 Maria Castella1,2,3*
Maria Castella1,2,3* Miguel Caballero-Baños4,5†
Miguel Caballero-Baños4,5† Valentín Ortiz-Maldonado1†
Valentín Ortiz-Maldonado1† Europa Azucena González-Navarro4Guillermo Suñé1,2Asier Antoñana-Vidósola1,2
Europa Azucena González-Navarro4Guillermo Suñé1,2Asier Antoñana-Vidósola1,2 Anna Boronat2,4Berta Marzal4Lucía Millán4
Anna Boronat2,4Berta Marzal4Lucía Millán4 Beatriz Martín-Antonio1,2
Beatriz Martín-Antonio1,2 Joan Cid2,6Miquel Lozano2,6Enric García3,7Jaime Tabera3,8
Joan Cid2,6Miquel Lozano2,6Enric García3,7Jaime Tabera3,8 Esteve Trias8
Esteve Trias8 Unai Perpiña9
Unai Perpiña9 Josep Ma Canals2,9,10Tycho Baumann1Daniel Benítez-Ribas2,4Elías Campo2,10,11,12,13
Josep Ma Canals2,9,10Tycho Baumann1Daniel Benítez-Ribas2,4Elías Campo2,10,11,12,13 Jordi Yagüe2,4,10Álvaro Urbano-Ispizua1,2,10,14,15Susana Rives16,17‡
Jordi Yagüe2,4,10Álvaro Urbano-Ispizua1,2,10,14,15Susana Rives16,17‡ Julio Delgado1,2,10,12‡
Julio Delgado1,2,10,12‡ Manel Juan2,3,4,5,10,15*‡
Manel Juan2,3,4,5,10,15*‡- 1Department of Hematology, Institut Clínic de Malalties Hematològiques i Oncològiques, Hospital Clínic de Barcelona, Barcelona, Spain
- 2Institut D'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain
- 3Blood and Tissue Bank (BST), Barcelona, Spain
- 4Department of Immunology, Centro de Diagnóstico Biomédico, Hospital Clínic de Barcelona, Barcelona, Spain
- 5Hospital Sant Joan de Déu, Barcelona, Spain
- 6Department of Hemotherapy and Hemostasis, Institut Clínic de Malalties Hematològiques i Oncològiques, Hospital Clínic de Barcelona, Barcelona, Spain
- 7Apheresis Unit, Hospital Sant Joan de Déu de Barcelona, Barcelona, Spain
- 8Unit of Advanced Therapies, Hospital Clínic de Barcelona, Barcelona, Spain
- 9Stem Cells and Regenerative Medicine Laboratory, Department of Biomedical Sciences, Production and Validation Center of Advanced Therapies (Creatio), Universitat de Barcelona, Barcelona, Spain
- 10Universitat de Barcelona, Barcelona, Spain
- 11Department of Pathology, Hospital Clínic de Barcelona, IDIBAPS, Barcelona, Spain
- 12Centro de Investigación Biomedical en Red de Cancer, Barcelona, Spain
- 13Institució Catalana de Recerca i Estudis Avancats, Barcelona, Spain
- 14Department of Biomedicine, School of Medicine, Josep Carreras Leukemia Research Institute, Universitat de Barcelona, Barcelona, Spain
- 15Immunotherapy Unit Blood and Tissue Bank-Hospital Clínic de Barcelona, Barcelona, Spain
- 16Department of Pediatric Hematology and Oncology, Hospital Sant Joan de Déu, Barcelona, Spain
- 17Institut de Recerca Sant Joan de Déu, Barcelona, Spain
Development of semi-automated devices that can reduce the hands-on time and standardize the production of clinical-grade CAR T-cells, such as CliniMACS Prodigy from Miltenyi, is key to facilitate the development of CAR T-cell therapies, especially in academic institutions. However, the feasibility of manufacturing CAR T-cell products from heavily pre-treated patients with this system has not been demonstrated yet. Here we report and characterize the production of 28 CAR T-cell products in the context of a phase I clinical trial for CD19+ B-cell malignancies (NCT03144583). The system includes CD4-CD8 cell selection, lentiviral transduction and T-cell expansion using IL-7/IL-15. Twenty-seven out of 28 CAR T-cell products manufactured met the full list of specifications and were considered valid products. Ex vivo cell expansion lasted an average of 8.5 days and had a mean transduction rate of 30.6 ± 13.44%. All products obtained presented cytotoxic activity against CD19+ cells and were proficient in the secretion of pro-inflammatory cytokines. Expansion kinetics was slower in patient's cells compared to healthy donor's cells. However, product potency was comparable. CAR T-cell subset phenotype was highly variable among patients and largely determined by the initial product. TCM and TEM were the predominant T-cell phenotypes obtained. 38.7% of CAR T-cells obtained presented a TN or TCM phenotype, in average, which are the subsets capable of establishing a long-lasting T-cell memory in patients. An in-depth analysis to identify individual factors contributing to the optimal T-cell phenotype revealed that ex vivo cell expansion leads to reduced numbers of TN, TSCM, and TEFF cells, while TCM cells increase, both due to cell expansion and CAR-expression. Overall, our results show for the first time that clinical-grade production of CAR T-cells for heavily pre-treated patients using CliniMACS Prodigy system is feasible, and that the obtained products meet the current quality standards of the field. Reduced ex vivo expansion may yield CAR T-cell products with increased persistence in vivo.
Introduction
Adoptive T-cell transfer (ACT) immunotherapy is a field in continuous expansion especially during the last three decades. ACT involves ex vivo expansion of tumor-specific cells and reinfusion into the patient. Among these therapies, the use of Chimeric Antigen Receptor (CAR) T-cells for the treatment of several hematologic malignancies has shown unprecedented efficacy rates. Consequently, the development of these therapies from bench to bedside has been done in an impressively short amount of time (1–3).
CAR T-cells are genetically modified cells whose cytotoxic activity has been redirected toward cancer cells with the aim of eliminating the tumor. CARs are chimeric proteins composed of an extracellular region responsible for binding to a particular antigen and an intracellular region that promotes T-cell cytotoxic activity and proliferation. CAR binding to the selected antigen is usually mediated by a single chain variable fragment (scFv) of a monoclonal antibody. The scFv-derived region results in a MHC-independent interaction of the CAR with its ligand. This scFv is combined with one or more intracellular co-stimulatory domains (usually CD28 or 4-1BB) and a pro-activator cytotoxic domain (CD3ζ) (4–6).
First great successes in the CAR T-cell field came from studies using a 2nd generation CAR (including a co-stimulatory domain) targeting the pan-B-cell lineage marker CD19. In this studies by National Cancer Institute and University of Pennsylvania, one patient with follicular lymphoma experienced partial remission (7) and two out of three patients with advanced refractory chronic lymphocytic leukemia (CLL) obtained complete responses (8). Afterwards, these and several other institutions including Memorial Sloan Kettering Cancer Center and the Fred Hutchinson Cancer Research Center pioneered several studies using slightly different CAR19 constructs, and confirmed response rates between 50 and 80% in CLL, non-Hodgkin's lymphoma (NHL) and acute lymphoblastic leukemia (ALL) (9–14).
Currently, a lot of effort is being put in developing new CARs to target other types of tumors. Anti-BCMA CAR has also shown impressive response rates (around 90%) for multiple myeloma (MM) (15–17). Also, other antigens such as CD30 and CD22 are currently being explored for the treatment of hematologic malignancies (18–20). In the field of solid tumors, CAR T-cell therapies have proven to be more challenging, so far. Initial clinical trials in solid tumors have shown limited efficacy and high toxicity (21–30). This fact can be attributed to several factors: difficulty in finding tumor-specific antigens to target, poor T cell infiltration in the tumor and immunosuppressive tumor microenvironment, among others. The scientific community is currently working in finding ways to overcome these challenges (31, 32). As a result, dozens of clinical trials using 4th generation CARs (Armored CARs, CAR T-cells containing suicide genes and antibody-producing CARTs) for solid tumors are ongoing (33–36).
The different clinical applications and the number of patients waiting to receive a treatment are exponentially growing. Accordingly, the number of institutions and centers that are in need of being capable of performing CAR T-cell therapies is also growing. In this context, developing systems that can facilitate CAR T-cell production is crucial to help CAR T-cell field move faster, thereby finding effective therapies for all kinds of malignances and other diseases.
