 Jinxin Qi
Jinxin Qi Chengchao Ding
Chengchao Ding Xian Jiang
Xian Jiang Yong Gao
Yong Gao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 March 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00361
This article is part of the Research Topic Immunotherapy of Chronic Viral Infections that Cause Cancer View all 6 articles
Acquired immune deficiency syndrome (AIDS), which is caused by HIV infection, is an epidemic disease that has killed millions of people in the last several decades. Although combination antiretroviral therapy (cART) has enabled tremendous progress in suppressing HIV replication, it fails to eliminate HIV latently infected cells, and infected individuals remain HIV positive for life. Lifelong antiretroviral therapy is required to maintain control of virus replication, which may result in significant problems, including long-term toxicity, high cost, and stigma. Therefore, novel therapeutic strategies are urgently needed to eliminate the viral reservoir in the host for HIV cure. In this review, we compare several potential strategies regarding HIV cure and focus on how we might utilize chimeric antigen receptor-modified T cells (CAR T) as a therapy to cure HIV infection.
According to UNAIDS, more than 70 million people have been infected with the human immunodeficiency virus (HIV) and about 35 million people have died of HIV infection since this epidemic was first identified in the early 1980s. Globally, 37.9 million people were living with HIV at the end of 2018. The most effective and powerful therapy for HIV infection currently is combination antiretroviral therapy (cART), which has remarkably reduced morbidity and mortality and achieves durable suppression of plasma viremia below the limit of detection (1). The therapy has greatly extended life expectancy, turning HIV into a chronic disease that can be controlled instead of a death sentence, and has helped HIV-infected individuals live an almost normal life.
However, cART fails to cure HIV infection because of the existence of the latent viral reservoir, which is mainly a group of latently infected resting memory CD4+ T cells containing replication-competent HIV. All the cell types bearing the CD4 and its co-receptor (CCR5 or CXCR4) can be infected and become HIV latent reservoirs, including monocytes, macrophages, and dendritic cells (2). The HIV reservoir can exist in various compartments, such as peripheral blood, lymph nodes, the central nervous system, gut-associated lymphoid tissue (GALT), the genital tract, and any other tissues that contain HIV-infected cells (3). This latency does not express virus under the suppression of cART, but it can cause virus rebound once the therapy is interrupted (4). Thus, the persistence of HIV latency is regarded as the major obstacle to viral eradication (5–7). Meanwhile, as the half-life of HIV under effective cART is 44 months long (8, 9), more than 70 years of treatment is needed to achieve viral eradication. Hence, cART alone cannot get rid of the HIV reservoir, no matter how effective the drugs might be in controlling viral replication (10). Certainly, long-term use of cART brings up problems such as accumulative toxicity (11), drug resistance (12, 13), patient compliance, high cost, and even social problems like stigma. Additionally, chronic low-grade inflammation due to HIV infection continues even under the control of cART, which can accelerate aging, causing frailty syndrome at a younger age and a higher rate of comorbidity (11). So far, only two cases of HIV remission have been achieved in the past decades. These are Berlin and London patients who suffered from both hematologic malignancies and HIV infection, received CCR5Δ32/Δ32 allogeneic hematopoietic stem-cell transplantation (allo-HSCT), and achieved functional cure of both diseases. However, their success cannot be applied widely to HIV patients because of the high level of risk associated with marrow transplantation and there being limited suitable donors with CCR5Δ32/Δ32(14). Therefore, new strategies for HIV therapy that can achieve sterilization or functional cure need to be investigated. Treatment with chimeric antigen receptor-modified T cells (CAR T), a type of adoptive immunotherapy, has shown promising prospects for the therapy of B-cell malignancies (15–17). In parallel, HIV-specific CAR T cells have been designed for the treatment of HIV/AIDS. The first generation of HIV-specific CD4 receptor-based CAR was developed more than 20 years ago but was aborted because the resultant CAR T cell was susceptible to HIV infection and had negligible efficacy (18, 19). With the discovery of numerous potent anti-HIV broadly neutralizing antibodies (bNAbs) in recent years, bNAb-based CAR T therapy has been viewed as a potential strategy to cure HIV infection. Here, we provide an overview of recent studies on possible strategies for achieving HIV cure and mainly focus on the development, barriers, and future direction of CAR T therapy for HIV cure.
