 Anje Cauwels
Anje Cauwels Jan Tavernier
Jan Tavernier- 1VIB-UGent Center for Medical Biotechnology, Ghent University, Ghent, Belgium
- 2Orionis Biosciences, Ghent, Belgium
Autoimmune diseases such as multiple sclerosis (MS), type I diabetes (T1D), inflammatory bowel diseases (IBD), and rheumatoid arthritis (RA) are chronic, incurable, incapacitating and at times even lethal conditions. Worldwide, millions of people are affected, predominantly women, and their number is steadily increasing. Currently, autoimmune patients require lifelong immunosuppressive therapy, often accompanied by severe adverse side effects and risks. Targeting the fundamental cause of autoimmunity, which is the loss of tolerance to self- or innocuous antigens, may be achieved via various mechanisms. Recently, tolerance-inducing cellular therapies, such as tolerogenic dendritic cells (tolDCs) and regulatory T cells (Tregs), have gained considerable interest. Their safety has already been evaluated in patients with MS, arthritis, T1D, and Crohn’s disease, and clinical trials are underway to confirm their safety and therapeutic potential. Cell-based therapies are inevitably expensive and time-consuming, requiring laborious ex vivo manufacturing. Therefore, direct in vivo targeting of tolerogenic cell types offers an attractive alternative, and several strategies are being explored. Type I IFN was the first disease-modifying therapy approved for MS patients, and approaches to endogenously induce IFN in autoimmune diseases are being pursued vigorously. We here review and discuss tolerogenic cellular therapies and targeted in vivo tolerance approaches and propose a novel strategy for cell-specific delivery of type I IFN signaling to a cell type of choice.
Tolerance-Inducing Cells
Dendritic cells (DC) are best known for their antigen (Ag) processing and presenting functions, driving immunological responses directed against pathogens and malignant cells. Nevertheless, they are also crucial for coordinating immunological tolerance and preventing autoimmunity. Several types of DCs exist: conventional (cDC), plasmacytoid (pDC), and monocyte-derived (moDC). They all originate from CD34+ hematopoietic progenitor cells in the bone marrow. For a long time, it was generally believed that differentiation via macrophage/DC progenitors (MDC) gave rise to either the monocyte/macrophage lineage or to common DC progenitors (CDP), which further differentiated into either pDCs or pre-cDCs (1, 2). Recently, however, single-cell analysis formally demonstrated that pDCs do not develop from myeloid but from lymphoid progenitors, indicating an early divergence of pDC and myeloid-derived cDC lineages (3). Monocyte-derived DCs (moDCs) differentiate from monocytes during inflammation, induced by cytokines such as GM-CSF, IL-4, and TNF.
Vaccination with or induction of tolerogenic DCs (tolDC) could constitute a powerful therapy for autoimmune diseases. As many studies do not separate cDCs from moDCs in their analysis, it is not unequivocally clear whether endogenous moDCs also contribute to immune tolerance, besides cDCs and pDCs (4). In humans, DC research and experimental therapy by necessity focuses on moDCs, generated ex vivo by cytokine treatment of peripheral blood monocytes obtained via leukapheresis. To what extent these artificially produced moDCs really resemble primary endogenous DCs is not clear. It has been shown that they share some functional features with cDCs, but their overall gene expression patterns are much closer to monocytes than to any DC subset (2).
In mice, pDCs have been identified to be crucial for tolerance in several autoimmune disease models. Although most cells in the body are able to produce type I interferon (IFN-I), pDCs have been termed natural IFN-I-producing cells because of their unique adaptations to nucleic acid-sensing, which result in rapid and robust IFN-I release. Nevertheless, their in vivo contribution to antiviral and other infectious immune responses is probably less crucial than originally assumed (5). In Experimental Autoimmune Encephalomyelitis (EAE, the mouse model for MS), αPDCA1-induced pDC depletion or selective abrogation of MHCII expression on pDCs exacerbates EAE from the onset on (6, 7), while cDC depletion in cDC11-iDTR mice worsens disease during the later effector phase (8). In addition, PDCA1+ or SiglecH+ CD11cint pDCs differentiated ex vivo from bone marrow-derived cells induce recovery (9). Also in acute graft-versus-host-disease (GvHD, induced via allogeneic bone marrow transplantation) and cardiac allograft models (10, 11), as well as in RA, asthma, T1D, and even atherosclerosis (12–15), pDCs have well-demonstrated tolerogenic functions, predominantly dependent on IDO (indoleamine-2,3-dioxygenase) and resulting in Treg induction and expansion (2, 4, 16).
In addition, type 1 and/or type 2 conventional DCs (CD8+ DEC205+ cDC1, C11b+ DCIR2+ cDC2) may also contribute to peripheral Treg differentiation and/or expansion and hence tolerance, both in homeostasis (17) and in certain autoimmune diseases such as EAE (4, 18–20). Also, in GvHD, host CD11c+ cDCs were shown not to be required for the induction of disease but rather to restrict alloreactive T cell expansion (21). In addition, protection against GvHD was recently revealed to involve the tolerogenic action of both CD8+ cDC1 and CD11b+ cDC2 (22, 23). In T1D, however, there is preclinical evidence for a predominant tolerogenic role for DCIR2+ cDC2, driving Treg expansion rather than differentiation (2, 24).
The mechanism by which tolDC instigate tolerance clearly involves the induction and expansion of Tregs. These are CD4+ Foxp3+ and may be generated in the thymus as natural Tregs or induced in the periphery as iTregs. Tregs are known to exert their immunosuppressive effect mainly via IL-10 and TGFβ production, which have well-established inhibitory effects on effector T cells (Teff) and positive effects on regulatory B cells (Bregs). Furthermore, Tregs may spread peripheral tolerance by generating tolDC from DC progenitors or by maintaining cDCs in an immature state (25–28). While most studies have reported no differences in the numbers of circulating Tregs in MS, RA, or T1D patients, defects in Treg phenotype and suppressive and migratory capacity have been demonstrated (29–32). Bregs represent a small population of B lymphocytes participating in immune suppression. Many of the different B cells with suppressive characteristics are CD5+ (33). A particular population, which is CD5+ CD1d+, are very potent producers of IL-10 and are hence often referred to as B10 lymphocytes. Like Tregs, Bregs perform their regulatory functions primarily via the production of IL-10 and TGFβ as well as IL-35 (34). They have recently been recognized as very important immune modulators in various autoimmune diseases, including MS, RA, T1D, and IBD, offering novel potential strategies for therapeutic interventions (35–39).
