 Courtney M. Kloske
Courtney M. Kloske Donna M. Wilcock
Donna M. Wilcock- Department of Physiology, Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY, United States
Alzheimer’s disease (AD) is the most prevalent form of neurodegenerative disease, currently affecting over 5 million Americans with projections expected to rise as the population ages. The hallmark pathologies of AD are Aβ plaques composed of aggregated beta-amyloid (Aβ), and tau tangles composed of hyperphosphorylated, aggregated tau. These pathologies are typically accompanied by an increase in neuroinflammation as an attempt to ameliorate the pathology. This idea has pushed the field toward focusing on mechanisms and the influence neuroinflammation has on disease progression. The vast majority of AD cases are sporadic and therefore, researchers investigate genetic risk factors that could lead to AD. Apolipoprotein E (ApoE) is the largest genetic risk factor for developing AD. ApoE has 3 isoforms-ApoE2, ApoE3, and ApoE4. ApoE4 constitutes an increased risk of AD, with one copy increasing the risk about 4-fold and two copies increasing the risk about 15-fold compared to those with the ApoE3 allele. ApoE4 has been shown to play a role in Aβ deposition, tau tangle formation, neuroinflammation and many subsequent pathways. However, while we know that ApoE4 plays a role in these pathways and virtually all aspects of AD, the exact mechanism of how ApoE4 impacts AD progression is murky at best and therefore the role ApoE4 plays in these pathways needs to be elucidated. This review aims to discuss the current literature regarding the pathways and mechanisms of ApoE4 in AD progression with a focus on its role in neuroinflammation.
Introduction
Alzheimer’s Disease
Alzheimer’s disease (AD) is one of the most common neurodegenerative diseases and the 6th leading cause of death in the United States. AD affects more than 5.7 million Americans and by 2050, it is projected to affect over 13 million. Not only is AD a growing health concern, it is also an extreme financial burden costing nearly 290 billion dollars, annually, not taking into account the thousands of unpaid caregivers (1, 2). Clinically, AD is characterized by progressive learning and memory deficits that ultimately impede a patient’s ability to perform daily activities. The hallmark plaque and tangle pathology associated with AD were originally described in 1907 by Alois Alzheimer. It is now known these plaques are composed of aggregated beta-amyloid (Aβ), and the tangles are composed of hyperphosphorylated, aggregated tau, present typically within the neurons. While these two hallmark pathologies together lead to the neurodegeneration seen in AD, (3–5) Aβ pathology deposition typically begins decades before tangles and tangles have been shown to be better indicators of cognitve decline (2, 6). Going back to Alois Alzheimer’s first description of the disease in 1907, he also noted activation of microglia and astrocytes in response to the pathology (5, 7–9), providing further areas in AD to study. In recent years, one of the major themes in AD research has been understanding neuroinflammation and the role it plays in AD progression using both animal models and human tissue.
While all cases of AD have Aβ plaques and tau tangles, the mechanism leading to pathology is believed to differ between cases. Exceptionally few cases (<1%) develop solely due to genetics, with mutations in genes involved in Aβ processing being the clear cause. Three genes where mutations are known to cause AD are the amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) (10–12). Inheritance of any of these genetic mutations will lead to the accumulation of Aβ and ultimately AD. These cases are characterized as early onset AD (EOAD), affecting patients between 30 and 60 years of age (13, 14). The remaining 99% of AD cases are sporadic and are often associated with late onset AD (LOAD). Apolipoprotein E4 (ApoE4) stands out as the largest genetic risk factor for developing LOAD (15–17). Apolipoprotein (ApoE) has three isoforms with varying risk for developing AD. ApoE4 confers an increased risk of AD relative to ApoE3 with E4 homozygotes showing the greatest risk of AD with an odds ratio of 10–15-fold increase. ApoE4 is present in about 14% of the general population and 37% in the AD population (16–18). ApoE3 is the most common allele is typically used as the baseline comparison in AD studies. ApoE3 is present in about 78% of the general population and 59% in the AD population (16). The ApoE2 allele has been shown to be protective for AD compared to the ApoE3 allele, being present in about 5% of all AD cases and about 9% in the general population (19). It is important to note, studies have shown ApoE4 has an increased risk in AD and ApoE2 has been protective when comparing these changes to ApoE3, as it is used as the control allele (20).
ApoE in the Central Nervous System
ApoE is the primary transporter of lipids and cholesterol in the brain and is mainly generated by astrocytes; however, microglia and neurons can generate ApoE in times of stress (21). ApoE works to reduce cholesterol levels as well as promoting lipoprotein clearance. ApoE binds to lipoproteins and provides clearance through the low-density lipoprotein receptor (LDLR). The ApoE isoforms impact how lipoproteins are cleared and the extent in which it is executed. ApoE3 has been shown to bind to LDLR allowing for lipid uptake and, again, it is characterized as the control phenotype for comparing function of the other two alleles. ApoE2 has a decreased affinity to LDLR and therefore leads to a type III hyperlipoproteinemia associated with ApoE2 patients. ApoE4 has an increased lipid binding ability but decreased proteolytic activity leading to an increase in lipoproteins and cholesterol (21–24). On astrocytes, ApoE interacts with plasma membrane ATP-binding cassette transporter A1 (ABCA1) and becomes loaded with lipids and cholesterol to provide the brain with needed nutrients (25–27). In addition to ApoE4 having an impaired interaction with receptors, ApoE4 has been shown to have a reduced interaction with ABCA1 and therefore is typically found in a hypolipidated state compared to ApoE3 (28–30).
ApoE4 has been suggested to decrease insulin signaling by impairing recycling of the insulin receptor which in turn could be leading to the decreased glucose metabolism seen in AD patients (31, 32). ApoE4 has also been shown to directly impact ApoER2 receptor recycling. This receptor works in conjunction with Reelin and is critical for synaptic plasticity in the aging brain. ApoE4 impairs recycling of ApoER2 and therefore impairs synaptic plasticity (33, 34). APP recycling has also been shown to be influenced by ApoE4 which can lead to an increase the amyloidogenic pathway leading to an increase in Aβ (35). In both post-mortem AD human brains and in mouse models of AD, ApoE4 has been shown to play a significant intracellular role in the movement and trafficking of receptors and intracellular vesicles (36).
The role of ApoE4 in the brain as a whole has been studied in metabolic approaches as well as through gross anatomy. These studies have implicated ApoE4 in increased regional cortical atrophy in the presence of AD compared to ApoE3. Specifically, there is a decrease in gray matter volume in both the medial temporal lobe and the anterior temporal lobes in AD cases (37–39). ApoE4 has also been implicated in overall brain energy and health as previously mentioned. Studies have shown ApoE status plays a role in cerebral glucose metabolism in an aging brain, regardless of the pathology present. At least one copy of ApoE4 significantly decreases glucose metabolism in comparison to non-ApoE4 carrying patients (40, 41).
ApoE has been shown to play integral roles in overall brain health and impact the development of Alzheimer’s disease. ApoE4 additionally plays many roles that are directly pertinent to the progression and development of AD specially through affecting inflammation. This will be discussed at length in this review.
Inflammation in Alzheimer’s Disease
Neuroinflammation in AD has been shown to contribute significantly to the onset, progression, and pathogenesis of AD. The main sources of inflammatory mediators in the brain are microglia and astrocytes. Microglia and astrocytes can release cytokines which can play both pro-inflammatory and anti-inflammatory roles in the brain depending on the stimulus and microenvironment (7, 42, 43). This fluctuation between the pro- and anti-inflammatory profiles has be seen in patients with early AD. One study showed patients with early AD have a bias toward either a pro-inflammatory or anti-inflammatory phenotype and, as disease progresses, the phenotype is more homogeneous, with both sides of inflammation elevated relative to age-matched, non-disease controls (44). These findings demonstrate the complex, and dynamic nature of inflammation in AD. Neuroinflammation is also impacted through normal aging, however, this review will focus on the impact on AD. For more information on neuroinflammation and aging see the review from Rea et.al (45).
