 Joyutpal Das1,2*
Joyutpal Das1,2* Atta Gill3
Atta Gill3 Christine Lo2,4
Christine Lo2,4 Natalie Chan-Lam3Siân Price2Stephen B. Wharton4,5Helen Jessop3
Natalie Chan-Lam3Siân Price2Stephen B. Wharton4,5Helen Jessop3 Basil Sharrack2,4
Basil Sharrack2,4 John A. Snowden3
John A. Snowden3- 1Department of Neurology, Salford Royal NHS Foundation Trust, Manchester, United Kingdom
- 2Department of Neurology, Royal Hallamshire Hospital, Teaching Hospitals NHS Foundation Trust, Sheffield, United Kingdom
- 3Department of Haematology, Royal Hallamshire Hospital, Teaching Hospitals NHS Foundation Trust, Sheffield, United Kingdom
- 4Sheffield Institute of Translational Neuroscience, University of Sheffield, Sheffield, United Kingdom
- 5Department of Histopathology, Royal Hallamshire Hospital, Teaching Hospitals NHS Foundation Trust, Sheffield, United Kingdom
Complications involving the central nervous system (CNS) occur in 9–14% of patients following allogeneic hematopoietic stem cell transplantation (HSCT), including stroke-like episodes, demyelination, encephalitis, and nonspecific neurological symptoms. Here we report a case of multiple sclerosis (MS) like relapsing remitting encephalomyelitis following allogeneic HSCT, which did not respond to disease modifying therapies (DMTs) and “domino” autologous HSCT. A 53-year-old male was treated with allogeneic HSCT for lymphoid blast transformation of chronic myeloid leukemia. Ten months later he presented with confusion, slurred speech, left sided facial weakness and ataxia. A magnetic resonance imaging brain scan showed multiple enhancing tumefactive lesions. Neuromyelitis optica (NMO) and myelin oligodendrocyte glycoprotein (MOG) antibodies were negative. After extensive investigations for infections, autoimmune disorders and recurrence of malignancy, he underwent brain biopsy, which showed a macrophage rich lesion with severe myelin loss but axonal preservation indicating a demyelinating pathology. Although his symptoms improved with corticosteroids, he relapsed five months later. In the absence of any systemic features suggesting graft versus host disease (GvHD), his presentation was thought to be compatible with MS. The illness followed an aggressive course that did not respond to glatiramer acetate and natalizumab. He was therefore treated with “domino” autologous HSCT, which also failed to induce long-term remission. Despite further treatment with ocrelizumab, he died of progressive disease. An autopsy limited to the examination of brain revealed multifocal destructive leukoencephalopathy with severe myelin and axonal loss. Immunohistochemistry showed macrophage located in the perivascular area, with no T or B lymphocytes. The appearance was unusual and not typical for chronic MS plaques. Reported cases of CNS demyelination following allogeneic HSCT are very limited in the literature, especially in relation to histopathological examination. Although the clinical disease course of our patient following allogeneic HSCT resembled an “MS-like” relapsing remitting encephalomyelitis, the autopsy examination did not show any evidence of active inflammation. The impact of DMTs and HSCT on the histological appearance of “MS-like” CNS pathologies is unknown. Therefore, reporting this and similar cases will improve our awareness and understanding of underlying disease mechanisms.
Introduction
Complications involving the central nervous system (CNS) occur in 9–14% of patients following allogeneic hematopoietic stem cell transplantation (HSCT) (1). These include drug toxicities, infections, and metabolic disturbances (2, 3). Graft versus host disease (GvHD) associated with allogeneic HSCT usually affects skin, gut, and liver. GvHD rarely affects the CNS, but when it is involved there is often a significant systemic GvHD elsewhere (4). Patients with chronic GvHD affecting the CNS may present with stroke-like episodes, transverse myelitis, multiple sclerosis (MS) or acute disseminated encephalomyelitis (ADEM) like disorders, encephalitis, and other nonspecific neurological symptoms (5).
Secondary autoimmune diseases, particularly thyroid and other endocrine disorders are recognized complications following allogeneic HSCT, but MS-like presentation has rarely been reported in the literature (5–8). In some cases, there is apparent adoptive transfer of specific autoimmune diseases or autoimmune diathesis (9). Here we report a case of “MS-like” relapsing remitting encephalomyelitis following allogeneic HSCT, which did not respond to three disease modifying therapies (DMTs) and “domino” autologous HSCT.
Case Presentation
A 53-year-old male with chronic myeloid leukemia (CML), who failed to respond to imatinib (tyrosine kinase inhibitor), underwent allogeneic HSCT for lymphoid blast transformation eighteen months after his initial presentation. Following lymphoid blast transformation, imatinib was also switched to nilotinib (tyrosine kinase inhibitor). The allogeneic HSCT was performed using a conditioning regimen consisting of fludarabine, busulphan and anti-thymocyte globulin (ATG) with ciclosporin A as GvHD prophylaxis. An unrelated male donor matched for HLA A, B, C, and DR loci was used (Supplementary Table 1). Neither donor nor patient had the HLA-DRB1*15:01 genotype associated with increased risk of MS (10). Engraftment of neutrophils and platelets occurred promptly, but the patient had routine transplant related toxicities, including post-transplant cytomegalovirus (CMV) reactivation and possible posterior reversible encephalopathy syndrome for which GvHD prophylaxis was switched from ciclosporin A to tacrolimus. There were no changes suggestive of demyelination on brain magnetic resonance imaging (MRI). Tacrolimus was successfully weaned off without GvHD. In view of the high relapse risk, he was maintained on nilotinib post-transplant. Subsequent bone marrow examinations along with peripheral blood monitoring showed full donor chimerism and molecular negativity for BCR-ABL transcripts confirming ongoing molecular remission of leukemia consistent with cure.