Ex Vivo Tolerance-Inducing Cellular Therapies in Clinical Trials
The number of patients suffering from autoimmune diseases and allergies is rising dramatically (40). To avoid or dampen the aberrant harmful immune response against a specific (auto)Ag, immunological tolerance is warranted. Dampening of the immune response is also required for people receiving organ or stem cell transplants. This is currently achieved by administering “all-purpose” immunosuppressive drugs, which cause both immediate and late side effects, including increased risk for life-threatening infections and malignancies.
With the identification of tolerance-inducing cell types, significant progress has recently been made in the manufacturing and usage of tolerance-inducing cells. However, as these autologous cells are generated and manipulated ex vivo, this personalized therapy is very laborious and expensive, with many challenges, pitfalls, and safety issues (41, 42). In addition, it remains unclear whether these artificially engineered cells adequately resemble their endogenous primary counterparts in vivo.
Amongst the different tolerogenic cell types, the application of tolDC is most advanced (Figure 1A). The first clinical study on tolDC therapy was performed in 2011 in adult T1D patients. Since then, phase I and II clinical trials have been conducted for T1D, RA, Crohn’s disease, and MS. TolDC therapy is safe and shows signs of causing clinical improvement in certain patients (43, 44). In addition, tolDCs have also proven their immune dampening and thus protective potential in animal models of transplantation and allergic asthma, and clinical trials in kidney and liver transplant recipients are being set up (45–47). Once injected, tolDCs are expected to induce tolerance through various mechanisms, including the induction of Tregs and Bregs, and the stimulation of autoreactive T cell anergy and apoptosis (43).
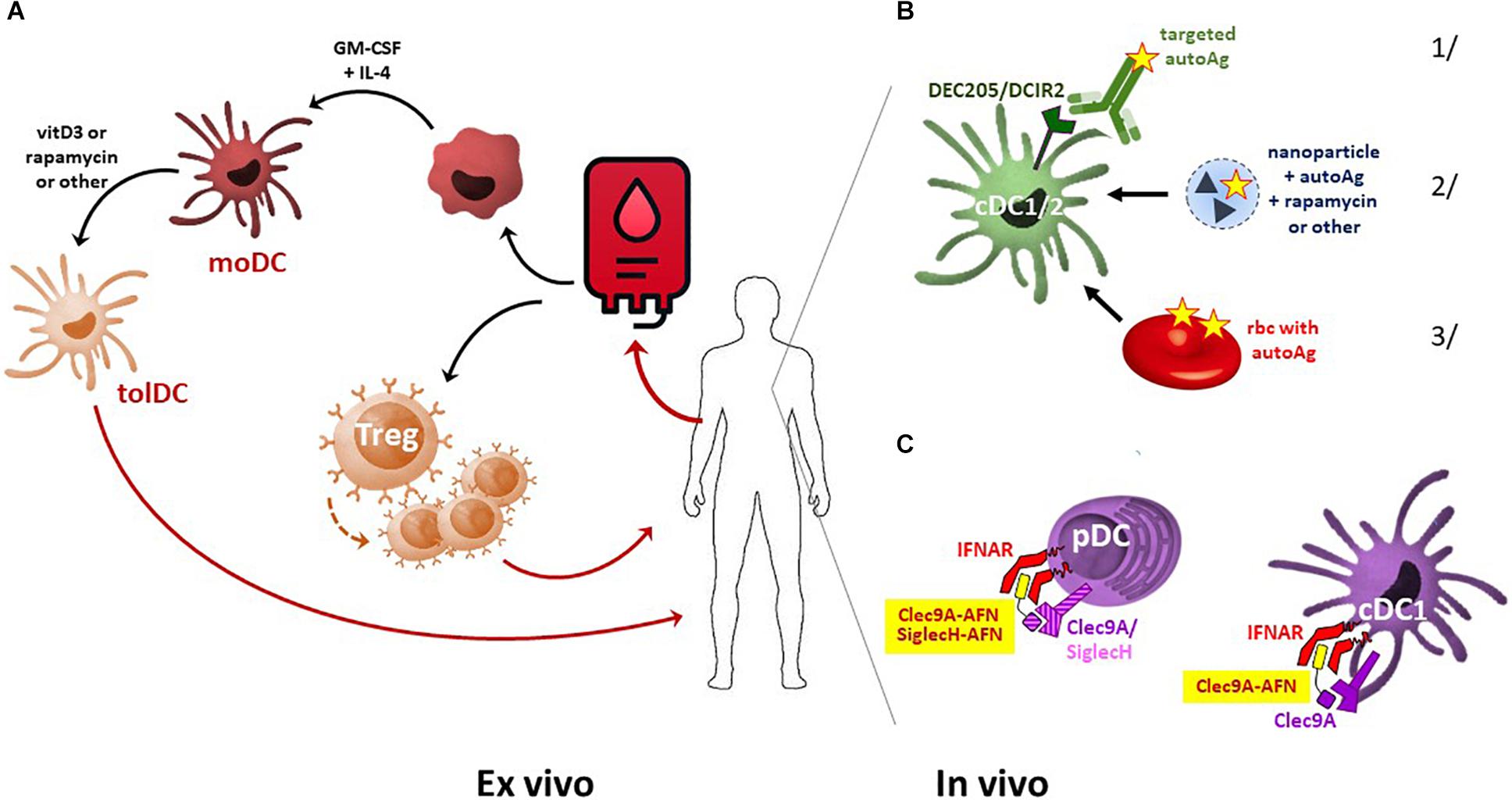
Figure 1. Comparison of ex vivo- and in vivo-generated tolDC. (A) For cellular tolDC therapy, monocytes are isolated from patient-derived peripheral blood, driven into moDC development using cytokine therapy, and subsequently tolerized by immunosuppressive agents such as vitamin D3 or rapamycin. These autologous tolDCs are then used for patient-specific treatment. From peripheral blood, Tregs may also be sorted and further expanded ex vivo. Once injected back into the patient, these Tregs dampen the immune system via multiple pathways, including the suppression of DC maturation. (B) In vivo induction of tolDC may be achieved by several approaches. Examples include delivering autoAg to DCs specifically via 1/antibody-mediated targeting of DC surface markers, 2/encapsulation in nanoparticles, microparticles, or liposomes, loaded (or not) with an immunosuppressive agent, or 3/infusion of Ag-carrying erythrocytes that will be cleared via phagocytosis predominantly by DCs and macrophages. (C) Delivery of self-Ag may add to disease development in a pro-inflammatory microenvironment, and autoAg patterns are not always uniform or stable over time. Alternatively, selective delivery of IFN-I signaling in pDC and cDC1 by AcTaferons (AFNs, targeted using SiglecH or Clec9A single-domain antibodies) may safely and cell-specifically induce systemic tolerance.
