 Lucia S. Borges
Lucia S. Borges David P. Richman
David P. Richman
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 May 2020
Sec. Multiple Sclerosis and Neuroimmunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00707
This article is part of the Research TopicAdvances in Autoimmune Myasthenia GravisView all 17 articles
Thirty to fifty percent of patients with acetylcholine receptor (AChR) antibody (Ab)-negative myasthenia gravis (MG) have Abs to muscle specific kinase (MuSK) and are referred to as having MuSK-MG. MuSK is a 100 kD single-pass post-synaptic transmembrane receptor tyrosine kinase crucial to the development and maintenance of the neuromuscular junction. The Abs in MuSK-MG are predominantly of the IgG4 immunoglobulin subclass. MuSK-MG differs from AChR-MG, in exhibiting more focal muscle involvement, including neck, shoulder, facial and bulbar-innervated muscles, as well as wasting of the involved muscles. MuSK-MG is highly associated with the HLA DR14-DQ5 haplotype and occurs predominantly in females with onset in the fourth decade of life. Some of the standard treatments of AChR-MG have been found to have limited effectiveness in MuSK-MG, including thymectomy and cholinesterase inhibitors. Therefore, current treatment involves immunosuppression, primarily by corticosteroids. In addition, patients respond especially well to B cell depletion agents, e.g., rituximab, with long-term remissions. Future treatments will likely derive from the ongoing analysis of the pathogenic mechanisms underlying this disease, including histologic and physiologic studies of the neuromuscular junction in patients as well as information derived from the development and study of animal models of the disease.
Myasthenia gravis (MG) is an autoimmune disease of the neuromuscular junction synapse (NMJ) characterized by weakness that worsens with continued muscle work and improves with resting of the involved muscle(s). Non-immune genetic diseases of this synapse, referred to as congenital myasthenic syndromes, produce similar symptoms (1, 2). For MG, the distribution of weakness is distinctive, involving primarily the extraocular muscles. In ocular MG, involvement is limited to these muscles. In more severe cases (generalized MG), the pontine- and bulbar-innervated muscles and the respiratory muscles are commonly also affected. Least frequently involved are the extremity muscles.
Most MG patients have circulating antibodies (Abs) to the NMJ postsynaptic neurotransmitter receptor, nicotinic acetylcholine receptor (AChR), AChR-MG (3, 4). The pathogenic role of these Abs has been demonstrated by induction of MG in experimental animals by both passive transfer of MG serum Abs (5) or anti-AChR monoclonal Abs (mAbs) (6–8) and by active immunization with purified AChR (9). For both AChR-MG and its experimental models, the Abs induce a destructive inflammatory attack on the AChR-containing postsynaptic membrane (10–13). In generalized MG, AChR Abs are present in 90 percent of patients. The remaining cases were initially designated as seronegative MG.
The earliest studies of these AChR Ab-negative MG cases failed to identify clinical or electrophysiologic features that distinguished it from AChR-MG. In 2001, Hoch and coworkers identified Abs to a different postsynaptic membrane protein, muscle-specific kinase (MuSK), present in the sera of 30–50 percent of seronegative MG patients (14). Once this group of MuSK-Ab-positive MG patients (MuSK-MG) was identified, clinical characteristics of MuSK-MG were discerned that distinguish it from AChR-MG, suggesting that MuSK-MG is a distinct autoimmune disease. Most striking is muscle wasting in many of the affected muscles. Although MuSK-MG is also an Ab-mediated disease, inflammatory damage to the NMJ does not occur. In fact, the majority of the Abs are of the IgG4 immunoglobulin subclass, which is characterized partly by inability to activate complement or bind to Fc receptors. The proposed mode of action of these auto-Abs is blockade of the normal function of MuSK.
Many of the standard treatments of AChR-MG are of limited effectiveness in MuSK-MG, including thymectomy and cholinesterase inhibitors. Therefore, current treatment involves immunosuppression, primarily by corticosteroids or B cell depletion agents.
Since the initial identification of MuSK-MG, a number of experimental animal models of this disease have been developed. As is the case in AChR-MG, careful analyses of both the human disease and the animal models have led to the determination of the pathogenic mechanisms underlying this disease. Such information has the potential for the development of improved treatments of MuSK-MG and similar diseases.
Muscle specific kinase was identified as a postsynaptic integral membrane protein playing a pivotal role in the development of the NMJ (15–19). This synapse begins to form when the axon growth cone of a developing motor neuron encounters a developing myotube and begins to secrete agrin, a glycoprotein with a laminin-binding domain that anchors it to the extracellular matrix (20–25). Prior to the arrival of the axon, AChRs, which initially are spread diffusely along the myotube, begin to cluster in the central region of the myotube (26, 27). When the axon growth cone eventually encounters this region and secretes agrin (Figure 1) (9, 10), the agrin induces more extensive dense clustering of the AChRs in the postsynaptic endplate membrane, which is the first step in the elaboration of this structure into its adult architecture (Figure 2), including a pretzel-like topographic profile (Figure 3A) and marked folding and specialization of that membrane at the ultrastructural level (Figure 3B) (20–25, 28–34).
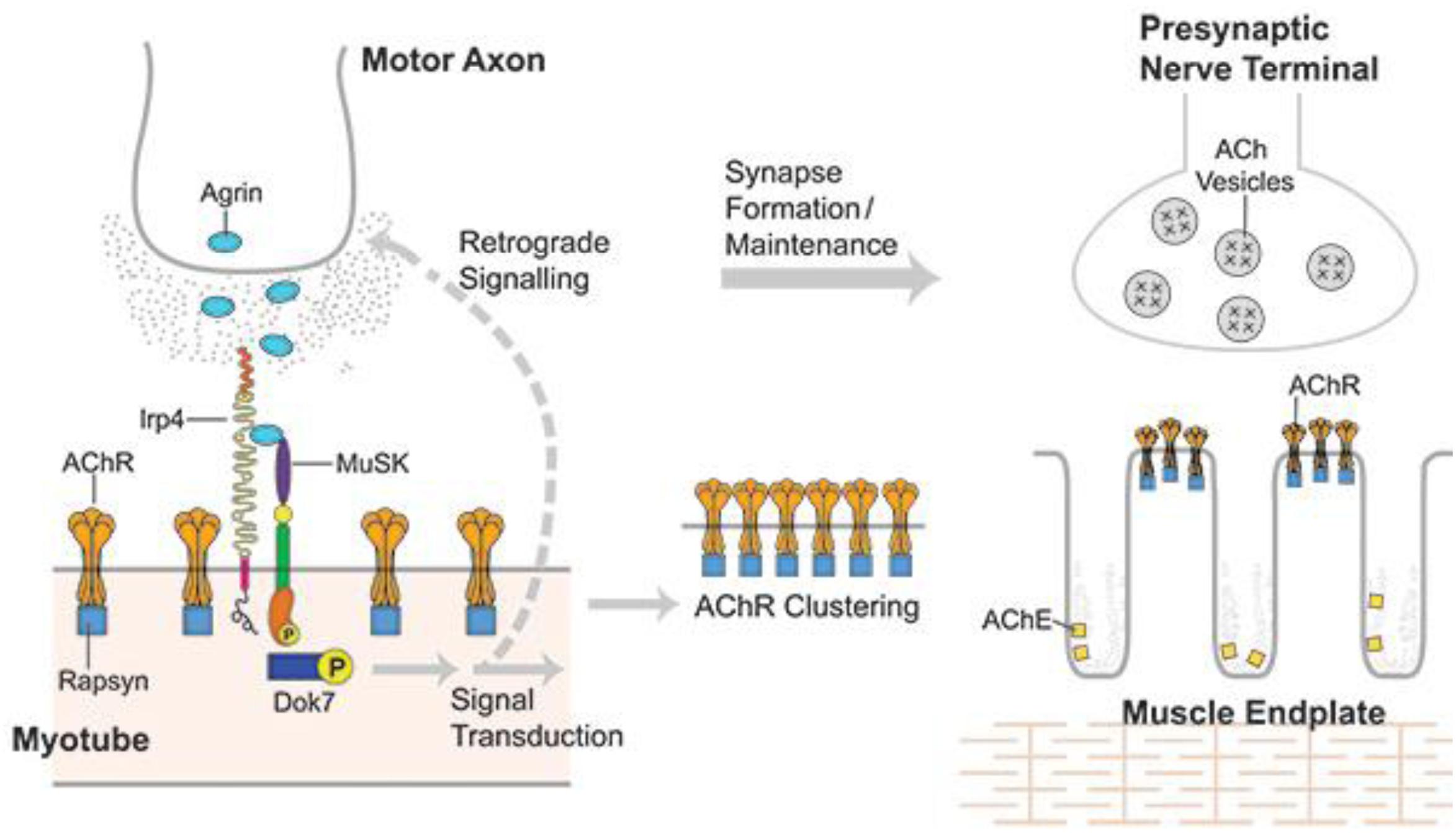
Figure 1. Developing NMJ: The motor axon growth cone releases agrin into the intercellular matrix when it reaches a developing myotube. Agrin binds lrp4 and the complex binds MuSK resulting in activation of MuSK, which self-phosphorylates and then initiates a series of phosphorylations beginning with Dok7 and ending with rapsyn and 8 subunit of AChR. This process induces dense AChR clustering, the first step in the development of both the postsynaptic and presynaptic portions of the mature NMJ. From Richman (66) with permission.