Despite a good initial recovery, 10 months later the patient was admitted with confusion, slurred speech, left sided facial weakness, and ataxia (Figure 1A). He had no systemic features suggestive of GvHD, such as rash, deranged liver function, or gastrointestinal disturbance. A brain MRI showed contrast enhancing tumefactive lesions in the left peri-insular area, both corona radiatae and brainstem (Figures 1B,C). Screenings for human immunodeficiency virus (HIV), Borrelia burgdorferi, syphilis and toxoplasmosis in serum were negative. Analysis of the cerebrospinal fluid (CSF) showed a white cell count of 3 × 106 /L with marginally elevated protein of 0.76 g/L and normal CSF to serum glucose ratio. He had matching bands in CSF and serum (type 4). Screenings for herpes simplex virus (type 1, 2, 6, and 7), varicella zoster virus, adenovirus, enteroviruses, Epstein-Barr virus (EBV), CMV and John Cunningham (JC) virus in CSF were negative. Immunophenotyping of CSF cells and a computed tomography (CT) scan of thorax, abdomen and pelvis did not detect any new or recurrent malignancy. Autoantibody screening for connective tissue diseases and systemic vasculitis were negative.
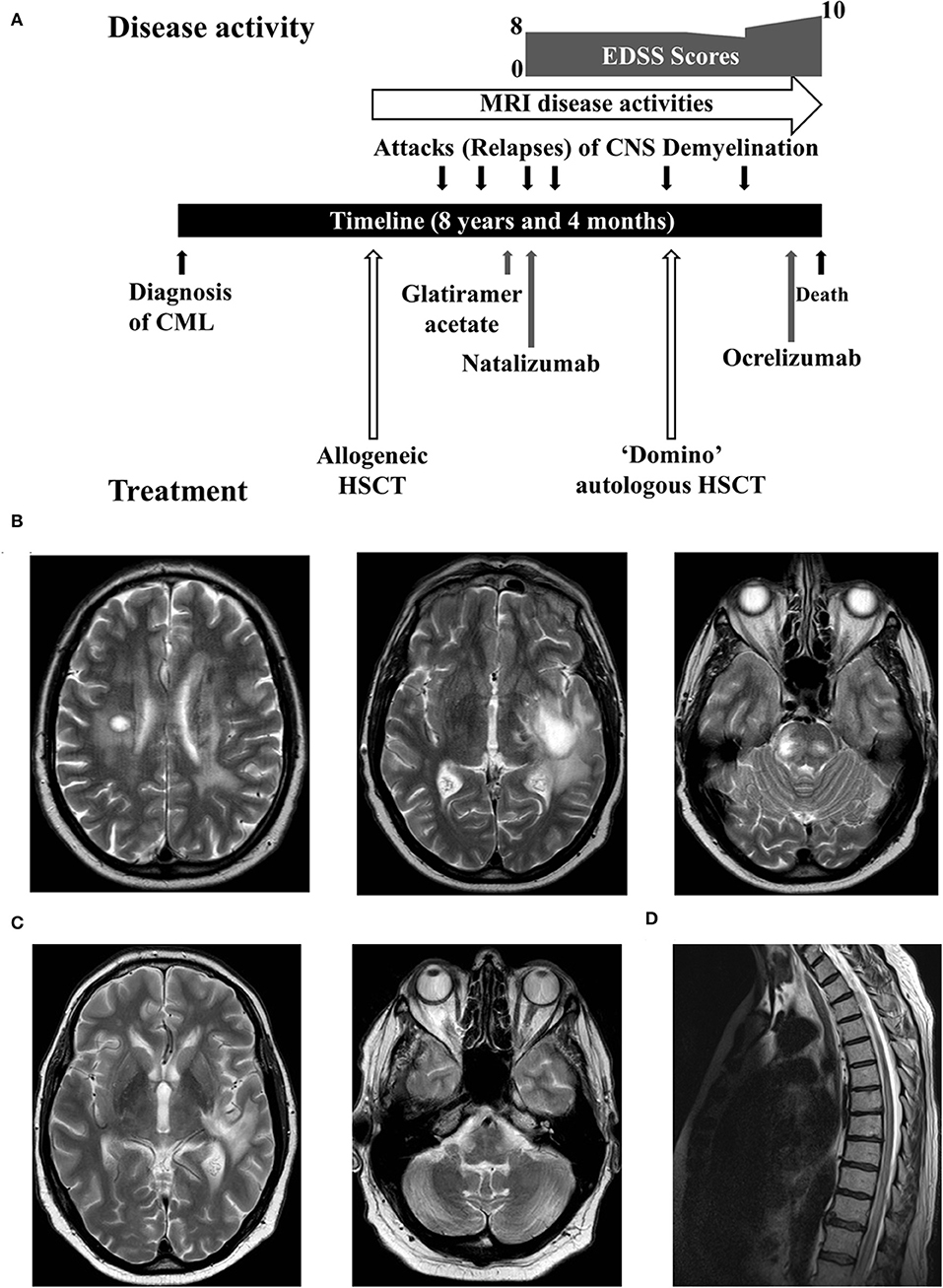
Figure 1. (A) A summary of the disease course. (B) Axial sections of the brain MRI showed tumefactive lesions. (C) Improvement of the edema around tumefactive lesions. (D) The spine MRI demonstrated a lesion extending from the thoracic cord to conus.
The patient underwent a brain biopsy, which showed a heavy infiltration of macrophages (CD68-positive), with isolated T (CD3-positive), and B (CD79a-positive) lymphocytes. A luxol fast blue stain revealed severe myelin loss whilst immunohistochemistry to neurofilament protein revealed preserved axons, although some were showing damage and swellings. Reactive astrocytes were present. There was no frank necrosis. Apoptotic cells were not conspicuous and there was no neoplastic infiltration. Special stains for bacteria and fungi were negative, as was immunohistochemistry for JC virus (SV40 antigen). The biopsy findings of a macrophage rich lesion with severe myelin loss but relative axonal preservation suggested a demyelinating pathology (Figure 2). Culture, 16s rRNA gene detection test, screening for pan-fungal and Aspergillus sp. were negative in the biopsy sample.
