 Nobuo Kanazawa
Nobuo Kanazawa- Department of Dermatology, Wakayama Medical University, Wakayama, Japan
“Autoinflammatory disease (AiD)” has first been introduced in 1999 when the responsible gene for the familial Hibernean fever or autosomal dominant-type familial Mediterranean fever-like periodic fever syndrome was reportedly identified as tumor necrosis factor receptor superfamily 1. Linked with the rapid research progress in the field of innate immunity, “autoinflammation” has been designated for dysregulated innate immunity in contrast to “autoimmunity” with dysregulated acquired immunity. As hereditary periodic fever syndromes represent the prototype of AiD, monogenic systemic diseases are the main members of AiD. However, skin manifestations provide important clinical information and there are even some AiDs originating from skin diseases. Recently, AiD showing psoriasis and related keratinization diseases have specifically been designated as “autoinflammatory keratinization diseases (AiKD)” and CARD14-associated psoriasis and deficiency of interleukin-36 receptor antagonist previously called as generalized pustular psoriasis are included. Similarly, a number of autoinflammatory skin diseases can be proposed; autoinflamatory urticarial dermatosis (AiUD) such as cryopyrin-associated periodic syndrome; autoinflammatory neutrophilic dermatosis (AiND) such as pyogenic sterile arthritis, pyoderma gangrenosm, and acne syndrome; autoinflammatory granulomatosis (AiG) such as Blau syndrome; autoinflammatory chilblain lupus (AiCL) such as Aicardi-Goutieres syndrome; autoinflammatory lipoatrophy (AiL) such as Nakajo-Nishimura syndrome; autoinflammatory angioedema (AiAE) such as hereditary angioedema; and probable autoinflammatory bullous disease (AiBD) such as granular C3 dermatosis. With these designations, skin manifestations in AiD can easily be recognized and, even more importantly, autoinflammatory pathogenesis of common skin diseases are expected to be more comprehensive.
Introduction
“Autoinflammatory disease (AiD)” has first been introduced in 1999 when the responsible gene for the familial Hibernean fever or autosomal dominant-type familial Mediterranean fever (FMF)-like periodic fever syndrome was reportedly identified as tumor necrosis factor receptor superfamily 1 (TNFRSF1) (1). Linked to the rapid progress in the research field of innate immunity, “autoinflammation” has been applied for dysregulated innate immunity and inflammation, in contrast to “autoimmunity” with dysregulated acquired immunity. Notably, while “auto” in autoimmunity clearly means the “self,” the “auto” in autoinflammation might originate from “automatic” or “autonomous” and, in this context, AiD has been expanded to a number of inflammatory diseases with genetic or idiopathic origin (2).
Although monogenic systemic diseases such as FMF and related periodic fever syndromes are the main members of AiD, some of the AiD members originate from skin diseases, in which characteristic skin manifestations provide clinically important information. For example, the mildest form of cryopyrin-associated periodic syndrome (CAPS) had been originally called as familial cold urticaria and, actually, the skin eruption of CAPS is hard to be distinguished from urticaria. Recent identification of CARD14 as the psoriasis susceptible gene 2 and IL36RN mutations in generalized pustular psoriasis have defined new entities, CARD14-associated psoriasis (CAMPS) and deficiency of interleukin-36 receptor antagonist (DITRA), respectively. Accordingly, “autoinflammatory keratinization diseases (AiKD)” has specifically been designated for AiD showing psoriasis and related keratinization diseases (3).
From a dermatological point of view, designation of AiKD has given a new concept of autoinflammation in so far-called inflammatory keratinization disease, which is one of the major category of chronic skin diseases. Especially, psoriasis has been a great challenge for dermatologists and there was a debate whether psoriasis is an immune disease or a keratinization disease. Application of cyclosporine revealed the major role of T cells and confirmed that psoriasis was an immune disease. Subsequent development of highly effective anti-cytokine therapy has revealed the critical role of specific cytokine cascades in psoriasis. The concept of AiD can support such clinical evidence which might not be explained by antigen-specific helper T cell-mediated autoimmunity.