Microglia in Alzheimer’s Disease
Microglia are the largest player in neuroinflammation in the CNS. Microglia have an important role in surveying the brain in order to detect and clear debris while maintaining an optimal microenvironment. Microglia can respond to virtually all foreign factors in the brain typically described as danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) (46–50). Of particular importance to AD, microglia respond to Aβ. Reports have suggested that in the presence of Aβ, microglia become activated and surround the plaque forming a barrier with the ultimate goal of preventing further spread while also attempting to clear the Aβ (51, 52). Upon activation around the Aβ plaque, the microglia can phagocytose Aβ (52, 53). However, if there is a buildup of Aβ in the microglia, this can subsequently lead to microglial cell death and an increase in inflammation and recruitment of more microglia, thus continuing this inflammatory cascade (54). Additionally, the activated microglia can respond with a pro-inflammatory response, releasing cytokines such as tumor necrosis factor-α (TNFα) and interleukin 1β (IL-1β) as well as other factors to potentially induce damage to surrounding tissue (50, 55–59).
Once a microglial receptor binds to a given ligand, microglia are able to become activated and work to ameliorate the situation. In regards to the clearance of Aβ, Aβ receptors have been shown to be present and have the ability to clear Aβ in early AD (60). In later stages of AD, however, this overall expression of both Aβ receptors and Aβ degradation enzymes are significantly downregulated. This downregulation of the Aβ receptors and Aβ degradation enzymes has been shown to be a direct response to the increase in inflammatory cytokines in AD (57, 61). One study looking at APP/PS1 mice found a twofold to fivefold decrease in Aβ receptors and a 2.5-fold increase in IL-1β and TNFα in older aged mice. This increase in pro-inflammatory cytokines can impair the surrounding neurons leading to neuronal degeneration (57). There are many microglial receptors that are able to bind Aβ and work to either phagocytize or create an inflammatory response to recruit other microglia in the presence of Aβ (62–65). Without control over their inflammatory response, the overall response of the microglia could become detrimental and cause problems such as neurodegeneration.
Microglial activation, in addition to phagocytosis and recruitment, can lead to induction of the inflammasome which can increase inflammatory cytokines as well as possibly increase Aβ deposition (59, 66). Inflammasomes help regulate the release of IL-1β and act as sensors of signals in the brain. IL-1β is incredibly potent, affecting the expression of adhesion molecules, immune cell infiltration and the overall increase of more cytokines. Due to its many functions, IL-1β requires several checkpoints before it is fully activated and, therefore, is made initially as an inactive molecule that the inflammasome cleaves and activates (67). The components needed for this activation include the inflammasome sensor molecule, adaptor molecule, and adapter protein apoptosis associated speck-like protein containing a CARD (ASC). Recent studies have shown, the ASC has the ability to help seeding of Aβ and by blocking the inflammasome it can decrease seeding of Aβ in the brain of mouse models (59, 67–71).
Upon debris clearance, microglia rapidly work to counteract their previously pro-inflammatory response (72–75). This can be done through the induction of anti-inflammatory responses which work to downregulate the potent, pro-inflammatory response. This allows for the microglia to revert to a resting state (50). In the case of AD, the microglia can become chronically activated and be in a perpetually activated state, leading to detrimental effects such as an increase in Aβ production and neurodegeneration (74, 76). In AD, both pro- and anti-inflammatory microglia states can be found in the same region, and this co-existence is likely detrimental; however, deciding which state the microglia should be in at a given instance remains unclear (77).
Recently, a microglial receptor, triggering receptor expressed on myeloid cells 2 (TREM2) has brought the importance of microglia in AD to the forefront of inflammatory research since mutations in TREM2 increase the risk for AD. As its name states, TREM2 is expressed on myeloid-derived cells such as microglia, macrophages, and osteoclasts. Rare mutations in TREM2 confer an increased risk of AD with an odds ratio of 4.5 (78–80). Individuals homozygous for mutations in TREM2 develop a rare disorder characterized by bone fractures and presenile dementia called Nasu-Hakola disease (81, 82). TREM2 responds to a wide range of stimuli including apoptotic cells, Aβ, and lipoproteins. Without AD pathology, TREM2 is expressed to clear damaged or apoptotic neurons and clear cellular debris through phagocytosis while downregulating the pro-inflammatory response to these stimuli (83–85). In AD, TREM2 has been shown to be highly expressed on microglia surrounding neuritic plaques and is important in clearance of Aβ (86, 87). In mouse models lacking TREM2, the microglia are unable to migrate toward an Aβ plaque and do not cluster when compared to a model with functional TREM2. While the overall impact of TREM2 is still up for debate, evidence suggests that the timing of TREM2 expression is key. Studies show that in early AD pathology, TREM2 is needed for clearance of early Aβ plaques and slowing of cognitive decline, while expression of TREM2 later in disease progression could lead to detrimental long-term consequences (88–92).
Astrocytes in Neuroinflammation
Astrocytes also play a role in the inflammation seen in AD (93). In the brain, astrocytes interact with both neurons and the cerebrovascular in order to maintain nutrients and chemical gradients in the brain. They additionally work to maintain calcium levels and potassium homeostasis, as well as provide overall neuronal support. In AD, normal functions of astrocytes become affected and contribute to astrocytic dysfunction and inflammation. This dysfunction includes astrocytes losing their ability to maintain calcium levels and potassium homeostasis (94–96). Additionally, activated astrocytes typically lose their ability to deliver nutrients to neurons leading to impairments in neuronal function (97, 98). Upon activation, studies have shown astrocytes release cytokines that can lead to increased neuronal toxicity as well as a decreased outgrowth of neuronal processes and an overall decreased activity rate (99). Recent studies suggest an interaction between microglia and astrocytes finding that once microglia become activated, they can lead to activation of astrocytes, resulting in a feed-forward loop which is detrimental to the surrounding environment. The mechanism showed that when activated, microglia release IL-1α, TNFα and C1q and astrocytes become activated. Additionally, in the presence of damaged blood vessels, the astrocytes promote tissue repair and neuronal survival rather than microglia (99). This provides a strong link showing that both microglia and astrocytes work in conjunction to increase the neuroinflammation seen in AD (Figure 1).
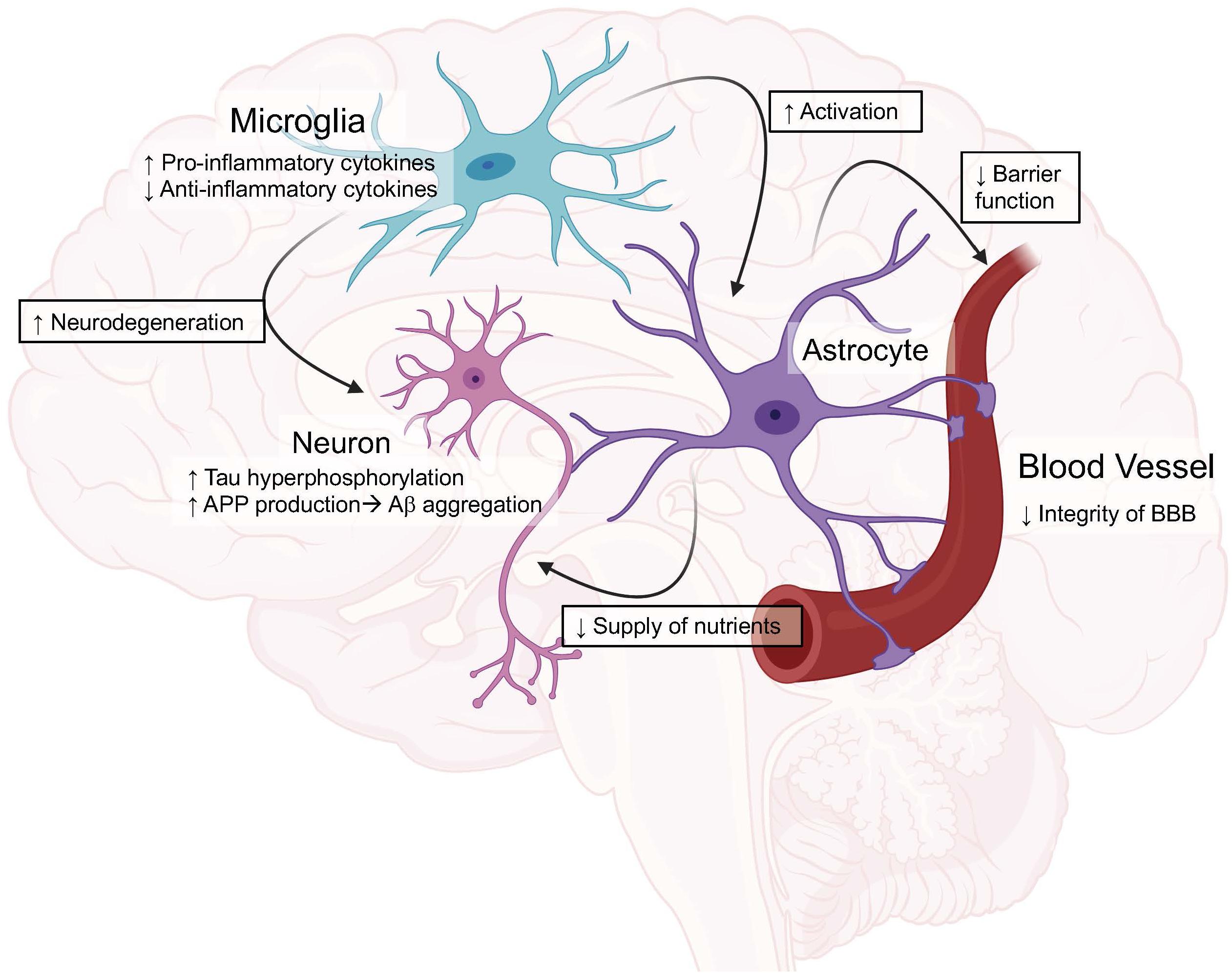
Figure 1. Graphical view of the impact of ApoE4 in the brain compared to ApoE3. These roles will be discussed in depth throughout the review. Created with BioRender.
