi: 10.1002/eji.201546047
23. Cimen Bozkus C, Elzey BD, Crist SA, Ellies LG, Ratliff TL. Expression of cationic amino acid transporter 2 is required for myeloid-derived suppressor cell-mediated control of T cell immunity. J Immunol. (2015) 195:5237–50. doi: 10.4049/jimmunol.1500959
24. Closs EI, Simon A, Vékony N, Rotmann A. Plasma membrane transporters for arginine. J Nutr. (2004) 1341:2752S−9S. doi: 10.1093/jn/134.10.2752S
25. Fotiadis D, Kanai Y, Palacín M. The SLC3 and SLC7 families of amino acid transporters. Mol Asp Med. (2013) 34:139–58. doi: 10.1016/j.mam.2012.10.007
26. Griswold A, Chen YM, Snyder JA, Burne RA. Characterization of the arginine deiminase operon of Streptococcus rattus FA-1. Appl Environ Microbiol. (2004) 70:1321–7. doi: 10.1128/AEM.70.3.1321-1327.2004
27. Lindgren JK, Thomas VC, Olson ME, Chaudhari SS, Nuxoll AS, Schaeffer CR, et al. Arginine deiminase in Staphylococcus epidermidis functions to augment biofilm maturation through pH homeostasis. J Bacteriol. (2014) 196:2277–89. doi: 10.1128/JB.00051-14
28. Burne RA, Marquis RE. Alkali production by oral bacteria and protection against dental caries. FEMS Microbiol Lett. (2000) 193:1–6. doi: 10.1111/j.1574-6968.2000.tb09393.x
29. Shore AC, Rossney AS, Brennan OM, Kinnevey PM, Humphreys H, Sullivan DJ, et al. Characterization of a novel arginine catabolic mobile element (ACME) and staphylococcal chromosomal cassette mec composite island with significant homology to Staphylococcus epidermidis ACME type II in methicillin-resistant Staphylococcus aureus genotype ST22-MRSA-IV. Antimicrob Agents Chemother. (2011) 55:1896–905. doi: 10.1128/AAC.01756-10
30. Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. (2006) 367:731–9. doi: 10.1016/S0140-6736(06)68231-7
31. Thurlow LR, Joshi GS, Clark JR, Spontak JS, Neely CJ, Maile R, et al. Functional modularity of the arginine catabolic mobile element contributes to the success of USA300 methicillin-resistant Staphylococcus aureus. Cell Host Microbe. (2013) 13:100–7. doi: 10.1016/j.chom.2012.11.012
32. Jenkinson CP, Grody WW, Cederbaum SD. Comparative properties of arginases. Comp Biochem Physiol B Biochem Mol Biol. (1996) 114:107–32. doi: 10.1016/0305-0491(95)02138-8
33. Morris SM Jr. Regulation of enzymes of the urea cycle and arginine metabolism. Ann Rev Nutr. (2002) 22:87–105. doi: 10.1146/annurev.nutr.22.110801.140547
34. Dizikes GJ, Grody WW, Kern RM, Cederbaum SD. Isolation of human liver arginase cDNA and demonstration of nonhomology between the two human arginase genes. Biochem Biophys Res Commun. (1986) 141:53–9. doi: 10.1016/S0006-291X(86)80333-3
35. Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol. (2009) 158:638–51. doi: 10.1111/j.1476-5381.2009.00291.x
36. Gotoh T, Sonoki T, Nagasaki A, Terada K, Takiguchi M, Mori M. Molecular cloning of cDNA for nonhepatic mitochondrial arginase (arginase II) and comparison of its induction with nitric oxide synthase in a murine macrophage-like cell line. FEBS Lett. (1996) 395:119–22. doi: 10.1016/0014-5793(96)01015-0
37. Haraguchi Y, Takiguchi M, Amaya Y, Kawamoto S, Matsuda I, Mori M. Molecular cloning and nucleotide sequence of cDNA for human liver arginase. Proc Natl Acad Sci USA. (1987) 84:412–5. doi: 10.1073/pnas.84.2.412
38. Chen F, Lucas R, Fulton D. The subcellular compartmentalization of arginine metabolizing enzymes and their role in endothelial dysfunction. Front Immunol. (2013) 4:184. doi: 10.3389/fimmu.2013.00184
39. Iyer RK, Yoo PK, Kern RM, Rozengurt N, Tsoa R, O'Brien WE, et al. Mouse model for human arginase deficiency. Mol Cell Biol. (2002) 22:4491–8. doi: 10.1128/MCB.22.13.4491-4498.2002
40. Kasten J, Hu C, Bhargava R, Park H, Tai D, Byrne JA, et al. Lethal phenotype in conditional late-onset arginase 1 deficiency in the mouse. Mol Gene Metab. (2013) 110:222–30. doi: 10.1016/j.ymgme.2013.06.020
41. Xiong Y, Yepuri G, Montani JP, Ming XF, Yang Z. Arginase-II Deficiency Extends Lifespan in Mice. Front Physiol. (2017) 8:682. doi: 10.3389/fphys.2017.00682
42. Shi O, Morris SM Jr, Zoghbi H, Porter CW, O'Brien WE. Generation of a mouse model for arginase II deficiency by targeted disruption of the arginase II gene. Mol Cell Biol. (2001) 21:811–3. doi: 10.1128/MCB.21.3.811-813.2001
43. Crombez EA, Cederbaum SD. Hyperargininemia due to liver arginase deficiency. Mol Gene Metab. (2005) 84:243–51. doi: 10.1016/j.ymgme.2004.11.004
44. Grody WW, Kern RM, Klein D, Dodson AE, Wissman PB, Barsky SH, et al. Arginase deficiency manifesting delayed clinical sequelae and induction of a kidney arginase isozyme. Hum Genet. (1993) 91:1–5. doi: 10.1007/BF00230212
45. Murray PJ Amino acid auxotrophy as a system of immunological control nodes. Nat Immunol. (2016) 17:132–9. doi: 10.1038/ni.3323
46. Mussai F, Egan S, Hunter S, Webber H, Fisher J, Wheat R, et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. (2015) 75:3043–53. doi: 10.1158/0008-5472.CAN-14-3443
47. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
48. Lewis ND, Asim M, Barry DP, de Sablet T, Singh K, Piazuelo MB, et al. Immune evasion by helicobacter pylori is mediated by induction of macrophage arginase II. J Immunol. (2011) 186:3632–41. doi: 10.4049/jimmunol.1003431
49. Elahi S, Ertelt JM, Kinder JM, Jiang TT, Zhang X, Xin L, et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature. (2013) 504:158–62. doi: 10.1038/nature12675
50. Ming XF, Rajapakse AG, Yepuri G, Xiong Y, Carvas JM, Ruffieux J, et al. Arginase II promotes macrophage inflammatory responses through mitochondrial reactive oxygen species, contributing to insulin resistance and atherogenesis. J Am Heart Assoc. (2012) 1:e000992. doi: 10.1161/JAHA.112.000992
51. Ashley JW, Hancock WD, Nelson AJ, Bone RN, Tse HM, Wohltmann M, et al. Polarization of macrophages toward M2 phenotype is favored by reduction in iPLA2β (Group VIA phospholipase A2). J Biol Chem. (2016) 291:23268–81. doi: 10.1074/jbc.M116.754945
52. Qie Y, Yuan H, von Roemeling CA, Chen Y, Liu X, Shih KD, et al. Surface modification of nanoparticles enables selective evasion of phagocytic clearance by distinct macrophage phenotypes. Sci Rep. (2016) 6:26269. doi: 10.1038/srep26269
53. Caldwell RB, Toque HA, Narayanan SP, Caldwell RW. Arginase: an old enzyme with new tricks. Trends Pharmacol Sci. (2015) 36:395–405. doi: 10.1016/j.tips.2015.03.006
54. Caldwell RW, Rodriguez PC, Toque HA, Narayanan SP, Caldwell RB. Arginase: a Multifaceted enzyme important in health and disease. Physiol Rev. (2018) 98:641–65. doi: 10.1152/physrev.00037.2016
55. Lee J, Ryu H, Ferrante RJ, Morris SM Jr, Ratan RR. Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc Natl Acad Sci USA. (2003) 100:4843–8. doi: 10.1073/pnas.0735876100
56. Albina JE, Mills CD, Henry WL Jr, Caldwell MD. Temporal expression of different pathways of 1-arginine metabolism in healing wounds. J Immunol. (1990) 144:3877–80.
57. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
58. Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. (2016) 18:1090–101. doi: 10.1038/ncb3410
59. Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife. (2019) 8. doi: 10.7554/eLife.44235
60. Gannon PO, Godin-Ethier J, Hassler M, Delvoye N, Aversa M, Poisson AO, et al. Androgen-regulated expression of arginase 1, arginase 2 and interleukin-8 in human prostate cancer. PLoS ONE. (2010) 5:e12107. doi: 10.1371/journal.pone.0012107
61. Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, R.M. McEwen-Smith, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. (2013) 122:749–58. doi: 10.1182/blood-2013-01-480129
62. Bron L, Jandus C, Andrejevic-Blant S, Speiser DE, Monnier P, Romero P, et al. Prognostic value of arginase-II expression and regulatory T-cell infiltration in head and neck squamous cell carcinoma. Int J Cancer. (2013) 132:E85–93. doi: 10.1002/ijc.27728
63. Ino Y, Yamazaki-Itoh R, Oguro S, Shimada K, Kosuge T, Zavada J, et al. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS ONE. (2013) 8:e55146. doi: 10.1371/journal.pone.0055146
64. Czystowska-Kuzmicz M, Sosnowska A, Nowis D, Ramji K, Szajnik M, Chlebowska-Tuz J, et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat Commun. (2019) 10:3000. doi: 10.1038/s41467-019-10979-3
65. Ma Z, Lian J, Yang M, Wuyang J, Zhao C, Chen W, et al. Overexpression of Arginase-1 is an indicator of poor prognosis in patients with colorectal cancer. Pathol Res Pract. (2019) 215:152383. doi: 10.1016/j.prp.2019.03.012
66. Obiorah IE, Chahine J, Ko K, Park BU, deGuzman J, Kallakury B. Prognostic implications of arginase and cytokeratin 19 expression in hepatocellular carcinoma after curative hepatectomy: correlation with recurrence-free survival. Gastroenterol Res. (2019) 12:78–87. doi: 10.14740/gr1156
67. You J, Chen W, Chen J, Zheng Q, Dong J, Zhu Y. The oncogenic role of ARG1 in progression and metastasis of hepatocellular carcinoma. BioMed Res Int. (2018) 2018:2109865. doi: 10.1155/2018/2109865
68. Mao H, Gao W, Lu G, Fang F, Teng L. Clinicopathological and prognostic implications of arginase expression in hepatocellular carcinoma. Clin Lab. (2013) 59:37–43. doi: 10.7754/Clin.Lab.2012.120210
69. Porembska Z, Luboinski G, Chrzanowska A, Mielczarek M, Magnuska J, Baranczyk-Kuzma A. Arginase in patients with breast cancer. Clin Chim Acta. (2003) 328:105–11. doi: 10.1016/S0009-8981(02)00391-1
70. Straus B, Cepelak I, Festa G. Arginase, a new marker of mammary carcinoma. Clin Chim Acta. (1992) 210:5–12. doi: 10.1016/0009-8981(92)90040-W
71. Polat MF, Taysi S, Polat S, Boyuk A, Bakan E. Elevated serum arginase activity levels in patients with breast cancer. Surg Today. (2003) 33:655–61. doi: 10.1007/s00595-002-2563-2
72. Avtandilyan N, Javrushyan H, Petrosyan G, Trchounian A. The involvement of arginase and nitric oxide synthase in breast cancer development: arginase and NO synthase as therapeutic targets in cancer. Biomed Res Int. (2018) 9:8696923. doi: 10.1155/2018/8696923
73. Vissers YL, Dejong CH, Luiking YC, Fearon KC, von Meyenfeldt MF, Deutz NE. Plasma arginine concentrations are reduced in cancer patients: evidence for arginine deficiency? Am J Clin Nutr. (2005) 81:1142–46. doi: 10.1093/ajcn/81.5.1142
74. M.P.Engelen KJ, Klimberg VS, Allasia A, N.Deutz EP. Major surgery diminishes systemic arginine availability and suppresses nitric oxide response to feeding in patients with early stage breast cancer. Clin Nutr. (2018) 37:1645–53. doi: 10.1016/j.clnu.2017.07.019
75. Gokmen SS, Aygit AC, Ayhan MS, Yorulmaz F, Gulen S. Significance of arginase and ornithine in malignant tumors of the human skin. J Lab Clin Med. (2001) 137:340–4. doi: 10.1067/mlc.2001.114543
76. Bedoya AM, Tate DJ, Baena A, Cordoba CM, Borrero M, Pareja R, et al. Immunosuppression in cervical cancer with special reference to arginase activity. Gynecol Oncol. (2014) 135:74–80. doi: 10.1016/j.ygyno.2014.07.096
77. Cerutti JM, Delcelo R, Amadei MJ, Nakabashi C, R.Maciel MB, Peterson B, et al. A preoperative diagnostic test that distinguishes benign from malignant thyroid carcinoma based on gene expression. J Clin Invest. (2004) 113:1234–42. doi: 10.1172/JCI19617
78. Akiba J, Nakashima O, Hattori S, Naito Y, Kusano H, Kondo R, et al. The expression of arginase-1, keratin (K) 8 and K18 in combined hepatocellular-cholangiocarcinoma, subtypes with stem-cell features, intermediate-cell type. J Clin Pathol. (2016) 69:846–51. doi: 10.1136/jclinpath-2015-203491
79. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. (2011) 60:1419–30. doi: 10.1007/s00262-011-1028-0
80. Harris BE, Pretlow TP, Bradley EL Jr., Whitehurst GB, Pretlow TG. II. Arginase activity in prostatic tissue of patients with benign prostatic hyperplasia and prostatic carcinoma. Cancer Res. (1983) 43:3008–12.
