 Lun Cai1Leidy D. Caraballo Galva1,2Yibing Peng1Xiaobing Luo3
Lun Cai1Leidy D. Caraballo Galva1,2Yibing Peng1Xiaobing Luo3 Wei Zhu1†Yihong Yao3
Wei Zhu1†Yihong Yao3 Yun Ji3
Yun Ji3 Yukai He1,2,4,5*
Yukai He1,2,4,5*- 1Georgia Cancer Center, Medical College of Georgia, Augusta, GA, United States
- 2The Graduate School, Augusta University, Augusta, GA, United States
- 3Cellular Biomedicine Group (CBMG), Gaithersburg, MD, United States
- 4Department of Medicine, Medical College of Georgia, Augusta University, Augusta, GA, United States
- 5Department of Biochemistry and Molecular Biology, Medical College of Georgia, Augusta University, Augusta, GA, United States
Autologous T cells engineered with T receptor genes (TCR) are being studied to treat cancers. We have recently identified a panel of mouse TCRs specific for the HLA-A0201/alpha fetoprotein epitope (AFP158) complex and have shown that human T cells engineered with these TCR genes (TCR-Ts) can eradicate hepatocellular carcinoma (HCC) xenografts in NSG mice. However, due to TCR’s promiscuity, their off-target cross-reactivity must be studied prior to conducting clinical trials. In this study, we conducted in vitro X-scan assay and in silico analysis to determine the off-target cross-reactivity of 3 AFP158-specific TCR-Ts. We found that the 3 AFP158-specific TCR-Ts could be cross-activated by ENPP1436 peptide and that the TCR3-Ts could also be activated by another off-target peptide, RCL1215. However, compared to AFP158, it requires 250 times more ENPP1436 and 10,000 times more RCL1215 peptides to achieve the same level of activation. The EC50 of ENPP1436 peptide for activating TCR-Ts is approximately 17–33 times higher than AFP158. Importantly, the ENPP1+ tumor cells did not activate TCR1-Ts and TCR2-Ts, and only weakly activated TCR3-Ts. The IFNγ produced by TCR3-Ts after ENPP1+ cell stimulation was >22x lower than that after HepG2 cells. And, all TCR-Ts did not kill ENPP1 + tumor cells. Furthermore, ectopic over-expression of ENPP1 protein in HLA-A2+ tumor cells did not activate TCR-Ts. In silico analysis showed that the ENPP1436 peptide affinity for HLA-A0201 was ranked 40 times lower than AFP158 and the chance of ENPP1436 peptide being processed and presented by HLA-A0201 was 100 times less likely than AFP158. In contrast, the two off-targets (Titin and MAGE-A3) that did cause severe toxicity in previous trials have the same or higher MHC-binding affinity and the same or higher chance of being processed and presented. In conclusion, our data shows that TCR-Ts can be activated by off-target ENPP1436 peptide. But, compared to target AFP158, it requires at least 250 times more ENPP1436 to achieve the same level of activation. Importantly, ENPP1436 peptide in human cells is not processed and presented to a sufficient level to activate the AFP158-specific TCR-Ts. Thus, these TCR-Ts, especially the TCR1-Ts and TCR2-Ts, will unlikely cause significant off-target toxicity.
Introduction
With 840,000 new diagnoses and 781,000 deaths annually, liver cancer is the 6th most common cancer, and the 3rd most common cause of cancer deaths due to the lack of effective treatment (1). The majority of liver cancer is hepatocellular carcinoma (HCC). Recently, several immunotherapies are being developed for HCC (2). The PD1 blockade has significantly increased the overall response rate (3). But its effect may depend on the presence of tumor reactive T cells (4), which are not always present in most HCC lesions. Redirecting autologous T cells with tumor antigen-specific T cell receptor (TCR) genes will provide the tumor-specific T cells, and thus has a great potential for cancer immunotherapy (5). The feasibility of TCR gene transfer to render T cell specificity was published in 1986 (6). And the first evidence that TCR gene engineered T cells (TCR-Ts) generated antitumor effect in treating human cancers was reported 20 years later in 2006 (7). Since then, a number of human tumor antigen specific TCRs derived from both mouse and human sources, including the TCRs specific for MART1 (human TCR) (8), GP100 (mouse TCR) (8), CEA (mouse TCR) (9), NY-ESO1 (human TCR) (10, 11), and MAGE-A3 (mouse and human TCR) (12, 13), have been tested in clinical trials. Adoptive transfer of TCR-Ts has generated significant antitumor effect in several cancers (14). The clinical trial data from NY-ESO1 specific TCR-Ts in treating melanoma, multiple myeloma, and synovial carcinoma is very promising with great safety profile (10, 11). Recently, we (15) and others (16) identified the human alpha fetoprotein (AFP)- specific TCR genes from mouse and human and showed that human T cells genetically modified with the AFP-specific TCRs could effectively kill HCC tumor cells and eliminated HCC xenografts in immune compromised NSG mice (15), demonstrating the potential of the TCR-Ts for HCC immunotherapy.
While the antitumor potency of TCR-Ts is evident, many of the TCR-Ts trials, such as those against CEA, GP100, MART, and MAGE-A3 antigens, also showed significant toxicity including patient death. The toxicity of TCR-Ts can come from three aspects: (1) the on-target/off tumor toxicity due to the low level expression of shared-tumor antigen in normal tissues (8, 9); (2) the off-target toxicity due to the TCR’s promiscuous recognition of unrelated epitopes derived from normal proteins (12, 13, 17); and (3) the alloreactivity of TCR-Ts recognizing different HLA presented random peptides. All three aspects of the TCR-T’s toxicity must be properly evaluated prior to conducting clinical trials. As investigations of TCR-Ts in animal models offers little value in evaluating their toxicity in human, an in vitro preclinical toxicity study strategy was proposed to assess the TCR-T’s risk (18). Ideally, tumor-specific or relatively tumor-specific antigens should be selected as the TCR-T’s target to reduce on-target/off-tumor reactivity. However, even with highly tumor-specific targets, the off-target cross-reactivity of TCR-Ts in recognition of MHC-peptide complex may still cause severe toxicity.
In this report and the accompanying study, we determined the optimal TCRs out of the 7 AFP158-specific TCRs based on their preclinical antitumor efficacy and toxicities. The selection of optimal TCR-Ts for HCC immunotherapy and the on-target/off-tumor toxicity and alloreactivity of the AFP148-specific TCR-T’s were reported in the accompanying paper (Luo et al). In this study, we investigated the off-target cross reactivity of 3 potent TCR-Ts by using X-scan. We found that TCR3-Ts could be cross-activated by 2 synthetic peptides, the ENPP1436 and RCL1215, while the TCR1-Ts and TCR2-Ts were activated by only ENPP1436. The EC50 of ENPP1436 peptide for activating AFP148-specific TCR-Ts was 17–33 times higher than the EC50 of AFP158. And it required 250–400 times more of ENPP1436 and 1000 times of RCL1215 peptide to achieve the same level of TCR-T activation as AFP158 peptide. Importantly, the HLA-A020 + ENPP1 + human cells do not activate TCR1-Ts and TCR2-Ts. In addition, in silico analysis showed that the ENPP1436 peptide’s MHC binding affinity and its chance of being processed and presented by HLA-A0201 were significantly lower than that of AFP158. In contrast, the two off-targets (Titin and MAGE-A3) that indeed caused severe toxicity in previous trials had the same MHC binding affinity and the same or higher chance of being processed and presented by host cells. Altogether, we conclude that the AFP158-specific TCR-Ts, especially TCR1-Ts and TCR2-Ts, will not cause significant off-target toxicity.
Materials and Methods
Cells
The cell lines of TAP–/– T2 (19), HepG2, and HEK293 were purchased from American Type Culture Collection (ATCC, Manassas, VA, United States). Breast cancer cell lines of MCF7, MDA-MB231, and brain tumor U87MG cells were purchased from ATCC. MDA-MB231-Luc cells were derived from MDA-MB231 by transfecting with luciferase gene and kindly provided by Dr. Hasan Korkaya of Georgia Cancer Center. The cells were cultured in standard DMEM or RPMI1640 media for no more than 8–10 passages to maintain their authenticity. Mycoplasma test was conducted according to manufacturer instructions (Thermo Fisher, MA, United States). Primary normal adult human hepatocytes were purchased from Lonza (Walkersville, MD, United States) and Novabiosis (Research Triangle Park, NC, United States).
