 Clotilde Lauro
Clotilde Lauro Cristina Limatola
Cristina Limatola
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 20 March 2020
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00493
This article is part of the Research TopicCellular Stress and Inflammation: How the Immune System Drives Tissue HomeostasisView all 11 articles
Microglia sustain normal brain functions continuously monitoring cerebral parenchyma to detect neuronal activities and alteration of homeostatic processes. The metabolic pathways involved in microglia activity adapt at and contribute to cell phenotypes. While the mitochondrial oxidative phosphorylation is highly efficient in ATP production, glycolysis enables microglia with a faster rate of ATP production, with the generation of intermediates for cell growth and cytokine production. In macrophages, pro-inflammatory stimuli induce a metabolic switch from oxidative phosphorylation to glycolysis, a phenomenon similar to the Warburg effect well characterized in tumor cells. Modification of metabolic functions allows macrophages to properly respond to a changing environment and many evidence suggest that, similarly to macrophages, microglial cells are capable of a plastic use of energy substrates. Neuroinflammation is a common condition in many neurodegenerative diseases and the metabolic reprograming of microglia has been reported in neurodegeneration. Here we review the existing data on microglia metabolism and the connections with neuroinflammatory diseases, highlighting how metabolic changes contribute to module the homeostatic functions of microglia.
Microglia are the resident immune cells of the central nervous system (CNS) and, depending on the brain region, they can represent from 5 to 12% of total cell population (1). Microglial cells continuously monitor the surrounding parenchyma to sense alteration of brain functions (2, 3) and are involved in controlling neuronal excitability, synaptic activity, neurogenesis, and clearance of apoptotic cells in the healthy adult brain (4). Microglia interact with the cerebral microenvironment through different molecules such as chemokines, cytokines, and trophic factors which, in turn, modulate microglia activities converting the homeostatic microglia into reactive microglia and viceversa (5). Alterations of functional phenotype are accompanied by dynamic changes of shape of cell body and processes, although no unique correlation among microglial cell morphology and functional phenotype has been identified (6). However, in early stages of brain development, and upon in vitro activation with pro-inflammatory stimuli, such as bacterial lipopolysaccharide (LPS), microglial cells display an ameboid profile, with large and round cell bodies, short and thick branches; this morphology is often accompanied by an increased phagocytic activity, production of specific molecules and gene expression signatures. At more mature stages of development, microglia have usually a highly ramified morphology, dynamically reacting to brain parenchymal alterations and injuries (3) and changing phenotype from surveillant to pro- or anti-inflammatory in response to pathological conditions (7, 8). Under pathological conditions, it was shown that microglia comprise cells with diverse phenotypes (9). In fact, microglial-activated cells can be roughly divided into classically activated M1 cells, with cytotoxic and pro-inflammatory properties and alternatively activated M2 cells, with phagocytic activities. The M2 condition can be further divided into three classes: M2a, involved in repair and regeneration; M2b, an immune-regulatory phenotype; M2c, an acquired-deactivating phenotype (10, 11). Indeed, more recent transcriptomic analysis of microglia in different brain area and different disease conditions, reveal a much higher complexity, with several overlapping genes and few signature genes specifically expressed by microglia subgroups (12, 13). Upon aging, microglia phenotype changes further, and it was recently demonstrated an age-related senescent microglial phenotype in humans, possibly involved in pathological processes associated with brain aging (14). Like other cells, in order to perform their functions, microglia require a large amount of energy and it has been recently shown that different microglia phenotypes are associated with distinct metabolic pathways (15–18). Under normal oxygen supply, cells produce energy in the mitochondria, in the glycolytic pathway, through the oxidative phosphorylation (19); in hypoxic conditions, the anaerobic glycolysis converts pyruvate into lactate in the cytoplasm (20, 21). The bioinformatics analysis of a transcriptome database of mouse brain cells (22) showed that microglia express all the genes required for the glycolytic and the oxidative energy metabolism (16). It has been proposed that glucose metabolism exerts transcriptional control over microglial activation, and that the homeostatic phenotype of (cultured) microglia preferentially utilize oxidative metabolism (23–26). An essential fuel for microglia is glucose, which enters the cell through different transporters (GLUTs) (27). Microglia predominantly express GLUT3 (28) and the fructose transporter GLUT5 (29, 30), but under inflammatory conditions, GLUT1 expression is upregulated to increase glucose uptake and promote glycolysis (31). In the absence of glucose, microglia are able to use free fatty acids as alternative energy source, as also suggested by the accumulation of lipid droplets in glucose-deprived microglial cells (32). Microglia also express the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase NOX2 and the superoxide is used to kill pathogens (33, 34). Glucose metabolism controls NOX activation by the NADH-dependent transcriptional co-repressor C-terminal binding protein (CtBP) that affects nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) signaling and the expression of inducible nitric oxide synthase (iNOS) (35, 36). Interestingly, microglia also express the monocarboxylic transporter (MCT) 1 and 2 and absorb lactate and ketons (37) and it has been demonstrated that a ketogenic diet is correlated with a suppression of microglia activation (38–40) likely due to the inhibition of histone deacetylases (HDACs) by ketonic bodies, which decreases NK-kB signaling (41–43). Moreover, silencing HDAC activity affects microglia during development and in adulthood, as a function of the activation state, suggesting that epigenetic changes affect cellular metabolism in activated microglia, modulating microglia function (44). Microglial activity, together with glucose availability and glycolytic rate, influences pro-inflammatory gene and protein expression (45). The oxidative phosphorylation occurs within the mitochondria and produces more ATP molecules; on the other hand, glycolysis permits a faster ATP production in activated microglia (46) allowing a rapid metabolism for cell growth, and the production of cytokines and reactive oxygen species (47). These pathways of energy production are both of primary importance for microglia to maintain their homeostatic functions and are critical for the progression and repair mechanisms upon CNS injury and neurodegeneration.