To this end, we have previously reported the development and pre-clinical evaluation of a new anti-CD19 CAR, based on the A3B1 antibody (37). Concomitantly, we have also established a CAR T-cell (ARI-0001) production system at our institution (Hospital Clinic de Barcelona). Our system is based on the use of a closed semi-automatic bioreactor (CliniMACS Prodigy®) for ex vivo selection, transduction and expansion of CAR T-cells. We are currently conducting a phase I clinical trial using our novel anti-CD19 CAR for CD19+ B-cell malignancies (NCT03144583). We have successfully prepared 28 ARI-0001 cell products in the context of this clinical trial. Here, we present the results and characteristics of the products obtained, thereby demonstrating, for the first time, the feasibility of CAR T-cell production in a relatively wide series of heavily pre-treated patients using CliniMACS Prodigy system.
Materials and Methods
Patients and Samples
At the time of submitting this manuscript, 28 products from 27 patients enrolled in phase I clinical trial for CD19+ B-cell malignancies (NCT03144583) have been produced. Among the 27 patients, 22 had ALL (14 adult and eight pediatric patients), four had NHL and 1 CLL. All patients included in the clinical trial had relapsed of their disease. Patients' pretreatment regimens are summarized in Table 1.
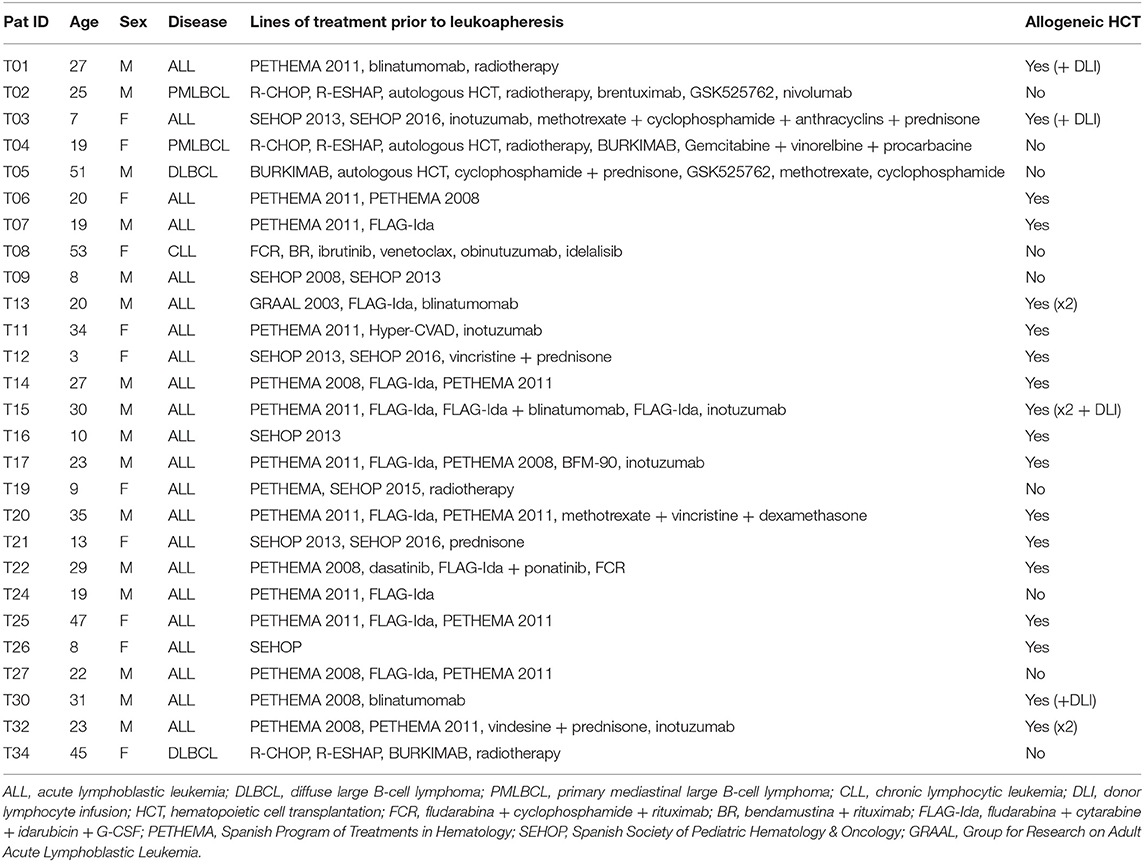
Table 1. Patients' pretreatment regimens.
Adult patients were subjected to leukocytapheresis at the Apheresis Unit, Hospital Clínic, and pediatric patients at the Apheresis Unit of Hospital Sant Joan de Déu/BST, after signing an informed consent. Apheresis procedures were performed using Amicus device (Fresenius Kabi, Lake Zurich, IL). A minimum of 1 × 108 total T-cells diluted in 50 ml of plasma were required. This study has been approved by the Research Ethics Comitee (CeIm) of Hospital Clinic. HCB/2017/0001. Clinical trial: CART19-BE-01. Eudra: 2016-002972-29.
ARI-0001 Cell Production
Apheresis products were connected to CliniMACS Prodigy® system (Miltenyi Biotec) tubing set. Erythrocytes and platelets were removed by density gradient centrifugation in the Centricult unit. The remaining cells were selected using CD4 and CD8 coated magnetic beads. Selected cells were eluted in the “Reapplication Bag.” After selection, 1 × 108 T-cells (from reapplication bag) were used to initiate cell culture. The remaining cells were cryopreserved in bags and vials to be used as control cells for product quality assays and as a backup in case of production failure. Cells were cultured using TexMACS® media supplemented with 3% human AB serum (obtained from the blood bank. BST) and with 155 IU/mL IL-7 and 290 IU/mL IL-15 (Miltenyi Biotec #170-076-111 and #170-076-114, respectively). Cells were immediately activated using TransACT GMP Grade (Miltenyi Biotec, Cat. N. 170-076-156) and transduced 24h later using CAR19-containing lentivirus at MOI = 10. A cell culture wash was programmed 48 h after transduction. The cells were then maintained in culture with increasing shaking until the desired cell number was reached (typically 7–10 days after cell culture initiation). Cells were finally eluted in 100 ml 0.9% NaCl + 1%HSA, aliquoted according to the desired ARI-0001 cell dose and cryopreserved until infusion.
The aim was to achieve two doses of ARI-0001 cells/patient. The planned target cell dose varied depending on the patient's disease. Typically, 1 × 106 ARI-0001 cells/kg for patients with ALL and CLL, and 5 × 106 ARI-0001 cells/kg for NHL patients.
Monoclonal Antibodies
CAR19 expression was detected with an APC-conjugated AffiniPureF(ab')2-fragment goat-anti-mouse IgG (Jackson ImmunoResearch Laboratories, 115-136-072). ARI-0001 cell product composition was determined by flow cytometry using staining with the following antibodies (all from BD): CD45-APC, CD3-BV421, CD4-FITC, CD8-PerCPCy5.5, CD19-PECy7, CD16-PE, CD56-PE.
For the T cell subset characterization experiments, CAR+ cells were detected using a CD19-Fc recombinant protein chimera (R&D, Cat. N. 9269-CD-050) and a secondary antibody FITC-Goat F(ab)2 anti-human IgG (Life Technologies, Cat. N. H10101C). This staining was combined with the following monoclonal antibodies (all from BD): CD3-BV421, CD8-APC.Cy7, CD45RA-PECy7, CD45RO-APC, CCR7-PerCPCy5.5, CD28-BV510, and CD95-PE (or CD27-PE). T cell subpopulations were defined as follows: TN: CD45RA+, CCR7+; TSCM: CD45RA+, CCR7+,CD95+; TCM: CD45RA–, CCR7+; TEM: CD45RA-, CCR7- and TEFF: CD45RA+, CCR7−.
For intracellular cytokine measurement, the following antibodies were used, all from BD: CD3-BV450, CD8-APC.H7, CD4-BV500, IFNγ-PerCP.Cy5.5, TNFα-PE.
For repeated challenges experiment, the antibodies used were the following, all from BD: CD3-APC, CD4-BV510, CD8-APC.Cy7, CD19-PE.
For flow cytometry analyses, cells were acquired using a FACS Canto II, BD and subsequently analyzed using FlowJo Software.
Product Quality Controls
Product potency assay was performed by flow cytometry as described in Castella et al. (37). Real-time PCR was used to measure number of copies/cell and to assess the presence of replication-competent lentivirus (RCL) in the final product. Primer sequences and PCR protocol has been described previously (37). Product sterility, absence of mycoplasma, endotoxin and adventitious virus was determined by a certified laboratory using the technique specified in Table S1. Adventitious virus included the determination of HIV virus presence among others. Since conventional HIV detection methods detect also the presence of the lentiviral transgene used to transduce the cells, an alternative PCR assay based on the detection of Env gene was used to discriminate between HIV infection and lentiviral transduction. The primers used to amplify Env gene are: Env_F: 5′CAATGTACACATGGAATTAGGCCA 3′ and Env_R: 5′ TTCTGGGTCCCCTCCTGAGGA 3′.