Not only tolDC but also other myeloid regulatory/immunosuppressive cell types are currently being explored, including immature myeloid-derived suppressor cells (MDSC) and activation-induced regulatory macrophages (Mregs) (48, 49). The latter are monocytes matured through adherence to plastic surfaces and exposure to various serum factors and/or cytokines and acting through IDO, IL-10, and TGFβ. In vitro, human Mregs are capable of deleting activated T cells, suppressing T-cell proliferation, and driving naive T cells to become Tregs, and the protective capacity of donor-derived Mregs is being explored in kidney transplant recipients (50).
The ex vivo expansion of autologous blood-derived Tregs has also been a clinical focus for inducing tolerance in autoimmune diseases such as GvHD, T1D, MS, Crohn’s disease, SLE, autoimmune hepatitis and uveitis, and in kidney transplant patients (43, 47). The outcomes of the completed trials indicated that Treg therapy is feasible and safe. However, like tolDC generation, this strategy requires personalized, complex, and expensive manufacturing processes. In addition, current techniques lack specificity as they expand polyclonal rather than Ag-specific Tregs and also carry the risk of expanding so-called unstable Tregs that may lose their tolerogenic function and undergo transformation to pathogenic T cells, exacerbating disease.
Still another cell type with tolerogenic capacity is the mesenchymal stromal cell (MSC) population, a non- hematopoietic, multipotent, and self-renewing population found in bone marrow as well as in other tissues such as umbilical cord, muscle, and adipose tissue, that has a proven potential to modulate anti-inflammatory monocytes and macrophages, DCs, B and T lymphocytes, and NK cells (51, 52). Clinical trials with ex vivo-expanded MSC have been successfully conducted, showing good tolerability and therapeutic potential in MS, RA, Crohn’s disease, SLE, and GvHD. A significant advantage of MSC therapy over other cell-based tolerogenic therapies is their lack of MHC expression, expanding the source of cells from autologous to allogeneic. In addition, MSC sources are multiple, including umbilical cord tissue and lipo-aspirate (43).
Generation and Mechanisms of tolDC
Human autologous tolDCs are generated ex vivo, starting from peripheral blood monocytes obtained via leukapheresis and cell sorting (Figure 1A). After culturing in the presence of GM-CSF and IL-4 to drive their development into moDCs, tolerization is usually achieved by treating with immunomodulatory agents such as vitamin D3, rapamycin, dexamethasone, corticosteroids, or specific cytokines (IL-10, TGFβ, IFNβ) (Figure 1A). Depending on the nature of the tolerizing protocol, the exact mechanisms involved in inducing systemic tolerance may diverge (53). Interestingly, whatever the tolerization protocol, this ex vivo approach will automatically lead to the generation of moDCs, which have gene expression patterns closer to monocytes than to DCs (2). As already mentioned, the endogenous DC subset that is typically found to be involved and necessary for protection in various autoimmune diseases is primarily the pDCs. In view of the recent finding that pDCs are not myeloid-derived, as was thought for decades, but are rather lymphoid-derived (3), the efficacy/efficiency of myeloid-derived moDCs as tolDCs can be questioned. Also in the cancer immunotherapy field, the disappointing performance of moDCs has been suggested to be due to an intrinsic lack of biological potency as compared to endogenous cDCs (54).
Both pDCs and cDCs induce tolerance by promoting immunosuppressive Treg differentiation and function. Important endogenous signaling agents for these processes include IL-10, TGFβ, retinoic acid (RA), and kynurenine produced by IDO (55). IDO is not expressed constitutively in DCs and requires induction by various pro- and anti-inflammatory mediators such as IFNs and TGFβ. Tolerance induced by IDO may even result in so-called “infectious” tolerance, spreading from one cell to another due to kynurenines acting as activating ligands for the aryl hydrocarbon receptor (AhR) and as such for the induction of IDO expression in other cells (56). In addition, IDO activity results in tryptophan catabolism and hence metabolic stress, negatively affecting Teff proliferation and survival. Furthermore, pDCs and cDCs can also induce peripheral tolerance by inducing Teff cell anergy, i.e. functional inactivation due to checkpoint inhibitions (18).
Type I Interferon in Autoimmune Diseases
At least 80 different forms of autoimmune diseases exist. Together, approximately 8% of the world’s population suffers from an autoimmune disease, and prevalence is sharply increasing (40). Autoimmune diseases mainly afflict women (>80%), strike at the prime of life, and cause significant debilitation, morbidity, and even mortality. In many of the most prevalent autoimmune diseases, various roles for type I IFN (IFN-I) have been described. Type I IFNs consist of a large group of structurally similar cytokines and include 13–14 subtypes of IFNα along with IFNβ, IFNε, IFNκ, IFNω, IFNδ, IFNζ, and IFNτ, all signaling through the same receptor composed of two subunits, IFNAR1 and IFNAR2. In most autoimmune models, both pathogenic and protective roles have been described, primarily for IFNα and/or IFNβ, probably depending on the disease state and the microenvironment. In general, it is important to realize that cytokines such as IFNα and IFNβ may exert different functions depending on the inflammatory context, location, and activation status of the responsive cell types.
IFNβ was the first disease-modifying therapy approved for the treatment of MS patients. Despite its therapeutic use for more than a quarter of a century, its precise mode(s) of action and specific target cells are still not completely understood. Most MS patients benefit from IFNβ therapy, but some exhibit no response or even a worsening (57). This may be due to differential effects on different cell types. In addition, side effects due to systemic toxicity preclude dose escalation and trigger therapy drop out.
In mice, triggering endogenous IFN-I release via TLR therapies can protect against IBD induced by DSS or IL-10 deficiency (58–60). Next to the activation of TLR7 or TLR9, endogenous IFN-I may also be induced by the ER-associated protein STING (stimulator of IFN genes), activated by cyclic dinucleotides. STING was shown to be important for maintaining intestinal homeostasis, and it was hence proposed that modulating the STING pathway may be of benefit in IBD (61). However, endogenous STING signaling induces both pro- and anti-inflammatory cytokines, and indeed, STING agonists were recently shown to exacerbate colitis (62). Collectively, these reports suggest that the beneficial effect of IFN-I in IBD is probably local and/or cell-specific.