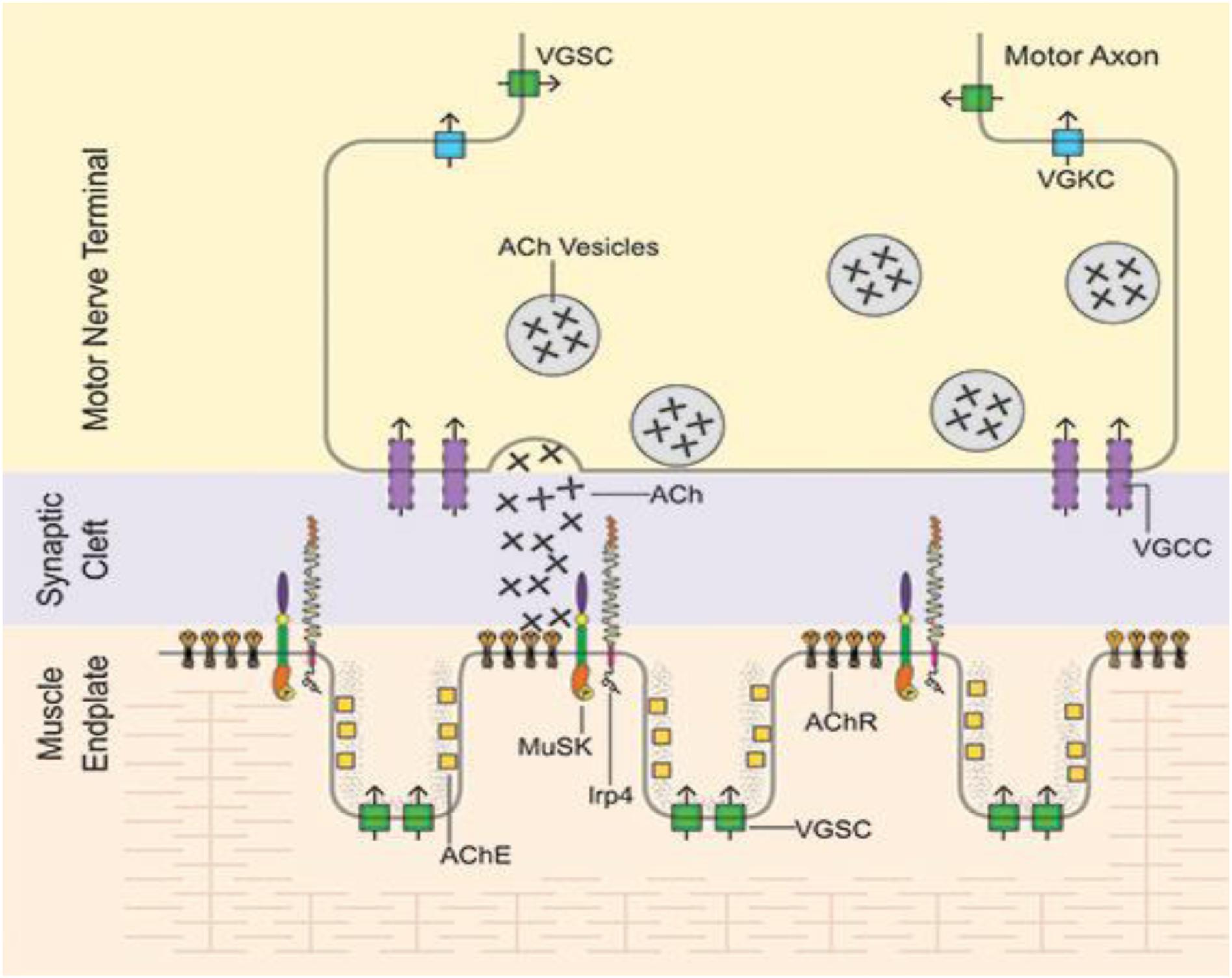
Figure 2. Mature NMJ: Motor axon action potentials reach the motor nerve terminal leading to release of vesicles of acetylcholine (ACh), which diffuses across the synaptic clef to bind to the tightly packed acetylcholine receptors (AChR) located on the peaks of the folds of the endplate membrane. After AChR activation, ACh is then hydrolyzed by acetylcholinesterase (AChE) in the muscle basal lamina. VGSC, voltage-gated sodium channels; VGKC, voltage-gated potassium channels; VGCC, voltage-gated calcium channels. From Richman (66) with permission.
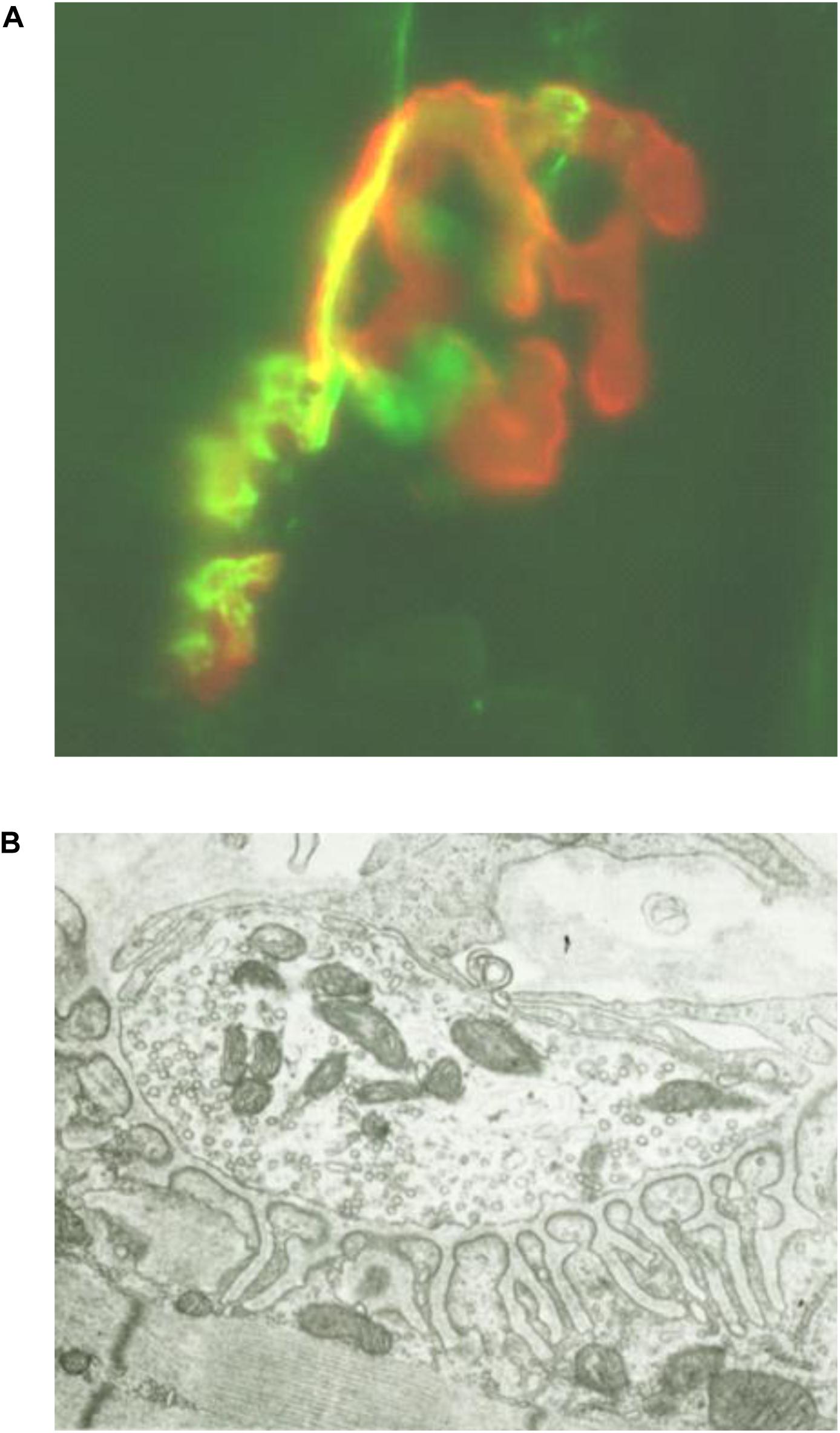
Figure 3. (A) Photomicrograph (x80) of longitudinal frozen section of diaphragm muscle stained immunohistochemically with alpha-bungarotoxin to label AChR (red) and anti-synapsin plus anti-neurofilament Abs to label presynaptic nerve terminals and axons (green), demonstrating pretzel appearance of endplate membrane. (B) Electron micrograph (×5000) of transverse section of diaphragm muscle neuromuscular junction demonstrating highly folded endplate membrane. [Modified from (63) with permission].
Both the initial spontaneous AChR clustering and the agrin-induced effects require the presence of MuSK (23, 35, 36). The paradoxical observation that agrin and MuSK do not bind in vitro led to a search for a third (intermediary) protein required for their interaction, which was eventually found and identified as the postsynaptic transmembrane protein low density lipoprotein receptor-related protein 4 (lrp4) (37–39).
The agrin-lrp4-MuSK interaction leads first to MuSK dimerization and then self-phosphorylation. The latter effect initiates a series of intracellular protein phosphorylations mediated through a downstream signal transduction pathway beginning with Dok7 and ending with rapsyn and the β subunit of AChR (40–43). Activation of this pathway results in dense AChR clustering, the first step in the elaboration of the postsynaptic components of the synapse (Figure 2) (44, 45). The AChR clustering also includes MuSK and lrp4 and the other components of the MuSK-associated signaling pathway (21, 46).