Cytokine Measurement
Cytokine level was measured using Milliplex MAP Human Cytokine/Chemokine Magnetic Bead panels (Millipore). A 10-plex kit for IFNγ, IL-10, IL-1β, IL-6, TNFα, IL-12(P40), IL-17, IL-2, IL-4, and IP-10, a 3-plex kit for IL-8, IL-15 and MIP1A (Cat N. HCYTOMAG-60K) and a 1-plex kit for GranzymeB (Cat. N. HCD8MAG-15K) were used. The assay was performed following manufacturer's instructions. Samples were run in a Luminex 200 system.
Alternatively, intracellular cytokine production (IFNγ and TNFα) was measured by flow cytometry. Briefly, cells were first labeled for extracellular markers CD4, CD8, and CD3 and incubated 15 min. Cells were then fixed using 1X BD lysing solution (Cat. N. 349202) and incubated for an additional 15 min. After 2 washes, cells were permeabilized using FACS buffer + 0.1% saponin, and incubated for 15 min. Cells were then incubated with anti-IFNγ and anti-TNFα, for 30 min at 4°C. After that, cells were washed in PBS and analyzed.
Small-Scale T Cell Expansions
0.5 × 106 T-cells were cultured with X-Vivo 15 Cell Medium (Cultek, Cat. N. BE02-060Q), 5% AB human serum (Sigma, Cat. N. H4522), penicillin-streptomycin (100 μg/ml) and the indicated cytokine: 50 IU/ml IL-2 (Miltenyi Biotec) or 155 IU/mL IL-7 and 290 IU/mL IL-15 (Miltenyi Biotec). Cytokines were added to the media every 48 h. Twenty-seven hours after thawing cells were activated with Dynabeads Human T-Activator CD3/CD28 (Gibco, Cat. N. 11131D) according to the manufacturer's instructions. Cells were transduced after an additional 24 h with an MOI of 10 and then expanded for 11 days at a concentration of 0.5 × 106 to 1.5 × 106 T-cells/ml.
T-Cell Expansion After Repeated Challenges With Target Cells
To analyze T-cell proliferation capacity after antigen encounter, we seeded a co-culture of CAR-T cells and NALM6 cells at 1:1 ratio (250,000 cells each). After 4 days of incubation, an aliquot of the culture was taken and analyzed to determine T-cell number. Cells were labeled with CD3, CD4, CD8, and CD19, and then 20 μl of beads (CountBright, Cat. N. C36950, Invitrogen) was added to the sample to determine absolute cell number. This process was repeated 3 times.
Statistics
Statistical significance was assessed using SPSS software. Unpaired T-test was used unless otherwise specified. U-Mann Whitney was used for comparison of variables with non-normal distributions. Statistical significance was considered when p-value ≤ 0.05.
Results
CAR T-Cell Expansion
Twenty-eight apheresis products were obtained from 27 patients included in the clinical trial. For one patient, the apheresis product was obtained twice due to ARI-0001 cell production failure (T10 and T13 products belong to the same patient). Description of apheresis products is presented in Table 2. Patients' apheresis products were subjected to CD4+ and CD8+ magnetic selection using the CliniMACS Prodigy system. In all cases except for one (Patient T27), the minimum number of T-cells (100 × 106) was obtained (Table 2). In Patient T27, cell culture was initiated with 50 × 106 cells.
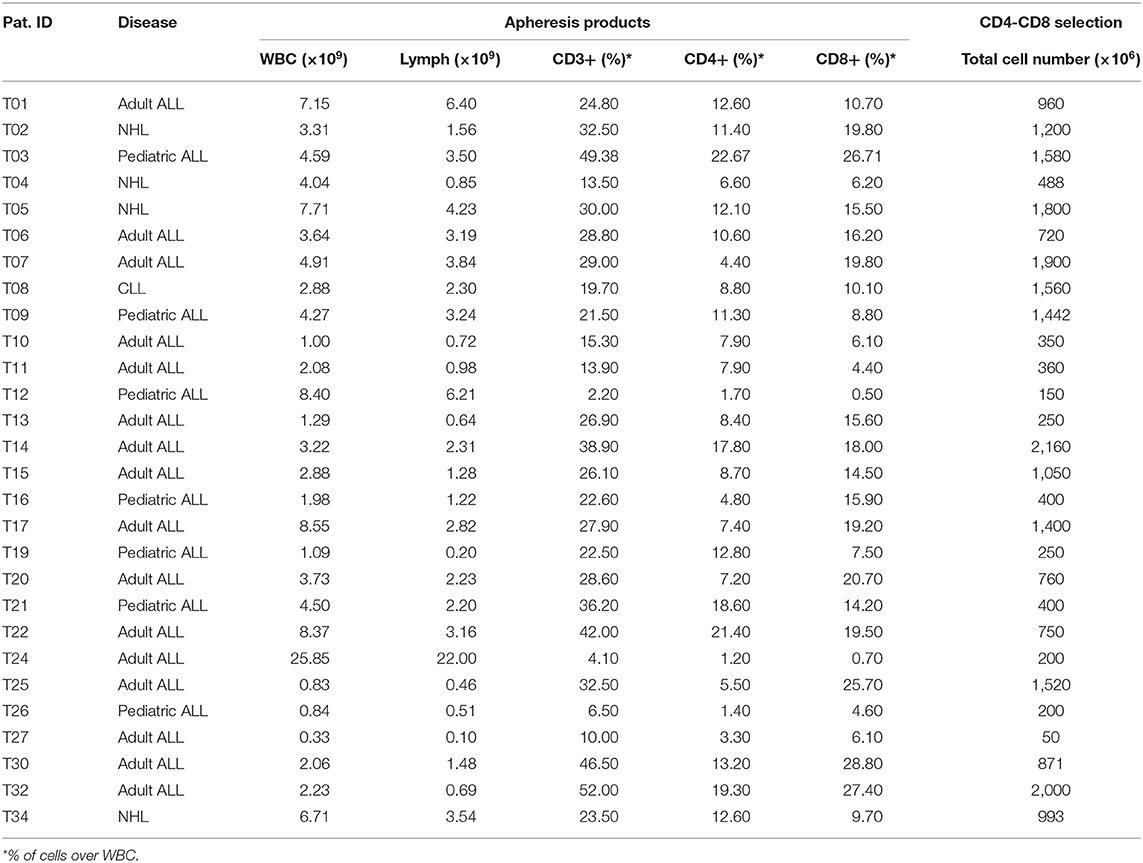
Table 2. Apheresis products composition and CD4-CD8 cell selection.
Results of cell expansion in CliniMACS Prodigy for the 27 products are presented in Figures 1A,B and Table S2. Cells were expanded for an average of 8.5 days, range 7–10. Average total cell number obtained in the final product was 2,548 × 106, range 600 × 106 to 5,200 × 106. In one patient where cell culture was started with 50 × 106 cells, the final product also met acceptance criteria. In this particular case, cell culture was maintained for 13 days, finally obtaining 3,300 × 106 cells. When compared to healthy donors (used in three previous validation runs), patient cells seem to expand more slowly, even if the number of runs performed with healthy donors is limited (Figure 1C).