Peptides
Peptides were synthesized by GenScript (Piscataway, NJ, United States) and Chinapeptides (Shanghai, China) to a purity of >95%. The stock peptides are dissolved in DMSO at 5 mg/ml and aliquoted and stored at −20°C.
T Cell Isolation and TCR Transduction
Buffy coat was obtained from local Shepard blood center. PBMCs were harvested by centrifugation on a Lymphoprep Ficoll gradient, diluted to 1 × 107 cells/ml, aliquoted, and frozen. Lentiviral vectors expressing TCRs were prepared by 4 a plasmid co-transfection as previously described (20–22). Total T cells were isolated from PBMC by negative isolation kit (STEMCELL Technologies, Vancouver, BC, Canada) and then transduced by lentiviral vectors at MOI of 20–40 as described (15). Between 12–15 days after transduction, tetramer (NIH Tetramer Core Facility) staining was conducted to measure the percentage of TCR+ T cells. The TCR-Ts were then aliquoted and frozen.
X-Scan Assay
X-Scan assay was performed as described (23). Briefly, one vial of TCR-Ts were thawed and recovered 2–3 days before use. For each well of the 96 well plate, 15,000 T cells (∼5,000 TCR-Ts) were cultured with 20,000 T2 cells in the presence of 10 ng/ml of X-peptides, which is equal to the EC90 of the index AFP158 peptide. After co-culture overnight, the media was collected and assayed for IFNγ by ELISA. The absolute amount of IFNγ after X-peptides was then compared to the IFNγ level after AFP158 peptide stimulation, and the ratios were calculated and presented.
Immune Analysis
IFN-γ ELISA kits and antibodies were from Biolegend (San Diego, CA, United States). HLA-A0201/AFP158 Tetramer was synthesized by NIH Tetramer Core facility. Antibody staining was done according to each antibody’s instructions. Flow cytometry was done on BD LSRII (San Jose, CA, United States). Data was analyzed using the FCS express software (De Novo Software, Pasadena, CA, United States).
Ectopic Over-Expression of Ectonucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1) Protein
MCF7 cells were transduced by ENPP1 gene (Addgene, Watertown, MA, United States) by utilizing Lipofectamine 2000 (Invitrogen, Carlsbad, CA, United States). The expression of ENPP1 was detected by Western blot (WB) analysis (Figure 5A).
WB Analysis
Protein expression in cell lines was examined by WB analysis. Briefly, cells was homogenized in RIPA lysis buffer containing 25 mmol/L Tris–HCl pH 7.6, 150 mmol/L NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail (Sigma Aldrich, St. Louis, MO, United States). Protein samples were resolved on 8% SDS polyacrylamide gel. The protein was transferred onto nitrocellulose membrane, which was blocked by using 5% (w/v) non-fat dried milk in Tris–buffered saline containing 25 mM Tris–HCl (pH 7. 4), 130 mM NaCl, 2.7 mM KCl and 0.1% Tween 20. The blot membranes were probed with anti-ENPP1 antibody (Genscript, Piscataway, NJ, United States) and the horseradish peroxidase conjugated anti-IgG secondary antibody (Cell Signaling, Danvers, MA, United States), followed by lightning ECL (PerkinElmer, Waltham, MA, United States).
In silico Analysis
The online software of NetMHC4.0 (24, 25), IEDB (26), and NetCTLpan (27) were used to analyze the ranks of peptide’s MHC binding affinity and peptide’s chance of being processed and presented. The rank of MHC binding affinity by NetMHC4.0 is based on a total of 400,000 random natural peptides in its databank and the rank of peptide’s chance of being processed and presented by HLA-A0201 is based on 200,000 random natural peptides in the NetCTLpan databank.
Statistics
Statistical analysis was done with Prism software using either ANOVA or student t-test.
Results
We have identified 7 unique AFP158-specific TCR sequences. A functional screening (Refer to Figure 1 of the accompanying paper, Luo et al) identified that human T cells engineered with TCR1, 2, and 3 genes (TCR1- TCR2-, and TCR3-Ts) generated strong cytotoxicity and produced high level of cytokines, which were consistent with our previous report (15). Thus, in the study of off-target cross-reactivity, only the TCR1-, 2-, and 3-Ts were used.
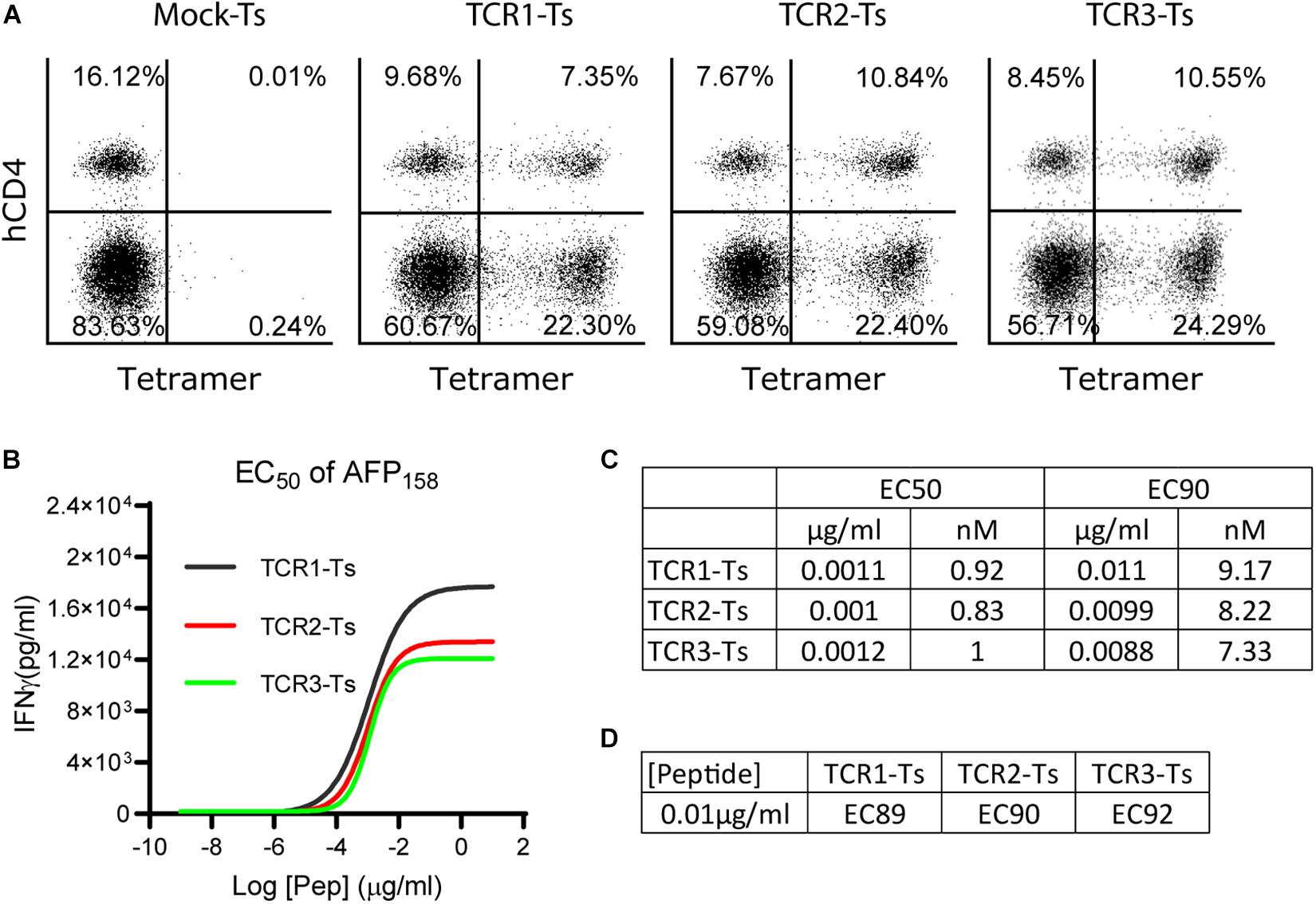
Figure 1. The EC50 and EC90 of AFP158 peptide for activating TCR-Ts. (A) TCR-Ts were generated by transducing primary human T cells with TCR genes. Representative dot plots showed the% of TCR + T cells 12–15 days after transduction. (B,C) The EC50 of AFP158 peptide for activating TCR-Ts were presented. The EC90 was calculated using Prism software. (D) The ECXX value corresponding to the 10 ng/ml of AFP158 peptide for activating different TCR-Ts was calculated using Prism software. The experiments were repeated three times with similar data.
