 Abdella M. Habib
Abdella M. Habib Karim Nagi
Karim Nagi Nagendra Babu Thillaiappan
Nagendra Babu Thillaiappan VijayaKumar Sukumaran
VijayaKumar Sukumaran Saghir Akhtar
Saghir Akhtar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 May 2020
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00820
This article is part of the Research TopicImmune-modulatory Effects of Vitamin DView all 14 articles
About 50 million of the U.S. adult population suffer from chronic pain. It is a complex disease in its own right for which currently available analgesics have been deemed woefully inadequate since ~20% of the sufferers derive no benefit. Vitamin D, known for its role in calcium homeostasis and bone metabolism, is thought to be of clinical benefit in treating chronic pain without the side-effects of currently available analgesics. A strong correlation between hypovitaminosis D and incidence of bone pain is known. However, the potential underlying mechanisms by which vitamin D might exert its analgesic effects are poorly understood. In this review, we discuss pathways involved in pain sensing and processing primarily at the level of dorsal root ganglion (DRG) neurons and the potential interplay between vitamin D, its receptor (VDR) and known specific pain signaling pathways including nerve growth factor (NGF), glial-derived neurotrophic factor (GDNF), epidermal growth factor receptor (EGFR), and opioid receptors. We also discuss how vitamin D/VDR might influence immune cells and pain sensitization as well as review the increasingly important topic of vitamin D toxicity. Further in vitro and in vivo experimental studies will be required to study these potential interactions specifically in pain models. Such studies could highlight the potential usefulness of vitamin D either alone or in combination with existing analgesics to better treat chronic pain.
The newly proposed definition of pain by the International Association for the Study of Pain states—“An aversive sensory and emotional experience typically caused by, or resembling that caused by, actual or potential tissue injury1”. Pain represents the body's alarm system and serves as an alert to danger. Chronic pain is a complex disease in its own right for which currently available analgesics have been deemed woefully inadequate since ~20% of the sufferers derive no benefit. This is thought to be due to the complexity and plasticity of chronic pain, and the underpinning mechanisms are not clear as yet. About 50 million of the U.S. adult population suffer from chronic pain, and regardless of the reason, the current treatments primarily manage the condition rather than provide a cure (1).
Vitamin D, commonly identified as a fat-soluble vitamin, is known for its role in calcium homeostasis and bone metabolism. Recent studies have linked vitamin D status and its receptor activity with several health conditions, including development of chronic pain,1 the leading cause of disability and disease burden globally (2). Studies on the association between vitamin D status and incidence of chronic pain have been contradictory (3, 4). There are potentially many reasons for the variations in the clinical trial studies. However, a recent well-controlled study in Europeans has shown that reduced vitamin D levels were significantly associated with painful diabetic peripheral neuropathy (5). A strong correlation is also shown for hypovitaminosis D and bone pain (6). Indeed, several other studies have now reported a progressive exacerbation of pain with decreasing serum vitamin D levels and conversely, by increasing serum vitamin D levels through appropriate vitamin D supplementation, especially in vitamin D deficient patients, leads to an improvement in pain-relief (see Table 1). However, the potential underlying mechanisms by which vitamin D might exert its analgesic effects are poorly understood. In this review, we discuss pathways involved in pain sensing and processing primarily at the level of DRG neurons and the potential interplay between vitamin D/VDR and known specific pain signaling pathways including nerve growth factor (NGF), glial-derived neurotrophic factor (GDNF), epidermal growth factor receptor (EGFR), and opioid receptors. We also discuss how vitamin D might influence immune cells and pain sensitization as well as include a section on the increasingly important topic of vitamin D toxicity.
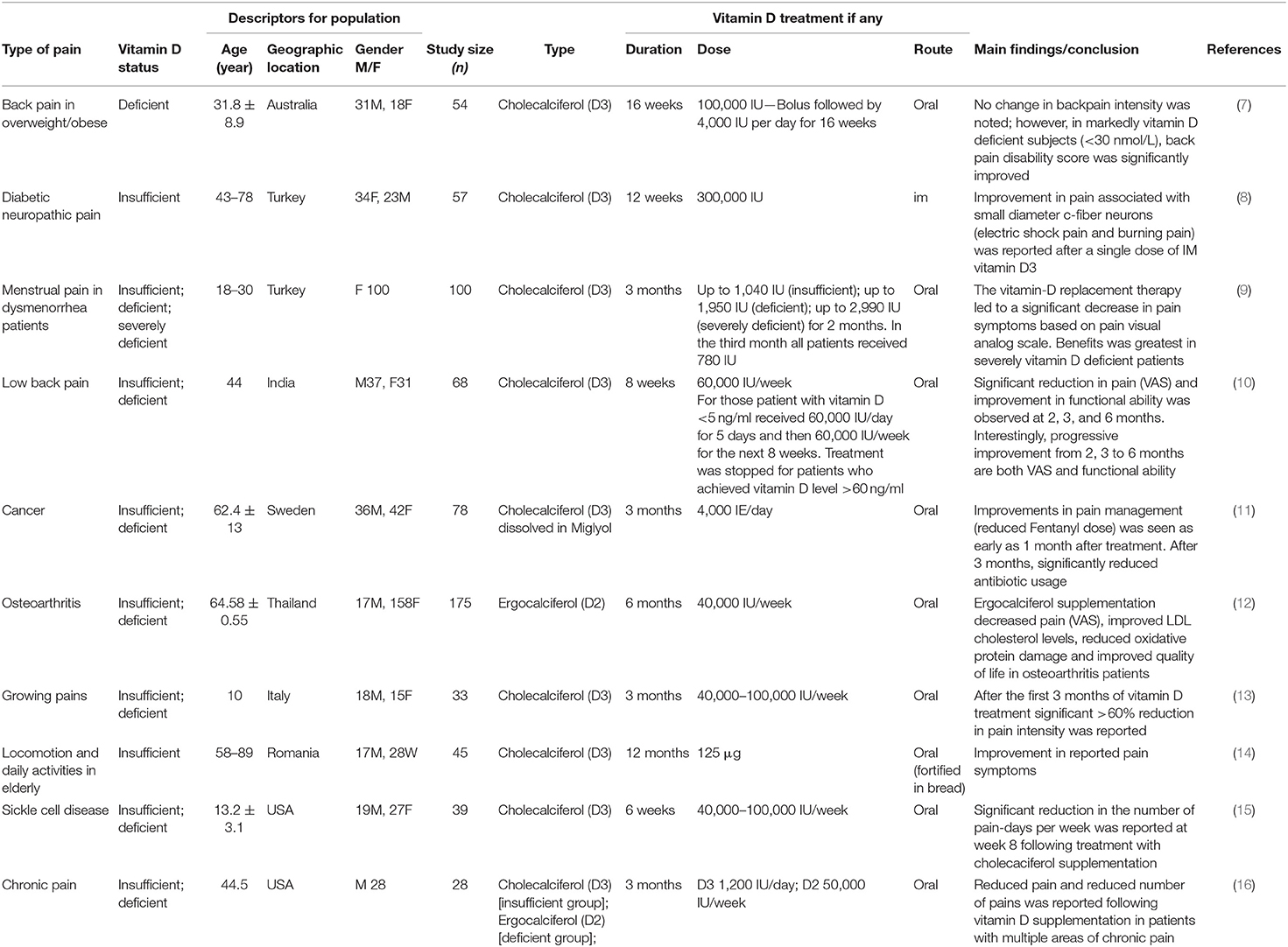
Table 1. Examples of clinical studies showing benefit in pain-relief following vitamin D supplementation.
Vitamin D, although identified as a fat-soluble vitamin, is increasingly being recognized as a prohormone. Traditionally, vitamin D was considered to be essential for calcium and phosphate homeostasis and thereby, bone health, and its deficiency causes rickets and osteomalacia. Recent evidence, however, advocates a role for vitamin D that extends beyond bone metabolism (6). Our body obtains vitamin D both directly from the diet, albeit from a narrow range of food sources, and through biosynthesis in the skin. Vitamin D3 is the natural form of vitamin D and is produced from 7-dehydrocholesterol in the skin. UVB irradiation helps convert 7-dehydrocholesterol, the precursor in the skin, into pre-vitamin D3 and then to vitamin D3. Synthesis in skin serves as an essential source of vitamin D3 and depends crucially on season and geographical latitude (17). Studies involving human and pig models indicate that vitamin D3 is predominantly distributed in fat tissue (about three quarters) and in smaller amounts in muscles, liver, and skin. Distribution of 25-(OH)D3 was somewhat similar within compartments (serum, 30%; muscle, 20%; fat, 35% and 15% in other tissues) (18). The body fat content has also been reported to inversely correlate with serum levels of 25-(OH)D3, which could indicate that those with high body fat content (obese populations) might be at risk of vitamin D3 deficiency (19, 20).
Vitamin D3, by itself, is not biologically active. 1,25-dihydroxy vitamin D3 (1,25-(OH)2D3) is the biologically active form of vitamin D and is produced by hydroxylation of vitamin D3 in two distinct steps and locations. Once synthesized (or absorbed from diet), vitamin D and its metabolites are bound to vitamin D-binding proteins (DBPs), which allow their transport in the blood. DBPs transport vitamin D3 to the liver where the first hydroxylation reaction occurs at C-25 position to produce 25-(OH)D3. 25-(OH)D3 is the major circulating form of vitamin D and hence is used as a biomarker of vitamin D status in the body. CYP2R1 is the key 25-hydroxylase of vitamin D in the liver (21). 25-(OH)D3 is then transported to the kidney where it is filtered and taken up into proximal renal tubule epithelial cells by the action of cell surface receptors for DBP, and megalin/cubulin complex (22, 23). In proximal renal tubules, 25-(OH)D3 is hydroxylated at the C-1 position by renal 25-(OH)D3 1α hydroxylase (CYP27B1), to produce 1,25-(OH)2D3, which is also known as calcitriol and is biologically active (24, 25). The circulating levels of 1,25-(OH)2D3 is regulated by the action of CYP24A1, another hydroxylase of C-24, to generate 24,25-(OH)2D3, which can allow its excretion or reduce the pool of 1,25-(OH)2D3 available for C-1 hydroxylation (26). CYP24A1 also catalyzes conversion of 1,25-(OH)2D3 and 25-(OH)D3 into 1,25-(OH)2D3-26,23 lactone and 25-(OH)D3-26,23 lactone, respectively. Presence of CYP24A1 in most cells expressing vitamin D receptors (VDR), suggests that CYP24A1 can regulate not only the circulating concentrations of 1,25-(OH)2D3 but also modulate its cellular levels locally (27).
Vitamin D mediates gene expression through interaction with its receptor, VDR. The VDR has been shown to recognize the sequence for vitamin D binding response element (VDRE) in the DNA and also forms a heterodimer with retinoid X receptor molecule. The effect of VDR is dependent on several other, as yet unknown, factors including enzymatic activities such as, demethyltransferase and RNA polymerase that mediate the promotion or inhibition of gene expression. Interestingly some of these target genes include those that bind 1,25(OH)2D3 to activate it or that mark it for its degradation, suggesting a self-regulating mechanism (28) and perhaps restricting the possibility for vitamin D toxicity due to over-exposure to sunlight or dietary intake. Other implicated target genes are those encoding for proteins involved in pain signaling pathways, such as growth factor receptors, ion channels, neurotrophic factors, and proteins involved in modulating neuronal axon growth, thereby implicating that vitamin D has a role in pain sensing and processing (see below).
Pain is a subjective sensory experience that involves multiple signaling pathways (29–31). However, there are several categories of pain. In the proceeding sections we will discuss the interplay between vitamin D and different types of pain.
The ability to feel acute “normal pain,” protecting the injured tissue of the body to facilitate recovery and the amelioration of the resulting pain from an injury, is one of mother nature's gift to mankind and the rest of animal kingdom. We evolved to feel pain because the sensation and pain-induced responses serve as an alarm system that is necessary for self-preservation, and this evolutionary advantage is maintained in every animal with a sensory nervous system.
However, the “normal pain” can change to “abnormal pain” and thereby the benefit as a necessary alarm to self-preservation becomes instead incapacitating to the individual and is then seen as nature's curse. This change is often due to damage to the nervous system, resulting in a lowering of the pain threshold to otherwise innocuous stimuli such as warmth and gentle touch and is now recognized as painful (allodynia). Or, when the degree of pain from a painful stimulus becomes intensified/exaggerated (hyperalgesia). These “abnormal pain” conditions often fail to resolve within 3 months (chronic pain), long after the acute injury, and are generally intractable to conventional analgesia and become debilitating. Chronic pain is broadly defined as “pain caused by a lesion or disease of the somatosensory nervous system” (www.iaps.org). The lesion could be due to damage to the nervous system (as observed in neuropathic pain) or inflammation (as found in arthritis). There are several types of chronic pain- [chronic primary pain, chronic postsurgical and posttraumatic pain, chronic neuropathic pain, chronic headache and orofacial pain, chronic visceral pain, and finally chronic musculoskeletal pain (32)] of which lower back pain probably being one of the most frequently experienced chronic pain conditions, and has a clear association between deficiency in vitamin D levels and pain as a result of softening of the bone (6).
Similar to nociception where the stimuli are sensed by the skin (see section below on Pain Signaling), the lining of the gut is also an “inside out” interface to our external environment and is essential in visceral nociception. Visceral pain is defined as that which originates from the tissues of the internal organs in the body, and pathologically affects more than 20% of the world's population. There is some homology between visceral nociceptors and those in the skin. For example, similar to the skin, the DRG nerve fibers that innervate the serosa and the muscle layer in the intestine also synapse in the spinal cord (Figure 1). Most visceral nociceptors are highly sensitive to pro-inflammatory signals, ischemia, and to chemicals released from the ischemic tissues.