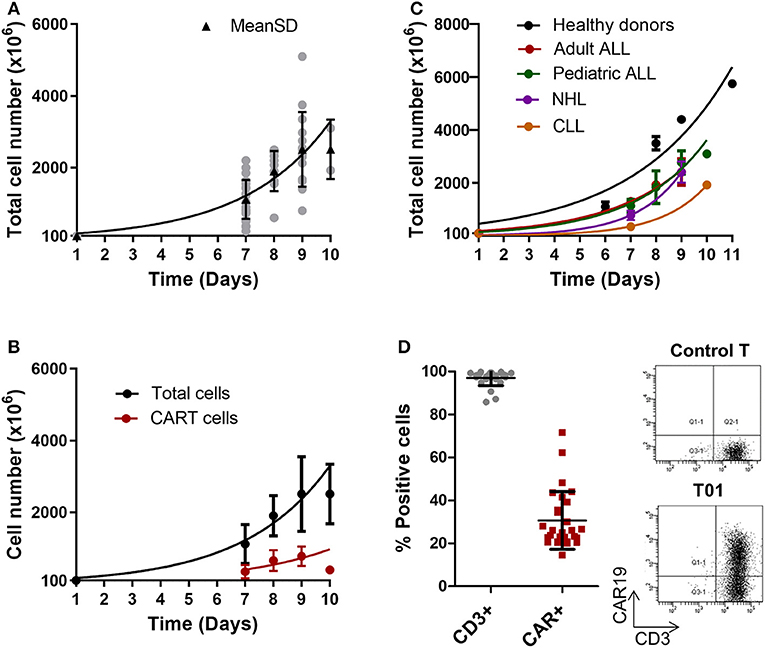
Figure 1. ARI-0001-cell expansion in CliniMACS Prodigy. (A) Expansion kinetics of ARI-0001-cell products (Total cell number). Gray points indicate individual products. Black triangles indicate Mean ± SD and adjusting curve. (B) Expansion kinetics of CAR19+ cells (red) and total cell number (black). Mean ± SD is represented. (C) Expansion kinetics of ARI-0001 cells (Total cell number) comparing healthy controls and different types of disease. Mean ± SEM is represented. (D) Percentage of CD3 and CAR19 positive cells as determined by flow cytometry. Mean ± SD is also indicated. Panels on the right show flow cytometry representative image corresponding to CAR19 and CD3 staining in ARI-0001-cell final products and Control T cells (Untransduced).
Products were analyzed in terms of appearance, quantity, identity, purity, safety, and potency. A complete list of product specifications is provided in Table S1.
Product Purity and Transduction Efficiency
The final product was characterized in terms of cell viability, percentage of CD3+ cells and percentage of CAR+ cells. This data is summarized in Table 3. All products met acceptance criteria for cell viability and percentage of CD3+ cells (>70% for both parameters). The lowest value detected for cell viability was 91 and 85.7% for CD3+ cells (Figure 1D).
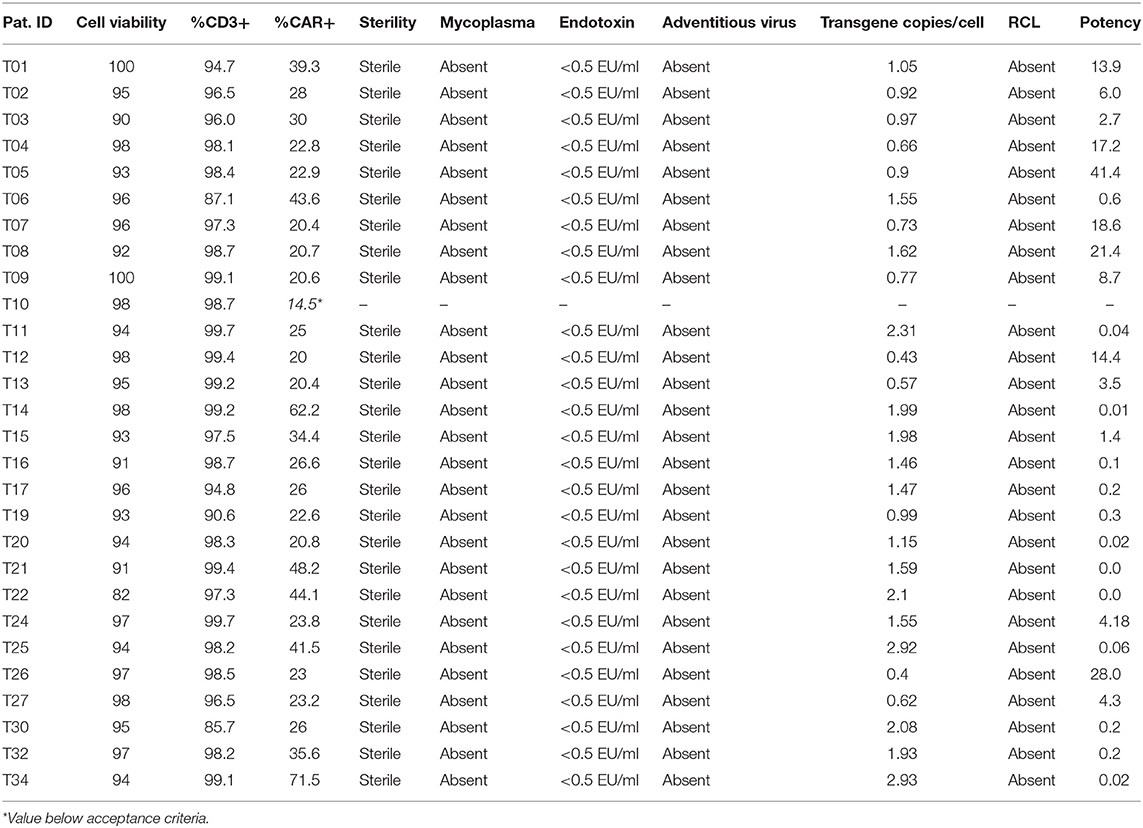
Table 3. ARI-0001 product characterization.
To analyze the percentage of CAR+ cells, we first validated our detection method based on the use of an APC-conjugated F(ab')2 anti-mouse antibody. To this end, we engineered a vector where CAR19 and GFP were co-expressed. As shown in Figure S2, the correlation between GFP+APC+ or GFP–APC– cells was of 93.5%, thereby indicating that the detection method had a good sensitivity and specificity.
Using this detection system, we assessed the percentage of CAR+ (ARI-0001) cells in the patients' products. All products except one met the specification of >20% ARI-0001 cells. In one product (T10) only 14.5% ARI-0001 cells were detected. Consequently, this product was considered a production failure. CAR T-cell production was repeated for this patient from a 2nd apheresis (T13). This time, a valid product could be obtained. Mean (±SD) of percentage of CAR+ cells in this series was 30.6 ± 13.44 (Figures 1B–D), slightly lower than transduction efficiencies achieved in small-scale expansions (45.3%) (Figure S1A). No significant differences in efficiency of transduction were observed between healthy donors and patients (35.8 vs. 30.6%), or among the different diseases (Figure S1B). Percentage of CAR+ cells over time during cell expansion was also investigated. A high degree of variability was detected among patients, with the percentage of CAR+ cells increasing in some patients while decreasing in others (Figure S3). In terms of number of cell doses obtained per patient, considering a standard weight of 70 kg for adults and 25 kg for pediatric patients, a minimum of two cell doses were quickly obtained (by day 7) for all ALL patients (dose 1 × 106 ARI-0001 cells/kg). For NHL patients (dose 5 × 106 ARI-0001 cells/kg), two cell doses were obtained for three out of four patients, by day 9. Indeed, the number of cell doses obtained for ALL far exceeded the need (nine cell doses for adult patients and 25.4 for pediatric patients), indicating that the time of ex vivo cell expansion could be reduced if necessary in these groups of patients. For NHL, the average number of ARI-0001 cell doses obtained was 2.5. Only one CLL patient has been produced so far. T-cells from this patient grew slower and required 10 days of expansion, finally obtaining 398 × 106 ARI-0001 cells.
CAR19 transduction was also assessed in terms of DNA copies/cell. As shown in Table 3 CAR19 was detected in all products, within a range of 0.4–2.9 copies/cell (all below the limit considered safe of <10 copies/cell). As expected, a positive correlation between percentage of CAR+ cells and DNA copies/cell was obtained, further validating both techniques (Figure S4).
Product Potency
Cytotoxic potential was analyzed in vitro for each product before infusion. A co-culture of the final product with NALM6 cell line was initiated at different E:T ratios. Percentage of alive-CD19+ cells was measured by flow cytometry after 4 h. As a control, the cytotoxic activity of non-transduced CD4+CD8+ cells from the same patient was also measured. Products were considered valid when the CD19+ cell surviving fraction with ARI-0001 cells, at ratio 1:1, was lower than 70%. Results are presented in Table 3 and Figure 2A. All products obtained met the specification of <70% CD19+ surviving fraction at E:T ratio 1:1, indicating that all products prepared had the intrinsic capacity of eliminating CD19+ cells.