81. Elgun S, Keskinege A, Yilmaz E, Baltaci S, Beduk Y. Evaluation of serum arginase activity in benign prostatic hypertrophy and prostatic cancer. Int Urol Nephrol. (1999) 31:95–9. doi: 10.1023/A:1007132109061
82. Mumenthaler SM, Yu H, Tze S, Cederbaum SD, Pegg AE, Seligson DB, et al. Expression of arginase II in prostate cancer. Int J Oncol. (2008) 32:357–65. doi: 10.3892/ijo.32.2.357
83. Rotondo R, Mastracci L, Piazza T, Barisione G, Fabbi M, Cassanello M, et al. Arginase 2 is expressed by human lung cancer, but it neither induces immune suppression, nor affects disease progression. Int J Cancer. (2008) 123:1108–16. doi: 10.1002/ijc.23437
84. Ascierto PA, Scala S, Castello G, Daponte A, Simeone E, Ottaiano A, et al. Pegylated arginine deiminase treatment of patients with metastatic melanoma: results from phase I and II studies. J Clin Oncol. (2005) 23:7660–8. doi: 10.1200/JCO.2005.02.0933
85. Yoon CY, Shim YJ, Kim EH, Lee JH, Won NH, Kim JH, et al. Renal cell carcinoma does not express argininosuccinate synthetase and is highly sensitive to arginine deprivation via arginine deiminase. Int J Cancer. (2007) 120:897–905. doi: 10.1002/ijc.22322
86. Ochocki JD, Khare S, Hess M, Ackerman D, Qiu B, Daisak JI, et al. Arginase 2 suppresses renal carcinoma progression via biosynthetic cofactor pyridoxal phosphate depletion and increased polyamine toxicity. Cell Metab. (2018) 27:1263–80. doi: 10.1016/j.cmet.2018.04.009
87. Schultze JL, Mass E, Schlitzer A. Emerging principles in myelopoiesis at homeostasis and during infection and inflammation. Immunity. (2019) 50:288–301. doi: 10.1016/j.immuni.2019.01.019
88. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
89. Messmer MN, Netherby CS, Banik D, Abrams SI. Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy. Cancer Immunol Immunother. (2015) 64:1–13. doi: 10.1007/s00262-014-1639-3
90. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
91. Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget. (2017) 8:3649–65. doi: 10.18632/oncotarget.12278
92. Al Sayed MF, Amrein MA, Bührer ED, A.-Huguenin L, Radpour R, Riether C, et al. T-cell–Secreted TNFα induces emergency myelopoiesis and myeloid-derived suppressor cell differentiation in cancer. Cancer Res. (2019) 79:346–59. doi: 10.1158/0008-5472.CAN-17-3026
93. Wu CW, Sun HW, Chen HT, Liang J, Yu J, Wu C, et al. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc Natl Acad Sci USA. (2014) 111:4221–6. doi: 10.1073/pnas.1320753111
94. Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. (2007) 67:10019–26. doi: 10.1158/0008-5472.CAN-07-2354
95. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. (2019) 50:1317–34. doi: 10.1016/j.immuni.2019.03.009
96. Zhao F, Obermann S, von Wasielewski R, Haile L, Manns MP, Korangy F, et al. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology. (2009) 128:141–9. doi: 10.1111/j.1365-2567.2009.03105.x
97. Ma P, Beatty PL, McKolanis J, Brand R, Schoen RE, Finn OJ. Circulating Myeloid Derived Suppressor Cells (MDSC) that accumulate in premalignancy share phenotypic and functional characteristics with MDSC in cancer. Front Immunol. (2019) 10:1401. doi: 10.3389/fimmu.2019.01401
98. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
99. Bronte V, Brandau S, S.-Chen H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
100. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
101. Kapanadze T, Gamrekelashvili J, Ma C, Chan C, Zhao F, Hewitt S, et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J Hepatol. (2013) 59:1007–13. doi: 10.1016/j.jhep.2013.06.010
102. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
103. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. (2009) 69:1553–60. doi: 10.1158/0008-5472.CAN-08-1921
104. Thomas AC, Mattila JT. “Of mice and men”: arginine metabolism in macrophages. Front Immunol. (2014) 5:479. doi: 10.3389/fimmu.2014.00479
105. Munder M, Mollinedo F, Calafat J, Canchado J, Gil-Lamaignere C, Fuentes JM, et al. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood. (2005) 105:2549–56. doi: 10.1182/blood-2004-07-2521
106. Gundra UM, Girgis NM, Ruckerl D, Jenkins S, Ward LN, Kurtz ZD, et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood. (2014) 123:e110–22. doi: 10.1182/blood-2013-08-520619
107. Scotton CJ, Martinez FO, Smelt MJ, Sironi M, Locati M, Mantovani A, et al. Transcriptional profiling reveals complex regulation of the monocyte IL-1β system by IL-13. J Immunol. (2005) 174:834–45. doi: 10.4049/jimmunol.174.2.834
108. Raes G, van den Bergh R, de Baetselier P, Ghassabeh GH, Scotton C, Locati M, et al. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J Immunol. (2005) 174:6561. doi: 10.4049/jimmunol.174.11.6561
109. Eldredge LC, Creasy RS, Tanaka S, Lai JF, Ziegler SF. Imbalance of Ly-6Chi and Ly-6Clo monocytes/macrophages worsens hyperoxia-induced lung injury and is rescued by IFN-γ. J Immunol. (2019) 202:2772–81. doi: 10.4049/jimmunol.1801374
110. Rouzaut A, Subirá ML, de Miguel C, Domingo-de-Miguel E, González A, Santiago E, et al. Co-expression of inducible nitric oxide synthase and arginases in different human monocyte subsets. Apoptosis regulated by endogenous NO. Biochim Biophys Acta. (1999) 1451:319–33. doi: 10.1016/S0167-4889(99)00106-8
111. Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem Biophys Res Commun. (1995) 206:667–73. doi: 10.1006/bbrc.1995.1094
112. Munder M, Eichmann K, Morán JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. (1999) 163:3771–7.
113. Debats IB, Wolfs TG, Gotoh T, Cleutjens JP, Peutz-Kootstra CJ, van der Hulst RR. Role of arginine in superficial wound healing in man. Nitric Oxide. (2009) 21:175–83. doi: 10.1016/j.niox.2009.07.006
114. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS–) vs. alternatively activated macrophages. Front Immunol. (2019) 11:234. doi: 10.3389/fimmu.2020.00234
115. Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ, et al. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics. (2018) 8:5842–54. doi: 10.7150/thno.26888
116. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. (2019) 35:588–602. doi: 10.1016/j.ccell.2019.02.009
117. Miret JJ, Kirschmeier P, Koyama S, Zhu M, Li YY, Naito Y, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a NSCLC mouse model by activating anti-tumor immunity. J Immunother Cancer. (2019) 7:32. doi: 10.1186/s40425-019-0504-5
118. Soncin I, Sheng J, Chen Q, Foo S, Duan K, Lum J, et al. The tumour microenvironment creates a niche for the self-renewal of tumour-promoting macrophages in colon adenoma. Nat Commun. (2018) 9:582. doi: 10.1038/s41467-018-02834-8
119. Neamah WH, Singh NP, Alghetaa H, Abdulla OA, Chatterjee S, Busbee PB, et al. AhR activation leads to massive mobilization of myeloid-derived suppressor cells with immunosuppressive activity through regulation of CXCR2 and microRNA miR-150-5p and miR-543-3p that target anti-Inflammatory genes. J Immunol. (2019) 203:1830–44. doi: 10.4049/jimmunol.1900291
120. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-Induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity. (2010) 32:790–802. doi: 10.1016/j.immuni.2010.05.010
121. Monu NR, Frey AB. Myeloid-derived suppressor cells and anti-tumor T cells: a complex relationship. Immunol Invest. (2012) 41:595–613. doi: 10.3109/08820139.2012.673191