Activation of the agrin/lrp4/MuSK pathway leads, as well, to increased expression/synthesis of the components of the pathway and other endplate-specific proteins (by subsynaptic muscle nuclei) (22, 47–49). The induced AChR clustering, and the eventual elaboration of the entire adult postsynaptic endplate structure, involves polymerization of actin leading to the production of an intracellular scaffolding, comprised of a number of proteins, upon which the mature structure of the muscle endplate is formed. This process results in tight packing of the phosphorylated AChRs on the peaks of the synaptic folds opposite the specialized nerve terminal (Figure 3B) (44, 45, 50). This actin/cytoskeletal remodeling is contributed to by a number of other proteins in the MuSK signaling pathway, most prominently cortactin, which when phosphorylated directly enhances further actin polymerization (44, 51). Extracellularly, ColQ, the collagen-like portion of the NMJ enzyme acetylcholinesterase, binds to the extracellular portion of concentrated (clustered) MuSK (52, 53) and also to the extracellular matrix protein perlecan, leading to anchoring of the enzyme to the extracellular matrix at the clustering sites (53).
The agrin/lrp4-induced activation (phosphorylation) of MuSK is also associated with development of the presynaptic portion of the NMJ. MuSK activation initiates a separate (less well understood) retrograde pathway, resulting first in a stop signal terminating the travels of the motor axon (Figure 1) (54, 55). The increased concentration (clustering) of lrp4 at the developing NMJ induced by activation of the MuSK transduction pathway is required for the further development of the axon growth cone into the adult specialized presynaptic nerve terminal. The concentrated lrp4 binds the nerve terminal, but the presynaptic “receptor” for lrp4 and the subsequent developmental steps have not yet been identified (56) (21).
The further maturation of the NMJ and, in particular, the mechanisms involved in the maintenance of the mature NMJ, are even less well understood (33, 55, 57, 58). Maintenance of the NMJ does appear to require MuSK functionality, as demonstrated by the dissolution of the synapse in adult animals (in the absence of inflammation) both in (1) experimental MuSK-MG induced by either passive or active immunization with MuSK (59–63) and (2) in adult animals in which MuSK has been inactivated or knocked down (64, 65).
Muscle specific kinase is a 100 kD single-pass transmembrane receptor tyrosine kinase with an N-terminal extracellular domain followed by a short transmembrane domain and then a C-terminal cytoplasmic domain (Figure 4) (15, 16, 18, 19). The extracellular domain of MuSK, which is required for interaction with agrin and lrp4, comprises three immunoglobulin (Ig)-like domains (37, 39, 67) followed by a cysteine-rich frizzled-like region (labeled C6-box in Figure 4) (15, 16, 18, 45). The cytoplasmic domain contains the kinase activity and signaling components of the molecule that lead to the development of the postsynaptic apparatus (see above) (45).
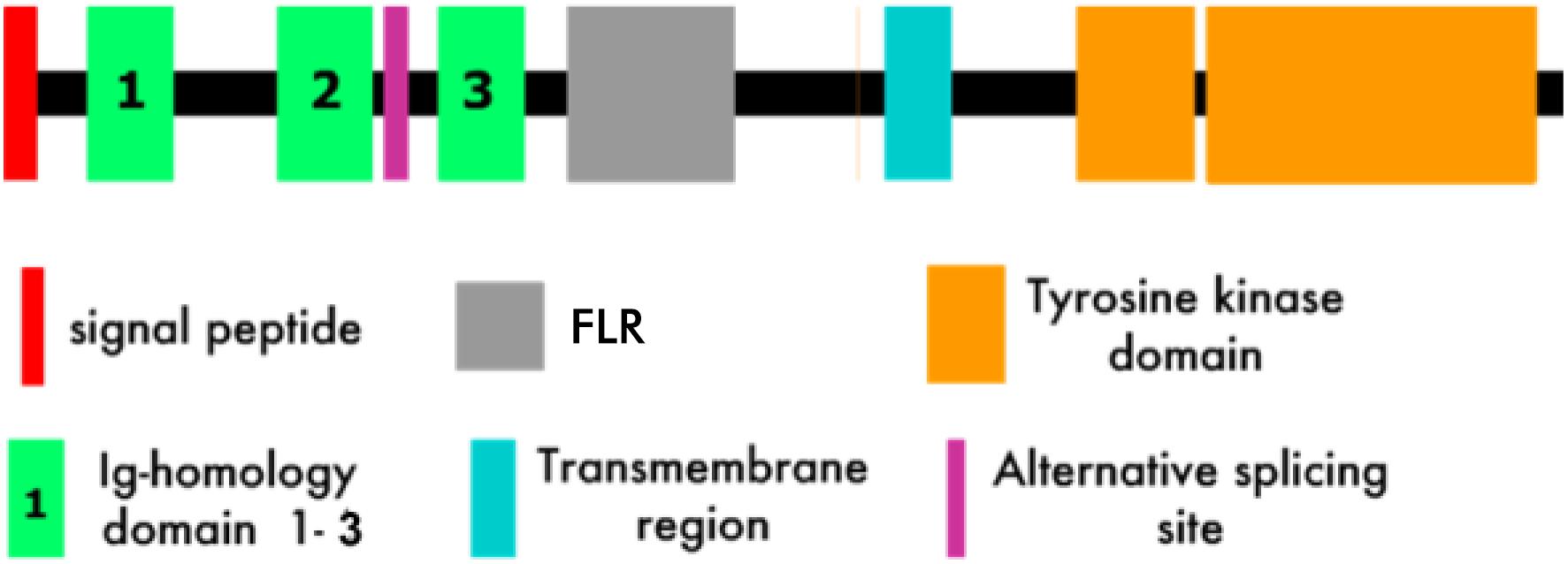
Figure 4. MuSK Structure (Modified from 15). FLR, Frizzled-like region.
The first two extracellular Ig-like domains, which are rigidly joined in a linear array (67), appear to play a dual role in activation of MuSK signaling. First, Ig-1 is crucial for binding to the MuSK ligand, i.e., agrin-associated lrp4 (68). Second, it is the substrate for the dimerization of two MuSK molecules (67). Dimerization is required for MuSK (trans) autophosphorylation, the first step in the activation of the MuSK-associated signaling pathway (69, 70). Autophosphorylation, along with binding of Dok7, an intracellular MuSK target, results in full activation of the MuSK kinase activity (71, 72). It is the combination of ligand (agrin/lrp4) binding and the full establishment of its kinase activity that results in the sequence of protein phosphorylations by MuSK that comprise the MuSK-associated signaling pathway that leads first to AChR clustering and subsequently to formation of the mature NMJ. The role of the frizzled-like region, which functions as a receptor for the wnt family of intracellular signaling proteins, is not yet well understood (73–76).
Early studies employing both rat and human MuSK have determined that it is only the extracellular domain of the molecule that is the target of the MuSK Abs in MuSK-MG (see below) (14, 77).
The MuSK Ab-positive subgroup of “seronegative patients,” anti-MuSK MG, does have clinical similarities to AChR-MG but tends to differ significantly in exhibiting more focal involvement than AChR-MG, frequently with severe involvement of neck, shoulder, facial and bulbar-innervated muscles, although there is considerable variability from patient to patient (78–82). When the extremities are involved, proximal muscles are more affected than distal ones (83). Unlike AChR-MG, many patients have wasting of these muscles (78, 79, 81, 84–86), and data suggest that this represents a direct myopathy and is not the result of denervation, a point that remains somewhat controversial (86–91).
The demographic characteristics of MuSK-MG differ from AChR MG. In the latter disease, the age/incidence curve is bimodal with a peak in the early 20’s, which is majority female, and with a second peak in the 60’s and 70’s, which is majority male (92–94). In contrast, MuSK-MG tends to occur in the 30’s with very strong female predominance (95). Also, MuSK-MG is highly associated with the HLA DR14-DQ5 haplotype (96).
The restricted HLA (MHC) class II association in MuSK-MG suggests a role for T helper cells in this Ab-mediated disease. A recent study observed antigen (MuSK)-specific T cell responses in cultures of circulating mononuclear cells (MNC) from MuSK-MG patients. These anti-MuSK responses utilized a somewhat limited number of T cell receptor variable region genes (97–99), consistent with a genetically influenced disease-specific T cell response. Also, in contrast to AChR-MG, only rare MuSK MG patients have been found to have thymic lymphoid hyperplasia (97–101).
Remarkably, data concerning NMJ histology and microphysiology in MuSK-MG are very limited. In the three histologic studies available (102–104), only relatively mild abnormalities of NMJ morphology/function were observed. The changes were all postsynaptic, including partially denervated postsynaptic membrane and moderate degeneration of postsynaptic folds (102). One of two microelectrode studies found only postsynaptic abnormalities, marked decrease in miniature endplate potential (MEPP) amplitudes (103). However, the other study found both postsynaptic abnormalities, mild decrease in MEPP amplitudes, and presynaptic abnormalities, reduced levels of presynaptic acetylcholine release (102).
Anti-MuSK Abs are detected in 1–10% of patients with MG, 40% of the AChR Ab-negative patients (14, 77, 78, 80, 81, 105). Most of the anti-MuSK Abs belong to the IgG4 immunoglobulin subclass (77, 106), which is unable to either activate complement or induce antigenic modulation (107). However, passive transfer of the IgG4 component of MuSK-MG serum is especially effective in inducing the experimental disease (108).