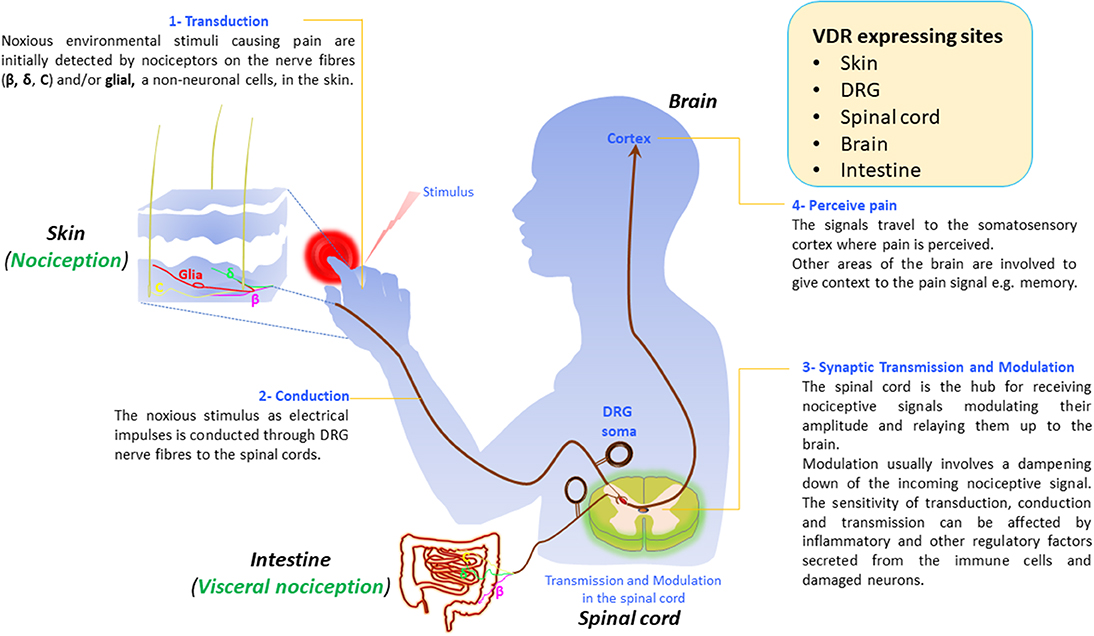
Figure 1. Vitamin D and its receptor in pain signaling pathways. Vitamin D is synthesized in the skin. Vitamin D receptor (VDR) is expressed in neurons in the skin, dorsal root ganglia (DRG), spinal cord, brain, and the intestine. Nociceptors, from the specific area of the periphery and viscera respond to environmental nociceptive stimuli such as thermal, mechanical, or chemicals by converting the stimuli into a chemoelectrical signal (1). The signal travels as a nerve impulse through dorsal root ganglia (DRG) nerve fibers (c, β, and δ) from the skin to the spinal cord (2) where it gets transmitted to secondary neurons in the dorsal horns of the spinal cord (3). After modulating the signal, the secondary neurons relay it to the cortex where the message gets decoded and pain is perceived (4).
Thus, visceral nociceptors are exquisite sensors of local changes in blood flow (ischemia) and inflammation within an organ, both of which can induce chronic visceral pain. Visceral pain can also emanate from factors in the lumen of the gut, including the microbiota and products derived from them.
The luminal microbiota aids digestion and generates short-chain fatty acids (SCFAs) that are an important energy source for the host. In addition, some of these SCFAs also serve as signaling molecules in host immunity and pain signaling. For example, we have shown, in mice, that SCFA can induce the release of glucagon-like peptide-1 (GLP-1) from intestinal L-cells (33). GLP-1 is a pleiotropic hormone mainly known for stimulating insulin release and appetite suppression. However, recent studies show that the GLP-1 receptor is also expressed in the sensory nerve fibers (34) and GLP-1 agonists have an anti-inflammatory action and can ameliorate pain (35, 36).
Several gastrointestinal neurotransmitters, in addition to their physiological role, also impact on pain sensing mechanisms. For example, the neurotransmitter serotonin is involved in motility of the gut, to propel the content in the lumen. However, it also modulates pain-sensing through potentiating the transient receptor potential cation channel subfamily V member 1 (TRPV1) channel (37, 38). Interestingly, the synthesis of serotonin in the enterochromaffin cell in the intestine is regulated in response to metabolites generated from gut microbiota (39). The importance of the gut microbiota in regulating gut motility was recently highlighted by Obata et al. (40). In this animal study it was shown that microbial colonization of the gut led to upregulation of several genes in the enteric neurons of the colon, including aryl hydrocarbon receptor, a transcription factor that regulates intestinal motility through neuronal programming (40). Furthermore, neuronal GPCRs that are involved in nociception are activated by SCFAs and other metabolites secreted or degraded from the gut microbiome (41, 42).
Under pathological conditions such as in irritable bowel syndrome, or in chemotherapy drug-induced mechanical hyperalgesia, a heightened pro-inflammatory state of the intestines is observed and results in exaggerated visceral pain. Thus, it is likely that the gut microbiota is an important regulator for the expression of pain-genes not only locally but also in distal organs, such as expression of Toll-like receptors (TLRs) and cytokines in the spinal cord that mediate visceral pain response, and are vital to the development of mechanical hyperalgesia (43, 44).
It is, therefore, perhaps unsurprising that dysbiosis of the gut microbiome results in chronic intestinal inflammation and other diseases. For example, the pathogenic bacteria- Salmonella enterica- commonly known to cause food poisoning, can disable the host's defenses against inflammation through activation of the TLR4. It also degrades intestinal alkaline phosphatase, an enzyme that is important in detoxifying endotoxin (lipopolysaccharide-phosphate) and thereby inducing gut microbiota-mediated autoimmunity and chronic inflammation (45).
Several studies indicate that vitamin D supplementation and/or deficiency changes the gut microbiota profile (46, 47); as such, this can potentially modulate visceral pain. Emerging data suggest vitamin D/VDR play a role in pain-sensing through modulating key pain-genes. Some of these pain-genes are common to both nociception and visceral nociception, for example, TRPV1, Toll-like receptor, trophic factors, such as NGF, BDNF and GPCR. The interplay between vitamin D and these pain-genes are further discussed below.
Spinal sensitization involves interactions between several neuronal and glial cells. The glial cells in the spinal cord play a significant role by responding to various cytokines and neurochemicals released from infiltrating macrophages, neutrophils and from the damaged peripheral nerve fibers. This response can be considered as part of protective measures, akin to acute pain, and vitamin D is thought to have a role in regulating the synthesis of cytokines (6).
There is growing evidence indicating that a family of TLR play a role in activating glial cells in the spinal cord and DRG neurons and thereby influence the sensitization process following nerve injury (48, 49). In a mouse neuropathic pain model of TLR2 knockout mice, the activity of the immune cells following the peripheral nerve injury is attenuated. Furthermore, reduced sensitivity to pain was observed in a behavior assay, suggesting that TLR2 could be a factor in neuropathic pain and in the activation of the glia in the spinal cord (50, 51). A similar result was also observed in TLR4 knockout mice (52). Vitamin D is known to be anti-inflammatory and inhibits the release of several cytokines and TLRs (53, 54). Expression of both receptors, TLR2 and TLR4, was shown to be modulated by vitamin D in human studies (55, 56). Identifying the molecular and neurochemical basis that cause/modulate the sensitization process may help enable us to identify therapeutic targets for the treatment of pain. In the next sections, we will focus on the potential ways by which vitamin D might play a role in the pain system.
Skin is unique in that, apart from it being the most substantial organ system that protects the body from damage, it is also the largest damage-sensing organ and the only site for vitamin D synthesis in the body. Nociceptors, the specialized mammalian sensory neurons that react to tissue injury are critical components of the somatosensory nervous system, pain-sensing machinery, proposed initially by Charles Sherrington a 100 years ago (57). These neurons with their nuclei in the dorsal root ganglia (DRG) and their fibers stretching into the skin, the gut, and other organs in the body convey the information about potential harm to the central nervous system. However, a more recent study indicates that the ability to initiate the signal for sensing damage to the tissue is no longer exclusive to nociceptive free nerve endings but can also occur through glial cell signaling (58). Nonetheless, the role of nociceptors in pain signaling has been validated by several studies over the years, and their healthy development is a prerequisite for sensing pain.
Glial and Schwann cells, together with nociceptors, constitute the peripheral nervous system. These neuron and non-neuronal cell complexes require a healthy vascular system that supplies oxygenated blood and nutrients through small blood vessels. Vitamin D supplementation is known to significantly improve vascular functions in type 2 diabetic patients with vitamin D deficiency (59). Vitamin D supplementation was also shown to enhance myelination of DRG neurons and to regulate expression of genes involved in axon growth in an animal model of nerve damage (60). More recently, the existence of a previously unknown sensory organ: the skin glial cell that is intimately associated with unmyelinated small thin fibers which sense pain, has been reported. These nociceptive glio-neural complexes have been shown to evoke pain signaling, as evidenced by whole-cell current recordings in response to mechanical pressure (58) and therefore are essential to our ability to sense damage to the skin. However, further validation of this ground-breaking finding in humans is required. Several studies have reported an association between vitamin D insufficiency with pain in general and or following injury (61–63); however, knowledge of the underpinning molecular mechanisms is limited.
There now exists several clinical studies (see Table 1) and some animal studies, mostly in rodents (see Table 2), that show that vitamin D deficiency leads to a worsening of pain whereas appropriate vitamin D supplementation leads to better outcomes relating to pain. However, there are no definitive studies with knockout mice lacking VDR or one of the key vitamin D metabolizing enzymes that have been performed which directly links vitamin D/VDR to pain pathways. This is most likely as a result of these knockout mice being infertile (68).
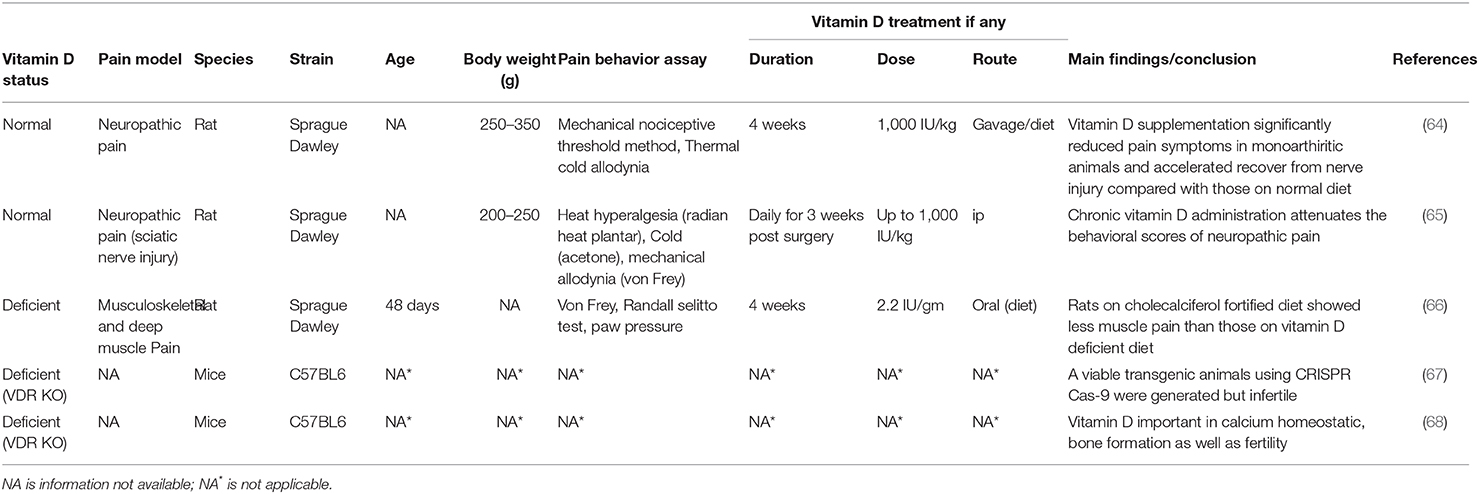
Table 2. Selective examples of in vivo studies with Vitamin D receptor (VDR) KO mice and those examining the effect of Vitamin D and/or VDR on pain using animal models.
Nonetheless, the hypothesis that vitamin D perhaps influences pain-signaling pathways is biologically plausible because vitamin D and/or vitamin D receptor gene expression has been shown in relevant tissues such as skin (pain signal transduction), DRG neurons (conduction), spinal cord (transmission/modulation), and brain (pain perception) (see also Figure 1).
Vitamin D receptor expression has been reported in the peripheral and central neurons involved in pain-sensing and processing. Indeed, there appears to be a local cell or tissue-specific processing of vitamin D in the neuronal system. Expressions of the transcript for nuclear vitamin D receptor and/or the enzymes regulating the active form of vitamin D levels have been shown in DRG neuron bare nerve fiber endings in the skin and the soma (69–71), in the spinal cord neurons (71, 72), and the brain (71, 73–75). The level of VDR transcript in DRG neurons is higher when compared to the different regions in the brain in humans, and interestingly, this expression pattern is inversed in the mouse (71). The reason for this is unclear, but it is noteworthy as mouse models of pain are widely used in studying pain signaling. Vitamin D activity is determined by two enzymes, CYP27B1 (activates vitamin D in the kidney) and CYP24A1 (inactivates the active vitamin D) (70). These two enzymes, along with VDR, are also expressed in nociceptor neurons as well as in the brain.
Transduction of the pain signal involves the activation of ion channels that are expressed in nociceptive neurons in the skin in response to external stimuli, such as thermal, mechanical, or chemical inputs (see also Figure 1). Channels that are represented in the dermal and epidermal layers of the skin include the TRPV1, TRPV2, TRPV3, TRPV4 (for detecting heat); TRPM8, TRPA1 (for detecting cold), and TRPV4, TRPC (for detecting mechanical pressure sensing). The activation of these channels in response to stimuli leads to a transient change in cellular Ca2+ and Na+ concentration in the nociceptive neurons and thereby affecting their intrinsic electrical properties. The sensitivity of the skin to pain, touch or temperature differs depending on the sensory nerve ending whose sensitivity determines the kind of sensation evoked from the spot. The external stimulus detected by these receptor channels is then converted into an electrical signal and is transmitted along the axon by voltage-gated sodium channels. Therefore, for example, we and others have shown that a mutation in sodium channel Nav1.7 results in a complete loss of pain sensation (29, 76) or in extreme pain phenotypes (77, 78). Vitamin D, which is synthesized in the skin is thought to interact with nociceptive neuron nerve endings in the skin to directly sense noxious proinflammatory stimuli (79), and also to control TRPV1 channel activity in T cells (38, 80).
Taken together, the above findings suggest that VDR may play a role in modulating the expression of pain-genes, for example, those involved in the development of neurons and Schwann cells (60, 81) as well as ion channels expressed in nociceptor neurons that innervate the skin as discussed above. The presence of a tissue-specific “vitamin D enzyme system” that regulates vitamin D activity locally in specific tissues or cells including neuronal cells implies that the active vitamin D levels might be different from those in the general circulation. Therefore, alterations in the expression or function of vitamin D regulating enzymes, VDR expression, VDR targets in the skin and/or in sensory neurons or the associated glial cells could likely impact on chronic pain disorders listed above including neuropathic pain (82) and painful diabetic neuropathy (83).
Studies in vivo and in vitro have shown that vitamin D increases nerve growth factor (NGF) expression from the DRG neurons innervating the skin in rats (84), as well as in hippocampal neurons (85). NGF is a necessary neurotrophic factor for the development and maturity of the nociceptors. It is composed of three subunits, 2-α, 2-γ and 1-β, and primarily exists in its precursor proNGF form. Cells that produce NGF include macrophages, mast cells, and bone marrow-derived macrophages and keratinocytes. The levels of NGF in disease conditions seems to increase in response to inflammation (86, 87).
NGF's role in pain signaling has been clearly demonstrated in families with a hereditary mutation in NGF or its receptor, tropomyosin kinase receptor type A (TrkA) (which is a typical tyrosine kinase receptor), resulting in a pain-free phenotype and impairment in the sensing of temperature (88, 89). NGF also stimulates the release of calcitonin gene-related peptide (CGRP) from the DRG peripheral endings (90). CGRP is thought to promote and maintain sensitized nociceptive neurons which implicates its role in chronic pain. The sensitization is also enhanced by increasing the insertion of TRPV1, the heated gated ion channel into the cell surface membrane, which is facilitated by NGF (91).