From experiments performed in diabetic mice and rats, the role of IFN-I in T1D pathogenesis was originally believed to be beneficial (16). Later, this protective role was questioned, as IFNα produced by pancreatic β-cells or by pDC was shown to hasten murine diabetes progression (63, 64), and a detrimental role for pDC-derived IFNα in the initiation of T1D was eventually concluded from experiments in NOD mice (65). Nevertheless, ingestion of low-dose IFNα preserved β-cell function in recent-onset T1D patients (66), and additional clinical trials have since shown protective effects of ingested IFNα in patients suffering from MS (67).
Also in arthritis models and human RA, various roles for IFN-I have been proposed, ranging from detrimental to protective. Several experiments performed in both mice and monkeys, as well as pilot studies in RA patients, clearly suggest clinically meaningful improvement due to IFNβ treatment (68). Interestingly, a protective role has also been demonstrated for pDC, and clinical trials with tolDC are ongoing (47, 69). The controversial results using systemic IFN-I could possibly indicate local and/or cell-specific differential effects of IFN-I.
Using a murine GvHD model, TLR7 agonists were found to protect IFNAR1-dependently, involving the tolerogenic action of cDCs and increased Treg responses (22). Furthermore, selective activation of IFN-I pathways prior to hematopoietic stem cell transplantation was shown to be dependent on IFN-I signaling in CD11c+ DC, reducing their ability to stimulate allogeneic T cells (23).
In addition, it has been suggested that the lack of IFN-I secretion by pDCs contributes to the development of a TH2 response in allergic asthma and that treatment of chronic allergic diseases with IFN-I may be a promising way to induce tolerance (70).
Strategies to Induce tolDC In Vivo
Ex vivo-generated tolDCs are well-tolerized and may have protective effects, but they also have several disadvantages, as they represent a personalized, laborious, and expensive therapy that raises many safety and economic concerns. To overcome these limitations, new approaches to induce tolDCs in vivo are being vigorously explored (Table 1). Examples include selective self-Ag targeting toward the DC receptor DEC205 before or after EAE immunization (19, 71, 72), or toward the pDC receptor SiglecH or the cDC2 receptor DCIR2 before EAE immunization, to promote immunological tolerance (20, 71, 73) (Figure 1B). Other successful approaches include injection of autoAg-containing nano- or microparticles or liposomes. These are taken up via phagocytosis or endocytosis, predominantly by antigen-presenting cells (APC, including DCs and myeloid cells), and disease-relevant peptides or proteins can be co-encapsulated with immunosuppressive agents such as rapamycin, IL-10, NFκB inhibitors, or AhR ligands (74–79). Most of these reported studies were performed in EAE and T1D models, but their efficacy has also been illustrated in other autoimmune diseases such as arthritis and IBD (76, 80, 81) and in various transplantation models (82) (Table 1). In addition, transfusion with autoAg-decorated red blood cells (rbc) (Figure 1B), which are known to be preferentially phagocytosed by DCs and macrophages, has recently proven its efficacy in both EAE and NOD diabetic mice (83). Importantly, maturation or activation signals for DCs, present under inflammatory conditions, may abrogate the tolerogenic protection conveyed by autoAg delivery (20), and as such endogenous inflammation could turn a self-Ag-based DC tolerizing therapy into one further exacerbating disease (44). In addition, self-Ag patterns are not always uniform or stable during disease development and progression.

Table 1. Summary of approaches to inducing tolDC in vivo.
Not only DCs and myeloid cells are being targeted; efforts are also underway to selectively stimulate the in vivo expansion of Tregs. So far, most trials have concentrated on the use of low-dose IL-2 to achieve this, since IL-2 is crucial for T-cell proliferation and its receptor is most abundant on natural Tregs, but an optimal and long-lasting regime has not yet been found and agreed upon (30, 43). Furthermore, no pharmacological approaches are currently available to selectively expand autoAg- or disease-specific Tregs in vivo.
Given the possible protective role of IFN-I in autoimmune diseases, especially in MS, we decided to apply our targeted AcTaferons in the EAE model. AcTaferons (AFNs) are IFN-I based AcTakines (Activity-on-Target cytokines). Basically, AcTakines are a novel class of engineered immunocytokines, the key difference between AcTakines and immunocytokines being the exclusive use of mutant (engineered) e-cytokines with severely reduced receptor affinity instead of wild-type (WT) cytokines (84) (Figure 2). While immunocytokines, where WT cytokines are fused to targeting antibodies or antibody fragments, can still bind with great affinity to their ubiquitous receptors while traveling through the body, causing residual side effects and their systemic removal [the so-called “sink” effect (85)], AcTakines cannot signal when administered systemically except in those cells that express a surface molecule specifically recognized by the targeting moiety linked to the mutant cytokine. As a result, AcTakines do not cause the multiple toxic side effects usually accompanying cytokine therapies. In addition, they provide unique research tools for dissecting the in vivo cell-specific functions of pleiotropic cytokines under normal or pathophysiological conditions. Thanks to a convenient “plug-and-play” assembly of modular building blocks, AcTakines can be engineered easily by coupling various mono- or multimeric e-cytokines to targeting moieties such as camelid-derived single-domain antibodies (sdAb, VHH), peptide motifs specifically recognized by receptor isoforms, or ligands interacting with their cell-specific cognate receptors. During recent years, we have successfully and safely employed various cell-specific AcTakines as potential anti-tumor therapies (86–88). Recently, we also obtained evidence in EAE that DC-targeted AFN can be used to specifically target IFN-I signaling to DCs as an in vivo method to induce tolerance (89) (Figure 1C). Systemic tolerance was evident in pDCs (increased numbers and an enhanced tolerogenic signature including IDO and TGFβ production) as well as in Tregs and Bregs, both of which produced significantly more IL-10 and TGFβ. Interestingly, pDC targeting was superior to cDC1 targeting during early progression of EAE, but cDC1 targeting later during disease progression significantly added to the protection. In contrast to therapy with autologous ex vivo-generated moDCs derived from a cell lineage that may not be optimal (myeloid), AFNs deliver the IFN-I signaling potential specifically to endogenous pDCs and cDC1s in vivo. Furthermore, cell-specific targeting not only limits the possibility of aspecific toxic side effects but also avoids signaling in unwanted cell types. The relevance of the latter becomes clear when comparing the protective capacities of untargeted WT IFN-I with CD8- or DC-targeted AFN. While WT IFN-I can delay disease onset and DC-targeted AFN provides profound protection, CD8-targeted AFN worsens disease (89). This strategy still leaves many more options open, such as selective targeting to B lymphocytes, specific myeloid subsets, and more.
