 Kristy Fu1†
Kristy Fu1† Kylie March
Kylie March Aikaterini Alexaki
Aikaterini Alexaki Giulia Fabozzi
Giulia Fabozzi Constantinos Petrovas
Constantinos Petrovas- 1Tissue Analysis Core, Immunology Laboratory, Vaccine Research Center, NIAID, National Institutes of Health (NIH), Bethesda, MD, United States
- 2Center for Biologics Evaluation and Research, Food and Drug Administration, Silver Spring, MD, United States
The continuous development of molecular biology and protein engineering technologies enables the expansion of the breadth and complexity of protein therapeutics for in vivo administration. However, the immunogenicity and associated in vivo development of antibodies against therapeutics are a major restriction factor for their usage. The B cell follicular and particularly germinal center areas in secondary lymphoid organs are the anatomical sites where the development of antibody responses against pathogens and immunogens takes place. A growing body of data has revealed the importance of the orchestrated function of highly differentiated adaptive immunity cells, including follicular helper CD4 T cells and germinal center B cells, for the optimal generation of these antibody responses. Understanding the cellular and molecular mechanisms mediating the antibody responses against therapeutics could lead to novel strategies to reduce their immunogenicity and increase their efficacy.
Introduction
Protein therapeutics is a new class of drugs that, unlike small molecule drugs, are not chemically synthesized; instead, they are produced within living cells or organisms. Remarkable developments in molecular biology and protein engineering methodologies in the last few decades have enabled the generation of several new biotherapeutics for a wide range of diseases. Despite the potential of protein therapeutics, a drawback, often associated with them, is the generation of antidrug antibodies (ADAs), which diminishes the bioactivity and effectiveness of the therapeutic (1). The anatomical sites where the development of ADA occurs are the secondary lymphoid organs, including lymph nodes and spleen, which are central for humoral responses to immunogens and pathogens (2–4). The organogenesis (5, 6), architecture (7, 8), and cellular composition (9, 10) of lymph nodes as well as the immune reactions to pathogens mediating the development of humoral responses (11, 12) are well-studied and understood. Here, we review these principles with a focus on ADA development and potential strategies aiming to minimize the immunogenicity of biotherapeutics.
Historical Background
The history of protein therapeutics starts probably with diphtheria antitoxin derived from horse serum. The extraction of insulin from bovine pancreas, a few decades later, was a milestone in the treatment of diabetes. The development and approval of recombinant insulin, a human insulin expressed in Escherichia coli, in 1982 (13) resulted in the increased accessibility of insulin. Around the same time, murine monoclonal antibodies were being considered as therapeutic agents, with Orthoclone OKT3 (muromonab-CD3) being the first licensed monoclonal antibody, in 1986 (14). Many of the first-generation monoclonal antibodies were significantly immunogenic, because of their murine origin. Orthoclone OKT3 was eventually discontinued from the market in 2010 owing to the development of ADA (14–17). New generations of monoclonal antibodies are primarily being developed as humanized or human antibodies and are far less immunogenic. The use of a fully humanized IgG1 mAb against TNFα (adalimumab) triggered the development of ADA in patients with plaque psoriasis (18) and rheumatoid arthritis (19–22), indicating that ADA development to therapeutics can vary depending on factors like preexisting activation of the immune system and chronic inflammation.
Monoclonal antibodies constitute a large fraction of biotherapeutics and target a specific protein, usually to inhibit or modulate its function; in some cases, they may have a diagnostic role, or they may deliver a drug. The rest of protein therapeutics are primarily replacement therapies for proteins that are deficient owing to genetic or other reasons. Such protein therapeutics include coagulation factors, hormones, growth factors, and enzymes. Recently, fusion proteins are becoming an important class of biotherapeutics (Table 1). There are several examples of albumin fusion, Fc fusion, and antibody drug conjugates available in the market (33). The breadth and the complexity of protein therapeutics increase the potential for immune response generation. Protein therapeutic immunogenicity poses a great challenge in the field; and a better understanding of the risks, development, and mechanisms of ADA is needed to allow for strategies to reduce immunogenicity.
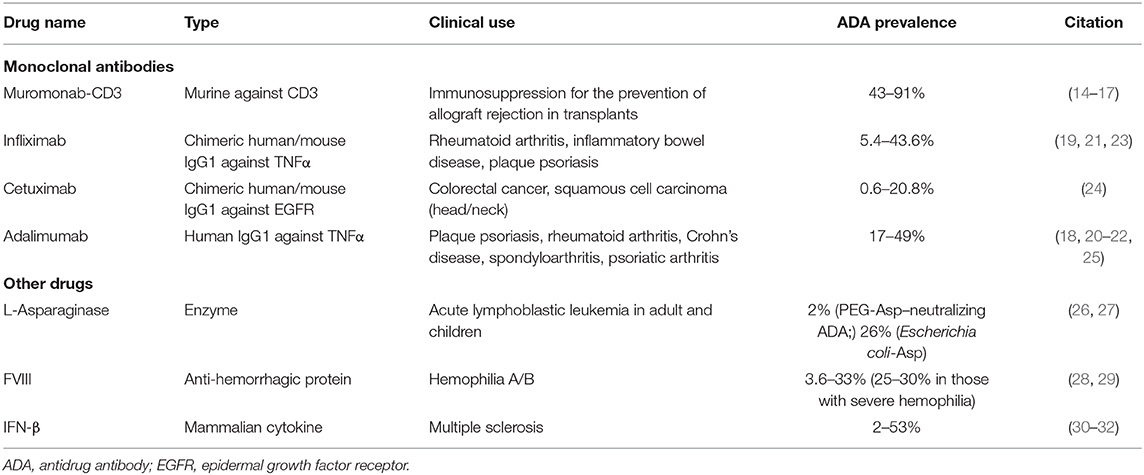
Table 1. Reported ADA development for licensed therapeutics.
Generation and Mechanisms of Action of Antidrug Antibodies
Several factors contributing to the immunogenicity of biotherapeutics have been identified and in many cases eliminated to generate better drugs. For example, animal proteins have been for the most part phased out of the market, as these were very often associated with strong immune responses. Initially, many recombinant proteins were generated through bacterial systems (especially E. coli), which are effective and simple to use but lack the higher mechanisms for glycosylation and therefore are often immunogenic in humans. Mammalian expression systems are now commonly used for biotherapeutics to allow for glycosylation. However, even within mammalian species, glycosylation may differ contributing to the development of ADA. For example, cetuximab, a monoclonal antibody against epidermal growth factor receptor (EGFR) inhibitor (24, 34, 35) generated in a mouse myeloma cell line SP2/0 was associated with development of ADA owing to its glycosylation profile (24, 34–36). Introduction of alternative mammalian cell lines [Chinese hamster ovary (CHO)] has greatly contributed to overcoming immunogenicity due to glycosylation patterns (34, 35). L-Asparaginase, a highly immunogenic enzyme, is effective in its own native, E. coli derived form (37, 38). However, allergic reactions due to multiple doses caused silent hypersensitivity that in turns generates ADA. Use of a pegylated form (26) or increasing the enzyme binding to erythrocytes (39) was able to reduce the development of ADA during multiple doses of E. coli asparaginase.