It is well-established that peripheral immune cells, such as macrophages and dendritic cells (DCs), switch from the oxidative phosphorylation to the aerobic glycolytic pathway when activated (48–50), similarly to what described in tumor cells (Warburg effect) (51–53), to foster cell proliferation. Even if microglia originate from a distinct embryological lineage, they share many characteristics with macrophages (54), as concern cell plasticity and the adaptable use of energy substrates. Several reports recently marked the metabolic similarity of microglia with DCs and macrophages: microglia exposed to inflammatory stimuli exhibit a transient upregulation of specific metabolic pathway's genes (45), indicating that energy metabolism is modulated during brain inflammation. Many studies have been performed with microglia cell lines: in particular, it was observed that upon activation, microglia alter the mitochondrial metabolism in a nitric oxide (NO)-dependent manner (24, 25). Another study demonstrated that lysophosphatidic acid (LPA) stimulates alteration in glycolysis, morphology and motility of C13NJ microglia cells (23). Furthermore, lipopolysaccharide (LPS) stimulation of the murine microglial cell line BV-2 increased lactate production, reduced the mitochondrial oxygen consumption and ATP production, with the resulting increase of glycolysis and decrease of oxidative phosphorylation (15), ultimately increasing nucleic acid production for gene transcription (55). It has also been observed that treatment of primary microglia with Deoxy-D-glucose (2-DG), a blocker of glycolytic pathway, reduced tumor necrosis factor α (TNFα) and interleukin-6 (IL-6) production through NF-kB inhibition, leading to microglia death (56, 57). On the other hand, primary rat microglia cultured with increasing glucose concentration (from 10 to 50 mM) boosted TNFα secretion (58, 59). More recently, Rubio-Araiz et al. showed that primary microglia exposure to LPS and amyloid-β (Aβ) induced an inflammatory state associated with the increase of the glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), with a boost in extracellular acidification rate (ECAR) (60). IFNγ and Aβ also increased microglia glycolysis together with an increase in PFKFB3, hexokinase II and Pyruvate kinase isozymes M2 (PKM2) (61), suggesting that inflammation affects microglia metabolism, driving the glycolysis pathway through increased PFKFB3 activation. Consistently, classic anti-inflammatory stimuli, such as interleukin-4 (IL-4), decreased glucose consumption and lactate production (55) in BV2, and this was confirmed in primary microglia, where IL-4 increased oxygen consumption rate (OCR), basal respiration and ATP production (62); in addition, IL-4/IL-13 stimulation maintained an oxidative metabolic state (16), suggesting that this metabolic shift was associated with a reduced need for anabolic reactions. Pro-inflammatory activation of microglia leads to changes in mitochondrial dynamics and in particular to the metabolic switch from oxidative phosphorylation to glycolysis. It has been recently demonstrated that in inflammatory conditions, microglia upregulate GLUT1 to facilitate glucose uptake and promote glycolysis and that the blockade of GLUT1 reprogrammed back microglia from glycolysis to mitochondrial oxidative phosphorylation, thus altering microglial activation and reducing retinal neurodegeneration in a mouse model (31). These changes represent an adaptive mechanism, since the conversion of microglia from surveying to reactive is accompanied by increased energy consumption. In line with this view, Nair et al. showed that LPS-treated primary microglia increased mitochondrial fragmentation together with a reduction in oxidative phosphorylation and an increase in both oxygen consumption rate, glycolysis and cytokine production (63). In fact, fragmented mitochondria represent the preferred morphofunctional state when the respiratory activity is low (64). Moreover, when mitochondrial fragmentation increases, due to overmuch fission, it can increase the inflammatory response of microglia modulating DRP1 de-phosphorylation and ROS elimination, as already demonstrated for macrophages (65, 66). The same authors also demonstrated that normalizing mitochondrial membrane potential and ROS production with a putative mitochondrial division inhibitor (Mdivi-1) abolished the release of pro- and anti- inflammatory cytokines and chemokines (63). It fact, it has been shown that LPS induces an increase in proton leak and in membrane potential of primary microglia, partially mediated by the uncoupling proteins (UCPs) present in the mitochondrial inner membrane (67).