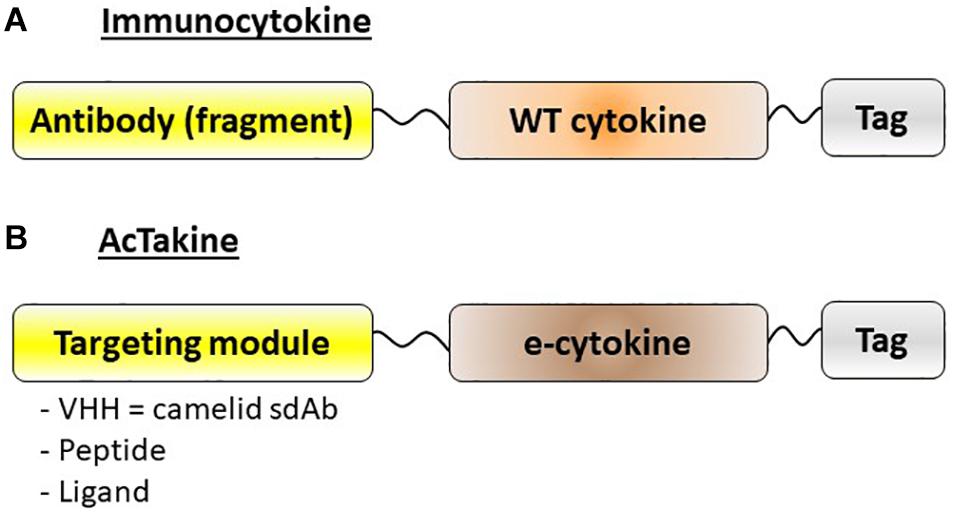
Figure 2. Schematic representation of immunocytokines and AcTakines. (A) Immunocytokines are typically engineered by coupling a wild-type (WT) cytokine to a targeting module, usually an antibody or antibody fragment. (B) AcTakines consist of a mutated (engineered) e-cytokine with reduced cognate receptor affinity, coupled C-terminally via a 20xGGS linker to a targeting moiety. In general, a camelid-derived single domain antibody (sdAb = VHH) is used for the latter, although peptides or ligands can also be employed. For purification purposes, AcTakines are decorated with a C-terminal affinity tag.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the Charcot Foundation and by a Sponsored Research Collaboration with Orionis Biosciences.
Conflict of Interest
JT was employed by company Orionis Biosciences, who provided funding for the study.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. (2018) 154:3–20. doi: 10.1111/imm.12888
2. Price JD, Tarbell KV. The role of dendritic cell subsets and innate immunity in the pathogenesis of Type 1 diabetes and other Autoimmune diseases. Front Immunol. (2015) 6:288. doi: 10.3389/fimmu.2015.00288
3. Dress RJ, Dutertre CA, Giladi A, Schlitzer A, Low I, Shadan NB, et al. Plasmacytoid dendritic cells develop from Ly6D(+) lymphoid progenitors distinct from the myeloid lineage. Nat Immunol. (2019) 20:852–64. doi: 10.1038/s41590-019-0420-3
4. Audiger C, Rahman MJ, Yun TJ, Tarbell KV, Lesage S. the importance of dendritic cells in maintaining immune tolerance. J Immunol. (2017) 198:2223–31. doi: 10.4049/jimmunol.1601629
5. Ali S, Mann-Nüttel R, Schulze A, Richter L, Alferink J, Scheu S. Sources of Type I interferons in infectious immunity: plasmacytoid dendritic cells not always in the driver’s seat. Front Immunol. (2019) 10:778. doi: 10.3389/fimmu.2019.00778
6. Bailey-Bucktrout SL, Caulkins SC, Goings G, Fischer JA, Dzionek A, Miller SD. Cutting edge: central nervous system plasmacytoid dendritic cells regulate the severity of relapsing experimental autoimmune encephalomyelitis. J Immunol. (2008) 180:6457–61.
7. Irla M, Küpfer N, Suter T, Lissilaa R, Benkhoucha M, Skupsky J, et al. MHC class II-restricted antigen presentation by plasmacytoid dendritic cells inhibits T cell-mediated autoimmunity. J Exp Med. (2010) 207:1891–905. doi: 10.1084/jem.20092627
8. Yogev N, Frommer F, Lukas D, Kautz-Neu K, Karram K, Ielo D, et al. Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor(+) regulatory T cells. Immunity. (2012) 37:264–75. doi: 10.1016/j.immuni.2012.05.025
9. Duraes FV, Lippens C, Steinbach K, Dubrot J, Brighouse D, Bendriss-Vermare N, et al. pDC therapy induces recovery from EAE by recruiting endogenous pDC to sites of CNS inflammation. J Autoimmun. (2016) 67:8–18. doi: 10.1016/j.jaut.2015.08.014
10. Hadeiba H, Sato T, Habtezion A, Oderup C, Pan J, Butcher EC. CCR9 expression defines tolerogenic plasmacytoid dendritic cells able to suppress acute graft-versus-host disease. Nat Immunol. (2008) 9:1253–60. doi: 10.1038/ni.1658
11. Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat Immunol. (2006) 7:652–62.
12. Jongbloed SL, Benson RA, Nickdel MB, Garside P, McInnes IB, Brewer JM. Plasmacytoid dendritic cells regulate breach of self-tolerance in autoimmune arthritis. J Immunol. (2009) 182:963–8.
13. Kool M, van Nimwegen M, Willart MA, Muskens F, Boon L, Smit JJ, et al. An anti-inflammatory role for plasmacytoid dendritic cells in allergic airway inflammation. J Immunol. (2009) 183:1074–82. doi: 10.4049/jimmunol.0900471
14. Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD. The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J Immunol. (2007) 179:5041–53.