In patients receiving replacement therapy, a significant factor affecting their risk to ADA development is the levels of endogenous protein, with patients expressing no or very little protein being at a much higher risk, presumably owing to compromised central tolerance induction (40). Even a few amino acid sequence changes between the endogenous protein and the administered biotherapeutic may lead to an increased risk in immunogenicity. Substitution of just three amino acids in the recombinant activated factor VII (rFVIIa) (1, 41) was shown to significantly increase immunogenicity of the therapeutic protein. In addition, dosing (42), protein folding/aggregation, route of administration, storage conditions, and excipients may also affect the development of ADA (43, 44). It has been proposed that even codon usage of the recombinant protein may affect protein conformation and modulate immunogenicity (45). The inhibitory activity of ADA can be mediated by several mechanisms. Development of anti-idiotypic antibodies against the therapeutic could lead to in vivo formation of immune complexes (ICs), which can diminish therapeutic efficacy by reducing the half-life of the therapeutic or engaging the complement cascade (46, 47). Larger ICs are removed from circulation faster than smaller ICs owing to engagement of FcR on macrophages, reducing drug levels and requiring more frequent administration (47, 48). Complement cascade activation (as seen with administration of therapeutic IFN-β for multiple sclerosis) enhances inflammatory responses (46, 47). Alternatively, generation of neutralizing antibodies (i.e., adalimumab and infliximab, anti-TNFα, and monoclonal Abs) could directly block the action of the administered antibody or modulate its in vivo half-life (18, 25, 49, 50). In rare cases, ADA generation may lead to anaphylactic shock and death (51).
Lymph Nodes: Primary Sites for the Development of Immune Responses Against Pathogens
Structure
Lymph node positioning along lymphatic vessels enables the efficient draining and detection of pathogens and immunogens (Figure 1). The number of human LNs varies depending on age and disease status (52–56). The LN architecture is characterized by well-organized, distinct anatomical areas: cortex, paracortex, follicles, germinal centers (GCs), high endothelial venules (HEVs), medulla, and fibroblastic reticular cells (FRCs) (57, 58) (Figure 1). The formation of distinct LN areas contributes to the compartmentalization of cellular and molecular mechanisms involved in the generation of antigen-specific humoral responses. This compartmentalization further contributes to the control of relevant immune interactions and reduction of unwanted B cell responses. The cortex consists of many lymphocytes, mainly naive B cells (sIgD+IgM+) packed into primary follicles (absence of GC) or secondary follicles that are characterized by the formation of GC (58, 59). GCs are the areas where B cells proliferate in response to T cell-dependent antigen and create memory cells and plasma cells (57). Two major GC areas have been characterized, dark zone (DZ) and light zone (LZ), with different cellularities and roles for the development of B cell responses (60, 61). The deeper cortex, also known as the paracortex, contains HEVs, which are specialized blood vessels that allow circulating lymphocytes, such as T cells, and innate immunity cells to directly enter the LN (58). The local interaction between T and dendritic cell (DC) subsets initiates a cascade of immune reactions critical to the formation of mature GCs (57). The medulla, located on the efferent side where the lymph drains out of the LN, contains blood vessels and medullary cords enriched in B cells, macrophages, and plasma cells (Figure 1). Finally, the backbone of the LN architecture is the FRCs. The FRCs form a network that allow DCs and T cells to travel throughout the LN (62).
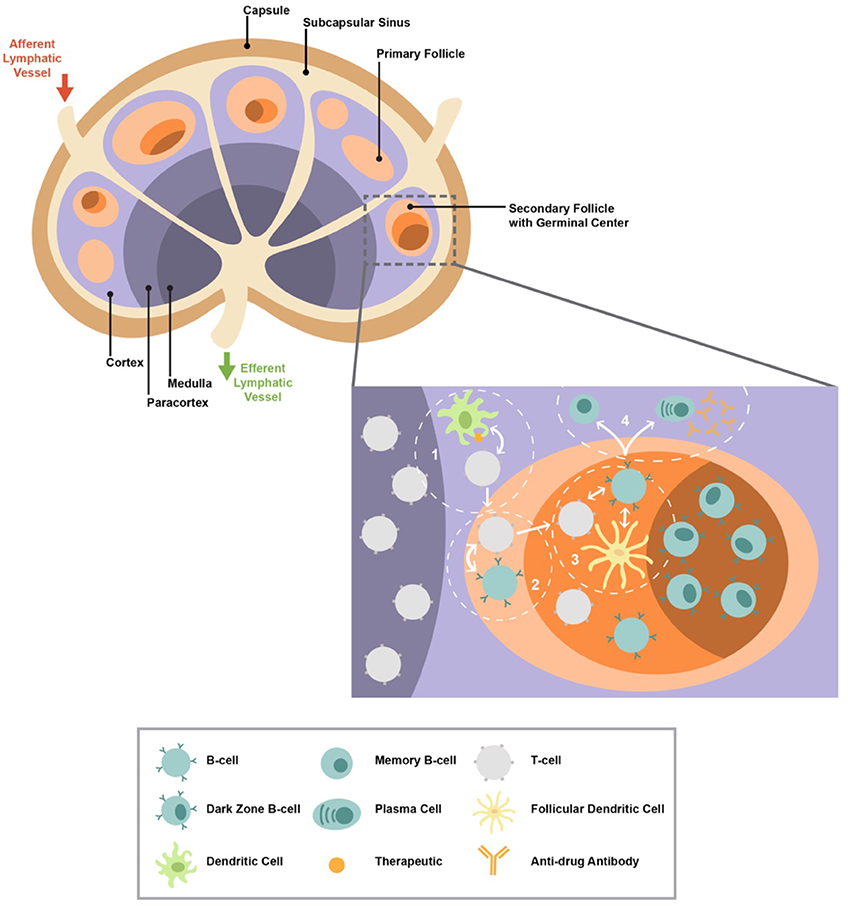
Figure 1. The lymph node structure/organization is shown. A zoomed T cell/follicular area with the major cell types involved in the development of antibody responses is shown. The presence of therapeutic within the lymph node can initiate a cascade of immune reactions ultimately leading to T cell-dependent germinal center (GC) activity and the generation of plasma cells and memory B cells that can produce antibodies. The cascade begins with (1) dendritic cells that present the therapeutic interaction with CD4 T cells resulting in their activation and differentiation; (2) activated CD4 T cells begin interacting with B cells, ultimately leading to further differentiation of both cell types and therefore trafficking into follicles/GCs; (3) within the GC, follicular CD4 T cells interact with GC B cells and follicular dendritic cell (FDC); (4) helping B cells promotes their maturation to memory and plasma cells.
Major Cell Populations
T cell zone (paracortex) is populated with innate immunity cells (DC, monocytes, macrophages, and granulocytes), adaptive immunity cells (CD4 and CD8), and stromal cells (FRCs). Subcapsular sinus macrophages is the first lymph node population encountering pathogens from the lymph (63) that controls the pathogen dissemination and inflammation and affects B cell responses to subsequent infections (64). These cells can trigger responses to lipid antigens, a mechanism mediated by activation of LN invariant natural killer cells (iNKTs) (65). Recirculating monocytes can traffic to LNs and either keep their classical status (66) or further differentiate to macrophages or DCs and initiate adaptive responses (67, 68). T cell zone macrophages can also function as scavengers for apoptotic cells (69). DC and monocyte in LNs are main producers of IL-6, an important cytokine for the differentiation development of Tfh cells (70, 71). FRCs provide a vital network for (i) recruitment of naive T cells and DCs through CCL21 and CCL19, the CCR7 ligands (72), a major chemokine receptor mediating tissue trafficking of several cell types (73, 74); (ii) T cell survival through IL-7, a survival factor particularly for naive T cells (75, 76); and (iii) trafficking of CD4 T cells toward the GC (62). When an antigen is present, major rearrangements take place within this area. Studies using mouse models have shown that the presence of antigen triggers the activation and repositioning of DC cells (77, 78), which have an important effect on CD4 T cell activation as well as the initiation of CD4 T and B cell interaction. B cells will follow a CCR7 gradient toward the follicle/T-zone (T–B) boundary where they could bind to multiple helper CD4 T cells at once, whereas T cells would only bind one at a time (79). The interaction of CD4 T and B cells in the T cell zone will activate a cascade of immune dynamics associated with major changes in differentiation status (phenotypes, transcriptome profile, and trafficking) (80) of both CD4 and B cells, ultimately enabling their trafficking into the GC area. In fact, the interaction between CD4, B, and antigen-presenting cells (APCs) in the T cell zone and T–B borders is required for optimal differentiation of CD4 T cells to Tfh cells (81, 82). These early interactions are also critical to the further development of B cell responses (83, 84).