When exerting homeostatic activities, microglia rely on several membrane proteins: the Pattern Recognition Receptors (PRRs) and immune receptors such as the triggering receptor expressed on myeloid cells-2 (TREM2), the signal regulatory protein 1A (SIRP1A), the fractalkine receptor (CX3CR1), the cell surface transmembrane glycoprotein receptor CD200 (CD200R) and the colony stimulating factor 1 receptor (CSF-1R) (68–70) that recognize Damage-associated molecular patterns (DAMPs) or Neurodegeneration-associated molecular patterns (NAMPs) (71). Upon stimulation by potentially dangerous molecules, microglia assume a neurodegenerative phenotype (MGnD) or disease-associated microglia (DAM), also recently identified as “dark microglia” (72) in several neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), and Alzheimer's disease (AD) (71, 73, 74). It was shown that an aberrant microglia activation may result in a loss or alteration of their physiological functions with possible implications on the emergence or maintenance of pathological conditions; moreover, neuro-inflammation caused by microglia hyperactivity has been associated with several neurodegenerative diseases (12, 75–77) and many evidence support a metabolic reprograming of microglia in neurodegeneration (17). A possible mechanism explaining this microglial metabolic reprogramming has been described in a mouse model of AD, where Aβ directly triggers microglial inflammation together with a metabolic reprogramming from oxidative phosphorylation to glycolysis, in mTOR-HIF-1α pathway-dependent manner (78). Upon activation, microglia enter in a tolerant state with defects in cellular metabolism and reduced responses to inflammatory stimuli, including cytokine secretion and phagocytosis, suggesting that Aβ-induced microglial tolerance might represent a critical cue for AD progression (78). Nonetheless, when microglial glycolytic metabolism was reactivated by interferon-γ (IFN-γ) treatment, which is a known regulator of the mTOR (79) and glycolysis pathway (80), the phagocytic activity of microglia was restored, Aβ plaques and neuronal losses were reduced and cognitive impairment was rescued (78) indicating a (close) relation between the cellular metabolic pathways and functional phenotypes of microglia. The involvement of mTOR pathway in modulating microglial metabolism in AD was also previously suggested by Ulland et al., that identified TREM2 and the downstream mTOR signaling as mediators in maintaining microglial metabolic homeostasis (17). In particular they found that in AD patients carrying a TREM risk variant (81, 82) and in TREM2-deficient mice with AD-like pathology, microglia have an anomalous autophagy activity due to defective mTOR signaling. They demonstrated that upon AD development, TREM2 deficiency affects the mTOR pathway and the energetic metabolism in microglia: TREM2 deficiency was associated with decreased expression of genes for glucose transporters, glycolytic enzymes, and the transcription factor HIF1α, all involved in glycolysis (17). The role of TREM2 in microglial metabolic function was also confirmed in microglia produced by patient-derived iPSC expressing loss of function variants of TREM2: TREM2 variants could not perform the immune-metabolic switch toward glycolysis due to altered PPARg-p38MAPK-PFKFB3 signaling (83). Of note, in AD as well as in other diseases such as traumatic brain injury and ischemia, microglia phenotype changes from anti- to pro-inflammatory upon disease progression (84–86). In particular, in brain ischemia, a phenotypic change is well-documented (87, 88): few minutes after the ischemic attack, resident microglial cells, mainly in the peri-infarct region, acquire an anti-inflammatory phenotype in order to restrain brain damage. Few days after the ischemic insult, pro-inflammatory microglia predominate in the region adjacent the infarct zone (89, 90) and release ROS and pro-inflammatory cytokines that induce the activation of cerebrovascular endothelial cells and sustain the adhesion and transmigration of leukocytes into the injured tissue, contributing to further brain damage (91–94). It was recently demonstrated that upon permanent middle cerebral artery occlusion (pMCAO), the expression profiles of anti- and pro-inflammatory genes in microglia correlates with the expression of genes related to the oxidative and glycolytic pathway, respectively (18), suggesting that a targeted modulation of microglia could be used to reduce the extent of tissue damage in brain ischemia. All these data indicate that a metabolic reprogramming is crucial for microglial function in several neuropathologies and the identification of tools to modulate microglial bioenergetics pathways might be a promising therapeutic strategy.