The mechanism of formation of HIV latency in memory CD4+ T cells is still unclear but is likely to involve viral tropism and activation of CD4+ T cells. When resting CD4+ T cells are exposed to chemokines in tissue sites, they might become permissive to being infected by HIV (20). However, these cells are usually not easily infected due to their low expression of the co-receptor CCR5 unless they are activated to upregulate this essential element for viral entry (21). On the other hand, HIV replication in infected cells usually leads to cell death (22, 23). However, after infection by HIV, the activated CD4+ T cell could revert to a resting state, which results in minimal viral gene expression (24) and enables the host cell to escape from the immune attack and survive the viral cytopathic effects.
It is important to note that, in HIV infection, very few CD4+ T cells can transit from the activated state to the memory state, as most infected and activated CD4+ T cells are killed either by cytopathic effects or host immune targeting (24). It was determined that latently infected cells have an extremely low frequency in HIV+ individuals and that the virus yield is about 0.03–3.00 IU (infectious units) per million resting CD4+ T cells (9). However, during acute infection, the HIV replication level is very high, which allows for latency to develop shortly after infection (e.g., within 3 days of infection) (25).
One important characteristic of the HIV latent reservoir is its remarkable stability due to the persistence of HIV in the long-lived resting memory CD4+ T cells, which can undergo antigen-driven clonal expansion (5, 26). Evidence for clonal expansion of infected resting memory CD4+ T cells is provided by the predominant clones discovered in plasma, which remain unchanged over months to years in HIV patients under effective cART (27, 28). Analyzing the integration sites of HIV proviral DNA provides more direct evidence because HIV randomly integrates into transcriptionally active regions of the host genome of each infected resting CD4+ T cell clone (29–31). Several studies have identified identical HIV integration sites in multiple CD4+ T cells in HIV patients receiving effective cART (32–35). In addition, full-length proviral sequencing (32, 36) and ex vivo culture systems also provided evidence for clonal expansion of infected cells. Furthermore, three recent studies have all shown that 50–60% of the latent reservoir is made up of expanded clones at any given time (36–38).
Importantly, infected cells carrying defective proviruses appear to expand more than infected cells with active provirus, suggesting that defective proviruses produce fewer viral proteins inducing cytopathic effects or immune response (32). However, some studies show that clonal expansion also occurs in cells carrying replication-competent proviruses (34, 36–38), even though it could possibly lead to HIV gene expression in the cells and consequent viral cytopathic effects.
As mentioned above, cART cannot cure HIV infection due to the existence of the HIV latent reservoir. A number of strategies, including gene therapy, “block and lock,” and “shock and kill,” have been developed and tested in order to eradicate the HIV reservoir. However, despite inducing detectable latency reversal, these strategies have not yet been able to eliminate the latent reservoir completely.
There are mainly two strategies to cure HIV infection by using gene-editing tools, which are also commonly used for other diseases. The first is to remove the latent reservoir directly by excising the provirus (Figure 1A). Ebina et al. designed a CRISPR/Cas9 system targeting the HIV long terminal repeat (LTR) region to excise integrated HIV provirus from the latently infected resting CD4+ T cells. The result showed efficient editing in target sites and great loss of LTR-driven expression (39). Furthermore, the latest report indicated that HIV could be eliminated from cell and tissue reservoirs in sequential long-acting slow effective release ART (LASER ART) and CRISPR/Cas9-treated humanized mice (40). This first successful experiment using an animal model shows that gene therapy should be combined with precisely targeted treatment delivery to effectively block HIV viral growth and provirus integration. However, the safety of CRISPR-based gene editing in the context of the human gene therapy is largely unknown, and the ethical issues involving human genome manipulation must also be taken into account.