The EC50 and EC90 of Target AFP158 and the Peptide Concentration for X-Scan
To comprehensively study the TCR-T’s off-target reactivity, X-scan assay was recently developed (23). To properly conduct an X-scan, we first need to decide the right peptide concentration for the assay. To this end, the EC50 of index AFP158 peptide for activating AFP158-specific TCR-Ts was determined. TCR-Ts were prepared by transducing primary human T cells with lentiviral vectors. Approximately 30–35% of T cells were stained positive by HLA-A0201/AFP158 tetramer, and 2/3 of the TCR + T cells were CD8 (Figure 1A). TCR-Ts were co-cultured with T2 cells in the presence of different concentrations of AFP158 peptide. The IFNγ in the media was measured 20 h later. Using this approach, we determined the EC50 of AFP158 peptide for activating the TCR1-Ts, TCR2-Ts, and TCR3-Ts were 1.1 ng/ml (0.92 nM), 1 ng/ml (0.83 nM), and 1.2 ng/ml (1.0 nM), respectively (Figures 1B,C). This AFP158 EC50 value for activating our TCR-Ts is similar to the EC50 of AFP158 peptide for activating human TCR-Ts recently reported by Docta et al. (16), but is slightly higher than several EC50 previously reported for other affinity-enhanced TCR-Ts. For example, the EC50 of cognate peptide for activating the MAGE-A10 specific TCR-Ts was 0.3–0.5 nM (23), and the EC50 of NY-ESO1157 peptide for activating cognate TCR-Ts was around 0.2 nM (28). In contrast, the EC50 of cognate peptide for activating the MAGE-C2 specific TCR6-T cells was 3.3 nM (18). Next, based on the EC50, the EC90 of AFP158 peptide was calculated as 10 ng/ml (Figures 1C,D). As recently reported by Border et al. (23), the EC90 of AFP158 was selected as the peptide concentration for conducting X-scan assay.
X-Scan Assay Determines the Peptide Motifs That Are Potentially Recognized by TCR-Ts
To conduct X-scan, each amino acid residue of the index AFP158 peptide was replaced with every other possible amino acids to create a library of X-peptide (Figure 2A). A total of 171 X-peptides and the original index AFP158 peptide were used to stimulate TCR-Ts. An amino acid substitution in the X-peptide was defined as tolerant if the TCR-T response stimulated by this peptide was >10% of original AFP158 peptide. X-scan allowed us to identify all tolerant amino acid replacements in each position (Figure 2B). The complete X-scan data was shown in Supplementary Figure S1. Based on the X-scan data, a peptide motif for each TCR-T was generated (Figure 2C). We also conducted the Alanine (A)-scan and Glycine (G)-scan according to previous report (17) (replacing each residue in the target epitope with Alanine or Glycine) (Supplementary Figure S2). While the data from A- or G- Scan can only indicate either 1 (intolerant) or 20 (tolerant), the motif obtained from the X-scan reveals more precise amino acid replacements (Figures 2C,D).
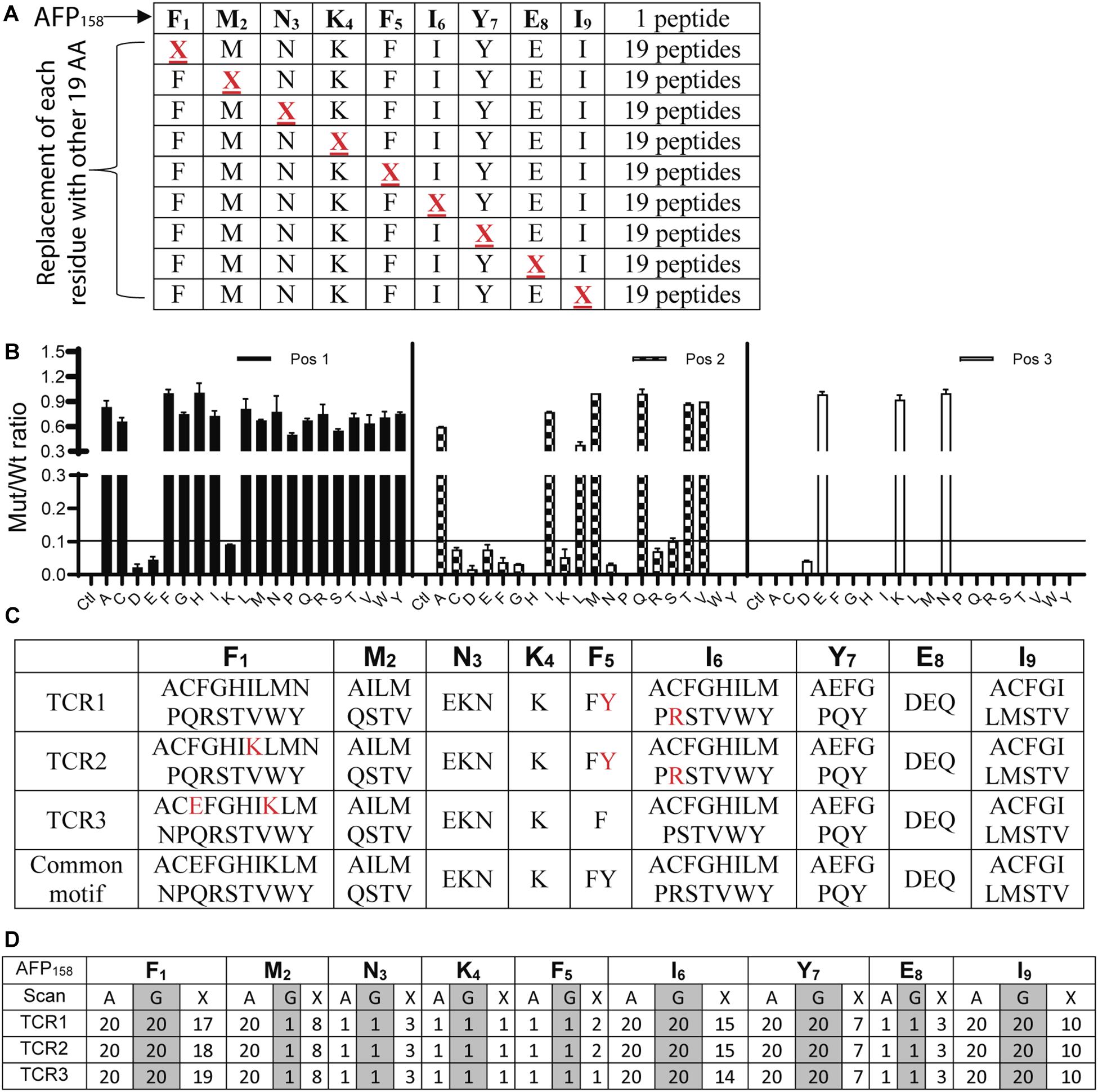
Figure 2. Identification of peptide motifs that can be recognized by TCR-Ts. (A) The scheme of X-Peptides. “X” in each position represents any of the other 19 amino acids. (B) A representative data (position 1–3 of TCR1-T) of X-Scan assay showed the response ratio of X-peptide vs. AFP158 peptide in stimulating TCR-Ts to produce IFNγ. The Mean ± SD from 3 wells was shown. The X-scan assay for each peptide was repeated 3–5 times and consistent observation was obtained. (C) X-Scan identified the peptide motifs potentially recognized by TCR-Ts. The colored letters indicate the difference among 3 TCR-Ts. A common motif that cover all 3 TCR-Ts is also shown. (D) Comparison of A-, G-, and X-Scan to reveal the number of the tolerant amino acids at each position AFP158 epitope.