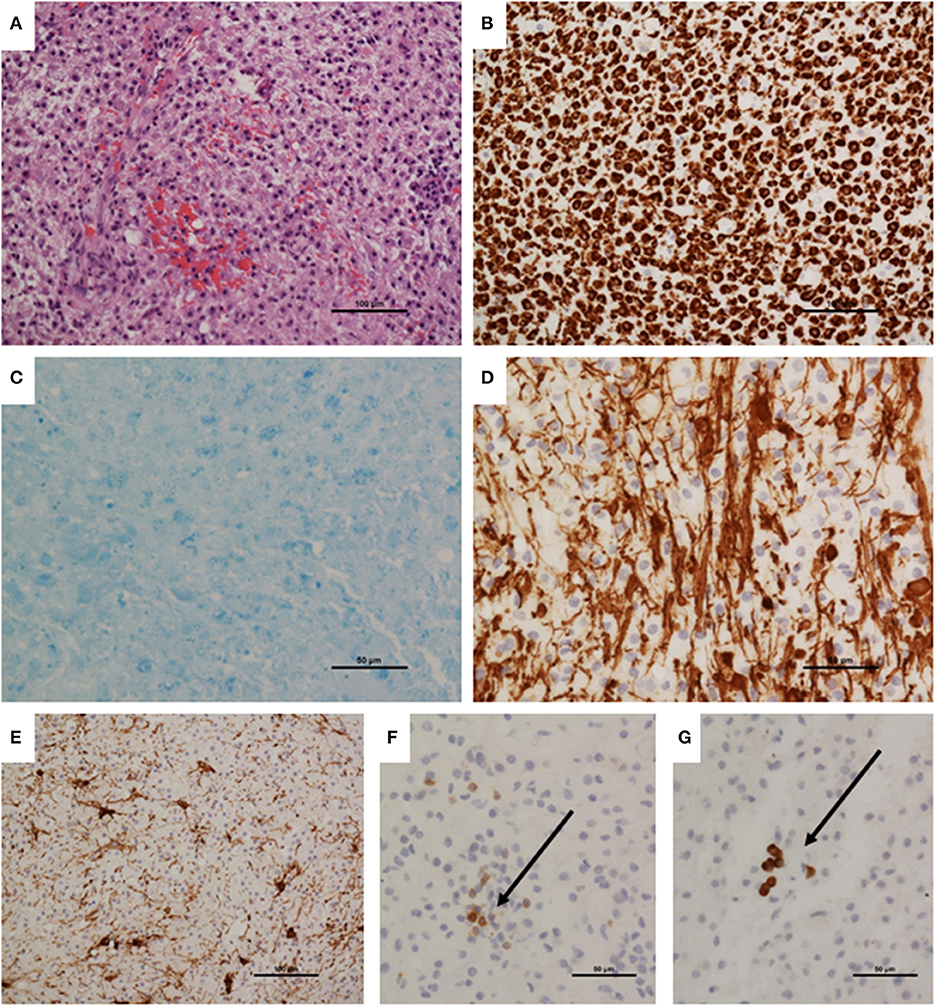
Figure 2. Brain biopsy neuropathology. (A) Biopsy appeared hypercellular, with a dense macrophage infiltrate. (B) Infiltrating population was confirmed as macrophages by immunohistochemistry to CD68. (C) Luxol fast blue stain demonstrating virtually total myelin loss. (D) Immunohistochemistry demonstrated relative preservation of axons; these were separated by infiltrating macrophages, and show irregularity, representing damage. (E) Immunohistochemistry to GFAP showing reactive astrocytes. (F) Immunohistochemistry to CD3 demonstrated sparse T cells (arrow). (G) A few CD79a-positive B cells were also present. Magnifications as shown on scale bars.
In the absence of systemic features suggesting GvHD, infections and relapse of CML, his clinical presentation and investigation results were considered to be in keeping with possible ADEM. He was treated with a 3-day course of intravenous methylprednisolone followed by tapering doses of oral prednisolone. His neurological symptoms gradually resolved, and he was walking 3−5 miles daily without assistance.
Six months later, he was re-admitted with a 3-week history of paraesthesia and weakness of lower limbs and urinary retention. A brain MRI revealed a new enhancing lesion in the occipital horn adjacent to the left lateral ventricle and a spine MRI showed a contrast enhancing lesion in the thoracic cord extending to the conus (Figure 1D). Similar to previous lumbar puncture results, CSF analysis showed mildly elevated protein with matching bands in CSF and serum. Neuromyelitis optica (NMO) antibody in serum was negative. He was treated with another course of intravenous methylprednisolone and a tapering dose of oral prednisolone. He made a full recovery within several weeks and continued to walk 3–5 miles a day. An enquiry to the donor registry confirmed that the donor remained fit and healthy without neurological or autoimmune diseases.
This case therefore posed a unique diagnostic challenge of differentiating between CNS demyelinating disorders and “pure” CNS GvHD. This presentation neither fulfilled the international consensus diagnostic criteria for NMO spectrum disorder nor the Grauer et al. criteria for the CNS manifestations of GvHD due to the lack of systemic involvement (4, 11). The relapsing remitting disease course was unlikely to be caused by fludarabine or nilotinib toxicity. As the clinical and radiological presentations together with brain biopsy results were thought to be more compatible with relapsing remitting MS, he was commenced on glatiramer acetate and continued on oral prednisolone 5 mg daily.
Two months later, he had another episode of severe myelitis, which was treated with a further course of intravenous methylprednisolone. As the CSF was negative for JC virus, glatiramer acetate was switched to natalizumab, but his disability did not improve and his expanded disability status scale (EDSS) score remained 8.0.
The patient continued to deteriorate requiring another admission with left sided facial and arm weakness five months after starting natalizumab. In view of the diagnostic uncertainty the patient was continued to be investigated for an alternative diagnosis. A spine MRI showed an enhancing lesion in the cervical cord. A third lumbar puncture did not show any new changes and JC virus in CSF remained negative. He was treated with another course of intravenous methylprednisolone. Over the ensuing 15 months, serial MRI scans identified multiple new enhancing lesions in the brain and spinal cord. Visual evoked potentials did not show any evidence of optic nerve involvement on two separate occasions. NMO and myelin oligodendrocyte glycoprotein (MOG) antibodies in serum remained negative.
As he had highly active disease clinically and radiologically despite treatment with a high efficacy DMT, autologous HSCT was considered. As his hematopoietic system was entirely derived from the matched unrelated donor used for the allogeneic HSCT, this was a “domino” autologous HSCT where the patient, a recipient of the previous allogeneic transplant, served as a “donor” for his second transplant. Natalizumab was discontinued and his peripheral blood hematopoietic stem cells were mobilized with cyclophosphamide 2 g/m2 and granulocyte-colony stimulating factor. This admission was complicated by confusion secondary to hyponatremia and a urinary tract infection from which he fully recovered.
