 Andrei Miclea
Andrei Miclea Maud Bagnoud
Maud Bagnoud Andrew Chan
Andrew Chan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 06 May 2020
Sec. Multiple Sclerosis and Neuroimmunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00781
This article is part of the Research Topic Vitamin D in Neurological Diseases: From Pathophysiology to Therapy View all 9 articles
Multiple sclerosis (MS) is characterized as an autoimmune disease affecting the central nervous system. It is one of the most common neurological disorders in young adults. Over the past decades, increasing evidence suggested that hypovitaminosis D is a contributing factor to the risk of developing MS. From different risk factors contributing to the development of MS, vitamin D status is of particular interest since it is not only a modifiable risk factor but is also associated with MS disease activity. MS patients with lower serum vitamin D concentrations were shown to have higher disease activity. However, this finding does not demonstrate causality. In this regard, prospective vitamin D supplementation studies missed statistical significance in its primary endpoints but showed promising results in secondary outcome measures or post hoc analyses. An explanation for missed primary endpoints may be underpowered trials. Besides vitamin D supplementation as a potential add-on to long-term immunotherapeutic treatment, a recent laboratory study of our group pointed toward a beneficial effect of vitamin D to improve the efficacy of glucocorticoids in relapse therapy. In the following article, we will briefly review the effects of vitamin D on MS by outlining its effects on the immune and nervous system and by reviewing the association between vitamin D and MS risk as well as MS disease activity. We will also review the effects of vitamin D supplementation on MS risk and MS disease activity.
The exact pathophysiological mechanisms leading to the development of multiple sclerosis (MS) are not fully understood (1, 2). Nonetheless, certain genetic and environmental factors influencing not only MS risk but also MS disease activity have been identified (1, 3–6). One of the identified factors is vitamin D status (1, 4). In this article, we will briefly review the effects of vitamin D on MS. First, we will review the metabolism (summarized in Figure 1), the biological and safety features, and the intake guidelines of vitamin D. Second, we will outline the effects of vitamin D on cells of the innate and adaptive immune system and cells of the nervous system (summarized in Figure 1). Third, the association of vitamin D and MS risk as well as MS disease activity will be laid out (summarized in Table 1). Lastly, the evidence of the effects of vitamin D supplementation on MS risk, MS disease activity, and as a potential add-on for relapse therapy will be outlined (summarized in Table 1).

Table 1. Vitamin D and its association with MS.
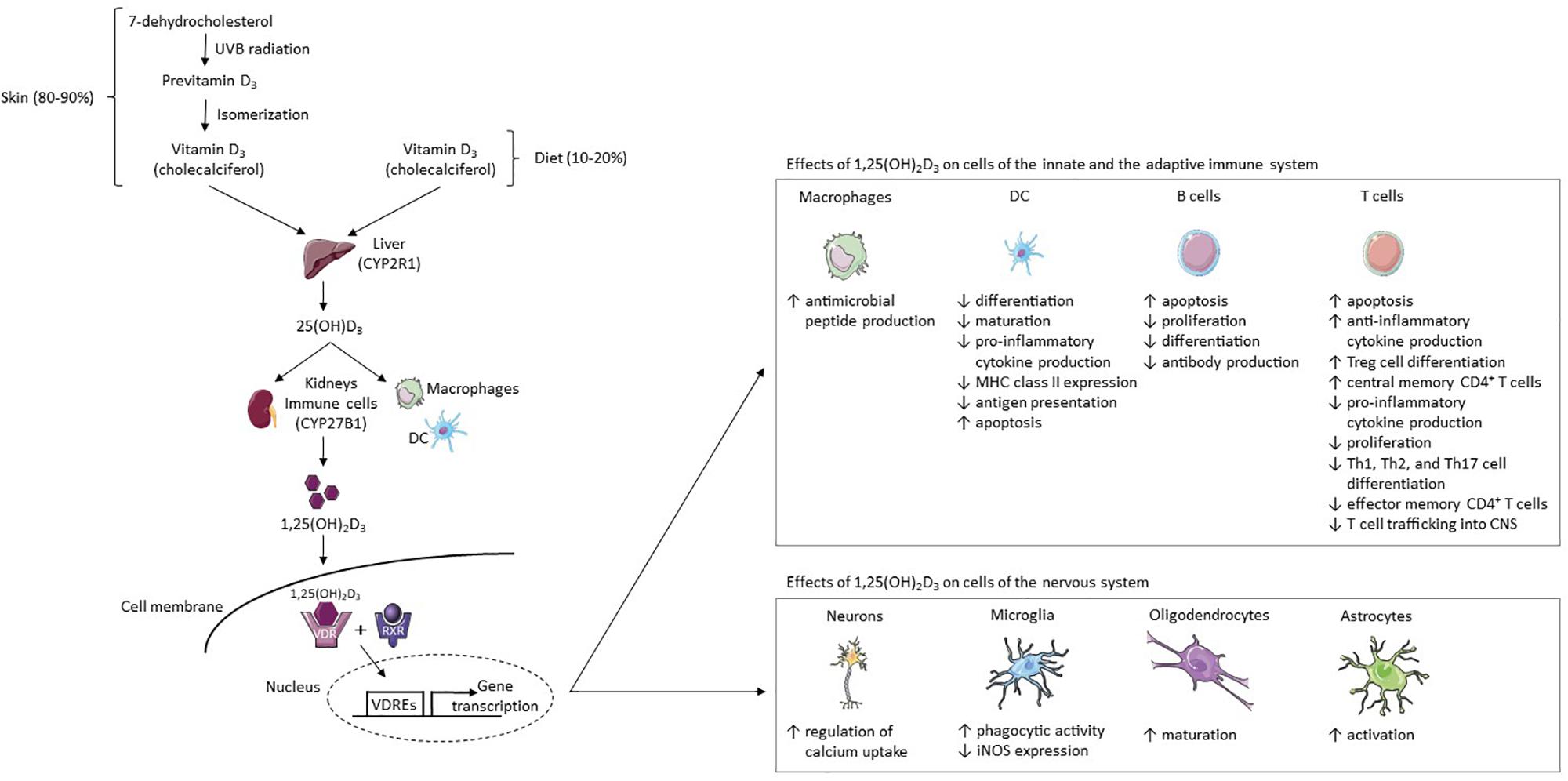
Figure 1. Vitamin D3 metabolism and its effects on cells of the immune and nervous system. 80–90% of the body’s vitamin D supply is produced by the skin’s exposure to UVB radiation and 10–20% is acquired through diet (7). Fatty fish contain high amounts of vitamin D3 (cholecalciferol) (8). In the skin, the vitamin D3 precursor 7-dehydrocholesterol converts to previtamin D3 after UVB exposure (10, 11). Previtamin D3 then isomerizes to cholecalciferol (10, 11). This physiologically inactive form of vitamin D3 is hydroxylated in the liver to 25(OH)D3 by CYP2R1 (13). It is then hydroxylated by the enzyme CYP27B1 in the kidneys or at inflammatory sites by immune cells such as DCs and macrophages, resulting in the fully-active metabolite 1,25(OH)2D3 (12, 14). In target cells, 1,25(OH)2D3 binds to the VDR, thereafter forming a complex with the RXR-γ (15, 16). The 1,25(OH)2D3-VDR-RXR-γ complex binds certain DNA sequences (VDREs), thereby modulating gene transcription (15–17). 1,25(OH)2D3 increases the production of antimicrobial peptides from macrophages, while in the DC line it inhibits (I) monocyte differentiation into DCs, (II) DC maturation, (III) production of pro-inflammatory cytokine IL-12, (IV) MHC class II expression, (V) and antigen presentation (46, 47, 52–55). DCs are induced to undergo apoptosis (55). 1,25(OH)2D3 increases (I) T cell apoptosis, (II) anti-inflammatory cytokine production, (III) Treg cell differentiation, and (IV) the proportion of central memory CD4 + T cells (55–58, 67, 77, 78). In addition, it decreases (I) pro-inflammatory cytokine production, (II) T cell proliferation, (III) Th1, Th2, and Th17 cell differentiation, (IV) the proportion of effector memory CD4 + T cells, and (V) T cell trafficking into the CNS (55–57, 67, 78, 88). In B cells, 1,25(OH)2D3 increases apoptosis and reduces proliferation, differentiation and antibody production (44, 59–61). 1,25(OH)2D3 increases regulation of calcium uptake in neurons and phagocytic activity in microglia but reduces iNOS expression in microglia (97, 98). Lastly, 1,25(OH)2D3 stimulates oligodendrocyte maturation and astrocyte activation (99). Abbreviations: CYP2R1, vitamin D3 25-hydroxylase; CYP27B1, 25(OH)D3-1α-hydroxylase; DC, dendritic cell; iNOS, inducible nitric acid synthase; MHC, major histocompatibility complex; RXR, retinoid x receptor; UVB, ultraviolet B; VDR, vitamin D receptor; VDREs, vitamin D response elements; 1,25(OH)2D3, 1,25-dihydroxyvitamin D3; 25(OH)D3, 25-hydroxyvitamin D3. This figure was created using Servier Medical Art templates licensed under a Creative Commons License (https://creativecommons.org/licenses/by/3.0/).