Abs of the IgG4 subclass behave as if they are functionally monovalent (109). Because of single amino acid differences in the IgG4 heavy chain constant region, the inter-heavy chain disulfide bonds that join the two halves of the immunoglobulin molecule are markedly weakened, leading to frequent separation of the two heavy chains. The resultant “half molecules,” consisting of a single light chain covalently bound to a single heavy chain, can readily bind to a half molecule from another IgG4 Ab to reform a complete Ab molecule, so called “Fab arm exchange” (109, 110). The new Ab molecule is now bispecific, that is each Ab arm binds a different antigen. Such Abs, including IgG4 MuSK Abs, cannot crosslink single antigens and, therefore cannot induce antigenic modulation (which requires antigen crosslinking), a mechanism important in AChR-MG. Also as noted above, these IgG4 Abs are minimally interactive with the innate immune system, in that they are deficient in complement activation and in binding to cell surface Fc receptors (107). Anti-MuSK Abs of the IgG1 subclass, a subclass capable of engaging these components of the innate immune system, are also present in most MuSK-MG patients, but at much lower levels than the IgG4 Abs (111, 112). The role the IgG1 Abs play in MuSK-MG is not known.
Hence, the pathogenic mode of action of the auto-Abs in MuSK-MG differs from that of the AChR Abs in AChR-MG. Rather than inducing destructive damage to the NMJ or antigenic modulation, the anti-MuSK Abs mask the binding sites on MuSK that interact with its binding proteins (ligands), including lrp4/agrin and ColQ, thereby blocking MuSK function (106, 111). Blockade of MuSK ligand binding leads to a reduced postsynaptic density of AChRs and impairs their alignment in the postsynaptic membrane (60).
Most anti-MuSK Abs bind to the Ig-like domains of the extracellular portion of MuSK (Figure 4) (14, 77, 106, 113, 114). In one study of 53 MuSK-MG patients, all had Abs to Ig-like domain 1 and about 50 percent also had Abs to Ig-like domain 2. For female patients, it was rare to have Ab reactivity to domains other than Ig-like domain 1 (113). However, Abs to the frizzled-like domain have been observed in MuSK MG (115).
As noted above, few human studies have addressed directly the pathogenesis of MuSK-MG (102–104). None have observed complement-mediated injury or cellular infiltration of the NMJ. In fact, initially there was controversy concerning the role of anti-MuSK Abs in MuSK-MG pathogenesis (103, 116, 117), in spite of the ability of these Abs to act as MuSK antagonists in vitro. On the other hand, experimental studies, involving the induction of experimental models of MuSK-MG, have provided the strongest evidence concerning the pathogenic mechanisms underlying this disease. The data from MuSK-MG animal models induced by both passive and active immunization with MuSK demonstrate the role of the anti-MuSK Abs in the induction of both the weakness and the morphological and physiological NMJ changes observed in MuSK-MG (59, 60, 117–123).
A number of studies have assessed the effect of daily intraperitoneal injections into immunosuppressed mice of very large amounts of IgG (usually 35–50 mg per day) purified from (human) MuSK-MG serum. In one study, injections for 5 days produced reduced neuromuscular transmission but without clinical weakness (118). A second study made use of IgG purified from a severely affected patient injected for 14 days (total of 0.68 g), which resulted in weakness and weight loss. Histologic analysis of these animals found patchy reduction in NMJ AChR staining, reduced registration between nerve terminals and motor endplates (59) and reduced phosphorylation of the downstream components of the MuSK signaling pathway (60). The Abs also produce internalization of MuSK with more rapid degradation leading to reduced endplate MuSK concentrations (60, 117). The marked effectiveness of passive transfer of the IgG4 component of MuSK-MG serum is consistent with the role of these mechanisms in inducing the experimental disease (108). These observations, along with the absence of observed complement-mediated injury, support the hypothesis that the MuSK Abs induce the disease by blocking MuSK signaling in vivo with the resultant postsynaptic changes described above, as well as damage to the nerve terminals (see below).
In rabbits (117) and, to a lesser extent in mice (118–123), repeatedly immunized with MuSK protein (of human or rat origin) over extended periods of time, induces mild weakness along with mild electrophysiologic evidence of disordered neuromuscular transmission and varying degrees of reduction in motor endplate size. For mice there has been considerable variability among strains in the susceptibility to the active induction of experimental MuSK-MG (118–123). For C57BL6, the distribution of weakness and wasting follows the gradient of normal MuSK expression in individual muscles (120), which somewhat mimics the distribution of muscle involvement in MuSK-MG (see above).
In contrast to the variability in response and the requirement for repeated immunizations in mice, the experimental disease in inbred Lewis rats is highly stereotyped. A single immunization of mouse MuSK ectodomain in adjuvants results in reproducible severe weakness (death within 4 weeks), along with muscle wasting and electrodiagnostic abnormalities typical of MuSK-MG. Histologically, there are extensive postsynaptic and presynaptic changes. The NMJ morphologic findings include fragmented NMJs with varying degrees of postsynaptic muscle end plate destruction, along with abnormal nerve terminals. The presynaptic changes are characterized by reduced terminal size, ongoing terminal degeneration and lack of registration between endplate and nerve terminals. In addition, there is local axon sprouting, and extrajunctional dispersion of cholinesterase activity (61–63).
Data from analysis of both MuSK-MG and its animal models described above have contributed to the information concerning the pathogenesis of the human disease. With the identification of MuSK Abs in “seronegative MG” patients, the initial hypothesis was that the MuSK Abs indeed induce the disease. As noted above, at the time, the hypothesis was not universally accepted (103, 116, 117). The alternative hypothesis advanced was that these Abs are an epiphenomenon occurring in parallel with the disease or even occurring as a result of the disease (103, 116). The dual observations (see above) of (1) the ability of IgG isolated from patient serum, when injected into immunosuppressed mice, to induce a disease similar to human MuSK-MG, and (2) immunization of normal animals with purified MuSK protein, leading to the production of MuSK Abs, also results in a disease that is highly similar to human MuSK-MG, together strongly support the hypothesis that the MuSK Abs are the etiologic agents in this disease.
Multiple observations of neuromuscular junction histology from the various animal models, along with the few histologic observations available from MuSK-MG patients, all demonstrating the absence of inflammatory damage, suggests that the innate immune system, especially the complement cascade, does not play a role in this disease. This characteristic distinguishes MuSK-MG from AChR-MG. It should be noted, however, that the data from the mouse passive transfer models employing human anti-MuSK IgG are somewhat confounded by both the necessary pretreatment with immunosuppressive agents, the extremely high doses of human Ig required to induce disease and the necessity in this model for interaction between the injected human IgG and the recipient mouse’s innate immune system. This heterologous interaction, theoretically, may not be strong enough to induce a vigorous inflammatory reaction. On the other hand, for the active immunization model, immunosuppressive agents are not employed and the Ab and the innate immune system components, e.g., complement proteins and inflammatory cells expressing Fc receptors, are autologous (syngeneic) and hence capable of inducing a strong inflammatory response. At least in humans, the absence of observed inflammation in the NMJ, may be the result of the high proportion of IgG4 anti-MuSK Abs in MuSK MG (see above).
Despite the lack of inflammatory damage to the neuromuscular junction in MuSK-MG and its animal models and the theoretical inability of the human anti-MuSK IgG4 to induce antigenic modulation (i.e., increased MuSK turnover), the concentration of AChR in the postsynaptic membrane is reduced, and, to a lesser extent, so is the MuSK concentration (60, 117). The current hypothesis is that the MuSK Abs act as antagonists to the MuSK function as a receptor kinase, with its natural ligand being the agrin/lrp4 complex. In vitro, MuSK Abs from MuSK-MG patients and from actively immunized animals block agrin-induced AChR clustering and downstream phosphorylation in muscle cells in tissue culture (14, 59, 106, 111, 124, 125). The latter observations and the development of the above abnormalities in the NMJs in adult animals (with fully developed neuromuscular junctions) in which experimental MuSK-MG is either actively or passively induced, suggests that MuSK plays a crucial role not only in the development of the NMJ, but also plays a role in the maintenance of this synapse in adult animals. Subsequent studies of adult animals in which MuSK has been inactivated or knocked down demonstrated similar NMJ changes (64, 65).
As noted above, presynaptic involvement is another characteristic of MuSK-MG that distinguishes it from AChR-MG. In the latter disease presynaptic dysfunction is absent except in the setting of severe NMJ inflammation (12). Otherwise, presynaptic activity, in fact, is increased (126). Similar to the human disease, the experimental models of MuSK-MG noted above exhibit non-inflammatory structural and functional abnormalities of both presynaptic and postsynaptic portions of the NMJ. The abnormal presynaptic function is most readily observed in the severe form of experimental MuSK-MG that occurs in the active-immunization rat model (63). In that disease model, both the cross-sectional area of individual nerve terminals is reduced as is the total nerve terminal cross-sectional area across the entire NMJ. In addition, the actively induced rat model and the passive transfer mouse model have both demonstrated abnormal registration between the nerve terminals and the muscle endplates. Whether the observed reduction in NMJ cholinesterase activity in MuSK-MG plays a role in the muscle weakness is yet to be determined.
Early on, the treatment of seronegative MG simply followed the protocols developed for seropositive MG, i.e., AChR-MG, including use of cholinesterase inhibitors, thymectomy, corticosteroids, plasma exchange and intravenous immunoglobulin and cytotoxic immunosuppressants. However, with the clear identification of the MuSK-MG subset of “seronegative” patients, data began to accumulate that a number of these treatments were ineffective, or even detrimental, in these patients. Thymectomy appears not to play a role in the treatment of this disease (78, 80, 96, 105, 127, 128). Some studies suggest other differences in response to treatment, including observations that MuSK-MG patients may respond poorly to intravenous immunoglobulin (78, 80, 96, 105, 129, 130). There is also a striking absence of improvement with cholinesterase inhibitors (101, 130–133). In fact, a number of MuSK-MG patients worsen in response these agents (101, 131). The latter observation may derive from diminished acetylcholinesterase concentrations at the NMJ, perhaps as a result of MuSK Ab-mediated blockade of ColQ binding to MuSK or Ab-mediated reduction in MuSK concentration (52, 53).