Similarly, each characteristic skin manifestation of various AiD can be linked to a specific category of chronic inflammatory skin diseases and, therefore, a number of autoinflammatory skin diseases other than AiKD can be proposed as shown in Table 1; autoinflamatory urticarial dermatosis (AiUD) such as CAPS; autoinflammatory neutrophilic dermatosis (AiND) such as pyogenic sterile arthritis, pyoderma gangrenosm, and acne (PAPA) syndrome; autoinflammatory granulomatosis (AiG) such as Blau syndrome; autoinflammatory chilblain lupus (AiCL) such as Aicardi-Goutieres syndrome (AGS); autoinflammatory lipoatrophy (AiL) such as Nakajo-Nishimura syndrome (NNS); autoinflammatory angioedema (AiAE) such as hereditary angioedema (HAE); and probable autoinflammatory bullous disease (AiBD) such as granular C3 dermatosis (GCD). With these designations, skin manifestations in AiD can easily be distinguished by all physicians. In addition, even more importantly, autoinflammatory pathogenesis of common skin diseases is expected to be more comprehensive like AiKD for dermatologists.
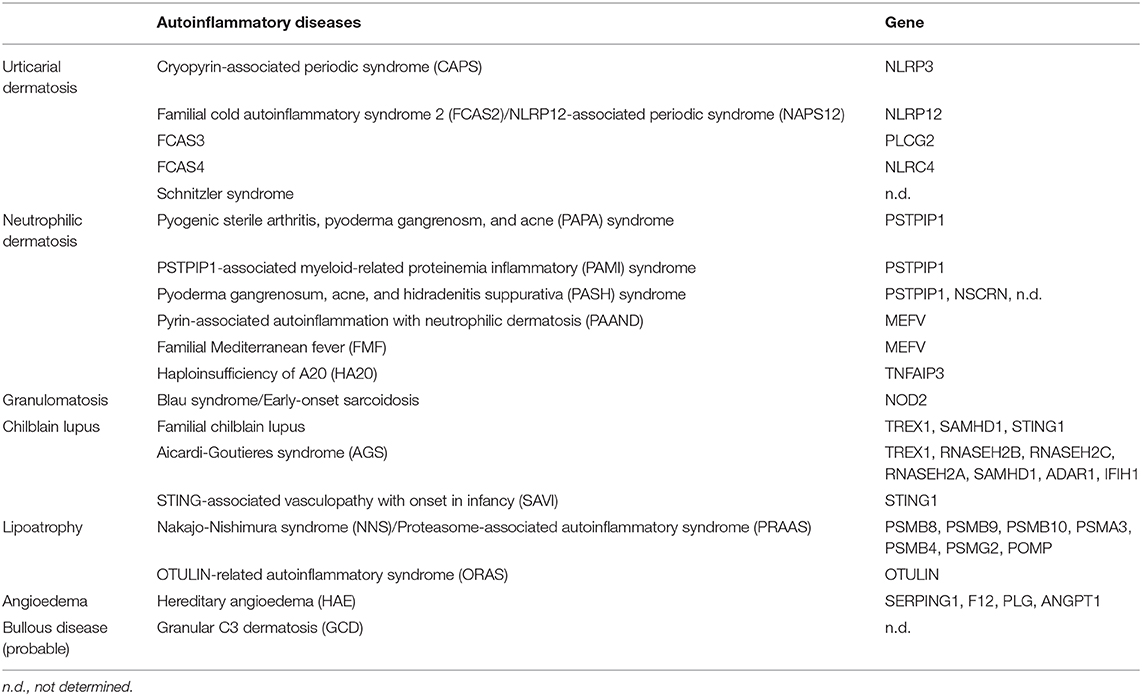
Table 1. Categorized autoinflammatory skin manifestations.