Furthermore, the transcription level of various isoforms of sodium channels (e.g., Nav1.6, Nav1.7, Nav1.8, and Nav1.9) is modulated by NGF and ultimately results in an increase in sodium current density and the floodgates to nociception, primarily through Nav1.8. The development of hyperalgesia during inflammation is also thought to arise from an increase in Nav1.7 expression promoted by NGF (92, 93). Since Nav1.7 expression is restricted to DRG neurons only, selective drugs modulating its channel activity have the potential to be a useful therapy for chronic pain (29). In a more recent study, lack of NGF is reported to cause retraction of nociceptors from the skin (94), hence severely affecting pain signaling pathways.
These studies suggest that NGF is critical for nociceptor neuron development and in pain processing; however, it is not clear whether this is a direct effect of vitamin D on NGF or if the outcome is indirect via extranuclear or nuclear signaling pathways.
The glial cell line-derived neurotrophic factor (GDNF) is yet another neurotrophic factor that is expressed in a small population of DRG neurons (95, 96) and is implicated in promoting the survival and activity of large cutaneous sensory, proprioceptive neurons (97). In an animal model of neuropathic pain (spinal nerve ligation), GDNF has been shown to reverse the sensory abnormality induced by nerve damage and thereby ameliorate the pain, possibly thorough a tetrodotoxin (TTX) sensitive channel inhibition (98, 99). The neuropathic pain, following nerve damage is thought to be due to ectopic activity in the damaged myelinated TTX-sensitive neurons, i.e., those nociceptor neurons expressing, for example, fast-acting and fast-inactivating Nav1.7 channels. The utility of this channel has been exemplified in conditional deletion of Nav1.7 in nociceptive neurons, animals becoming pain-free in response to painful mechanical and inflammatory stimuli (100). Subsequent work by Minett et al. (101) showed a loss of pain-sensation to a noxious thermal stimulus. A similar pain phenotype has been documented in humans with inherited loss of function mutations in Nav1.7 (76). These findings demonstrate a central role for GDNF in pain signaling. Interestingly, a recent study shows that GDNF and its receptor, C-Ret are directly regulated by vitamin D (102). It is tempting to speculate that perhaps both vitamin D and its receptor could play a role in sodium channel-mediated neuropathic pain through modulating GDNF expression; however, experimental verification is needed.
The epidermal growth factor receptor (EGFR) and its downstream effectors have been recently identified as novel signaling pathways involved in pain processing (103) and their expression is also known to be regulated by the vitamin D pathway (104).
EGFR is a 175 KDa glycoprotein that is a member of the larger EGFR (also known as ErbB or HER) family of receptor tyrosine kinases which are important regulators of vital cellular processes such as cell growth, proliferation, differentiation, and apoptosis (105). The three other members of this family are known as EGFR2 (ErbB2, HER2), EGFR3 (ErbB3 or HER3), and EGFR 4 or (HER4) (106). EGFR signaling can be activated by either ligand-dependent or ligand-independent pathways such as via G-protein coupled receptor (GPCR) mediated transactivation that includes Angiotensin II (Ang II)-type 1 (AT1) receptors which are known to be involved in mediating several types of pain (107). In addition, EGFR transactivation has also been reported to occur via Beta-adrenergic receptors (108) as well as opioid and cannabinoid receptors (109–112) that also have important functions in regulating pain (see Figure 2).
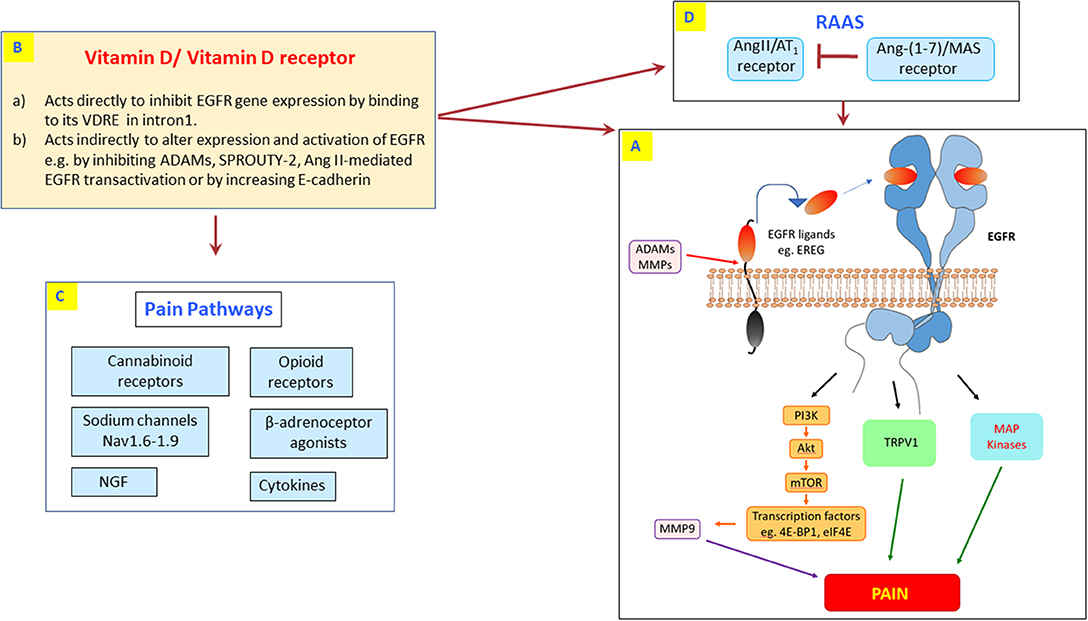
Figure 2. The potential cross-talk between vitamin D and EGFR pain signaling. (A) Highlights the known EREG/EGFR signaling pathways involved in pain sensing and processing including downstream PI3K/AKT, TRPV1, MAP kinases, and MMP-9 effector molecules. Vitamin D might exert its analgesic effect through direct inhibition of EGFR mRNA expression via binding to its VDRE in intron 1 (B). Alternatively, vitamin D can act indirectly through modulation of other intermediary signaling cascades that lead to inhibition/modulation of EGFR (B–D). This includes vitamin D-mediated regulation of known pain signals via opioid receptors, cannabinoid receptors, beta-adrenoceptors, sodium channels, cytokines, and NGF (C) as well as inhibiting the Ang II/AT1 receptor-mediated transactivation by the renin-angiotensin-aldosterone system (RAAS; D). In the RAAS, activation of the Ang-(1-7)/Mas receptor can counter-regulate the actions of Ang II/AT1 receptor and also inhibit EGFR signaling (113, 114) (D). Additionally, vitamin D can also inhibit EGFR by inhibiting ADAMs, SPROUTY-2, or by increasing E-cadherin (B; see main text for details).
For ligand-dependent EGFR activation, at least seven ligands have been described that bind to the mammalian receptor: Epidermal growth factor (EGF), transforming growth factor-alpha (TGF-α), heparin-binding EGF-like growth factor (HBEGF), betacellulin (BTC), amphiregulin (AREG), epiregulin (EREG), and epigen (EPGN). All EGFR ligands are synthesized as precursors that are tethered to the plasma membrane and require extracellular domain matrix metalloproteinases (MMPs) or members of the disintegrin and metalloprotease (ADAM) family (115). The soluble ligands released can then bind to and activate the EGFR (see also Figure 2). Autophosphorylation at specific tyrosine sites within the intracellular tyrosine kinase domain induces EGFR homo- or hetero-dimerization with other EGFR family members. This results in subsequent activation of downstream signaling pathways including MAP kinases and PI3K/AKT/mTOR (106), both of which are also known to be involved in pain regulation (116, 117).
EGFR is expressed widely in the cells of the body including epithelial cells, pain-transmitting neurons and keratinocytes of the skin—a primary source of vitamin D for the body (118, 119).
Dysregulated EGFR signaling is well-known in leading to several cancers but there is now strong evidence for its importance in other pathologies that result in pain as well as in the underlying mechanisms of pain sensing and processing. For example, in diabetes, microvascular dysfunction/microangiopathy is critically involved in the etiology of disease-associated neuropathy and subsequent neuropathic pain (120, 121). In this regard, we were the first to show that increased EGFR signaling was a critical mediator of diabetes-induced microvascular dysfunction (122–124). Mechanistic studies from our group identified that EGFR signaling via multiple pathways including those already known to be involved in pain signaling, such as MAP kinases and PI3K/AKT, were critical mediators of diabetes-induced microvascular dysfunction (125–128). Further we showed that EGFR inhibition could prevent vascular dysfunction by normalizing up to 90% of the 1,100+ gene changes observed during the development of diabetes-induced microvascular dysfunction and that upregulation of EGFR appears to be a key early change leading to vascular pathology associated with diabetes (125, 129, 130). Thus, our work implied that treatment of diabetes-induced vascular dysfunction- a precursor to the development of disease associated neuropathy- with EGFR inhibitors might also be potentially useful in preventing or at least delaying diabetic neuropathy and attenuating neuropathic pain. Vitamin D has been shown to prevent vascular dysfunction in diabetes patients and is known to attenuate the development of neuropathic pain (5, 59) suggesting that common pathways might be responsible in mediating vascular dysfunction and pain. Indeed, there appears to be a significant overlap of EGFR signaling pathways involved in microvascular dysfunction and the more recent studies on the role of EGFR and its ligands in pain processing including chronic neuropathic pain (118, 131, 132).
Martin et al. (103) reported that increased EGFR phosphorylation and upregulation of its ligand epiregulin (EREG) occurred in mouse models of chronic pain. In an elegant study conducted in human subjects, mice, and Drosophila, it was shown that activation of EGFR, specifically by EREG (and not other ligands such as EGF), activates DRG neurons and pain behaviors via mechanisms involving the TRPV1-a non-selective cation channel involved in nociception (discussed above), the PI3K/AKT/mTOR pathway as well as the matrix metalloproteinase, MMP-9 (103)-a molecule previously known to have a role in mediating inflammation and the early stages of neuropathic pain (133). In several mouse models of pain, intrathecal administration of EREG promoted nociception and led to pain hypersensitivity whereas treatment with preclinical (AG1478) or clinically used EGFR tyrosine kinase inhibitors (Gefitinib and Lapatinib) resulted in analgesia with efficacies and potencies comparable to that of morphine- an opioid analgesic. All EGFR inhibitors were effective in blocking inflammatory pain in two different animal models. They inhibited tonic inflammatory pain induced by formalin and completely attenuated thermal hypersensitivity induced by carrageenan- an inflammatory mediator. EGFR inhibitors were also completely and dose-dependently effective in reversing allodynia in three models of chronic pain including neuropathic pain. Knockdown of the EGFR ortholog specifically in peripheral neurons expressing pickpocket (ppk)—a nociceptive marker- in Drosophila resulted in prolonged withdrawal latencies in response to a heated probe at 46°C. This effect was reversed when EGFR gene was expressed in the neurons implying that the role of EGFR in pain processing is conserved across species. This study also showed that a single nucleotide polymorphism (rs1140475) in EGFR gene and one of its ligands (rs1563826 in epiregulin) was associated with development of chronic pain in humans suffering from temporomandibular disorder (103). Thus, this study demonstrated for the first time the direct association of EGFR and EREG with chronic pain in a clinical cohort of patients and further advocates the use of EREG/EGFR inhibitors as potential analgesics in the management of non-cancer pain in humans. These findings are supported by a recent study of lumbar radicular pain after disc herniation in rats, in which EREG was also thought to be released from herniated disc and mediated pain via PI3K/AKT pathway (134).
The analgesic effects of EGFR inhibition in several other human clinical studies have also been reported where diminished reporting of pain and an overall improvement in quality of life of patients was observed (131, 132, 135–137). In contrast to opioid analgesics, EGFR inhibitors showed no drug tolerance over the treatment period and the main side effect appeared to be skin rashes (131).
Taken together these studies suggest that EGFR is a key player in pain processing and sensation. Since it is already known that EGFR acts as an important hub and relay of signals from a variety of stimuli, its new role in processing pain signals would only provide added value to its proposed appellation as a “master hub or relay” of cell signaling (138) (see also Figure 2). Thus, inhibition of this important EGFR signaling hub by vitamin D might explain the latter's analgesic effects. Indeed, there are several studies suggesting that vitamin D inhibits EGFR gene expression either directly or indirectly via other intermediary signaling pathways (104, 107, 139–141).
Vitamin D through its nuclear receptor can directly regulate expression of EGFR mRNA by binding to VDRE identified in intron 1 of the EGFR gene (104). Interestingly, this study identified EGFR as the primary vitamin D/VDR target gene in these cells (104). Indirect actions of vitamin D in regulating EGFR signaling have also been reported. 1α,25(OH)2D3 increases the level of E-cadherin protein at the plasma membrane, which downregulates EGFR (142, 143), or by decreasing levels of SPROUTY-2, a cytosolic protein that normally inhibits EGFR ubiquitination, internalization and degradation (144, 145). A vitamin D3 derivative increased nerve growth factor levels in streptozotocin-diabetic rats (84) that is known to inhibit EGFR gene expression (146). Vitamin D supplements such as paricalcitol (a vitamin D analog) have also been reported to decrease EGFR signaling via inhibition of ADAM17 (141)-a key cleaving enzyme that mediates the release of EGFR ligands from the plasma membrane. In renal tubular cells, paricalcitol inhibited aldosterone-induced EGFR transactivation, and its resulting proinflammatory effects via inhibition of TGF-α/ADAM-17 gene expression (141). The fact that anti-inflammatory actions of paricalcitol in tubular cells were dependent on the inhibition of TGF-α/ADAM17/EGFR pathway may explain why both EGFR inhibitors and vitamin D are effective in models of inflammatory pain (64, 103). Vitamin D can also inhibit GPCR-mediated EGFR transactivation by blocking the actions of members of the renin-angiotensin system such as Ang II (107). The presence of these direct and indirect regulatory mechanisms of vitamin D in reducing EGFR signaling in pain neurons is not known but if reproduced in neuronal tissue, they might account for how vitamin D exerts its pain relief.
As a recent genetic study has implicated polymorphisms in EGFR and its ligand, epiregulin, to the development of chronic pain (103), it might be noteworthy that low 25(OH)D3 levels are associated with EGFR mutations in pulmonary adenocarcinoma (147). Whether hypovitaminosis D is responsible for EGFR polymorphisms in pain models is not known and is worthy of further study.
As EGFR is differentially expressed (129), it should also be noted that vitamin D regulation of EGFR signaling is also likely to be cell or tissue specific. In contrast to the above studies showing vitamin D-induced inhibition of EGFR in several cell types, in BT-20 breast cancer cells 1,25(OH)2D3 increases EGFR expression (148). Also, vitamin D supplementation with calcitriol led to the beneficial increase in neuregulin (a ligand for EGFR3 and 4 receptors) levels in the heart (149). Incidentally, Neuregulin is also required for remyelination and regeneration after nerve injury (150) and this may represent another mechanism by which vitamin D exerts its beneficial effects in models of neuropathic pain.