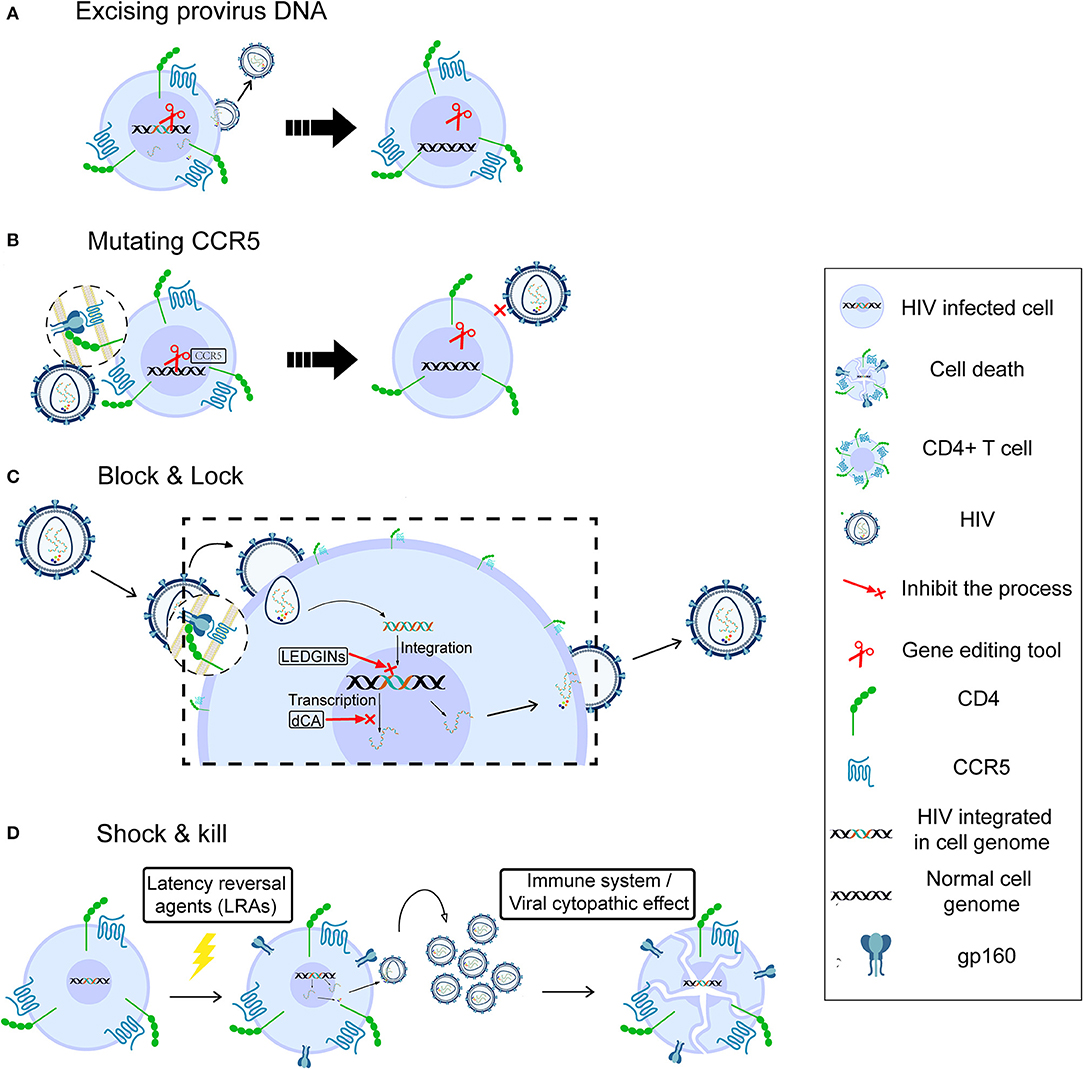
Figure 1. Possible strategies for HIV cure. Gene therapy for HIV cure by excising provirus DNA (A), mutating CCR5 (B), “block and lock” through silencing latent reservoir permanently (C), and “shock and kill,” through activating HIV latently infected cells followed by immune destruction or viral cytopathic effects (D).