A cautious approach was adopted, with a decision to observe the neurological response following cyclophosphamide-primed stem cells mobilization, before pursuing with the “domino” autologous HSCT. However, the patient was re-admitted with dysarthria and dysphagia three months later and a brain MRI showed four new enhancing lesions. A decision was therefore made to proceed with the “domino” autologous HSCT following immunoablation with cyclophosphamide and ATG. Similar to the first transplant, the engraftment of neutrophils and platelets occurred promptly. He had routine transplant related toxicities and also made an unhindered recovery following discharge from hospital. Nilotinib was discontinued after the transplant. Three months later his EDSS score was 7.0.
Twelve months after the transplant, he was admitted with confusion, swallowing difficulties and aspiration pneumonia. A brain MRI revealed new enhancing lesions in the left occipital lobe. He was re-investigated for opportunistic infections and malignancies. Sputum culture, throat swabs, galactomannan, and β-d-glucan tests, multiple blood cultures and viral screenings for HIV, hepatitis B and C, syphilis, toxoplasmosis and cryptococcus were negative. Autoimmune screening for connective tissue disease and vasculitis were also negative. A lumbar puncture showed CSF protein of 0.99 g/L and white cell count of 56 × 106 /L with lymphocytosis. CSF to serum glucose ratio was normal. CSF culture and screening for JC virus, EBV, CMV, toxoplasmosis, Cryptococcus, and acid-fast bacilli were negative. On this occasion, there were matching bands in serum and CSF as well as additional monoclonal bands. Neither immunophenotyping of cells in CSF nor CT scan of chest, abdomen and pelvis showed any evidence of recurrence or new malignancy. A second brain biopsy was offered but declined by the patient.
Over the next ten months, he had several brain and spine MRIs, which continued to show radiologically active disease with new T2 lesions and contrast enhancements. He continued oral prednisolone 5 mg daily. He was started on ocrelizumab but died of progressive disease four months later.
The patient underwent a post-mortem limited to the examination of the brain which weighed 1,208 g and was examined after fixation. Coronal slices of the brain revealed multifocal, irregular white matter lesions in frontal, temporal, and occipital lobes, measuring up to 30 mm in diameter. The lesions had a yellowish granular appearance with a tendency to cavitation. Subcortical arcuate (U-) fibers were not spared. These lesions did not have the appearance of classical MS plaques. In places lesions were sharply circumscribed but elsewhere had more diffuse margins. Myelin stains demonstrated virtually total myelin loss and neurofilament immunohistochemistry showed severe axonal loss with a few remained axonal threads at the lesion margins in contrast to the biopsy. Beta amyloid precursor proteins was upregulated in axons and pyramidal cells at the lesion margins. Ameboid macrophages were highlighted by CD68, particularly in a perivascular location but immunohistochemistry to CD3 and CD20 did not reveal T or B cell infiltration. Occasional vessels showed perivascular sclerosis, but immunohistochemistry to smooth muscle actin showed preservation of vascular media suggesting that there was not a vasculopathy process. S100 labeled small round nuclei in the lesions, suggesting some preservation of oligodendrocytes. No oligodendroglial inclusions were seen and immunohistochemistry for JC virus (SV40 antigen) was negative, as were stains for bacteria and fungi. Focal brown pigment was present, staining with both the Perl's and Masson Fontana methods, suggesting that haemosiderin and some melanin was present. The findings were of a multifocal leukoencephalopathy, with severe loss of both axons and myelin (Figure 3).
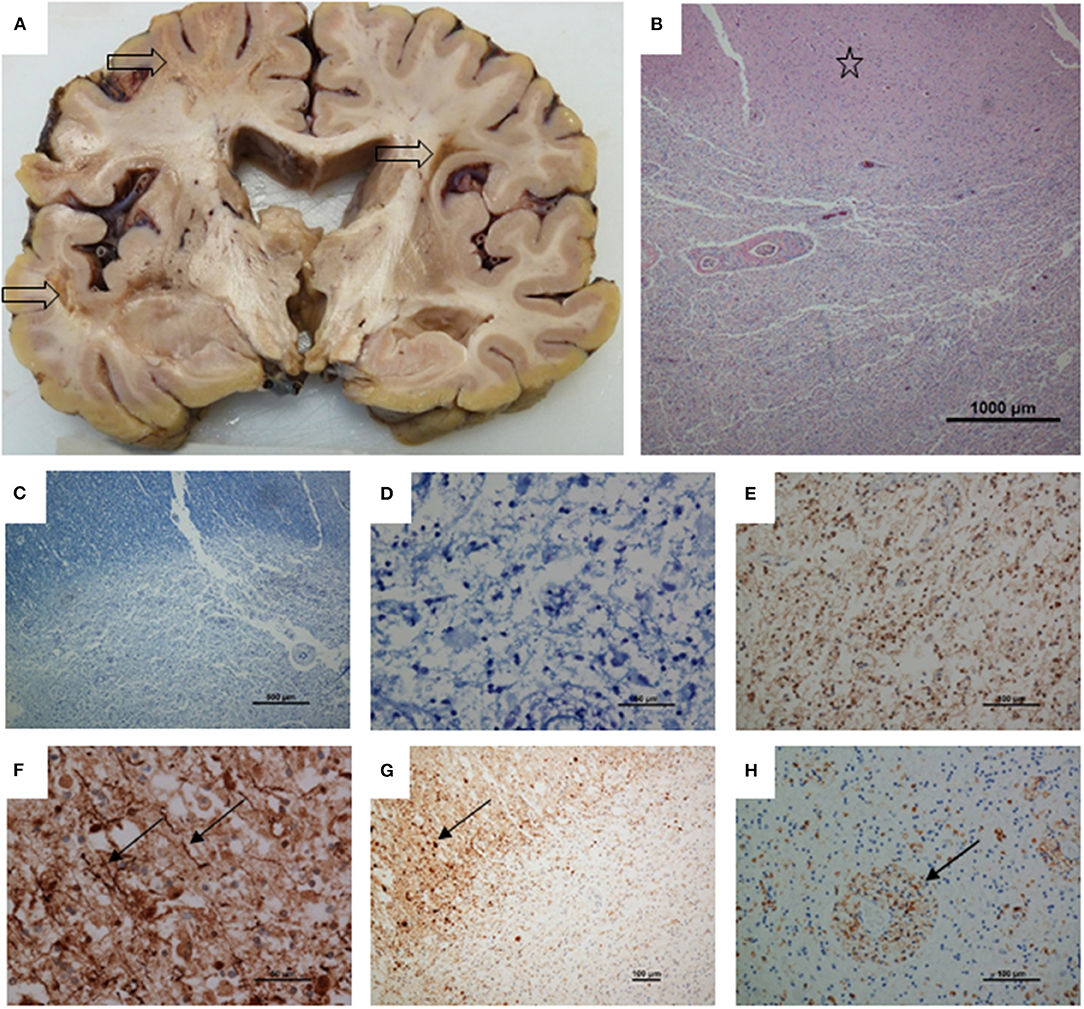
Figure 3. Post-mortem neuropathology. (A) Coronal slice of cerebrum showing three irregular, cavitating white matter lesions (arrows). There is some dilatation ex vacuo of the ventricles, presumably secondary to white matter loss. (B) H and E stained section showing part of a lesion with preserved cortex above (star). (C) Luxol fast blue showing loss of myelin. (D) High power view of lesion stained with luxol fast blue shows total loss of myelin. (E) Neurofilament immunohistochemistry showing total axon loss in the center of a lesion. (F) Toward the lesion margin a few axons remain (arrows). (G) Up-regulation of amyloid precursor protein, upper left, with multiple axonal spheroids (arrow). (H) CD68 staining showing ameboid microglia, particularly in a perivascular location. Magnifications as shown on scale bars.