Like vitamin D, existing and newly discovered analgesic agents might also exert their pharmacological effects, at least in part, via EGFR inhibition. For example, opioids and cannabinoids are also known to regulate EGFR activity (109–112). Indeed, a novel role of cannabinoid receptor 2 in inhibiting EGF/EGFR pathway in breast cancer has recently been reported (111). Thus, the addition of vitamin D to the list of reagents modulating pain pathways via regulating EGFR signaling (see Figure 2) further implies that EGFR acts as a key convergent pathway in pain and as such might represent a novel pharmacological target for the development of new drugs and/or repurposing of existing anti-cancer EGFR inhibitors for pain therapy.
Despite the recent harrowing crisis of opioid dependency and its adverse sequalae in patients, opioids remain among the most widely used and indispensable analgesics for the treatment of moderate to severe pain. At present, no other oral therapeutic drug provides instant and effective relief of severe pain (151, 152). In rats, a vitamin D-deficiency diet was able to modulate the analgesic effect of the opioid, morphine- an effect that was blunted following administration of cholecalciferol (153). Cholecalciferol supplementation also induced dysregulation of opioid peptide genes, specifically POMC, PDYN, and PENK that code for endogenous opioids in the cerebrum (64, 154). In addition to modulating the expression of opioid agonists that are involved in pain perception, cholecalciferol supplementation did not affect the expression levels of opioid receptors. However, this supplementation was shown to dysregulate transcription levels of G protein subunits (Gαo) and effectors like subunits of the voltage-gated calcium channels (Cav2) and the adenylyl cyclase type 5 (AC5) enzymes as well as many other genes involved in opioid receptor signaling (64).
Voltage-gated Cav2 channels are effectors of opioid receptors that initiate their activity through direct binding of G protein subunits. Following receptor activation, the Gβγ dimer released from Gαi/o protein binds to the intracellular loop between the transmembrane (TM) TM I and TM II of Cav2α1 channel subunits (155, 156). This binding shifts channel activation to more depolarized potentials (157) and decreases the activation kinetics of the calcium currents (158, 159), resulting in reduced synaptic vesicle fusion with the plasma membrane for neurotransmitter release (160). This opioid modulation of channel activity is thus involved in the analgesic effect of opioids.
Functional studies showed that expression of delta opioid receptors (DORs) in sensory neurons from rat trigeminal (161) or dorsal root ganglia (162) inhibited voltage-gated calcium channels following stimulation with DOR agonists. In addition, characterizing the different subtypes of calcium channels subject to DOR inhibition was elucidated using specific toxins and nominated Cav2.1 (P/Q-type) and Cav2.2 (N-type) channels as notable effectors of DORs (163–165) in DRG sensory neurons. Both Cav2.1 and Cav2.2 are localized in axon presynaptic terminals and play an important role in synaptic transmission between somatosensory neurons and dorsal horn neurons (166). However, while Cav2.1 is found in large-diameter DRG neurons, Cav2.2 is more expressed in small-diameter neurons (167–169). Surprisingly, similar to opioid receptors, vitamin D receptors have a widespread expression within the rat central nervous system and are found in high levels within distinct portions of the sensory, motor and limbic systems (72). Vitamin D receptors, like Cav2 channels, are also expressed in DRG neurons (69, 70, 170) and whether those receptors could affect opioid signaling by dimerizing with opioid receptors or by directly modulating the expression levels of Cav2 channels or other effectors, like the G protein-coupled inwardly rectifying potassium (GIRK or Kir3) channels involved in the analgesic effects of opioids (171–174), is still unknown.
The cAMP signaling pathway regulated via the stimulation of AC enzymes is one of the key mediators of opioid actions (172, 175, 176). Membrane-bound AC isoforms are highly expressed in the central nervous system and have overlapping expression patterns with opioid receptors. In the striatum, stimulation of DORs or mu opioid receptors (MORs) which are found to be co-expressed with AC5 (177–179), inhibits cAMP production and causes locomotor activation (176, 180). Those responses were attenuated in mice lacking AC5 (AC5−/−), making this enzyme an important component of the signal transduction mechanisms induced by opioid receptors.
cAMP signaling mediated by DORs may also contribute to the analgesic effects of opioids. In AC5−/− mice, the analgesic effect of acute opioid stimulation was reduced compared to wild-type mice (180). Moreover, those results were specific for the AC5 enzymes since acute pain responses did not differ in mice lacking either AC1, AC8 or both isoforms compared to wild-type animals (181). However, like DOR and AC5, the vitamin D receptor is also expressed in the striatum and the prefrontal cortex (72). In addition, cholecalciferol supplementation was shown to induce down-regulation of AC5 gene in the cerebrum (64) and might than define a potential effect of vitamin D on the analgesic effects of opioids via the cAMP pathway.
Low serum vitamin D levels have been linked to the development of opioid side effects and dependency. Among opioid-dependent patients recruited from the methadone maintenance treatment (MMT), more than half recorded a low vitamin D status (182). This common association deserves attention since high prevalence of chronic pain has also been observed in drug-dependent populations (183, 184). Chronic muscle and joint pain are common features of opioid withdrawal and are usually attributed to the effect of methadone (185). But, whether these conditions or other forms of chronic pain are affected by altered nutritional or metabolic factors related to vitamin D deficiency in drug-dependent populations is still unknown. Also, it should be noted that long-term therapy with some medications commonly used to treat chronic pain, such as steroids, antiepileptic drugs, and anticonvulsants can decrease levels of vitamin D (186–188). Therefore, it could be appropriate to recommend vitamin D supplements when levels are either deficient or insufficient with regards to the patient's medical conditions, current medications and skin exposure to sunlight. Nonetheless, vitamin D supplementation was recently shown to improve cognitive function and mental health in patients undergoing the MMT program (189).
Moreover, it has been suggested, using animal models, that ultraviolet B (UVB) light exposure induces strong analgesic effects due to elevation of endogenous opioids in the skin (190). This elevation in the POMC-derived peptide, β-endorphin, generated in response to UV radiation (191–193) was suggested to produce an opioid receptor-mediated addiction that leads to an increase in the nociceptive thresholds with pain relieving actions. However, since UVB light is responsible for the formation of vitamin D (194), it would be of interest to know if vitamin D plays a role in releasing β-endorphins to develop such addiction and if signaling pathways involved in this addiction are similar to those involved in exogenous opioid addiction.
It is known that sustained activation of opioid receptors results in compensatory mechanisms that increase cAMP production (195), referred to as AC superactivation- one of the key mechanisms leading to the development of physical dependence (196–198) and tolerance to opioids (172, 199–202). However, since cholecalciferol supplementation was shown to dysregulate AC5 gene expression (64), it might also be believed to play a critical role in modulating the development of opioid dependence and tolerance by affecting the AC superactivation mechanism. This correlation was partially proved by demonstrating the involvement of vitamin D deficiency in quickening the development of tolerance to opioid analgesics (153). In this study, rats raised for 3–4 weeks on a vitamin D-deficient rachitogenic diet developed tolerance to morphine faster than control rats, an effect that was prevented by cholecalciferol treatment.
Association between vitamin D levels and opioid use has also been investigated in human subjects. In an observational study, palliative cancer patients with 25-OHD levels <50 nmol/L required higher opioid (fentanyl) dose for pain relief compared to those with 25-OHD levels above 50 nmol/L (203). In a follow-up study, palliative cancer patients with 25-OHD levels <75 nmol/L were given vitamin D with a dose of 4,000 IU daily. After 1 month of supplementation, a decrease in fentanyl doses was observed compared to untreated controls (11). Together, these observations describe a significant correlation between low vitamin D levels and increased opioid consumption.
Both hypovitaminosis D (low levels of vitamin D) and hypervitaminosis D (high Levels; also known as vitamin D toxicity) are linked with unwanted effects and symptoms. Low levels of serum 25(OH)D3 concentration typically below 12 ng ml−1 (30 nmol l−1) are associated with an increased risk of rickets/osteomalacia (204), however the precise serum levels of 25(OH)D3 that cause toxicity are not known. Those patients with serum levels >100 ng/ml are defined as being “at risk of toxicity” whereas serum values >150 ng/ml are considered “toxic” (205–207). However, another report suggests that serum levels above 50 ng/ml may be “at risk of toxicity” (208).
Hypervitaminosis D is usually diagnosed from presenting symptoms (see below), high serum 25(OH)D3 levels as well as elevated serum and urine level of calcium and reduced serum level of intact parathyroid hormone. Vitamin D toxicity is more likely in the modern age where excessive screening and media attention have led to widespread (mis)use of vitamin D supplements including as a result of over-prescribing by physicians (209, 210). It is thought to arise from chronic use of high dose vitamin D supplements and not from abnormally high exposure of skin to the sun or from eating a regular diet (211, 212). Data from National Health and Nutrition Examination Survey (NHANES) of US residents gathered between 1999 and 2014 found a 3% increase in the number of subjects taking potentially unsafe amounts of vitamin D—more than 4,000 international units (IU) per day- and a nearly 18% increase in the number of people taking more than 1,000 IU of vitamin D daily, well exceeding the Institute of Medicine recommended dose of 600 to 800 IU (213). These findings are consistent with another report showing concentrations of 25(OH)D3 have also modestly increased over this time frame from a similar population data-set (214). As a result of increasing use of vitamin D usage, a substantial increase in the number of reports of vitamin D intoxication, with the majority (75%) of reports published since 2010 (207). Hypervitaminosis D may also result from medication/prescription errors as well as formulation errors such as under reporting (wrong strength) on labels by unlicensed manufacturers of supplements and fortified foods (207, 215, 216).
Hypervitaminosis D, by definition, arises from elevated plasma concentrations of 25(OH)D3 and its metabolites such as 24,25(OH), 25,26(OH)2D3, and 25(OH)D3-26,23-lactone (217). Although the exact role of the individual metabolites is not well-understood, vitamin D toxicity is thought to arise from a saturation of its catabolizing enzymes such as CYP24A1 and hyperactivation of the vitamin D/VDR signaling pathways especially those regulating calcium levels (210, 218, 219).
It appears that short excursions into very high vitamin D serum levels such as after a single megadose of 300,000 IU might be safe (220) whereas chronic high doses of vitamin D in human subjects results in the following clinical manifestations: GI abnormalities >90%, Azotaemia ~80%, Polydipsia/Polyurea ~50% Encephalopathy 40%, Nephrocalcinosis 40%, Pancreatitis ~10% (207, 221, 222). Persistent vomiting as a result of vitamin D toxicity has also been recently reported (223). These clinical effects are largely thought to result from the vitamin-D-induced hypercalcemia that also leads to fatigue, generalized weakness and anorexia. The main mechanism of increased calcium in serum is due to vitamin D-induced bone resorption that can be blocked by bisphosphonates- known specific inhibitors of bone resorption- that are used clinically to treat hypercalcemia (224). In addition, hypervitaminosis D can also lead to increased absorption of dietary calcium from the GI tract, though this likely represents a minor contribution to the raised calcium levels in serum (224).
Hypervitaminosis D has been associated with increased risk of falls and bone fractures in post-menopausal women receiving high dose supplements (209). In extreme cases, vitamin D toxicity results in acute kidney injury (AKI). Chronic administration of mega doses (600,000 IU) has been shown to cause AKI and symptoms associated with hypercalcemia (210, 221, 225–227). In one study, 51 out of 62 patients receiving unusually high doses of vitamin D supplements developed AKI (226). The exact mechanisms for vitamin D associated renal complications are poorly understood but one rational explanation for the renal injury is hypercalcemia-induced severe dehydration. Renal toxicity may also be precipitated by and/or exacerbated by nephrocalcinosis (kidney stones) due to increased calcium deposition in renal tubules (228).
Vitamin D toxicity may also result from increased vitamin D/VDR signaling and/or decreased levels of catabolizing enzymes that could conceivably lead to hypervitaminosis D and subsequent toxic pharmacological effects (211, 212). However, as to what role is played by the “local vitamin D system” in cell/tissue specific toxicity remains poorly understood. For example, could overactivity of the “local vitamin D system” lead to a state of “local hypervitaminosis D” that then results in local cell/tissue toxicity? This would be important not only for its effects on the kidney but in other tissues such as in neurons involved in pain sensing and processing. Although, the contribution of the local vitamin D enzyme system to specific local tissue injury or damage is not well-understood, recent data suggests that the kidneys may be particularly prone to vitamin D toxicity. In addition to the normal uptake of vitamin D, the kidney proximal tubules also have the capability for re-uptake of DPB bound-vitamin D (DBP-D) through a Megalin and cubilin complex (229, 230). Thus, renal uptake of vitamin D by these multiple mechanisms could potentially result in higher accumulation and toxicity of vitamin D in the kidneys during hypervitaminosis D.
It should be noted, however, that vitamin D toxicity is rare and not all individuals with high serum 25(OH)D3 levels (even those with >150 ng/mL) manifest with clinical symptoms of vitamin D toxicity (24, 217, 218, 231, 232). Indeed, a recent study suggests that despite 167 patients having serum 25(OH)D3 levels >150 ng/mL, only 19 patients went on to develop vitamin D toxicity with AKI (210).
Vitamin D toxicity is more likely in extremes of age (children and elderly), in patients with impaired renal function, and those on certain prescription drugs such as thiazide diuretics (that decrease urinary calcium excretion) as their co-administration with vitamin D (that enhances intestinal calcium absorption) may theoretically result in hypercalcemia (233, 234).
Hypervitaminosis D and associated toxicity is largely preventable as it arises mainly from over-administration of high doses of vitamin D either by the patient, given by the physician/health care professional or because of manufacturer/formulation errors (207). Nonetheless, it is clear that further studies are needed to achieve a better understanding and definition of the precise cut-off levels for vitamin D toxicity in serum as well as in understanding the role of the local vitamin D system in mediating cell-specific toxicities (“local hypervitaminosis D”) that might arise even when circulating serum 25(OH)D3 levels are not abnormally high.
One in 5 of the adult population in the US is reported to suffer from chronic pain and 20% of these patients do not gain significant pain relief from currently available analgesic therapies. Vitamin D is speculated to provide clinical benefit in patients with chronic pain and several observational studies exist that have shown pain relief with vitamin D supplements (see Table 1). However, systematic reviews do not conclusively show patients benefit from vitamin D use in chronic pain (3, 235). There are multiple reasons for this discrepancy between the different clinical trials on vitamin D in pain which are discussed in more detail elsewhere [e.g., (3, 4)].