In humans, between 80 and 90% of the body’s vitamin D supply is produced by the skin’s exposure to ultraviolet B (UVB) radiation and only 10–20% is acquired through diet (7). Fatty fish, e.g., salmon, sardines, and tuna, contain relatively high amounts of vitamin D3 (cholecalciferol), while plants provide vitamin D2 (ergocalciferol) (8, 9). In the skin, the vitamin D3 precursor 7-dehydrocholesterol converts to previtamin D3 after UVB exposure (10, 11). Previtamin D3 then isomerizes to cholecalciferol (10, 11). Ergocalciferol and cholecalciferol are physiologically inactive forms of vitamin D, which are hydroxylated in the liver to 25-hydroxyvitamin D2 [25(OH)D2] and 25-hydroxyvitamin D3 [25(OH)D3], respectively (12, 13). Because the human body’s vitamin D supply is largely provided by the skin’s production of cholecalciferol, the major circulatory form of 25(OH)D is 25(OH)D3 being produced in the liver by the key enzyme vitamin D3 25-hydroxylase (CYP2R1) (7, 13). 25(OH)D3 is then hydroxylated by the enzyme 25(OH)D3-1α-hydroxylase (CYP27B1) in the kidneys or at inflammatory sites by immune cells such as dendritic cells and macrophages, resulting in the fully-active metabolite 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] (calcitriol) (12, 14). In target cells, 1,25(OH)2D3 binds to the vitamin D receptor (VDR), thereafter forming a complex with the retinoid x receptor γ (RXR-γ) (15, 16). The 1,25(OH)2D3-VDR-RXR-γ complex then binds certain DNA sequences called vitamin D response elements (15–17). In consequence, transcription rates of genes involved in a whole array of different functions of the human body are modified, including regulation of the immune system as well as cellular proliferation and differentiation (18). The VDR genotype is associated with different autoimmune disorders such as type 1 diabetes mellitus, Grave’s disease, Addison’s disease, and rheumatoid arthritis (19–23). 1,25(OH)2D3 is inactivated via a third hydroxylation by the enzyme CYP24A1 to 1,24,25(OH)3D3, which is present in target cells expressing the VDR (12, 24). The VDR is present in many human tissues explaining the various effects of 1,25(OH)2D3 on the human body, yet it is predominantly expressed in the intestines, the pancreas, the kidneys, and osteoblasts (25). In addition to 1,25(OH)2D3, also 25(OH)D3 binds to the VDR (26). However, 1,25(OH)2D3 has a higher affinity to the VDR and is, therefore, the more active metabolite (26).
1,25(OH)2D has a relatively short half-life (4 h) compared to 25(OH)D (2–3 weeks) (27, 28). Due to its relatively stable properties, serum 25(OH)D concentration serves as the common surrogate of the body’s vitamin D status (29). In the general population, the Endocrine Society considers serum 25(OH)D concentrations <20 ng/mL (50 nmol/L) to signal vitamin D deficiency, concentrations between ≥20 ng/mL and <30 ng/mL (75 nmol/L) to be insufficient, ≥30 ng/mL to be sufficient, and concentrations ≤100 ng/mL (250 nmol/L) to be safe (conversion factor: 1 ng/mL = 2.5 nmol/L) (30–32). However, recommendations on optimal serum 25(OH)D concentrations differ between medical societies (31, 33, 34). Since 40% of adults have 25(OH)D concentrations of <20 ng/mL (50 nmol/L), it is evident that the “western” lifestyle and diet are not sufficient for an adequate vitamin D supply (35). In vitamin D deficient adults, the Endocrine Society suggests to supplement with 6,000 IU/d for 8 weeks and afterward to maintain serum 25(OH)D concentrations >30 ng/mL by prescribing 1,500 – 2,000 IU/d of cholecalciferol or ergocalciferol, which is the required maintenance dose of desirable 25(OH)D concentrations (>30 ng/mL) (31). The upper tolerable intake limit is set at 10,000 IU/d (31). Nonetheless, in a randomized clinical trial (N = 229) comparing placebo and 14,007 IU/d of cholecalciferol over 48 weeks in MS patients receiving beta interferons, the occurrence of adverse events was similar in the cholecalciferol plus interferons group and in the placebo plus interferons group (36). However, since patient numbers are low, the occurrence of side effects caused by such a vitamin D dose cannot be ruled out (36). Vitamin D intoxication might become clinically relevant in persons using very high doses (mostly >50,000 IU/d), resulting in serum 25(OH)D concentrations ≥150 ng/mL (375 nmol/L) (37). These doses and serum concentrations can lead to hypercalcemia, hypercalciuria, and hyperphosphatemia, which can manifest as nausea and emesis, muscle weakness, polyuria, calcification of the kidneys, and in extreme cases kidney failure (37). Therefore, ultra-high-dose vitamin D regimens such as the “Coimbra-protocol” in MS with suggested doses of up to 400,000 IU/d pose a considerable safety hazard for patients (38). In this regard, Häusler et al. demonstrated in a rodent animal model of MS, experimental autoimmune encephalitis (EAE), that prolonged high-dose vitamin D supplementation can lead to disease exacerbation if serum 25(OH)D concentrations >80 ng/mL (200 nmol/L) were reached (39). However, disease exacerbation seemed to be mediated primarily by vitamin D induced hypercalcemia rather than 1,25(OH)2D3 itself because hypercalcemia induced the activation of T cells leading to the migration of activated myeloid, Th1, and Th17 cells into the central nervous system (CNS) (39). Similar 25(OH)D concentrations in humans (>64 ng/mL, or 160 nmol/L) lead only in approximately 10% of patients to hypercalcemia (40). Therefore, the translational significance of autoimmune disease exacerbation through high-dose vitamin D supplementation remains unclear.
Not only MS but also several other autoimmune disorders are associated with vitamin D deficiency (1, 41–43). Accordingly, studies performed in vitro and in vivo have shown that 1,25(OH)2D3 has anti-inflammatory effects by suppressing the innate as well as the adaptive immune system (44).
Regarding the innate immune system, after phagocytosis of microbes through macrophages, Toll-like receptors are activated, resulting in an up-regulation of VDR and CYP27B1 expression in macrophages and monocytes (45). In macrophages, 1,25(OH)2D3 then activates cathelicidins, which are antimicrobial peptides (46, 47). Another anti-inflammatory mechanism of action of 1,25(OH)2D3 is exerted through its various effects on glucocorticoids, including an increased stimulation of monocytes by glucocorticoids to produce mitogen-activated kinase phosphatase 1, which reduces the pro-inflammatory activity of mitogen-activated protein kinases (48, 49).
Addressing immune cells, which are part of the innate and the adaptive immune system, 1,25(OH)2D3 increases the differentiation of hematopoietic stem cells into natural killer cells and inhibits the function of the dendritic cell line (50, 51). Regarding the dendritic cell line, 1,25(OH)2D3 inhibits (I) the differentiation of monocytes into dendritic cells, (II) the maturation of dendritic cells, (III) the production of pro-inflammatory cytokine IL-12, (IV) the expression of the major histocompatibility complex class II, and (V) the presentation of antigens (52–55). Furthermore, dendritic cells are induced to undergo apoptosis (55).