AiUD (Autoinflamatory Urticarial Dermatosis)
Urticaria is a kind of skin rash with red, raised and mostly itchy wheal. Each rash last for a few hours to days but disappear without any skin changes. The rashes move around in the whole body and completely disappear after several days. Skin mast cell-derived histamine and other mediators such as prostaglandins and cytokines are responsible and allergic (IgE-mediated) and non-allergic (mechanical, solar light, cold, water, stress, etc.) origins are related. Histologically, various stages of change can be observed from just dermal edema to vasculitis with inflammatory cell infiltration. Whereas, infection-induced urticaria is accompanied with fever and upregulation of inflammatory biomarkers, most urticaria does not show systemic inflammation other than airway and intestinal edema and anaphylaxis. Notably, <5% of the patients develop idiopathic chronic urticaria which lasts for longer than 4 weeks. In this type, urticaria appears spontaneously almost every day without apparent trigger and significantly decreases the patient's quality of life. For intractable cases, anti-IgE therapy with omalizumab is effective. Although the mechanism how omalizumab exerts its effect is not fully understood, a role of IgE on the maintenance and enhancement of mast cell activities has been suggested (4).
AiUD is linked to CAPS, including familial cold-induced autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS) and chronic infantile neurological cutaneous and articular (CINCA) syndrome, with NLRP3 mutations, FCAS2 with NLRP12 mutations, FCAS3 with PLCG2 mutations, FCAS4 with NLRC4 mutations, and Schnitzler syndrome without monogenic background (5). Interestingly, cold can be a trigger in all diseases, while “evaporative” cold is specifically involved in FCAS3. Except for PLCG2 mutations which directly affect mast cell activation through dysregulated phospholipase Cγ, mutations in NOD-like receptor (NLR) family molecules cause inflammasome activation in monocytes and secreted IL-1β/IL-18 lead to fever, vascular leakage and neutrophilia. The fact that NLR molecules react to various pathogen-associated molecular patterns may explain the clinical similarity of AiUD and infection-induced neutrophilic urticaria.
AiND (Autoinflammatory Neutrophilic Dermatosis)
Neutrophilic dermatosis is another category of inflammatory skin diseases characterized by aseptic accumulation of neutrophils in the skin. The representative Sweet's syndrome is also called as acute febrile neutrophilic dermatosis and can be secondary to myelodysplastic syndrome. Another major disease, pyoderma gangrenosum, typically shows intractable skin ulceration, which mostly occurs secondary to Crohn's disease, ulcerative colitis, Takayasu's arteritis, rheumatoid arthritis, and systemic lupus erythematosus. Thus, neutrophilic dermatosis can be developed with hyperactivation of neutrophils accompanied with myeloproliferative disorders, chronic inflammatory diseases or rheumatic diseases. Pathergy can be commonly observed and is considered diagnostic.
This category can be expanded to include pustular psoriasis, palmoplantar pustulosis, subcorneal pustulosis, hidradenitis suppurativa, severe acne, folliculitis decalvans, erosive pustular dermatosis of the scalp, neutrophilic urticarial dermatosis, erythema elevatum diutinum, and Behcet's disease. However, hidradenitis suppurativa and severe acne are associated with abnormal follicular keratinization and are rather categorized in AiKD (6), like pustular psoriasis and palmoplantar pustulosis.
Specific AiND is linked to PAPA syndrome with PSTPIP1 mutations. PAPA syndrome mostly starts with pyogenic sterile arthritis in childhood and develops cystic acne and pyoderma gangrenosum in adolescence. Recently, severer cases showing growth failure, pancytopenia, hepatosplenomegaly with hyperzincemia and hypercalprotectinemia have been identified to be associated with particular PSTPIP1 mutations and designated as PSTPIP1-associated myeloid-related proteinemia inflammatory (PAMI) syndrome (7). On the other hand, cases with pyoderma gangrenosum, acne, and hidradenitis suppurativa without arthritis have been reported as PASH syndrome and PSTPIP1 mutations have been identified in some cases (8). Thus, hidradenitis suppurativa can be included in AiND.