One of the concerns is the precise definition for serum 25(OH)D3 levels for its deficiency, for normal range and cut-off for toxicity. The difficulty in establishing pathophysiologically deficient 25(OH)D3 levels has been attributed to variations in methodological (statistical tools), the difference in experimental assays used (technical), and in the geographical latitude or other variations in the individuals being studied (236). Thus, it is argued that the so called “normal” range for serum 25(OH)D3 levels should be defined on an individual basis and in the clinical context (236). Serum variations can also arise from genetic polymorphisms in vitamin D processing enzymes and changes in vitamin D pharmacokinetics and pharmacodynamics. Another layer of complexity can arise from the specific variations in the individuals' disease state and this is particularly important in chronic pain that exhibits extreme heterogeneity amongst individuals and its perception can be very personalized. This leads to great challenges in the accurate assessment of pain especially when relying on self-reporting of pain by sufferers. Thus, there is an urgent need for the development of reliable biological markers of pain that can be accurately used in assessing pain in clinical trials. There is therefore a need for well-controlled large randomized clinical trials that account for the many variations, including having reliable biomarkers of pain, to conclusively determine the analgesic benefit of vitamin D in chronic pain.
The underlying molecular mechanism by which vitamin D/VDR modulates pain are not fully understood. In this review we have highlighted the potential interaction of pain signaling genes/pathways that are regulated by vitamin D/VDR. The reviewed literature suggests that vitamin D and/or its receptor can play a key role in modulating pain. Known pain genes such as NGF, GDNF, and EGFR, and opioid signaling are important components in pain signaling. We present here literature evidence for how vitamin D/VDR signaling might cross-talk with and regulate these genes that are critical in pain signaling. Further experimental studies in vitro and vivo are necessary to study these potential interactions specifically in pain models. Such studies could highlight the potential usefulness of vitamin D either alone or in combination with existing analgesics to better treat chronic pain.
AH and SA conceived the idea for the review. AH, SA, KN, NT, and VS wrote and edited the final draft of the manuscript.
Research in the laboratories of SA (QUCG-CMED-19/20-3) and AMH (QUCG-CMED-19/20-4) are funded by grants from Qatar University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ^www.iasp.org (accessed March 20, 2020).
1. Dahlhamer J, Lucas J, Zelaya C, Nahin R, Mackey S, DeBar L, et al. Prevalence of chronic pain and high-impact chronic pain among adults - United States, 2016. MMWR Morb Mortal Wkly Rep. (2018) 67:1001–6. doi: 10.15585/mmwr.mm6736a2
2. Vos T, Allen C, Arora M. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. (2017) 390:1211–59. doi: 10.1016/S0140-6736(17)32154-2
3. Straube S, Derry S, Straube C, Moore RA. Vitamin D for the treatment of chronic painful conditions in adults. Cochrane Database Syst Rev. (2015) 2015:CD007771. doi: 10.1002/14651858.CD007771.pub3
4. Wu Z, Malihi Z, Stewart AW, Lawes CM, Scragg R. Effect of vitamin D supplementation on pain: a systematic review and meta-analysis. Pain Phys. (2016) 19:415–27.
5. Shillo P, Selvarajah D, Greig M, Gandhi R, Rao G, Wilkinson ID, et al. Reduced vitamin D levels in painful diabetic peripheral neuropathy. Diabet Med. (2019) 36:44–51. doi: 10.1111/dme.13798
7. Brady SRE, Naderpoor N, de Courten MPJ, Scragg R, Cicuttini F, Mousa A, et al. Vitamin D supplementation may improve back pain disability in vitamin D deficient and overweight or obese adults. Steroid Biochem Mol Biol. (2019) 185:212–7. doi: 10.1016/j.jsbmb.2018.09.005
8. Sari A, Akdogan Altun Z, Arifoglu Karaman C, Bilir Kaya B, Durmus B. Does vitamin D affect diabetic neuropathic pain and balance? J Pain Res. (2020) 13:171–9. doi: 10.2147/JPR.S203176
9. Kucukceran H, Ozdemir O, Kiral S, Berker DS, Kahveci R, Ozkara A, et al. The impact of circulating 25-hydroxyvitamin and oral cholecalciferol treatment on menstrual pain in dysmenorrheic patients. Gynecol Endocrinol. (2019) 35:53–7. doi: 10.1080/09513590.2018.1490407
10. Ghai B, Bansal D, Kanukula R, Gudala K, Sachdeva N, Dhatt SS, et al. Vitamin D Supplementation in patients with chronic low back pain: an open label, single arm clinical trial. Pain Phys. (2017) 20:E99–105. doi: 10.36076/ppj.2017.1.E99
11. Helde-Frankling M, Höijer J, Bergqvist J, Björkhem-Bergman L. Vitamin D supplementation to palliative cancer patients shows positive effects on pain and infections-results from a matched case-control study. PLoS ONE. (2017) 12:e0184208. doi: 10.1371/journal.pone.0184208
12. Manoy P, Yuktanandana P, Tanavalee A, Anomasiri W, Ngarmukos S, Tanpowpong T, et al. Vitamin D supplementation improves quality of life and physical performance in osteoarthritis patients. Nutrients. (2017) 9:E799. doi: 10.3390/nu9080799
13. Morandi G, Maines E, Piona C, Monti E, Sandri M, Gaudino R, et al. Significant association among growing pains, vitamin D supplementation, and bone mineral status: results from a pilot cohort study. J Bone Miner Metab. (2015) 33:201–6. doi: 10.1007/s00774-014-0579-5
14. Costan AR, Vulpoi C, Mocanu V. Vitamin D fortified bread improves pain and physical function domains of quality of life in nursing home residents. J Med Food. (2014) 17:625–31. doi: 10.1089/jmf.2012.0210
15. Osunkwo I, Ziegler TR, Alvarez J, McCracken C, Cherry K, Osunkwo CE, et al. High dose vitamin D therapy for chronic pain in children and adolescents with sickle cell disease: results of a randomized double blind pilot study. Br J Haematol. (2012) 159:211–5. doi: 10.1111/bjh.12019
16. Huang W, Shah S, Long Q, Crankshaw AK, Tangpricha V. Improvement of pain, sleep, and quality of life in chronic pain patients with vitamin D supplementation. Clin J Pain. (2013) 29:341–7. doi: 10.1097/AJP.0b013e318255655d
17. Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: Metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. (2016) 96:365–408. doi: 10.1152/physrev.00014.2015
18. Heaney RP, Horst RL, Cullen DM, Armas LA. Vitamin D3 distribution and status in the body. J Am Coll Nutr. (2009) 28:252–6. doi: 10.1080/07315724.2009.10719779
19. Oliai Araghi S, van Dijk SC, Ham AC, Brouwer-Brolsma EM, Enneman AW, Sohl E, et al. BMI and body fat mass is inversely associated with vitamin D levels in older individuals. J Nutr Health Aging. (2015) 19:980–5. doi: 10.1007/s12603-015-0657-y
20. Arunabh S, Pollack S, Yeh J, Aloia JF. Body fat content and 25-hydroxyvitamin D levels in healthy women. J Clin Endocrinol Metab. (2003) 88:157–61. doi: 10.1210/jc.2002-020978
21. Cheng JB, Motola DL, Mangelsdorf DJ, Russell DW. De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxilase. J Biol Chem. (2003) 278:38084–93. doi: 10.1074/jbc.M307028200
22. Chun RF, Peercy BE, Orwoll ES, Nielson CM, Adams JS, Hewison M. Vitamin D and DBP: the free hormone hypothesis revisited. J Steroid Biochem Mol Biol. (2014) 144:132–7. doi: 10.1016/j.jsbmb.2013.09.012
23. Nykjaer A, Fyfe JC, Kozyraki R, Leheste JR, Jacobsen C, Nielsen MS, et al. Cubilin dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D3. Proc Natl Acad Sci USA. (2001) 98:13895–900. doi: 10.1073/pnas.241516998
24. Jones G, Prosser DE, Kaufmann M. Cytochrome P450-mediated metabolism of vitamin. D J Lipid Res. (2014) 55:13–31. doi: 10.1194/jlr.R031534
25. Jones G, Prosser DG. The activating enzymes of vitamin D. Vitamin D. (2011) 45:23–42. doi: 10.1016/B978-0-12-381978-9.10003-4
26. Shinki T, Jin CH, Nishimura A, Nagai Y, Ohyama Y, Noshiro M, et al. Parathyroid hormone inhibits 25-hydroxyvitamin D3-24-hydroxylase mRNA expression stimulated by 1 alpha,25-dihydroxyvitamin D3 in rat kidney but not in intestine. J Biol Chem. (1992) 267:13757–62.
27. Jones G, Prosser DE, Kaufmann M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): its important role in the degradation of vitamin D. Arch Biochem Biophys. (2012) 523:9–18. doi: 10.1016/j.abb.2011.11.003
28. Pike JW, Meyer MB. The vitamin D receptor: new paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D(3). Endocrinol Metab Clin North Am. (2010) 39:255–69. doi: 10.1016/j.ecl.2010.02.007
29. Habib AM, Wood JN, Cox JJ. Sodium channels and pain. Handb Exp Pharmacol. (2015) 227:39–56. doi: 10.1007/978-3-662-46450-2_3
30. Habib AM, Okorokov AL, Hill MN, Bras JT, Lee MC, Li S, et al. Microdeletion in a FAAH pseudogene identified in a patient with high anandamide concentrations and pain insensitivity. Br J Anaesth. (2019) 123:e249–53. doi: 10.1016/j.bja.2019.02.019
31. Habib AM, Matsuyama A, Okorokov AL, Santana-Varela S, Bras JT, Aloisi AM, et al. A novel human pain insensitivity disorder caused by a point mutation in ZFHX2. Brain. (2018) 141:365–76. doi: 10.1093/brain/awx326
32. Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, et al. A classification of chronic pain for ICD-11. Pain. (2015) 156:1003–7. doi: 10.1097/j.pain.0000000000000160
33. Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. (2012) 61:364–71. doi: 10.2337/db11-1019
34. Anand U, Yiangou Y, Akbar A, Quick T, MacQuillan A, Fox M, et al. Glucagon-like peptide 1 receptor (GLP-1R) expression by nerve fibres in inflammatory bowel disease and functional effects in cultured neurons. PLoS ONE. (2018) 13:e0198024. doi: 10.1371/journal.pone.0198024
35. Que Q, Guo X, Zhan L, Chen S, Zhang Z, Ni X, et al. The GLP-1 agonist, liraglutide, ameliorates inflammation through the activation of the PKA/CREB pathway in a rat model of knee osteoarthritis. J Inflamm. (2019) 16:13. doi: 10.1186/s12950-019-0218-y
36. Lee YS, Jun HS. Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediat Inflamm. (2016) 2016:3094642. doi: 10.1155/2016/3094642
37. Loyd DR, Weiss G, Henry MA, Hargreaves KM. Serotonin increases the functional activity of capsaicin-sensitive rat trigeminal nociceptors via peripheral serotonin receptors. Pain. (2011) 152:2267–76. doi: 10.1016/j.pain.2011.06.002
38. Bertin S, Aoki-Nonaka Y, de Jong PR, Nohara LL, Xu H, Stanwood SR, et al. (2014). The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4+ T cells. Nat Immunol. (2014) 15:1055–63. doi: 10.1038/ni.3009.