Considering the heterogeneity of microglia phenotypes present in specific time windows in different CNS regions in pathophysiological conditions (87, 88, 95), studies based on general microglial depletion cannot be considered effective therapeutic strategies to eliminate potentially dangerous microglia phenotypes. Accordingly, since a given microglia subpopulation can plastically modify its phenotype and function in response to signals from the microenvironment (10), the targeting of specific microglial phenotypes in a proper time window could represent a more selective and efficacious approach and represent the current challenge of this field of research. One recent experimental approach proposes to induce a ketogenic state in microglia, suppressing glucose utilization to reduce inflammation, tissue loss and functional impairment after brain injury (41–43). The activation of the G-protein-coupled receptor 109A (GPR109A) with b-hydroxybutyrate (41, 43, 96) on microglial cells attenuates the NF-kB signaling and the production of pro-inflammatory cytokines, promoting a microglial neuroprotective phenotype in a mouse model of PD (42). Also, a metabolic switch toward oxidative metabolism might contribute to promote a protective microglia in some pathophysiological conditions, resulting in the production of metabolites beneficial for neurons (18, 97). Starting from the observation that in animal models of cerebral ischemia the increased anti-inflammatory polarization of microglia is associated with a smaller infarct area and the resolution of inflammation (98) it could be useful to identify a number of factors able to induce a metabolic switch in favor of an anti-inflammatory state of microglia. Among the possible candidates is CX3CL1, a chemokine released from neurons in response to ischemic insult that has neuroprotective properties in permanent focal cerebral ischemia (99), able to modulate the activation state of microglia and its metabolism, down-modulating the expression of several pro-inflammatory and glycolytic pathway-related genes and inducing an increase in the expression of several anti-inflammatory and oxidative pathway-related genes after the ischemic insult (18). CX3CL1 thus acts potentiating the anti-inflammatory function of microglia, prolonging this phenotype to limit neuro-inflammation and gaining time used by parenchymal cells to organize a neuroprotective response. Another possibility could be to regulate the dynamic of microglial mitochondria to prevent neurological disorders caused by aberrant microglial activation: as discussed above, microglia mitochondrial functions correlate with neuronal survival, as a function of microglial ROS production, but also indirectly affecting the activation state and cytokine production (63, 65). Therefore, targeting cytokines that promote the anti-inflammatory phenotype of microglia may result in protecting mitochondrial homeostasis and, on the other hand, direct approaches to enhance microglial mitochondrial function may promote the activation of the microglia anti-inflammatory state (Figure 1).
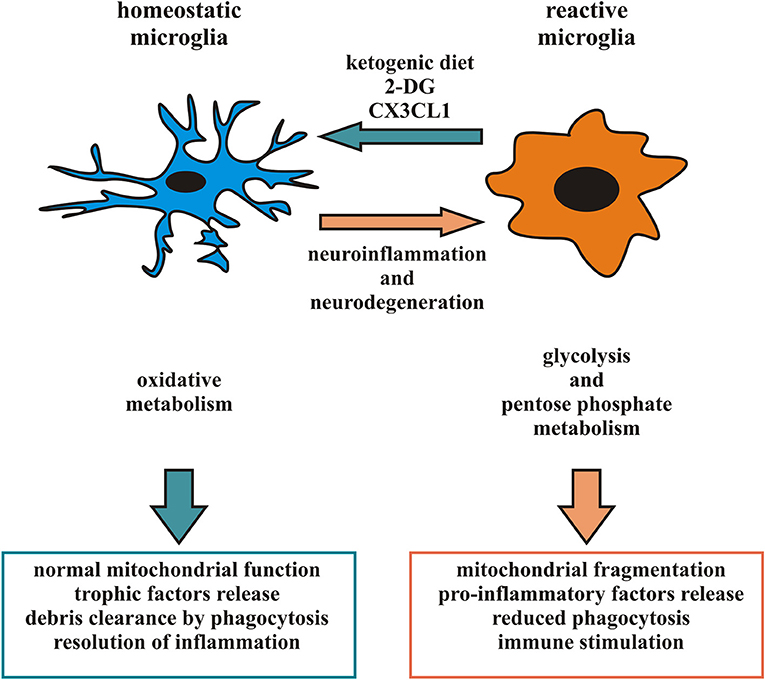
Figure 1. Microglia phenotype and metabolic state: in response to appropriate signals, reactive microglia can switch from a pro-inflammatory to an anti-inflammatory phenotype and vice versa, reorganizing their structure and functions. In particular, pro-inflammatory microglia release cytokines and free radicals that impair brain repair and regeneration while anti-inflammatory microglia resolve cerebral inflammation and promote brain repair increasing phagocytosis and release of trophic factors. Different phenotypes of microglia are associated to distinct metabolic pathways, in order to perform their different functions and their activation leads to changes in mitochondrial dynamics and switch among oxidative phosphorylation and glycolytic metabolism. Several neurodegenerative diseases have been associated with neuro-inflammation related to microglia hyperactivity or mitochondrial dysfunction. Factors able to promote an anti-inflammatory microglia, such as a ketogenic diet, 2-DG and CX3CL1, may represent an intriguing approach to counteract some aspect of neurodegenerative diseases.
Many brain disorders are accompanied by changes in energy metabolism (100–105). While the mechanisms connecting inflammation to cell energy metabolism have been addressed (106), few information are available on how energy metabolism affects the inflammatory responses. Since microglia represent the sentries of the CNS, consistently, they respond to changes in brain metabolism; however, very little is known about their own metabolism, especially because most of the metabolic studies in microglia were conducted in dissociated populations of primary cultures, which do not mirror the complexity and diversity of multiple cell types which interact with other cells and external cues to adapt to bioenergetics changes. For this reason, it is essential to identify experimental approaches to study microglia metabolism in in vivo systems, in pathophysiological conditions. Moreover, most of our knowledge on microglia biology derives from rodents and, even if some in vitro studies suggest that polarization of human microglia might resemble that observed in rodents cells (107), there are several important differences between rodent microglia and their human counterparts (108) and additional studies using human biological systems, such as induced pluripotent stem cells, will be useful in the effort to translate the studies on microglia phenotype into preclinical biomedical research. However, despite these limitations, microglia represent an intriguing target for the treatment of neurodegenerative diseases and targeting their metabolism in order to change their immunological phenotype could represent a promising future therapeutic approach.