122. Sippel TR, Shimizu T, Strnad F, Traystman RJ, Herson PS, Waziri A. Arginase I release from activated neutrophils induces peripheral immunosuppression in a murine model of stroke. J Cerebr Blood Flow Metab. (2015) 35:1657–63. doi: 10.1038/jcbfm.2015.103
123. Munder M, Schneider H, Luckner C, Giese T, Langhans CD, Fuentes JM, et al. Suppression of T-cell functions by human granulocyte arginase. Blood. (2006) 108:1627–34. doi: 10.1182/blood-2006-11-010389
124. Elpek KG, Cremasco V, Shen H, Harvey CJ, Wucherpfennig KW, Goldstein DR, et al. The tumor microenvironment shapes lineage, transcriptional, and functional diversity of infiltrating myeloid cells. Cancer Immunol Res. (2014) 2:655–67. doi: 10.1158/2326-6066.CIR-13-0209
125. Dunand-Sauthier I, Irla M, Carnesecchi S, Seguín-Estévez Q, Vejnar CE, Zdobnov EM, et al. Repression of arginase-2 expression in dendritic cells by microRNA-155 is critical for promoting T cell proliferation. J Immunol. (2014) 193:1690–700. doi: 10.4049/jimmunol.1301913
126. Keeley T, Costanzo-Garvey DL, Cook LM. Unmasking the many faces of tumor-associated neutrophils and macrophages: considerations for targeting innate immune cells in cancer. Trends Cancer. (2019) 5:789–98. doi: 10.1016/j.trecan.2019.10.013
127. Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, A.-Gaston T, Clement M, et al. Macrophage plasticity in experimental atherosclerosis. PLoS ONE. (2010) 5:e8852. doi: 10.1371/journal.pone.0008852
128. Kung JT, Brooks SB, Jakway JP, Leonard LL, Talmage DW. Suppression of in vitro cytotoxic response by macrophages due to induced arginase. J Exp Med. (1977) 146:665–72. doi: 10.1084/jem.146.3.665
129. Schneider E, Dy M. The role of arginase in the immune response. Immunol Today. (1985) 6:136–40. doi: 10.1016/0167-5699(85)90081-7
130. Currie GA. Activated macrophages kill tumour cells by releasing arginase. Nature. (1978) 273:758–9. doi: 10.1038/273758a0
131. Currie GA, Gyure L, Cifuentes L. Microenvironmental arginine depletion by macrophages in vivo. Br J Cancer. (1979) 39:613–20. doi: 10.1038/bjc.1979.112
132. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
133. Sahin E, Haubenwallner S, Kuttke M, Kollmann I, Halfmann A, Dohnal AB, et al. Macrophage PTEN regulates expression and secretion of arginase I modulating innate and adaptive immune responses. J Immunol. (2014) 193:1717–27. doi: 10.4049/jimmunol.1302167
134. Tsui SM, Lam WM, Lam TL, Chong HC, So PK, Kwok SY, et al. Pegylated derivatives of recombinant human arginase (rhArg1) for sustained in vivo activity in cancer therapy: preparation, characterization and analysis of their pharmacodynamics in vivo and in vitro and action upon hepatocellular carcinoma cell (HCC). Cancer Cell Int. (2009) 9:9. doi: 10.1186/1475-2867-9-9
135. Wang Y, Tian J, Tang X, Rui K, Tian X, Ma J, et al. Exosomes released by granulocytic myeloid-derived suppressor cells attenuate DSS-induced colitis in mice. Oncotarget. (2016) 7:15356–68. doi: 10.18632/oncotarget.7324
136. Jost MM, Ninci E, Meder B, Kempf C, Van Royen N, Hua J, et al. Divergent effects of GM-CSF and TGFbeta1 on bone marrow-derived macrophage arginase-1 activity, MCP-1 expression, and matrix metalloproteinase-12: a potential role during arteriogenesis. FASEB J. (2003) 17:2281–3. doi: 10.1096/fj.03-0071fje
137. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol. (2008) 9:1399–406. doi: 10.1038/ni.1671
138. Corraliza IM, Modolell M, Ferber E, Soler G. Involvement of protein kinase A in the induction of arginase in murine bone marrow-derived macrophages. Biochim Biophys Acta. (1997) 1334:123–8. doi: 10.1016/S0304-4165(96)00081-5
139. Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. (2002) 169:2253–63. doi: 10.4049/jimmunol.169.5.2253
140. Pesce J, Kaviratne M, Ramalingam TR, Thompson RW, Urban JF Jr, et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest. (2006) 116:2044–55. doi: 10.1172/JCI27727
141. Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. (2003) 170:270–8. doi: 10.4049/jimmunol.170.1.270
142. Liu Y, Van Ginderachter JA, Brys L, De Baetselier P, Raes G, Geldhof AB. Nitric oxide-independent CTL suppression during tumor progression: association with arginase-producing (M2) myeloid cells. J Immunol. (2003) 170:5064–74. doi: 10.4049/jimmunol.170.10.5064
143. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. (2005) 65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505
144. Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. (2007) 13:721s−26s. doi: 10.1158/1078-0432.CCR-06-2197
145. Chai E, Zhang L, Li C. LOX-1+ PMN-MDSC enhances immune suppression which promotes glioblastoma multiforme progression. Cancer Manag Res. (2019) 11:7307–15. doi: 10.2147/CMAR.S210545
146. Nan J, Y.-Xing F, Hu B, J.-Tang X, H.-Dong M, Y.-He M, et al. Endoplasmic reticulum stress induced LOX-1(+) CD15(+) polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology. (2018) 154:144–55. doi: 10.1111/imm.12876
147. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Bossman SA, Ter Laan M, et al. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. (2016) 18:1253–64. doi: 10.1093/neuonc/now034
148. Mao FY, Zhao YL, Lv YP, Teng YS, Kong H, Liu YG, et al. CD45(+)CD33(low)CD11b(dim) myeloid-derived suppressor cells suppress CD8(+) T cell activity via the IL-6/IL-8-arginase I axis in human gastric cancer. Cell Death Dis. (2018) 9:763. doi: 10.1038/s41419-018-0803-7
149. Zhang J, Xu X, Shi M, Chen Y, Yu D, Zhao C, et al. CD13(hi) neutrophil-like myeloid-derived suppressor cells exert immune suppression through arginase 1 expression in pancreatic ductal adenocarcinoma. Oncoimmunology. (2017) 6:e1258504. doi: 10.1080/2162402X.2016.1258504
150. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. (2008) 181:5791–802. doi: 10.4049/jimmunol.181.8.5791
151. Si Y, Merz SF, Jansen P, Wang B, Bruderek K, Altenhoff P, et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci Immunol. (2019) 4:9159. doi: 10.1126/sciimmunol.aaw9159
152. Nakamura K, Kassem S, Cleynen A, Chretien ML, Guillerey C, Putz EM, et al. Dysregulated IL-18 Is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell. (2018) 33:634–648. doi: 10.1016/j.ccell.2018.02.007
153. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. (2008) 68:5439–49. doi: 10.1158/0008-5472.CAN-07-6621
154. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. (2008) 135:234–43. doi: 10.1053/j.gastro.2008.03.020
155. Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol. (2012) 91:167–81. doi: 10.1189/jlb.0311177
156. Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. (2008) 222:162–79. doi: 10.1111/j.1600-065X.2008.00602.x
157. Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, et al. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity. (2014) 41:389–401. doi: 10.1016/j.immuni.2014.08.015
158. Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. (2015) 75:275–83. doi: 10.1158/0008-5472.CAN-14-1491
159. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. (2013) 123:1580–9. doi: 10.1172/JCI60083
160. Hossain DM, Pal SK, Moreira D, Duttagupta P, Zhang Q, Won H, et al. TLR9-Targeted STAT3 silencing abrogates immunosuppressive activity of myeloid-derived suppressor cells from prostate cancer patients. Clin Cancer Res. (2015) 21:3771–82. doi: 10.1158/1078-0432.CCR-14-3145
161. Mohammadpour H, MacDonald CR, Qiao G, Chen M, Dong B, Hylander BL, et al. beta2 adrenergic receptor-mediated signaling regulates the immunosuppressive potential of myeloid-derived suppressor cells. J Clin Invest. (2019) 129:5537–52. doi: 10.1172/JCI129502
162. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. (2005) 202:931–9. doi: 10.1084/jem.20050715
163. Talmadge JE, Hood KC, Zobel LC, Shafer LR, Coles M, Toth B. Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int Immunopharmacol. (2007) 7:140–51. doi: 10.1016/j.intimp.2006.09.021
164. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
165. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. (2019) 30:36–50. doi: 10.1016/j.cmet.2019.06.001
166. Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter J, Sarukhan A. Mechanisms driving macrophage diversity and specialization in distinct tumor microenvironments and parallelisms with other tissues. Front Immunol. (2014) 5:127. doi: 10.3389/fimmu.2014.00127
167. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
168. Gray MJ, Poljakovic M, Kepka-Lenhart D, Morris SM. Jr. Induction of arginase I transcription by IL-4 requires a composite DNA response element for STAT6 and C/EBPbeta. Gene. (2005) 353:98–106. doi: 10.1016/j.gene.2005.04.004
169. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. (2017) 8:93. doi: 10.3389/fimmu.2017.00093
170. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
171. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE. (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
172. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci USA. (2012) 109:2491–6. doi: 10.1073/pnas.1113744109
173. Laviron M, Boissonnas A. Ontogeny of tumor-associated macrophages. Front Immunol. (2019) 10:1799. doi: 10.3389/fimmu.2019.01799
174. Loyher PL, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med. (2018) 215:2536–53. doi: 10.1084/jem.20180534
175. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. (2017) 47:323–38. doi: 10.1016/j.immuni.2017.07.014
176. Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. (2015) 6:588. doi: 10.3389/fimmu.2015.00588
177. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. (2010) 70:7465–75. doi: 10.1158/0008-5472.CAN-10-1439
178. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
179. Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N, et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. (2018) 19:1319–29. doi: 10.1038/s41590-018-0226-8
180. El-Kenawi A, Gatenbee C, Robertson-Tessi M, Bravo R, Dhillon J, Balagurunathan Y, et al. Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer. Br J Cancer. (2019) 121:556–66. doi: 10.1038/s41416-019-0542-2
181. Cho H, Seo Y, Loke KM, Kim SW, Oh SM, Kim JH, et al. Cancer-stimulated CAFs enhance monocyte differentiation and protumoral TAM activation via IL6 and GM-CSF secretion. Clin Cancer Res. (2018) 24:5407–21. doi: 10.1158/1078-0432.CCR-18-0125
182. Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest. (2013) 123:3231–42. doi: 10.1172/JCI67655
183. Tong Y, Zhou L, Yang L, Guo P, Cao Y, Qin FX, et al. Concomitant type I IFN and M-CSF signaling reprograms monocyte differentiation and drives pro-tumoral arginase production. EBiomedicine. (2019) 39:132–44. doi: 10.1016/j.ebiom.2018.11.062
184. Van Ginderachter JA, Meerschaut S, Liu Y, Brys L, De Groeve K, Hassanzadeh Ghassabeh G, et al. Peroxisome proliferator-activated receptor γ (PPARγ) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood. (2006) 108:525–35. doi: 10.1182/blood-2005-09-3777
185. Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res. (2001) 61:1100–6. Available online at: https://cancerres.aacrjournals.org/content/61/3/1100.long
186. Hayes CS, Shicora AC, Keough MP, Snook AE, Burns MR, Gilmour SK. Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol Res. (2014) 2:274–85. doi: 10.1158/2326-6066.CIR-13-0120-T
187. Alexander ET, Minton A, Peters MC, Phanstiel OT, Gilmour SK. A novel polyamine blockade therapy activates an anti-tumor immune response. Oncotarget. (2017) 8:84140–152. doi: 10.18632/oncotarget.20493
188. Rosales C. Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol. (2018) 9:113. doi: 10.3389/fphys.2018.00113
189. Ocana A, Nieto-Jimenez C, Pandiella A, Templeton AJ. Neutrophils in cancer: prognostic role and therapeutic strategies. Mol Cancer. (2017) 16:137. doi: 10.1186/s12943-017-0707-7
190. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TA. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
191. Jacobsen LC, Theilgaard-Monch K, Christensen EI, Borregaard N. Arginase 1 is expressed in myelocytes/metamyelocytes and localized in gelatinase granules of human neutrophils. Blood. (2007) 109:3084–7. doi: 10.1182/blood-2006-06-032599
192. Rotondo R, Bertolotto M, Barisione G, Astigiano S, Mandruzzato S, Ottonello L, et al. Exocytosis of azurophil and arginase 1-containing granules by activated polymorphonuclear neutrophils is required to inhibit T lymphocyte proliferation. J Leukoc Biol. (2011) 89:721–7. doi: 10.1189/jlb.1109737
193. Rotondo R, Barisione G, Mastracci L, Grossi F, Orengo AM, Costa R, et al. IL-8 induces exocytosis of arginase 1 by neutrophil polymorphonuclears in nonsmall cell lung cancer. Int J Cancer. (2009) 125:887–93. doi: 10.1002/ijc.24448
194. Hurt B, Schulick R, Edil B, El Kasmi KC, Barnett C. Jr. Cancer-promoting mechanisms of tumor-associated neutrophils. Am J Surg. (2017) 214:938–44. doi: 10.1016/j.amjsurg.2017.08.003
195. Sippel TR, White J, Nag K, Tsvankin V, Klaassen M, B.K. Kleinschmidt-DeMasters, et al. Neutrophil degranulation and immunosuppression in patients with GBM: restoration of cellular immune function by targeting arginase I. Clin Cancer Res. (2011) 17:6992–7002. doi: 10.1158/1078-0432.CCR-11-1107
196. Oberlies J, Watzl C, Giese T, Luckner C, Kropf P, Muller I, et al. Regulation of NK cell function by human granulocyte arginase. J Immunol. (2009) 182:5259–67. doi: 10.4049/jimmunol.0803523
197. Oberg HH, Wesch D, Kalyan S, Kabelitz D. Regulatory interactions between neutrophils, tumor cells and T Cells. Front Immunol. (2019) 10:1690. doi: 10.3389/fimmu.2019.01690
198. Berry RS, Xiong MJ, Greenbaum A, Mortaji P, Nofchissey RA, Schultz F, et al. High levels of tumor-associated neutrophils are associated with improved overall survival in patients with stage II colorectal cancer. PLoS ONE. (2017) 12:e0188799. doi: 10.1371/journal.pone.0188799
199. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. (2006) 311:17–58. doi: 10.1007/3-540-32636-7_2
200. Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. (2019) 19:89–103. doi: 10.1038/s41577-018-0088-1
201. DeVito NC, Plebanek MP, Theivanthiran B, Hanks BA. Role of tumor-mediated dendritic cell tolerization in immune evasion. Front Immunol. (2019) 10:2876. doi: 10.3389/fimmu.2019.02876
202. Liu Q, Zhang C, Sun A, Zheng Y, Wang L, Cao X. Tumor-Educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J Immunol. (2009) 182:6207–16. doi: 10.4049/jimmunol.0803926
203. Norian LA, Rodriguez PC, O'Mara LA, Zabaleta J, Ochoa AC, Cella M, et al. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res. (2009) 69:3086–94. doi: 10.1158/0008-5472.CAN-08-2826
204. Panfili E, Mondanelli G, Orabona C, Bianchi R, Gargaro M, Fallarino F, et al. IL-35Ig-expressing dendritic cells induce tolerance via Arginase 1. J Cell Mol Med. (2019) 23:3757–61. doi: 10.1111/jcmm.14215
205. Chang J, Thangamani S, Kim MH, Ulrich B, Morris SM Jr, et al. Retinoic acid promotes the development of Arg1-expressing dendritic cells for the regulation of T-cell differentiation. Eur J Immunol. (2013) 43:967–78. doi: 10.1002/eji.201242772
206. McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature. (2017) 546:662–6.
207. Cobbold SP, Adams E, Farquhar CA, Nolan KF, Howie D, Lui KO, et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci USA. (2009) 106:12055–60. doi: 10.1073/pnas.0903919106
208. Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, Zanetti M. Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming. PLoS ONE. (2012) 7:e51845. doi: 10.1371/journal.pone.0051845
209. Herbert DR, Orekov T, Roloson A, Ilies M, Perkins C, O'Brien W, et al. Arginase I suppresses IL-12/IL-23p40-driven intestinal inflammation during acute schistosomiasis. J Immunol. (2010) 184:6438–46. doi: 10.4049/jimmunol.0902009
210. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. (2017) 5:101. doi: 10.1186/s40425-017-0308-4
211. Menjivar RE, Halbrook C, Velez A, Lima F, Espinoza C, Galban S, et al. Abstract A31: Investigating the effect of myeloid Arg1 deletion on tumor growth and CD8+ T-cell infiltration and activation in pancreatic cancer. Cancer Res. (2019) 79:A31. doi: 10.1158/1538-7445.PANCA19-A31
212. Hornyák L, Dobos N, Koncz G, Karányi Z, Páll D, Szabó Z, et al. The role of indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front Immunol. (2018) 9:151. doi: 10.3389/fimmu.2018.00151
213. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. (2007) 117:1147–54. doi: 10.1172/JCI31178
214. Munder M, Engelhardt M, Knies D, Medenhoff S, Wabnitz G, Luckner-Minden C, et al. Cytotoxicity of tumor antigen specific human T cells is unimpaired by arginine depletion. PloS one. (2013) 8:e63521. doi: 10.1371/journal.pone.0063521
215. Chuang JC, Yu CL, Wang SR. Modulation of lymphocyte proliferation by enzymes that degrade amino acids. Clin Exp Immunol. (1990) 82:469–472. doi: 10.1111/j.1365-2249.1990.tb05473.x
216. van de Velde, Murray PJ. Proliferating helper T cells require rictor/mTORC2 complex to integrate signals from limiting environmental amino acids. J Biol Chem. (2016) 291:25815–22. doi: 10.1074/jbc.C116.763623
217. Yu R, C.-Tsai C, L.-Chang S, H.-Huang C, H.-Cheng H, J.-Wang Y, et al. L-arginine-dependent epigenetic regulation of interleukin-10, but not transforming growth factor-β, production by neonatal regulatory T lymphocytes. Front Immunol. (2017) 8:487. doi: 10.3389/fimmu.2017.00487
218. Zea AH, Rodriguez PC, Culotta KS, Hernandez CP, DeSalvo J, Ochoa JB, et al. L-Arginine modulates CD3zeta expression and T cell function in activated human T lymphocytes. Cell Immunol. (2004) 232:21–31. doi: 10.1016/j.cellimm.2005.01.004
219. Modolell M, Choi BS, Ryan RO, Hancock M, Titus RG, Abebe T, et al. Local suppression of T cell responses by arginase-induced L-arginine depletion in nonhealing leishmaniasis. PLoS Negl Trop Dis. (2009) 3:e480. doi: 10.1371/journal.pntd.0000480
220. Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol. (2006) 176:2085–94. doi: 10.4049/jimmunol.176.4.2085
221. Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T cell responses: naive to memory and everything in between. Adv Physiol Educ. (2013) 37:273–83. doi: 10.1152/advan.00066.2013
222. Rohr JC, Gerlach C, Kok L, Schumacher TN. Single cell behavior in T cell differentiation. Trends Immunol. (2014) 35:170–7. doi: 10.1016/j.it.2014.02.006
223. Cao Y, Feng Y, Zhang Y, Zhu X, Jin F. L-Arginine supplementation inhibits the growth of breast cancer by enhancing innate and adaptive immune responses mediated by suppression of MDSCs in vivo. BMC Cancer. (2016) 16:343. doi: 10.1186/s12885-016-2376-0
224. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–842.e13. doi: 10.1016/j.cell.2016.09.031
225. Lowe MM, Boothby I, Clancy S, Ahn RS, Liao W, Nguyen DN, et al. Regulatory T cells use arginase 2 to enhance their metabolic fitness in tissues. JCI insight. (2019) 4. doi: 10.1172/jci.insight.129756
226. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. (2009) 30:832–44. doi: 10.1016/j.immuni.2009.04.014
227. Shi H, Chapman NM, Wen J, Guy C, Long L, Dhungana Y, et al. Amino acids license kinase mTORC1 activity and treg cell function via small G proteins rag and rheb. Immunity. (2019) 51:1012–27. doi: 10.1016/j.immuni.2019.10.001
228. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. (2013) 499:485–90. doi: 10.1038/nature12297
229. Wu H, Zhen Y, Ma Z, Li H, Yu J, Xu ZG, et al. Arginase-1-dependent promotion of TH17 differentiation and disease progression by MDSCs in systemic lupus erythematosus. Sci Trans Med. (2016) 8:331ra40. doi: 10.1126/scitranslmed.aae0482
230. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J Biol Chem. (2002) 277:21123–9. doi: 10.1074/jbc.M110675200
231. Feldmeyer N, Wabnitz G, Leicht S, Luckner-Minden C, Schiller M, Franz T, et al. Arginine deficiency leads to impaired cofilin dephosphorylation in activated human T lymphocytes. Int Immunol. (2012) 24:303–13. doi: 10.1093/intimm/dxs004
232. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. (2007) 109:1568–73. doi: 10.1182/blood-2006-06-031856
233. Taheri F, Ochoa JB, Faghiri Z, Culotta K, Park HJ, Lan MS, et al. L-Arginine regulates the expression of the T-cell receptor zeta chain (CD3zeta) in Jurkat cells. Clin Cancer Res. (2001) 7:958s–65s.
234. Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. (1991) 64:891–901. doi: 10.1016/0092-8674(91)90314-O
235. Liu H, Rhodes M, Wiest DL, D.Vignali AA. On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity. (2000) 13:665–75. doi: 10.1016/S1074-7613(00)00066-2
236. Valitutti S, Muller S, Salio M, Lanzavecchia A. Degradation of T cell receptor (TCR)-CD3-zeta complexes after antigenic stimulation. J Exp Med. (1997) 185:1859–64. doi: 10.1084/jem.185.10.1859
237. Rabinowich H, Reichert TE, Kashii Y, Gastman BR, Bell MC, Whiteside TL. Lymphocyte apoptosis induced by Fas ligand- expressing ovarian carcinoma cells. implications for altered expression of T cell receptor in tumor-associated lymphocytes. J Clin Invest. (1998) 101:2579–88. doi: 10.1172/JCI1518
238. Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol. (2003) 171:1232–9. doi: 10.4049/jimmunol.171.3.1232
239. Kono K, Salazar-Onfray F, Petersson M, Hansson J, Masucci G, Wasserman K, et al. Hydrogen peroxide secreted by tumor-derived macrophages down-modulates signal-transducing zeta molecules and inhibits tumor-specific T cell-and natural killer cell-mediated cytotoxicity. Eur J Immunol. (1996) 26:1308–313. doi: 10.1002/eji.1830260620
240. Rodriguez PC, Hernandez CP, Morrow K, Sierra R, Zabaleta J, Wyczechowska DD, et al. L-arginine deprivation regulates cyclin D3 mRNA stability in human T cells by controlling HuR expression. J Immunol. (2010) 185:5198–204. doi: 10.4049/jimmunol.1001224
241. Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell. (2003) 4:451–61. doi: 10.1016/S1535-6108(03)00301-5
242. Laphanuwat P, Jirawatnotai S. Immunomodulatory roles of cell cycle regulators. Front Cell Dev Biol. (2019) 7:23. doi: 10.3389/fcell.2019.00023
243. Huppa JB, Davis MM. T-cell-antigen recognition and the immunological synapse. Nat Rev Immunol. (2003) 3:973–83. doi: 10.1038/nri1245
244. Eibert SM, Lee KH, Pipkorn R, Sester U, Wabnitz GH, Giese T, et al. Cofilin peptide homologs interfere with immunological synapse formation and T cell activation. Proc Natl Acad Sci USA. (2004) 101:1957–62. doi: 10.1073/pnas.0308282100
245. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. (2005) 5:844–52. doi: 10.1038/nri1710
246. Marti i Lindez AA, Dunand-Sauthier I, Conti M, Gobet F, Nunez N, Hannich JT, et al. Mitochondrial arginase-2 is a cellautonomous regulator of CD8+ T cell function and antitumor efficacy. JCI insight. (2019) 4. doi: 10.1172/jci.insight.132975
247. Garcia-Navas R, Munder M, Mollinedo F. Depletion of L-arginine induces autophagy as a cytoprotective response to endoplasmic reticulum stress in human T lymphocytes. Autophagy. (2012) 8:1557–76. doi: 10.4161/auto.21315
248. Saxton RA, Chantranupong L, Knockenhauer KE, Schwartz TU, Sabatini DM. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature. (2016) 536:229–33. doi: 10.1038/nature19079
249. de Jonge WJ, Kwikkers KL, te Velde AA, S.van Deventer JH, Nolte MA, Mebius RE, et al. Arginine deficiency affects early B cell maturation and lymphoid organ development in transgenic mice. J Clin Invest. (2002) 110:1539–48. doi: 10.1172/JCI0216143
250. Kobayashi T, Yamamoto M, Hiroi T, McGhee J, Takeshita Y, Kiyono H. Arginine enhances induction of T helper 1 and T helper 2 cytokine synthesis by Peyer's patch alpha beta T cells and antigen-specific mucosal immune response. Biosci Biotechnol Biochem. (1998) 62:2334–40. doi: 10.1271/bbb.62.2334
251. Lelis FJN, Jaufmann J, Singh A, Fromm K, Teschner AC, Poschel S, et al. Myeloid-derived suppressor cells modulate B-cell responses. Immunol Lett. (2017) 188:108–15. doi: 10.1016/j.imlet.2017.07.003
252. Jaufmann J, F.Lelis JN, Teschner AC, Fromm K, Rieber N, Hartl D, et al. Human monocytic myeloid-derived suppressor cells impair B-cell phenotype and function in vitro. Eur J Immunol. (2020) 50:33–47. doi: 10.1002/eji.201948240
253. Choi BS, Martinez-Falero IC, Corset C, Munder M, Modolell M, Muller I, et al. Differential impact of L-arginine deprivation on the activation and effector functions of T cells and macrophages. J Leuko Biol. (2009) 85:268–77. doi: 10.1189/jlb.0508310
254. Yang Q, Wei J, Zhong L, Shi M, Zhou P, Zuo S, et al. Cross talk between histone deacetylase 4 and STAT6 in the transcriptional regulation of arginase 1 during mouse dendritic cell differentiation. Mol Cell Biol. (2015) 35:63–75. doi: 10.1128/MCB.00805-14
255. Yurdagul A, Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. (2020) 31:518–33.e10 doi: 10.1016/j.cmet.2020.01.001
256. Lamas B, Vergnaud-Gauduchon J, Goncalves-Mendes N, Perche O, Rossary A, M.-Vasson P, et al. Altered functions of natural killer cells in response to L-Arginine availability. Cell Immunol. (2012) 280:182–90. doi: 10.1016/j.cellimm.2012.11.018
257. Suer Gokmen S, Yoruk Y, Cakir E, Yorulmaz F, Gulen S. Arginase and ornithine, as markers in human non-small cell lung carcinoma. Cancer Biochem Biophys. (1999) 17:125–31.
258. Susskind BM, Chandrasekaran J. Inhibition of cytolytic T lymphocyte maturation with ornithine, arginine, and putrescine. J Immunol. (1987) 139:905–12.
259. Dröge W, Männel D, Falk W, Lehmann V, Schmidt H, Nick S, et al. Suppression of cytotoxic T lymphocyte activation by L-ornithine. J Immunol. (1985) 134:3379–83.
260. Casero RA Jr, Murray Stewart T, Pegg AE. Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat Rev Cancer. (2018) 18:681–95. doi: 10.1038/s41568-018-0050-3
261. Igarashi K, Kashiwagi K. The functional role of polyamines in eukaryotic cells. Int J Biochem Cell Biol. (2019) 107:104–15. doi: 10.1016/j.biocel.2018.12.012
262. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
263. Hukelmann JL, Anderson KE, Sinclair LV, Grzes KM, Murillo AB, Hawkins PT, et al. The cytotoxic T cell proteome and its shaping by the kinase mTOR. Nat Immunol. (2016) 17:104–12. doi: 10.1038/ni.3314
264. Yang Q, Zheng C, Cao J, Cao G, Shou P, Lin L, et al. Spermidine alleviates experimental autoimmune encephalomyelitis through inducing inhibitory macrophages. Cell Death Diff. (2016) 23:1850–61. doi: 10.1038/cdd.2016.71
265. Chaturvedi R, Asim M, Hoge S, Lewis ND, Singh K, Barry DP, et al. Polyamines impair immunity to helicobacter pylori by inhibiting l-arginine uptake required for nitric oxide production. Gastroenterology. (2010) 139:1686-98. doi: 10.1053/j.gastro.2010.06.060
266. Hasko G, Kuhel DG, Marton A, Nemeth ZH, Deitch EA, Szabo C. Spermine differentially regulates the production of interleukin-12 p40 and interleukin-10 and suppresses the release of the T helper 1 cytokine interferon-gamma. Shock. (2000) 14:144–9. doi: 10.1097/00024382-200014020-00012
267. Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, et al. A relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity. (2017) 46:233–44. doi: 10.1016/j.immuni.2017.01.005
268. Ovsepian SV, O'Leary VB. Can arginase inhibitors be the answer to therapeutic challenges in alzheimer's disease? Neurotherapeutics. (2018) 15:1032–5. doi: 10.1007/s13311-018-0668-6
269. Iniesta V, Carcelén J, Molano I, Peixoto PM, Redondo E, Parra P, et al. Arginase I induction during leishmania major infection mediates the development of disease. Infect Immun. (2005) 73:6085–90. doi: 10.1128/IAI.73.9.6085-6090.2005
270. Pudlo M, Demougeot C, Girard-Thernier C. Arginase inhibitors: a rational approach over one century. Med Res Rev. (2017) 37:475–513. doi: 10.1002/med.21419
271. Abdelkawy KS, Lack K, Elbarbry F. Pharmacokinetics and pharmacodynamics of promising arginase inhibitors. Eur J Drug Metab Pharmacokinet. (2017) 42:355–70. doi: 10.1007/s13318-016-0381-y
272. Di Costanzo L, Ilies M, Thorn KJ, Christianson DW. Inhibition of human arginase I by substrate and product analogues. Arch Biochem Biophys. (2010) 496:101–8. doi: 10.1016/j.abb.2010.02.004
273. Baggio R, Emig FA, Christianson DW, Ash DE, Chakder S, Rattan S. Biochemical and functional profile of a newly developed potent and isozyme-selective arginase inhibitor. J Pharmacol Exp Ther. (1999) 290:1409–16.