Discussion
This is an unusual case of MS-like relapsing remitting encephalomyelitis following allogeneic HSCT, which did not respond to DMTs and “domino” autologous HSCT. This patient's disease course run an initial relapsing remitting phase with complete neurological recovery which was followed by a progressive disease phase with superimposed acute episodic neurological dysfunction. There were corresponding MRI disease activities in brain and spine throughout the disease course.
The initial brain biopsy showed a macrophage rich lesion with loss of myelin, but relative preservation of axons, which would be consistent with a demyelinating pathology. It is a rare histological feature of hematological neoplasm and may be erroneously diagnosed as inflammatory demyelination if corticosteroid therapy is used prior to the brain biopsy (20). Acute plaques of demyelination usually have T cells, which tend to be localized perivascularly and are less prevalent than macrophages (21). In tumefactive MS, T cells are detected at lower levels than in biopsies that subsequently turn out to be lymphoma (20). However in a small biopsy sample, it is possible that these changes may not be well represented, which is a ubiquitous problem independent of disease studied (21). In the absence of clinical features suggestive of GvHD and CML recurrence, his brain biopsy was thought to be consistent with a primary inflammatory demyelinating plaque.
The use of fludarabine has previously been associated with monophasic diffuse necrotizing leukoencephalopathy (22). Although five cases of CNS demyelination had also been reported in patients receiving treatment with nilotinib or imatinib for various malignancies, it was not clear if tyrosine kinase inhibitors were the causal agents, particularly as two out of those five patients recovered without discontinuing these drugs (23–25). Furthermore, tyrosine kinase inhibitor had therapeutic benefit in people with progressive forms of MS and drug toxicities are unlikely to present with a relapsing remitting disease course (26).
There was further discordance between the clinical presentation and the autopsy findings, which showed a destructive leukoencephalopathy. Although the initial biopsy showed demyelination, lymphocytic infiltration was not a feature of either the biopsy or the white matter lesions at autopsy. It is not known how DMTs and HSCT modulate the histological appearance of CNS demyelination. Although the innate immune system recovers within weeks after HSCT, the reconstitution of adoptive immune system occurs over several years. Immunohistochemistry to CD3 and CD20 did not show any T or B cell infiltration during autopsy examination. In particular, ATG was used for in vivo T cell purging during domino autologous HSCT. Furthermore, the patient received two doses of ocrelizumab prior to his death. This humanized anti-CD20 monoclonal antibody targets B lymphocytes. The lack of inflammatory cells in the autopsy histology samples could be related to primary underlying pathology or the effect of ocrelizumab and / or the HSCT received earlier.
To our knowledge this was the first patient with MS-like neuroinflammation following allogeneic HSCT, who was treated with a “domino” autologous HSCT. Our patient experienced an aggressive disease course and rapidly became disabled. His failure to respond to glatiramer acetate and natalizumab left his neurologists with limited treatment options. Although the use of alemtuzumab was not completely contraindicated, caution was exercised, as it could cause a prolonged period of lymphopenia potentially making it a less appropriate choice given his immunosuppressed state following allogeneic HSCT (27). Autologous HSCT has been increasingly used to treat patients with MS, who have highly active disease clinically and radiologically, as the safety and efficacy of this procedure has increased over the years through improvement of patient selection, optimization of transplant technique and increased center experience (28). This was therefore thought to be the best treatment option. Although the procedure was associated with routine and well-tolerated toxicities, the response was only transient and failed to achieve long-term remission. In this case, we chose a clinical decision pathway directed at MS, with the use of three DMTs and HSCT, whereas the management of chronic GvHD would have been significantly different. Calcineurin inhibitors, higher doses of steroids, mycophenolate and even extracorporeal photopheresis could have been used for GvHD. We can only speculate whether GvHD management would have made a greater impact on the course of his CNS inflammation compared with a DMT-based, MS-directed approach, even though systemic GvHD was not present.
Reported cases of CNS demyelinating disorders following allogeneic HSCT are very limited. Tables 1, 2 summarize 20 such cases that have been reported in the literature (5–8, 12–19). The median age of receiving allogeneic HSCT was 45.5 (range, 17–65) years and the median interval between HSCT and the onset of CNS demyelination was 1 (range, 0.1–8) year. Twelve of these patients presented with neurological symptoms within 1 year of allogeneic HSCT and remaining eight patients developed neurological symptoms after 2 years or more. Male to female ratio was 3: 1. There was evidence of GvHD in 12 patients and peripheral nerves involvement was reported in 13 patients. Inflammation less frequently affected brainstem, cerebellum and meninges. CSF analysis was normal in only 6 patients and oligoclonal bands were present in 7 patients.