Germinal Center: The Laboratory for the Development of B Cell Responses
The role of the Tfh cells is to help create high-affinity memory B and plasma cells (85); thus, this subset of CD4 T cells is crucial in the immune response. GCs, the structures found in mature, secondary follicles (59), are populated with activated B cells, follicular DCs (FDCs), Tfh cells, and macrophages (tingible body macrophages) (59). Upon antigen stimulation, naive B cells traffic to the T–B border following a CCR7 gradient (79). Further interaction with CD4 T cells and receipt of co-stimulatory signals (86) trigger a rigorous proliferation of B cells and the formation of a tight cluster within the follicle, which becomes the GC. Within the GC, the B cells undergo somatic hypermutation, affinity maturation, class switch recombination, and plasma/memory B cell production (87–89). Formation of GC is mediated by help from FDCs and the function of G-protein-coupled receptors (GPCR) like S1PR2 and P2RY8 (59, 90, 91). In primary follicles, FDCs help B cells form a follicle (92), whereas in secondary follicles, FDCs support GC B cell survival (93–97). B cell survival was impaired when FDCs were exposed to HIV-1 (98), smaller GCs, formed and lower antibody titers were obtained when FDC activation was blocked through TLR4 (99). FDCs modulate antigen availability by cycling the antigens between the FDC surface and other endosomal compartments (100) or accumulating ICs bound to Fc receptors on their cell surface (100), a process critical to the affinity maturation of B cells (101, 102). Tfh cells are a subset of CD4 T cells that are specialized to help B cells. They are located inside B cell follicles of secondary lymphoid organs and are responsible for activation, isotype switching, affinity maturation, and differentiation of B cells (103–105). Tfh cells express a unique phenotype and transcriptome signature (106–109). A mutual regulation through modulation of Bcl-6 between Tfh and GC B cells has been proposed (110–112). Tfh cells produce cytokines like IL-21 and IL-4, which are important for the GC B cell dynamics (105, 113–115) and maintenance of Tfh cells. Two distinct GC areas have been identified; the DZ, where B cell proliferation and somatic hypermutation occurs, and the LZ, where B cells interact with Tfh cells and FDCs (60, 116). Expression of the chemokine receptor CXCR4, which is highly expressed on Tfh and GC B cells (117, 118), and local production of its ligand CXCL12 (SDF-1) in DZ by CXCL12-expressing reticular cells (CRC) (119, 120) play a critical role for the organization of DZ and LZ (116, 121, 122). LZ is less compact and more diverse than is DZ. B cells continuously move between the DZ and LZ, helping in the further differentiation and affinity maturation of GC B cells (123–126). Within the LZ, B cells continuously interact with Tfh, FDC, and antigen, interactions that dictate their survival and clonal selection (84, 127–129).
Germinal Center Reactivity Against Therapeutics
Several studies have investigated GC dynamics after drug administration. Understanding how the LN and GC react to therapeutics (antibodies, recombinant proteins, cytokines, vaccines, enzymes, etc.) is important to reduce/eliminate ADA development. ADA can be generated by T cell-dependent (Td) and T cell-independent (Ti) pathways (130–132). The Td pathway involves an antigen-activated T cell that then stimulates B cell activation and differentiation to plasma cells. Neutralizing IgG4 ADA against FVIII (6, 133) was triggered by a Th2 CD4 T cell response (134), whereas initiation of Th1 responses was found to induce IgG1 and IgG2 ADA against FVIII when patients were on immunosuppressive therapy (134), which may sometimes be non-neutralizing (133, 134). Administration of IFN-β was found to induce either non-neutralizing and transient neutralizing ADA, mainly of low titers and affinity IgG1 and IgG3 subclasses, or persistent neutralizing ADA, which had mostly IgG2 and IgG4 antibodies (135). ADA binding affinity was positively correlated with IgG4 production and neutralizing ADA titers but negatively correlated with IgG3 production. Similarly, generation of high-affinity antibodies to biopharmaceuticals is CD4 T cell dependent (136, 137). In fact several studies have shown the development of neutralizing antibodies ADA (138–142) mainly of IgG4 subtype (143). Similar polyclonal IgG1 responses consisting of neutralizing and non-neutralizing specificities have also been detected in response to natalizumab (NZM) administration in multiple sclerosis patients. Neutralizing antibodies in these patients carry a higher load of somatic mutations in the complementary determining regions (CDRs) and have a higher affinity than have non-neutralizing, binding antibody specificities, which is consistent with LN-specific antigen-driven selection (144).
These considerations indicate that alternative cytokine milieu and initial programing of CD4 T cells in the LN can affect the outcome of the GC B cell responses to a given therapeutic in a way that parallels the LN-associated changes seen in vaccine-specific or pathogen-associated antibody production. The Ti pathway is triggered when the B cell is activated directly by the antigen. In general, polyvalent antigens, such as an aggregated biologic (145, 146), are more likely to induce Ti-B cell responses (147, 148). Ti responses lead to IgM or low affinity IgG ADA owing to lack of T cell help (130). Because most ADAs are IgG, the possible role of complement activation by ADA needs further investigation (130). Neutralizing antibodies, particularly the broad neutralizing antibodies (bNABs), are characterized by high levels of somatic mutations (149) and are indicative of GC maturation. The mechanisms leading to such maturation process in the GC are not well-understood. Presumably, antigen concentration within LN/GC and the co-evolution of Tfh and GC B cells (selection of TCR and BCR clones) are major biological factors affecting this process. Studies using mouse models have shown that the quality of Tfh help to GC B cell is an important biological factor for the development of high-affinity antibodies (127, 129, 150). Specifically, Tfh helps regulate the metabolic programming of LZ B cells that support their proliferation in DZ (129). Furthermore, this helps prolong the duration of B cell cycle in the DZ, a process associated with the generation of high-affinity GC B cells (151). Shuttling/binding of antigen to FDC (152) as well as the amount of antigen presented to Tfh by GC B cells (150) can have a significant impact on the development of high-affinity B cell responses. Dynamics and factors in the follicular, non-GC area can also affect the maturation of B cell responses. High-affinity B cell clones can be selected during early interaction between CD4 T and B cells at the T–B cell border (84), whereas class switch recombination can be started outside GC (153). Furthermore, recently identified populations like the CD25+FoxP3+ T follicular IL-10-producing cells (154) and Tbet+ B cells, mainly localized around the GC (155), could be important regulators for the development of neutralizing antibodies. Host genetics are also relevant. The presentation of MHC class II-restricted drug-specific peptides on CD4+ T cells can further contribute to the emergence and maintenance of polyclonal drug-specific B cell responses in lymphoid localities (144). Conversely, elimination of specific drug-associated T cell epitopes in mice treated with recombinant immunotoxins curtails the development of high-affinity antidrug IgG responses in primary as well as anamnestic responses (156). Taken together, these findings suggest that at least for some types of biologic pharmaceuticals, T-dependent pathways in LNs are central in the induction of neutralizing ADAs. Therefore, understanding in more detail the nature, trafficking/distribution of each biopharmaceutical into LN and its availability/sustainability on FDCs is warranted, as these are factors could direct the cellular and molecular mechanisms immobilized in the LN, which lead to the development of specific types of antibody responses, especially in the absence of adjuvants, which trigger innate immunity.