PSTPIP1 directly associates with pyrin, whose mutations are responsible for familial Mediterranean fever (FMF), and PAPA/PAMI-causing PSTPIP1 mutations increase pyrin activity to cause autoinflammation. The fact that other cases with the same triads as PAPA syndrome have been identified to be associated with particular MEFV mutations indicates that PSTPIP1 and pyrin are located on the same signaling pathway. They are designated as pyrin-associated autoinflammation with neutrophilic dermatosis (PAAND) (9). Even FMF itself, in which erysipelas-like rash can be rarely but characteristically seen, can be linked to AiND.
More recently, in cases with familial Behcet's disease, heterozygous TNFAIP3 mutations have been identified and the designation haploinsufficiency of A20 (HA20) has been given (10). A20 is a negative regulator of TNFα signaling with dual activities of ubiquitin ligase and deubiquitinase. A20 also regulates B cell receptor and T cell receptor signaling and HA20 can additionally show autoimmune disorders.
AiG (Autoinflammatory Granulomatosis)
Sarcoidosis has been defined as a systemic granulomatosis with unknown etiology. Although histopathological finding of sarcoid-type non-caseating epithelioid cell granuloma is characteristic, the most recent diagnostic criteria does not necessarily require biopsy. The concept that latent infection with Propionibacterium acnes has a causative role is widely accepted at least in Japan (11). Cutaneous sarcoidosis shows various manifestations, including maculopapular, plaque, nodular, subcutaneous, lupus pernio, nodular erythema-like, scar, and rare variants such as angiolipoid, psoriasiform, lichenoid, ichthyosiform, erythrodermic, ulcerative, and non-specific nodular erythema.
Sarcoid-type granuloma with Langhans-type giant cell formation is considered as T cell-mediated super delayed-type hypersensitivity reaction to indigestible antigen. It has been reported that sarcoid-type granuloma can be induced by metal allergy, even after usual closed patch test (12). In contrast, granuloma annulare with palisading granuloma is caused by a reaction to denatured collagen and in case of annular elastolytic giant cell granuloma, undigested elastin can be found in foreign body-type giant cells.
Specific AiG is linked to Blau syndrome with NOD2 mutations. Blau syndrome show sarcoid-type granuloma with characteristic skin, eye and joint involvement, but without hilar lymphadenopathy. Sporadic early-onset sarcoidosis shows the same clinical triad with NOD2 mutations and are considered the same disease as Blau syndrome (13). It is understandable that Blau syndrome mimics intracellular bacterial infection because NOD2 acts as an intracellular sensor for muramyl dipeptide, the common component of bacterial cell wall peptidoglycan. However, it remains unclear whether any indigestible antigen and T cells are required for granuloma formation and why skin, eye and joint are specifically affected in this disease.
AiCL (Autoinflammatory Chilblain Lupus)
Chilblain lupus is a kind of cutaneous lupus erythematosus and shows pernio (chilblain)-like eruption with vasculopathy in cold seasons. Among chilblain lupus patients, familial occurrence is present and heterozygous mutations in TREX1 (14), SAMHD1, and STING1 have been identified to be responsible. Three prime repair exonuclease 1 (TREX1) has a 3′-5′ exonuclease activity and its mutations are associated with systemic lupus erythematosus (15).
The most typical AiCL is linked to AGS, a hereditary early-onset encephalitis accompanied by basal ganglia calcification and pernio-like rash. This disease is classified to 7 types based on their responsible genes; TREX1, RNASEH2B, RNASEH2C, RNASEH2A, SAMHD1, ADAR1, and IFIH1. Most of them have DNA/RNA degrading or modifying activities and their dysfunction leads to accumulation of intracellular DNA/RNA. While intracellular dsRNA is recognized by MDA5 encoded by IFIH1, dsDNA activates cyclic GMP-AMP synthase (cGAS) and synthesized cGMP-cAMP (cGAMP) binds to an adaptor molecule, STING (stimulator of interferon genes). Heterozygous STING1 mutations cause another severer AiCL, STING-associated vasculopathy with onset in infancy (SAVI) showing interstitial lung disease and ulcerating skin lesions. MDA5 or STING stimulation activates IFNα/β expression through IRF (IFN-regulatory factor) and secreted IFNα/β binds to its receptor to induce expression of IFN-stimulated genes through Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway in the autocrine or paracrine manner. Recently, effect of JAK1/2 inhibitor baricitinib on these autoinflammatory interferonopathies has been reported (16).