CLa made substantial contributions to conception and design of the review. CLi contributed to the manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. (1990) 39:151–70. doi: 10.1016/0306-4522(90)90229-W
2. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
3. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
4. Lenz KM, McCarthy MM. A starring role for microglia in brain sex differences. Neuroscientist. (2015) 21:306–21. doi: 10.1177/1073858414536468
5. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. (2018) 18:225–42. doi: 10.1038/nri.2017.125
6. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. (2011) 91:461–553. doi: 10.1152/physrev.00011.2010
7. Greter M, Lelios I, Croxford AL. Microglia versus myeloid cell nomenclature during brain inflammation. Front Immunol. (2015) 26:249. doi: 10.3389/fimmu.2015.00249
8. Tay TL, Savage JC, Hui CW, Bisht K, Tremblay MÈ. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J Physiol. (2017) 595:1929–45. doi: 10.1113/JP272134
9. Schwartz M, Butovsky O, Brück W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. (2006) 29:68–74. doi: 10.1016/j.tins.2005.12.005
10. Chhor V, Le Charpentier T, Lebon S, Oré M-V, Celador IL, Josserand J, et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav Immun. (2013) 32:70–85. doi: 10.1016/j.bbi.2013.02.005
11. Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. (2013) 39:3–18. doi: 10.1111/nan.12011
12. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. (2017) 23:1018–27. doi: 10.1038/nm.4397
13. Tan YL, Yuan Y, Tian L. Microglial regional heterogeneity and its role in the brain. Mol Psychiatry. (2020) 25:351–67. doi: 10.1038/s41380-019-0609-8
14. Fyfe I. Neurodegenerative disease: gene expression in aged microglia is related to neurodegenerative disease. Nat Rev Neurol. (2018) 14:193. doi: 10.1038/nrneurol.2018.21
15. Voloboueva LA, Emery JF, Sun X, Giffard RG. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. (2013) 587:756–62. doi: 10.1016/j.febslet.2013.01.067
16. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. (2016) 173:649–65. doi: 10.1111/bph.13139
17. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in Alzheimer's disease. Cell. (2017) 70:649–63.e13. doi: 10.1016/j.cell.2017.07.023
18. Lauro C, Chece G, Monaco L, Antonangeli F, Peruzzi G, Rinaldo S, et al. Fractalkine modulates microglia metabolism in brain ischemia. Front Cell Neurosci. (2019) 13:414. doi: 10.3389/fncel.2019.00414
19. Dashty M. A quick look at biochemistry: carbohydrate metabolism. Clin Biochem. (2013) 46:1339–52. doi: 10.1016/j.clinbiochem.2013.04.027
20. Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol. (2006) 6:379–86. doi: 10.1016/j.coph.2006.01.010
21. Hardie DG. Biochemistry. Balancing cellular energy. Science. (2007) 315:1671–2. doi: 10.1126/science.1140737
22. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
23. Bernhart E, Kollroser M, Rechberger G, Reicher H, Heinemann A, Schratl P, et al. Lysophosphatidic acid receptor activation affects the C13NJ microglia cell line proteome leading to alterations in glycolysis, motility, and cytoskeletal architecture. Proteomics. (2010) 10:141–58. doi: 10.1002/pmic.200900195
24. Chénais B, Morjani H, Drapier JC. Impact of endogenous nitric oxide on microglial cell energy metabolism and labile iron pool. J Neurochem. (2002) 81:615–23. doi: 10.1046/j.1471-4159.2002.00864.x
25. Moss DW, Bates TE. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Eur J Neurosci. (2001) 13:529–38. doi: 10.1046/j.1460-9568.2001.01418.x
26. Won SJ, Yoo BH, Kauppinen TM, Choi BY, Kim JH. Recurrent/moderate hypoglycemia induces hippocampal dendritic injury, microglial activation, and cognitive impairment in diabetic rats. J Neuroinflammation. (2012) 9:182. doi: 10.1186/1742-2094-9-182
27. Aldana BI. Microglia-specific metabolic changes in neurodegeneration. J Mol Biol. (2019) 431:1830–42. doi: 10.1016/j.jmb.2019.03.006