Mediated by its effects on dendritic cells, 1,25(OH)2D3’s influence on the adaptive immune system has been attributed to its various effects on T cells, including the altered production of cytokines and selective induction of T cells into apoptosis (55–58). Thus far, the effect of 1,25(OH)2D3 on B cells remains inconclusive as some of the in vitro experiments could not be replicated in vivo (59). In vitro, B cell proliferation and B cell differentiation into plasma cells are inhibited and B cell apoptosis is induced, resulting in the reduced production of antibodies (44, 60, 61). Accordingly, higher serum immunoglobulin G concentrations were associated with lower serum 25(OH)D concentrations in patients with cystic fibrosis (62). In a study of 40 MS patients, however, immunoglobulin G (IgG) concentrations and 25(OH)D concentrations did not significantly correlate in CSF or serum (63). This could be another indicator that the response of MS patients to vitamin D is reduced as shown by Bhargava et al. (64).
Furthermore, in the presence of 1,25(OH)2D3, the stimulation of T cells by B cells is impaired in vitro (65). In reverse, 1,25(OH)2D3 is produced by T cells as these express CYP27B1 (66). Most experimental studies found that 1,25(OH)2D3 acts on CD4 + T cells (Th1, Th2, Th17, Treg cells) by inhibiting their proliferation and their secretion of pro-inflammatory cytokines (IL-2, IL-17, IFN-γ) and by stimulating their secretion of anti-inflammatory cytokines (IL-4, IL-10) (57, 67–76). In consequence, the cytokine profile is skewed from a Th1 (decrease of IFN-γ) to a Th2 mediated profile (increase of IL-4) (75, 76). Furthermore, the differentiation of Th1, Th2, and Th17 cells is inhibited and Treg cell differentiation is induced by 1,25(OH)2D3 (67, 77). A double-blind prospective study in MS patients confirmed the finding that vitamin D supplementation reduces IL-17 production by CD4 + T cells (78). Additionally, it demonstrated an increased proportion of central memory CD4 + T cells and naive CD4 + T cells but a decrease in the proportion of effector memory CD4 + T cells (78).
Neural cells express the VDR, 1,25(OH)2D3 is synthesized by neurons and microglia, and cerebrospinal fluid (CSF) 25(OH)D concentration significantly correlates with its concentration in the plasma (79–83). In contrast to a study performed by Balabanova et al. in the 1980s, a more recent (2009) study by Holmøy et al. found a substantially lower ratio of CSF to serum 25(OH)D concentration (0.57:1 vs. 0.006:1) (83). Nonetheless, both studies showed a correlation between 25(OH)D concentrations in CSF and serum (82, 83). 1,25(OH)2D3 modulates neurotrophic factors and regulates the influx of calcium into neurons through the interaction with L-type calcium channels (84–87). The ability of 1,25(OH)2D3 to suppress the progression of EAE is attributed to its modulation of T cell trafficking into the CNS, its inhibition of Th1 cells, and its stimulation of IL-10 production (79, 88–95). 1,25(OH)2D3 induces Indoleamine 2,3-dioxygenase-positive (IDO+) tolerogenic dendrocytes and Treg in the periphery and concomitantly reduces the number of autoreactive T cells in the CNS, thereby reducing the severity of guinea pig MBP73–86 EAE in lewis rats (96). Demyelination is reduced via 1,25(OH)2D3’s activation of microglia resulting in the clearance of myelin debris, phagocytosis of pathological proteins such as amyloid-β peptides, and the reduced expression of inducible nitric acid synthase, which is a pro-inflammatory enzyme (97, 98). Lastly, 1,25(OH)2D3 might induce remyelination by stimulating the maturation of oligodendrocytes and the activation of astrocytes in female C57Bl/6 mice demyelinated with cuprizone (99).
An important clinical association between MS and vitamin D is that populations located farther from the equator and, therefore, receiving less exposure to UVB radiation face more frequently vitamin D deficiency and simultaneously a higher risk of MS (100, 101). In observational studies, the distinction of the effects of UVB radiation and the effects of vitamin D on MS risk is only insufficiently adjusted for as also UVB radiation is able to suppress the development of MOG35–55 EAE (102). However, since Ramagopalan et al. found the MS susceptibility gene HLA-DRB1∗1501 to be regulated by a vitamin D dependent promotor, a clinically relevant UVB independent effect of vitamin D on MS risk appears feasible (54). To attempt to answer the question whether low 25(OH)D causes MS or MS causes low 25(OH)D, different research groups performed Mendelian randomization studies (103–105). Concordantly, these studies found a higher likelihood of developing MS if patients’ genes predetermined them to have lower 25(OH)D concentrations leading to the conclusion that 25(OH)D concentrations indeed influence MS risk (103–105). A Scandinavian study reported an almost two-fold risk of developing MS in the offspring of mothers with 25(OH)D concentrations <12.02 ng/mL (30.05 nmol/L) during early pregnancy (106). Accordingly, neonates with serum 25(OH)D concentrations in the bottom quintile (<8.28 ng/mL, or 20.7 nmol/L) had the highest likelihood of developing MS and neonates in the upper quintile (≥19.56 ng/mL, or 48.9 nmol/L) the lowest likelihood (107). In the 24-month period prior to the development of clinically isolated syndrome (CIS), our research group demonstrated that patients not only showed significantly lower 25(OH)D concentrations in comparison to healthy controls but that they also showed a gradual decrease in 25(OH)D concentrations as the incident of the first clinical manifestation of MS approached (108). 1,25(OH)2D3 and the vitamin D receptor were also shown to interact with Epstein-Barr virus (EBV) nuclear antigens (EBNA), which are thought of as key contributors to MS pathogenesis (3, 109–111). It is hypothesized that hypovitaminosis D increases the autoimmune effects of EBV infection, thereby increasing the risk of developing MS because of the following reasons (111). First, in young MS patients, antibody reactivity against EBNA-1 increases with lower 25(OH)D levels (109). Second, anti-EBNA 1 protein and fragment antibody concentrations decrease after vitamin D supplementation in comparison to placebo (112, 113). Third, EBNA 2 and the VDR have common DNA binding sites associated with MS (114). Lastly, the activation of VDR target genes is blocked by EBNA 3 binding to the VDR (115).
Not only is MS risk associated with low 25(OH)D concentration but also certain parameters of MS disease activity (116–121). In a post hoc analysis of patients with CIS included in a randomized placebo-controlled clinical trial originally designed to investigate the effects of early versus delayed treatment with interferon beta-1b (BENEFIT), patients with a 20 ng/mL (50 nmol/L) higher serum 25(OH)D concentration had subsequently a 57% lower relapse rate and a 57% lower rate of new active lesions (116). Conflictingly, in an analysis of MS patients, included in a randomized placebo-controlled clinical trial to investigate two different doses of interferon beta-1b (BEYOND), serum 25(OH)D concentrations were not significantly associated with subsequent relapse rates (119). Regarding CNS lesions, however, the analysis of BEYOND patients showed that a 20 ng/mL (50 nmol/L) higher serum 25(OH)D concentration was associated with a 31% lower risk of new lesions (119). Next to relapse rate, conflicting evidence also exists regarding disability progression as the post hoc analysis of BENEFIT reported an inverse correlation between 25(OH)D concentration and subsequent disability progression, yet the analysis of BEYOND patients could not confirm this finding (116, 119). In populations with MS, initial findings of an inverse correlation between serum 25(OH)D concentration and depression as well as fatigue were non-significant after adjusting for UVB exposure as a confounder (122). The authors concluded that sunlight exposure was more robustly associated with depression scores and fatigue than 25(OH)D concentrations (122).
In a large observational study including >187,000 women, 173 of whom developed MS, Munger et al. demonstrated that women using vitamin D supplements had a 40% reduced risk of developing MS in the follow-up period (1980–2001) compared to non-users (123). However, because of the observational character, it is possible that relevant confounding factors existed even though the authors adjusted for known MS risk factors such as age, smoking, and latitude of residence at birth (123).