AFP158-Specific TCR-Ts Can Be Cross-Activated by the ENPP1436 Peptide
The peptide recognition motifs of TCR1, 2 and 3 are slightly different (Figure 2C). To make sure that we would not miss any potential reactive peptides, we used a common peptide motif (Figure 2C) that cover all 3 TCRs to search the SwissProtein databank using the ScanProsite program (29). A total of 93 peptides with the potential capability of activating the TCR-Ts were identified and synthesized (Supplementary Table S1). In the 1st experiment, we used high peptide concentration (1 μg/ml) in the stimulation assay to catch all potential peptides that may activate TCR-Ts. By using 10% of the AFP158 response as cut-off, we found that TCR1-Ts and TCR2-Ts could be cross-activated by ENPP1436 and FL2D189 peptide. TCR3-Ts could be cross-activated by four peptides, the ENPP1436, FL2D189, EPG51033, and RCL1215 peptides (Figure 3A and Supplementary Figure S3). But, at a lower peptide concentration (10 ng/ml), TCR1-T and TCR2-T could be cross-activated by ENPP1436 only, while TCR3-Ts were cross-activated by both ENPP1436 and RCL1215 peptides. A peptide titration study showed that it required high concentration (1 μg/ml) of FL2D189, EPG51033, and RCL1215 to activate TCR-Ts. At 10 ng/ml, the RCL1215 could weakly cross-activate TCR3-Ts. The data also showed that it required 10,000 times more RCL1215 peptide to achieve the same level of activation as AFP158. Even for the more reactive ENPP1436, it would require 2 log more (250–400 times) of peptide to generate the same level of response as AFP158 (Figure 3B). Furthermore, the EC50 of ENPP1436 for cross-activating the AFP158-specific TCR-Ts were 17–33 times higher than AFP158 (Figures 3C,D).
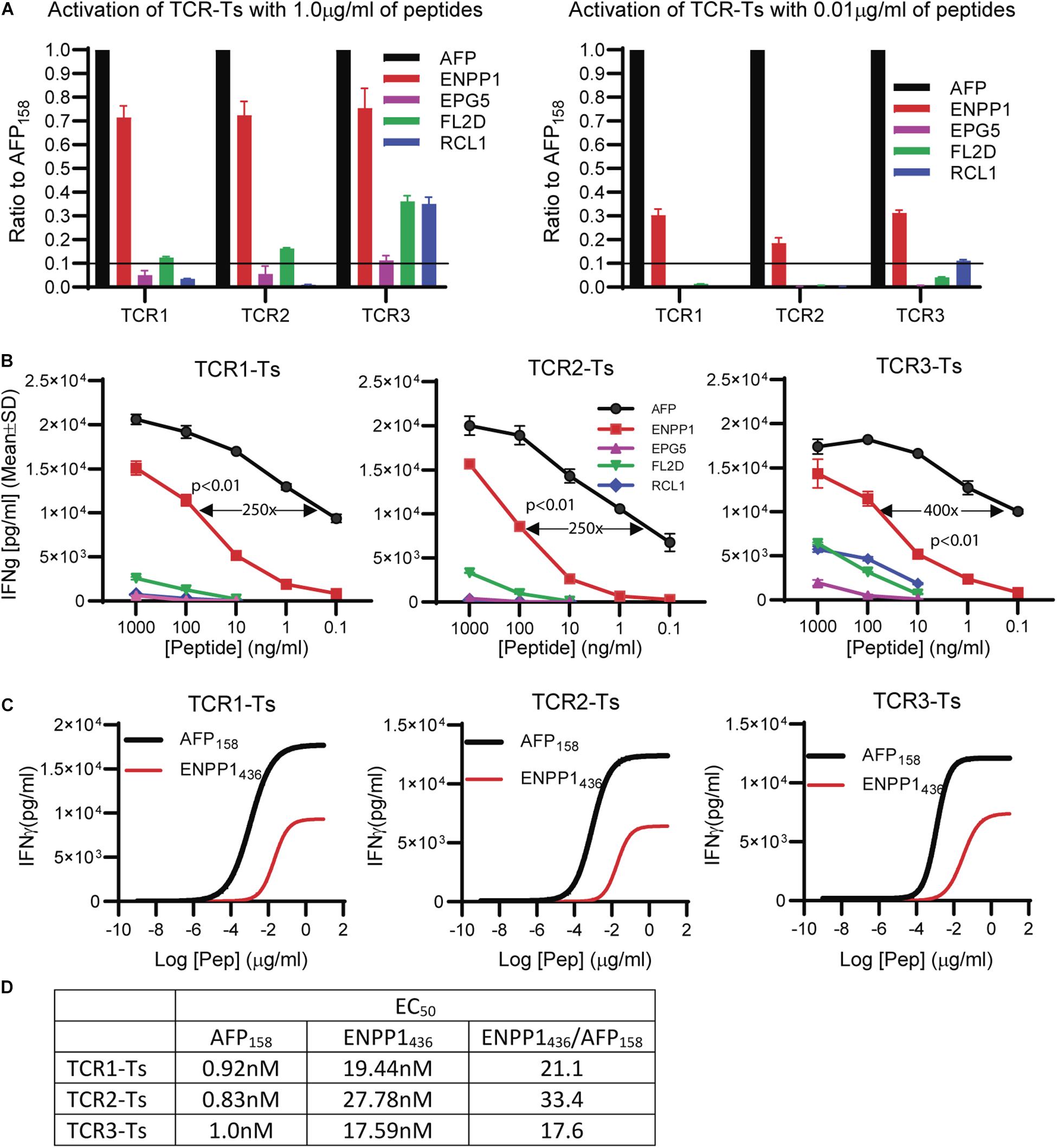
Figure 3. Synthetic off-target peptides cross-activate TCR-Ts. (A) The activation of TCR-Ts to produce IFNγ by 5 peptides at high (1 μg/ml) and low (0.01 μg/ml) concentrations is presented. The data shown was the response ratios of off-target peptides vs. AFP158. (B) Shown is the dose-dependent production of IFNγ by TCR-Ts after stimulation with different peptides. ANOVA was used for statistical analysis. (C,D) The EC50 of AFP158 and ENPP1436 was measured and compared.
ENPP1 Expressing Cells Do Not Activate TCR-Ts
A number of human tissues express ENPP11. Thus, we studied whether the ENPP1 expressing cells could cross-activate AFP158-specific TCR-Ts. To this end, we detected ENPP1 in several cell lines and found out that MB231 and MCF7 are ENPP1 + (Figure 4A) and HLA-A2 +. The MDA-MB231 cells and MB231-luc (derived from MB231 and expressing luciferase) have a very high level of HLA-A2 (10 times more A2 than 293T cells) (Figure 4B). Consistent with the highest level of HLA-A2, MB231 pulsed with ENPP1 peptide stimulated the highest activation of TCR-Ts (Figure 4C). However, even though the MB231 and MB231-luc express high level of ENPP1 and HLA-A2, they did not activate TCR-Ts (Figure 4D). A detail analysis revealed that, while TCR1-Ts and TCR2-Ts did not produce more IFNγ than Mock-T cells after MB231 stimulation, the TCR3-Ts produced slightly more IFNγ (∼300 pg/ml by TCR3-Ts vs. 100 pg/ml by Mock-Ts) (Figure 4E). But the IFNγ level produced by TCR3-Ts after MB231 stimulation was 22x lower than that after HepG2 stimulation (Figures 4D,E). The in vitro CTL assay showed that all 3 TCR-Ts were unable to kill MB231 tumor cells (Figure 4F). To further test whether TCR-Ts could be activated by ENPP1 expressing cells, we then used U87MG brain tumor cells, which express higher level of ENPP1 than MB231 and similar level of HLA-A2. Again, all TCR-Ts were not cross-activated by and did not kill the U87MG tumor cells (Supplementary Figure S4). In addition, normal primary hepatocytes also express ENPP1 (Supplementary Figure S5A). The hepatocytes from 2 donors were also HLA-A2+ (refer to Figure 3C of the accompanied paper, Luo et al). The data showed that hepatocytes did not activate TCR-Ts (Supplementary Figure S5B), consistent with our previous report (15).