28. Kalsbeek MJ, Mulder L, Yi CX. Microglia energy metabolism in metabolic disorder. Mol Cell Endocrinol. (2016) 438:27–35. doi: 10.1016/j.mce.2016.09.028
29. Payne J, Maher F, Simpson I, Mattice L, Davies P. Glucose transporter Glut 5 expression in microglial cells. Glia. (1997) 21:327–31. doi: 10.1002/(sici)1098-1136(199711)21:3<327::aid-glia7>3.0.co;2-1
30. Douard V, Ferraris RP. Regulation of the fructose transporter GLUT5 in health and disease. Am J Physiol Endocrinol Metab. (2008) 295:E227–37. doi: 10.1152/ajpendo.90245.2008
31. Wang L, Pavlou S, Du X, Bhuckory M, Xu H, Chen M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol Neurodegener. (2019) 14:2. doi: 10.1186/s13024-019-0305-9
32. Churchward MA, Tchir DR, Todd KG. Microglial function during glucose deprivation: inflammatory and neuropsychiatric implications. Mol Neurobiol. (2018) 55:1477–87. doi: 10.1007/s12035-017-0422-9
33. Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, Swanson RA. Zinc triggers microglial activation. J Neurosci. (2008) 28:5827–35. doi: 10.1523/JNEUROSCI.1236-08.2008
34. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. (2014) 24:R453–62. doi: 10.1016/j.cub.2014.03.034
35. Ghosh S, George S, Roy U, Ramachandran D, Kolthur-Seetharam U. NAD: a master regulator of transcription. Biochim Biophys Acta. (2010) 1799:681–93. doi: 10.1016/j.bbagrm.2010.08.002
36. Shen Y, Kapfhamer D, Minnella AM, Kim JE, Won SJ, Chen Y, et al. Bioenergetic state regulates innate inflammatory responses through the transcriptional co-repressor CtBP. Nat Commun. (2017) 8:624. doi: 10.1038/s41467-017-00707-0
37. Moreira TJ, Pierre K, Maekawa F, Repond C, Cebere A, Liljequist S, et al. Enhanced cerebral expression of MCT1 and MCT2 in a rat ischemia model occurs in activated microglial cells. J Cereb Blood Flow Metab. (2009) 29:1273–83. doi: 10.1038/jcbfm.2009.50
38. Gasior M, Rogawski MA, Hartman AL. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav Pharmacol. (2006) 17:431–9. doi: 10.1097/00008877-200609000-00009
39. Fann DY, Santro T, Manzanero S, Widiapradja A, Cheng YL, Lee SY, et al. Intermittent fasting attenuates inflammasome activity in ischemic stroke. Exp Neurol. (2014) 257:114–9. doi: 10.1016/j.expneurol.2014.04.017
40. Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. (2014) 19:181–92. doi: 10.1016/j.cmet.2013.12.008
41. Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. (2014) 25:42–52. doi: 10.1016/j.tem.2013.09.002
42. Fu SP, Wang JF, Xue WJ, Liu HM, Liu BR, Zeng YL, et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson's disease models are mediated by GPR109A-dependent mechanisms. J Neuroinflammation. (2015) 12:9. doi: 10.1186/s12974-014-0230-3
43. Huang C, Wang P, Xu X, Zhang Y, Gong Y, Hu W, et al. The ketone body metabolite β-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia. (2018) 66:256–78. doi: 10.1002/glia.23241
44. Datta M, Staszewski O, Raschi E, Frosch M, Hagemeyer N, Tay TL, et al. Histone deacetylases 1 and 2 regulate microglia function during development, homeostasis, and neurodegeneration in a context-dependent manner. Immunity. (2018) 48:514–29. doi: 10.1016/j.immuni.2018.02.016
45. Ghosh S, Castillo E, Frias ES, Swanson RA. Bioenergetic regulation of microglia. Glia. (2018) 66:1200–12. doi: 10.1002/glia.23271
46. Schuster S, Boley D, Möller P, Stark H, Kaleta C. Mathematical models for explaining the warburg effect: a review focussed on ATP and biomass production. Biochem Soc Trans. (2015) 43:1187–94. doi: 10.1042/BST20150153
47. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol. (2014) 15:323–32. doi: 10.1038/ni.2833
48. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
49. Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. (2015) 25:771–84. doi: 10.1038/cr.2015.68
50. De Santa F, Vitiello L, Torcinaro A, Ferraro E. The role of metabolic remodeling in macrophage polarization and its effect on skeletal muscle regeneration. Antioxid Redox Signal. (2018). doi: 10.1089/ars.2017.7420. [Epub ahead of print].