ApoE, Inflammation, and Alzheimer’s Disease
Early Investigations of ApoE Isoforms’ Role on Inflammation
While microglia and astrocytes are the main modulators of inflammation in the brain, they are also the major sources of ApoE in the brain. Early studies suggested that in glial cultures, the presence of ApoE helped suppress glial activation in response to lipopolysaccharide (LPS), showing ApoE provides a protective anti-inflammatory response (100, 101). Studies showed that this anti-inflammatory response could be through the inhibition of the c-Jun N-terminal kinase (JNK) cascade (102). Another study showed that APP was capable of activating microglia and producing neurotoxic molecules in the presence of ApoE4, however ApoE3 was able to prevent this activation (49).
Moving into animal studies, researchers then began to investigate how ApoE as a whole, as well as the isoforms, impacted neuroinflammation (49, 103, 104). In one study, APP mice with and without ApoE were chronically administered LPS and it was found that in the presence of ApoE, the mice had increased gliosis and Aβ deposition suggesting a role for ApoE to increase inflammation in AD models (104). When looking at the ApoE isoforms specific impact on the inflammatory response, a study from Lynch et.al. used humanized ApoE3 and ApoE4 mice and chronically administered LPS. They found a significant increase in TNFα and IL6 present in the brains of ApoE4 mice. The authors then examined the impact of a small ApoE-mimetic peptide on inflammation and saw a significant reduction in inflammation. These results together show ApoE isoforms impact inflammation in the brain and that exogenous ApoE has the potential to reverse these effects (105). Another study using humanized ApoE3 and ApoE4 models showed ApoE isoforms play a regulatory role in nitric oxide (NO) production from microglia. They show that ApoE4 microglia release significantly more NO compared to ApoE3 microglia, which could cause the detrimental effects seen in the brains of ApoE4 patients (106). The results from this study were later confirmed and further research was done showing ApoE4 increased NO production in mice as well as in humans. Additionally, the authors showed resting microglia from the ApoE4 targeted replacement mice had an increased proinflammatory profile which led to an altered microglial phenotype compared to ApoE3 (Figure 1) (107).
ApoE and Aβ
Evidence indicates that the three common ApoE isoforms impact the clearance and aggregation of Aβ (16, 35, 108–112). Studies have shown that ApoE is essential for deposition of Aβ in animal models (29, 111, 113–115). ApoE4 has been shown to have an increased binding ability to Aβ compared to ApoE3 which contributes to the increased aggregation of Aβ and decreased clearance. In addition to increased binding, ApoE4 has been shown to have increased rate of Aβ oligomerization and increased Aβ plaque generation (116, 117). This increase has been confirmed in human autopsy tissue as ApoE4 patients show an increase in both vascular and parenchymal Aβ plaques (37, 118–120). While the exact mechanism of how ApoE4 carriers exhibit increases in Aβ plaques is not fully understood, it may be due to the increased ability of ApoE4 to bind Aβ and inability to fully remove Aβ from the brain.
There are several mechanisms in which Aβ could be cleared and where ApoE4 could impact clearance (Figure 2). Typically, Aβ is removed through either proteomic degradation, lysosomal clearance or through the blood brain barrier (BBB) (121). ApoE4 has been shown to have an impact on these pathways and could contribute to the decrease in Aβ removal seen with the ApoE4 isoform. In addition to clearance of Aβ, ApoE has also been shown to play a role in phagocytosis of apoptotic cells which is necessary in AD due to neurodegeneration. Surprisingly, one study showed ApoE4 has an increased phagocytosis of apoptotic neuronal cells while having a decreased clearance of Aβ in vitro. This increase in phagocytosis could be contributing to the switch in phenotype from resting microglia to an altered phenotype, leading to the detrimental phenotype associated with ApoE4 (122).
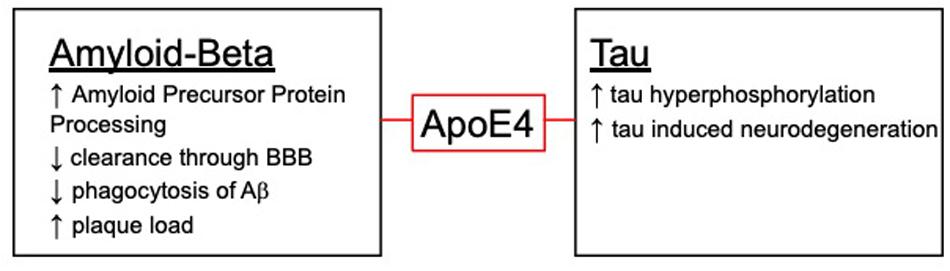
Figure 2. The role ApoE4 plays with the hallmark AD pathologies.
APP processing and recycling has also been shown to be influenced by ApoE4, leading to an increase in Aβ (35). A recent study showed ApoE isoforms differently regulated APP processing through altered MAPK signaling. They showed that ApoE4 can function as a signaling molecule to enhanced activity of this cascade leading to an increase of APP. This study gives an ApoE dependent mechanism in which ApoE4 could directly impact disease progression through alteration in a signaling cascade (123).
In addition to ApoE4 playing a role in Aβ generation, ApoE4 and Aβ can also have a significant role in the ApoE-inflammatory cascade. As previously mentioned, ApoE4 has a role in increasing the proinflammatory response of glia in response to inflammatory stimuli. Researchers then moved into transgenic mice expressing human ApoE in various AD models to determine (1) the role of ApoE and (2) the role of ApoE isoforms in disease progression. In a study investigating the impact of ApoE using APP/PS1 mouse models, researchers found ApoE had a direct impact on the deposition of Aβ through its influence on microglial activation. These results show ApoE is required to stimulate the innate immune response to Aβ (124). A study using human ApoE mice with familial AD mutations (EFAD model), found ApoE4 significantly impacted Aβ plaque morphology and showed increased glial activity. The increased glial activity was measured by IL1β levels and showed a negative impact on microglial morphology (114).
ApoE and Tau
ApoE4 has been shown to have varying inflammatory effects not only on Aβ but, it has been shown to have differential effects on tau induced neuroinflammation (Figure 2). ApoE4 has been shown to exacerbate neurodegeneration through a tau-mediated mechanism using P301S-ApoE TR models. The authors showed ApoE4 microglia generate significantly more TNFα and led to impaired neuronal viability compared to other ApoE isoforms (125). Another study described how microglia could be playing a vital role in neurodegeneration in a similar tau model through an ApoE dependent method. This study suggests microglia could be a key target in therapeutics when it comes to tauopathies (126). This idea of microglia and ApoE being the driving factor in the tau deposition is paralleled in another study using a model of microglial ablation. This study showed microglia were the key player in promoting plaque formation and neurodegeneration in the animal models (127). These studies show the importance of investigating both the detrimental and beneficial roles of microglia, especially with ApoE isoforms (Figure 1).