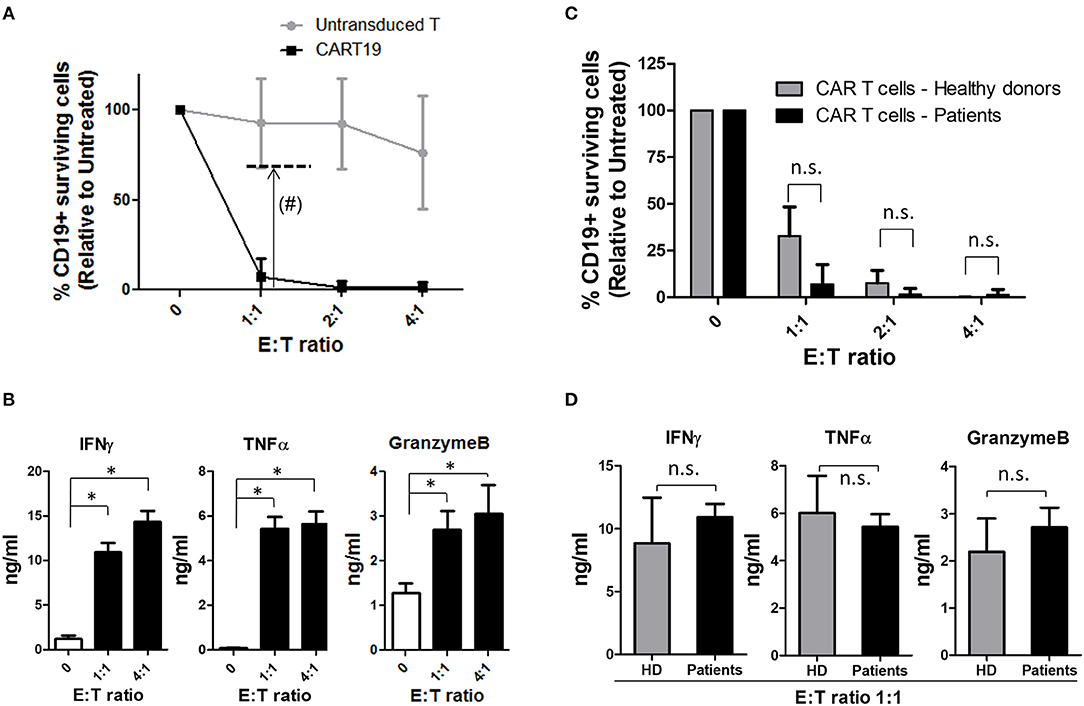
Figure 2. ARI-0001-cell potency. (A) Cytotoxicity assay after 4 h of ARI-0001 co-culture with NALM6 cells, at the indicated ratios. Mean ± SD of all 27 CAR T cell products is indicated. (#) Dashed line indicates minimum of ARI-0001-cell cytotoxicity level for a product to be considered valid. (B) IFNγ, TNFα, and GranzymeB levels measured in the supernatants of the cytotoxicity assays. E:T ratio 0 indicates no target cells. (*) indicates statistical significance, p < 0.05. (C) Comparison of ARI-0001 cytotoxic potential after 4 h of co-culture with NALM6 cells, at the indicated ratios. Mean ± SD is shown. “n.s.” indicates not statistically significant (Non parametric test). (D) Comparison of IFNγ, TNFα, and GranzymeB levels measured in the supernatants of the cytotoxicity assay at E:T ratio 1:1. “HD” indicates Healthy donors. “n.s.” indicates not statistically significant (Parametric test applied to IFNγ and TNFα and non-parametric test applied to GranzymeB).
Cytokine level was also measured in the supernatant of cytotoxicity assays. As expected, increased levels of pro-inflammatory cytokines such as IFNγ and TNFα was observed when ARI-0001 cells were co-cultured with NALM6, compared to ARI-0001 cells alone. The level of GranzymeB was also significantly increased (Figure 2B) consistent with the cytotoxic activity of ARI-0001 cells. The complete set of analyzed cytokines is provided in Figure S5.
CAR T-cells produced from patients were compared to those obtained from healthy controls in terms of cytotoxic activity and cytokine production. As shown in Figure 2C, patients' and healthy donors' CAR T-cells showed similar cytotoxic potential (even slightly higher for patient's cells although this was not statistically significant). Production of pro-inflammatory cytokines (IFNγ and TNFα) and GranzymeB was also comparable (Figure 2D).
T Cell Subset Characterization
Product composition was further analyzed in terms of CD4/CD8 ratio and TN, TSCM, TCM, TE, and TEM subsets. Consistent with previous reports (38), CD4/CD8 ratio was inverted (CD4/CD8 ratio <1) in a large subset of patients that were candidate for a CAR T-cell therapy (Figure 3A). Average CD4/CD8 ratio was 0.93 ± 0.67 in the apheresis products. This ratio was not significantly altered after CD4 and CD8 cell selection in the vast majority of patients. However, a significant increase in the proportion of CD4 cells was detected during cell expansion. CD4/CD8 ratio increased from 0.64 ± 0.61 after CD4-CD8 cell selection, to 1.61 ± 1.04 in the final product. A deeper analysis of this data reveled that in patients starting with a CD4/CD8 ratio <1, the proportion of CD4+ cells tended to increase during cell expansion, while in patients where a CD4/CD8 ratio > 1 was obtained after cell selection, the proportion of CD4+ cells tended to decrease (Figure 3B). Therefore, the difference in CD4/CD8 ratio (ΔCD4/CD8) before and after cell expansion was significantly different depending on the initial ratio (Figure S6A). The efficiency of transduction differed between CD4+ and CD8+ subsets, as CD4 showed a significantly higher percentage of CAR+ cells (Figure 3C).
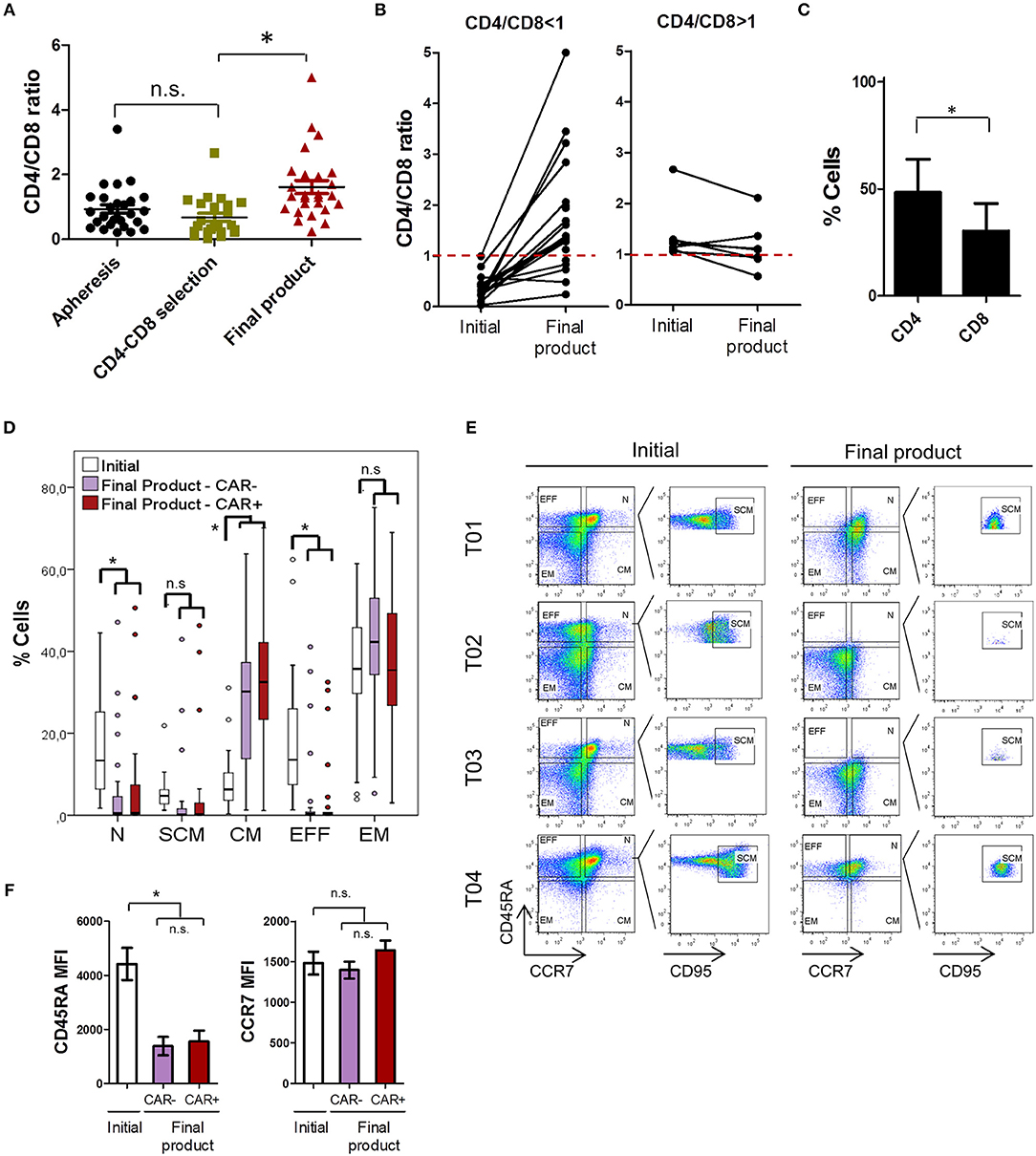
Figure 3. ARI-0001 cell subset characterization. (A) CD4/CD8 ratio of apheresis products, after CD4-CD8 cell selection and of the final product. (B) CD4/CD8 ratio variation during cell expansion. Left panel corresponds to products with an initial ratio < 1. Right panel corresponds to products with an initial ratio > 1. (C) CAR19 transduction efficiency in CD4 and CD8 cells. Mean ± SD is shown. (D) Percentage of T-cell subpopulations within initial (CD4-CD8 cell selection) and final products (CAR– and CAR+ cells). (E) Representative flow cytometry plots of three different patients showing T cell populations in initial and final products. (F) Differences in MFI for CD45RA and CCR7 in initial and final products. Lower panel shows paired analysis for CCR7 MFI. (*) indicates statistical significance, p < 0.05. n.s. indicates not statistically significant.