51. Warburg O. On respiratory impairment in cancer cells. Science. (1956) 124:269–70. doi: 10.1126/science.124.3215.267
52. Griffin JL, Shockcor JP. Metabolic profiles of cancer cells. Nat Rev Cancer. (2004) 4:551–61. doi: 10.1038/nrc1390
53. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
54. Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. (2018) 19:622–35. doi: 10.1038/s41583-018-0057-5
55. Gimeno-Bayón J, López-López A, Rodríguez MJ, Mahy N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci Res. (2014) 92:723–31. doi: 10.1002/jnr.23356
56. Vilalta A, Brown GC. Deoxyglucose prevents neurodegeneration in culture by eliminating microglia. J Neuroinflammation. (2014) 11:58. doi: 10.1186/1742-2094-11-58
57. Wang Q, Zhao Y, Sun M, Liu S, Li B, Zhang L, et al. 2-Deoxy-d-glucose attenuates sevoflurane-induced neuroinflammation through nuclear factor-kappa B pathway in vitro. Toxicol in Vitro. (2014) 28:1183–9. doi: 10.1016/j.tiv.2014.05.006
58. Quan Y, Jiang CT, Xue B, Zhu SG, Wang X. High glucose stimulates TNFα and MCP-1 expression in rat microglia via ROS and NF-κB pathways. Acta Pharmacol Sin. (2011) 32:188–93. doi: 10.1038/aps.2010.174
59. Zhang X, Dong H, Zhang S, Lu S, Sun J, Qian Y. Enhancement of LPS-induced microglial inflammation response via TLR4 under high glucose conditions. Cell Physiol Biochem. (2015) 35:1571–81. doi: 10.1159/000373972
60. Rubio-Araiz A, Finucane OM, Keogh S, Lynch MA. Anti-TLR2 antibody triggers oxidative phosphorylation in microglia and increases phagocytosis of β-amyloid. J Neuroinflammation. (2018) 15:247. doi: 10.1186/s12974-018-1281-7
61. McIntosh A, Mela V, Harty C, Minogue AM, Costello DA, Kerskens C, et al. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. (2019) 29:606–21. doi: 10.1111/bpa.12704
62. Holland R, McIntosh AL, Finucane OM, Mela V, Rubio-Araiz A, Timmons G, et al. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav Immun. (2018) 68:183–96. doi: 10.1016/j.bbi.2017.10.017
63. Nair S, Sobotka KS, Joshi P, Gressens P, Fleiss B, Thornton C, et al. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia. (2019) 67:1047–61. doi: 10.1002/glia.23587
64. Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta. (2012) 1817:1833–8. doi: 10.1016/j.bbabio.2012.02.033
65. Park J, Choi H, Kim B, Chae U, Lee DG, Lee SR, et al. Peroxiredoxin 5 (Prx5) decreases LPS-induced microglial activation through regulation of Ca2+/calcineurin-Drp1-dependent mitochondrial fission. Free Radic Biol Med. (2016) 99:392–404. doi: 10.1016/j.freeradbiomed.2016.08.030
66. Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. (2012) 148:228–43. doi: 10.1016/j.cell.2011.11.030
67. De Simone R, Ajmone-Cat MA, Pandolfi M, Bernardo A, De Nuccio C, Minghetti L, et al. The mitochondrial uncoupling protein-2 is a master regulator of both M1 and M2 microglial responses. J Neurochem. (2015) 135:147–56. doi: 10.1111/jnc.13244
68. Wright GJ, Cherwinski H, Foster-Cuevas M, Brooke G, Puklavec MJ, Bigler M, et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol. (2003) 171:3034–46. doi: 10.4049/jimmunol.171.6.3034
69. Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci. (2013) 7:44. doi: 10.3389/fncel.2013.00044
70. Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. (2018) 69:437–49. doi: 10.1146/annurev-med-050715-104343
71. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. (2018) 173:1073–81. doi: 10.1016/j.cell.2018.05.003
72. Bisht K, Sharma KP, Lecours C, Sánchez MG, El Hajj H, Milior G, et al. Dark microglia: a new phenotype predominantly associated with pathological states. Glia. (2016) 64:826–39. doi: 10.1002/glia.22966
73. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. (2017) 47:566–81.e9. doi: 10.1016/j.immuni.2017.08.008
74. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. Unique microglia type associated with restricting development of Alzheimer's disease. Cell. (2017) 169:1276–90.e17. doi: 10.1016/j.cell.2017.05.018
75. Cartier N, Lewis CA, Zhang R, Rossi FM. The role of microglia in human disease: therapeutic tool or target? Acta Neuropathol. (2014) 128:363–80. doi: 10.1007/s00401-014-1330-y
76. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
77. Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. (2017) 20:136–44. doi: 10.1038/nn.4475
78. Baik SH, Kang S, Lee W, Choi H, Chung S, Kim J-IL, et al. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer's disease. Cell Metab. (2019) 30:493–507.e6. doi: 10.1016/j.cmet.2019.06.005
79. Kroczynska B, Rafidi RL, Majchrzak-Kita B, Kosciuczuk EM, Blyth GT, Jemielity J, et al. Interferon γ (IFNγ) signaling via mechanistic target of rapamycin complex 2 (mTORC2) and regulatory effects in the generation of type II interferon biological responses. J Biol Chem. (2016) 291:2389–96. doi: 10.1074/jbc.M115.664995
80. Cheng SC, Scicluna BP, Arts RJ, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. (2016) 17:406–13. doi: 10.1038/ni.3398
81. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. (2013) 368:117–27. doi: 10.1056/NEJMoa1211851
82. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. (2013) 368:107–16. doi: 10.1056/NEJMoa1211103
83. Piers TM, Cosker K, Mallach A, Johnson GT, Guerreiro R, Hardy J, et al. A locked immunometabolic switch underlies TREM2 R47H loss of function in human iPSC-derived microglia. FASEB J. (2020) 34:2436–50. doi: 10.1096/fj.201902447R
84. Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp. (2012) 60:251–66. doi: 10.1007/s00005-012-0181-2
85. Wang H, Guo W, Liu H, Zeng R, Lu M, Chen Z, et al. Inhibition of inflammatory mediator release from microglia can treat ischemic/hypoxic brain injury. Neural Regen Res. (2013) 8:1157–68. doi: 10.3969/j.issn.1673-5374.2013.13.001
86. Suenaga J, Hu X, Pu H, Shi Y, Hassan SH, Hassan SH, et al. White matter injury and microglia/macrophage polarization are strongly linked with age-related long-term deficits in neurological function after stroke. Exp Neurol. (2015) 272:109–19. doi: 10.1016/j.expneurol.2015.03.021
87. Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni MG. The ischemic environment drives microglia and macrophage function. Front Neurol. (2015) 8:6–81. doi: 10.3389/fneur.2015.00081
88. Ma Y, Wang J, Wang Y, Yang GY. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. (2017) 157:247–72. doi: 10.1016/j.pneurobio.2016.01.005
89. Schroeter M, Jander S, Huitinga I, Witte OW, Stoll G. Phagocytic response in photochemically induced infarction of rat cerebral cortex. Stroke. (1997) 28:382–6. doi: 10.1161/01.STR.28.2.382
90. Perego C, Fumagalli S, De Simoni MG. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation. (2011) 8:174. doi: 10.1186/1742-2094-8-174
91. Kriz J. Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol. (2006) 18:145–57. doi: 10.1615/CritRevNeurobiol.v18.i1-2.150
92. Ceulemans AG, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J Neuroinflammation. (2010) 7:74. doi: 10.1186/1742-2094-7-74
93. Jin AY, Tuor UI, Rushforth D, Kaur J, Muller RN, Petterson JL, et al. Reduced blood brain barrier breakdown in P-selectin deficient mice following transient ischemic stroke: a future therapeutic target for treatment of stroke. BMC Neurosci. (2010) 11:12. doi: 10.1186/1471-2202-11-12
94. Grønberg NV, Johansen FF, Kristiansen U, Hasseldam H. Leukocyte infiltration in experimental stroke. J Neuroinflammation. (2013) 10:115. doi: 10.1186/1742-2094-10-115
95. Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation. (2013) 10:4. doi: 10.1186/1742-2094-10-4
96. Rahman M, Muhammad S, Khan MA, Chen H, Ridder DA, Müller-Fielitz H, et al. The β-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat Commun. (2014) 5:3944. doi: 10.1038/ncomms4944
97. Cherry JD, Olschowka JA, O'Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. (2014) 11:98. doi: 10.1186/1742-2094-11-98
98. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. (2011) 17:796–808. doi: 10.1038/nm.2399
99. Cipriani R, Villa P, Chece G, Lauro C, Paladini A, Micotti E, et al. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J Neurosci. (2011) 31:16327–35. doi: 10.1523/JNEUROSCI.3611-11.2011
100. Beal MF. Metabolic disorders and neurotoxicology. Curr Opin Neurol. (1995) 8:467–8. doi: 10.1097/00019052-199512000-00013
101. Bergsneider M, Hovda DA, Shalmon E, Kelly DF, Vespa PM, Martin NA, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg. (1997) 86:241–51. doi: 10.3171/jns.1997.86.2.0241
102. Ding F, Yao J, Rettberg JR, Chen S, Brinton RD. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer's mouse brain: implication for bioenergetic intervention. PLoS ONE. (2013) 8:e79977. doi: 10.1371/journal.pone.0079977
103. Dusick JR, Glenn TC, Lee WN, Vespa PM, Kelly DF, Lee SM, et al. Increased pentose phosphate pathway flux after clinical traumatic brain injury: a [1,2-13C2]glucose labeling study in humans. J Cereb Blood Flow Metab. (2007) 27:1593–602. doi: 10.1038/sj.jcbfm.9600458
104. Orth M, Schapira AH. Mitochondrial involvement in Parkinson's disease. Neurochem Int. (2002) 40:533–41. doi: 10.1016/S0197-0186(01)00124-3
105. Robbins NM, Swanson RA. Opposing effects of glucose on stroke and reperfusion injury: acidosis, oxidative stress, and energy metabolism. Stroke. (2014) 45:1881–6. doi: 10.1161/STROKEAHA.114.004889
106. Hotamisligil GS. Inflammation and metabolic disorders. Nature. (2006) 444:860–7. doi: 10.1038/nature05485
107. Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC, et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. (2012) 60:717–27. doi: 10.1002/glia.22298
Keywords: microglia, metabolism, neuroinflammation, neurodegeneration, homeostasis
Citation: Lauro C and Limatola C (2020) Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front. Immunol. 11:493. doi: 10.3389/fimmu.2020.00493
Received: 28 December 2019; Accepted: 04 March 2020;
Published: 20 March 2020.
Edited by:
Ola Grimsholm, University of Gothenburg, SwedenReviewed by:
Kyoungho Suk, Kyungpook National University, South KoreaCopyright © 2020 Lauro and Limatola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clotilde Lauro, Y2xvdGlsZGUubGF1cm9AdW5pcm9tYTEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.