39. Yano JM, Yu K, Donaldson GP, Shastri GG, Ann P, Ma L, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. (2015) 161:264–76. doi: 10.1016/j.cell.2015.02.047
40. Obata Y, Castaño Á, Boeing S, Bon-Frauches AC, Fung C, Fallesen T, et al. Neuronal programming by microbiota regulates intestinal physiology. Nature. (2020) 578:284–9. doi: 10.1038/s41586-020-1975-8
41. Torres-Fuentes C, Golubeva AV, Zhdanov AV, Wallace S, Arboleya S, Papkovsky DB, et al. Short-chain fatty acids and microbiota metabolites attenuate ghrelin receptor signaling. FASEB J. (2019) 33:13546–59. doi: 10.1096/fj.201901433R
42. Suez J, Elinav E. The path towards microbiome-based metabolite treatment. Nat Microbiol. (2017) 2:17075. doi: 10.1038/nmicrobiol.2017.75
43. O'Mahony SM, Dinan TG, Cryan JF. The gut microbiota as a key regulator of visceral pain. Pain. (2017) 158(Suppl 1):S19–S28. doi: 10.1097/j.pain.0000000000000779
44. Shen S, Lim G, You Z, Ding W, Huang P, Ran C, et al. Gut microbiota is critical for the induction of chemotherapy-induced pain. Nat Neurosci. (2017) 20:1213–6. doi: 10.1038/nn.4606
45. Yang WH, Heithoff DM, Aziz PV, Sperandio M, Nizet V, Mahan MJ, et al. Recurrent infection progressively disables host protection against intestinal inflammation. Science. (2017) 358:eaao5610. doi: 10.1126/science.aao5610
46. Waterhouse M, Hope B, Krause L, Morrison M, Protani MM, Zakrzewski M, et al. Vitamin D and the gut microbiome: a systematic review of in vivo studies. Eur J Nutr. (2019) 58:2895–910. doi: 10.1007/s00394-018-1842-7
47. Zhu W, Yan J, Zhi C, Zhou Q, Yuan X. 1,25(OH)2D3 deficiency-induced gut microbial dysbiosis degrades the colonic mucus barrier in Cyp27b1 knockout mouse model. Gut Pathog. (2019). 11:8. doi: 10.1186/s13099-019-0291-z
48. Lacagnina MJ, Watkins LR, Grace PM. Toll-like receptors and their role in persistent pain. Pharmacol Ther. (2018) 184:145–58. doi: 10.1016/j.pharmthera.2017.10.006
49. Bruno K, Woller SA, Miller YI, Yaksh TL, Wallace M, Beaton G, et al. Targeting Toll-Like receptor-4 (TLR4)-an emerging therapeutic target for persistent pain states. Pain. (2018) 159:1908–15. doi: 10.1097/j.pain.0000000000001306
50. Kim D, Kim MA, Cho IH, Kim MS, Lee S, Jo EK, et al. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem. (2007) 282:14975–83. doi: 10.1074/jbc.M607277200
51. Shi XQ, Zekki H, Zhang J. The role of TLR2 in nerve injury-induced neuropathic pain is essentially mediated through macrophages in peripheral inflammatory response. Glia. (2011) 59:231–41. doi: 10.1002/glia.21093
52. Stokes JA, Cheung J, Eddinger K, Corr M, Yaksh TL. Toll-like receptor signaling adapter proteins govern spread of neuropathic pain and recovery following nerve injury in male mice. J Neuroinflamm. (2013) 10:148. doi: 10.1186/1742-2094-10-148
53. Deluca HF, Cantorna MT. Vitamin D: its role and uses in immunology. FASEB J. (2001) 15:2579–85. Review. doi: 10.1096/fj.01-0433rev
54. Saul L, Mair I, Ivens A, Brown P, Samuel K, Campbell JDM, et al. 1,25-Dihydroxyvitamin D 3 restrains CD4 + T cell priming ability of CD11c + dendritic cells by upregulating expression of CD31. Front Immunol. (2019) 10:600. doi: 10.3389/fimmu.2019.00600
55. Ojaimi S, Skinner NA, Strauss BJ, Sundararajan V, Woolley I, Visvanathan K. Vitamin D deficiency impacts on expression of Toll-like receptor-2 and cytokine profile: a Pilot Study. J Transl Med. (2013) 11:176. doi: 10.1186/1479-5876-11-176
56. Do JE, Kwon SY, Park S, Lee E-S. Effects of vitamin D on expression of Toll-like receptors of monocytes from patients with Behcet's disease. Rheumatology. (2008) 47:840–8. doi: 10.1093/rheumatology/ken109
57. Foulkes T, Wood JN. Pain genes. PLoS Genet. (2008) 4:e1000086. doi: 10.1371/journal.pgen.1000086
58. Abdo H, Calvo-Enrique L, Lopez JM, Song J, Zhang M-D, Usoskin D, et al. Specialized cutaneous schwann cells initiate pain sensation. Science. (2019) 365:695–9. doi: 10.1126/science.aax6452
59. Al-Mheid I, Patel R, Murrow J, Morris A, Rahman A, Fike L, et al. Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J Am Coll Cardiol. (2011) 58:186–92. doi: 10.1016/j.jacc.2011.02.051
60. Chabas JF, Stephan D, Marqueste T, Garcia S, Lavaut M-N, Nguyen C, et al. Cholecalciferol (Vitamin D3) improves myelination and recovery after nerve injury. PLoS ONE. (2013) 8:e65034. doi: 10.1371/journal.pone.0065034
61. Hirani V. Vitamin D status and pain: analysis from the health survey for England among English adults aged 65 years and over. Br J Nutr. (2012) 107:1080–4. doi: 10.1017/S0007114511003965
62. Mauck MC, Hu J, Sefton C, Swor RA, Peak DA, Jones JS, et al. Obesity increases the risk of chronic pain development after motor vehicle collision. Pain. (2019) 160:670–5. doi: 10.1097/j.pain.0000000000001446
63. Dzik KP, Skrobot W, Kaczor KB, Flis DJ, Karnia MJ, Libionka W, et al. Vitamin D deficiency is associated with muscle atrophy and reduced mitochondrial function in patients with chronic low back pain. Oxid Med Cell Longev. (2019) 2019:6835341. doi: 10.1155/2019/6835341
64. Poisbeau P, Aouad M, Gazzo G, Lacaud A, Kemmel V, Landel V, et al. Cholecalciferol (Vitamin D3) reduces rat neuropathic pain by modulating opioid signaling. Mol Neurobiol. (2019) 56:7208–21. doi: 10.1007/s12035-019-1582-6
65. Banafshe HR, Khoshnoud MJ, Abed A, Saghazadeh M, Mesdaghinia A. Vitamin D supplementation attenuates the behavioral scores of neuropathic pain in rats. Nutr Neurosci. (2019) 22:700–5. doi: 10.1080/1028415X.2018.1435485
66. Tague SE, Clarke GL, Winter MK, McCarson KE, Wright DE, Smith PG. Vitamin D deficiency promotes skeletal muscle hypersensitivity and sensory hyperinnervation. J Neurosci. (2011) 31:13728–38. doi: 10.1523/JNEUROSCI.3637-11.2011
67. Zhang T, Yin Y, Liu H, Du W, Ren C, Wang L, et al. Generation of VDR knock-out mice via Zygote injection of CRISPR/Cas9 system. PLoS ONE. (2016) 11:e0163551. doi: 10.1371/journal.pone.0163551
68. Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. (1997) 16:391–6. doi: 10.1038/ng0897-391
69. Filipović N, Ferhatović L, Marelja I, Puljak L, Grković I. Increased vitamin D receptor expression in dorsal root Ganglia neurons of diabetic rats. Neurosci Lett. (2013) 549:140–5. doi: 10.1016/j.neulet.2013.05.023
70. Tague SE, Smith PG. Vitamin D receptor and enzyme expression in dorsal root ganglia of adult female rats: modulation by ovarian hormones. J Chem Neuroanat. (2011) 41:1–12. doi: 10.1016/j.jchemneu.2010.10.001
71. Ray P, Torck A, Quigley L, Wangzhou A, Neiman M, Rao C, et al. Comparative transcriptome profiling of the human and mouse dorsal root Ganglia: an RNA-seq-based resource for pain and sensory neuroscience research. Pain. (2018) 159:1325–45. doi: 10.1097/j.pain.0000000000001217
72. Prufer K, Veenstra TD, Jirikowski GF, Kumar R. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the rat brain and spinal cord. J Chem Neuroanat. (1999) 16:135–45. doi: 10.1016/S0891-0618(99)00002-2
73. Eyles DW, Smith S, Kinobe R, Hewison M, McGrath JJ. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. (2005) 29:21–30. doi: 10.1016/j.jchemneu.2004.08.006
74. Garcion E, Wion-Barbot N, Montero-Menei CN, Berger F, Wion D. New clues about vitamin D functions in the nervous system. Trends Endocrinol Metab. (2002) 13:100–5. doi: 10.1016/S1043-2760(01)00547-1
75. Eyles DW, Liu PY, Josh P, Cui X. Intracellular distribution of the vitamin D receptor in the brain: comparison with classic target tissues and redistribution with development. Neuroscience. (2014) 268:1–9. doi: 10.1016/j.neuroscience.2014.02.042
76. Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. (2006) 444:894–8. doi: 10.1038/nature05413
77. Emery EC, Habib AM, Cox JJ, Nicholas AK, Gribble FM, Woods CG, et al. Novel SCN9A mutations underlying extreme pain phenotypes: unexpected electrophysiological and clinical phenotype correlations. J Neurosci. (2015) 35:7674–81. doi: 10.1523/JNEUROSCI.3935-14.2015
78. McDermott LA, Weir GA, Themistocleous AC, Segerdahl AR, Blesneac I, Baskozos G. Defining the functional role of Na V 1.7 in human nociception. Neuron. (2019) 101:905–19 e8. doi: 10.1016/j.neuron.2019.01.047
79. Cohen JA, Edwards TN, Liu AW, Hirai T, Ritter Jones M, Wu J, et al. Cutaneous TRPV1 + neurons trigger protective innate type 17 anticipatory immunity. Cell. (2019) 178:919–32.e14. doi: 10.1016/j.cell.2019.06.022
80. Long W, Fatehi M, Soni S, Panigrahi R, Rees Kelly G, Philippaert K, et al. LIGHT. 258-LB: vitamin D directly regulates TRPV1 channel activity may play an important role in the autoimmune disease of type 1 diabetes. Diabetes. (2019) 68(Supplement 1). doi: 10.2337/db19-258-LB
81. Chabas JF, Alluin O, Rao G, Garcia S, Lavaut M-N, Jacques Risso J, et al. Vitamin D2 potentiates axon regeneration. J Neurotrauma. (2008) 25:1247–56. doi: 10.1089/neu.2008.0593
82. Harty BL, Monk KR. Unwrapping the unappreciated: recent progress in remak Schwann cell biology. Curr Opin Neurobiol. (2017) 47:131–7. doi: 10.1016/j.conb.2017.10.003
83. Basit A, Basit KA, Fawwad A, Shaheen F, Fatima N, Petropoulos IN, et al. Vitamin D for the treatment of painful diabetic neuropathy. BMJ Open Diabetes Res Care. (2016) 4:e000148. doi: 10.1136/bmjdrc-2015-000148
84. Riaz S, Malcangio M, Miller M, Tomlinson DR. A vitamin D(3) derivative (CB1093) induces nerve growth factor and prevents neurotrophic deficits in streptozotocin-diabetic rats. Diabetologia. (1999) 42:1308–13. doi: 10.1007/s001250051443
85. Gezen-Ak D, Dursun E, Yilmazer S. The effect of vitamin D treatment on nerve growth factor (NGF) release from hippocampal neurons. Noro Psikiyatr Ars. (2014) 51:157–62. doi: 10.4274/npa.y7076
86. Woolf CJ, Allchorne A, Safieh-Garabedian B, Poole S. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor alpha. Br J Pharmacol. (1997) 121:417–24. doi: 10.1038/sj.bjp.0701148
87. Mizumura K, Murase S. Role of nerve growth factor in pain. Handb Exp Pharmacol. (2015) 227:57–77. doi: 10.1007/978-3-662-46450-2_4
88. Einarsdottir E, Carlsson A, Minde J, Toolanen G, Svensson O, Solders G, et al. A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum Mol Genet. (2004) 13:799–805. doi: 10.1093/hmg/ddh096
89. Carvalho OP, Thornton GK, Hertecant J, Houlden H, Nicholas AK, Cox JJ, et al. A novel NGF mutation clarifies the molecular mechanism and extends the phenotypic spectrum of the HSAN5 neuropathy. J Med Genet. (2011) 48:131–5. doi: 10.1136/jmg.2010.081455
90. Lyengar S, Ossipov MH, Johnsona KW. The role of calcitonin gene–related peptide in peripheral and central pain mechanisms including migraine. Pain. (2017) 158:543–59. doi: 10.1097/j.pain.0000000000000831
91. Zhan X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. (2005) 24:4211–23. doi: 10.1038/sj.emboj.7600893
92. Gould HJ, Gould TN, England JD, Paul D, Liu ZP, Levinson SR. A possible role for nerve growth factor in the augmentation of sodium channels in models of chronic pain. Brain Res. (2000) 854:19–29. doi: 10.1016/S0006-8993(99)02216-7
93. Schaefer I, Prato V, Arcourt A, Taberner FJ, Lechner SG. Differential modulation of voltage-gated sodium channels by nerve growth factor in three major subsets of TrkA-expressing nociceptors. Mol Pain. (2018) 14:1744806918814640. doi: 10.1177/1744806918814640
94. Nocchi L, Portulano C, Franciosa F, Doleschall B, Panea M, Roy N, et al. Nerve growth factor-mediated photoablation of nociceptors reduces pain behavior in mice. Pain. (2019) 160:2305–15. doi: 10.1097/j.pain.0000000000001620
95. Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. (2002) 3:383–94. doi: 10.1038/nrn812
96. Brumovsky P, Villar MJ, Hökfelt T. Tyrosine hydroxylase is expressed in a subpopulation of small dorsal root Ganglion neurons in the adult mouse. Exp Neurol. (2006) 200:153–65. doi: 10.1016/j.expneurol.2006.01.023
97. Buj-Bello A, Buchman VL, Horton A, Rosenthal A, Davies AM. GDNF is an age-specific survival factor for sensory and autonomic Neurons Neuron. (1995) 15:821–8. doi: 10.1016/0896-6273(95)90173-6
98. Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB. Potent analgesic effects of GDNF in neuropathic pain states. Science. (2000) 290:124–7. doi: 10.1126/science.290.5489.124
99. Boucher TJ, McMahon SB. Neurotrophic factors and neuropathic pain. Curr Opin Pharmacol. (2001) 1:66–72. doi: 10.1016/S1471-4892(01)00010-8
100. Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH, et al. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci USA. (2004) 101:12706–11. doi: 10.1073/pnas.0404915101
101. Minett MS, Nassar MA, Clark AK, Passmore G, Dickenson AH, Wang F, et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun. (2012) 3:791. doi: 10.1038/ncomms1795
102. Pertile RAN, Cui X, Hammond L, Eyles DW. Vitamin D regulation of GDNF/Ret signaling in dopaminergic neurons. FASEB J. (2018) 32:819–28. doi: 10.1096/fj.201700713R
103. Martin LJ, Smith SB, Khoutorsky A, Magnussen CA, Samoshkin A, Sorge RE, et al. Epiregulin and EGFR interactions are involved in pain processing. J Clin Invest. (2017) 127:3353–66. doi: 10.1172/JCI87406
104. Shen Z, Zhang X, Tang J, Kasiappan R, Jinwal U, Li P, et al. The coupling of epidermal growth factor receptor down regulation by 1alpha,25-dihydroxyvitamin D3 to the hormone-induced cell cycle arrest at the G1-S checkpoint in ovarian cancer cells. Mol Cell Endocrinol. (2011) 338:58–67. doi: 10.1016/j.mce.2011.02.023
105. Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. (2014) 79:34–74. doi: 10.1016/j.phrs.2013.11.002
106. Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers. (2017) 9:52. doi: 10.3390/cancers9050052
107. Dougherty U, Mustafi R, Sadiq F, Almoghrabi A, Mustafi D, Kreisheh M, et al. The renin-angiotensin system mediates EGF receptor-vitamin d receptor cross-talk in colitis-associated colon cancer. Clin Cancer Res. (2014) 20:5848–59. doi: 10.1158/1078-0432.CCR-14-0209