15. Yun TJ, Lee JS, Machmach K, Shim D, Choi J, Wi YJ, et al. Indoleamine 2,3-dioxygenase-expressing aortic plasmacytoid dendritic cells protect against atherosclerosis by induction of regulatory T cells. Cell Metab. (2016) 23:852–66.
16. Guery L, Hugues S. Tolerogenic and activatory plasmacytoid dendritic cells in autoimmunity. Front Immunol. (2013) 4:59. doi: 10.3389/fimmu.2013.00059
17. Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, et al. CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol. (2008) 181:6923–33.
18. Hasegawa H, Matsumoto T. Mechanisms of tolerance induction by dendritic cells in vivo. Front Immunol. (2018) 9:350. doi: 10.21614/chirurgia.112.1.25
19. Ring S, Maas M, Nettelbeck DM, Enk AH, Mahnke K. Targeting of autoantigens to DEC205(+) dendritic cells in vivo suppresses experimental allergic encephalomyelitis in mice. J Immunol. (2013) 191:2938–47. doi: 10.4049/jimmunol.1202592
20. Tabansky I, Keskin DB, Watts D, Petzold C, Funaro M, Sands W, et al. Targeting DEC-205(−)DCIR2(+) dendritic cells promotes immunological tolerance in proteolipid protein-induced experimental autoimmune encephalomyelitis. Mol Med. (2018) 24:17. doi: 10.1186/s10020-018-0017-6
21. Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, Don AL, et al. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat Med. (2011) 18:135–42. doi: 10.1038/nm.2597
22. Gaignage M, Marillier RG, Cochez PM, Dumoutier L, Uyttenhove C, Coutelier JP, et al. The TLR7 ligand R848 prevents mouse graft-versus-host disease and cooperates with anti-interleukin-27 antibody for maximal protection and regulatory T-cell upregulation. Haematologica. (2019) 104:392–402. doi: 10.3324/haematol.2018.195628
23. Fischer JC, Bscheider M, Göttert S, Thiele Orberg E, Combs SE, Bassermann F, et al. Type I interferon signaling before hematopoietic stem cell transplantation lowers donor T cell activation via reduced allogenicity of recipient cells. Sci Rep. (2019) 9:14955. doi: 10.1038/s41598-019-51431-2
24. Price JD, Hotta-Iwamura C, Zhao Y, Beauchamp NM, Tarbell KV. DCIR2+ cDC2 DCs and Zbtb32 restore CD4+ T-cell tolerance and inhibit diabetes. Diabetes. (2015) 64:3521–31. doi: 10.2337/db14-1880
25. Darrasse-Jèze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med. (2009) 206:1853–62. doi: 10.1084/jem.20090746
26. Min WP, Zhou D, Ichim TE, Strejan GH, Xia X, Yang J, et al. Inhibitory feedback loop between tolerogenic dendritic cells and regulatory T cells in transplant tolerance. J Immunol. (2003) 170:1304–12.
27. Maldonado RA, von Andrian UH. How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol. (2010) 108:111–65. doi: 10.1161/ATVBAHA.116.308464
28. Probst HC, Muth S, Schild H. Regulation of the tolerogenic function of steady-state DCs. Eur J Immunol. (2014) 44:927–33. doi: 10.1002/eji.201343862
29. Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation. (2017) 14:117. doi: 10.1186/s12974-017-0892-8
30. Hull CM, Peakman M, Tree TIM. Regulatory T cell dysfunction in type 1 diabetes: what’s broken and how can we fix it? Diabetologia. (2017) 60:1839–50. doi: 10.1007/s00125-017-4377-1
31. Malemud CJ. Defective T-cell apoptosis and T-regulatory cell dysfunction in rheumatoid arthritis. Cells. (2018) 7:223. doi: 10.3390/cells7120223
32. Xufré C, Costa M, Roura-Mir C, Codina-Busqueta E, Usero L, Pizarro E, et al. Low frequency of GITR+ T cells in ex vivo and in vitro expanded Treg cells from type 1 diabetic patients. Int Immunol. (2013) 25:563–74. doi: 10.1093/intimm/dxt020
33. Klinker MW, Lundy SK. Multiple mechanisms of immune suppression by B lymphocytes. Mol Med. (2012) 18:123–37. doi: 10.2119/molmed.2011.00333
34. Baba Y, Saito Y, Kotetsu Y. Heterogeneous subsets of B-lineage regulatory cells (Breg cells). Int Immunol. (2019) 32:155–62.
35. Boldison J, Da Rosa LC, Davies J, Wen L, Wong FS. Dendritic cells license regulatory B cells to produce IL-10 and mediate suppression of antigen-specific CD8 T cells. Cell Mol Immunol. (2019). [Epub ahead of print]. doi: 10.1038/s41423-019-0324-z
36. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. (2002) 3:944–50.
37. Kim Y, Kim G, Shin HJ, Hyun JW, Kim SH, Lee E, et al. Restoration of regulatory B cell deficiency following alemtuzumab therapy in patients with relapsing multiple sclerosis. J Neuroinflammation. (2018) 15:300. doi: 10.1186/s12974-018-1334-y
38. Mauri C, Menon M. Human regulatory B cells in health and disease: therapeutic potential. J Clin Invest. (2017) 127:772–9. doi: 10.1172/JCI85113
39. Oleinika K, Rosser EC, Matei DE, Nistala K, Bosma A, Drozdov I, et al. CD1d-dependent immune suppression mediated by regulatory B cells through modulations of iNKT cells. Nat Commun. (2018) 9:684. doi: 10.1038/s41467-018-02911-y
40. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. (2002) 347:911–20.
41. Gross CC, Jonuleit H, Wiendl H. Fulfilling the dream: tolerogenic dendritic cells to treat multiple sclerosis. Eur J Immunol. (2012) 42:569–72. doi: 10.1002/eji.201242402
42. Kim SH, Jung HH, Lee CK. Generation, characteristics and clinical trials of ex vivo generated tolerogenic dendritic cells. Yonsei Med J. (2018) 59:807–15. doi: 10.3349/ymj.2018.59.7.807
43. Mosanya CH, Isaacs JD. Tolerising cellular therapies: what is their promise for autoimmune disease? Ann Rheum Dis. (2019) 78:297–310. doi: 10.1136/annrheumdis-2018-214024
44. Fucikova J, Palova-Jelinkova L, Bartunkova J, Spisek R. Induction of tolerance and immunity by dendritic cells: mechanisms and clinical applications. Front Immunol. (2019) 10:2393. doi: 10.3389/fimmu.2019.02393
45. de Aragao-Franca LS, Aragão-França LS, Rocha VCJ, Rocha VCJ, Cronemberger-Andrade A, da Costa FHB, et al. Tolerogenic dendritic cells reduce airway inflammation in a model of dust mite triggered allergic inflammation. Allergy Asthma Immunol Res. (2018) 10:406–19.