Patients with MuSK-MG respond especially well to high dose corticosteroids (129, 134–136). This appears to be most often true for patients with rapidly progressive or aggressive disease. The effectiveness of steroid treatment, along with the absence of a role for thymectomy and the limited or absent role for cholinesterase inhibitors, make corticosteroids the foundation of the current treatment of this disease. However, about 15 percent of patients treated with high dose corticosteroids do not adequately respond, so called refractory disease. This figure is somewhat higher than the comparable data in AChR-MG (137, 138). Similar to the case in AChR-MG, the significant side effects of these agents often limit their effectiveness. In both diseases, the treatment protocols aim at inducing a remission with high doses, followed by slow tapering of the dose to the lowest effective dose (139). An additional issue in MuSK-MG is the muscle wasting that occurs uniquely in this form of autoimmune MG. In many patients, in spite of the early effectiveness of corticosteroids in inducing a clinical remission, the muscle wasting appears to continue to progress (135).
Cytotoxic agents effective in preventing and treating solid organ transplant rejection have been used as single immunosuppressive agents in the treatment of AChR-MG, including azathioprine, mycophenolate mofetil and cyclosporine, with some efficacy in inducing remission. Their major role in that disease, however, has been as steroid-sparing treatments, that is, effective in facilitating corticosteroid dose tapering (140). Anecdotal data for MuSK-MG suggest that these agents are even less effective in inducing remission than in AChR-MG but, similarly, may be useful as steroid-sparing medications (135, 140, 141).
The B cell depleting agent rituximab, a chimeric anti-CD20 monoclonal Ab, has been especially effective in MuSK-MG. Because of its toxicity profile, including a 1:10,000 risk of the induction of progressive multifocal leukoencephalopathy, the drug has been used primarily in treatment of (steroid)-refractory patients. A significant number of reported studies, all limited by relatively small numbers of subjects, have shown efficacy in this disease (142–146). In a number of cases, treatment led to eventual elimination of the need for other immune-directed treatments, e.g., steroids, and without the necessity for repeated rituximab infusions (143–145). A recent set of clinical guidelines has supported earlier use of this agent when an initial standard treatment does not induce rapid remission (140). One successful protocol is to use two courses of rituximab at a dose of 375 mg per meter squared body surface area weekly for four doses, each course separated by 6 months. A 4-infusion course can then be repeated as needed (143). It appears that adding an infusion 1 month later and another 2 months later improves efficacy even further (147).
Plasma exchange has been a rapidly effective treatment for active AChR-MG. Intravenous immunoglobulin infusions have been equally effective and somewhat safer (148). Initial studies of plasma exchange in acute MuSK-MG demonstrated its efficacy in this disease (129, 134, 149). Unlike in AChR-MG, the efficacy of intravenous immunoglobulin in MuSK-MG appears to be less than that of plasma exchange (130, 134, 136, 150), but the data supporting the latter statement are much less robust (151, 152).
As noted above, the various immune-directed treatments currently in use have been reasonably effective in MuSK-MG. However, these treatments are all limited by their broad effect on the immune system: on both the pathogenic (autoimmune) components and the normally functioning components. As in all other autoimmune diseases, the treatment paradigm is to adjust drug dosages and timing to maximize the effect on the autoimmune portion of the immune response while reducing the “off-target” effect on the remainder of the immune system.
A theoretical means of focusing the treatment on the autoimmune portion is to employ an antigen-specific therapy, that is, one only targeting the attack on the auto-antigen. For an Ab-mediated disease such as MG, this would involve targeting the auto-Abs. One possibly means to accomplish this therapeutic effect involves physical removal of the auto-Abs, for example, by immunoadsorption plasmapheresis. For AChR-MG, such antigen-specific Ab removal employing an affinity column containing AChR antigen as the affinity agent has been examined. To date, this approach has been no more effective than gross removal of all Abs by plasma exchange (153, 154).
An alternative antigen-specific approach is to target the B cells that are secreting the auto-Abs. This approach is currently under study in both AChR-MG and in MuSK-MG, through the use of either genetically engineered Abs or genetically engineered T cells that target the pathogenic autoimmune B cells (155, 156).
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
LB and DR have received research funding from NINDS (1R21NS104516), Myasthenia Gravis Foundation of America, and Cabaletta Bio Inc.
LB and DR have received research funding from NINDS (1R21NS104516), Myasthenia Gravis Foundation of America, and Cabaletta Bio Inc. DR is a member of the Advisory Board of Cabaletta Bio Inc.
1. Engel AG. Congenital myasthenic syndromes in 2018. Curr Neurol Neurosci Rep. (2018) 18:46. doi: 10.1007/s11910-018-0852-4
2. Rodriguez Cruz PM, Palace J, Beeson D. The neuromuscular junction and wide heterogeneity of congenital myasthenic syndromes. Int J Mol Sci. (2018) 19:1667. doi: 10.3390/ijms19061677
4. Richman DP, Agius MA. Myasthenia gravis: pathogenesis and treatment. Semin Neurol. (1994) 14:106–10.
5. Toyka KV, Brachman DB, Pestronk A, Kao I. Myasthenia gravis: passive transfer from man to mouse. Science. (1975) 190:397–9.
6. Lennon VA, Lambert EH. Myasthenia gravis induced by monoclonal antibodies to acetylcholine receptors. Nature. (1980) 285:238–40.
7. Richman DP, Gomez CM, Berman PW, Burres SA, Fitch FW, Arnason BG. Monoclonal anti-acetylcholine receptor antibodies can cause experimental myasthenia. Nature. (1980) 286:738–9.
8. Tzartos SJ, Lindstrom JM. Monoclonal antibodies used to probe acetylcholine receptor structure: localization of the main immunogenic region and detection of similarities between subunits. Proc Natl Acad Sci USA. (1980) 77:755–9.
9. Patrick J, Lindstrom J. Autoimmune response to acetylcholine receptor. Science. (1973) 180:871–2.
10. Richman DP, Agius MA, Kirvan CA, Gomez CM, Fairclough RH, Dupont BL, et al. Antibody effector mechanisms in myasthenia gravis. The complement hypothesis. Ann N Y Acad Sci. (1998) 841:450–65.
11. Cetin H, Vincent A. Pathogenic mechanisms and clinical correlations in autoimmune myasthenic syndromes. Semin Neurol. (2018) 38:344–54. doi: 10.1055/s-0038-1660500
12. Maselli RA, Mass DP, Distad BJ, Richman DP. Anconeus muscle: a human muscle preparation suitable for in-vitro microelectrode studies. Muscle Nerve. (1991) 14:1189–92.
13. Engel AG, Tsujihata M, Lindstrom JM, Lennon VA. The motor end plate in myasthenia gravis and in experimental autoimmune myasthenia gravis. A quantitative ultrastructural study. Ann N Y Acad Sci. (1976) 274:60–79. doi: 10.1111/j.1749-6632.1976.tb47676.x
14. Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. (2001) 7:365–8.
15. Hopf C, Hoch W. Tyrosine phosphorylation of the muscle-specific kinase is exclusively induced by acetylcholine receptor-aggregating agrin fragments. Eur J Biochem. (1998) 253:382–9.
16. Jennings CG, Dyer SM, Burden SJ. Muscle-specific trk-related receptor with a kringle domain defines a distinct class of receptor tyrosine kinases. Proc Natl Acad Sci USA. (1993) 90:2895–9.
17. Stiegler AL, Burden SJ, Hubbard SR. Crystal structure of the frizzled-like cysteine-rich domain of the receptor tyrosine kinase MuSK. J Mol Biol. (2009) 393:1–9. doi: 10.1016/j.jmb.2009.07.091
18. Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, et al. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. (1995) 15:573–84.
19. Vincent A, Newland C, Croxen R, Beeson D. Genes at the junction–candidates for congenital myasthenic syndromes. Trends Neurosci. (1997) 20:15–22. doi: 10.1016/s0166-2236(96)10066-7
20. Burden SJ, Huijbers MG, Remedio L. Fundamental molecules and mechanisms for forming and maintaining neuromuscular synapses. Int J Mol Sci. (2018) 19:490. doi: 10.3390/ijms19020490
21. Burden SJ, Yumoto N, Zhang W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb Perspect Biol. (2013) 5:a009167. doi: 10.1101/cshperspect.a009167
23. Burden SJ, Fuhrer C, Hubbard SR. Agrin/MuSK signaling: willing and Abl. Nat Neurosci. (2003) 6:653–4.
24. Hughes BW, Kusner LL, Kaminski HJ. Molecular architecture of the neuromuscular junction. Muscle Nerve. (2006) 33:445–61.
25. Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. (2001) 2:791–805. doi: 10.1038/35097557
26. Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. (2001) 410:1057–64.