Conclusions
The cellular and molecular mechanisms governing the development of ADA responses in humans are not well-understood. This is a highly coordinated process taking place in secondary lymphoid organs where the nature of the “antigen,” tissue structure, and spatial positioning of relevant cell populations, particularly in the follicular/GC area, play a critical role for the host–therapeutic interplay leading to the differentiation of adaptive immune cells to enable the generation of antibody-secreting B cells. Today, the link between ADA and changes in LN function is still not well-studied. Major aspects related to ADA development that need further investigation include the following:
1. The trafficking and sustainability of a particular therapeutic in the LN areas, particularly the GC. Formulation for the in vivo delivery of an immunogen could significantly change its dynamics in the GCs with major impact on the B cell response development (157, 158).
2. The activation of specific innate immunity cells and the concomitant changes in the local cytokine/chemokine milieu are factors regulating the degree of CD4 T cell help for the B cell responses (78).
3. The possible association between ADA titer and affinity maturation and particular GC dynamics (i.e., magnitude of Tfh cell responses and expansion of particular Tfh subsets).
4. The possible role of preexisting immune activation and inflammation within the lymphoid organs.
Of particular interest is the investigation of ADA development in aging where the GC dynamics are different compared with those in young individuals (159–161) as well as in chronic inflammatory diseases like HIV and autoimmunity. For example, altered antibody responses are expected in HIV-infected individuals where chronic infection is associated with LN inflammation, architecture damage (fibrosis), and dramatic changes to GC dynamics (100, 155, 162, 163). Such LN changes could have a major impact in the ADA development to a specific therapeutic. Despite the limited predictive value for the drug immunogenicity in humans based on non-human primate (NHP) studies (164), NHP represents the only model for testing antibody development under such conditions. However, we need to keep in mind that compared with that in humans, ADA in NHP is mainly directed against the Fc region of the monoclonal antibody, causing loss of efficacy and adverse effects. Supplemental to human studies, investigation of therapeutic immunogenicity, when it occurs, in NHPs could lead to identification of particular cell types, molecules, and molecular pathways driving the responses to a particular therapeutic. The wide range of titers, subtypes, and function of ADA induced by different therapeutics argues for the need for identification of “LN molecular/cellular signatures” specific to certain therapeutic, which could lead to targets for “individualized” in vivo manipulation of ADA development.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This research was supported by the Intramural Research Program of the Vaccine Research Center, NIAID, National Institutes of Health; and a CAVD grant (#OP1032325) from the Bill and Melinda Gates Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with one of the authors AA at time of review.
References
1. Pratt KP. Anti-drug antibodies: emerging approaches to predict, reduce or reverse biotherapeutic immunogenicity. Antibodies. (2018) 31:7. doi: 10.3390/antib7020019
2. Kuka M, Iannacone M. Viral subversion of B cell responses within secondary lymphoid organs. Nat Rev Immunol. (2018) 18:255–65. doi: 10.1038/nri.2017.133
3. Bar-Ephraim YE, Mebius RE. Innate lymphoid cells in secondary lymphoid organs. Immunol Rev. (2016) 271:185–99. doi: 10.1111/imr.12407
4. Magrone T, Jirillo E. Development and organization of the secondary and tertiary lymphoid organs: influence of microbial and food antigens. Endocr Metab Immune Disord Drug Targets. (2019) 19:128–35. doi: 10.2174/1871530319666181128160411
5. Onder L, Morbe U, Pikor N, Novkovic M, Cheng HW, Hehlgans T, et al. Lymphatic endothelial cells control initiation of lymph node organogenesis. Immunity. (2017) 47:80–92. doi: 10.1016/j.immuni.2017.05.008
6. Baker MP, Reynolds HM, Lumicisi B, Bryson CJ. Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self Nonself . (2010) 1:314–22. doi: 10.4161/self.1.4.13904
7. Dimopoulos Y, Moysi E, Petrovas C. The lymph node in HIV pathogenesis. Curr HIV/AIDS Rep. (2017) 14:133–40. doi: 10.1007/s11904-017-0359-7
8. Denton AE, Linterman MA. Stromal networking: cellular connections in the germinal centre. Curr Opin Immunol. (2017) 45:103–11. doi: 10.1016/j.coi.2017.03.001
9. Ruddle NH, Akirav EM. Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J Immunol. (2009) 183:2205–12. doi: 10.4049/jimmunol.0804324
10. Hansen KC, D'Alessandro A, Clement CC, Santambrogio L. Lymph formation, composition and circulation: a proteomics perspective. Int Immunol. (2015) 27:219–27. doi: 10.1093/intimm/dxv012
11. Pape KA, Catron DM, Itano AA, Jenkins MK. The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity. (2007) 26:491–502. doi: 10.1016/j.immuni.2007.02.011
12. Gatto D, Brink R. The germinal center reaction. J Allergy Clin Immunol. (2010) 126:898–907. doi: 10.1016/j.jaci.2010.09.007
13. Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. (2008) 7:21–39. doi: 10.1038/nrd2399
14. Sgro C. Side-effects of a monoclonal antibody, muromonab CD3/orthoclone OKT3: bibliographic review. Toxicology. (1995) 105:23–9. doi: 10.1016/0300-483X(95)03123-W
15. Reichert JM. Marketed therapeutic antibodies compendium. MAbs. (2012) 4:413–5. doi: 10.4161/mabs.19931
16. Niaudet P, Jean G, Broyer M, Chatenoud L. Anti-OKT3 response following prophylactic treatment in paediatric kidney transplant recipients. Pediatr Nephrol. (1993) 7:263–7. doi: 10.1007/BF00853215
17. Thistlethwaite JR Jr, Stuart JK, Mayes JT, Gaber AO, Woodle S, Buckingham MR, et al. Complications and monitoring of OKT3 therapy. Am J Kidney Dis. (1988) 11:112–9. doi: 10.1016/S0272-6386(88)80192-6
18. Menting SP, van Lumig PP, de Vries AC, van den Reek JM, van der Kleij D, de Jong EM, et al. Extent and consequences of antibody formation against adalimumab in patients with psoriasis: one-year follow-up. JAMA Dermatol. (2014) 150:130–6. doi: 10.1001/jamadermatol.2013.8347
19. Radstake TR, Svenson M, Eijsbouts AM, van den Hoogen FH, Enevld C, van Riel PL, et al. Formation of antibodies against infliximab and adalimumab strongly correlates with functional drug levels and clinical responses in rheumatoid arthritis. Ann Rheum Dis. (2009) 68:1739–45. doi: 10.1136/ard.2008.092833
20. Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. (2011) 305:1460–8. doi: 10.1001/jama.2011.406
21. Moots RJ, Xavier RM, Mok CC, Rahman MU, Tsai WC, Al-Maini MH, et al. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with adalimumab, etanercept, or infliximab: results from a multinational, real-world clinical practice, non-interventional study. PLoS ONE. (2017) 12:e0175207. doi: 10.1371/journal.pone.0175207
22. Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis. (2007) 66:921–6. doi: 10.1136/ard.2006.065615