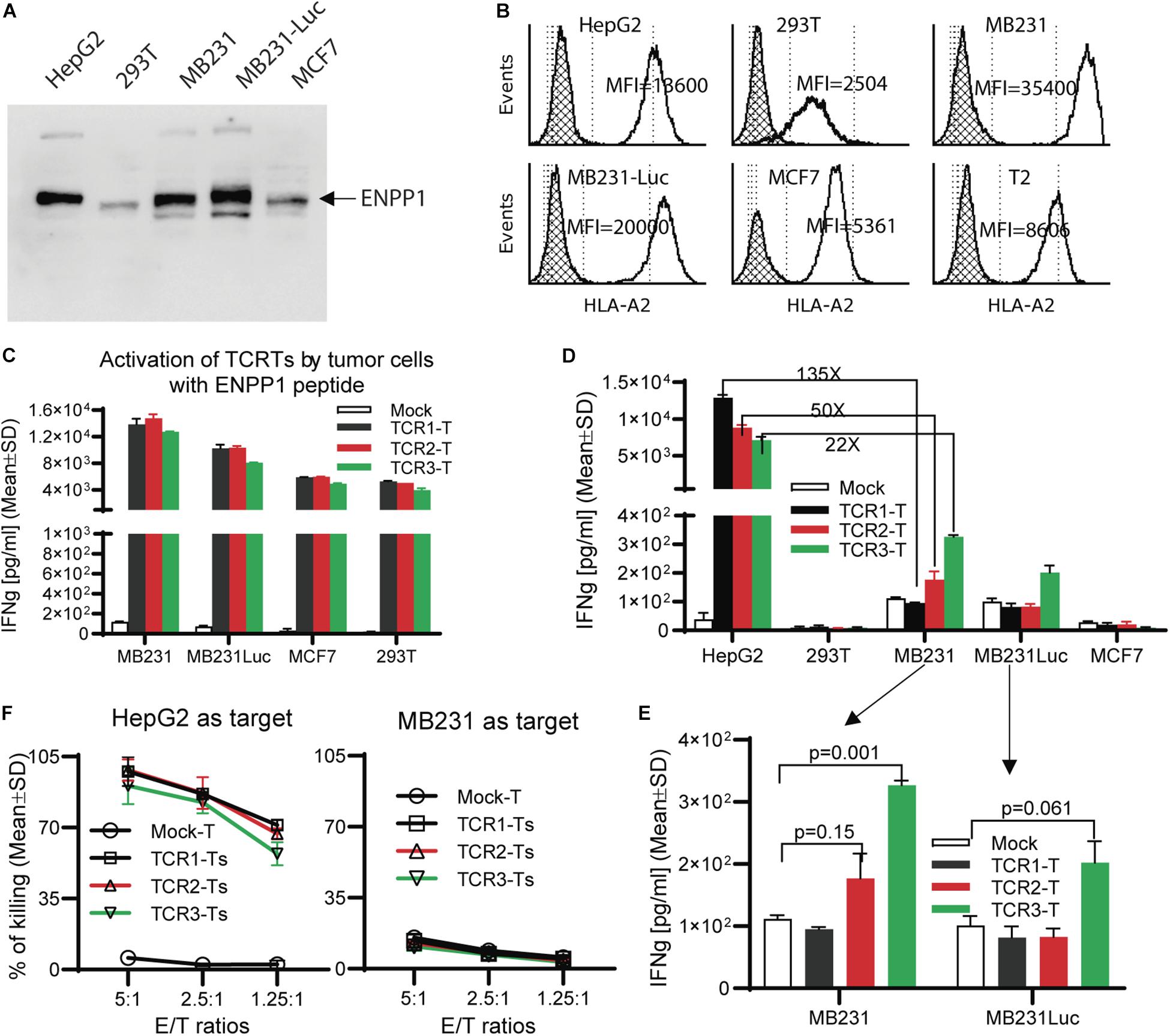
Figure 4. TCR-Ts do not recognize and kill ENPP1 + tumor cells. (A) Western Blot analysis showed ENPP1 expression in different cell lines. MB231-Luc was derived from MDA231 by transfecting Luciferase gene. (B) Flow cytometry data demonstrated the HLA-A2 expression level on the surface of different cell lines by anti-HLA-A2 antibody staining. The hatched lines indicate the isotype staining controls. (C) ELISA assay showed the level of IFNγ production by TCR-Ts after stimulation with indicated cell lines in the presence of 1 μg/ml ENPP1 peptide. (D,E). The activation of TCR-Ts by different cell lines in the absence of ENPP1 peptides was presented. Only the TCR3-Ts were weakly activated, while TCR1-Ts and TCR2-Ts could not be activated by ENPP1 + MB231 and MCF7 cells. (F) LDH assay showed that TCR-Ts do not kill the ENPP1 + MB231 cells. HepG2 cells were used as positive control. Student t-test was used for statistical analysis. This experiment was repeated 4 times with similar data.
Ectopic Over-Expression of ENPP1 in MCF7 Did Not Activate TCR-Ts
In this experiment, we studied whether ectopic over-expression of ENPP1 in MCF7 tumor cells could cross-reactivate TCR-Ts. We found that while all 3 TCR-Ts could be activated by and kill the MCF7 pulsed with ENPP1436 peptide, they were not activated by and did not kill the ENPP1 overexpressing MCF7 tumor cells (Figures 5A–C). In conclusion, all the data indicates that it is unlikely that the ENPP1 expressing cells in the patients will cross-activate AFP158-specific TCR-Ts.
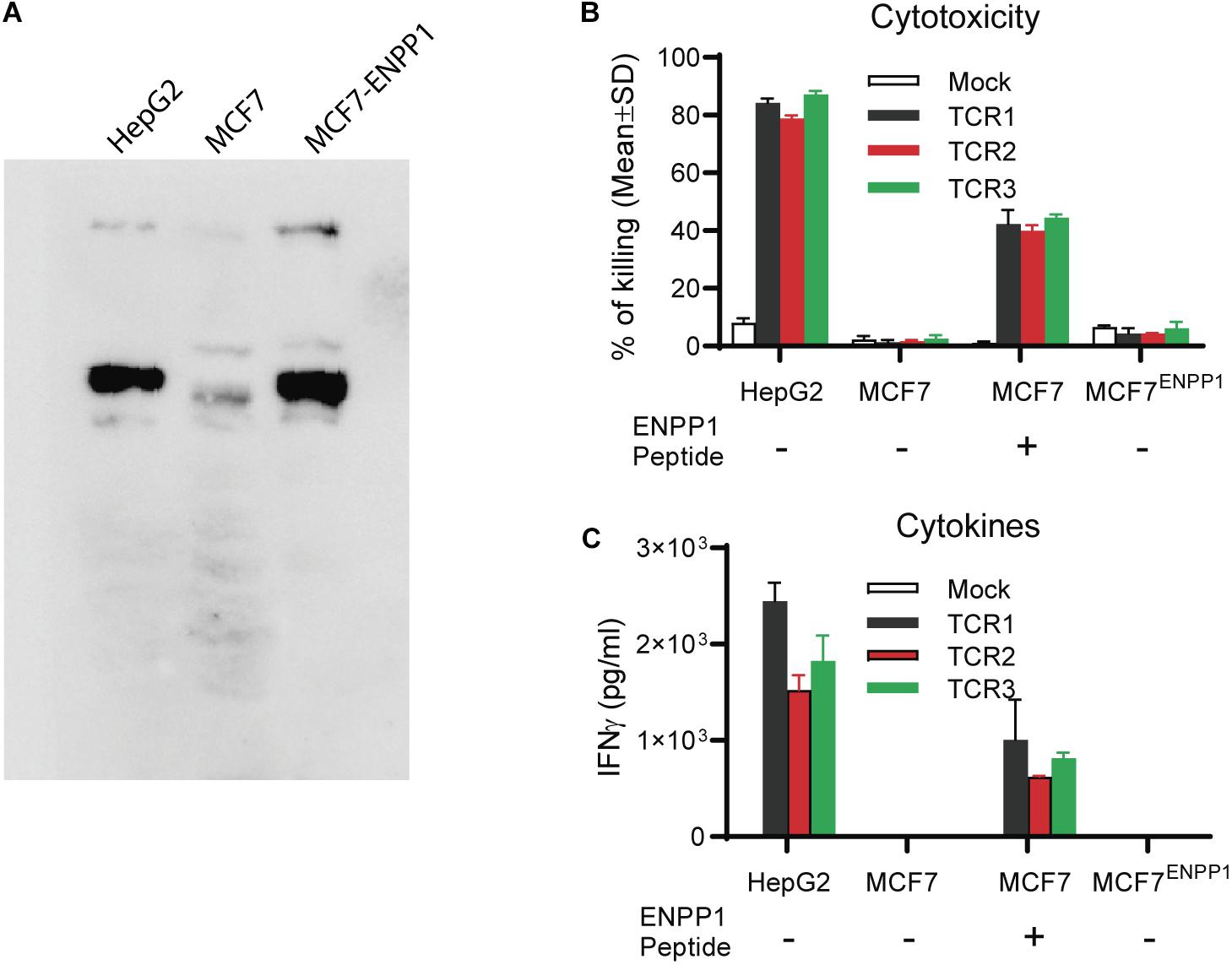
Figure 5. Tumor cells overexpressing ENPP1 do not activate TCRTs. (A) Western Blot analysis showed the ectopic overexpression of ENPP1 in MCF7 cells after transient gene transfer. (B) LDH assay showed that TCR-Ts did not kill the MCF7 cells that overexpress ENPP1 protein. (C) ELISA assay indicated that overexpression of ENPP1 in MCF7 did not activate TCR-Ts to produce IFNγ.