ApoE and Inflammatory Signaling Cascades
ApoE has been shown to interact with TREM2 leading to activation of TREM2’s signaling cascade (91, 128–130). Recent studies have suggested that, in the presence of neurodegeneration, ApoE and TREM2 are both required for microglia to become activated and assume a Disease Associated Microglia (DAM) phenotype. This phenotype allows for microglia clustering around the Aβ plaque and clearance of apoptotic neurons. Microglia with a DAM phenotype are highly localized around neuritic plaques and activation requires a step wise transition (131–134). First is an increase in Apoe and downregulation of homeostatic genes such as Purinergic Receptor P2Y12 (P2ry12), followed by an upregulation of Trem2. This DAM phenotype has helped bring together ApoE and TREM2 through a disease specific pathway as well as how mulitple risk factors for AD play a role in this cascade (131, 134). Further studies are needed to examine the role of ApoE isoforms on this cascade as well as looking into the proteomics of these cascades.
ApoE and Autophagy
Autophagy is a mechanism to eliminate aggregated proteins and internal structures that become dysfunctional. Studies have shown this pathway specifically becomes dysfunctional early in disease states and can lead to neurodegeneration. With the role of ApoE4 in neurodegeneration, researchers have suggested that ApoE4 likely has an impact on clearance of neuronal degenerative products after injury since ApoE is upregulated post insult, thus potentially linking ApoE4 with autophagy dysfunction (135–138). Recent studies suggest a potential interaction with ApoE4 and an autophagy pathway that may account for the decreased autophagy in ApoE4 carriers (139). This study showed a direct interaction with ApoE4 and coordinated lysosomal expression and regulation (CLEAR), suggesting ApoE4 has a direct impact on autophagy transcription leading to impaired autophagy (140). This allows for further studies in ApoE4 as a transcription factor as well as other potential mechanisms in which ApoE4 impacts autophagy.
ApoE Fragmentation
The ApoE4 protein has been shown to have a decreased expression compared to the other isoforms but the exact mechanism of this is unknown. One possibility of this downregulation is through the rate of degradation of ApoE. Studies have suggested that ApoE can be fragmented by high-temperature requirement serine peptidase A1 (HtrA1). Of the ApoE isoforms, ApoE4 is the least stable protein confirmation and therefore most susceptible to this fragmentation. These fragments could bind to Aβ or cell surface receptors to prevent clearance, possibly contributing to the progression of the disease through neurotoxicity and neuroinflammation (141–144). ApoE3 also can become fragmented and studies suggest the primary fragment produced is neuroprotective (105, 145). More studies are needed to examine the exact mechanism in which ApoE4 fragments can contribute to inflammation and disease progression and potential therapeutics with ApoE3 fragments.
ApoE and Blood Brain Barrier Dysfunction
The importance of ApoE on the BBB has been a growing topic in the field of AD as well as other brain injury research due to the fact that brain trauma leads to a disruption in the BBB (146). Studies have shown patients with ApoE4 have worsened outcomes to brain injuries including traumatic brain injury (TBI) and stroke compared to ApoE3. Additionally, TBI earlier in life has been shown to contribute to an increased risk of AD later in life. TBI patients with ApoE4 typically experience increased coma length, mortality and decreased prognosis (147–150). These suggest ApoE isoforms play a role in BBB regulation and ApoE4 has a clear impact on the overall strength and integrity of the BBB (Figure 1).
The mechanism as to how ApoE isoforms could be involved in BBB dysfunction is necessary to understand why ApoE4 causes detrimental outcomes when the brain becomes compromised. One crucial component of the BBB are the tight junctions, as they provide a barrier that allows selective molecules to flow between blood and brain, maintaining homeostasis and keeping out unwanted molecules. Nishitsuji et al. showed that these tight junctions are important in BBB integrity and are significantly impaired in the presence of ApoE4. The authors suggest that this could be due to changes in matrix-metalloproteinase-9 (MMP9) or other molecules that would degrade or impact the BBB integrity (151). MMP9 has been shown to have multiple targets present in the BBB, but one specifically is tight junctions. MMP9 can work to decrease the bond in the tight junction leading to a leaky BBB (152). A study from Bell et.al built upon the idea that ApoE4 triggers BBB breakdown and found that cyclophilin A (CypA) is the likely culprit in this breakdown. They showed that an increase in CypA led to activation of NF-kB, followed by a significant increase in MMP9, specifically in the pericytes found surrounding and supporting the vasculature in the brain (153). Recently, Main et.al showed that ApoE4 had an impact on the BBB integrity and the activation of the MMP9 pathway in a comprehensive study using a TBI model. This study once again confirmed that ApoE4 with TBI worsens disease outcomes. They also found that after injury in both ApoE3 and ApoE4, the BBB must become stabilized which leads to an increase in ApoE. After the ApoE increase, there is pericyte loss, decreased tight junction expression and an increase in MMP9 expression. This appears in both ApoE3 and ApoE4 models, however, ApoE4 is much slower at the resolution of this progression. ApoE4 has clear detrimental effects on BBB integrity by activating the MMP9 system and activating other inflammatory cascades (154). In the presence of ApoE4, the pericytes significantly lose their supportive capabilities, leading to an increase in leaky vessels (155). A culmination of these studies implicates ApoE4 in decreasing BBB integrity through weakening tight junctions, increases in MMP9 and a loss of support needed for the BBB.
The evidence shows an increase in MMP9 with ApoE4 leads to the loss of BBB integrity and increase in inflammation. ApoE4 has also been associated to an increase in cerebral amyloid angiopathy (CAA) which can trigger BBB dysfunction due to the deposition of Aβ in the vessel walls and impaired clearance through the BBB compared to ApoE3 (118). Anti-Aβ immunotherapies for animal models and in clinical trials have been shown to increase MMP9 expression leading to an increase in microhemorrhages, showing a decrease in BBB integrity (156). While the majority of the anti-Aβ trials failed due to adverse effects, these adverse events were more robust in ApoE4 carriers. This connection between MMP9 activation due to the anti-Aβ therapies and the effect of ApoE4 on MMP9 expression can explain the ApoE4 involvement in BBB dysregulation that in turn, leads to neurodegeneration. More information on ApoE4 in clinical trials will be provided later in the review.
In addition to the disruption of the BBB integrity upon brain injury, there has been shown to be an interesting connection between TBI and development of AD later in life. In ApoE TR models of ApoE3 and ApoE4, the mice were given repeated TBI over 1 month. This showed a significant increase in microgliosis, via IBA1 immunoreactivity, in ApoE3 mice but not ApoE4 mice. ApoE4 mice showed significant increases in phospholipids and LDLR but not in inflammation suggesting that ApoE4 plays a role in many detrimental pathways but also shows an impaired inflammatory phenotype (157). This finding suggests that something more than just a decrease in BBB integrity is occurring in the presence of ApoE4.
ApoE and the Glymphatic System
As previously discussed, ApoE4 constitutes a significant impairment in the clearance of Aβ. This leads to the buildup of Aβ in the vessels of ApoE4 AD patients, contributing to an inflammatory response. This impairment in clearance has been hypothesized to be directly related to ApoE4 forming globular structures in conjunction with Aβ, potentially impairing the availability of receptors necessary for the clearance of Aβ into the blood vessels. Due to the involvement of ApoE in clearance of Aβ from the brain to the vasculature, it is logical to hypothesize that ApoE4 could also be involved in the impairments seen in the glymphatic system due to a similar mechanism. Studies thus far have examined glymphatic system impairments with Aβ removal without examining ApoE isoforms. This study also examined human autopsy tissue and found impairments and buildup of Aβ in the glymphatic system but again, ApoE isoforms were not considered (158, 159). More studies are needed to specifically examine human tissue and consider ApoE isoforms to determine the potential role of ApoE in the glymphatic system.
ApoE and Other Neurodegenerative Diseases
Studies have implicated ApoE4 in other forms of dementia other than AD, including Lewy Body Dementia (LBD). LBD is one of the most prevalent forms of dementia and is found in about 40% of Parkinson’s Disease patients. Two independent studies published in 2020 investigated the effect of ApoE4 on α-synuclein (α-syn), the defining pathology seen in LBD, and showed an increase in pathology was associated with ApoE4. Both studies used transgenic mice expressing human ApoE isoforms with differing routes of α-syn production (transgenic model and AAV- α-syn). The results from the animal studies both showed ApoE4 lead to an increase in α-syn pathology, a decrease in cognitive decline and an increase in gliosis around the pathology (160, 161). In addition to animals, human patients were also investigated, and it was shown that ApoE4 patients with Parkinson’s disease had an increased rate of cognitive decline (161). Human neuropathology data showed that ApoE4 patients had an increase in α-syn pathology as well (160). Together these studies show a clear impact of ApoE4 in other neurological diseases as well as the impact of ApoE4 on neuroinflammation is not limited to AD pathology.