46. Obregon C, Kumar R, Pascual MA, Vassalli G, Golshayan D. Update on dendritic cell-induced immunological and clinical tolerance. Front Immunol. (2017) 8:1514. doi: 10.3389/fimmu.2017.01514
47. Ten Brinke A, Martinez-Llordella M, Cools N, Hilkens CMU, van Ham SM, Sawitzki B, et al. Ways forward for tolerance-inducing cellular therapies- an AFACTT perspective. Front Immunol. (2019) 10:181. doi: 10.3389/fimmu.2019.00181
48. Consonni FM, Porta C, Marino A, Pandolfo C, Mola S, Bleve A, et al. Myeloid-derived suppressor cells: ductile targets in disease. Front Immunol. (2019) 10:949. doi: 10.3389/fimmu.2019.00949
49. Ten Brinke A, Hilkens CM, Cools N, Geissler EK, Hutchinson JA, Lombardi G, et al. Clinical use of tolerogenic dendritic cells-harmonization approach in european collaborative effort. Mediators Inflamm. (2015) 2015:471719. doi: 10.1155/2015/471719
50. Amodio G, Cichy J, Conde P, Matteoli G, Moreau A, Ochando J, et al. Role of myeloid regulatory cells (MRCs) in maintaining tissue homeostasis and promoting tolerance in autoimmunity, inflammatory disease and transplantation. Cancer Immunol Immunother. (2019) 68:661–72. doi: 10.1007/s00262-018-2264-3
51. Weiss ARR, Dahlke MH. Immunomodulation by mesenchymal stem cells (MSCs): mechanisms of action of living, apoptotic, and dead MSCs. Front Immunol. (2019) 10:1191. doi: 10.3389/fimmu.2019.01191
52. Mizukami A, Swiech K. Mesenchymal stromal cells: from discovery to manufacturing and commercialization. Stem Cells Int. (2018) 2018:4083921. doi: 10.1155/2018/4083921
53. Navarro-Barriuso J, Mansilla MJ, Naranjo-Gómez M, Sánchez-Pla A, Quirant-Sánchez B, Teniente-Serra A, et al. Comparative transcriptomic profile of tolerogenic dendritic cells differentiated with vitamin D3, dexamethasone and rapamycin. Sci Rep. (2018) 8:14985. doi: 10.1038/s41598-018-33248-7
54. Osugi Y, Vuckovic S, Hart DN. Myeloid blood CD11c(+) dendritic cells and monocyte-derived dendritic cells differ in their ability to stimulate T lymphocytes. Blood. (2002) 100:2858–66.
55. Mayer CT, Berod L, Sparwasser T. Layers of dendritic cell-mediated T cell tolerance, their regulation and the prevention of autoimmunity. Front Immunol. (2012) 3:183. doi: 10.3389/fimmu.2012.00183
56. Wu H, Gong J, Liu Y. Indoleamine 2, 3-dioxygenase regulation of immune response (Review). Mol Med Rep. (2018) 17:4867–73.
57. Axtell RC, Raman C. Janus-like effects of type I interferon in autoimmune diseases. Immunol Rev. (2012) 248:23–35. doi: 10.1136/bmjopen-2017-020904
58. Bleich A, Janus LM, Smoczek A, Westendorf AM, Strauch U, Mähler M, et al. CpG motifs of bacterial DNA exert protective effects in mouse models of IBD by antigen-independent tolerance induction. Gastroenterology. (2009) 136:278–87. doi: 10.1053/j.gastro.2008.09.022
59. Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. (2005) 115:695–702.
60. Sainathan SK, Bishnupuri KS, Aden K, Luo Q, Houchen CW, Anant S, et al. Toll-like receptor-7 ligand Imiquimod induces type I interferon and antimicrobial peptides to ameliorate dextran sodium sulfate-induced acute colitis. Inflamm Bowel Dis. (2012) 18:955–67. doi: 10.1002/ibd.21867
61. Ahn J, Son S, Oliveira SC, Barber GN. STING-dependent signaling underlies IL-10 controlled inflammatory colitis. Cell Rep. (2017) 21:3873–84. doi: 10.1016/j.celrep.2017.11.101
62. Martin GR, Blomquist CM, Henare KL, Jirik FR. Stimulator of interferon genes (STING) activation exacerbates experimental colitis in mice. Sci Rep. (2019) 9:14281. doi: 10.1038/s41598-019-50656-5
63. Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA. (2008) 105:12439–44. doi: 10.1073/pnas.0806439105
64. Stewart TA, Hultgren B, Huang X, Pitts-Meek S, Hully J, MacLachlan NJ. Induction of type I diabetes by interferon-alpha in transgenic mice. Science. (1993) 260:1942–6.
65. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. (2013) 19:65–73. doi: 10.1038/nm.3042
66. Rother KI, Brown RJ, Morales MM, Wright E, Duan Z, Campbell C, et al. Effect of ingested interferon-alpha on beta-cell function in children with new-onset type 1 diabetes. Diabetes Care. (2009) 32:1250–5. doi: 10.2337/dc08-2029
67. Brod SA, Ingested Type I. Interferon-state of the art as treatment for autoimmunity part 2. Pharmaceuticals (Basel). (2010) 3:1108–21.
68. van Holten J, Plater-Zyberk C, Tak PP. Interferon-beta for treatment of rheumatoid arthritis? Arthritis Res. (2002) 4:346–52.