274. Kim NN, Cox JD, Baggio RF, Emig FA, Mistry SK, Harper SL, et al. Probing erectile function: S-(2-boronoethyl)-L-cysteine binds to arginase as a transition state analogue and enhances smooth muscle relaxation in human penile corpus cavernosum. Biochemistry. (2001) 40:2678–88. doi: 10.1021/bi002317h
275. Secondini C, Coquoz O, Spagnuolo L, Spinetti T, Peyvandi S, Ciarloni L, et al. Arginase inhibition suppresses lung metastasis in the 4T1 breast cancer model independently of the immunomodulatory and anti-metastatic effects of VEGFR-2 blockade. Oncoimmunology. (2017) 6:e1316437. doi: 10.1080/2162402X.2017.1316437
276. Blaszczyk R, Brzezinska J, Dymek B, Stanczak PS, Mazurkiewicz M, Olczak J, et al. Discovery and pharmacokinetics of sulfamides and guanidines as potent human arginase 1 inhibitors. ACS Med Chem Lett. (2020) 11:433–438. doi: 10.1021/acsmedchemlett.9b00508
277. Stanczak PS, Wolska P, Zdziarska AM, Mazurkiewicz M, Blaszczyk R, Nowicka J, et al. Development of OAT-1746, a novel arginase 1 and 2 inhibitor for cancer immunotherapy. In: 42nd ESMO Congress (ESMO 2017). Barcelona (2017). doi: 10.1093/annonc/mdx376.045
278. Tangphao O, Grossmann M, Chalon S, Hoffman BB, Blaschke TF. Pharmacokinetics of intravenous and oral L-arginine in normal volunteers. Br J Clin Pharmacol. (1999) 47:261–6. doi: 10.1046/j.1365-2125.1999.00883.x
279. Sin YY, Ballantyne LL, Mukherjee K, St Amand T, Kyriakopoulou L, Schulze A, et al. Inducible arginase 1 deficiency in mice leads to hyperargininemia and altered amino acid metabolism. PLoS ONE. (2013) 8:e80001. doi: 10.1371/journal.pone.0080001
280. Ballantyne LL, Sin YY, Al-Dirbashi OY, Li X, Hurlbut DJ, Funk CD. Liver-specific knockout of arginase-1 leads to a profound phenotype similar to inducible whole body arginase-1 deficiency. Mol Genet Metab Rep. (2016) 9:54–60. doi: 10.1016/j.ymgmr.2016.10.003
281. Yu H, Iyer RK, Yoo PK, Kern RM, Grody WW, Cederbaum SD. Arginase expression in mouse embryonic development. Mech Dev. (2002) 115:151–5. doi: 10.1016/S0925-4773(02)00089-8
282. Sin YY, Baron G, Schulze A, Funk CD. Arginase-1 deficiency. J Mol Med. (2015) 93:1287–96. doi: 10.1007/s00109-015-1354-3
283. Naing TB, Papadopoulos KP, Rahma O, Tsai F, Garralda E, Naidoon J, et al. Phase I study of the arginase inhibitor INCB (1158) 001158:alone and in combination with pembrolizumab (PEM) in patients (Pts) with advanced/metastatic (adv/met) solid tumours. In: 44th ESMO Congress (ESMO 2019). Barcelona (2019). doi: 10.1093/annonc/mdz244.002
284. Phillips MM, Sheaff MT, Szlosarek PW. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res Treat. (2013) 45:251–62. doi: 10.4143/crt.2013.45.4.251
285. Izzo F, Marra P, Beneduce G, Castello G, Vallone P, De Rosa V, et al. Pegylated arginine deiminase treatment of patients with unresectable hepatocellular carcinoma: results from phase I/II studies. J Clin Oncol. (2004) 22:1815–22. doi: 10.1200/JCO.2004.11.120
286. Yau T, Cheng PN, Chan P, Chan W, Chen L, Yuen J, et al. A phase 1 dose-escalating study of pegylated recombinant human arginase 1 (Peg-rhArg1) in patients with advanced hepatocellular carcinoma. Invest New Drugs. (2013) 31:99–107. doi: 10.1007/s10637-012-9807-9
287. Yau T, Cheng PN, Chan P, Chen L, Yuen J, Pang R, et al. Preliminary efficacy, safety, pharmacokinetics, pharmacodynamics and quality of life study of pegylated recombinant human arginase 1 in patients with advanced hepatocellular carcinoma. Invest New Drugs. (2015) 33:496–504. doi: 10.1007/s10637-014-0200-8
288. Zou S, Wang X, Liu P, Ke C, Xu S. Arginine metabolism and deprivation in cancer therapy. Biomed Pharmacother. (2019) 118:109210. doi: 10.1016/j.biopha.2019.109210
289. Miller EF, Maier RJ. Ammonium metabolism enzymes aid Helicobacter pylori acid resistance. J Bacteriol. (2014) 196:3074–81. doi: 10.1128/JB.01423-13
290. Zabaleta J, McGee DJ, Zea AH, Hernández CP, Rodriguez PC, Sierra RA, et al. Helicobacter pylori arginase inhibits T cell proliferation and reduces the expression of the TCR -Chain (CD3ζ). Oncol Lett. (2004) 173:586–93. doi: 10.4049/jimmunol.173.1.586
291. Yachimovich-Cohen N, Even-Ram S, Shufaro Y, Rachmilewitz J, Reubinoff B. Human embryonic stem cells suppress T cell responses via arginase i-dependent mechanism. J Immunol. (2010) 184:1300–8. doi: 10.4049/jimmunol.0804261
292. Kropf P, Baud D, Marshall SE, Munder M, Mosley A, Fuentes JM, et al. Arginase activity mediates reversible T cell hyporesponsiveness in human pregnancy. Eur J Immunol. (2007) 37:935–45. doi: 10.1002/eji.200636542
293. Petrone AB, O'Connell GC, Regier MD, Chantler PD, Simpkins JW, Barr TL. The role of arginase 1 in post-stroke immunosuppression and ischemic stroke severity. Transl Stroke Res. (2016) 7:103–10. doi: 10.1007/s12975-015-0431-9
294. Xu L, Hilliard B, Carmody RJ, Tsabary G, Shin H, Christianson DW, et al. Arginase and autoimmune inflammation in the central nervous system. Immunology. (2003) 110:141–8. doi: 10.1046/j.1365-2567.2003.01713.x
295. Suwanpradid J, Shih M, Pontius L, Yang B, Birukova A, Guttman-Yassky E, et al. Arginase1 deficiency in monocytes/macrophages upregulates inducible nitric oxide synthase to promote cutaneous contact hypersensitivity. J Immunol. (2017) 199:1827–34. doi: 10.4049/jimmunol.1700739
296. Schleicher U, Paduch K, Debus A, Obermeyer S, König T, Kling JC, et al. TNF-mediated restriction of arginase 1 expression in myeloid cells triggers type 2 NO synthase activity at the site of infection. Cell Rep. (2016) 15:1062–75. doi: 10.1016/j.celrep.2016.04.001
Keywords: arginase, arginine, immunosuppression, tumor immunology, immunotherapy, T lymphocyte, T-cell metabolism
Citation: Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D and Golab J (2020) Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 11:938. doi: 10.3389/fimmu.2020.00938
Received: 19 February 2020; Accepted: 22 April 2020;
Published: 15 May 2020.
Edited by:
Sergei Kusmartsev, University of Florida, United StatesReviewed by:
Markus Munder, Johannes Gutenberg University Mainz, GermanyIlaria Marigo, Istituto Oncologico Veneto (IRCCS), Italy
Copyright © 2020 Grzywa, Sosnowska, Matryba, Rydzynska, Jasinski, Nowis and Golab. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominika Nowis, dominika.nowis@wum.edu.pl; Jakub Golab, jakub.golab@wum.edu.pl