The largest study investigating the effects of vitamin D supplementation on MS disease activity is the SOLAR trial, which was a randomized, double-blind, placebo-controlled trial investigating supplementation with 14,007 IU/d of cholecalciferol for 48 weeks in 229 relapsing-remitting MS patients treated with interferon beta-1a (36). The number of new gadolinium-enhancing or new/enlarging T2 lesions was significantly reduced by 32% in patients supplemented with cholecalciferol in comparison to supplementation with placebo (p = 0.0045), yet no significant results were reported regarding annualized relapse rate (ARR) and disability progression (36). Nonetheless, a non-significant trend toward a lower ARR in patients treated with cholecalciferol became evident (0.28 vs. 0.41, p = 0.17) (36). Significant results in radiological but non-significant results in clinical disease parameters could be due to higher incidence rates of new/enlarging lesions than relapses or sustained disability progression (124). However, the main reason for missed significance may have been the violation of the power calculation (36). Due to difficulties in patient recruitment, the study duration was shortened from 96 weeks to 48 weeks and the randomized total patient number was reduced by a third from 348 to 232 (36). Furthermore, the primary endpoint was changed from the mean number of active T2 lesions and the patient proportion with no relapses to the patient proportion with no evidence of disease activity (NEDA) 3 defined as no relapses, no disability progression, and no new gadolinium-enhancing or new/enlarging T2 lesions, which was nonetheless missed (36). Camu et al. also investigated in a randomized clinical trial the effects of vitamin D supplementation in patients receiving interferons (125). In comparison to the SOLAR trial, this study was performed over a longer duration (96 weeks) but with fewer patients (N = 129) and an overall smaller dose of cholecalciferol (equivalent of 7,143 IU/d) (125). In the intention to treat population (N = 129), vitamin D supplementation did not show a significant reduction in the ARR [rate ratio (rR) = 0.799, p = 0.38], which was the primary outcome measure. In patients who completed the study (N = 90), however, vitamin D supplementation led after 96 weeks to a significant reduction in the ARR (rR = 0.395, p = 0.01), in new T1 lesions (rR = 0.494, p = 0.03), and in disability progression as measured by the EDSS (−0.06 vs. 0.32, respectively, p = 0.03) in comparison to placebo (125). Since in both studies, SOLAR and CHOLINE, cholecalciferol was used as an add-on to interferon-beta, it is important to mention that in patients being treated with interferon-beta who received repeated MRI scans and measurements of serum 25(OH)D concentration, an inverse correlation between 25(OH)D and MRI activity was found before but not during treatment with interferon-beta (126). Other vitamin D supplementation studies investigating relapse risk, CNS lesions, and/or disability progression were either substantially smaller, or of shorter duration, or were not prospectively randomized and are therefore only listed in the reference section [see (127–130)]. Regarding the effects of vitamin D supplementation on fatigue and depression scores, Rolf et al. showed in a prospectively studied cohort of 40 MS patients that patients receiving cholecalciferol for 48 weeks did not improve significantly in fatigue or depression scores in comparison to placebo (131). In contrast, Achiron et al. found in a larger (N = 158), yet shorter (24 weeks of supplementation) prospective study a significant reduction of fatigue scores through the supplementation with alfacalcidol (132). Therefore, the effect of vitamin D supplementation on fatigue remains uncertain. To conclude, the vitamin D supplementation studies published so far were mostly insufficiently powered to detect significant differences in clinical disease parameters, especially since MS patients show a reduced serological and metabolic response to vitamin D supplementation and, therefore, may need higher doses to demonstrate clinically relevant effects (64, 133). Ongoing trials such as VIDAMS, PrevANZ, and D-Lay-MS, might shed new light on the efficacy of vitamin D supplementation in MS and CIS (134–137).
The immunological mechanism by which 1,25(OH)2D3 increases the effect of corticosteroids have led us to investigate whether 1,25(OH)2D3 increases the efficacy of methylprednisolone pulse therapy for the treatment of MS relapses (138). We demonstrated in human and murine CD3 + T cells that 1,25(OH)2D3 increases glucocorticoid receptor protein expression and consequently upregulates methylprednisolone induced apoptosis (138). In vivo, the combination therapy led to a significant decrease in active MOG35–55 EAE disease severity (138). Effects appear to be mediated via the glucocorticoid receptor because no difference was observed in animals specifically lacking glucocorticoid receptor expression in CD3 + T cells (138). Furthermore, in two different independent cohorts, patients with a steroid-resistant MS relapse had significantly lower 25(OH)D concentrations (138). The cellular pathway most plausible to explain this finding is the inhibition of mTOR because in mTORc1-deficient animals no synergistic effects were found, whereas treatment of wild type animals with mTOR inhibitors led to synergistic glucocorticoid effects. These findings may lead to a prospective clinical evaluation of 1,25(OH)2D3 in MS relapses because so far sufficient evidence for its use in this specific setting is lacking.
Although certain parameters of radiological MS disease activity were significantly reduced by vitamin D supplementation as shown in randomized double-blind placebo-controlled trials, the evidence accumulated so far is not sufficient to allow drawing a definite conclusion on the effects of vitamin D supplementation on clinical parameters. The VIDAMS trial may provide further insights as it aims to investigate vitamin D supplementation in the yet largest (N = 172) prospectively randomized MS patient population followed up over ≥96 weeks (134). In addition, basic scientific research may increase our knowledge about the effects of vitamin D on the immune system. Considering the interactions between vitamin D and glucocorticosteroids, this knowledge may provide us with new therapeutic strategies for vitamin D administration in MS.
AM and RH contributed to the research of the literature and the writing and revision of the manuscript. MB and AC contributed to the writing and revision of the manuscript.
AC has received personal compensation for activities with Bayer, Biogen, Genzyme, Merck, Novartis, Roche, and Teva; received research support from the Swiss National Funds (SNF, No. 310030_172952), Genzyme, and UCB; and serves in the Editorial Board for Clinical and Translational Neuroscience and the Journal of International medical research. RH received research and travel grants from Novartis and Biogen Idec and received speaker’s honoraria from Biogen, Novartis, Merk, and Almirall.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. (2016) 13:26–36. doi: 10.1038/nrneurol.2016.187
2. Antel J, Antel S, Caramanos Z, Arnold DL, Kuhlmann T. Primary progressive multiple sclerosis: part of the MS disease spectrum or separate disease entity? Acta Neuropathol. (2012) 123:627–38. doi: 10.1007/s00401-012-0953-0
3. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol. (2007) 61:288–99. doi: 10.1002/ana.21117
4. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part II: noninfectious factors. Ann Neurol. (2007) 61:504–13. doi: 10.1002/ana.21141
5. Healy BC, Ali EN, Guttmann CR, Chitnis T, Glanz BI, Buckle G, et al. Smoking and disease progression in multiple sclerosis. Arch Neurol. (2009) 66:858–64. doi: 10.1001/archneurol.2009.122
6. Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. J Am Med Assoc. (2006) 296:2832–8. doi: 10.1001/jama.296.23.2832
7. Pilz S, Tomaschitz A, Ritz E, Pieber TR. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol. (2009) 6:621–30. doi: 10.1038/nrcardio.2009.135
8. Wacker M, Holick MF. Vitamin D-effects on skeletal and extraskeletal health and the need for supplementation. Nutrients. (2013) 5:111–48. doi: 10.3390/nu5010111
9. Smolders J, Torkildsen Ø, Camu W, Holmøy T. An update on vitamin D and disease activity in multiple sclerosis. CNS Drugs. (2019) 33:1187–99. doi: 10.1007/s40263-019-00674-8
10. Holick MF, MacLaughlin JA, Clark MB, Holick SA, Potts JT Jr, Anderson RR, et al. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science. (1980) 210:203–5. doi: 10.1126/science.6251551
11. Sassi F, Tamone C, D’amelio P. Vitamin D: nutrient, hormone, and immunomodulator. Nutrients. (2018) 10:1656. doi: 10.3390/nu10111656
12. Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. (2015) 96:365–408. doi: 10.1152/physrev.00014.2015
13. Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci USA. (2004) 101:7711–5. doi: 10.1073/pnas.0402490101
14. Prietl B, Treiber G, Pieber TR, Amrein K. Vitamin D and immune function. Nutrients. (2013) 5:2502–21. doi: 10.3390/nu5072502
15. McCarty DE, Chesson AL, Jain SK, Marino AA. The link between vitamin D metabolism and sleep medicine. Sleep Med Rev. (2014) 18:311–9. doi: 10.1016/j.smrv.2013.07.001
16. Rowe A. Retinoic X receptors. Int J Biochem Cell Biol. (1997) 29:275–8. doi: 10.1016/S1357-2725(96)00101-X
17. Kang SW, Kim SH, Lee N, Lee WW, Hwang KA, Shin MS, et al. 1,25-Dihyroxyvitamin D 3 promotes FOXP3 expression via binding to vitamin D response elements in its conserved noncoding sequence region. J Immunol. (2012) 188:5276–82. doi: 10.4049/jimmunol.1101211
18. Sintzel MB, Rametta M, Reder AT. Vitamin D and multiple sclerosis: a comprehensive review. Neurol Ther. (2018) 7:59–85. doi: 10.1007/s40120-017-0086-4
19. Gallone G, Haerty W, Disanto G, Ramagopalan SV, Ponting CP, Berlanga-Taylor AJ. Identification of genetic variants affecting vitamin D receptor binding and associations with autoimmune disease. Hum Mol Genet. (2017) 26:2164–76. doi: 10.1093/hmg/ddx092
20. Pani MA, Knapp M, Donner H, Braun J, Baur MP, Usadel KH, et al. Vitamin D receptor allele combinations influence genetic susceptibility to 1 diabetes in Germans. Diabetes. (2000) 49:504–7. doi: 10.2337/diabetes.49.3.504
21. Pani MA, Seissler J, Badenhoop K. Vitamin D receptor genotype is associated with Addison’s disease. Eur J Endocrinol. (2002) 147:635–40. doi: 10.1530/eje.0.1470635
22. Ban Y, Taniyama M, Ban Y. Vitamin D receptor gene polymorphism is associated with Graves’ disease in the Japanese population1. J Clin Endocrinol Metab. (2000) 85:4639–43. doi: 10.1210/jcem.85.12.7038
23. Rass P, Pákozdi A, Lakatos P, Zilahi E, Sipka S, Szegedi G, et al. Vitamin D receptor gene polymorphism in rheumatoid arthritis and associated osteoporosis. Rheumatol Int. (2006) 26:964–71. doi: 10.1007/s00296-006-0106-7
24. Jones G, Prosser DE, Kaufmann M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): its important role in the degradation of vitamin D. Arch Biochem Biophys. (2012) 523:9–18. doi: 10.1016/j.abb.2011.11.003
25. Wang Y, Zhu J, DeLuca HF. Where is the vitamin D receptor? Arch Biochem Biophys. (2012) 523:123–33. doi: 10.1016/j.abb.2012.04.001
26. Marshall TG. Are statins analogues of vitamin D? Lancet. (2006) 368:1234. doi: 10.1016/S0140-6736(06)69509-3
27. Jones KS, Assar S, Vanderschueren D, Bouillon R, Prentice A, Schoenmakers I. Predictors of 25(OH)D half-life and plasma 25(OH)D concentration in the Gambia and the UK. Osteoporos Int. (2015) 26:1137–46. doi: 10.1007/s00198-014-2905-0
28. Zerwekh JE. Blood biomarkers of vitamin D status. Am J Clin Nutr. (2008) 87:10875S–91S. doi: 10.1093/ajcn/87.4.1087s
30. Yin L, Grandi N, Raum E, Haug U, Arndt V, Brenner H. Meta-analysis: serum vitamin D and breast cancer risk. Eur J Cancer. (2010) 46:2196–205. doi: 10.1016/j.ejca.2010.03.037
31. Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2011) 96:1911–30. doi: 10.1210/jc.2011-0385
32. Holick MF. The vitamin D deficiency pandemic: approaches for diagnosis, treatment and prevention. Rev Endocr Metab Disord. (2017) 18:153–65. doi: 10.1007/s11154-017-9424-1
33. Aspray TJ, Bowring C, Fraser W, Gittoes N, Javaid MK, Macdonald H, et al. National osteoporosis society vitamin D guideline summary. Age Ageing. (2014) 43:592–5. doi: 10.1093/ageing/afu093
34. Rosen CJ, Abrams SA, Aloia JF, Brannon PM, Clinton SK, Durazo-Arvizu RA, et al. IOM committee members respond to endocrine society vitamin D guideline. J Clin Endocrinol Metab. (2012) 97:1146–52. doi: 10.1210/jc.2011-2218
35. Cashman KD, Dowling KG, Škrabáková Z, Gonzalez-Gross M, Valtueña J, De Henauw S, et al. Vitamin D deficiency in Europe: pandemic? Am J Clin Nutr. (2016) 103:1033–44. doi: 10.3945/ajcn.115.120873
36. Hupperts R, Smolders J, Vieth R, Holmøy T, Marhardt K, Schluep M, et al. Randomized trial of daily high-dose vitamin D 3 in patients with RRMS receiving subcutaneous interferon β-1a. Neurology. (2019) 93:e1906–16.
37. Galior K, Grebe S, Singh R. Development of vitamin D toxicity from overcorrection of vitamin D deficiency: a review of case reports. Nutrients. (2018) 10:953. doi: 10.3390/nu10080953
38. Breuer J, Loser K, Mykicki N, Wiendl H, Schwab N. Does the environment influence multiple sclerosis pathogenesis via UVB light and/or induction of vitamin D? J Neuroimmunol. (2019) 329:1–8. doi: 10.1016/j.jneuroim.2018.05.006
39. Häusler D, Torke S, Peelen E, Bertsch T, Djukic M, Nau R, et al. High dose vitamin D exacerbates central nervous system autoimmunity by raising T-cell excitatory calcium. Brain. (2019) 142:2737–55. doi: 10.1093/brain/awz190
40. Pérez-Barrios C, Hernández-Álvarez E, Blanco-Navarro I, Pérez-Sacristán B, Granado-Lorencio F. Prevalence of hypercalcemia related to hypervitaminosis D in clinical practice. Clin Nutr. (2016) 35:1354–8. doi: 10.1016/j.clnu.2016.02.017
41. Ananthakrishnan AN, Khalili H, Higuchi LM, Bao Y, Korzenik JR, Giovannucci EL, et al. Higher predicted vitamin D status is associated with reduced risk of crohn’s disease. Gastroenterology. (2012) 142:482–9. doi: 10.1053/j.gastro.2011.11.040
42. Hyppönen E, Läärä E, Reunanen A, Järvelin MR, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. (2001) 358:1500–3. doi: 10.1016/S0140-6736(01)06580-1
43. Wang J, Lv S, Chen G, Gao C, He J, Zhong H, et al. Meta-analysis of the association between vitamin D and autoimmune thyroid disease. Nutrients. (2015) 7:2485–98. doi: 10.3390/nu7042485
44. Colotta F, Jansson B, Bonelli F. Modulation of inflammatory and immune responses by vitamin D. J Autoimmun. (2017) 85:78–97. doi: 10.1016/j.jaut.2017.07.007
45. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. (2006) 311:1770–3. doi: 10.1126/science.1123933
46. Rode AKO, Kongsbak M, Hansen MM, Lopez DV, Levring TB, Woetmann A, et al. Vitamin D counteracts Mycobacterium tuberculosis-induced cathelicidin downregulation in dendritic cells and allows Th1 differentiation and IFNγ secretion. Front Immunol. (2017) 8:656. doi: 10.3389/fimmu.2017.00656
47. Gauzzi MC. Vitamin D-binding protein and multiple sclerosis: evidence, controversies, and needs. Mult Scler J. (2018) 24:1526–35. doi: 10.1177/1352458518792433
48. Czaja AJ, Montano-Loza AJ. Evolving role of vitamin D in immune-mediated disease and its implications in autoimmune hepatitis. Dig Dis Sci. (2019) 64:324–44. doi: 10.1007/s10620-018-5351-6
49. Zhang Y, Leung DYM, Goleva E. Vitamin D enhances glucocorticoid action in human monocytes: involvement of granulocyte-macrophage colony-stimulating factor and mediator complex subunit 14. J Biol Chem. (2013) 288:14544–53. doi: 10.1074/jbc.M112.427054