108. Nackley-Neely AG, Tan KS, Karamarie F, Patrick F, Luda D, William M. Catechol-O-methyltransferase inhibition increases pain sensitivity through activation of Both β2 and β3 adrenergic receptors. Pain. (2007) 128:199–208. doi: 10.1016/j.pain.2006.09.022
109. Araldi D, Ferrari LF, Levine JD. Role of GPCR (mu-opioid)-receptor tyrosine kinase (epidermal growth factor) crosstalk in opioid-induced hyperalgesic priming (type II). Pain. (2018) 159:864–75. doi: 10.1097/j.pain.0000000000001155
110. Weingaertner IR, Koutnik S, Ammer H. Chronic morphine treatment attenuates cell growth of human BT474 breast cancer cells by rearrangement of the ErbB signalling network. PLoS ONE. (2013) 8:e53510. doi: 10.1371/journal.pone.0053510
111. Elbaz M, Ahirwar D, Ravi J, Nasser MW, Ganju RK. Novel role of cannabinoid receptor 2 in inhibiting EGF/EGFR and IGF-I/IGF-IR pathways in breast cancer. Oncotarget. (2017) 8:29668–78. doi: 10.18632/oncotarget.9408
112. Ravi J, Elbaz M, Wani NA, Nasser MW, Ganju RK. Cannabinoid receptor-2 agonist inhibits macrophage induced EMT in non-small cell lung cancer by downregulation of EGFR pathway. Mol Carcinog. (2016) 55:2063–76. doi: 10.1002/mc.22451
113. Akhtar S, Yousif MH, Dhaunsi GS, Chandrasekhar B, Al-Farsi O, Benter IF. Angiotensin-(1-7) inhibits epidermal growth factor receptor transactivation via a Mas receptor-dependent pathway. Br J Pharmacol. (2012) 165:1390–400. doi: 10.1111/j.1476-5381.2011.01613.x
114. Akhtar S, Chandrasekhar B, Attur S, Dhaunsi GS, Yousif MHM, Benter IF. Transactivation of ErbB family of receptor tyrosine kinases is inhibited by angiotensin-(1-7) via its Mas receptor. PLoS ONE. (2015) 10:e0141657. doi: 10.1371/journal.pone.0141657
115. Singh B, Carpenter G, Coffey RJ. EGF receptor ligands: recent advances version 1. F1000Res. (2016) 5:F1000 Faculty Rev-2270. doi: 10.12688/f1000research.9025.1
116. Popiolek-Barczyk K, Mika J. Targeting the microglial signaling pathways: new insights in the modulation of neuropathic pain. Curr Med Chem. (2016) 23:2908–28. doi: 10.2174/0929867323666160607120124
117. Cheng JK, Ru-Rong J. Intracellular signaling in primary sensory neurons and persistent pain. Neurochem Res. (2008) 33:1970–8. doi: 10.1007/s11064-008-9711-z
118. Bikle DD. Vitamin D and the skin: Physiology and pathophysiology. Rev Endocr Metab Disord. (2012) 13:3–19. doi: 10.1007/s11154-011-9194-0
119. Ahrens JM, Jones JD, Nieves NJ, Mitzey AM, DeLuca HF, Clagett-Dame M. Differential activity of 2-methylene-19-nor vitamin D analogs on growth factor gene expression in rhino mouse skin and comparison to all-trans retinoic acid. PLoS ONE. (2017) 12:e0188887. doi: 10.1371/journal.pone.0188887
120. Tesfaye S, Chaturvedi N, Eaton SE, Ward JD, Manes C, Ionescu-Tirgoviste C, et al. Vascular risk factors and diabetic neuropathy. N Engl J Med. (2005) 352:341–50. doi: 10.1056/NEJMoa032782
121. Malik RA, Tesfaye S, Thompson SD, Veves A, Sharma AK, Boulton AJ, et al. Endoneurial localisation of microvascular damage in human diabetic neuropathy. Diabetologia. (1993) 36:454–9. doi: 10.1007/BF00402283
122. Benter IF, Yousif MH, Griffiths SM, Benboubetra M, Akhtar S. Epidermal growth factor receptor tyrosine kinase-mediated signalling contributes to diabetes-induced vascular dysfunction in the mesenteric bed. Br J Pharmacol. (2005) 145:829–36. doi: 10.1038/sj.bjp.0706238
123. Benter IF, Yousif MH, Hollins AJ, Griffiths SM, Akhtar S. Diabetes-induced renal vascular dysfunction is normalized by inhibition of epidermal growth factor receptor tyrosine kinase. J Vasc Res. (2005) 42:284–91. doi: 10.1159/000085904
124. Yousif MH, Benter IF, Akhtar S. The role of tyrosine kinase-mediated pathways in diabetes-induced alterations in responsiveness of rat carotid artery. Autonomic Autacoid Pharmacol. (2005) 25:69–78. doi: 10.1111/j.1474-8673.2004.00333.x
125. Benter IF, Benboubetra M, Yousif M, Hollins AJ, Canatan H, Akhtar S. Early inhibition of EGFR signaling prevents diabetes-induced up-regulation of multiple gene pathways in the mesenteric vasculature. Vasc Pharmacol. (2009) 51:236–45. doi: 10.1016/j.vph.2009.06.008
126. Benter IF, Sarkhou F, Al-Khaldi AT, Chandrasekhar B, Attur S, Dhaunsi GS, et al. The dual targeting of EGFR and ErbB2 with the inhibitor Lapatinib corrects high glucose-induced apoptosis and vascular dysfunction by opposing multiple diabetes-induced signaling changes. J Drug Target. (2015) 23:506–18. doi: 10.3109/1061186X.2015.1057150
127. Akhtar S, Yousif MHM, Dhaunsi GS, Sarkhouh F, Chandrasekhar B, Attur S, et al. Activation of ErbB2 and downstream signalling via rho kinases and ERK1/2 contributes to diabetes-induced vascular dysfunction. PLoS ONE. (2013) 8:e67813. doi: 10.1371/journal.pone.0067813
128. Yousif MH, Benter IF, Hares N, Canatan H, Akhtar S. Phosphoinositide 3-kinase mediated signalling contributes to development of diabetes-induced abnormal vascular reactivity of rat carotid artery. Cell Biochem Funct. (2006) 24:13–22. doi: 10.1002/cbf.1278
129. Akhtar S, Benter IF. The role of epidermal growth factor receptor in diabetes-induced cardiac dysfunction. BioImpacts. (2013) 3:5–9. doi: 10.5681/bi.2013.008
130. Akhtar S, Chandrasekhar B, Yousif MHM, Renno W, Benter IF, El-Hashim AZ. Chronic administration of nano-sized PAMAM dendrimers in vivo inhibits EGFR-ERK1/2-ROCK signaling pathway and attenuate diabetes-induced vascular remodeling and dysfunction. Nanomedicine. (2019) 18:78–89. doi: 10.1016/j.nano.2019.02.012
131. Kersten C, Cameron MG, Laird B, Mjåland S. Epidermal growth factor receptor-inhibition (EGFR-I) in the treatment of neuropathic pain. Br J Anaesth. (2015) 115:761–7. doi: 10.1093/bja/aev326
132. Kersten C, Cameron MG, Bailey AG, Fallon MT, Laird BJ, Paterson V, et al. Relief of neuropathic pain through epidermal growth factor receptor inhibition: a randomized proof-of-concept trial. Pain Med. (2019) 20:2495–505. doi: 10.1093/pm/pnz101
133. Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. (2008) 14:331–6. doi: 10.1038/nm1723
134. Kongstorp M, Schjølberg T, Jacobsen DP, Fred H, Gjerstad J. Epiregulin is released from intervertebral disks and induces spontaneous activity in pain pathways. Pain Rep. (2019) 4:718. doi: 10.1097/PR9.0000000000000718
135. Kersten C, Cameron MG, Mjåland S. Epithelial growth factor receptor (EGFR)-inhibition for relief of neuropathic pain-a case series. Scand J Pain. (2013) 4:3–7. doi: 10.1016/j.sjpain.2012.11.011
136. Erschbamer M, Pernold K, Olson L. Inhibiting epidermal growth factor receptor improves structural, locomotor, sensory, and bladder recovery from experimental spinal cord injury. J Neurosci. (2007) 27:6428–35. doi: 10.1523/JNEUROSCI.1037-07.2007
137. Kersten C, Cameron MG. Cetuximab alleviates neuropathic pain despite tumour progression. BMJ Case Rep. (2012) 2012:bcr1220115374. doi: 10.1136/bcr.12.2011.5374
138. Klapper LN, Kirschbaum MH, Sela M, Yarden Y. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors. Adv Cancer Res. (2000) 77:25–79. doi: 10.1016/S0065-230X(08)60784-8
139. Tong WM, Kállay E, Hofer H, Hulla W, Manhardt T, Peterlik M, et al. Growth regulation of human colon cancer cells by epidermal growth factor and 1,25-dihydroxyvitamin D3 is mediated by mutual modulation of receptor expression. Eur J Cancer. (1998) 34:2119–25. doi: 10.1016/S0959-8049(98)00267-6
140. Tong WM, Hofer H, Ellinger A, Peterlik M, Cross HS. Mechanism of antimitogenic action of vitamin D in human colon carcinoma cells: relevance for suppression of epidermal growth factor-stimulated cell growth. Oncol Res. (1999) 11:77–84.
141. Arcidiacono MV, Yang J, Fernandez E, Dusso A. The induction of C/EBPβ contributes to vitamin D inhibition of ADAM17 expression and parathyroid hyperplasia in kidney disease. Nephrol Dial Transplant. (2015) 30:423–33. doi: 10.1093/ndt/gfu311
142. Pálmer HG, González-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. (2001) 154:369–87. doi: 10.1083/jcb.200102028
143. Andl CD, Rustgi AK. No one-way street: cross-talk between e-cadherin and receptor tyrosine kinase (RTK) signaling: a mechanism to regulate RTK activity. Cancer Biol Ther. (2005) 4:28–31. doi: 10.4161/cbt.4.1.1431
144. Cabrita MA, Christofori G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis. (2008) 11:53–62. doi: 10.1007/s10456-008-9089-1
145. Barbáchano A, Ordóñez-Morán P, García JM, Sánchez A, Pereira F, Larriba MJ, et al. SPROUTY-2 and E-cadherin regulate reciprocally and dictate colon cancer cell tumourigenicity. Oncogene. (2010) 29:4800–13. doi: 10.1038/onc.2010.225
146. Cohen G, Ettinger K, Lecht S, Lelkes PI, Lazarovici P. Transcriptional down-regulation of epidermal growth factor (EGF) receptors by nerve growth factor (NGF) in PC 12 cells. J Mol Neurosci. (2014) 54:574–85. doi: 10.1007/s12031-014-0388-2
147. Shin DY, Kim S, Park S, Koh JS, Kim CH, Baek H, et al. Serum 25-hydroxyvitamin D levels correlate with EGFR mutational status in pulmonary adenocarcinoma. Endocr Relat Cancer. (2014) 21:715–21. doi: 10.1530/ERC-14-0259
148. Desprez PY, Poujol D, Falette N, Lefebvre MF, Saez S. 1,25-Dihydroxyvitamin D3 increases epidermal growth factor receptor gene expression in BT-20 breast carcinoma cells. Biochem Biophys Res Commun. (1991) 176:1–6. doi: 10.1016/0006-291X(91)90880-G
149. Dang R, Guo Y, Zhu Y, Yang R, Cai H, Jiang P. Chronic administration of calcitriol enhanced neuregulin-1/ErbB signaling in rat myocardium. Pharmazie. (2016) 71:192–5.
150. Fricker FR, Lago N, Balarajah S, Tsantoulas C, Tanna S, Zhu N, et al. Axonally derived neuregulin-1 is required for remyelination and regeneration after nerve injury in adulthood. J Neurosci. (2011) 31:3225–33. doi: 10.1523/JNEUROSCI.2568-10.2011
151. Szigethy E, Knisely M, Drossman D. Opioid misuse in gastroenterology and non-opioid management of abdominal pain. Nat Rev Gastroenterol Hepatol. (2018) 15:168–80. doi: 10.1038/nrgastro.2017.141
152. Chen Q, Larochelle MR, Weaver DT, Lietz AP, Mueller PP, Mercaldo S, et al. Prevention of prescription opioid misuse and projected overdose deaths in the United States. JAMA Netw Open. (2019) 2:e187621. doi: 10.1001/jamanetworkopen.2018.7621
153. Bazzani C, Arletti R, Bertolini A. Pain threshold and morphine activity in vitamin D-deficient rats. Life Sci. (1984) 34:461–6. doi: 10.1016/0024-3205(84)90501-0
154. Rossier J. Opioid peptides have found their roots. Nature. (1982) 298:221–2. doi: 10.1038/298221a0
155. De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature. (1997) 385:446–50. doi: 10.1038/385446a0
156. Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. (1997) 385:442–6. doi: 10.1038/385442a0
157. Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. (1989) 340:153–6. doi: 10.1038/340153a0
158. Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. (1996) 380:258–62. doi: 10.1038/380258a0
159. Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature. (1996) 380:255–8. doi: 10.1038/380255a0
160. Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. (2004) 27:509–47. doi: 10.1146/annurev.neuro.26.041002.131412
161. Pettinger L, Gigout S, Linley JE, Gamper N. Bradykinin controls pool size of sensory neurons expressing functional delta-opioid receptors. J Neurosci. (2013) 33:10762–71. doi: 10.1523/JNEUROSCI.0123-13.2013
162. Bardoni R, Tawfik VL, Wang D, François A, Solorzano C, Shuster SA, et al. Delta opioid receptors presynaptically regulate cutaneous mechanosensory neuron input to the spinal cord dorsal horn. Neuron. (2014) 81:1443. doi: 10.1016/j.neuron.2014.03.006
163. Acosta CG, Lopez HS. Delta opioid receptor modulation of several voltage dependent Ca(2+) currents in rat sensory neurons. J Neurosci. (1999) 19:8337–48. doi: 10.1523/JNEUROSCI.19-19-08337.1999
164. Khasabova IA, Harding-Rose C, Simone DA, Seybold VS. Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. J Neurosci. (2004) 24:1744–53. doi: 10.1523/JNEUROSCI.4298-03.2004
165. Wu ZZ, Chen SR, Pan HL. Distinct inhibition of voltage-activated Ca2+ channels by delta-opioid agonists in dorsal root ganglion neurons devoid of functional T-type Ca2+ currents. Neuroscience. (2008) 153:1256–67. doi: 10.1016/j.neuroscience.2008.03.031
166. Simms BA, Zamponi GW. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron. (2014) 82:24–45. doi: 10.1016/j.neuron.2014.03.016
167. Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron. (2004) 41:127–38. doi: 10.1016/S0896-6273(03)00801-8
168. Murali SS, Napier IA, Mohammadi SA, Alewood PF, Lewis RJ, Christie MJ. High-voltage-activated calcium current subtypes in mouse DRG neurons adapt in a subpopulation-specific manner after nerve injury. J Neurophysiol. (2015) 113:1511–9. doi: 10.1152/jn.00608.2014
169. Yusaf SP, Goodman J, Pinnock RD, Dixon AK, Lee K. Expression of voltage-gated calcium channel subunits in rat dorsal root ganglion neurons. Neurosci Lett. (2001) 311:137–41. doi: 10.1016/S0304-3940(01)02038-9
170. Johnson JA, Grande JP, Windebank AJ, Kumar R. 1,25-Dihydroxyvitamin D(3) receptors in developing dorsal root ganglia of fetal rats. Brain Res Dev Brain Res. (1996) 92:120–4. doi: 10.1016/0165-3806(95)00204-9
171. Richard-Lalonde M, Nagi K, Audet N, Sleno R, Amraei M, Hogue M, et al. Conformational dynamics of Kir3.1/Kir3.2 channel activation via δ-opioid receptors. Mol Pharmacol. (2013) 83:416–28. doi: 10.1124/mol.112.081950
172. Nagi K, Pineyro G. Kir3 channel signaling complexes: focus on opioid receptor signaling. Front Cell Neurosci. (2014) 8:186. doi: 10.3389/fncel.2014.00186