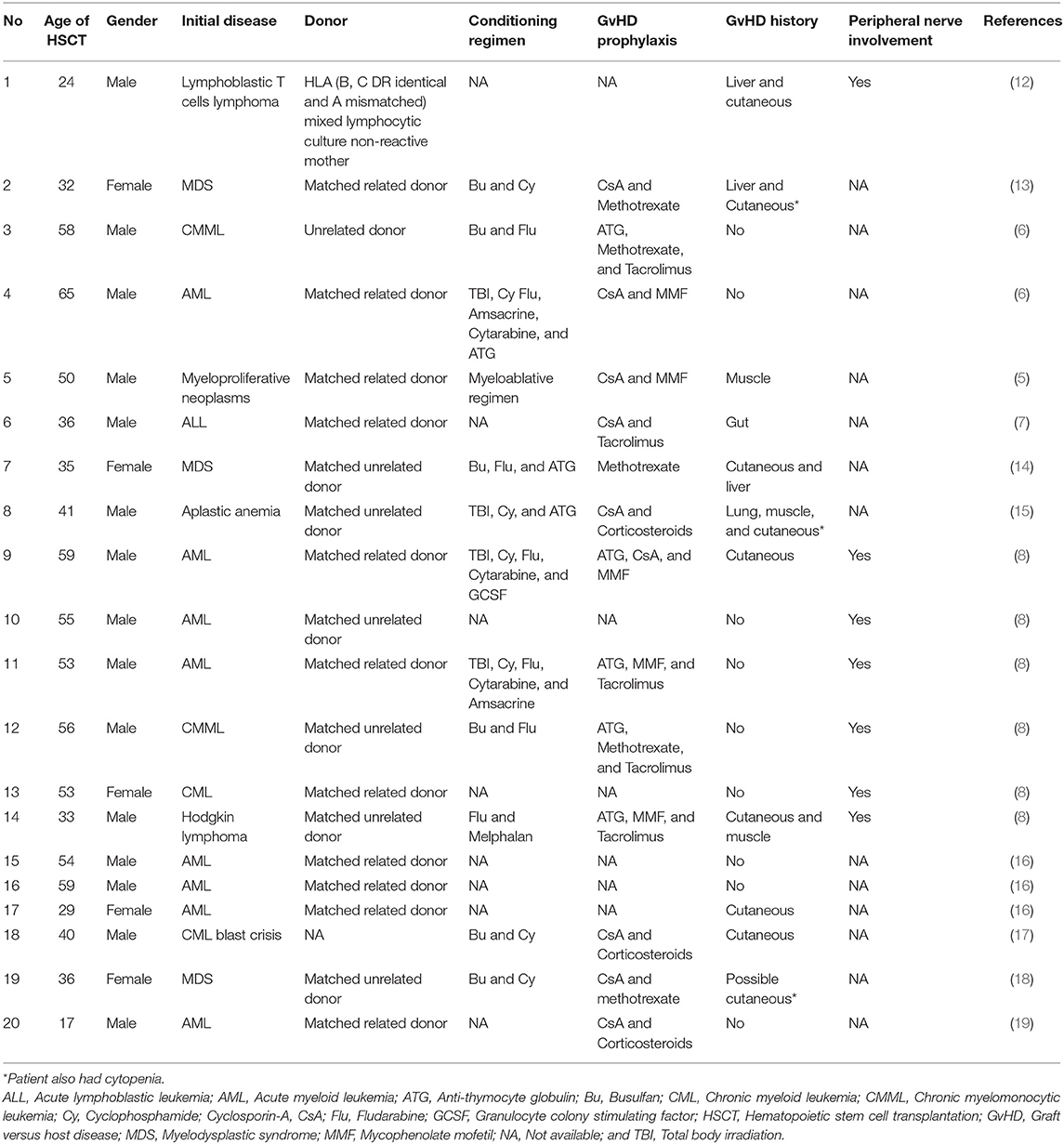
Table 1. Demographic details, allogeneic HSCT procedures, GvHD and other immune mediated complications of post-transplant CNS demyelinating disorders.
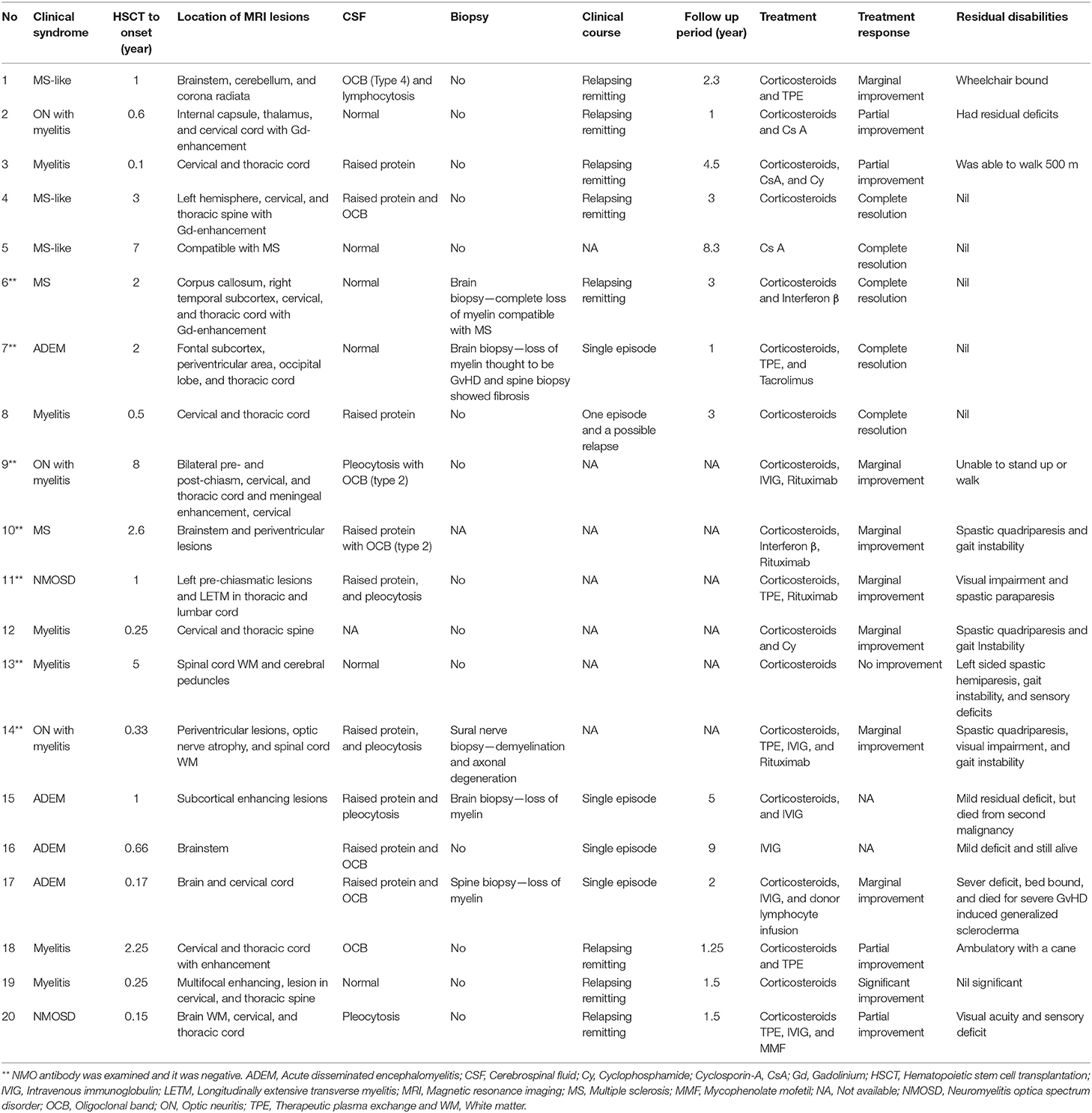
Table 2. Clinical features, investigation results and treatment outcomes of post-transplant CNS demyelinating disorders.