Impact of ApoE on Clinical Trials
Due to the growing impact of AD, the need for therapeutics to either prevent or delay the onset of AD is imperative. In 2012, the National Alzheimer’s Project Act was established to help prevent future cases of AD and related dementias. The group came up with five major goals that includes optimizing care quality for patients, improving support for those with AD and their families, enhancing public awareness, tracking progress, and preventing and effectively treating AD by 2025 (162). Having a prevention or treatment option by 2025 is a lofty goal but the field is working hard to achieve this goal. While we have had many drug trials in the past 20 years, the failure rate is high which is pushing the field in a direction of a more personalized medicine approach to AD. Using this idea in clinical trials and therapeutic treatments, the likelihood of a single drug helping everyone with the disease is not likely. As this review has showed, ApoE4 plays a large and varied role in AD, and thus is necessary to take into consideration upon selection for clinical trials.
Targeting Aβ through anti-Aβ immunotherapy has been a constant focus of trials and is still being actively perused. Bapineuzumab was one of the initial anti-Aβ immunotherapies designed to increase clearance of Aβ. In a phase II clinical trial, bapineuzumab treated ApoE4 non-carriers had significant benefits on cognition and function at the endpoints of the study while this was not seen in ApoE4 patients. This trial also showed that ApoE4 patients had an increase in amyloid related imaging abnormalities (ARIA) which occurred 3–7x more in ApoE4 patients depending on copy number. These findings led to the phase III trial where ApoE4 patients received a lower dose to offset ARIA (163–165). This study ultimately failed to meet the endpoints and Aducanumab moved in as a newer anti-Aβ immunotherapy for trial. Aducanumab can be tolerated at a higher concentration and was dosed every 4 weeks. This trial was focused on patients with AD before cognitive symptoms had occurred. While the drug showed an effect on cognition, ARIA was the main adverse effect in ApoE4 patients. Moving into the phase III trial for Aducanumab, patients were segregated out into ApoE status to dose according (166). These two studies have clearly shown that while the goal is to find a treatment for AD, it is important to consider ApoE status into the trial design and study outcomes.
Currently, very few clinical trials take ApoE status into account while grouping or dosing for trials. One current ongoing trial, “A Study of CAD106 and CNP520 Versus Placebo in Participants at Risk for the Onset of Clinical Symptoms of Alzheimer’s Disease” (NCT02565511), is investigating the effects of CAD106 and CNP520, which both target amyloid, on the impact of cognition, clinical status and pathology. They will investigate ApoE4/4 patients due to their high risk of progression to MCI and AD and is expected to reach completion in 2024. Another upcoming trial is directly targeting ApoE4 patients by using an adeno-associated virus in order to convert ApoE4 patients to ApoE2 in hopes to delay AD onset (NCT03634007). Studies incorporating ApoE status into their inclusion and dosing criteria will be imperative in the growing field of AD trials and research.
One of the most multi-faceted clinical trials going on right now is the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), which now has branches in the United States as well as around the world. The original FINGER study showed that after lifestyle modifications, ApoE4 carriers showed no significant cognitive improvements to those without ApoE4 and that E4 carriers with lifestyle modifications had greater cognitive and physical improvements than those without the modifications (167, 168). This study emphasized the importance of preventative strategies for ApoE4 patients, specifically through lifestyle modifications such as physical activity.
Conclusion
As shown through this review, ApoE4 plays a role in virtually all aspects of AD ranging from clearance of hallmark pathologies, to disruption of intracellular pathways, to impacts on whole brain metabolism. While these pathways can seem unrelated, they can be connected through the overarching theme of neuroinflammation. In moving forward, the impact of ApoE4 needs to be considered in studies, both human and animals. In conclusion, ApoE4 plays a negative role in AD through both gain of misfunction and loss of function mechanisms and further studies are needed to elucidate these pathways and push the field toward new therapeutics.
Author Contributions
CK wrote and developed much of the manuscript content. DW wrote the portions, edited, and reviewed for content.
Funding
NIA grant RF1AG057754 funded CK, a graduate student.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. CDC A Public Health Appoach to Alzheimer’s and other Dementias. Atlanta, GA: Centers for Disease Control and Prevention. (2017).
2. Alzheimer’s AssociationAlzheimer’s disease facts and figures. Alzheimers Dement. (2019).15:321–87.
3. Thal DR, Walter J, Saido TC, Fandrich M. Neuropathology and biochemistry of Abeta and its aggregates in Alzheimer’s disease. Acta Neuropathol. (2015) 129:167–82. doi: 10.1007/s00401-014-1375-y
4. Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. (1995) 16:271–8; discussion 278–84. doi: 10.1016/0197-4580(95)00021-6
5. Maurer K, Volk S, Gerbaldo H. Auguste D and Alzheimer’s disease. Lancet. (1997) 349:1546–9. doi: 10.1016/S0140-6736(96)10203-8
6. Beason-Held LL, Goh JO, An Y, Kraut MA, O’Brien RJ, Ferrucci L, et al. Changes in brain function occur years before the onset of cognitive impairment. J Neurosci. (2013) 33:18008–14. doi: 10.1523/JNEUROSCI.1402-13.2013
7. Stephenson J, Nutma E, van der Valk P, Amor S. Inflammation in CNS neurodegenerative diseases. Immunology. (2018) 154:204–19. doi: 10.1111/imm.12922
8. Laurent C, Buee L, Blum D. Tau and neuroinflammation: what impact for Alzheimer’s disease and tauopathies? Biomed J. (2018) 41:21–33. doi: 10.1016/j.bj.2018.01.003
9. Navarro V, Sanchez-Mejias E, Jimenez S, Munoz-Castro C, Sanchez-Varo R, Davila JC, et al. Microglia in Alzheimer’s disease: activated, dysfunctional or degenerative. Front Aging Neurosci. (2018) 10:140. doi: 10.3389/fnagi.2018.00140
10. Lanoiselee HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Med. (2017) 14:e1002270. doi: 10.1371/journal.pmed.1002270
11. Rademakers R, Cruts M, Van Broeckhoven C. Genetics of early-onset Alzheimer dementia. Sci World J. (2003) 3:497–519. doi: 10.1100/tsw.2003.39
12. Wu L, Rosa-Neto P, Hsiung GY, Sadovnick AD, Masellis M, Black SE, et al. Early-onset familial Alzheimer’s disease (EOFAD). Can J Neurol Sci. (2012) 39:436–45. doi: 10.1017/s0317167100013949
13. Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. (1999) 65:664–70. doi: 10.1086/302553
14. Koedam EL, Lauffer V, van der Vlies AE, van der Flier WM, Scheltens P, Pijnenburg YA. Early-versus late-onset Alzheimer’s disease: more than age alone. J Alzheimers Dis. (2010) 19:1401–8. doi: 10.3233/JAD-2010-1337
15. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. (1993) 261:921–3. doi: 10.1126/science.8346443
16. Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high avidity binding to B-amyloid and increase frequency of type 4 allele in late-onset familial Alzheimer Disease. Proc Natl Acad Sci USA. (1993) 90:1977–81.
17. Saunders AMP, Strittmatter WJM, Schmechel DM, George-Hyslop PH, Pericak-Vance MAP, Joo SH, et al. Association of apolipoprotein E allele E4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. (1993) 43:1467–72.