In silico Analyses May Predict TCR-T’s Off-Target Reactivity
Here we studied whether in silico analysis would help predict TCR-T’s off-target reactivity. First, we used the NetMHC4.0 program to rank the 93 peptide’s MHC binding affinity based on databank of 400,000 random natural peptides. The results were then correlated to the experimental data of peptide’s TCR-T activation. The data was summarized in Table 1 and Supplementary Table S1. According to the default criteria of 0.5% set by the software, only AFP158 (rank: 0.01%) and ENPP1436 (rank: 0.4%) peptides are strong binders of HLA-A0201. Consistent with in silico analysis, the experimental data showed that these two peptides could activate all 3 TCR-Ts at physiological concentration (10 ng/ml). In contrast, RCL1215 (rank: 0.8%) is a weak binder. The experimental data showed that RCL1215 cross-activated only TCR3-Ts (Figure 3A). FL2D189 (rank: 17%) is a non-binder. It weakly cross-activated TCR-Ts only at high concentration. The EPG1033 peptide (rank: 29%) is also a non-binder. It activated only TCR3-Ts at high concentration. The MHC binding affinity of peptides was also analyzed by IEDB software and its rank was similar to NetMHC4.0 (Table 1). Secondly, we used the NetCTLpan program to rank a peptide’s chance of being processed and presented by MHC molecule. Based on the data of 200,000 random natural peptides, RCL1215 and ENPP1436 peptides are ranked at 0.8% and 1% (Table 1), which means they are the top 1600 and 2000 peptide of being processed and presented, respectively. In contrast, the AFP158 is ranked at 0.01% (Table 1), which means it is the top 20 out of 200,000 peptides that are processed and presented. Thus, AFP158 has much higher probability of being processed and presented. In conclusion, the in silico analysis data of peptide’s MHC binding affinity and their chance of being processed and presented correlate well with the experimental data of cross-reactivity.
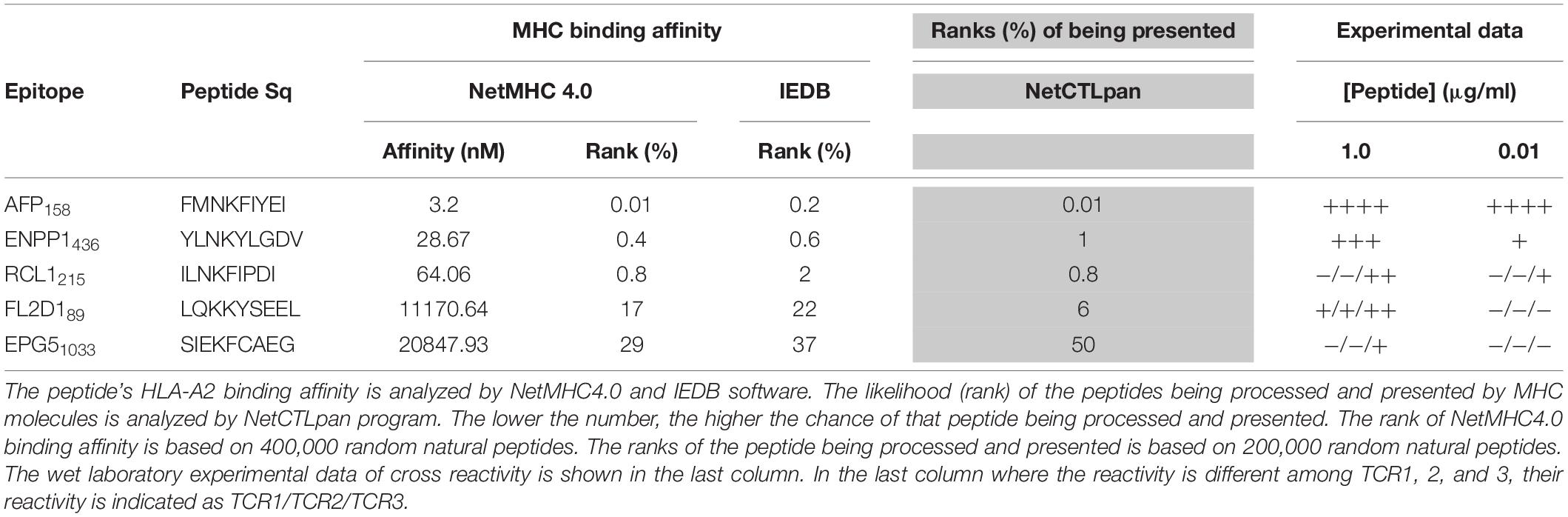
Table 1. Correlation of the in silico analysis data with experimental data of cross-reactivity.
In addition, we did retrospective in silico analysis of the peptides that were known to cause severe toxicity in previous trials. It was reported that the HLA-A01/MAGE-A3 specific TCR-Ts (17) and HLA-A0201/MAGE-A3 specific TCR-Ts (30, 31) caused severe cardiac and neurological toxicity by cross-reacting with off-target peptides of Titin in the heart and MAGE-A12 in the brain, respectively. We compared the peptides of MAGE-A3 vs. Titin and MAGE-A3 vs. MAGE-A12 regarding to their MHC binding affinity and chance of being processed and presented by in silico analysis. The MAGE-A3 and Titin peptides have the same ranking of binding to HLA-A01 (0.01% vs. 0.01% by NetMHC4.0; 0.12% vs. 0.17% by IEDB software) (Table 2). Both epitopes also have the same chance (0.05%) of being processed and presented (Table 2). In the second example, MAGE-A12 epitope (rank 0.01%) has an even higher affinity than index MAGE-A3 peptide (rank 0.03%) for binding HLA-A0201. The MAGE-A12 epitope also has higher chance of being processed and presented by HLA-A0201 than the index MAGE-A3 peptide (0.2% vs. 0.01%) (Table 2). Thus, in both cases that TCR-Ts caused severe toxicity, in silico analysis showed that the off-target peptide’s MHC binding affinity and chance of being processed and presented were at least at the same level as the intended index epitopes.
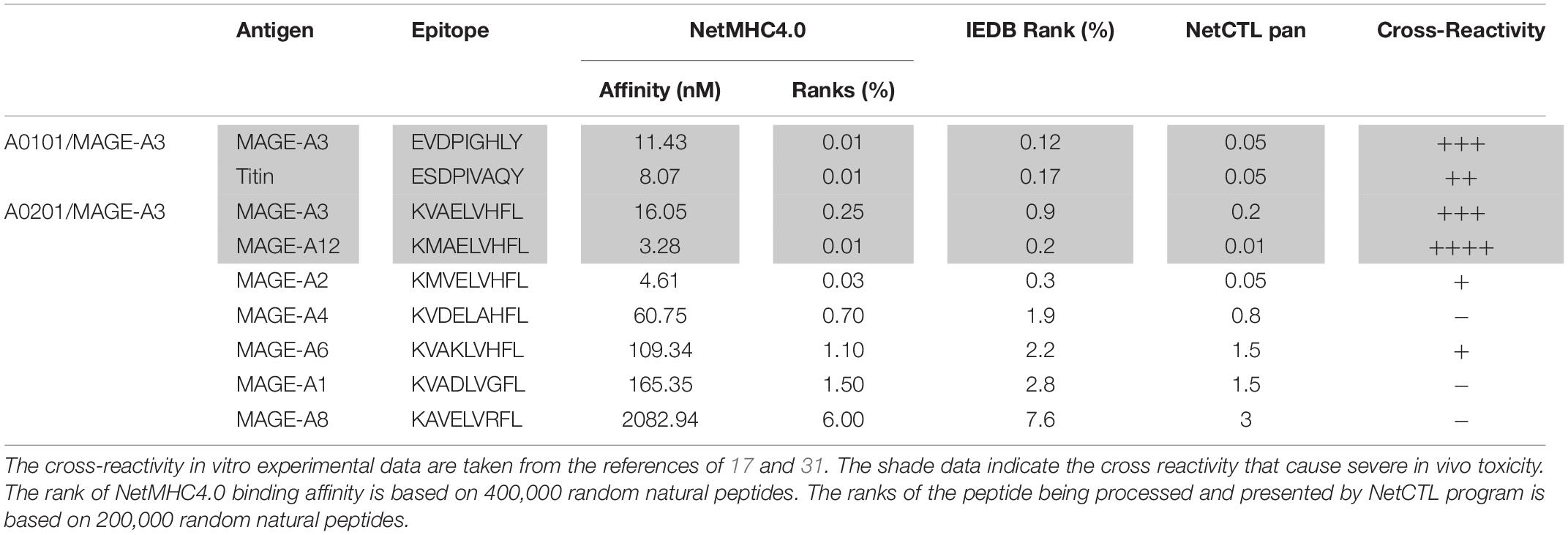
Table 2. In silico analysis of two previous TCR-Ts that showed severe toxicity in clinical trials.