A second strategy for gene therapy is to stop new infection, aiming at functional cure. HIV enters a target cell with the help of CD4 and the CCR5 (41) or CXCR4 (42) co-receptor. A homozygous 32-bp deletion in the CCR5 gene can make individuals naturally resistant to CCR5-tropic HIV infection (43, 44) though still susceptible to virus targeting CXCR4 tropism (45). The success of the “Berlin patient,” the first case in which HIV sterilizing cure was achieved by transplantation of allogeneic donor CCR5Δ32 hematopoietic stem progenitor cells (HSPCs) (46), demonstrated that disruption of the CCR5 gene to prevent new infection could be a potential cure (47). However, it is unclear which part of the treatment of this case, the total body irradiation before each HSCT or the HSCT itself, contributed more to this long-term HIV remission (14). The second case, the “London patient,” also achieved HIV remission after a single allo-HSCT with homozygous CCR5Δ32 donor cells but did not receive any irradiation (14). This strongly supports the strategy of deleting the CCR5 receptor on the cell surface to cure HIV infection. Tebas et al. made CCR5 gene permanently dysfunctional in autologous CD4+ T cells through ZFN modification (Figure 1B), then reinfused the modified T cells into patients. During treatment interruption and resultant viremia, the decline in circulating CCR5-modified cells was significantly less than the decline in unmodified cells, and the blood level of HIV DNA decreased in most patients (48). Recently, Xu et al. reported successful transplantation and long-term engraftment of CRISPR/Cas9-edited, CCR5-ablated HSPCs in a patient with HIV infection and acute lymphoblastic leukemia (49). However, the percentage of CCR5 ablation in lymphocytes was only ~5%. Moreover, a recent study showed that the mortality rate of homozygosity for CCR5-Δ32 mutation is higher (~21%) than for the other genotypes before age 76 (50). Hence, it is necessary to pay more attention to the safety and risks of gene editing and the potential deleterious effect of CCR5 mutation at the individual level.
Although cART cannot suppress HIV replication completely, it reveals the possibility of curing HIV through silencing the latent reservoir permanently, known as the block & lock strategy (Figure 1C). The whole process, from entry to virus release, can be the target of this strategy. Lentiviruses, including HIV, prefer to integrate into the active transcriptional regions of host DNA (29, 51), indicating that integrase inhibitors may help block the HIV reservoir. Vranckx et al. used LDGEF/p75 inhibitors (LEDGINs), a kind of integrase inhibitor that can inhibit HIV integrase from interacting with LEDGF/p75 host cofactor, to retarget HIV integration, resulting in the provirus becoming more refractory to reactivation even by using latency-reversing agents (LRAs) (52). Mousseau et al. showed that the Trans-activator of transcription (Tat) protein inhibitor didehydro-cortistatin A (dCA), an analog of the natural product cortistatin A, could bind to the trans-activating response (TAR)-binding domain of Tat and selectively inhibited Tat transactivation of the HIV promoter. Importantly, dCA abrogated viral production from stable reservoirs, reduced residual viremia during cART (53), and greatly diminished the capacity for virus reactivation (54). According to these results, the inclusion of a Tat inhibitor in current cART regimens may also contribute to the functional HIV cure.
Initial attempts at viral eradication involved global T-cell activators such as IL-2 and IL-2 + anti-CD3 antibodies (55, 56). However, despite inducing detectable latency reversal, these strategies ultimately failed to reduce the latent reservoir size and were associated with significant side effects due to massive cytokine release (57). In the last few years, HIV cure research has focused on the “shock and kill” strategy, which is aimed at attacking the HIV reservoir directly. This strategy could effectively expose viral reservoirs to a combination of cART and immune-mediated destruction or even eliminate the latently infected cells through viral cytopathic effects (vCPE). The goal of the “shock” is to reactivate viral replication in infected resting CD4+ T cells by LRAs in order to induce “kill” through attack by the immune system or active viral production (Figure 1D). There are many kinds of LRAs, for example, Protein kinase C (PKC) agonists (58), Histone Deacetylase inhibitors (HDACi) (59), and Histone Methylation inhibitors (HMTi) (60, 61). The most promising LRAs at this time are non-specific Histone Deacetylase inhibitors (HDACi), as they can acetylate the histone of integrated proviral promoters in vitro. Vorinostat, Disulfiram, and Romidepsin have been tested in clinical studies as candidate HDACi to induce viral replication, while the activated immune response would be expected to “kill” cells producing HIV (62–65).