18. Qian J, Wolters FJ, Beiser A, Haan M, Ikram MA, Karlawish J, et al. APOE-related risk of mild cognitive impairment and dementia for prevention trials: an analysis of four cohorts. PLoS Med. (2017) 14:e1002254. doi: 10.1371/journal.pmed.1002254
19. Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC Jr., et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. (1994) 7:180–4. doi: 10.1038/ng0694-180
20. Pitas RE, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. (1987) 262:14352–60.
21. de Chaves EP, Narayanaswami V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol. (2008) 3:505–30. doi: 10.2217/17460875.3.5.505
22. Phillips MC. Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life. (2014) 66:616–23. doi: 10.1002/iub.1314
23. Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. (2009) 50:S183–8. doi: 10.1194/jlr.R800069-JLR200
24. Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, Permanne B, et al. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid β peptides. Biochem J. (2000) 348:359–65. doi: 10.1042/bj3480359
25. Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. (2008) 118:671–82. doi: 10.1172/JCI33622
26. Fan J, Stukas S, Wong C, Chan J, May S, DeValle N, et al. An ABCA1-independent pathway for recycling a poorly lipidated 8.1 nm apolipoprotein E particle from glia. J Lipid Res. (2011) 52:1605–16. doi: 10.1194/jlr.M014365
27. Minagawa H, Gong JS, Jung CG, Watanabe A, Lund-Katz S, Phillips MC, et al. Mechanism underlying apolipoprotein E (ApoE) isoform-dependent lipid efflux from neural cells in culture. J Neurosci Res. (2009) 87:2498–508. doi: 10.1002/jnr.22073
28. Boehm-Cagan A, Bar R, Liraz O, Bielicki JK, Johansson JO, Michaelson DM. ABCA1 agonist reverses the ApoE4-driven cognitive and brain pathologies. J Alzheimers Dis. (2016) 54:1219–33. doi: 10.3233/JAD-160467
29. Boehm-Cagan A, Bar R, Harats D, Shaish A, Levkovitz H, Bielicki JK, et al. Differential effects of apoE4 and activation of ABCA1 on brain and plasma lipoproteins. PLoS One. (2016) 11:e0166195. doi: 10.1371/journal.pone.0166195
30. Rawat V, Wang S, Sima J, Bar R, Liraz O, Gundimeda U, et al. ApoE4 alters ABCA1 membrane trafficking in astrocytes. J Neurosci. (2019) 39:9611–22. doi: 10.1523/JNEUROSCI.1400-19.2019
31. Rhea EM, Raber J, Banks WA. ApoE and cerebral insulin: trafficking, receptors, and resistance. Neurobiol Dis. (2020) 137:104755. doi: 10.1016/j.nbd.2020.104755
32. Zhao N, Liu CC, Van Ingelgom AJ, Martens YA, Linares C, Knight JA, et al. Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron. (2017) 96:115–29.e5. doi: 10.1016/j.neuron.2017.09.003
33. Lane-Donovan C, Herz J. The ApoE receptors Vldlr and Apoer2 in central nervous system function and disease. J Lipid Res. (2017) 58:1036–43. doi: 10.1194/jlr.R075507
34. Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci USA. (2010) 107:12011–6. doi: 10.1073/pnas.0914984107
35. Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. (2009) 10:333–44. doi: 10.1038/nrn2620
36. Van Acker ZP, Bretou M, Annaert W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol Neurodegener. (2019) 14:20. doi: 10.1186/s13024-019-0323-7
37. Filippini N, Rao A, Wetten S, Gibson RA, Borrie M, Guzman D, et al. Anatomically-distinct genetic associations of APOE epsilon4 allele load with regional cortical atrophy in Alzheimer’s disease. Neuroimage. (2009) 44:724–8. doi: 10.1016/j.neuroimage.2008.10.003
38. Boccardi M, Sabattoli F, Testa C, Beltramello A, Soininen H, Frisoni GB. APOE and modulation of Alzheimer’s and frontotemporal dementia. Neurosci Lett. (2004) 356:167–70. doi: 10.1016/j.neulet.2003.11.042
39. Agosta F, Vossel KA, Miller BL, Migliaccio R, Bonasera SJ, Filippi M, et al. Apolipoprotein E epsilon4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc Natl Acad Sci USA. (2009) 106:2018–22. doi: 10.1073/pnas.0812697106
40. Jagust WJ, Landau SM. Alzheimer’s disease neuroimaging I. Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci. (2012) 32:18227–33. doi: 10.1523/JNEUROSCI.3266-12.2012
41. Wu L, Zhang X, Zhao L. Human ApoE isoforms differentially modulate brain glucose and ketone body metabolism: implications for Alzheimer’s disease risk reduction and early intervention. J Neurosci. (2018) 38:6665–81. doi: 10.1523/JNEUROSCI.2262-17.2018
42. Griffin WST, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, et al. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. (2006) 8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x
43. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
44. Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol Aging. (2013) 34:1051–9. doi: 10.1016/j.neurobiolaging.2012.09.012
45. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. (2018) 9:586. doi: 10.3389/fimmu.2018.00586
46. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
47. Vance RE, Isberg RR, Portnoy DA. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. (2009) 6:10–21. doi: 10.1016/j.chom.2009.06.007
48. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. (2008) 8:279–89. doi: 10.1038/nri2215
49. Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. (1997) 388:878–81. doi: 10.1038/42257
50. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. (2011) 91:461–553. doi: 10.1152/physrev.00011.2010
51. Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. (2016) 90:724–39. doi: 10.1016/j.neuron.2016.05.003
52. Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun. (2015) 6:6176. doi: 10.1038/ncomms7176
53. Yu Y, Ye RD. Microglial Abeta receptors in Alzheimer’s disease. Cell Mol Neurobiol. (2015) 35:71–83. doi: 10.1007/s10571-014-0101-6
54. Baik SH, Kang S, Son SM, Mook-Jung I. Microglia contributes to plaque growth by cell death due to uptake of amyloid beta in the brain of Alzheimer’s disease mouse model. Glia. (2016) 64:2274–90. doi: 10.1002/glia.23074
55. Veerhuis R, Janssen I, De Groot CJ, Van Muiswinkel FL, Hack CE, Eikelenboom P. Cytokines associated with amyloid plaques in Alzheimer’s disease brain stimulate human glial and neuronal cell cultures to secrete early complement proteins, but not C1-inhibitor. Exp Neurol. (1999) 160:289–99. doi: 10.1006/exnr.1999.7199
56. Li Y, Liu L, Kang J, Sheng JG, Barger SW, Mrak RE, et al. Neuronal–Glial Interactions Mediated by Interleukin-1 Enhance Neuronal Acetylcholinesterase Activity and mRNA Expression. The J Neurosci. (2000) 20:149–55. doi: 10.1523/jneurosci.20-01-00149.2000
57. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. (2008) 28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
58. Combs CK, Karlo JC, Kao S-C, Landreth GE. β-Amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. (2001) 21:1179–88. doi: 10.1523/jneurosci.21-04-01179.2001
59. Houtman J, Freitag K, Gimber N, Schmoranzer J, Heppner FL, Jendrach M. Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. EMBO J. (2019) 38:e99430. doi: 10.15252/embj.201899430
60. Jarosz-Griffiths HH, Noble E, Rushworth JV, Hooper NM. Amyloid-beta receptors: the good, the bad, and the prion protein. J Biol Chem. (2016) 291:3174–83. doi: 10.1074/jbc.R115.702704
61. Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. (2008) 28:4283–92. doi: 10.1523/JNEUROSCI.4814-07.2008
62. Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, et al. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. (2002) 277:47373–9. doi: 10.1074/jbc.M208788200
63. Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. (2012) 32:17321–31. doi: 10.1523/JNEUROSCI.1569-12.2012
64. Wang CY, Wang ZY, Xie JW, Cai JH, Wang T, Xu Y, et al. CD36 upregulation mediated by intranasal LV-NRF2 treatment mitigates hypoxia-induced progression of Alzheimer’s-like pathogenesis. Antioxid Redox Signal. (2014) 21:2208–30. doi: 10.1089/ars.2014.5845
65. Choi BR, Cho WH, Kim J, Lee HJ, Chung C, Jeon WK, et al. Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer’s disease. Exp Mol Med. (2014) 46:e75. doi: 10.1038/emm.2013.147
66. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
67. Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
68. Ahmed ME, Iyer S, Thangavel R, Kempuraj D, Selvakumar GP, Raikwar SP, et al. Co-localization of glia maturation factor with NLRP3 inflammasome and autophagosome markers in human Alzheimer’s disease brain. J Alzheimers Dis. (2017) 60:1143–60. doi: 10.3233/JAD-170634
69. Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, et al. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav Immun. (2017) 61:306–16. doi: 10.1016/j.bbi.2016.12.014
70. Kuwar R, Rolfe A, Di L, Xu H, He L, Jiang Y, et al. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J Neuroinflammation. (2019) 16:81. doi: 10.1186/s12974-019-1471-y
71. Yap JKY, Pickard BS, Chan EWL, Gan SY. The role of neuronal NLRP1 inflammasome in Alzheimer’s disease: bringing neurons into the neuroinflammation game. Mol Neurobiol. (2019) 56:7741–53. doi: 10.1007/s12035-019-1638-7
72. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
73. Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. (2005) 2:24. doi: 10.1186/1742-2094-2-24
74. Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. (2009) 4:399–418. doi: 10.1007/s11481-009-9164-4
75. Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. (2006) 29:68–74. doi: 10.1016/j.tins.2005.12.005
76. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. (2007) 10:1387–94. doi: 10.1038/nn1997
77. Dionisio-Santos DA, Olschowka JA, O’Banion MK. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J Neuroinflammation. (2019) 16:74. doi: 10.1186/s12974-019-1453-0
78. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. (2013) 368: 117–27.