The clinical course of these patients was monophasic or relapsing remitting. They had variable clinical presentations including optic neuritis with myelitis, pure myelitis, ADEM, and MS-like neuroinflammation (Table 2). The diagnostic criteria for MS and NMO spectrum disorder were satisfied in two patients each. NMO antibody was absent in all cases where it was examined (Table 2). Terminologies such as “central and peripheral nervous system immune-mediated demyelinating disease (CPID)” and “immune-mediated demyelinating disease (IMDD)” had been coined by some authors to refer these presentations as a rare late onset complication of allogeneic HSCT (8, 16). Some authors also suggested that these presentations may be CNS manifestation of GvHD (5). Three patients had brain biopsy, two had spine biopsy and another person had sural nerve biopsy. All of these biopsies showed loss of myelin and the brain biopsy of one patient was compatible with GvHD (Table 2). None of those case reports included post-mortem examination.
One possible pathophysiological mechanism of CNS demyelination following allogeneic HSCT could be immune mediated damage due to minor histocompatibility differences between brain tissues and the graft derived cells. Another possible mechanism is adoptive transfer of autoimmunity resulting in CNS inflammation in the host.
Complete resolution of symptoms or significant improvement was observed in 6 patients and partial improvement was reported in another 4 patients (5–7, 14, 18, 19). Eight of them had marginal or no improvement following treatment and data were not available for 2 patients (5–8, 13, 16). A range of therapies including corticosteroids, intravenous immunoglobulin, therapeutic plasma exchange, mycophenolate, tacrolimus, cyclosporine A, cyclophosphamide, rituximab, and donor lymphocyte infusion had been used in these 20 patients (Table 2). Interferon ß was used in both patients, whose diagnoses were compatible with MS. Five patients, who were treated with mycophenolate, tacrolimus or cyclosporine A, had complete resolution of symptoms or partial improvement following treatment. The size of the sample was too small for any statistical analysis, but normal CSF constitutes or absence of oligoclonal bands were appeared to be associated with better prognosis. Complete resolution of symptoms or significant clinical improvement was observed more frequently in those patients who had later onset of neurological symptoms following allogeneic HSCT (Table 2).
Conclusions
This case was noteworthy for two reasons. Firstly, the disease course which resembled an “MS-like” relapsing remitting encephalomyelitis although the exact etiology remained unknown even after autopsy. In such a case full autopsy examination may be helpful to confirm the absence of occult malignancy and also to investigate whether the disease was limited to the CNS tissue. Secondly, this case demonstrated the feasibility of using “domino” autologous HSCT in patients presenting with “MS-like” encephalomyelitis following allogeneic HSCT, but further studies are required to evaluate its safety and efficacy.
Ethics Statement
A written consent was obtained from the relative of the patient for the publication of this case report.
Author Contributions
JD summarized the case, reviewed the literature and drafted the manuscript. SP, SW, BS, and JS drafted the manuscript, AG, CL, NC-L, and HJ reviewed and summarized the case.
Conflict of Interest
JS declares speaker fees at educational events supported by Sanofi, Janssen, Jazz, Mallinckrodt and Gilead. JS is a member of a trial IDMC for Kiadis Pharma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors acknowledge Sheffield Hospitals Charity and NIHR Sheffield Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, the Department of Health and social Care or the Sheffield Hospitals Charity.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00668/full#supplementary-material
References
1. Syed FI, Couriel DR, Frame D, Srinivasan A. Central nervous system complications of hematopoietic stem cell transplant. Hematol Oncol Clin North Am. (2016) 30:887–98. doi: 10.1016/j.hoc.2016.03.009
2. Barba P, Piñana JL, Valcárcel D, Querol L, Martino R, Sureda A, et al. Early and late neurological complications after reduced-Intensity conditioning allogeneic stem cell transplantation. Biol Blood Marrow Transplant. (2009) 15:1439–46. doi: 10.1016/j.bbmt.2009.07.013
3. Avivi I, Wittmann T, Henig I, Kra-Oz Z, Szwarcwort Cohen M, Zuckerman T, et al. Development of multifocal leukoencephalopathy in patients undergoing allogeneic stem cell transplantation-can preemptive detection of john cunningham virus be useful? Int J Infect Dis. (2014) 26:107–9. doi: 10.1016/j.ijid.2014.03.1381
4. Grauer O, Wolff D, Bertz H, Greinix H, Kühl J-S, Lawitschka A, et al. Neurological manifestations of chronic graft-versus-host disease after allogeneic haematopoietic stem cell transplantation: report from the consensus conference on clinical practice in chronic graft-versus-host disease. Brain. (2010) 133:2852–65. doi: 10.1093/brain/awq245
5. Ruggiu M, Cuccuini W, Mokhtari K, Meignin V, Peffault de Latour R, Robin M, et al. Case report: central nervous system involvement of human graft versus host disease: report of 7 cases and a review of literature. Medicine. (2017) 96:e8303. doi: 10.1097/MD.0000000000008303