Discussion
The Reliability of X-Scan
T cell activation requires their TCR to specifically bind to cognate MHC/peptide complex. However, the TCR’s recognition of MHC/peptide complex is degenerate (32), which may cause lethal off-target cross-reactivity (12, 30). X-scan is a comprehensive approach to study TCR-T’s off-target reactivity (16, 23). But its reliability of X-scan may depend on the X-peptide concentration used for the assay. Conducting the assay with too high concentration of X-peptide will unnecessarily find many peptides that cross-activate TCR-Ts, but in reality will never do so because of their lower physiological level. On the other hand, too low of concentration will miss out the peptides that may do cause cross reactivity. In our study, we found that the EC50 of AFP158 peptide for activating AFP158-specific TCR-Ts was ∼1 nM. According to previous study (33), T2 cells pulsed with 1 nM of gp100 peptide and WT-1 peptides yielded 12–47 copies of MHC/peptide complex on cell surface. This number of MHC/peptide complex is similar to the naturally presented gp100 epitopes on the melanoma Mel526 and Mel624 cells (9–68 copies) (33) and the naturally processed MHC/NY-ESO1 peptide complex on tumor cells (34). Therefore, 1 nM is the relevant concentration for X-scan that generates physiological level of MHC/peptide complex on cell surface. Thus, we agree that the EC90 of index peptide used in a recent study (23) is the right concentration for X-scan assay in order to make sure that no potential peptides will be missed out.
The reliability of X-scan may also be affected by the fact that the X-peptide contains only one amino acid residue replacement while all other amino acid residues remain original. It does not consider the effect of other amino acid residues in the epitope that may also affect its capability of activating TCR-Ts. Thus, it is possible that peptides with more than one amino acids being simultaneously changed may not follow the rule concluded from X-scan assay. For example, the Y replacement at position 5 of AFP158 was intolerant for TCR3-T activation (even though it is tolerant for TCR1-T and TCR2-T) (Supplementary Figure S1). However, contradictory to this rule, the ENPP1436 peptide is capable of activating TCR3-Ts (tolerant) even it has Y at position 5. Compared to AFP158 (FMNKFIYEI), the ENPP1436 peptide (YLNKYLGDV) has Y at position 5, but also has other 7 amino acid replaced. Those additional amino acid changes may work together to enhance the peptide’s activation of TCR-Ts. To minimize the chance of missing any potential reactive peptides, for the three positions (E and K at Position 1; Y at Position 5; R at Position 6) in the peptide motifs that are different among the 3 TCR-Ts, we used a common peptide motif to cover all potential amino acids to search the protein databank. In this case, as long as one TCR-T is tolerant for one particular amino acid replacement, it will be included in the common motif.
In silico Analysis of Peptide’s MHC Binding Affinity Helps Predict TCR-T’s Off-Target Toxicity
The X-scan strategy may generate a long list of potential off-target peptides. In this study, 93 peptides were identified by X-scan that may potentially cross-activate AFP158-specific TCR-Ts. But, the experimental data showed that the peptides indeed cross-activated TCR-Ts at physiological concentration were within the top 1% of their MHC binding affinity ranked by NetMHC4.0 analysis (Supplementary Table S1). The default cut-offs of strong and weaker binder are 0.5 and 2%, respectively. Although more experimental data will be needed, it is reasonable to assume that, at physiological concentration, only the top 2% of peptides ranked by NetMHC4.0 have the potential to truly cross-activate TCR-Ts. Thus, the programs of calculating peptide’s MHC binding affinity, such as NetMHC4.0 and IEDB, may help shorten the peptide list so that wet laboratory experiments will not be too costly.
Secondly, the computer program of NetCTLpan (27), which integrates peptide’s proteasomal C terminal cleavage, TAP transport efficiency, and MHC binding affinity, may help predict the probability of a particular peptide being processed and presented by MHC. The NetCTLpan analysis showed that the two off-target peptides (Titin and MAGE-A12) that indeed cause severe toxicity in human trials had similar or higher chances of being processed and presented as the intended target of MAGE-A3 (Table 2). In contrast, the NetCTLpan ranks that the chance of ENPP1436 peptide being processed and presented by HLA-A0201 is 100 times lower than the target epitope AFP158 (top 1% vs. top 0.01% in Table 1). Thus, the data of in silico analysis is in agreement that the ENPP1436 peptide is not efficiently processed and presented.
Thirdly, will TCR bind the index and cross-reactive peptides with similar strength? The recent software to predict TCR and MHC/peptide interaction is highly immature (35). However, this may not be necessary as the ultimate test is the wet experiments. According to previous study (33), as low as 0.1 nM of peptides can stimulate TCR-T to produce IFNγ that can be detected, but it requires 1 nM peptides to generate sufficient level of MHC/Peptide complex on cell surface to be detected by microscope. And, it needs 10 nM of peptides to generate measurable MHC-peptide complex by flow cytometry. Thus, the TCR-T functional activation by measuring the IFNγ level is the most sensitive way of finding potential off-target peptides.
TCR-T’s Cross-Reactivity to Synthetic ENPP1436 Peptide Unlikely Causes Severe Off-Target Toxicity
The cross-activation of AFP158-specific TCR-Ts by synthetic ENPP1436 peptide raises the concern that AFP158-specific TCR-Ts may cause off-target toxicity. However, our experimental data indicates such off-target toxicity is highly unlikely. 1st, the cross-activation of AFP158-specific TCR-Ts by ENPP1436 peptide is significantly weaker than AFP158 peptide. The EC50 of ENPP1436 peptide for activating the TCR-Ts is 17–33 time higher than AFP158 peptide. It requires 250–400 times more of ENPP1436 peptide to achieve the same level of activation as AFP158 peptide. The significant weak activation of TCR-Ts by ENPP1436 peptide is further supported by in silico analysis showing that the MHC binding affinity of ENPP1436 peptide is ranked 40 times lower than AFP158 peptide. Thus, there is a wide concentration window to distinct the intended target from off-target activation. 2nd, the chance of ENPP1436 peptide being processed and presented is low. The activation assay of using peptide pulsed T2 cells to stimulate TCR-Ts omits the process of antigen processing which including protein degradation and TAP transport and MHC loading (36). Not all so-called epitopes identified by synthetic peptides can be truly processed and presented by cells (37, 38). In fact, the process of antigen processing and presentation is rather very inefficient, estimating only 1/10,000 peptides being processed and presented (39). The incapability of ENPP1+ MB231, MCF7, U87MG, and normal hepatocytes to activate TCR-Ts is in agreement with the computer prediction that ENPP1436 epitope is unlikely being processed and presented by HLA-A0201 to the level that is sufficient to activate TCR-Ts. Thus, we have strong evidence that the AFP158-specific TCR-Ts will unlikely cause severe off-target toxicity. That being said, the concern of causing off-target toxicity cannot be completely excluded. Further safety switches such as incorporation of suicide gene (40) and truncated EGFR (41) tag may be added in case of the TCRTs need to be removed.
Conclusion
In conclusion, our off-target toxicity study showed: (1) The TCR3-T has a broader cross-reactivity than TCR1-T and TCR2-T, highlighting the need of multiple TCRs to find a proper one; (2) The ENPP1436 peptide can weakly activate AFP158-specific TCR-Ts, but, it requires 250–400 times more of peptide to achieve the same level of activation as AFP158 peptide; (3) Importantly, the ENPP1 expressing cells do not activate TCR1-Ts and TCR2-Ts even though they may slightly activate TCR3-Ts; (4) In silico analysis show that ENPP1436 peptide has a much lower HLA-A0201 binding affinity than AFP158 and is less likely of being processed and presented. In contrast, the off-target peptides that indeed cause severe toxicity in previous studies have a similar HLA binding affinity and a similar or even higher chance of being processed and presented than the intended target peptides. Thus, we conclude that it is unlikely that the AFP158-specific TCRs will cause significant off-target toxicity.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
LC and YH designed the study, conducted majority of the experiments, summarized the data, and wrote the manuscript. LDC and YP prepared the recombinant DNA, lentiviral vectors, and the TCR-Ts. WZ did the A- and G- Scan experiments. XL, YY, and YJ conducted the Western Blot of ENPP1 in primary hepatocytes and the TCR-T activation by primary normal hepatocytes. All authors agreed the manuscript for publication.