AiL (Autoinflammatory Lipoatrophy)
NNS was originally reported as “secondary hyperperiostosis with pernio” in 1939 and had long been unique in Japan (17). Not only pernio-like rash and basal ganglia calcification, but progressive partial lipomuscular atrophy with long clubbed fingers are characteristic. In 2010, joint contractures, muscular atrophy, microcytic anemia, and panniculitis-induced lipodystrophy (JMP) syndrome was reported by experts of lipodystrophy and a homozygous PSMB8 mutation causing immunoproteasome dysfunction was identified as responsible. Now NNS, JMP syndrome and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome are collectively called as proteasome-associated autoinflammatory syndrome (PRAAS). In PRAAS, type I interferonopathy is considered responsible but the mechanism how immunoproteasome dysfunction causes interferonopathy has not been defined. Progressive lipoatrophy is observed in all PRAAS cases but not in AiCL cases. Although immunoproteasome dysfunction reportedly disturbs adipocyte differentiation (18), effect of JAK inhibitor on lipoatrophy has suggested that interferonopathy itself might be responsible for lipoatrophy (16).
Another AiL is linked to OTULIN-related autoinflammatory syndrome (ORAS) (19). OTU deubiquinase with linear linkage specificity (OTULIN) is a negative regulator of linear ubiquitin chain assembly complex (LUBAC)-mediated NF-κB activation. Lipoatrophy in ORAS is considered panniculitis-associated and similar to PRAAS.
AiAE (Autoinflammatory Angioedema)
Quinke's AE is a localized non-pitting edema which rapidly develops and lasts for several days. Eyelids, lips, throat and digestive tract are mainly affected. Long-lasting facial edema impairs the patient's quality of life and pharyngeal edema is life-threatening. Abdominal pain by digestive tract edema is difficult to diagnose and the patient can be misdiagnosed as acute abdomen to be applied for surgical treatment. Similar to urticaria, allergic and non-allergic origins are causable and, especially, angiotensin converting enzyme (ACE) inhibitor can be a trigger. Anti-histamines, and corticosteroids are ineffective and tranexamic acid is widely used.
Among AE cases, hereditary form has been designated as HAE and impairment of complement component 1 inhibitor (C1-INH) have been identified to be responsible (5). While cases with impaired C1-INH quantity and activity have been defined as HAE type 1, those with impaired C1-INH activity and its intact quantity has been defined as HAE type 2 and those with intact C1-INH activity have been classified as HAE type 3. In cases with type 1 and type 2 HAE, replacement of recombinant C1-INH is critical. While HAE type 1 and type 2 are associated with C1-INH-encoding SERPING1 mutations, type 3 HAE is not associated with these mutations and, by analyses of unrelated cases, F12 encoding coagulation Factor XII (20), and more recently, mutations in PLG encoding Plasminogen (21) and ANGPT1 encoding angiopoietin-1 (22) have been reportedly identified. C1-INH regulates not only the complement system, but also the coagulation and kallikrein-kinin systems. Among them, bradykinin have been identified as the final mediator causing angioedema. Factor XII is the first activator of the kallikrein-kinin system and ACE inhibitor inhibits bradykinin degradation. Therefore, the bradykinin B2 receptor antagonist icatibant has been developed as a new drug for HAE (5). Thus, HAE is no longer just a complement disorder, but defined as AiAE with bradykinin overactivation.