The discovery that some of the latently infected CD4+ T cells are HIV-specific inspired another approach to reverse HIV latency (66, 67). By using an HIV vaccine providing a near-complete representation of viral quasispecies, such HIV-specific latently infected cells might be reactivated (68), and at the same time, cytotoxic T Lymphocyte (CTL) priming as well as the “kill” part of “shock and kill” strategies could be enhanced (57). A latency-reversing intervention could induce HIV expression indirectly by involving other cells. For instance, the TLR-7 agonist GS-9620 shows the ability to indirectly induce HIV expression in CD4+ T cells, probably through IFN-γ release from plasmacytoid dendritic cells (69). However, some reports indicate that these strategies have been unable to significantly impact the HIV reservoir size in patients (70, 71). Additionally, there is evidence to suggest that certain strategies may also adversely affect immune responses (72, 73). Thus, extensive research is being conducted to build more powerful “kill” in order to improve the strategy, including broadly neutralizing antibodies and immune checkpoint inhibitors (74).
Chimeric antigen receptor (CAR) contains three domains: an extracellular domain to specifically bind antigens, a transmembrane portion to anchor the receptor, and an endo-domain to transfer signals (75). The extracellular domain is a single-chain fragment variant (scFv) derived from the variable domain of antibodies or receptors. The endo-domain being used is CD3ζ, a signal-transduction component of the T-cell antigen receptor (76). With these characteristics, researchers can design CAR to recognize specific antigens and activate CAR-expressed effector-cells (77). In practice, researchers could generate and expand CAR T cells from patients' blood, followed by reinfusion of CAR T cells into the patients (78). Take HIV-specific CAR T as example: CD8+ T cells are collected from HIV patients and transduced with CAR genes; after in vitro verification of the anti-HIV specificity and effectiveness, the functional HIV-specific CAR T cells are reinfused into patients to kill HIV-infected cells (Figure 2).
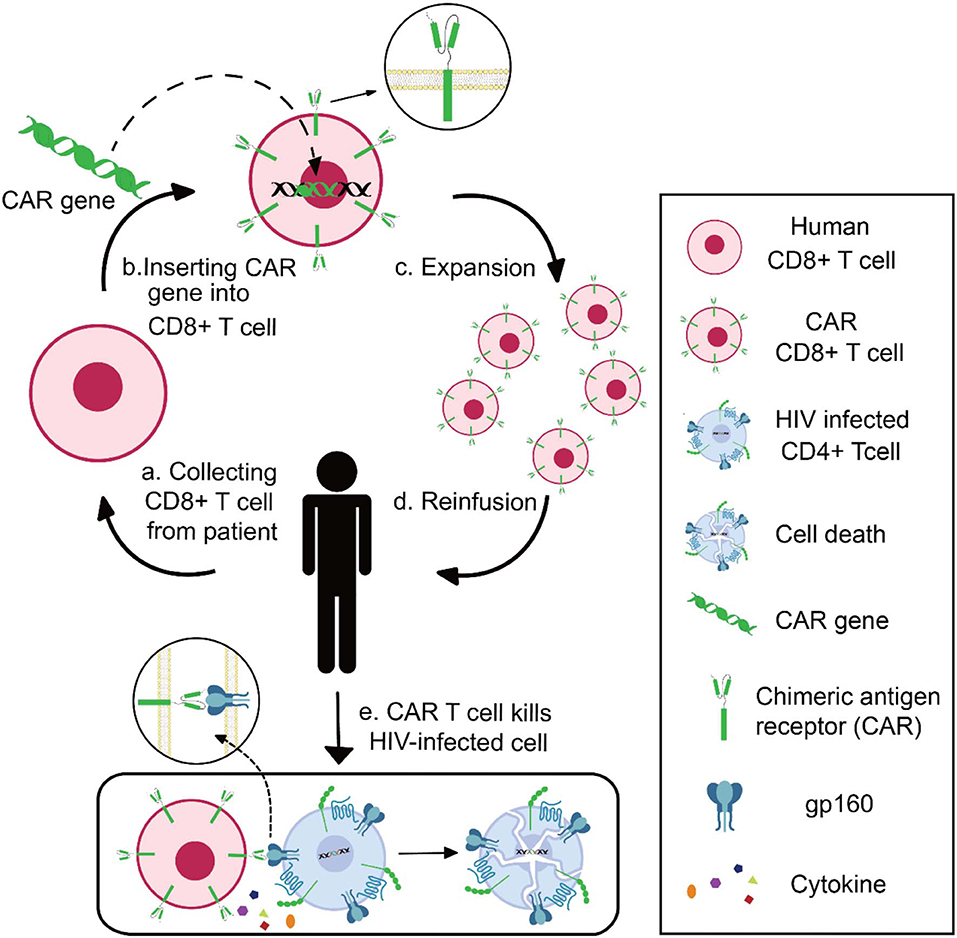
Figure 2. Schematics of CAR T-cell therapy for HIV infection. Collecting CD8+ T cell from HIV patient (a), inserting CAR genes into CD8+ T cells in vitro (b), expansion and functional identification of CAR T cells (c), reinfusing the HIV-specific CAR T cells into patients (d), and CAR T cells killing HIV-infected cells (e).