50. Lee KN, Kang HS, Jeon JH, Kim EM, Yoon SR, Song H, et al. VDUP1 is required for the development of natural killer cells. Immunity. (2005) 22:195–208. doi: 10.1016/j.immuni.2004.12.012
51. Barragan M, Good M, Kolls J. Regulation of dendritic cell function by vitamin D. Nutrients. (2015) 7:8127–51. doi: 10.3390/nu7095383
52. Piemonti L, Monti P, Sironi M, Fraticelli P, Leone BE, Dal Cin E, et al. Vitamin D 3 affects differentiation, maturation, and function of human monocyte-derived dendritic cells. J Immunol. (2000) 164:4443–51. doi: 10.4049/jimmunol.164.9.4443
53. D’Ambrosio D, Cippitelli M, Cocciolo MG, Mazzeo D, Di Lucia P, Lang R, et al. Inhibition of IL-12 production by 1,25-dihydroxyvitamin D3. Involvement of NF-κB downregulation in transcriptional repression of the p40 gene. J Clin Invest. (1998) 101:252–62. doi: 10.1172/JCI1050
54. Ramagopalan SV, Maugeri NJ, Handunnetthi L, Lincoln MR, Orton SM, Dyment DA, et al. Expression of the multiple sclerosis-associated MHC class II allele HLA-DRB1∗1501 is regulated by vitamin D. PLoS Genet. (2009) 5:e1000369. doi: 10.1371/journal.pgen.1000369
55. Penna G, Adorini L. 1α,25-Dihydroxyvitamin D 3 Inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol. (2000) 164:2405–11. doi: 10.4049/jimmunol.164.5.2405
56. Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, et al. DCs metabolize sunlight-induced vitamin D3 to “program” T cell attraction to the epidermal chemokine CCL27. Nat Immunol. (2007) 8:285–93. doi: 10.1038/ni1433
57. Xie Z, Chen J, Zheng C, Wu J, Cheng Y, Zhu S, et al. 1,25-dihydroxyvitamin D3-induced dendritic cells suppress experimental autoimmune encephalomyelitis by increasing proportions of the regulatory lymphocytes and reducing T helper type 1 and type 17 cells. Immunology. (2017) 152:414–24. doi: 10.1111/imm.12776
58. Van Halteren AGS, Tysma OM, Van Etten E, Mathieu C, Roep BO. 1α,25-Dihydroxyvitamin D3 or analogue treated dendritic cells modulate human autoreactive T cells via the selective induction of apoptosis. J Autoimmun. (2004) 23:233–9. doi: 10.1016/j.jaut.2004.06.004
59. Rolf L, Muris A-H, Hupperts R, Damoiseaux J. Illuminating vitamin D effects on B cells - the multiple sclerosis perspective. Immunology. (2016) 147:275–84. doi: 10.1111/imm.12572
60. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
61. Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory effects of 1,25-Dihydroxyvitamin D 3 on human B Cell differentiation. J Immunol. (2007) 179:1634–47. doi: 10.4049/jimmunol.179.3.1634
62. Pincikova T, Nilsson K, Moen IE, Karpati F, Fluge G, Hollsing A, et al. Inverse relation between vitamin D and serum total immunoglobulin G in the Scandinavian cystic fibrosis nutritional study. Eur J Clin Nutr. (2011) 65:102–9. doi: 10.1038/ejcn.2010.194
63. Holmøy T, Lossius A, Gundersen TE, Moen SM, Castellazzi M, Fainardi E, et al. Intrathecal levels of vitamin D and IgG in multiple sclerosis. Acta Neurol Scand. (2012) 125:e28–31. doi: 10.1111/j.1600-0404.2011.01577.x
64. Bhargava P, Steele SU, Waubant E, Revirajan NR, Marcus J, Dembele M, et al. Multiple sclerosis patients have a diminished serologic response to Vitamin D supplementation compared to healthy controls. Mult Scler J. (2016) 22:753–60. doi: 10.1177/1352458515600248
65. Drozdenko G, Scheel T, Heine G, Baumgrass R, Worm M. Impaired T cell activation and cytokine production by calcitriol-primed human B cells. Clin Exp Immunol. (2014) 178:364–72. doi: 10.1111/cei.12406
66. Kongsbak M, von Essen MR, Levring TB, Schjerling P, Woetmann A, Ødum N, et al. Vitamin D-binding protein controls T cell responses to vitamin D. BMC Immunol. (2014) 15:35. doi: 10.1186/s12865-014-0035-2
67. Pichler J, Gerstmayr M, Szépfalusi Z, Urbanek R, Peterlik M, Willheim M. 1α,25(OH)2D3 inhibits not only Th1 but also Th2 differentiation in human cord blood T cells. Pediatr Res. (2002) 52:12–8. doi: 10.1203/00006450-200207000-00005
68. Chang SH, Chung Y, Dong C. Vitamin D suppresses Th17 cytokine production by inducing C/EBP Homologous protein (CHOP) expression. J Biol Chem. (2010) 285:38751–5. doi: 10.1074/jbc.C110.185777
69. Ikeda U, Wakita D, Ohkuri T, Chamoto K, Kitamura H, Iwakura Y, et al. 1α,25-Dihydroxyvitamin D3 and all-trans retinoic acid synergistically inhibit the differentiation and expansion of Th17 cells. Immunol Lett. (2010) 134:7–16. doi: 10.1016/j.imlet.2010.07.002
70. Jeffery LE, Burke F, Mura M, Zheng Y, Qureshi OS, Hewison M, et al. 1,25-Dihydroxyvitamin D 3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J Immunol. (2009) 183:5458–67. doi: 10.4049/jimmunol.0803217
71. Cantorna M, Snyder L, Lin Y-D, Yang L. Vitamin D and 1,25(OH)2D regulation of T cells. Nutrients. (2015) 7:3011–21. doi: 10.3390/nu7043011
72. Peelen E, Knippenberg S, Muris AH, Thewissen M, Smolders J, Tervaert JW, et al. Effects of vitamin D on the peripheral adaptive immune system: a review. Autoimmun Rev. (2011) 10:733–43. doi: 10.1016/j.autrev.2011.05.002
73. Gregori S, Giarratana N, Smiroldo S, Uskokovic M, Adorini LA. 1α,25-dihydroxyvitamin D3 analog enhances regulatory T-cells and arrests autoimmune diabetes in NOD mice. Diabetes. (2002) 51:1367–74. doi: 10.2337/diabetes.51.5.1367
74. Gratz IK, Campbell DJ. Organ-specific and memory Treg cells: specificity, development, function, and maintenance. Front Immunol. (2014) 5:333. doi: 10.3389/fimmu.2014.00333
75. Mahon BD, Gordon SA, Cruz J, Cosman F, Cantorna MT. Cytokine profile in patients with multiple sclerosis following vitamin D supplementation. J Neuroimmunol. (2003) 134:128–32. doi: 10.1016/S0165-5728(02)00396-X
76. Sloka S, Silva C, Wang J, Yong VW. Predominance of Th2 polarization by Vitamin D through a STAT6-dependent mechanism. J Neuroinflammation. (2011) 8:56. doi: 10.1186/1742-2094-8-56
77. Zhou Q, Qin S, Zhang J, Zhon L, Pen Z, Xing T. 1,25(OH)2D3 induces regulatory T cell differentiation by influencing the VDR/PLC-γ1/TGF-β1/pathway. Mol Immunol. (2017) 91:156–64. doi: 10.1016/j.molimm.2017.09.006
78. Sotirchos ES, Bhargava P, Eckstein C, Van Haren K, Baynes M, Ntranos A, et al. Safety and immunologic effects of high-vs low-dose cholecalciferol in multiple sclerosis. Neurology. (2016) 86:382–90. doi: 10.1212/WNL.0000000000002316
79. Fernandes de Abreu DA, Eyles D, Féron F. Vitamin D, a neuro-immunomodulator: implications for neurodegenerative and autoimmune diseases. Psychoneuroendocrinology. (2009) 34(Suppl. 1):S265–77. doi: 10.1016/j.psyneuen.2009.05.023
80. Baas D, Prüfer K, Ittel ME, Kuchler-Bopp S, Labourdette G, Sarliève LL, et al. Rat oligodendrocytes express the vitamin D3 receptor and respond to 1,25-dihydroxyvitamin D3. Glia. (2000) 31:59–68.
81. Eyles DW, Smith S, Kinobe R, Hewison M, McGrath JJ. Distribution of the Vitamin D receptor and 1α-hydroxylase in human brain. J Chem Neuroanat. (2005) 29:21–30. doi: 10.1016/j.jchemneu.2004.08.006
82. Balabanova S, Richter HP, Antoniadis G, Homoki J, Kremmer N, Hanle J, et al. 25-hydroxyvitamin D, 24, 25-dihydroxyvitamin D and 1,25-dihydroxyvitamin D in human cerebrospinal fluid. Klin Wochenschr. (1984) 62:1086–90. doi: 10.1007/BF01711378
83. Holmøy T, Moen SM, Gundersen TA, Holick MF, Fainardi E, Castellazzi M, et al. 25-Hydroxyvitamin D in cerebrospinal fluid during relapse and remission of multiple sclerosis. Mult Scler. (2009) 15:1280–5. doi: 10.1177/1352458509107008
84. Neveu I, Naveilhan P, Baudet C, Brachet P, Metsis M. 1,25-Dihydroxyvitamin D3regulates NT-3, NT-4 but not BDNF mRNA in astrocytes. Neuroreport. (1994) 6:124–6. doi: 10.1097/00001756-199412300-00032
85. Cornet A, Baudet C, Neveu I, Baron-Van Evercooren A, Brachet P, Naveilhan P. 1,25-Dihydroxyvitamin D3 regulates the expression of VDR and NGF gene in Schwann cells in vitro. J Neurosci Res. (1998) 53:742–6.