AiBD (Autoinflammatory Bullous Disease)
Autoimmune bullous diseases are the major tissue-specific autoimmune diseases mediated by skin antigen-specific autoantibodies. In case of pemphigus, anti-desmoglein antibodies damage the inter-keratinocytes adhesion to develop intraepidermal blister. In case of pemphigoid and epidermal bullosa aquisita, anti-collagen antibodies damage the keratinocytes-dermis adhesion in the basement membrane zone (BMZ) to develop subepidermal blister. In contrast, in dermatitis herpetiformis patients, detection of dot-like IgA deposition just beneath the BMZ is characteristic (23). As this disease is associated with celiac disease with gluten allergy, cross-reactivity of tissue transglutaminase acting on gliadin in digestive tract and epidermal transglutaminase (eTG) is postulated. However, celiac disease is almost absent in Japan and it remains unclear how anti-eTG IgA antibodies are generated to form dot-like subepidermal deposition in Japanese dermatitis herpetiformis patients.
Recently, among patients with clinically bullous diseases, those who showed dot-like subepidermal deposition of only C3 without any antibodies by direct immunofluorescence of their biopsy specimens have been collected and the designation GCD has been proposed (24). Although precise pathomechanism needs further investigation, gluten/eTG components for dot formation and dysregulation of antibody-independent pathway for C3 activation might be involved. In case of C3 glomerulopathy, genetic or acquired dysfunction of regulatory factors of the complement alternative pathway is known to be associated and effect of anti-C5 antibody eculizumab has been reported (25). Accordingly, C3 glomerulopathy with genetic abnormality can be considered an AiD with dysregulated complement activation. Based on the disease similarity, probable AiBD is applied for GCD as a new frontier of AiD, while its genetic background remains to be elucidated.
Outlook
In this review, representative AiD has been introduced according to a classification by characteristic skin manifestations. Following the designation of AiKD as a breakthrough, specific skin manifestation has been adapted to some specific genetic background according to the phenotype-genotype correlation in AiD. Defining the affected specific signaling pathway would provide a clue to understand the pathogenesis of idiopathic or spontaneous skin diseases and give more choice on selecting treatments.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported in part by Grants from the Ministry of Health, Labour, and Welfare, Wakayama Medical University Special Grant-in-Aid for Research Projects, and Lydia O'leary Memorial Pias Dermatological Foundation.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. (1999) 97:133–44. doi: 10.1016/S0092-8674(00)80721-7
2. Kanazawa N, Tchernev G, Wollina U. Autoimmunity versus autoinflammation - friend or foe? Win Med Wochenschr. (2014) 164:274–77. doi: 10.1007/s10354-014-0290-0
3. Akiyama M, Takeichi T, McGrath JA, Sugiura K. Autoinflammatory keratinizeation diseases. J Allergy Clin Immunol. (2017) 140:1545–7. doi: 10.1016/j.jaci.2017.05.019
4. Chang TW, Chen C, Lin CJ, Metz M, Church MK, Maurer M. The potential pharmacologic mechanisms of omalizumab in patients with chronic spontaneous urticaria. J Allergy Clin Immunol. (2015) 135:337–42. doi: 10.1016/j.jaci.2014.04.036
5. Kanazawa N. Hereditary disorders presenting with urticaria. Immunol Allergy Clin North Am. (2014) 34:169–79. doi: 10.1016/j.iac.2013.08.001
6. Takeichi T, Matsumoto T, Nomura T, Takeda M, Niwa H, Kono M, et al. A novel NCSTN missense mutation in the signal peptide domain causes hidradenitis suppurativa, which has features characteristic of an autoinflammatory keratinization disease. Br J Dermatol. (2020) 182:491–3. doi: 10.1111/bjd.18445