27. Yang X, Arber S, William C, Li L, Tanabe Y, Jessell TM, et al. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. (2001) 30:399–410. doi: 10.1016/s0896-6273(01)00287-2
28. Chen F, Liu Y, Sugiura Y, Allen PD, Gregg RG, Lin W. Neuromuscular synaptic patterning requires the function of skeletal muscle dihydropyridine receptors. Nat Neurosci. (2011) 14:570–7. doi: 10.1038/nn.2792
29. Ghazanfari N, Fernandez KJ, Murata Y, Morsch M, Ngo ST, Reddel SW, et al. Muscle specific kinase: organiser of synaptic membrane domains. Int J Biochem Cell Biol. (2011) 43:295–8. doi: 10.1016/j.biocel.2010.10.008
30. Hallock PT, Xu CF, Park TJ, Neubert TA, Curran T, Burden SJ. Dok-7 regulates neuromuscular synapse formation by recruiting Crk and Crk-L. Genes Dev. (2010) 24:2451–61. doi: 10.1101/gad.1977710
31. Henriquez JP, Salinas PC. Dual roles for Wnt signalling during the formation of the vertebrate neuromuscular junction. Acta Physiol (Oxf). (2011) 204:128–36. doi: 10.1111/j.1748-1716.2011.02295.x
32. Hoch W. Molecular dissection of neuromuscular junction formation. Trends Neurosci. (2003) 26:335–7.
33. Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr Opin Neurobiol. (2006) 16:74–82. doi: 10.1016/j.conb.2005.12.003
34. Marques MJ, Conchello JA, Lichtman JW. From plaque to pretzel: fold formation and acetylcholine receptor loss at the developing neuromuscular junction. J Neurosci. (2000) 20:3663–75.
35. Flanagan-Steet H, Fox MA, Meyer D, Sanes JR. Neuromuscular synapses can form in vivo by incorporation of initially aneural postsynaptic specializations. Development. (2005) 132:4471–81.
36. Panzer JA, Gibbs SM, Dosch R, Wagner D, Mullins MC, Granato M, et al. Neuromuscular synaptogenesis in wild-type and mutant zebrafish. Dev Biol. (2005) 285:340–57.
37. Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, et al. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. (2008) 135:334–42. doi: 10.1016/j.cell.2008.10.002
38. Weatherbee SD, Anderson KV, Niswander LA. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. (2006) 133:4993–5000. doi: 10.1242/dev.02696
39. Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. (2008) 60:285–97. doi: 10.1016/j.neuron.2008.10.006
40. Borges LS, Yechikhov S, Lee YI, Rudell JB, Friese MB, Burden SJ, et al. Identification of a motif in the acetylcholine receptor beta subunit whose phosphorylation regulates rapsyn association and postsynaptic receptor localization. J Neurosci. (2008) 28:11468–76. doi: 10.1523/JNEUROSCI.2508-08.2008
41. Qu ZC, Moritz E, Huganir RL. Regulation of tyrosine phosphorylation of the nicotinic acetylcholine receptor at the rat neuromuscular junction. Neuron. (1990) 4:367–78.
42. Ramarao MK, Cohen JB. Mechanism of nicotinic acetylcholine receptor cluster formation by rapsyn. Proc Natl Acad Sci USA. (1998) 95:4007–12.
43. Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. (2006) 312:1802–5.
44. Dai Z, Luo X, Xie H, Peng HB. The actin-driven movement and formation of acetylcholine receptor clusters. J Cell Biol. (2000) 150:1321–34.
45. Strochlic L, Cartaud A, Cartaud J. The synaptic muscle-specific kinase (MuSK) complex: new partners, new functions. Bioessays. (2005) 27:1129–35.
46. Lin W, Dominguez B, Yang J, Aryal P, Brandon EP, Gage FH, et al. Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk5-dependent mechanism. Neuron. (2005) 46:569–79.
48. DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. (1996) 85:501–12.
49. Merlie JP, Sanes JR. Concentration of acetylcholine receptor mRNA in synaptic regions of adult muscle fibres. Nature. (1985) 317:66–8.
50. Cartaud A, Stetzkowski-Marden F, Maoui A, Cartaud J. Agrin triggers the clustering of raft-associated acetylcholine receptors through actin cytoskeleton reorganization. Biol Cell. (2011) 103:287–301. doi: 10.1042/BC20110018
51. Madhavan R, Gong ZL, Ma JJ, Chan AW, Peng HB. The function of cortactin in the clustering of acetylcholine receptors at the vertebrate neuromuscular junction. PLoS One. (2009) 4:e8478. doi: 10.1371/journal.pone.0008478
52. Cartaud A, Strochlic L, Guerra M, Blanchard B, Lambergeon M, Krejci E, et al. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J Cell Biol. (2004) 165:505–15.
53. Sigoillot SM, Bourgeois F, Lambergeon M, Strochlic L, Legay C. ColQ controls postsynaptic differentiation at the neuromuscular junction. J Neurosci. (2010) 30:13–23. doi: 10.1523/jneurosci.4374-09.2010
54. Dimitropoulou A, Bixby JL. Motor neurite outgrowth is selectively inhibited by cell surface MuSK and agrin. Mol Cell Neurosci. (2005) 28:292–302. doi: 10.1016/j.mcn.2004.09.013
55. Yumoto N, Kim N, Burden SJ. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature. (2012) 489:438–42. doi: 10.1038/nature11348
56. Wu H, Lu Y, Shen C, Patel N, Gan L, Xiong WC, et al. Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron. (2012) 75:94–107. doi: 10.1016/j.neuron.2012.04.033
57. Kummer TT, Misgeld T, Lichtman JW, Sanes JR. Nerve-independent formation of a topologically complex postsynaptic apparatus. J Cell Biol. (2004) 164:1077–87.
58. Smith CL, Mittaud P, Prescott ED, Fuhrer C, Burden SJ. Src, Fyn, and Yes are not required for neuromuscular synapse formation but are necessary for stabilization of agrin-induced clusters of acetylcholine receptors. J Neurosci. (2001) 21:3151–60.
59. Cole RN, Reddel SW, Gervasio OL, Phillips WD. Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction. Ann Neurol. (2008) 63:782–9.
60. Ghazanfari N, Morsch M, Reddel SW, Liang SX, Phillips WD. Muscle-specific kinase (MuSK) autoantibodies suppress the MuSK pathway and ACh receptor retention at the mouse neuromuscular junction. J Physiol. (2014) 592(Pt 13):2881–97. doi: 10.1113/jphysiol.2013.270207
61. Richman DP, Nishi K, Ferns MJ, Schnier J, Pytel P, Maselli RA, et al. Animal models of antimuscle-specific kinase myasthenia. Ann N Y Acad Sci. (2012) 1274:140–7. doi: 10.1111/j.1749-6632.2012.06782.x
62. Richman DP, Nishi K, Morell S, Maselli RA, Agius MA. Acute severe model of anti-muscle specific kinase (MuSK) myasthenia in lewis rats (Abstract). Neurology. (2008) 71:153.
63. Richman DP, Nishi K, Morell SW, Chang JM, Ferns MJ, Wollmann RL, et al. Acute severe animal model of anti-muscle-specific kinase myasthenia: combined postsynaptic and presynaptic changes. Arch Neurol. (2012) 69:453–60. doi: 10.1001/archneurol.2011.2200
64. Hesser BA, Henschel O, Witzemann V. Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK. Mol Cell Neurosci. (2006) 31:470–80.
65. Kong XC, Barzaghi P, Ruegg MA. Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference. EMBO Rep. (2004) 5:183–8.
66. Richman DP. The future of research in myasthenia. JAMA Neurol. (2015) 72:812–4. doi: 10.1001/jamaneurol.2014.4740
67. Stiegler AL, Burden SJ, Hubbard SR. Crystal structure of the agrin-responsive immunoglobulin-like domains 1 and 2 of the receptor tyrosine kinase MuSK. J Mol Biol. (2006) 364:424–33.
68. Zhang W, Coldefy AS, Hubbard SR, Burden SJ. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J Biol Chem. (2011) 286:40624–30. doi: 10.1074/jbc.M111.279307
69. Hopf C, Hoch W. Dimerization of the muscle-specific kinase induces tyrosine phosphorylation of acetylcholine receptors and their aggregation on the surface of myotubes. J Biol Chem. (1998) 273:6467–73.
70. Xie MH, Yuan J, Adams C, Gurney A. Direct demonstration of MuSK involvement in acetylcholine receptor clustering through identification of agonist ScFv. Nat Biotechnol. (1997) 15:768–71.
71. Bergamin E, Hallock PT, Burden SJ, Hubbard SR. The cytoplasmic adaptor protein Dok7 activates the receptor tyrosine kinase MuSK via dimerization. Mol Cell. (2010) 39:100–9. doi: 10.1016/j.molcel.2010.06.007
72. Inoue A, Setoguchi K, Matsubara Y, Okada K, Sato N, Iwakura Y, et al. Dok-7 activates the muscle receptor kinase MuSK and shapes synapse formation. Sci Signal. (2009) 2:ra7. doi: 10.1126/scisignal.2000113
73. Jing L, Lefebvre JL, Gordon LR, Granato M. Wnt signals organize synaptic prepattern and axon guidance through the zebrafish unplugged/MuSK receptor. Neuron. (2009) 61:721–33. doi: 10.1016/j.neuron.2008.12.025
74. Messeant J, Dobbertin A, Girard E, Delers P, Manuel M, Mangione F, et al. MuSK frizzled-like domain is critical for mammalian neuromuscular junction formation and maintenance. J Neurosci. (2015) 35:4926–41. doi: 10.1523/JNEUROSCI.3381-14.2015
75. Remedio L, Gribble KD, Lee JK, Kim N, Hallock PT, Delestree N, et al. Diverging roles for Lrp4 and Wnt signaling in neuromuscular synapse development during evolution. Genes Dev. (2016) 30:1058–69. doi: 10.1101/gad.279745.116
76. Rodriguez Cruz PM, Cossins J, Cheung J, Maxwell S, Jayawant S, Herbst R, et al. Congenital myasthenic syndrome due to mutations in MUSK suggests that the level of MuSK phosphorylation is crucial for governing synaptic structure. Hum Mutat. (2020) 41:619–31. doi: 10.1002/humu.23949
77. McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, Newsom-Davis J, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. (2004) 55:580–4.