In terms of TN, TSCM, TCM, TE, and TEM subsets, we observed a high degree of variability among patients' final products (Figure 3D). This high variability is exemplified by the different level of CD45RA and CCR7 expression in samples from different patients (Figures 3E,F) and cannot be attributed to the different diseases (Figure S1C). Within the CAR+ T-cells of the final product, memory phenotypes (CM and EM) predominated in the vast majority of patients. Average percentage and SD for each subpopulation in the CAR+ cells of the final product is as follows: TN: 7.71 ± 13.9, TSCM: 5.26 ± 12.0, TCM: 31.01 ± 16.7, TEM: 35.11 ± 17.7, and TE: 4.2 ± 9.5. Analysis of CD4 and CD8 cells separately showed that CD8 cells have a more TN, TSCM, and TCM phenotype than CD4 cells (Figure S6B). We also analyzed how these subsets varied during ex vivo cell expansion by comparing T-cell subsets in the initial (after CD4-CD8 cell selection) and in the final product, and if CAR expression influenced T-cell subpopulations (CAR– vs. CAR+ cells). As shown in Figure 3D, we observed a robust increase in the proportion of TCM during cell expansion while TN and TEFF cells decreased. These changes in T-cell subsets can be attributed to a decrease in CD45RA expression which is expected upon cell activation (Figures 3E,F).
No statistically significant changes in the T-cell subsets were detected between CAR– and CAR+ cells in the final product, although a further increase in TCM, and consequent decrease in TEFF cells, was observed in CAR+ cells compared to CAR– (Figure 3D). Consistently, a small increase in CCR7 expression was also detected in CAR+ cells vs. CAR- cells. (Figure 3F). The impact of CAR expression on CCR7 was further explored in independent small-scale expansions (see next section). The changes in expression of CD27, CD28, and CD95 were also assessed by flow cytometry. As shown in Figure S6C, CD95 was increased during cell expansion and CD27 decreased. CD28 did not show significant changes during expansion, although presented a higher expression in CAR+ compared to CAR- cells.
Small-Scale CAR T Cell Expansions
To further evaluate the impact of culture conditions or CAR expression on the proportion of CD4/CD8 ratio or T-cell phenotype, cell expansions from patients' selected cells were repeated in a small scale experiment, under different conditions. We selected six of the patients (three adult ALL and three NHL) from which frozen leftover cells after CD4-CD8 cell selection were available. We expanded patients' cells in four different conditions: (1a) IL2—Untransduced T-cells, (1b) IL2—CAR T-cells, (2a) IL7/IL15—Untransduced T-cells, (2b) IL7/IL15—CAR T-cells. As shown in Figure S7A, cells expanded between 17 and 100 times over a period of 11 days. CAR19 transduced T-cells expanded less (or slowly) compared to untransduced counterparts, and IL2 grown cells expanded more than IL7/IL15 (in both untransduced and CAR19 conditions) (Figure S7B). Cell transduction or cytokines used did not condition CD4/CD8 ratio in a consistent way. However, as detected previously in the products expanded using the Prodigy system, in patients starting with CD4/CD8 ratio>1 (T04 and T34), the ratio tended to decrease, while in patients starting with CD4/CD8 ratio <1 (T02, T15, T22, and T34), the ratio tended to increase (Figure S7C). Indeed, since the expansions were maintained for longer in the small-scale expansions than in the Prodigy system, we observed that the ratio CD4/CD8 may fluctuate in a more or less pronounced way, but it tends to CD4/CD8 = 1 if the cells are cultured for longer periods of time.
Interestingly, significant differences were found in terms of T-cell subsets depending on the culture conditions. The cytokines used in the growth media did not provide significant differences in terms of the different subsets in this series of patients. However, a significant and consistent difference was appreciated in CAR19 expressing cells vs. untransduced T-cells for almost all subsets. As shown in Figure S7D, CAR19 transduction resulted in a much higher percentage of TN, TSCM and TCM subsets independently of the cytokine used in the culture media. By contrary, TEM cells were decreased in the CAR19+ cells compared to the untransduced samples. In this case, no difference in CD45RA MFI between untransduced and CAR19+ cells was observed that can account for the decrease in TN and TSCM, since in both conditions, cells were activated and proliferated ex vivo. However, we observed a significant increase in CCR7 expression in CAR19+ cells compared to untransduced cells (Figure S7E). This increase explains a higher percentage of TN, TSCM, and TCM subsets and lower TEM. Increase in CCR7 expression upon 4-1BB activation has been previously described in monocytes (39) and also proposed for CAR T-cells (40). To test if 4-1BB activation is responsible for the increase in CCR7 we observe in the CAR+ cells, we modified our CAR construct by changing the co-stimulatory domain to CD28 (Figure S8A). T-cells from a healthy donor were then left untransduced or transduced with the 4-1BB- or CD28-containing CARs and expanded in vitro for 10 days. Again, we observed an increase in CCR7 expression in the CAR-positive fraction of the cells transduced with the 4-1BB-containing construct, compared to untransduced cells or CD28-containing CAR+ cells (Figure S8B). As expected, percentage of TCM cells is also higher in 4-1BB-containing CAR+ cells (Figure S8C).
Finally, the functionality of CAR T-cells manufactured with the Prodigy system and small-scale expansions was also compared. For this comparison, cells from 3 patients expanded with IL-7/IL-15 were used. The production of pro-inflammatory cytokines, cytotoxic potential and T-cell expansion was measured after adjusting for the same percentage of CAR+ cells. Production of IFNγ and TNFα was measured after co-culture of CAR T-cells with NALM6 at 1:1 ratio, at 4 h time-point. Level of these two cytokines was measured both by intracellular staining (Figure S9A) and cytokines present in the media (Figure S9B), yielding consistent results. Cells manufactured in the Prodigy system consistently produced slightly more IFNγ and TNFα than cells manufactured in small-scale expansions. However, these differences were not statistically significant. In terms of cytotoxic potential, cells produced with both methods showed comparable results (Figure S9C). Finally, T-cell expansion upon repeated challenges with fresh target cells (NALM6) was slightly higher in cells manufactured with the Prodigy system than with small-scale expansions, although it did not reach statistical significance (Figure S9D). Therefore, we conclude that cells manufactured with the Prodigy system are functionally comparable, or even slightly more active, than those produced in small-scale expansions.
Taking all this data together, we conclude that ex vivo cell expansion causes a loss of TN and TEFF, which is observed in both the Prodigy system and small-scale expansions. On the contrary, TCM cells are largely accumulated, both by ex vivo expansion and as a result of CAR expression (in CARs containing 4-1BB as a co-stimulatory domain). Cells produced in Prodigy system are functionally similar to those produced in small-scale expansions.
Discussion
CAR T-cells are complex medical products. Most of the approaches explored so far are based on the use of autologous CAR T-cells, thereby requiring the production of a personalized product for each patient. Moreover, CAR T-cells are gene-therapy products, involving clinical-grade preparation of vectors, and subsequent T-cell selection, transduction and expansion. Two different CAR T-products (Yescarta, Kite-Gilead and Kymriah, Novartis) have already been approved for the treatment of pediatric-ALL and NHL. Nonetheless, the development of new CAR T-products, including phase I and II clinical trials, for many different targets and diseases is primarily conducted in academic research centers. Manufacturing of clinical-grade CAR T-cells can pose a challenge for many medium-sized academic institutions. We have previously published the development of our own CAR19 product (based on the scFv sequence of A3B1 antibody) and the establishment of a CAR T-cell production protocol and infrastructure, based on the use of CliniMACS Prodigy, a semi-automated closed system by Miltenyi (37). We and others have previously demonstrated the feasibility of CAR T-cell manufacturing from healthy donors, using CliniMACS Prodigy (41–45). Now, we report the preparation and characterization of 27 CAR T-cell products for ALL, CLL, or NHL patients.