6. Voß M, Bischof F. Recurrent myelitis after allogeneic stem cell transplantation. Report of two cases. BMC Neurol. (2010) 10:76. doi: 10.1186/1471-2377-10-76
7. Yoon B-N, Ha CK, Lee K-W, Park S-H, Sung J-J. A confusing case of multiple sclerosis and central nervous system graft versus host disease. Korean J Intern Med. (2016) 31:995–8. doi: 10.3904/kjim.2015.065
8. Stefanou MI, Bischof F. Central and peripheral nervous system immune-mediated demyelinating disease after allogeneic hematopoietic stem cell transplantation. J Neuroimmunol. (2017) 307:74–81. doi: 10.1016/j.jneuroim.2017.04.005
9. Daikeler T, Tyndall A. Autoimmunity following haematopoietic stem-cell transplantation. Best Pract Res Clin Haematol. (2007) 20:349–60. doi: 10.1016/j.beha.2006.09.008
10. Parnell GP, Booth DR. The multiple sclerosis (MS) genetic risk factors indicate both acquired and innate immune cell subsets contribute to MS pathogenesis and identify novel therapeutic opportunities. Front Immunol. (2017) 8:425. doi: 10.3389/fimmu.2017.00425
11. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
12. Solaro C, Murialdo A, Giunti D, Mancardi G, Uccelli A. Central and peripheral nervous system complications following allogeneic bone marrow transplantation. Eur J Neurol. (2001) 8:77–80. doi: 10.1046/j.1468-1331.2001.00160.x
13. Matsuo Y, Kamezaki K, Takeishi S, Takenaka K, Eto T, Nonami A, et al. Encephalomyelitis mimicking multiple sclerosis associated with chronic graft-versus-host disease after allogeneic bone marrow transplantation. Intern Med. (2009) 48:1453–6. doi: 10.2169/internalmedicine.48.2003
14. Min G-J, Park S, Park S-S, Yoon J-H, Lee S-E, Cho B-S, et al. A case of central nervous system graft-versus-host disease following allogeneic stem cell transplantation. Int J Hematol. (2019) 110:635–9. doi: 10.1007/s12185-019-02702-1
15. Richard S, Fruchtman S, Scigliano E, Skerrett D, Najfeld V, Isola L. An immunological syndrome featuring transverse myelitis, evans syndrome and pulmonary infiltrates after unrelated bone marrow transplant in a patient with severe aplastic anemia. Bone Marrow Transplant. (2000) 26:1225–8. doi: 10.1038/sj.bmt.1702677
16. Delios AM, Rosenblum M, Jakubowski AA, DeAngelis LM. Central and peripheral nervous system immune mediated demyelinating disease after allogeneic hemopoietic stem cell transplantation for hematologic disease. J Neurooncol. (2012) 110:251–6. doi: 10.1007/s11060-012-0962-9
17. Openshaw H, Slatkin NE, Parker PM, Forman SJ. Immune-mediated myelopathy after allogeneic marrow transplantation. Bone Marrow Transplant. (1995) 15:633–6.
18. Sakai M, Ohashi K, Ohta K, Yamashita T, Akiyama H, Kisida S, et al. Immune-Mediated myelopathy following allogeneic stem cell transplantation. Int J Hematol. (2006) 84:272–5. doi: 10.1532/IJH97.06060
19. Baumer FM, Kamihara J, Gorman MP. Neuromyelitis optica in an adolescent after bone marrow transplantation. Pediatr Neurol. (2015) 52:119–24. doi: 10.1016/j.pediatrneurol.2014.07.007
20. Barrantes-Freer A, Engel AS, Rodríguez-Villagra OA, Winkler A, Bergmann M, Mawrin C, et al. Diagnostic red flags: steroid-treated malignant CNS lymphoma mimicking autoimmune inflammatory demyelination. Brain Pathol. (2018) 28:225–33. doi: 10.1111/bpa.12496
21. Kuhlmann T, Ludwin S, Prat A, Antel J, Brück W, Lassmann H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. (2017) 133:13–24. doi: 10.1007/s00401-016-1653-y
22. Beitinjaneh A, McKinney AM, Cao Q, Weisdorf DJ. Toxic leukoencephalopathy following fludarabine-Associated hematopoietic cell transplantation. Biol Blood Marrow Transplant. (2011) 17:300–8. doi: 10.1016/j.bbmt.2010.04.003
23. Rekhi E, Pryce A, Sohal M, Dassan P. CNS demyelination in patients on nilotinib treatment for CML. J Neurol Neurosurg Psychiatry. (2016) 87:e1.62-e1. doi: 10.1136/jnnp-2016-315106.154
24. Judge C, Moheb N, Melinosky C. Nilotinib-associated demyelinating disease (P2.2-093). Neurology. (2019) 92.
25. Rotstein DL, Sawicka K, Bharatha A, Montalban X, Lipton JH. CNS demyelination after initiating the tyrosine kinase inhibitor imatinib: a report of two cases. Mult Scler. (2019) 17:1–4. doi: 10.1177/1352458519892914
26. Vermersch P, Benrabah R, Schmidt N, Zéphir H, Clavelou P, Vongsouthi C, et al. Masitinib treatment in patients with progressive multiple sclerosis: a randomized pilot study. BMC Neurol. (2012) 12:36. doi: 10.1186/1471-2377-12-36
27. Bourdette D. Alemtuzumab and multiple sclerosis: is it safe? Neurology. (2014) 83:17–8. doi: 10.1212/WNL.0000000000000554
Keywords: multiple sclerosis, allogeneic hematopoietic stem cell transplantation, “domino” autologous hematopoietic stem cell transplantation, graft versus host disease, multifocal leukoencephalopathy
Citation: Das J, Gill A, Lo C, Chan-Lam N, Price S, Wharton SB, Jessop H, Sharrack B and Snowden JA (2020) A Case of Multiple Sclerosis—Like Relapsing Remitting Encephalomyelitis Following Allogeneic Hematopoietic Stem Cell Transplantation and a Review of the Published Literature. Front. Immunol. 11:668. doi: 10.3389/fimmu.2020.00668
Received: 25 November 2019; Accepted: 24 March 2020;
Published: 05 May 2020.
Edited by:
Jukka Partanen, Finnish Red Cross Blood Service, FinlandReviewed by:
Daniel Wolff, University Hospital Regensburg, GermanyPentti Tienari, University of Helsinki, Finland
Copyright © 2020 Das, Gill, Lo, Chan-Lam, Price, Wharton, Jessop, Sharrack and Snowden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joyutpal Das, ai5kYXNAZG9jdG9ycy5vcmcudWs=