78. Evoli A, Tonali PA, Padua L, Monaco ML, Scuderi F, Batocchi AP, et al. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain. (2003) 126(Pt 10):2304–11.
79. Farrugia ME, Kennett RP, Hilton-Jones D, Newsom-Davis J, Vincent A. Quantitative EMG of facial muscles in myasthenia patients with MuSK antibodies. Clin Neurophysiol. (2007) 118:269–77.
80. Sanders DB, El Salem K, Massey JM, McConville J, Vincent A. Clinical aspects of MuSK antibody positive seronegative MG. Neurology. (2003) 60: 1978–80.
81. Vincent A, Bowen J, Newsom-Davis J, McConville J. Seronegative generalised myasthenia gravis: clinical features, antibodies, and their targets. Lancet Neurol. (2003) 2:99–106. doi: 10.1016/s1474-4422(03)00306-5
82. Zhou L, McConville J, Chaudhry V, Adams RN, Skolasky RL, Vincent A, et al. Clinical comparison of muscle-specific tyrosine kinase (MuSK) antibody-positive and -negative myasthenic patients. Muscle Nerve. (2004) 30:55–60. doi: 10.1002/mus.20069
83. Wolfe GI, Oh SJ. Clinical phenotype of muscle-specific tyrosine kinase-antibody-positive myasthenia gravis. Ann N Y Acad Sci. (2008) 1132:71–5. doi: 10.1196/annals.1405.005
84. Farrugia ME, Bydder GM, Francis JM, Robson MD. Magnetic resonance imaging of facial muscles. Clin Radiol. (2007) 62:1078–86.
85. Farrugia ME, Robson MD, Clover L, Anslow P, Newsom-Davis J, Kennett R, et al. MRI and clinical studies of facial and bulbar muscle involvement in MuSK antibody-associated myasthenia gravis. Brain. (2006) 129(Pt 6): 1481–92.
86. Ishii W, Matsuda M, Okamoto N, Shimojima Y, Yazaki M, Motomura M, et al. Myasthenia gravis with anti-MuSK antibody, showing progressive muscular atrophy without blepharoptosis. Intern Med. (2005) 44:671–2.
87. Benveniste O, Jacobson L, Farrugia ME, Clover L, Vincent A. MuSK antibody positive myasthenia gravis plasma modifies MURF-1 expression in C2C12 cultures and mouse muscle in vivo. J Neuroimmunol. (2005) 170:41–8.
88. Boneva N, Frenkian-Cuvelier M, Bidault J, Brenner T, Berrih-Aknin S. Major pathogenic effects of anti-MuSK antibodies in myasthenia gravis. J Neuroimmunol. (2006) 177:119–31.
89. Finsterer J. Turn/amplitude analysis to assess bulbar muscle wasting in MuSK positive myasthenia. Clin Neurophysiol. (2007) 118:1173–4.
90. Martignago S, Fanin M, Albertini E, Pegoraro E, Angelini C. Muscle histopathology in myasthenia gravis with antibodies against MuSK and AChR. Neuropathol Appl Neurobiol. (2009) 35:103–10.
91. Cenacchi G, Valentina P, Marina F, Elena P, Corrado A. Comparison of muscle ultrastructure in myasthenia gravis with anti-MuSK and anti-AChR antibodies. J Neurol. (2011) 258:746–52. doi: 10.1007/s00415-010-5823-x
92. Mantegazza R, Baggi F, Antozzi C, Confalonieri P, Morandi L, Bernasconi P, et al. Myasthenia gravis (MG): epidemiological data and prognostic factors. Ann N Y Acad Sci. (2003) 998:413–23.
93. Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle Nerve. (2008) 37:141–9.
94. Osserman KE, Genkins G. Studies in myasthenia gravis: review of a twenty-year experience in over 1200 patients. Mt Sinai J Med. (1971) 38:497–537.
95. Niks EH, Kuks JB, Verschuuren JJ. Epidemiology of myasthenia gravis with anti-muscle specific kinase antibodies in the Netherlands. J Neurol Neurosurg Psychiatry. (2007) 78:417–8.
96. Niks EH, Kuks JB, Roep BO, Haasnoot GW, Verduijn W, Ballieux BE, et al. Strong association of MuSK antibody-positive myasthenia gravis and HLA-DR14-DQ5. Neurology. (2006) 66:1772–4.
97. Lauriola L, Ranelletti F, Maggiano N, Guerriero M, Punzi C, Marsili F, et al. Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology. (2005) 64:536–8.
98. Leite MI, Strobel P, Jones M, Micklem K, Moritz R, Gold R, et al. Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann Neurol. (2005) 57:444–8.
99. Saka E, Topcuoglu MA, Akkaya B, Galati A, Onal MZ, Vincent A. Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology. (2005) 65:782–3.
100. Marino M, Maiuri MT, Di Sante G, Scuderi F, La Carpia F, Trakas N, et al. T cell repertoire in DQ5-positive MuSK-positive myasthenia gravis patients. J Autoimmun. (2014) 52:113–21. doi: 10.1016/j.jaut.2013.12.007
101. Lavrnic D, Losen M, Vujic A, De Baets M, Hajdukovic LJ, Stojanovic V, et al. The features of myasthenia gravis with autoantibodies to MuSK. J Neurol Neurosurg Psychiatry. (2005) 76:1099–102.
102. Niks EH, Kuks JB, Wokke JH, Veldman H, Bakker E, Verschuuren JJ, et al. Pre- and postsynaptic neuromuscular junction abnormalities in musk myasthenia. Muscle Nerve. (2010) 42:283–8. doi: 10.1002/mus.21642
103. Selcen D, Fukuda T, Shen XM, Engel AG. Are MuSK antibodies the primary cause of myasthenic symptoms? Neurology. (2004) 62:1945–50.
104. Shiraishi H, Motomura M, Yoshimura T, Fukudome T, Fukuda T, Nakao Y, et al. Acetylcholine receptors loss and postsynaptic damage in MuSK antibody-positive myasthenia gravis. Ann Neurol. (2005) 57:289–93.
105. Evoli A, Batocchi AP, Lo MM, Servidei S, Padua L, Majolini L, et al. Clinical heterogeneity of seronegative myasthenia gravis. Neuromuscul Disord. (1996) 6:155–61.
106. Huijbers MG, Zhang W, Klooster R, Niks EH, Friese MB, Straasheijm KR, et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci USA. (2013) 110:20783–8. doi: 10.1073/pnas.1313944110
107. Tao MH, Smith RI, Morrison SL. Structural features of human immunoglobulin G that determine isotype-specific differences in complement activation. J Exp Med. (1993) 178:661–7.
108. Klooster R, Plomp JJ, Huijbers MG, Niks EH, Straasheijm KR, Detmers FJ, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain. (2012) 135(Pt 4):1081–101. doi: 10.1093/brain/aws025
109. Trampert DC, Hubers LM, van de Graaf SFJ, Beuers U. On the role of IgG4 in inflammatory conditions: lessons for IgG4-related disease. Biochim Biophys Acta Mol Basis Dis. (2018) 1864(4 Pt B):1401–9.
110. Koneczny I, Stevens JA, De Rosa A, Huda S, Huijbers MG, Saxena A, et al. IgG4 autoantibodies against muscle-specific kinase undergo Fab-arm exchange in myasthenia gravis patients. J Autoimmun. (2017) 77:104–15. doi: 10.1016/j.jaut.2016.11.005
111. Koneczny I, Cossins J, Waters P, Beeson D, Vincent A. MuSK myasthenia gravis IgG4 disrupts the interaction of LRP4 with MuSK but both IgG4 and IgG1-3 can disperse preformed agrin-independent AChR clusters. PLoS One. (2013) 8:e80695. doi: 10.1371/journal.pone.0080695
112. Leite MI, Jacob S, Viegas S, Cossins J, Clover L, Morgan BP, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain. (2008) 131(Pt 7):1940–52. doi: 10.1093/brain/awn092
113. Huijbers MG, Vink AF, Niks EH, Westhuis RH, van Zwet EW, de Meel RH, et al. Longitudinal epitope mapping in MuSK myasthenia gravis: implications for disease severity. J Neuroimmunol. (2016) 291:82–8. doi: 10.1016/j.jneuroim.2015.12.016
114. Otsuka K, Ito M, Ohkawara B, Masuda A, Kawakami Y, Sahashi K, et al. Collagen Q and anti-MuSK autoantibody competitively suppress agrin/LRP4/MuSK signaling. Sci Rep. (2015) 5:13928. doi: 10.1038/srep13928
115. Takamori M, Nakamura T, Motomura M. Antibodies against Wnt receptor of muscle-specific tyrosine kinase in myasthenia gravis. J Neuroimmunol. (2013) 254:183–6. doi: 10.1016/j.jneuroim.2012.09.001
116. Lindstrom J. Is “seronegative” MG explained by autoantibodies to MuSK? Neurology. (2004) 62:1920–1.
117. Shigemoto K, Kubo S, Maruyama N, Hato N, Yamada H, Jie C, et al. Induction of myasthenia by immunization against muscle-specific kinase. J Clin Invest. (2006) 116:1016–24.
118. Viegas S, Jacobson L, Waters P, Cossins J, Jacob S, Leite MI, et al. Passive and active immunization models of MuSK-Ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp Neurol. (2012) 234:506–12. doi: 10.1016/j.expneurol.2012.01.025
119. Jha S, Xu K, Maruta T, Oshima M, Mosier DR, Atassi MZ, et al. Myasthenia gravis induced in mice by immunization with the recombinant extracellular domain of rat muscle-specific kinase (MuSK). J Neuroimmunol. (2006) 175:107–17.