23. Hsu L, Armstrong AW. Anti-drug antibodies in psoriasis: a critical evaluation of clinical significance and impact on treatment response. Expert Rev Clin Immunol. (2013) 9:949–58. doi: 10.1586/1744666X.2013.836060
24. Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. (2008) 358:1109–17. doi: 10.1056/NEJMoa074943
25. Mok CC, van der Kleij D, Wolbink GJ. Drug levels, anti-drug antibodies, and clinical efficacy of the anti-TNFalpha biologics in rheumatic diseases. Clin Rheumatol. (2013) 32:1429–35. doi: 10.1007/s10067-013-2336-x
26. Avramis VI, Sencer S, Periclou AP, Sather H, Bostrom BC, Cohen LJ, et al. A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a children's cancer group study. Blood. (2002) 99:1986–94. doi: 10.1182/blood.V99.6.1986
27. Wetzler M, Sanford BL, Kurtzberg J, DeOliveira D, Frankel SR, Powell BL, et al. Effective asparagine depletion with pegylated asparaginase results in improved outcomes in adult acute lymphoblastic leukemia: cancer and leukemia group B study 9511. Blood. (2007) 109:4164–7. doi: 10.1182/blood-2006-09-045351
28. Ehrenforth S, Kreuz W, Scharrer I, Linde R, Funk M, Gungor T, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet. (1992) 339:594–8. doi: 10.1016/0140-6736(92)90874-3
29. Lillicrap D, Fijnvandraat K, Santagostino E. Inhibitors - genetic and environmental factors. Haemophilia. (2014) 20(Suppl. 4):87–93. doi: 10.1111/hae.12412
30. Haji Abdolvahab M, Mofrad MR, Schellekens H. Interferon beta: from molecular level to therapeutic effects. Int Rev Cell Mol Biol. (2016) 326:343–72. doi: 10.1016/bs.ircmb.2016.06.001
31. Nazarov VD, Lapin SV, Mazing AV, Evdoshenko EP, Totolian AA. Immunogenicity of human interferon-beta-containing pharmaceuticals. Biochemistry (Mosc). (2016) 81:1396–400. doi: 10.1134/S000629791611016X
32. Govindappa K, Sathish J, Park K, Kirkham J, Pirmohamed M. Development of interferon beta-neutralising antibodies in multiple sclerosis–a systematic review and meta-analysis. Eur J Clin Pharmacol. (2015) 71:1287–98. doi: 10.1007/s00228-015-1921-0
33. Lagasse HA, Alexaki A, Simhadri VL, Katagiri NH, Jankowski W, Sauna ZE, et al. Recent advances in (therapeutic protein) drug development. F1000Res. (2017) 6:113. doi: 10.12688/f1000research.9970.1
34. Buettner MJ, Shah SR, Saeui CT, Ariss R, Yarema KJ. Improving immunotherapy through glycodesign. Front Immunol. (2018) 9:2485. doi: 10.3389/fimmu.2018.02485
35. Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. J Immunol Res. (2016) 2016:1298473. doi: 10.1155/2016/1298473
36. Hilger C, Fischer J, Wolbing F, Biedermann T. Role and mechanism of galactose-alpha-1,3-galactose in the elicitation of delayed anaphylactic reactions to red meat. Curr Allergy Asthma Rep. (2019) 19:3. doi: 10.1007/s11882-019-0835-9
37. Peterson RG, Handschumacher RE, Mitchell MS. Immunological responses to L-asparaginase. J Clin Invest. (1971) 50:1080–90. doi: 10.1172/JCI106579
38. Woo MH, Hak LJ, Storm MC, Evans WE, Sandlund JT, Rivera GK, et al. Anti-asparaginase antibodies following E. coli asparaginase therapy in pediatric acute lymphoblastic leukemia. Leukemia. (1998) 12:1527–33. doi: 10.1038/sj.leu.2401162
39. Lorentz KM, Kontos S, Diaceri G, Henry H, Hubbell JA. Engineered binding to erythrocytes induces immunological tolerance to E. coli asparaginase. Sci Adv. (2015) 1:e1500112. doi: 10.1126/sciadv.1500112
40. Pandey GS, Sauna ZE. Pharmacogenetics and the immunogenicity of protein therapeutics. J Interferon Cytokine Res. (2014) 34:931–7. doi: 10.1089/jir.2012.0136
41. Mahlangu JN, Weldingh KN, Lentz SR, Kaicker S, Karim FA, Matsushita T, et al. Changes in the amino acid sequence of the recombinant human factor VIIa analog, vatreptacog alfa, are associated with clinical immunogenicity. J Thromb Haemost. (2015) 13:1989–98. doi: 10.1111/jth.13141
42. Bar-Yoseph H, Pressman S, Blatt A, Gerassy Vainberg S, Maimon N, Starosvetsky E, et al. Infliximab-tumor necrosis factor complexes elicit formation of anti-drug antibodies. Gastroenterology. (2019) 157:1338–51. doi: 10.1053/j.gastro.2019.08.009
43. Rossert J, Casadevall N, Eckardt KU. Anti-erythropoietin antibodies and pure red cell aplasia. J Am Soc Nephrol. (2004) 15:398–406. doi: 10.1097/01.ASN.0000107561.59698.42
44. Schellekens H. Immunologic mechanisms of EPO-associated pure red cell aplasia. Best Pract Res Clin Haematol. (2005) 18:473–80. doi: 10.1016/j.beha.2005.01.016
45. Alexaki A, Hettiarachchi GK, Athey JC, Katneni UK, Simhadri V, Hamasaki-Katagiri N, et al. Effects of codon optimization on coagulation factor IX translation and structure: implications for protein and gene therapies. Sci Rep. (2019) 9:15449. doi: 10.1038/s41598-019-51984-2
46. Sethu S, Govindappa K, Quinn P, Wadhwa M, Stebbings R, Boggild M, et al. Immunoglobulin G1 and immunoglobulin G4 antibodies in multiple sclerosis patients treated with IFNbeta interact with the endogenous cytokine and activate complement. Clin Immunol. (2013) 148:177–85. doi: 10.1016/j.clim.2013.05.008
47. Krishna M, Nadler SG. Immunogenicity to biotherapeutics - the role of anti-drug immune complexes. Front Immunol. (2016) 7:21. doi: 10.3389/fimmu.2016.00021
48. Rojko JL, Evans MG, Price SA, Han B, Waine G, DeWitte M, et al. Formation, clearance, deposition, pathogenicity, and identification of biopharmaceutical-related immune complexes: review and case studies. Toxicol Pathol. (2014) 42:725–64. doi: 10.1177/0192623314526475
49. Ducourau E, Mulleman D, Paintaud G, Miow Lin DC, Lauferon F, Ternant D, et al. Antibodies toward infliximab are associated with low infliximab concentration at treatment initiation and poor infliximab maintenance in rheumatic diseases. Arthritis Res Ther. (2011) 13:R105. doi: 10.1186/ar3386
50. van Schouwenburg PA, van de Stadt LA, de Jong RN, van Buren EE, Kruithof S, de Groot E, et al. Adalimumab elicits a restricted anti-idiotypic antibody response in autoimmune patients resulting in functional neutralisation. Ann Rheum Dis. (2013) 72:104–9. doi: 10.1136/annrheumdis-2012-201445
51. Chitlur M, Warrier I, Rajpurkar M, Lusher JM. Inhibitors in factor IX deficiency a report of the ISTH-SSC international FIX inhibitor registry (1997-2006). Haemophilia. (2009) 15:1027–31. doi: 10.1111/j.1365-2516.2009.02039.x
52. McDonald JR, Renehan AG, O'Dwyer ST, Haboubi NY. Lymph node harvest in colon and rectal cancer: current considerations. World J Gastrointest Surg. (2012) 4:9–19. doi: 10.4240/wjgs.v4.i1.9
53. Thompson RH, Carver BS, Bosl GJ, Bajorin D, Motzer R, Feldman D, et al. Body mass index is associated with higher lymph node counts during retroperitoneal lymph node dissection. Urology. (2012) 79:361–4. doi: 10.1016/j.urology.2011.04.050
54. Morikawa T, Tanaka N, Kuchiba A, Nosho K, Yamauchi M, Hornick JL, et al. Predictors of lymph node count in colorectal cancer resections: data from US nationwide prospective cohort studies. Arch Surg. (2012) 147:715–23. doi: 10.1001/archsurg.2012.353
55. McFadden C, McKinley B, Greenwell B, Knuckolls K, Culumovic P, Schammel D, et al. Differential lymph node retrieval in rectal cancer: associated factors and effect on survival. J Gastrointest Oncol. (2013) 4:158–63. doi: 10.3978/j.issn.2078-6891.2013.023