79. Hooli BV, Parrado AR, Mullin K, Yip WK, Liu T, Roehr JT, et al. The rare TREM2 R47H variant exerts only a modest effect on Alzheimer disease risk. Neurology. (2014) 83:1353–8. doi: 10.1212/WNL.0000000000000855
80. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. (2013) 368:107–16. doi: 10.1056/NEJMoa1211103
81. Paloneva J, Mandelin J, Kiialainen A, Bohling T, Prudlo J, Hakola P, et al. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med. (2003) 198:669–75. doi: 10.1084/jem.20030027
82. Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. (2002) 71:656–62. doi: 10.1086/342259
83. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. (2005) 201:647–57.
84. Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. (2009) 109:1144–56. doi: 10.1111/j.1471-4159.2009.06042.x
85. Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. (2017) 18:1186–98. doi: 10.15252/embr.201743922
86. Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, et al. TREM2 is upregualated in amyloid plaques-associated microglia in Aged APP23 transgenic mice. Glia. (2008) 56:1438–47.
87. Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, et al. Altered microglial response to Ab plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. (2014) 9:20. doi: 10.1186/1750-1326-9-20
88. Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatiory macrophages and ameliorates pathology in Alzheimers’s Disease mouse models. J Exp Med. (2015) 212:287–95.
89. Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. Neurobiol Dis. (2017) 37:637–47.
90. Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, et al. TREM-2 mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. (2016) 213:667–75.
91. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains microglia response in an Alzheimer’s disease model. Cell. (2015) 160:1061–71.
92. Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. (2019) 22:191–204. doi: 10.1038/s41593-018-0296-9
93. Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ. Astrocytes in Alzheimer’s disease. Neurotherapeutics. (2010) 7:399–412. doi: 10.1016/j.nurt.2010.05.017
94. Kalmár B, Kittel Á, Lemmens R, Környei Z, Madarász E. Cultured astrocytes react to LPS with increased cyclooxygenase activity and phagocytosis. Neurochem Int. (2001) 38:453–61. doi: 10.1016/s0197-0186(00)00090-5
95. Wallace MN, Geddes JG, Farquhar DA, Masson MR. Nitric oxide synthase in reactive astrocytes adjacent to beta-amyloid plaques. Exp Neurol. (1997) 144:266–72. doi: 10.1006/exnr.1996.6373
96. Gonzalez-Reyes RE, Nava-Mesa MO, Vargas-Sanchez K, Ariza-Salamanca D, Mora-Munoz L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci. (2017) 10:427. doi: 10.3389/fnmol.2017.00427
97. Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci. (2012) 32:6391–410. doi: 10.1523/JNEUROSCI.6221-11.2012
98. Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. (2016) 532:195–200. doi: 10.1038/nature17623
99. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
100. Laskowitz DT, Matthew WD, Bennett ER, Schmechel D, Herbstreith MH, Goel S, et al. Endogenous apolipoprotein E suppresses LPS-stimulated microglial nitric oxide production. Neuro Rep. (1998) 9:615–8.
101. Laskowitz DT, Goel S, Bennett ER, Matthew WD. Apolipoprotein E suppresses glial cell secretion of TNFα. J Neuroimmunol. (1997) 76:70–4. doi: 10.1016/s0165-5728(97)00021-0
102. Pocivavsek A, Rebeck GW. Inhibition of c-Jun N-terminal kinase increases apoE expression in vitro and in vivo. Biochem Biophys Res Commun. (2009) 387:516–20. doi: 10.1016/j.bbrc.2009.07.048
103. Bales K. Neuroinflammation and Alzheimer’s disease: critical roles for cytokine/Aβ-induced glial activation, NF-κB, and apolipoprotein E. Neurobiol Aging. (2000) 21:427–32. doi: 10.1016/s0197-4580(00)00143-3
104. Qiao X, Cummins DJ, Paul SM. Neuroinflammation-induced acceleration of amyloid deposition in the APPV717F transgenic mouse. Eur J Neurosci. (2001) 14:474–82. doi: 10.1046/j.0953-816x.2001.01666.x
105. Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, et al. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. (2003) 278:48529–33. doi: 10.1074/jbc.M306923200
106. Colton C, Brown C, Cook D, Needham L, Xu Q, Czapiga M, et al. APOE and the regulation of microglial nitric oxide production: a link between genetic risk and oxidative stress. Neurobiol Aging. (2002) 23:777–85. doi: 10.1016/s0197-4580(02)00016-7
107. Vitek MP, Brown CM, Colton CA. APOE genotype specific differences in the innate immune response. Neurobiol Aging. (2009) 30:1350–60.
108. Hashimoto T, Serrano-Pozo A, Hori Y, Adams KW, Takeda S, Banerji AO, et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J Neurosci. (2012) 32:15181–92. doi: 10.1523/JNEUROSCI.1542-12.2012
109. Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. (2012) 2:a006312. doi: 10.1101/cshperspect.a006312
110. Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA. (1993) 90:8098–102. doi: 10.1073/pnas.90.17.8098
111. Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, Saunders A, et al. Apolipoprotein E associates with beta amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J Clin Invest. (1994) 94:860–9. doi: 10.1172/JCI117407
112. Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW, et al. ApoE4 accelerates early seeding of amyloid pathology. Neuron. (2017) 96:1024–32e3. doi: 10.1016/j.neuron.2017.11.013
113. Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. (2011) 3:89ra57. doi: 10.1126/scitranslmed.3002156
114. Rodriguez GA, Tai LM, LaDu MJ, Rebeck GW. Human APOE4 increases microglia reactivity at Abeta plaques in a mouse model of Abeta deposition. J Neuroinflammation. (2014) 11:111. doi: 10.1186/1742-2094-11-111
115. Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. (1999) 96:15233–8. doi: 10.1073/pnas.96.26.15233
116. Wood SJ, Chan W, Wetzel R. Seeding of a beta fibril formation is inhibited by all three isotypes of apolipoprotein E. Biochemistry. (1996) 35:12623–8. doi: 10.1021/bi961074j
117. Harper JD, Lansbury PT Jr. Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. (1997) 66:385–407. doi: 10.1146/annurev.biochem.66.1.385
118. Nelson PT, Pious NM, Jicha GA, Wilcock DM, Fardo DW, Estus S, et al. APOE-epsilon2 and APOE-epsilon4 correlate with increased amyloid accumulation in cerebral vasculature. J Neuropathol Exp Neurol. (2013) 72:708–15. doi: 10.1097/NEN.0b013e31829a25b9
119. Ghebremedhin E, Schultz C, Thal DR, Rub U, Ohm TG, Braak E, et al. Gender and age modify the association between APOE and AD-related neuropathology. Neurology. (2001) 56:1696–701. doi: 10.1212/wnl.56.12.1696
120. Nagy ZS, Esiri MM, Jobst KA, Johnston C, Litchfield S, Sim E, et al. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience. (1995) 69:757–61. doi: 10.1016/0306-4522(95)00331-c
121. Yoon SS, Jo SA. Mechanisms of amyloid-beta peptide clearance: potential therapeutic targets for Alzheimer’s disease. Biomol Ther (Seoul). (2012) 20:245–55. doi: 10.4062/biomolther.2012.20.3.245
122. Muth C, Hartmann A, Sepulveda-Falla D, Glatzel M, Krasemann S. Phagocytosis of apoptotic cells is specifically upregulated in ApoE4 expressing microglia in vitro. Front Cell Neurosci. (2019) 13:181. doi: 10.3389/fncel.2019.00181
123. Huang YA, Zhou B, Wernig M, Sudhof TC. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and abeta secretion. Cell. (2017) 168:427–41.e21. doi: 10.1016/j.cell.2016.12.044
124. Ulrich JD, Ulland TK, Mahan TE, Nyström S, Nilsson KP, Song WM, et al. ApoE facilitates the microglial response to amyloid plaque pathology. J Exp Med. (2018) 215:1047–58. doi: 10.1084/jem.20171265
125. Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. (2017) 549:523–7. doi: 10.1038/nature24016
126. Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. (2019) 216:2546–61. doi: 10.1084/jem.20190980
127. Sosna J, Philipp S, Albay R III, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, et al. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener. (2018) 13:11. doi: 10.1186/s13024-018-0244-x
128. Bailey CC, DeVaux LB, Farzan M. The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J Biol Chem. (2015) 290:26033–42. doi: 10.1074/jbc.M115.677286
129. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ and thereby facilitates uptake of amyloid-beta by micrglia. Neuron. (2016) 91:328–40. doi: 10.1016/j.neuron.2016.06.015
130. Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem. (2015) 209:26043–50.
131. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. (2018) 173:1073–81. doi: 10.1016/j.cell.2018.05.003
132. Pimenova AA, Marcora E, Goate AM. A tale of two genes: microglial apoe and Trem2. Immunity. (2017) 47:398–400. doi: 10.1016/j.immuni.2017.08.015
133. Lee CYD, Daggett A, Gu X, Jiang LL, Langfelder P, Li X, et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron. (2018) 97:1032–48e5. doi: 10.1016/j.neuron.2018.02.002
134. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. (2017) 47:566–81.e9. doi: 10.1016/j.immuni.2017.08.008
135. Ignatius MJ, Gebicke-Harter PJ, Skene JH, Schilling JW, Weisgraber KH, Mahley RW, et al. Expression of apolipoprotein E during nerve degeneration and regeneration. Proc Natl Acad Sci USA. (1986) 83:1125–9. doi: 10.1073/pnas.83.4.1125
136. Fagan AM, Murphy BA, Patel SN, Kilbridge JF, Mobley WC, Bu G, et al. Evidence for normal aging of the septo-hippocampal cholinergic system in apoE. (-/-) mice but impaired clearance of axonal degeneration products following injury. Exp Neurol. (1998) 151:314–25. doi: 10.1006/exnr.1998.6818
137. Lane-Donovan C, Wong WM, Durakoglugil MS, Wasser CR, Jiang S, Xian X, et al. Genetic restoration of plasma apoe improves cognition and partially restores synaptic defects in ApoE-deficient mice. J Neurosci. (2016) 36:10141–50. doi: 10.1523/JNEUROSCI.1054-16.2016
138. Di Meco A, Curtis ME, Lauretti E, Pratico D. Autophagy dysfunction in Alzheimer’s disease: mechanistic insights and new therapeutic opportunities. Biol Psychiatry. (2019) 87:797–807. doi: 10.1016/j.biopsych.2019.05.008
139. Cortes CJ, La Spada AR. TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol Dis. (2019) 122:83–93. doi: 10.1016/j.nbd.2018.05.012
140. Parcon PA, Balasubramaniam M, Ayyadevara S, Jones RA, Liu L, Shmookler Reis RJ, et al. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement. (2018) 14:230–42. doi: 10.1016/j.jalz.2017.07.754
141. Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. (2008) 28:11445–53. doi: 10.1523/JNEUROSCI.1972-08.2008
142. Chu Q, Diedrich JK, Vaughan JM, Donaldson CJ, Nunn MF, Lee KF, et al. HtrA1 proteolysis of ApoE in vitro is allele selective. J Am Chem Soc. (2016) 138:9473–8. doi: 10.1021/jacs.6b03463
143. Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci USA. (2001) 98:8838–43. doi: 10.1073/pnas.151254698
144. Munoz SS, Garner B, Ooi L. Understanding the role of ApoE fragments in Alzheimer’s disease. Neurochem Res. (2019) 44:1297–305. doi: 10.1007/s11064-018-2629-1
145. Laskowitz DT, Thekdi AD, Thekdi SD, Han SK, Myers JK, Pizzo SV, et al. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp Neurol. (2001) 167:74–85. doi: 10.1006/exnr.2001.7541
146. Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: a matter of blood-brain barrier dysfunction? J Exp Med. (2017) 214:3151–69. doi: 10.1084/jem.20171406
147. Friedman G, Froom P, Sazbon L, Grinblatt I, Shochina M, Tsenter J, et al. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. (1999) 52:244–8. doi: 10.1212/wnl.52.2.244
148. Sorbi S, Nacmias B, Piacentini S, Repice A, Latorraca S, Forleo P, et al. ApoE as a prognostic factor for post-traumatic coma. Nat Med. (1995) 1:852. doi: 10.1038/nm0995-852
149. Teasdale GM, Nicoll JAR, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet. (1997) 350:1069–71. doi: 10.1016/s0140-6736(97)04318-3
150. Biffi A, Sonni A, Anderson CD, Kissela B, Jagiella JM, Schmidt H, et al. International stroke genetics C. variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann Neurol. (2010) 68:934–43. doi: 10.1002/ana.22134
151. Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem. (2011) 286:17536–42. doi: 10.1074/jbc.M111.225532
152. Turner RJ, Sharp FR. Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front Cell Neurosci. (2016) 10:56. doi: 10.3389/fncel.2016.00056
153. Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. (2012) 485:512–6. doi: 10.1038/nature11087
154. Main BS, Villapol S, Sloley SS, Barton DJ, Parsadanian M, Agbaegbu C, et al. Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol Neurodegener. (2018) 13:17. doi: 10.1186/s13024-018-0249-5
155. Yamazaki Y, Shinohara M, Yamazaki A, Ren Y, Asmann YW, Kanekiyo T, et al. ApoE (Apolipoprotein E) in brain pericytes regulates endothelial function in an isoform-dependent manner by modulating basement membrane components. Arterioscler Thromb Vasc Biol. (2020) 40:128–44. doi: 10.1161/ATVBAHA.119.313169
156. Wilcock DM, Morgan D, Gordon MN, Taylor TL, Ridnour LA, Wink DA, et al. Activation of matrix metalloproteinases following anti-Abeta immunotherapy; implications for microhemorrhage occurrence. J Neuroinflammation. (2011) 8:115. doi: 10.1186/1742-2094-8-115
157. Muza P, Bachmeier C, Mouzon B, Algamal M, Rafi NG, Lungmus C, et al. APOE Genotype specific effects on the early neurodegenerative sequelae following chronic repeated mild traumatic brain injury. Neuroscience. (2019) 404:297–313. doi: 10.1016/j.neuroscience.2019.01.049
158. Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. (2018) 560:185–91. doi: 10.1038/s41586-018-0368-8
159. Sweeney MD, Zlokovic BV. A lymphatic waste-disposal system implicated in Alzheimer’s disease. Nature. (2018) 560:172–4. doi: 10.1038/d41586-018-05763-0
160. Zhao N, Attrebi ON, Ren Y, Qiao W, Sonustun B, Martens YA, et al. APOE4 exacerbates alpha-synuclein pathology and related toxicity independent of amyloid. Sci Transl Med. (2020) 12:eaay1809. doi: 10.1126/scitranslmed.aay1809
161. Davis AA, Inman CE, Wargel ZM, Dube U, Freeberg BM, Galluppi A, et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med. (2020) 12:eaay3069. doi: 10.1126/scitranslmed.aay3069
162. ASPE National Plan to Address Alzheimer’s Disease: 2019 Update2019. Rosemont, IL: American Society of Plumbing Engineers. (2019).
163. Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. (2009) 73:2061–70. doi: 10.1212/WNL.0b013e3181c67808
164. Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. (2012) 11:241–9. doi: 10.1016/s1474-4422(12)70015-7
165. Doody RS, Farlow M, Aisen PS, Alzheimer’s Disease Cooperative Study Data Analysis and Publication Committee. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N Engl J Med. (2014) 370:1459–60. doi: 10.1056/NEJMc1402193
166. Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature. (2016) 537:50–6. doi: 10.1038/nature19323
167. Solomon A, Turunen H, Ngandu T, Peltonen M, Levalahti E, Helisalmi S, et al. Effect of the apolipoprotein E genotype on cognitive change during a multidomain lifestyle intervention: a subgroup analysis of a randomized clinical trial. JAMA Neurol. (2018) 75:462–70. doi: 10.1001/jamaneurol.2017.4365
168. Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. (2015) 385:2255–63. doi: 10.1016/s0140-6736(15)60461-5
Keywords: microglia, cytokines, dementia, apolipoprotein E allele, neuroinflammation
Citation: Kloske CM and Wilcock DM (2020) The Important Interface Between Apolipoprotein E and Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 11:754. doi: 10.3389/fimmu.2020.00754
Received: 02 February 2020; Accepted: 02 April 2020;
Published: 30 April 2020.
Edited by:
Sally Ann Frautschy, University of California, Los Angeles, United StatesReviewed by:
Jacob Raber, Oregon Health and Science University, United StatesWayne W. Poon, University of California, Irvine, United States
G. William Rebeck, Georgetown University, United States
Copyright © 2020 Kloske and Wilcock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Donna M. Wilcock, ZG9ubmEud2lsY29ja0B1a3kuZWR1