86. Zanatta L, Goulart PB, Gonçalves R, Pierozan P, Winkelmann-Duarte EC, Woehl VM, et al. 1α,25-Dihydroxyvitamin D3 mechanism of action: modulation of L-type calcium channels leading to calcium uptake and intermediate filament phosphorylation in cerebral cortex of young rats. Biochim Biophys Acta - Mol Cell Res. (2012) 1823:1708–19. doi: 10.1016/j.bbamcr.2012.06.023
87. Gezen-Ak D, Dursun E, Yilmazer S. Vitamin D inquiry in hippocampal neurons: consequences of vitamin D-VDR pathway disruption on calcium channel and the vitamin D requirement. Neurol Sci. (2013) 34:1453–8. doi: 10.1007/s10072-012-1268-6
88. Grishkan IV, Fairchild AN, Calabresi PA, Gocke AR. 1,25-Dihydroxyvitamin D3 selectively and reversibly impairs T helper-cell CNS localization. Proc Natl Acad Sci USA. (2013) 110:21101–6. doi: 10.1073/pnas.1306072110
89. Spach KM, Nashold FE, Dittel BN, Hayes CE. IL-10 signaling is essential for 1,25-Dihydroxyvitamin D 3 -mediated inhibition of experimental autoimmune encephalomyelitis. J Immunol. (2006) 177:6030–7. doi: 10.4049/jimmunol.177.9.6030
90. Waddell A, Zhao J, Cantorna MT. NKT cells can help mediate the protective effects of 1,25-dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis in mice. Int Immunol. (2015) 27:237–44. doi: 10.1093/intimm/dxu147
91. Pikor NB, Astarita JL, Summers-Deluca L, Galicia G, Qu J, Ward LA, et al. Integration of Th17- and lymphotoxin-derived signals initiates meningeal-resident stromal cell remodeling to propagate neuroinflammation. Immunity. (2015) 43:1160–73. doi: 10.1016/j.immuni.2015.11.010
92. Shirazi HA, Rasouli J, Ciric B, Wei D, Rostami A, Zhang GX. 1,25-Dihydroxyvitamin D3 suppressed experimental autoimmune encephalomyelitis through both immunomodulation and oligodendrocyte maturation. Exp Mol Pathol. (2017) 102:515–21. doi: 10.1016/j.yexmp.2017.05.015
93. Matías-Guíu J, Oreja-Guevara C, Matias-Guiu JA, Gomez-Pinedo U. Vitamin D and remyelination in multiple sclerosis. Neurologia. (2018) 33:177–86. doi: 10.1016/j.nrl.2016.05.001
94. Cantorna MT, Hayes CE, Deluca HF. 1,25-dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci USA. (1996) 93:7861–4. doi: 10.1073/pnas.93.15.7861
95. Molina-Holgado E, Vela JM, Arévalo-Martín A, Guaza C. LPS/IFN-γ cytotoxicity in oligodendroglial cells: role of nitric oxide and protection by the anti-inflammatory cytokine IL-10. Eur J Neurosci. (2001) 13:493–502. doi: 10.1046/j.0953-816X.2000.01412.x
96. Farias AS, Spagnol GS, Bordeaux-Rego P, Oliveira CO, Fontana AG, de Paula RF, et al. Vitamin D3 induces IDO+ tolerogenic DCs and enhances treg, reducing the severity of EAE. CNS Neurosci Ther. (2013) 19:269–77. doi: 10.1111/cns.12071
97. Masoumi A, Goldenson B, Ghirmai S, Avagyan H, Zaghi J, Abel K, et al. 1α,25-dihydroxyvitamin D3 interacts with curcuminoids to stimulate amyloid-β clearance by macrophages of alzheimer’s disease patients. J Alzheimers Dis. (2009) 17:703–17. doi: 10.3233/JAD-2009-1080
98. Garcion E, Nataf S, Berod A, Darcy F, Brachet P. 1,25-Dihydroxyvitamin D3 inhibits the expression of inducible nitric oxide synthase in rat central nervous system during experimental allergic encephalomyelitis. Mol Brain Res. (1997) 45:255–67. doi: 10.1016/S0169-328X(96)00260-4
99. Nystad AE, Wergeland S, Aksnes L, Myhr KM, Bø L, Torkildsen Ø. Effect of high-dose 1.25 dihydroxyvitamin D3on remyelination in the cuprizone model. APMIS. (2014) 122:1178–86. doi: 10.1111/apm.12281
100. Kimlin MG. Geographic location and vitamin D synthesis. Mol Aspects Med. (2008) 29:453–61. doi: 10.1016/j.mam.2008.08.005
101. Orton SM, Wald L, Confavreux C, Vukusic S, Krohn JP, Ramagopalan SV, et al. Association of UV radiation with multiple sclerosis prevalence and sex ratio in France. Neurology. (2011) 76:425–31. doi: 10.1212/WNL.0b013e31820a0a9f
102. Becklund BR, Severson KS, Vang SV, DeLuca HF. UV radiation suppresses experimental autoimmune encephalomyelitis independent of vitamin D production. Proc Natl Acad Sci USA. (2010) 107:6418–23. doi: 10.1073/pnas.1001119107
103. Mokry LE, Ross S, Ahmad OS, Forgetta V, Smith GD, Goltzman D, et al. Vitamin D and risk of multiple sclerosis: a mendelian randomization study. PLoS Med. (2015) 12:e1001866. doi: 10.1371/journal.pmed.1001866
104. Rhead B, Bäärnhielm M, Gianfrancesco M, Mok A, Shao X, Quach H, et al. Mendelian randomization shows a causal effect of low Vitamin D on multiple sclerosis risk. Neurol Genet. (2016) 2:e97. doi: 10.1212/NXG.0000000000000097
105. Gianfrancesco MA, Stridh P, Rhead B, Shao X, Xu E, Graves JS, et al. Evidence for a causal relationship between low Vitamin D, high BMI, and pediatric-onset MS. Neurology. (2017) 88:1623–9. doi: 10.1212/WNL.0000000000003849
106. Munger KL, Åivo J, Hongell K, Soilu-Hänninen M, Surcel HM, Ascherio A. Vitamin D status during pregnancy and risk of multiple sclerosis in offspring of women in the Finnish Maternity Cohort. JAMA Neurol. (2016) 73:515–9. doi: 10.1001/jamaneurol.2015.4800
107. Ueda P, Rafatnia F, Bäärnhielm M, Fröbom R, Korzunowicz G, Lönnerbro R, et al. Neonatal vitamin D status and risk of multiple sclerosis. Ann Neurol. (2014) 76:338–46. doi: 10.1002/ana.24210
108. Décard BF, von Ahsen N, Grunwald T, Streit F, Stroet A, Niggemeier P, et al. Low vitamin D and elevated immunoreactivity against Epstein-Barr virus before first clinical manifestation of multiple sclerosis. J Neurol Neurosurg Psychiatry. (2012) 83:1170–3. doi: 10.1136/jnnp-2012-303068
109. Salzer J, Nyström M, Hallmans G, Stenlund H, Wadell G, Sundström P. Epstein-Barr virus antibodies and vitamin D in prospective multiple sclerosis biobank samples. Mult Scler. (2013) 19:1587–91. doi: 10.1177/1352458513483888
110. Sundström P, Nyström M, Ruuth K, Lundgren E. Antibodies to specific EBNA-1 domains and HLA DRB1∗1501 interact as risk factors for multiple sclerosis. J Neuroimmunol. (2009) 215:102–7. doi: 10.1016/j.jneuroim.2009.08.004
111. Marcucci SB, Obeidat AZ. EBNA1, EBNA2, and EBNA3 link Epstein-Barr virus and hypovitaminosis D in multiple sclerosis pathogenesis. J Neuroimmunol. (2020) 339:577116. doi: 10.1016/j.jneuroim.2019.577116
112. Rolf L, Muris AH, Mathias A, Du Pasquier R, Koneczny I, Disanto G, et al. Exploring the effect of vitamin D3 supplementation on the anti-EBV antibody response in relapsing-remitting multiple sclerosis. Mult Scler J. (2018) 24:1280–7. doi: 10.1177/1352458517722646
113. Røsjø E, Lossius A, Abdelmagid N, Lindstrøm JC, Kampman MT, Jørgensen L, et al. Effect of high-dose Vitamin D3 supplementation on antibody responses against Epstein-Barr virus in relapsing-remitting multiple sclerosis. Mult Scler. (2017) 23:395–402. doi: 10.1177/1352458516654310