Funding
The project is funded in part by NIH grant CA235159. XL, YY, and YJ are employee of Cellular Biomedicine Group and YH is a part-time consultant of Cellular Biomedicine Group, Inc., which licensed the TCRs patent from Augusta University and is developing TCR-T based immunotherapy for liver cancers. The data and materials are open for anyone who is interested.
Conflict of Interest
XL, YY, and YJ are employees of Cellular Biomedicine Group and YH is a part-time consultant for Cellular Biomedicine Group, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The project is funded in part by NIH grant CA235159. XL, YY, and YJ are employees of Cellular Biomedicine Group and YH is a part-time consultant for Cellular Biomedicine Group, Inc., which licensed the TCRs patent from Augusta University and is developing TCR-T based immunotherapy for liver cancers. The data and materials are open for anyone who is interested.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00607/full#supplementary-material
Abbreviations
AFP, alpha fetoprotein; ENPP1, ectonucleotide pyrophosphatase/phosphodiesterase 1; HCC, hepatocellular carcinoma; RCL1, RNA 3’-terminal phosphate cyclase-like protein; TCR, T cell receptors.
Footnotes
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Mizukoshi E, Kaneko S. Immune cell therapy for hepatocellular carcinoma. J Hematol Oncol. (2019) 12:52. doi: 10.1186/s13045-019-0742-5
3. Hato T, Goyal L, Greten TF, Duda DG, Zhu AX. Immune checkpoint blockade in hepatocellular carcinoma: current progress and future directions. Hepatology. (2014) 60:1776–82. doi: 10.1002/hep.27246
4. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. (2015) 75:2139–45. doi: 10.1158/0008-5472.CAN-15-0255
6. Dembic Z, Haas W, Weiss S, McCubrey J, Kiefer H, von Boehmer H, et al. Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature. (1986) 320:232–8.
7. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314:126–9.
8. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. (2009) 114:535–46. doi: 10.1182/blood-2009-03-211714
9. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. (2011) 19:620–6. doi: 10.1038/mt.2010.272
10. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. (2015) 21:914–21. doi: 10.1038/nm.3910
11. Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. (2015) 21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708
12. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. (2013) 122:863–71. doi: 10.1182/blood-2013-03-490565
13. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
14. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. (2016) 16:566–81.
15. Zhu W, Peng Y, Wang L, Hong Y, Jiang X, Li Q, et al. Identification of alpha-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology. (2018) 68:574–89. doi: 10.1002/hep.29844
16. Docta RY, Ferronha T, Sanderson JP, Weissensteiner T, Pope GR, Bennett AD, et al. Tuning T-cell receptor affinity to optimize clinical risk-benefit when targeting alpha-fetoprotein-positive liver cancer. Hepatology. (2019) 69:2061–75.
17. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. (2013) 5:197ra03.
18. Kunert A, Obenaus M, Lamers CHJ, Blankenstein T, Debets R. T-cell receptors for clinical therapy: in vitro assessment of toxicity risk. Clin Cancer Res. (2017) 23:6012–20. doi: 10.1158/1078-0432.CCR-17-1012
19. Salter RD, Cresswell P. Impaired assembly and transport of HLA-A and -B antigens in a mutant TxB cell hybrid. EMBO J. (1986) 5:943–9.
20. He Y, Zhang J, Mi Z, Robbins P, Falo LD Jr. Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J Immunol. (2005) 174:3808–17.
21. Liu Y, Peng Y, Mi M, Guevara-Patino J, Munn DH, Fu N, et al. Lentivector immunization stimulates potent CD8 T cell responses against melanoma self-antigen tyrosinase-related protein 1 and generates antitumor immunity in mice. J Immunol. (2009) 182:5960–9. doi: 10.4049/jimmunol.0900008
22. He Y, Zhang J, Donahue C, Falo LD Jr. Skin-derived dendritic cells induce potent CD8(+) T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity. (2006) 24:643–56.
23. Border EC, Sanderson JP, Weissensteiner T, Gerry AB, Pumphrey NJ. Affinity-enhanced T-cell receptors for adoptive T-cell therapy targeting MAGE-A10: strategy for selection of an optimal candidate. Oncoimmunology. (2019) 8:e1532759. doi: 10.1080/2162402X.2018.1532759
24. Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. (2016) 32:511–7. doi: 10.1093/bioinformatics/btv639
25. Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. (2003) 12:1007–17.
26. Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. (2015) 43:D405–12. doi: 10.1093/nar/gku938
27. Stranzl T, Larsen MV, Lundegaard C, Nielsen M. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics. (2010) 62:357–68. doi: 10.1007/s00251-010-0441-4
28. Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria JC, et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol. (2010) 184:4936–46. doi: 10.4049/jimmunol.1000173
29. de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, et al. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. (2006) 34:W362–5.
30. doi: 10.1097/cji.0b013e3182829747Brichard VG, Louahed J, Clay TM. Cancer regression and neurological toxicity cases after anti-MAGE-A3 TCR gene therapy. J Immunother. (2013) 36:79–81.
31. Chinnasamy N, Wargo JA, Yu Z, Rao M, Frankel TL, Riley JP, et al. A TCR targeting the HLA-A∗0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J Immunol. (2011) 186:685–96. doi: 10.4049/jimmunol.1001775
32. Wilson DB, Wilson DH, Schroder K, Pinilla C, Blondelle S, Houghten RA, et al. Specificity and degeneracy of T cells. Mol Immunol. (2004) 40:1047–55.
33. doi: 10.4161/onci.26840Bossi G, Gerry AB, Paston SJ, Sutton DH, Hassan NJ, Jakobsen BK. Examining the presentation of tumor-associated antigens on peptide-pulsed T2 cells. Oncoimmunology. (2013) 2: e26840.
34. Purbhoo MA, Sutton DH, Brewer JE, Mullings RE, Hill ME, Mahon TM, et al. Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. J Immunol. (2006) 176:7308–16.
35. Jurtz VI, Olsen LR. Computational methods for identification of T Cell neoepitopes in tumors. Methods Mol Biol. (2019) 1878:157–72. doi: 10.1007/978-1-4939-8868-6_9
36. Akram A, Inman RD. Immunodominance: a pivotal principle in host response to viral infections. Clin Immunol. (2012) 143:99–115. doi: 10.1016/j.clim.2012.01.015
37. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. (2013) 31:443–73. doi: 10.1146/annurev-immunol-032712-095910
38. Purbhoo MA, Li Y, Sutton DH, Brewer JE, Gostick E, Bossi G, et al. The HLA A∗0201-restricted hTERT(540-548) peptide is not detected on tumor cells by a CTL clone or a high-affinity T-cell receptor. Mol Cancer Ther. (2007) 6:2081–91.
39. Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat Rev Immunol. (2003) 3: 952–61.
40. Greco R, Oliveira G, Stanghellini MT, Vago L, Bondanza A, Peccatori J, et al. Improving the safety of cell therapy with the TK-suicide gene. Front Pharmacol. (2015) 6:95. doi: 10.3389/fphar.2015.00095
Keywords: T cell receptors, T cell engineering, alpha fetoprotein, hepatocellular carcinoma, TCR cross-reactivity, off-target toxicity, immunotherapy
Citation: Cai L, Caraballo Galva LD, Peng Y, Luo X, Zhu W, Yao Y, Ji Y and He Y (2020) Preclinical Studies of the Off-Target Reactivity of AFP158-Specific TCR Engineered T Cells. Front. Immunol. 11:607. doi: 10.3389/fimmu.2020.00607
Received: 16 January 2020; Accepted: 17 March 2020;
Published: 27 April 2020.
Edited by:
Cameron Turtle, Fred Hutchinson Cancer Research Center, United StatesReviewed by:
Chrystal M. Paulos, Medical University of South Carolina, United StatesJan Joseph Melenhorst, University of Pennsylvania, United States
Copyright © 2020 Cai, Caraballo Galva, Peng, Luo, Zhu, Yao, Ji and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yukai He, eWhlQGF1Z3VzdGEuZWR1
†Present address: Wei Zhu, Department of Infectious Diseases, Liver Research Center, Nanfang Hospital, Southern Medical University, Guangzhou, China