Patients eligible for CAR T-cell therapies have previously received multiple lines of therapies. As a result, T-cells from these patients have a reduced proliferation capacity and abnormal CD4/CD8 ratio compared to healthy donors (46, 47). Nevertheless, we have been able to obtain 27 valid CAR T-cell products (according to the specifications indicated in Table S1) out of 28 attempts, proving the feasibility of CAR T-cell manufacturing for heavily pre-treated patients using the Prodigy system. Cell expansion was maintained for an average of 8.5 days. Indeed, patient-derived T-cells expand more slowly compared to the expansions performed from healthy T-cell donors. However, for ALL patients, where the infused dose is typically 1 × 106 CAR T-cells/kg, a minimum of 2 CAR T-cell doses were already achieved by day 7 in a 100% of patients, indicating that it is possible to reduce the expansion time in this subset of patients (actual expansion time was 8.4 days, due to pre-scheduled work plans). CLL and NHL patients required 9–10 days of expansion due to reduced intrinsic T-cell proliferation (48, 49) and higher CAR T-cell numbers needed, respectively. Reduced T-cell expansion time is highly desirable, not only to reduce the cost of batch production and to shorten the time until the patient can receive the treatment, but also for the quality of the product obtained, as discussed below. Despite reduced expansion capacity of heavily pretreated patient's T-cells compared to healthy donors, we have observed equivalent product potency in terms of cytotoxic potential and pro-inflammatory cytokine production. The method of CAR T-cell production (Prodigy vs. small-scale) yielded also comparable products in terms of product potency, or even slightly more active in cells obtained with Prodigy. We reasoned that the higher cell densities reached in the Prodigy system may be advantageous for T-cell fitness and potency.
Average transduction efficiency was 30.6% on average in our series and does not differ significantly between patients and healthy controls. Similar transduction efficiencies have been reported by other groups using CliniMACS Prodigy for production of CAR T-cells from healthy controls (41, 50–52), except one study that reports transduction efficiencies between 50–60% using very high MOI (MOI = 100) (44). Transduction efficiency in small-scale CAR-T cell productions is slightly higher (45.3 vs. 30.6%). We can speculate that having the possibility to put in direct contact cells and lentivirus, i.e., spinoculation, or adding reagents to the media that facilitate the entry of the lentivirus to the cells (such as polybrene) could help increase transduction efficiency in the Prodigy system.
We observed a preference of CD4+ cell expansion over CD8+ ex vivo. This is consistent with previous observations from other groups using different expansion systems and CAR constructs (53, 54). Gomes-Silva and co-workers reported a preferential expansion of CD4 cells when using a 4-1BB co-stimulatory domain compared to a CD28-containing CAR (55). Therefore, this effect may contribute partially to explain this phenomenon. Indeed, we detected a higher fraction of CD4-transduced cells than CD8. Nevertheless, we have clearly shown than CD4/CD8 ratios are highly dependent on the initial ratio and tend to equilibrium in longer ex vivo expansions, indicating that preferential CD4 expansion may be transitory and fluctuate also after CAR T-cell infusion.
TN, TSCM, and TCM phenotypes have been associated to longer persistence in vivo and higher anti-tumor efficacy (38, 56–58). We have dissected the factors that determine these T-cell populations with the aim of analyzing if our production system could be optimized. Indeed, there is a very high degree of variability in T-cell phenotypes among patients, which highly determines final product composition. In our experimental setting, both IL-7/IL-15 and IL-2 yielded comparable population phenotypes. In most patients, ex vivo cell culture led to a decrease in the fraction of CD45RA positive cells in the final product. Loss of CD45RA expression is expected upon cell activation and proliferation, and therefore intrinsic to ex vivo cell expansion, resulting in decreased numbers of TN cells (38, 40, 59, 60). In this regard, shortening the time of ex vivo cell expansion when possible may help preserve this population. TCM cells have also been shown to be able to establish persistent T-cell memory (58) and have superior anti-tumor effects (38). Therefore, the loss of TN cells may not have a major impact in anti-tumor efficacy, since most of them are converted to TCM cells. Actually, memory phenotypes are the most represented populations in our CAR T final products, consistent with previous reports (53). 4-1BB-containing CAR T-cells have been shown to have an increased proportion of TCM compared to CD28-containg CARs, which have a predominant TEM population (40, 55). We have shown that the increase in TCM cells is not only the result of CD45RA loss, but also due to increased CCR7 expression in 4-1BB-containing CAR-expressing cells [our data and Kawalekar et al. (40)]. This data correlates with a longer CAR T-cell persistence in vivo of 4-1BB- compared to CD28-containing CARs (11, 12, 49).
In conclusion, we have demonstrated for the first time the feasibility of CAR T-cell manufacturing for heavily pre-treated ALL, CLL, and NHL patients, using CliniMACS Prodigy. To our knowledge, this is the first report describing the characteristics of the products obtained with this system, including a medium-large cohort of patients. Our study shows that CAR T-cell manufacturing can be completed in as low as 7 days for ALL patients and that reduced ex vivo expansion time may yield CAR T-cell products with increased persistence in vivo. The products obtained show potent anti-tumor efficacy and are characterized by a predominance of TCM and TEM phenotypes.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
This study has been approved by the Research Ethics Comitee (CeIm) of Hospital Clinic. HCB/2017/0001. Clinical trial: CART19-BE-01. Eudra: 2016-002972-29. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
MC designed and performed experiments, analyzed data, and wrote the manuscript. MC-B and VO-M coordinated specific parts of the clinical trial and analyzed data. EG-N, GS, AA-V, AB, BM, and LM performed experiments and procedures. BM-A, JCi, ML, EG, JT, ET, UP, JCa, TB, and DB-R supervised procedures. EC critically read the manuscript. ÁU-I, JY SR, JD, and MJ coordinated and supervised the study.
Funding
This work was mainly funded by several public grants PI13/00676, PIE13/00033, PI17/01043, PICIS14/00122 and PI18/00775; included in the Plan Nacional de I + D + I and co-financed by the ISCIII—Subdirección General de Evaluación y Fomento de la Investigación Sanitaria—and the Fondo Europeo de Desarrollo Regional (FEDER), and a crowdfunding program, Projecte ARI.