7. Holzinger D, Fassl SK, de Jager W, Lohse P, Röhrig UF, Gattorno M, et al. Single amino acid charge switch defines clinically distinct proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1)-associated inflammatory diseases. J Allergy Clin Immunol. (2015) 136:1337–45. doi: 10.1016/j.jaci.2015.04.016
8. Saito N, Minami-Hori M, Nagahata H, Nozaki H, Iinuma S, Igawa S, et al. Novel PSTPIP1 gene mutation in pyoderma gangrenosum, acne and suppurative hidradenitis syndrome. J Dermatol. (2018) 45:e213–4. doi: 10.1111/1346-8138.14259
9. Masters SL, Lagou V, Jéru I, Baker PJ, Van Eyck L, Parry DA, et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med. (2016) 8:332ra45. doi: 10.1126/scitranslmed.aaf1471
10. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. (2016) 48:67–73. doi: 10.1038/ng.3459
11. Eishi Y. Etiologic link between sarcoidosis and Propionibacterium acnes. Respir Investig. (2013) 51:56–68. doi: 10.1016/j.resinv.2013.01.001
12. Casper C, Groth W, Hunzelmann N. Sarcoidal-type allergic contact granuloma: a rare complication of ear piercing. Am J Dermatopathol. (2004) 26:59–62. doi: 10.1097/00000372-200402000-00008
13. Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. (2005) 105:1195–7. doi: 10.1182/blood-2004-07-2972
14. Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. (2007) 80:811–5. doi: 10.1086/513443
15. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3-prime-5-prime DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. (2007) 39:1065–7. doi: 10.1038/ng2091
16. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. (2018) 128:3041–52. doi: 10.1172/JCI98814
17. Kanazawa N. Nakajo-Nishimura syndrome: an autoinflammatory disorder showing pernio-like rashes and progressive partial lipodystrophy. Allergol Int. (2012) 61:197–206. doi: 10.2332/allergolint.11-RAI-0416
18. Arimochi H, Sasaki Y, Kitamura A, Yasutomo K. Differentiation of preadipocytes and mature adipocytes requires PSMB8. Sci Rep. (2016) 6:26791. doi: 10.1038/srep26791
19. Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell. (2016) 166:1215–30. doi: 10.1016/j.cell.2016.07.019
20. Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. (2006) 343:1286–9. doi: 10.1016/j.bbrc.2006.03.092
21. Bork K, Wulff K, Steinmuller-Magin L, Braenne I, Staubach-Renz P, Witzke G, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy. (2018) 73:442–50. doi: 10.1111/all.13270
22. Bafunno V, Firinu D, D'Apolito M, Cordisco G, Loffredo S, Leccese A, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. (2018) 141:1009–17. doi: 10.1016/j.jaci.2017.05.020
23. Antiga E, Maglie R, Quintarelli L, Verdelli A, Bonciani D, Bonciolini V, et al. Dermatitis herpetiformis: novel perspectives. Front Immunol. (2019) 10:1290. doi: 10.3389/fimmu.2019.01290
24. Hashimoto T, Tsuruta D, Yasukochi A, Imanishi H, Sekine H, Fujita T, et al. Granular C3 dermatosis. Acta Derm Venereol. (2016) 96:748–53. doi: 10.2340/00015555-2379
Keywords: autoinflammatory skin manifestations, urticarial dermatosis, neutrophilic dermatosis, granulomatosis, chilblain lupus, lipoatrophy, angioedema, bullous disease
Citation: Kanazawa N (2020) Designation of Autoinflammatory Skin Manifestations With Specific Genetic Backgrounds. Front. Immunol. 11:475. doi: 10.3389/fimmu.2020.00475
Received: 14 January 2020; Accepted: 02 March 2020;
Published: 18 March 2020.
Edited by:
Masashi Akiyama, Nagoya University, JapanReviewed by:
Chao-Kai Hsu, National Cheng Kung University Hospital, TaiwanHideyuki Ujiie, Hokkaido University, Japan
Copyright © 2020 Kanazawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nobuo Kanazawa, bmthbmF6YXdAd2FrYXlhbWEtbWVkLmFjLmpw