56. Chou JF, Row D, Gonen M, Liu YH, Schrag D, Weiser MR. Clinical and pathologic factors that predict lymph node yield from surgical specimens in colorectal cancer: a population-based study. Cancer. (2010) 116:2560–70. doi: 10.1002/cncr.25032
57. Willard-Mack CL. Normal structure, function, and histology of lymph nodes. Toxicol Pathol. (2006) 34:409–24. doi: 10.1080/01926230600867727
58. Fossum S, Ford WL. The organization of cell populations within lymph nodes: their origin, life history and functional relationships. Histopathology. (1985) 9:469–99. doi: 10.1111/j.1365-2559.1985.tb02830.x
59. Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity. (2016) 45:471–82. doi: 10.1016/j.immuni.2016.09.001
60. Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood. (2012) 120:2240–8. doi: 10.1182/blood-2012-03-415380
61. Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, et al. The microanatomic segregation of selection by apoptosis in the germinal center. Science. (2017) 358:eaao2602. doi: 10.1126/science.aao2602
62. Bajenoff M, Glaichenhaus N, Germain RN. Fibroblastic reticular cells guide T lymphocyte entry into and migration within the splenic T cell zone. J Immunol. (2008) 181:3947–54. doi: 10.4049/jimmunol.181.6.3947
63. Louie DAP, Liao S. Lymph node subcapsular sinus macrophages as the frontline of lymphatic immune defense. Front Immunol. (2019) 10:347. doi: 10.3389/fimmu.2019.00347
64. Gaya M, Castello A, Montaner B, Rogers N, Reise Sousa C, Bruckbauer A, et al. Host response. inflammation-induced disruption of SCS macrophages impairs B cell responses to secondary infection. Science. (2015) 347:667–72. doi: 10.1126/science.aaa1300
65. Barral P, Polzella P, Bruckbauer A, van Rooijen N, Besra GS, Cerundolo V, et al. CD169(+) macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol. (2010) 11:303–12. doi: 10.1038/ni.1853
66. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
67. Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell. (2010) 143:416–29. doi: 10.1016/j.cell.2010.09.039
68. Chen Q, Snapper CM. Inflammatory monocytes are critical for induction of a polysaccharide-specific antibody response to an intact bacterium. J Immunol. (2013) 190:1048–55. doi: 10.4049/jimmunol.1202455
69. Baratin M, Simon L, Jorquera A, Ghigo C, Dembele D, Nowak J, et al. T cell zone resident macrophages silently dispose of apoptotic cells in the lymph node. Immunity. (2017) 47:349–62. doi: 10.1016/j.immuni.2017.07.019
70. Mohr E, Serre K, Manz RA, Cunningham AF, Khan M, Hardie DL, et al. Dendritic cells and monocyte/macrophages that create the IL-6/APRIL-rich lymph node microenvironments where plasmablasts mature. J Immunol. (2009) 182:2113–23. doi: 10.4049/jimmunol.0802771
71. Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J Immunol. (2013) 190:3049–53. doi: 10.4049/jimmunol.1203032
72. Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. (2008) 8:362–71. doi: 10.1038/nri2297
73. Hauser MA, Legler DF. Common and biased signaling pathways of the chemokine receptor CCR7 elicited by its ligands CCL19 and CCL21 in leukocytes. J Leukoc Biol. (2016) 99:869–82. doi: 10.1189/jlb.2MR0815-380R
74. Comerford I, Harata-Lee Y, Bunting MD, Gregor C, Kara EE, McColl SR. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. (2013) 24:269–83. doi: 10.1016/j.cytogfr.2013.03.001
75. Donovan GM, Lythe G. T cell and reticular network co-dependence in HIV infection. J Theor Biol. (2016) 395:211–20. doi: 10.1016/j.jtbi.2016.01.040
76. Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. (2007) 8:1255–65. doi: 10.1038/ni1513
77. Gerner MY, Casey KA, Kastenmuller W, Germain RN. Dendritic cell and antigen dispersal landscapes regulate T cell immunity. J Exp Med. (2017) 214:3105–22. doi: 10.1084/jem.20170335
78. Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, et al. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity. (2012) 37:1091–10. doi: 10.1016/j.immuni.2012.08.016
79. Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, et al. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol. (2005) 3:e150. doi: 10.1371/journal.pbio.0030150
80. Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. (2007) 179:5099–108. doi: 10.4049/jimmunol.179.8.5099
81. Ballesteros-Tato A, Leon B, Lund FE, Randall TD. CD4+ T helper cells use CD154-CD40 interactions to counteract T reg cell-mediated suppression of CD8+ T cell responses to influenza. J Exp Med. (2013) 210:1591–601. doi: 10.1084/jem.20130097
82. Eivazi S, Bagheri S, Hashemzadeh MS, Ghalavand M, Qamsari ES, Dorostkar R, et al. Development of T follicular helper cells and their role in disease and immune system. Biomed Pharmacother. (2016) 84:1668–78. doi: 10.1016/j.biopha.2016.10.083
83. Shih TA, Meffre E, Roederer M, Nussenzweig MC. Role of BCR affinity in T cell dependent antibody responses in vivo. Nat Immunol. (2002) 3:570–5. doi: 10.1038/ni803
84. Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, et al. A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J Exp Med. (2011) 208:1243–52. doi: 10.1084/jem.20102477
85. Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS, et al. Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity. (2013) 38:805–17. doi: 10.1016/j.immuni.2013.02.020
86. Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. (1998) 281:96–9. doi: 10.1126/science.281.5373.96
87. Suan D, Sundling C, Brink R. Plasma cell and memory B cell differentiation from the germinal center. Curr Opin Immunol. (2017) 45:97–102. doi: 10.1016/j.coi.2017.03.006
88. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. (2015) 15:137–48. doi: 10.1038/nri3804
89. Zhang Y, Garcia-Ibanez L, Toellner KM. Regulation of germinal center B-cell differentiation. Immunol Rev. (2016) 270:8–19. doi: 10.1111/imr.12396
90. Muppidi JR, Lu E, Cyster JG. The G protein-coupled receptor P2RY8 and follicular dendritic cells promote germinal center confinement of B cells, whereas S1PR3 can contribute to their dissemination. J Exp Med. (2015) 212:2213–22. doi: 10.1084/jem.20151250
91. Lu E, Cyster JG. G-protein coupled receptors and ligands that organize humoral immune responses. Immunol Rev. (2019) 289:158–72. doi: 10.1111/imr.12743
92. Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, et al. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med. (2011) 208:2497–510. doi: 10.1084/jem.20111449
93. Lindhout E, Mevissen ML, Kwekkeboom J, Tager JM, de Groot C. Direct evidence that human follicular dendritic cells (FDC) rescue germinal centre B cells from death by apoptosis. Clin Exp Immunol. (1993) 91:330–6. doi: 10.1111/j.1365-2249.1993.tb05904.x
94. Lindhout E, de Groot C. Follicular dendritic cells and apoptosis: life and death in the germinal centre. Histochem J. (1995) 27:167–83. doi: 10.1007/BF00177584
95. Liu YJ, Mason DY, Johnson GD, Abbot S, Gregory CD, Hardie DL, et al. Germinal center cells express bcl-2 protein after activation by signals which prevent their entry into apoptosis. Eur J Immunol. (1991) 21:1905–10. doi: 10.1002/eji.1830210819
96. Schwarz YX, Yang M, Qin D, Wu J, Jarvis WD, Grant S, et al. Follicular dendritic cells protect malignant B cells from apoptosis induced by anti-Fas and antineoplastic agents. J Immunol. (1999) 163:6442–7.