173. Nagi K, Charfi I, Pineyro G. Kir3 channels undergo arrestin-dependant internalization following delta opioid receptor activation. Cellular and molecular life sciences: CMLS. (2015) 72:3543–57. doi: 10.1007/s00018-015-1899-x
174. Gendron L, Nagi K, Zeghal M, Giguère PM, Pineyro G. Molecular aspects of delta opioid receptors. Vitam Horm. (2019) 111:49–90. doi: 10.1016/bs.vh.2019.06.001
175. Charfi I, Nagi K, Mnie-Filali O, Thibault D, Balboni G, Schiller PW, et al. Ligand- and cell-dependent determinants of internalization and cAMP modulation by delta opioid receptor (DOR) agonists. Cell Mol Life Sci. (2014) 71:1529–46. doi: 10.1007/s00018-013-1461-7
176. Gendron L, Cahill CM, von Zastrow M, Schiller PW, Pineyro G. Molecular pharmacology of delta-opioid receptors. Pharmacol Rev. (2016) 68:631–700. doi: 10.1124/pr.114.008979
177. Mansour A, Fox CA, Burke S, Meng F, Thompson RC, Akil H, et al. Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J Compar Neurol. (1994) 350:412–38. doi: 10.1002/cne.903500307
178. Mons N, Harry A, Dubourg P, Premont RT, Iyengar R, Cooper DM. Immunohistochemical localization of adenylyl cyclase in rat brain indicates a highly selective concentration at synapses. Proc Natl Acad Sci USA. (1995) 92:8473–7. doi: 10.1073/pnas.92.18.8473
179. Scherrer G, Tryoen-Tóth P, Filliol D, Matifas A, Laustriat D, Cao YQ, et al. Knockin mice expressing fluorescent delta-opioid receptors uncover G protein-coupled receptor dynamics in vivo. Proc Natl Acad Sci USA. (2006) 103:9691–6. doi: 10.1073/pnas.0603359103
180. Kim KS, Lee KW, Lee KW, Im JY, Yoo JY, Kim SW, et al. Adenylyl cyclase type 5 (AC5) is an essential mediator of morphine action. Proc Natl Acad Sci USA. (2006) 103:3908–13. doi: 10.1073/pnas.0508812103
181. Li S, Lee ML, Bruchas MR, Chan GC, Storm DR, Chavkin C. Calmodulin-stimulated adenylyl cyclase gene deletion affects morphine responses. Mol Pharmacol. (2006) 70:1742–9. doi: 10.1124/mol.106.025783
182. Kim TW, Alford DP, Holick MF, Malabanan AO, Samet JH. Low vitamin d status of patients in methadone maintenance treatment. J Addict Med. (2009) 3:134–8. doi: 10.1097/ADM.0b013e31819b736d
183. Larson MJ, Paasche-Orlow M, Cheng DM, Lloyd-Travaglini C, Saitz R, Samet JH. Persistent pain is associated with substance use after detoxification: a prospective cohort analysis. Addiction. (2007) 102:752–60. doi: 10.1111/j.1360-0443.2007.01759.x
184. Rosenblum A, Joseph H, Fong C, Kipnis S, Cleland C, Portenoy RK. Prevalence and characteristics of chronic pain among chemically dependent patients in methadone maintenance and residential treatment facilities. JAMA. (2003) 289:2370–8. doi: 10.1001/jama.289.18.2370
185. Karasz A, Zallman L, Berg K, Gourevitch M, Selwyn P, Arnsten JH. The experience of chronic severe pain in patients undergoing methadone maintenance treatment. J Pain Symptom Manage. (2004) 28:517–25. doi: 10.1016/j.jpainsymman.2004.02.025
186. Misra A, Aggarwal A, Singh O, Sharma S. Effect of carbamazepine therapy on vitamin D and parathormone in epileptic children. Pediatric neurology. (2010) 43:320–4. doi: 10.1016/j.pediatrneurol.2010.05.013
187. Pascussi JM, Robert A, Nguyen M, Walrant-Debray O, Garabedian M, Martin P, et al. Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J Clin Invest. (2005) 115:177–86. doi: 10.1172/JCI21867
188. Zhou C, Assem M, Tay JC, Watkins PB, Blumberg B, Schuetz EG, et al. Steroid and xenobiotic receptor and vitamin D receptor crosstalk mediates CYP24 expression and drug-induced osteomalacia. J Clin Invest. (2006) 116:1703–12. doi: 10.1172/JCI27793
189. Ghaderi A, Rasouli-Azad M, Farhadi MH, Mirhosseini N, Motmaen M, Pishyareh E, et al. Exploring the effects of vitamin D supplementation on cognitive functions and mental health status in subjects under methadone maintenance treatment. J Addict Med. (2020) 14:18–25. doi: 10.1097/ADM.0000000000000550
190. Fell GL, Robinson KC, Mao J, Woolf CJ, Fisher DE. Skin beta-endorphin mediates addiction to UV light. Cell. (2014) 157:1527–34. doi: 10.1016/j.cell.2014.04.032
191. Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. (2007) 128:853–64. doi: 10.1016/j.cell.2006.12.045
192. Skobowiat C, Dowdy JC, Sayre RM, Tuckey RC, Slominski A. Cutaneous hypothalamic-pituitary-adrenal axis homolog: regulation by ultraviolet radiation. Am J Physiol Endocrinol Metab. (2011) 301:E484–93. doi: 10.1152/ajpendo.00217.2011
193. Slominski A, Wortsman J. Neuroendocrinology of the skin. Endocrine Rev. (2000) 21:457–87. doi: 10.1210/er.21.5.457
194. Wacker M, Holick MF. Sunlight and vitamin D: a global perspective for health. Dermato Endocrinol. (2013) 5:51–108. doi: 10.4161/derm.24494
195. Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol. (2008) 154:384–96. doi: 10.1038/bjp.2008.100
196. Cao JL, Vialou VF, Lobo MK, Robison AJ, Neve RL, Cooper DC, et al. Essential role of the cAMP-cAMP response-element binding protein pathway in opiate-induced homeostatic adaptations of locus coeruleus neurons. Proc Natl Acad Sci USA. (2010) 107:17011–6. doi: 10.1073/pnas.1010077107
197. Han MH, Bolaños CA, Green TA, Olson VG, Neve RL, Liu RJ, et al. Role of cAMP response element-binding protein in the rat locus ceruleus: regulation of neuronal activity and opiate withdrawal behaviors. J Neurosci. (2006) 26:4624–9. doi: 10.1523/JNEUROSCI.4701-05.2006
198. Yang HY, Wu ZY, Wood M, Whiteman M, Bian JS. Hydrogen sulfide attenuates opioid dependence by suppression of adenylate cyclase/cAMP pathway. Antioxidants Redox Signal. (2013) 20:31–41. doi: 10.1089/ars.2012.5119
199. He L, Whistler JL. The biochemical analysis of methadone modulation on morphine-induced tolerance and dependence in the rat brain. Pharmacology. (2007) 79:193–202. doi: 10.1159/000100893
200. Nagi K, Pineyro G. Regulation of opioid receptor signalling: implications for the development of analgesic tolerance. Mol Brain. (2011) 4:25. doi: 10.1186/1756-6606-4-25
201. Shy M, Chakrabarti S, Gintzler AR. Plasticity of adenylyl cyclase-related signaling sequelae after long-term morphine treatment. Mol Pharmacol. (2008) 73:868–79. doi: 10.1124/mol.107.042184
202. Wang L, Gintzler AR. Morphine tolerance and physical dependence: reversal of opioid inhibition to enhancement of cyclic AMP formation. J Neurochem. (1995) 64:1102–6. doi: 10.1046/j.1471-4159.1995.64031102.x
203. Bergman P, Sperneder S, Höijer J, Bergqvist J, Björkhem-Bergman L. Low vitamin D levels are associated with higher opioid dose in palliative cancer patients–results from an observational study in Sweden. PLoS ONE. (2015) 10:e0128223. doi: 10.1371/journal.pone.0128223
204. Sempos CT, Heijboer AC, Bikle DD, Bollerslev J, Bouillon R, Brannon PM, et al. Vitamin D assays and the definition of hypovitaminosis D: results from the First International Conference on Controversies in Vitamin D. Br J Clin Pharmacol Rev. (2018) 84:2194–207. doi: 10.1111/bcp.13652
205. Scientific Committee on Food. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Vitamin D. Brussels: European Commission (2002).
206. Vieth R. Why the optimal requirement for Vitamin D3 is probably much higher than what is officially recommended for adults. J Steroid Biochem Mol Biol. (2004) 89–90:575–9. doi: 10.1016/j.jsbmb.2004.03.038
207. Taylor PN, Davies JS. A review of the growing risk of vitamin D toxicity from inappropriate practice. Br J Clin Pharmacol. (2018) 84:1121–7. doi: 10.1111/bcp.13573
208. Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. (2011) 96:53–8. doi: 10.1016/j.jada.2011.01.004
209. Sanders KM, Stuart AL, Williamson EJ, Simpson JA, Kotowicz MA, Young D, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA. (2010) 303:1815–22. doi: 10.1001/jama.2010.594
210. Chowdry AM, Azad H, Najar MS, Mir I. Acute kidney injury due to overcorrection of hypovitaminosis D: a tertiary center experience in the Kashmir Valley of India. Saudi J Kidney Dis Transpl. (2017) 28:1321–9. doi: 10.4103/1319-2442.220873
211. Marcinowska-Suchowierska E, Kupisz-Urbanska M, Łukaszkiewicz J, Płudowski P, Jones G. Vitamin D toxicity-a clinical perspective. Front Endocrinol. (2018) 9:550. doi: 10.3389/fendo.2018.00550
212. Marcinowska-Suchowierska E, Płudowski P, Witamina Z, Tałałaj M. Vitamin D toxicity. Post N Med. (2016) 29:756–9.
213. Rooney MR, Harnack L, Michos ED, Ogilvie RP, Sempos CT, Lutsey PL. Trends in use of high-dose vitamin D supplements exceeding 1000 or 4000 international units daily, 1999-2014. JAMA. (2017) 317:2448–50. doi: 10.1001/jama.2017.4392
214. Schleicher RL, Sternberg MR, Lacher DA, Sempos CT, Looker AC, Durazo-Arvizu RA, et al. The vitamin D status of the US population from 1988 to 2010 using standardized serum concentrations of 25-hydroxyvitamin D shows recent modest increases. Am J Clin Nutr. (2016) 104:454–61. doi: 10.3945/ajcn.115.127985
215. Klontz KC, Acheson DW. Dietary supplement-induced vitamin D intoxication. N Engl J Med. (2007) 357:308–9. doi: 10.1056/NEJMc063341
216. Koutkia P, Chen TC, Holick MF. Vitamin D intoxication associated with an over-the-counter supplement. N Engl J Med. (2001) 345:66–7. doi: 10.1056/NEJM200107053450115
217. Shephard RM, Deluca HF. Plasma concentrations of vitamin D3 and its metabolites in the rat as influenced by vitamin D3 or 25-hydroxyvitamin D3 intakes. Arch Biochem Biophys. (1980) 202:43–53. doi: 10.1016/0003-9861(80)90404-X
218. Gupta AK, Jamwal V, Sakul, Malhotra P. Hypervitaminosis D and systemic manifestations: a comprehensive review. JIMSA. (2014) 27:236–7.
219. Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr. (2008) 88:582S–6S. doi: 10.1093/ajcn/88.2.582S
220. Apaydin M, Can AG, Kizilgul M, Beysel S, Kan S, Caliskan M, et al. The effects of single high-dose or daily low-dosage oral colecalciferol treatment on vitamin D levels and muscle strength in postmenopausal women. BMC Endocr Disord. (2018) 18:48. doi: 10.1186/s12902-018-0277-8
221. Misgar RA, Sahu D, Bhat MH, Wani AI, Bashir MI. Vitamin D toxicity: a prospective study from a tertiary care centre in Kashmir Valley. Indian J Endocrinol Metab. (2019) 23:363–6. doi: 10.4103/ijem.IJEM_116_19
222. Alshahrani F, Aljohani N. Vitamin D: deficiency, sufficiency and toxicity. Forum Nutr. (2013) 5:3605–16. doi: 10.3390/nu5093605
223. Ghauri MI, Bareeqa SB, Riaz A, Kumar A. Redundancy is of no good; iatrogenic hypervitaminosis d: a rare case of persistent vomiting due to hypercalcemia. Clin Med Insights Case Rep. (2019) 12:1179547619828688. doi: 10.1177/1179547619828688
224. Selby PL, Davies M, Marks JS, Mawer EB. Vitamin D intoxication causes hypercalcaemia by increased bone resorption which responds to pamidronate. Clin Endocrinol. (1995) 43:531–6. doi: 10.1111/j.1365-2265.1995.tb02916.x
225. De Francesco Daher E, Mesquita Martiniano LV, Lopes Lima LL, Viana Leite Filho NC, de Oliveira Souza LE, Duarte Fernandes PH, et al. Acute kidney injury due to excessive and prolonged intramuscular injection of veterinary supplements containing vitamins A, D and E: a series of 16 cases. Nefrologia. (2017) 37:61–7. doi: 10.1016/j.nefro.2016.05.017
226. Wani M, Wani I, Banday K, Ashraf M. The other side of vitamin D therapy: a case series of acute kidney injury due to malpractice-related vitamin D intoxication. Clin Nephrol. (2016) 86:236–41. doi: 10.5414/CN108904
227. Kaur P, Mishra SK, Mithal A. Vitamin D toxicity resulting from overzealous correction of vitamin D deficiency. Clin Endocrinol. (2015) 83:327–31. doi: 10.1111/cen.12836
228. Ammenti A, Pelizzoni A, Cecconi M, Molinari PP, Montini G. Nephrocalcinosis in children: a retrospective multi-centre study. Acta Paediatr. (2009) 98:1628–31. doi: 10.1111/j.1651-2227.2009.01401.x
229. Bikle DD, Schwartz J. Vitamin D binding protein, total and free vitamin D levels in different physiological and pathophysiological conditions. Front Endocrinol. (2019) 10:317. doi: 10.3389/fendo.2019.00317
230. Nielsen R, Christensen EI, Birn H. Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int. (2016) 89:58–67. doi: 10.1016/j.kint.2015.11.007
231. Vogiatzi MG, Jacobson-Dickman E, DeBoer MD. Drugs, and therapeutics committee of the pediatric endocrine society. Vitamin D supplementation and risk of toxicity in pediatrics: a review of current literature. J Clin Endocrinol Metab. (2014) 99:1132–41. doi: 10.1210/jc.2013-3655
232. Maalouf J, Nabulsi M, Vieth R, Kimball S, El-Rassi R, Mahfoud Z. Short- and long-term safety of weekly high-dose vitamin D3 supplementation in school children. J Clin Endocrinol Metab. (2008) 93:2693–701. doi: 10.1210/jc.2007-2530
233. Robien K, Oppeneer SJ, Kelly JA, Hamilton-Reeves JM. Drug-vitamin D interactions: a systematic review of the literature. Nutr Clin Pract. (2013) 28:194–208. doi: 10.1177/0884533612467824
234. Hathcock JN, Shao A, Vieth R, Heaney R. Risk assessment for vitamin D. Am J Clin Nutr. (2007) 85:6–18. doi: 10.1093/ajcn/85.1.6
235. Manson JE, Bassuk SS. Vitamin D research and clinical practice: at a crossroads. JAMA. (2015) 313:1311–2. doi: 10.1001/jama.2015.1353
Keywords: nociception, DRG, VDR, NGF, EGFR, GDNF, opioids, vitamin D toxicity
Citation: Habib AM, Nagi K, Thillaiappan NB, Sukumaran V and Akhtar S (2020) Vitamin D and Its Potential Interplay With Pain Signaling Pathways. Front. Immunol. 11:820. doi: 10.3389/fimmu.2020.00820
Received: 31 December 2019; Accepted: 09 April 2020;
Published: 28 May 2020.
Edited by:
Abdulbari Bener, Istanbul University Cerrahpasa Faculty of Medicine, TurkeyReviewed by:
Makoto Makishima, Nihon University School of Medicine, JapanCopyright © 2020 Habib, Nagi, Thillaiappan, Sukumaran and Akhtar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdella M. Habib, YWhhYmliQHF1LmVkdS5xYQ==; Saghir Akhtar, cy5ha2h0YXJAcXUuZWR1LnFh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.