69. Nehmar R, Alsaleh G, Voisin B, Flacher V, Mariotte A, Saferding V, et al. Therapeutic modulation of plasmacytoid dendritic cells in experimental arthritis. Arthritis Rheumatol. (2017) 69:2124–35. doi: 10.1002/art.40225
70. Gonzales-van Horn SR, Farrar JD. Interferon at the crossroads of allergy and viral infections. J Leukoc Biol. (2015) 98:185–94. doi: 10.1189/jlb.3RU0315-099R
71. Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, et al. Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest. (2013) 123:844–54. doi: 10.1172/JCI65260
72. Stern JN, Keskin DB, Kato Z, Waldner H, Schallenberg S, Anderson A, et al. Promoting tolerance to proteolipid protein-induced experimental autoimmune encephalomyelitis through targeting dendritic cells. Proc Natl Acad Sci USA. (2010) 107:17280–5. doi: 10.1073/pnas.1010263107
73. Loschko J, Heink S, Hackl D, Dudziak D, Reindl W, Korn T, et al. Antigen targeting to plasmacytoid dendritic cells via Siglec-H inhibits Th cell-dependent autoimmunity. J Immunol. (2011) 187:6346–56. doi: 10.4049/jimmunol.1102307
74. LaMothe RA, Kolte PN, Vo T, Ferrari JD, Gelsinger TC, Wong J, et al. Tolerogenic nanoparticles induce antigen-specific regulatory T cells and provide therapeutic efficacy and transferrable tolerance against experimental autoimmune encephalomyelitis. Front Immunol. (2018) 9:281. doi: 10.3389/fimmu.2018.00281
75. Serra P, Santamaria P. Nanoparticle-based approaches to immune tolerance for the treatment of autoimmune diseases. Eur J Immunol. (2018) 48:751–6.
76. Capini C, Jaturanpinyo M, Chang HI, Mutalik S, McNally A, Street S, et al. Antigen-specific suppression of inflammatory arthritis using liposomes. J Immunol. (2009) 182:3556–65. doi: 10.4049/jimmunol.0802972
77. Cappellano G, Woldetsadik AD, Orilieri E, Shivakumar Y, Rizzi M, Carniato F, et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine. (2014) 32:5681–9. doi: 10.1016/j.vaccine.2014.08.016
78. Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci USA. (2015) 112:E156–65. doi: 10.1073/pnas.1408686111
79. Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2012) 109:11270–5. doi: 10.1073/pnas.1120611109
80. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. (2016) 530:434–40. doi: 10.1038/nature16962
81. Getts DR, Terry RL, Getts MT, Deffrasnes C, Müller M, van Vreden C, et al. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Transl Med. (2014) 6:219ra7. doi: 10.1126/scitranslmed.3007563
82. Ochando J, Ordikhani F, Jordan S, Boros P, Thomson AW. Tolerogenic dendritic cells in organ transplantation. Transpl Int. (2020) 33:113–27. doi: 10.1111/tri.13504
83. Pishesha N, Bilate AM, Wibowo MC, Huang NJ, Li Z, Deshycka R, et al. Engineered erythrocytes covalently linked to antigenic peptides can protect against autoimmune disease. Proc Natl Acad Sci USA. (2017) 114:3157–62. doi: 10.1073/pnas.1701746114
84. Garcin G, Paul F, Staufenbiel M, Bordat Y, Van der Heyden J, Wilmes S, et al. High efficiency cell-specific targeting of cytokine activity. Nat Commun. (2014) 5:3016. doi: 10.1038/ncomms4016
85. Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci USA. (2015) 112:3320–5. doi: 10.1073/pnas.1416159112
86. Cauwels A, Van Lint S, Garcin G, Bultinck J, Paul F, Gerlo S, et al. A safe and highly efficient tumor-targeted type I interferon immunotherapy depends on the tumor microenvironment. Oncoimmunology. (2018) 7:e1398876. doi: 10.1080/2162402X.2017.1398876
87. Cauwels A, Van Lint S, Paul F, Garcin G, De Koker S, Van Parys A, et al. Delivering type I interferon to dendritic cells empowers tumor eradication and immune combination treatments. Cancer Res. (2018) 78:463–74. doi: 10.1158/0008-5472.CAN-17-1980
88. Huyghe L, Van Parys A, Cauwels A, Van Lint S, De Munter S, Bultinck J, et al. Safe eradication of large established tumors using neovasculature-targeted tumor necrosis factor-based therapies. EMBO Mol Med. (2020) 12:e11223. doi: 10.15252/emmm.201911223
89. Cauwels A, Van Lint S, Catteeuw D, Pang S, Paul F, Rogge E, et al. Targeting interferon activity to dendritic cells enables in vivo tolerization and protection against EAE in mice. J Autoimmun. (2019) 97:70–6. doi: 10.1016/j.jaut.2018.10.010
90. Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, et al. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J Hepatol. (2015) 62:1349–56. doi: 10.1016/j.jhep.2015.01.006
91. Kontos S, Kourtis IC, Dane KY, Hubbell JA. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc Natl Acad Sci USA. (2013) 110:E60–8. doi: 10.1073/pnas.1216353110
92. Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, et al. Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci Signal. (2016) 9:ra61. doi: 10.1126/scisignal.aad0612
93. Tanriver Y, Ratnasothy K, Bucy RP, Lombardi G, Lechler R. Targeting MHC class I monomers to dendritic cells inhibits the indirect pathway of allorecognition and the production of IgG alloantibodies leading to long-term allograft survival. J Immunol. (2010) 184:1757–64. doi: 10.4049/jimmunol.0902987
94. Xu W, Ling P, Zhang T. Toward immunosuppressive effects on liver transplantation in rat model: tacrolimus loaded poly(ethylene glycol)-poly(D,L-lactide) nanoparticle with longer survival time. Int J Pharm. (2014) 460:173–80. doi: 10.1016/j.ijpharm.2013.10.035
95. Bahmani B, Uehara M, Jiang L, Ordikhani F, Banouni N, Ichimura T, et al. Targeted delivery of immune therapeutics to lymph nodes prolongs cardiac allograft survival. J Clin Invest. (2018) 128:4770–86. doi: 10.1172/JCI120923
Keywords: autoimmunity, dendritic cells, tolerance, type-I-IFN, tolerogenic dendritic cells, pDC, cDC
Citation: Cauwels A and Tavernier J (2020) Tolerizing Strategies for the Treatment of Autoimmune Diseases: From ex vivo to in vivo Strategies. Front. Immunol. 11:674. doi: 10.3389/fimmu.2020.00674
Received: 04 February 2020; Accepted: 25 March 2020;
Published: 14 May 2020.
Edited by:
Erwan Mortier, INSERM U1232 Centre de Recherche en Cancérologie et Immunologie Nantes Angers (CRCINA), FranceReviewed by:
Martina Severa, Istituto Superiore di Sanità (ISS), ItalyRodrigo Naves, University of Chile, Chile
Copyright © 2020 Cauwels and Tavernier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Tavernier, amFuLnRhdmVybmllckB2aWItdWdlbnQuYmU=