97. Qin D, Wu J, Burton GF, Szakal AK, Tew JG. Follicular dendritic cells mediated maintenance of primary lymphocyte cultures for long-term analysis of a functional in vitro immune system. J Immunol Methods. (1999) 226:19–27. doi: 10.1016/S0022-1759(99)00038-1
98. Sabri F, Prados A, Munoz-Fernandez R, Lantto R, Fernandez-Rubio P, Nasi A, et al. Impaired B cells survival upon production of inflammatory cytokines by HIV-1 exposed follicular dendritic cells. Retrovirology. (2016) 13:61. doi: 10.1186/s12977-016-0295-4
99. Garin A, Meyer-Hermann M, Contie M, Figge MT, Buatois V, Gunzer M, et al. Toll-like receptor 4 signaling by follicular dendritic cells is pivotal for germinal center onset and affinity maturation. Immunity. (2010) 33:84–95. doi: 10.1016/j.immuni.2010.07.005
100. Bronnimann MP, Skinner PJ, Connick E. The B-cell follicle in HIV infection: barrier to a cure. Front Immunol. (2018) 9:20. doi: 10.3389/fimmu.2018.00020
101. Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, et al. Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity. (2013) 38:1164–75. doi: 10.1016/j.immuni.2013.02.023
102. Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol. (2014) 14:495–504. doi: 10.1038/nri3689
103. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
104. Kim CH, Lim HW, Kim JR, Rott L, Hillsamer P, Butcher EC. Unique gene expression program of human germinal center T helper cells. Blood. (2004) 104:1952–60. doi: 10.1182/blood-2004-03-1206
105. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
106. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. (2000) 192:1545–52. doi: 10.1084/jem.192.11.1545
107. Kim CH, Rott LS, Clark-Lewis I, Campbell DJ, Wu L, Butcher EC. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J Exp Med. (2001) 193:1373–81. doi: 10.1084/jem.193.12.1373
108. Rasheed AU, Rahn HP, Sallusto F, Lipp M, Muller G. Follicular B helper T cell activity is confined to CXCR5(hi)ICOS(hi) CD4 T cells and is independent of CD57 expression. Eur J Immunol. (2006) 36:1892–903. doi: 10.1002/eji.200636136
109. Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE, et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest. (2015) 125:292–303. doi: 10.1172/JCI76238
110. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. (2009) 325:1006–10. doi: 10.1126/science.1175870
111. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science. (2009) 325:1001–5. doi: 10.1126/science.1176676
112. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. (2009) 31:457–68. doi: 10.1016/j.immuni.2009.07.002
113. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. (2010) 207:353–63. doi: 10.1084/jem.20091738
114. Zotos D, Coquet JM, Zhang Y, Light A, D'Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med. (2010) 207:365–78. doi: 10.1084/jem.20091777
115. Poholek AC, Hansen K, Hernandez SG, Eto D, Chandele A, Weinstein JS, et al. In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol. (2010) 185:313–26. doi: 10.4049/jimmunol.0904023
116. Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. (2007) 27:190–202. doi: 10.1016/j.immuni.2007.07.009
117. Weber TS. Cell cycle-Associated CXCR4 expression in germinal center B cells and its implications on affinity maturation. Front Immunol. (2018) 9:1313. doi: 10.3389/fimmu.2018.01313
118. Estes JD, Keele BF, Tenner-Racz K, Racz P, Redd MA, Thacker TC, et al. Follicular dendritic cell-mediated up-regulation of CXCR4 expression on CD4 T cells and HIV pathogenesis. J Immunol. (2002) 169:2313–22. doi: 10.4049/jimmunol.169.5.2313
119. Bannard O, Horton RM, Allen CD, An J, Nagasawa T, Cyster JG. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity. (2013) 39:912–24. doi: 10.1016/j.immuni.2013.08.038
120. Rodda LB, Bannard O, Ludewig B, Nagasawa T, Cyster JG. Phenotypic and morphological properties of germinal center dark zone Cxcl12-expressing reticular cells. J Immunol. (2015) 195:4781–91. doi: 10.4049/jimmunol.1501191
121. Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. (2004) 5:943–52. doi: 10.1038/ni1100
122. Barinov A, Luo L, Gasse P, Meas-Yedid V, Donnadieu E, Arenzana-Seisdedos F, et al. Essential role of immobilized chemokine CXCL12 in the regulation of the humoral immune response. Proc Natl Acad Sci USA. (2017) 114:2319–24. doi: 10.1073/pnas.1611958114
123. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. (2010) 143:592–605. doi: 10.1016/j.cell.2010.10.032
124. Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science. (2007) 315:528–31. doi: 10.1126/science.1136736
125. Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, et al. Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity. (2007) 26:655–67. doi: 10.1016/j.immuni.2007.04.008
126. Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, et al. In vivo imaging of germinal centres reveals a dynamic open structure. Nature. (2007) 446:83–7. doi: 10.1038/nature05573
127. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science. (2014) 345:1058–62. doi: 10.1126/science.1257861
128. van der Poel CE, Bajic G, Macaulay CW, van den Broek T, Ellson CD, Bouma G, et al. Follicular dendritic cells modulate germinal center B cell diversity through fcgammaRIIB. Cell Rep. (2019) 29:2745–55. doi: 10.1016/j.celrep.2019.10.086
129. Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, et al. Germinal center selection and affinity maturation require dynamic regulation of mTORC1 kinase. Immunity. (2017) 46:1045–58. doi: 10.1016/j.immuni.2017.06.005
130. Sethu S, Govindappa K, Alhaidari M, Pirmohamed M, Park K, Sathish J. Immunogenicity to biologics: mechanisms, prediction and reduction. Arch Immunol Ther Exp. (2012) 60:331–44. doi: 10.1007/s00005-012-0189-7
131. Winthrop KL, Korman N, Abramovits W, Rottinghaus ST, Tan H, Gardner A, et al. T-cell-mediated immune response to pneumococcal conjugate vaccine (PCV-13) and tetanus toxoid vaccine in patients with moderate-to-severe psoriasis during tofacitinib treatment. J Am Acad Dermatol. (2018) 78:1149–55. doi: 10.1016/j.jaad.2017.09.076
132. Roth A, Glaesener S, Schutz K, Meyer-Bahlburg A. Reduced number of transitional and naive B cells in addition to decreased BAFF levels in response to the T cell independent immunogen pneumovax(R)23. PLoS ONE. (2016) 11:e0152215. doi: 10.1371/journal.pone.0152215
133. Reding MT, Lei S, Lei H, Green D, Gill J, Conti-Fine BM. Distribution of Th1- and Th2-induced anti-factor VIII IgG subclasses in congenital and acquired hemophilia patients. Thromb Haemost. (2002) 88:568–75. doi: 10.1055/s-0037-1613257
134. Reding MT. Immunological aspects of inhibitor development. Haemophilia. (2006) 12(Suppl. 6):30–5. doi: 10.1111/j.1365-2516.2006.01363.x
135. Dujmovic I, Hegen H, Paz P, Croze E, Deisenhammer F. Persistency of neutralizing anti-interferon-beta antibodies in patients with multiple sclerosis treated with subcutaneous interferon-beta depends on antibody titers, IGG subclasses, and affinity maturation. J Interferon Cytokine Res. (2017) 37:317–24. doi: 10.1089/jir.2016.0080
136. Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS. T-cell dependent immunogenicity of protein therapeutics: preclinical assessment and mitigation. Clin Immunol. (2013) 149:534–55. doi: 10.1016/j.clim.2013.09.006
137. Pratt KP, Thompson AR. B-cell and T-cell epitopes in anti-factor VIII immune responses. Clin Rev Allergy Immunol. (2009) 37:80–95. doi: 10.1007/s12016-009-8120-7
138. Lenders M, Neusser LP, Rudnicki M, Nordbeck P, Canaan-Kuhl S, Nowak A, et al. Dose-dependent effect of enzyme replacement therapy on neutralizing antidrug antibody titers and clinical outcome in patients with fabry disease. J Am Soc Nephrol. (2018) 29:2879–89. doi: 10.1681/ASN.2018070740
139. Bloem K, Hernandez-Breijo B, Martinez-Feito A, Rispens T. Immunogenicity of therapeutic antibodies: monitoring antidrug antibodies in a clinical context. Ther Drug Monit. (2017) 39:327–32. doi: 10.1097/FTD.0000000000000404
140. Wu Y, Akhgar A, Li JJ, Yu B, Chen C, Lee N, et al. Selection of a ligand-binding neutralizing antibody assay for benralizumab: comparison with an antibody-dependent cell-mediated cytotoxicity (ADCC) cell-based assay. AAPS J. (2018) 20:49. doi: 10.1208/s12248-018-0207-8
141. Domachowske JB, Khan AA, Esser MT, Jensen K, Takas T, Villafana T, et al. Safety, tolerability and pharmacokinetics of medi8897, an extended half-life single-dose respiratory syncytial virus prefusion f-targeting monoclonal antibody administered as a single dose to healthy preterm infants. Pediatr Infect Dis J. (2018) 37:886–92. doi: 10.1097/INF.0000000000001916
142. van Schie KA, Hart MH, de Groot ER, Kruithof S, Aarden LA, Wolbink GJ, et al. The antibody response against human and chimeric anti-TNF therapeutic antibodies primarily targets the TNF binding region. Ann Rheum Dis. (2015) 74:311–4. doi: 10.1136/annrheumdis-2014-206237
143. Lenders M, Schmitz B, Brand SM, Foell D, Brand E. Characterization of drug-neutralizing antibodies in patients with fabry disease during infusion. J Allergy Clin Immunol. (2018) 141:2289–92. doi: 10.1016/j.jaci.2017.12.1001
144. Cassotta A, Mikol V, Bertrand T, Pouzieux S, Le Parc J, Ferrari P, et al. A single T cell epitope drives the neutralizing anti-drug antibody response to natalizumab in multiple sclerosis patients. Nat Med. (2019) 25:1402–7. doi: 10.1038/s41591-019-0568-2
145. Antonelli G, Dianzani F. Development of antibodies to interferon beta in patients: technical and biological aspects. Eur Cytokine Netw. (1999) 10:413–22.
146. Hermeling S, Crommelin DJ, Schellekens H, Jiskoot W. Structure-immunogenicity relationships of therapeutic proteins. Pharm Res. (2004) 21:897–903. doi: 10.1023/B:PHAM.0000029275.41323.a6
147. Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol. (2009) 9:15–27. doi: 10.1038/nri2454
148. Depoil D, Weber M, Treanor B, Fleire SJ, Carrasco YR, Harwood NE, et al. Early events of B cell activation by antigen. Sci Signal. (2009) 2:pt1. doi: 10.1126/scisignal.263pt1
149. Mascola JR, Haynes BF. HIV-1 neutralizing antibodies: understanding nature's pathways. Immunol Rev. (2013) 254:225–44. doi: 10.1111/imr.12075
150. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature. (2014) 509:637–40. doi: 10.1038/nature13300
151. Gitlin AD, Mayer CT, Oliveira TY, Shulman Z, Jones MJ, Koren A, et al. Humoral immunity. T cell help controls the speed of the cell cycle in germinal center B cells. Science. (2015) 349:643–6. doi: 10.1126/science.aac4919
152. Tokatlian T, Read BJ, Jones CA, Kulp DW, Menis S, Chang JYH, et al. Innate immune recognition of glycans targets HIV nanoparticle immunogens to germinal centers. Science. (2019) 363:649–54. doi: 10.1126/science.aat9120
153. Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, et al. Class-switch recombination occurs infrequently in germinal centers. Immunity. (2019) 51:337–50. doi: 10.1016/j.immuni.2019.07.001
154. Canete PF, Sweet RA, Gonzalez-Figueroa P, Papa I, Ohkura N, Bolton H, et al. Regulatory roles of IL-10-producing human follicular T cells. J Exp Med. (2019) 216:1843–56. doi: 10.1084/jem.20190493
155. Austin JW, Buckner CM, Kardava L, Wang W, Zhang X, Melson VA, et al. Overexpression of T-bet in HIV infection is associated with accumulation of B cells outside germinal centers and poor affinity maturation. Sci Transl Med. (2019) 27:11. doi: 10.1126/scitranslmed.aax0904
156. Mazor R, Crown D, Addissie S, Jang Y, Kaplan G, Pastan I. Elimination of murine and human T-cell epitopes in recombinant immunotoxin eliminates neutralizing and anti-drug antibodies in vivo. Cell Mol Immunol. (2017) 14:432–42. doi: 10.1038/cmi.2015.91
157. Cirelli KM, Carnathan DG, Nogal B, Martin JT, Rodriguez OL, Upadhyay AA, et al. Slow delivery immunization enhances HIV neutralizing antibody and germinal center responses via modulation of immunodominance. Cell. (2020) 180:206. doi: 10.1016/j.cell.2019.12.027
158. Moyer TJ, Kato Y, Abraham W, Chang JYH, Kulp DW, Watson N, et al. Engineered immunogen binding to alum adjuvant enhances humoral immunity. Nat Med. (2020) 26:430–40. doi: 10.1038/s41591-020-0753-3
159. Thompson HL, Smithey MJ, Surh CD, Nikolich-Zugich J. Functional and homeostatic impact of age-related changes in lymph node stroma. Front Immunol. (2017) 8:706. doi: 10.3389/fimmu.2017.00706
160. Nikolich-Zugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. (2018) 19:10–9. doi: 10.1038/s41590-017-0006-x
161. Shankwitz K, Pallikkuth S, Sirupangi T, Kirk Kvistad D, Russel KB, Pahwa R, et al. Compromised steady-state germinal center activity with age in nonhuman primates. Aging Cell. (2019) 19:e13087. doi: 10.1111/acel.13087
162. Estes JD. Pathobiology of HIV/SIV-associated changes in secondary lymphoid tissues. Immunol Rev. (2013) 254:65–77. doi: 10.1111/imr.12070
163. Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, Smith EC, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest. (2012) 122:3281–94. doi: 10.1172/JCI63039
Keywords: therapeutics, follicle, germinal center, B cells, ADA, Tfh cell
Citation: Fu K, March K, Alexaki A, Fabozzi G, Moysi E and Petrovas C (2020) Immunogenicity of Protein Therapeutics: A Lymph Node Perspective. Front. Immunol. 11:791. doi: 10.3389/fimmu.2020.00791
Received: 03 January 2020; Accepted: 07 April 2020;
Published: 14 May 2020.
Edited by:
Amy Rosenberg, United States Food and Drug Administration, United StatesReviewed by:
Wayne Robert Thomas, University of Western Australia, AustraliaSrinivasa Reddy Bonam, École Supérieure de Biotechnologie Strasbourg (ESBS), France
Copyright © 2020 Fu, March, Alexaki, Fabozzi, Moysi and Petrovas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Constantinos Petrovas, cGV0cm92YXNjQG1haWwubmloLmdvdg==
†These authors have contributed equally to this work