114. Ricigliano VA, Handel AE, Sandve GK, Annibali V, Ristori G, Mechelli R, et al. EBNA2 Binds to genomic intervals associated with multiple sclerosis and overlaps with vitamin D receptor occupancy. PLoS One. (2015) 10:e0119605. doi: 10.1371/journal.pone.0119605
115. Yenamandra SP, Hellman U, Kempkes B, Darekar SD, Petermann S, Sculley T, et al. Epstein-Barr virus encoded EBNA-3 binds to vitamin D receptor and blocks activation of its target genes. Cell Mol Life Sci. (2010) 67:4249–56. doi: 10.1007/s00018-010-0441-4
116. Ascherio A, Munger KL, White R, Köchert K, Simon KC, Polman CH, et al. Vitamin D as an early predictor of multiple sclerosis activity and progression. JAMA Neurol. (2014) 71:306–14. doi: 10.1001/jamaneurol.2013.5993
117. Stewart N, Simpson S Jr, van der Mei I, Ponsonby AL, Blizzard L, Dwyer T, et al. Interferon-β and serum 25-hydroxyvitamin D interact to modulate relapse risk in MS. Neurology. (2012) 79:254–60. doi: 10.1212/WNL.0b013e31825fded9
118. Simpson S Jr, Taylor B, Blizzard L, Ponsonby AL, Pittas F, Tremlett H, et al. Higher 25-hydroxyvitamin D is associated with lower relapse risk in multiple sclerosis. Ann Neurol. (2010) 68:193–203. doi: 10.1002/ana.22043
119. Fitzgerald KC, Munger KL, Köchert K, Arnason BG, Comi G, Cook S, et al. Association of vitamin D levels with multiple sclerosis activity and progression in patients receiving interferon Beta-1b. JAMA Neurol. (2015) 72:1458–65. doi: 10.1001/jamaneurol.2015.2742
120. Mowry EM, Waubant E, McCulloch CE, Okuda DT, Evangelista AA, Lincoln RR, et al. Vitamin D status predicts new brain magnetic resonance imaging activity in multiple sclerosis. Ann Neurol. (2012) 72:234–40. doi: 10.1002/ana.23591
121. Smolders J, Menheere P, Kessels A, Damoiseaux J, Hupperts R. Association of vitamin D metabolite levels with relapse rate and disability in multiple sclerosis. Mult Scler. (2008) 14:1220–4. doi: 10.1177/1352458508094399
122. Knippenberg S, Damoiseaux J, Bol Y, Hupperts R, Taylor BV, Ponsonby AL, et al. Higher levels of reported sun exposure, and not vitamin D status, are associated with less depressive symptoms and fatigue in multiple sclerosis. Acta Neurol Scand. (2014) 129:123–31. doi: 10.1111/ane.12155
123. Munger KL, Zhang SM, O’Reilly E, Hernán MA, Olek MJ, Willett WC, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology. (2004) 62:60–5. doi: 10.1212/01.WNL.0000101723.79681.38
124. Dobson R, Giovannoni G. Multiple sclerosis – a review. Eur J Neurol. (2019) 26:27–40. doi: 10.1111/ene.13819
125. Camu W, Lehert P, Pierrot-Deseilligny C, Hautecoeur P, Besserve A, Deleglise ASJ, et al. Cholecalciferol in relapsing-remitting MS: a randomized clinical trial (CHOLINE). Neurol Neuroimmunol Neuroinflamm. (2019) 6:e597. doi: 10.1212/nxi.0000000000000597
126. Løken-Amsrud KI, Holmøy T, Bakke SJ, Beiske AG, Bjerve KS, Bjørnarå BT, et al. Vitamin D and disease activity in multiple sclerosis before and during interferon-β treatment. Neurology. (2012) 79:267–73. doi: 10.1212/WNL.0b013e31825fdf01
127. Stein MS, Liu Y, Gray OM, Baker JE, Kolbe SC, Ditchfield MR, et al. A randomized trial of high-dose vitamin D2 in relapsing-remitting multiple sclerosis. Neurology. (2011) 77:1611–8. doi: 10.1212/WNL.0b013e3182343274
128. Soilu-Hänninen M, Åivo J, Lindström BM, Elovaara I, Sumelahti ML, Färkkilä M, et al. A randomised, double blind, placebo controlled trial with vitamin D 3 as an add on treatment to interferon β-1b in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry. (2012) 83:565–71. doi: 10.1136/jnnp-2011-301876
129. Laursen JH, Søndergaard HB, Sørensen PS, Sellebjerg F, Oturai AB. Vitamin D supplementation reduces relapse rate in relapsing-remitting multiple sclerosis patients treated with natalizumab. Mult Scler Relat Disord. (2016) 10:169–73. doi: 10.1016/j.msard.2016.10.005
130. Miclea A, Miclea M, Pistor M, Hoepner A, Chan A, Hoepner R. Vitamin D supplementation differentially affects seasonal multiple sclerosis disease activity. Brain Behav. (2017) 7:e00761. doi: 10.1002/brb3.761
131. Rolf L, Muris AH, Bol Y, Damoiseaux J, Smolders J, Hupperts R. Vitamin D3 supplementation in multiple sclerosis: symptoms and biomarkers of depression. J Neurol Sci. (2017) 378:30–5. doi: 10.1016/j.jns.2017.04.017
132. Achiron A, Givon U, Magalashvili D, Dolev M, Liraz Zaltzman S, Kalron A, et al. Effect of alfacalcidol on multiple sclerosis-related fatigue: a randomized, double-blind placebo-controlled study. Mult Scler. (2015) 21:767–75. doi: 10.1177/1352458514554053
133. Bhargava P, Fitzgerald KC, Calabresi PA, Mowry EM. Metabolic alterations in multiple sclerosis and the impact of vitamin D supplementation. JCI Insight. (2017) 2:e95302. doi: 10.1172/jci.insight.95302
134. Bhargava P, Cassard S, Steele SU, Azevedo C, Pelletier D, Sugar EA, et al. The vitamin D to ameliorate multiple sclerosis (VIDAMS) trial: study design for a multicenter, randomized, double-blind controlled trial of vitamin D in multiple sclerosis. Contemp Clin Trials. (2014) 39:288–93. doi: 10.1016/j.cct.2014.10.004
135. ClinicalTrials.gov Efficacy of Cholecalciferol (Vitamin D3) for Delaying the Diagnosis of MS After a Clinically Isolated Syndrome (D-Lay-MS). (2019). Available online at: https://clinicaltrials.gov/ct2/show/NCT01817166 (accessed November 16, 2019).
136. Ms Trials Preventing the Risk of MS Using Vitamin D in Patients with a First Demyelinating Event in Australia and New Zealand (PrevANZ). (2019). Available online at: https://mstrials.org.au/using-vitamin-d-in-patients-with-a-first-demyelinating-event/ (accessed November 16, 2019).
137. Smolders J, Hupperts R, Damoiseaux J. The way forward with vitamin D in multiple sclerosis. In: Minagar A editor. Neuroinflammation. Amsterdam: Elsevier. (2018). p. 175–91. doi: 10.1016/b978-0-12-811709-5.00009-0
Keywords: metabolism, guidelines, nervous system, MS risk, disease activity, innate adaptive immune system, calcitriol, cholecalciferol
Citation: Miclea A, Bagnoud M, Chan A and Hoepner R (2020) A Brief Review of the Effects of Vitamin D on Multiple Sclerosis. Front. Immunol. 11:781. doi: 10.3389/fimmu.2020.00781
Received: 22 January 2020; Accepted: 06 April 2020;
Published: 06 May 2020.
Edited by:
William Camu, Hôpital Gui De Chauliac, FranceReviewed by:
Trygve Holmøy, Akershus University Hospital, NorwayCopyright © 2020 Miclea, Bagnoud, Chan and Hoepner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrei Miclea, YW5kcmVpLm1pY2xlYUBpbnNlbC5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.