120. Punga AR, Lin S, Oliveri F, Meinen S, Ruegg MA. Muscle-selective synaptic disassembly and reorganization in MuSK antibody positive MG mice. Exp Neurol. (2011) 230:207–17. doi: 10.1016/j.expneurol.2011.04.018
121. Mori S, Kubo S, Akiyoshi T, Yamada S, Miyazaki T, Hotta H, et al. Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am J Pathol. (2012) 180:798–810. doi: 10.1016/j.ajpath.2011.10.031
122. Ulusoy C, Kim E, Tuzun E, Huda R, Yilmaz V, Poulas K, et al. Preferential production of IgG1, IL-4 and IL-10 in MuSK-immunized mice. Clin Immunol (Orlando, Fla). (2014) 151:155–63.
123. Patel V, Oh A, Voit A, Sultatos LG, Babu GJ, Wilson BA, et al. Altered active zones, vesicle pools, nerve terminal conductivity, and morphology during experimental MuSK myasthenia gravis. PLoS One. (2014) 9:e110571. doi: 10.1371/journal.pone.0110571
124. Cole RN, Ghazanfari N, Ngo ST, Gervasio OL, Reddel SW, Phillips WD. Patient autoantibodies deplete postsynaptic muscle specific kinase leading to disassembly of the ACh receptor scaffold and myasthenia gravis in mice. J Physiol. (2010) 588(Pt 17):3217–29. doi: 10.1113/jphysiol.2010.190298
125. Mori S, Yamada S, Kubo S, Chen J, Matsuda S, Shudou M, et al. Divalent and monovalent autoantibodies cause dysfunction of MuSK by distinct mechanisms in a rabbit model of myasthenia gravis. J Neuroimmunol. (2012) 244:1–7. doi: 10.1016/j.jneuroim.2011.12.005
126. Plomp JJ, Van Kempen GT, De Baets MB, Graus YM, Kuks JB, Molenaar PC. Acetylcholine release in myasthenia gravis: regulation at single end-plate level. Ann Neurol. (1995) 37:627–36.
127. Ponseti JM, Caritg N, Gamez J, Lopez-Cano M, Vilallonga R, Armengol M. A comparison of long-term post-thymectomy outcome of anti-AChR-positive, anti-AChR-negative and anti-MuSK-positive patients with non-thymomatous myasthenia gravis. Expert Opin Biol Ther. (2009) 9:1–8. doi: 10.1517/14712590802588831
128. Clifford KM, Hobson-Webb LD, Benatar M, Burns TM, Barnett C, Silvestri NJ, et al. Thymectomy may not be associated with clinical improvement in MuSK myasthenia gravis. Muscle Nerve. (2019) 59:404–10. doi: 10.1002/mus.26404
129. Guptill JT, Sanders DB. Update on muscle-specific tyrosine kinase antibody positive myasthenia gravis. Curr Opin Neurol. (2010) 23:530–5.
130. Pasnoor M, Wolfe GI, Nations S, Trivedi J, Barohn RJ, Herbelin L, et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a U.S. experience. Muscle Nerve. (2010) 41:370–4. doi: 10.1002/mus.21533
131. Punga AR, Flink R, Askmark H, Stalberg EV. Cholinergic neuromuscular hyperactivity in patients with myasthenia gravis seropositive for MuSK antibody. Muscle Nerve. (2006) 34:111–5.
132. Evoli A, Lindstrom J. Myasthenia gravis with antibodies to MuSK: another step toward solving mystery? Neurology. (2011) 77:1783–4.
133. Morren J, Li Y. Myasthenia gravis with muscle-specific tyrosine kinase antibodies: a narrative review. Muscle Nerve. (2018) 58:344–58. doi: 10.1002/mus.26107
134. Reddel SW, Morsch M, Phillips WD. Clinical and scientific aspects of muscle-specific tyrosine kinase-related myasthenia gravis. Curr Opin Neurol. (2014) 27:558–65. doi: 10.1097/WCO.0000000000000136
135. Evoli A, Alboini PE, Damato V, Iorio R, Provenzano C, Bartoccioni E, et al. Myasthenia gravis with antibodies to MuSK: an update. Ann N Y Acad Sci. (2018) 1412:82–9. doi: 10.1111/nyas.13518
136. Guptill JT, Sanders DB, Evoli A. Anti-musk antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve. (2011) 44:36–40. doi: 10.1002/mus.22006
137. Suh J, Goldstein JM, Nowak RJ. Clinical characteristics of refractory myasthenia gravis patients. Yale J Biol Med. (2013) 86:255–60.
138. Silvestri NJ, Wolfe GI. Treatment-refractory myasthenia gravis. J Clin Neuromuscul Dis. (2014) 15:167–78. doi: 10.1097/CND.0000000000000034
140. Sanders DB, Wolfe GI, Benatar M, Evoli A, Gilhus NE, Illa I, et al. International consensus guidance for management of myasthenia gravis: Executive summary. Neurology. (2016) 87:419–25. doi: 10.1212/WNL.0000000000002790
141. Sanders DB, Juel VC. MuSK-antibody positive myasthenia gravis: questions from the clinic. J Neuroimmunol. (2008) 20:85–9. doi: 10.1016/j.jneuroim.2008.05.032
142. Hehir MK, Hobson-Webb LD, Benatar M, Barnett C, Silvestri NJ, Howard JF Jr., et al. Rituximab as treatment for anti-MuSK myasthenia gravis: multicenter blinded prospective review. Neurology. (2017) 89:1069–77. doi: 10.1212/WNL.0000000000004341
143. Keung B, Robeson KR, DiCapua DB, Rosen JB, O’Connor KC, Goldstein JM, et al. Long-term benefit of rituximab in MuSK autoantibody myasthenia gravis patients. J Neurol Neurosurg Psychiatry. (2013) 84:1407–9.
144. Illa I, Diaz-Manera J, Rojas-Garcia R, Pradas J, Rey A, Blesa R, et al. Sustained response to Rituximab in anti-AChR and anti-MuSK positive Myasthenia Gravis patients. J Neuroimmunol. (2008) 20:90–4. doi: 10.1016/j.jneuroim.2008.04.039
145. Diaz-Manera J, Martinez-Hernandez E, Querol L, Klooster R, Rojas-Garcia R, Suarez-Calvet X, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. (2012) 78:189–93. doi: 10.1212/WNL.0b013e3182407982
146. Nowak RJ, Dicapua DB, Zebardast N, Goldstein JM. Response of patients with refractory myasthenia gravis to rituximab: a retrospective study. Ther Adv Neurol Disord. (2011) 4:259–66. doi: 10.1177/1756285611411503
147. Cortes-Vicente E, Rojas-Garcia R, Diaz-Manera J, Querol L, Casasnovas C, Guerrero-Sola A, et al. The impact of rituximab infusion protocol on the long-term outcome in anti-MuSK myasthenia gravis. Ann Clin Transl Neurol. (2018) 5:710–6. doi: 10.1002/acn3.564
148. Gajdos P, Chevret S, Toyka K. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev. (2008) 12:CD002277.
149. Deguchi K, Matsuzono K, Nakano Y, Kono S, Sato K, Deguchi S, et al. Anti-MuSK antibody-positive Myasthenia Gravis successfully treated with outpatient periodic weekly blood purification therapy. Intern Med. (2018) 57:1455–8. doi: 10.2169/internalmedicine.9466-17
150. Govindarajan R, Iyadurai SJ, Connolly A, Zaidman C. Selective response to rituximab in a young child with MuSK-associated myasthenia gravis. Neuromuscul Disord. (2015) 25:651–2. doi: 10.1016/j.nmd.2015.03.014
151. Takahashi H, Kawaguchi N, Nemoto Y, Hattori T. High-dose intravenous immunoglobulin for the treatment of MuSK antibody-positive seronegative myasthenia gravis. J Neurol Sci. (2006) 247:239–41.
152. Shibata-Hamaguchi A, Samuraki M, Furui E, Iwasa K, Yoshikawa H, Hayashi S, et al. Long-term effect of intravenous immunoglobulin on anti-MuSK antibody-positive myasthenia gravis. Acta Neurol Scand. (2007) 116:406–8.
153. Tzartos SJ, Bitzopoulou K, Gavra I, Kordas G, Jacobson L, Kostelidou K, et al. Antigen-specific apheresis of pathogenic autoantibodies from myasthenia gravis sera. Ann N Y Acad Sci. (2008) 1132:291–9. doi: 10.1196/annals.1405.017
154. Yeh JH, Chiu HC. Comparison between double-filtration plasmapheresis and immunoadsorption plasmapheresis in the treatment of patients with myasthenia gravis. J Neurol. (2000) 247:510–3.
155. Trinh VB, Foster AJ, Fairclough RH. Design, synthesis, and characterization of a 39 amino acid peptide mimic of the main immunogenic region of the Torpedo acetylcholine receptor. Mol Immunol. (2014) 59:79–90. doi: 10.1016/j.molimm.2014.01.002
Keywords: myasthenia gravis, muscle specific kinase, neuromuscular junction, pathogenesis, treatment, animal models, review
Citation: Borges LS and Richman DP (2020) Muscle-Specific Kinase Myasthenia Gravis. Front. Immunol. 11:707. doi: 10.3389/fimmu.2020.00707
Received: 11 November 2019; Accepted: 30 March 2020;
Published: 08 May 2020.
Edited by:
Anna Rostedt Punga, Uppsala University, SwedenReviewed by:
Ruksana Huda, The University of Texas Medical Branch at Galveston, United StatesCopyright © 2020 Borges and Richman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David P. Richman, ZHByaWNobWFuQHVjZGF2aXMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.