Conflict of Interest
SR declares speakers bureau and travel expenses: Novartis, Shire, JazzPharma, Erytech.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank patients and families who participated in this trial and to those who made it possible. The authors also want to thank Sara Varea responsible for quality control of this clinical trial. Carolina España, Juan Ramon Ortega, Carla Guijarro, and Libertad Heredia for their technical support in CAR T-cell production. Finally, the authors thank Cristina Llanos the clinical research nurse of the pediatric patients at Hospital Sant Joan de Déu. Finally, we also want to thank the support of many people and institutions that encouraged and helped us develop this proposal in Barcelona.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00482/full#supplementary-material
References
1. Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. (2015) 263:68–89. doi: 10.1111/imr.12243
2. Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. (2014) 65:333–47. doi: 10.1146/annurev-med-060512-150254
3. Dai H, Wang Y, Lu X, Han W. Chimeric antigen receptors modified T-cells for cancer therapy. JNCI J Natl Cancer Inst. (2016) 108:djv439. doi: 10.1093/jnci/djv439
4. Sadelain M. CAR therapy: the CD19 paradigm. J Clin Invest. (2015) 125:3392–400. doi: 10.1172/JCI80010
5. Jensen MC, Riddell SR. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol Rev. (2014) 257:127–44. doi: 10.1111/imr.12139
6. Chang ZL, Chen YY. CARs: synthetic immunoreceptors for cancer therapy and beyond. Trends Mol Med. (2017) 23:430–50. doi: 10.1016/j.molmed.2017.03.002
7. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. (2010) 116:4099–102. doi: 10.1182/blood-2010-04-281931
8. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
9. Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. (2016) 127:2980–90. doi: 10.1182/blood-2015-12-686725
10. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
11. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
12. Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. (2011) 118:4817–28. doi: 10.1182/blood-2011-04-348540
13. Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. (2013) 122:4129–39. doi: 10.1182/blood-2013-08-519413
14. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RPT, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
15. Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. (2018) 36:2267–80. doi: 10.1200/JCO.2018.77.8084
16. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. (2016) 128:1688–700. doi: 10.1182/blood-2016-04-711903
17. Zhao W-H, Liu J, Wang B-Y, Chen Y-X, Cao X-M, Yang Y, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. (2018) 11:141. doi: 10.1186/s13045-018-0681-6
18. Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest. (2017) 127:3462–71. doi: 10.1172/JCI94306
19. Wang C-M, Wu Z-Q, Wang Y, Guo Y-L, Dai H-R, Wang X-H, et al. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory hodgkin lymphoma: an open-label phase I trial. Clin Cancer Res. (2017) 23:1156–66. doi: 10.1158/1078-0432.CCR-16-1365
20. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. (2017) 24:20–8. doi: 10.1038/nm.4441
21. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183
22. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
23. Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res. (2015) 21:3149–59. doi: 10.1158/1078-0432.CCR-14-1421
24. Beatty GL, O'Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. (2018) 155:29–32. doi: 10.1053/j.gastro.2018.03.029
25. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
26. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
27. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
28. Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. (2013) 21:904–12. doi: 10.1038/mt.2013.17
29. Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. (2006) 24:e20–2. doi: 10.1200/JCO.2006.05.9964
30. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
31. Knochelmann HM, Smith AS, Dwyer CJ, Wyatt MM, Mehrotra S, Paulos CM. CAR T cells in solid tumors: blueprints for building effective therapies. Front Immunol. (2018) 9:1740. doi: 10.3389/fimmu.2018.01740
32. Long KB, Young RM, Boesteanu AC, Davis MM, Melenhorst JJ, Lacey SF, et al. CAR T cell therapy of non-hematopoietic malignancies: detours on the road to clinical success. Front Immunol. (2018) 9:2740. doi: 10.3389/fimmu.2018.02740
33. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. (2010) 24:1160–70. doi: 10.1038/leu.2010.75
34. Chmielewski M, Abken H. CAR T cells releasing IL-18 convert to T-Bethigh FoxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. (2017) 21:3205–19. doi: 10.1016/j.celrep.2017.11.063
35. Krenciute G, Prinzing BL, Yi Z, Wu M-F, Liu H, Dotti G, et al. Gottschalk S. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. (2017) 5:571–81. doi: 10.1158/2326-6066.CIR-16-0376
36. Chmielewski M, Abken H. CAR T cells transform to trucks: chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother. (2012) 61:1269–77. doi: 10.1007/s00262-012-1202-z
37. Castella M, Boronat A, Martín-Ibáñez R, Rodríguez V, Suñé G, Caballero M, et al. Development of a novel anti-CD19 chimeric antigen receptor: a paradigm for an affordable CAR T cell production at academic institutions. Mol Ther Methods Clin Dev. (2019) 12:134–44. doi: 10.1016/j.omtm.2018.11.010
38. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. (2016) 30:492–500. doi: 10.1038/leu.2015.247
39. Kwajah MMS, Schwarz H. CD137 ligand signaling induces human monocyte to dendritic cell differentiation. Eur J Immunol. (2010) 40:1938–49. doi: 10.1002/eji.200940105
40. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
41. Aleksandrova K, Leise J, Priesner C, Melk A, Kubaink F, Abken H, et al. Functionality and cell senescence of CD4/ CD8-selected CD20 CAR T cells manufactured using the automated CliniMACS Prodigy® platform. Transfus Med Hemotherapy. (2019) 46:47–54. doi: 10.1159/000495772
42. Marín Morales JM, Münch N, Peter K, Freund D, Oelschlägel U, Hölig K, et al. Automated clinical grade expansion of regulatory T cells in a fully closed system. Front Immunol. (2019) 10:38. doi: 10.3389/fimmu.2019.00038
43. Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. (2016) 3:16015. doi: 10.1038/mto.2016.15
44. Zhang W, Jordan KR, Schulte B, Purev E. Characterization of clinical grade CD19 chimeric antigen receptor T cells produced using automated CliniMACS prodigy system. Drug Des Devel Ther. (2018) 12:3343–56. doi: 10.2147/DDDT.S175113
45. Priesner C, Aleksandrova K, Esser R, Mockel-Tenbrinck N, Leise J, Drechsel K, et al. Automated enrichment, transduction, and expansion of clinical-scale CD62L+ T cells for manufacturing of gene therapy medicinal products. Hum Gene Ther. (2016) 27:860–9. doi: 10.1089/hum.2016.091
46. Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, et al. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood. (1997) 89:3700–7. doi: 10.1182/blood.V89.10.3700.3700_3700_3707
47. Mackall C, Fleisher T, Brown M, Magrath I, Shad A, Horowitz M, et al. Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood. (1994) 84:2221–8. doi: 10.1182/blood.V84.7.2221.2221
48. Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. (2016) 127:1117–27. doi: 10.1182/blood-2015-11-679134
49. Porter DL, Hwang W-T, Frey N V, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
50. Zhu F, Shah NN, Xu H, Schneider D, Orentas R, Dropulic B, et al. CAR-T cell production using the Clinimacs® Prodigy system. Blood. (2016) 128:5724. doi: 10.1182/blood.V128.22.5724.5724
51. Mock U, Nickolay L, Philip B, Cheung GWK, Zhan H, Johnston ICD, et al. Automated manufacturing of chimeric antigen receptor T cells for adoptive immunotherapy using CliniMACS prodigy. Cytotherapy. (2016) 18:1002–11. doi: 10.1016/j.jcyt.2016.05.009
52. Blaeschke F, Stenger D, Kaeuferle T, Willier S, Lotfi R, Kaiser AD, et al. Induction of a central memory and stem cell memory phenotype in functionally active CD4+ and CD8+ CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19+ acute lymphoblastic leukemia. Cancer Immunol Immunother. (2018) 67:1053–66. doi: 10.1007/s00262-018-2155-7
53. Blaeschke F, Kaeuferle T, Feucht J, Weber D, Lotfi R, Kaiser A, et al. Defined central memory and stem memory T cell phenotype of CD4 and CD8 CAR T cells for the treatment of CD19+ acute lymphoblastic leukemia in an automated closed system. Blood. (2016) 128:1053–66. doi: 10.1182/blood.V128.22.4558.4558
54. Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. (2009) 32:169–80. doi: 10.1097/CJI.0b013e318194a6e8
55. Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, et al. Tonic 4-1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector-dependent. Cell Rep. (2017) 21:17–26. doi: 10.1016/j.celrep.2017.09.015
56. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. (2011) 17:1290–7. doi: 10.1038/nm.2446
57. Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. (2011) 117:808–14. doi: 10.1182/blood-2010-05-286286
58. Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. (2008) 118:294–305. doi: 10.1172/JCI32103
59. Larbi A, Fulop T. From “truly naïve” to “exhausted senescent” T cells: when markers predict functionality. Cytom Part A. (2014) 85:25–35. doi: 10.1002/cyto.a.22351
Keywords: chimeric antigen receptor, CD19, leukemia, lymphoma, immunotherapy, CAR T-cell production, CliniMACS Prodigy
Citation: Castella M, Caballero-Baños M, Ortiz-Maldonado V, González-Navarro EA, Suñé G, Antoñana-Vidósola A, Boronat A, Marzal B, Millán L, Martín-Antonio B, Cid J, Lozano M, García E, Tabera J, Trias E, Perpiña U, Canals JM, Baumann T, Benítez-Ribas D, Campo E, Yagüe J, Urbano-Ispizua Á, Rives S, Delgado J and Juan M (2020) Point-Of-Care CAR T-Cell Production (ARI-0001) Using a Closed Semi-automatic Bioreactor: Experience From an Academic Phase I Clinical Trial. Front. Immunol. 11:482. doi: 10.3389/fimmu.2020.00482
Received: 27 September 2019; Accepted: 02 March 2020;
Published: 20 March 2020.
Edited by:
Rayne Rouce, Baylor College of Medicine, United StatesReviewed by:
Pappanaicken R. Kumaresan, University of Texas MD Anderson Cancer Center, United StatesSanjivan Gautam, National Cancer Institute, National Institutes of Health (NIH), United States
Copyright © 2020 Castella, Caballero-Baños, Ortiz-Maldonado, González-Navarro, Suñé, Antoñana-Vidósola, Boronat, Marzal, Millán, Martín-Antonio, Cid, Lozano, García, Tabera, Trias, Perpiña, Canals, Baumann, Benítez-Ribas, Campo, Yagüe, Urbano-Ispizua, Rives, Delgado and Juan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manel Juan, bWp1YW5AY2xpbmljLmNhdA==; Maria Castella, bWNhc3RlbGxhQGNsaW5pYy5jYXQ=
†These authors have contributed equally to this work
‡These authors share senior authorship