Four generations of CARs have been developed for the treatment of diseases since the chimeric antigen receptor was first presented in 1989. The first generation contains the CD3ζ-chain as the typical signal structure (75). The second generation adds additional costimulatory molecules like CD28, 4-1BB (CD137) to the signaling domain to improve the effector cells' proliferation, persistence, cytotoxicity, and sustained response (79–81). The third generation has two costimulatory molecules and outperforms the second generation in terms of cytotoxicity and long-term survival (82). The latest generation of CAR-T cells, also known as T cell redirected for universal cytokine-mediated killing (TRUCK), adds IL-12 gene into the CAR cassette; thus, CAR expression is accompanied by the release of IL-12. This structure enhances T-cell activation and attracts and activates innate immune cells to eliminate antigen-negative targeted cells in the targeted lesion (83).
Recently, by using autologous CD19-specific CAR-modified T cells, researchers have achieved complete remission in patients with hematologic malignancies like chronic lymphocytic leukemia (84), acute lymphoblastic leukemia (ALL) (85), diffuse large B-cell lymphoma, and follicular lymphoma (86) who were refractory to immunochemotherapy and transplantation or relapsed after the treatment. These results showed the safety, efficiency, feasibility, and durable effect of CAR T therapy. In the field of anti-ALL, CAR T therapy achieved complete remission in 27 out of 30 patients with relapsed or refractory ALL. At 6 months, the event-free survival rate was 67%, while the overall rate of survival is 78%. Durable remissions up to 24 months were also observed (85). While CAR T therapy worked well in hematologic cancers, it has been less effective in the treatment of solid tumors, likely due to lymphocyte trafficking problems (87). Fortunately, this might not be a limitation for HIV, as the main latency pool of HIV consists of CD4+ T cells (6, 11–13).
The CTL response is a key component of host immunity against HIV infection (88, 89). In elite controllers, a rare group of people who are able to control HIV replication by their immune system for a prolonged period without anti-HIV treatment (90), it is believed that their spontaneous viral control is mediated largely by CD8+ T-cell response (91, 92). In addition to its significant role in suppressing HIV replication during acute infection (93, 94), boosting HIV-specific CTL responses before viral activation by LRAs could lead to rapid and effective killing of infected cells (57). On the other hand, due to this robust selective pressure, HIV quickly obtains mutations to escape CTL recognition (93, 95). It is reported that unless cART is started in the early stages of HIV infection, the vast majority (>98%) of latent viruses will carry CTL escape mutations (95). Studies also revealed that elite controllers have higher functional avidity and broader variant cross-reactivity of CTL responses when compared with non-controllers, indicating the critical importance of dealing with viral escape mutations for controlling HIV infection (96). Therefore, equipping CD8+ T cells with a CAR that is able to recognize various HIV antigens is a key for HIV cure. At present, the CD4 receptor (97) and bNAbs (98, 99) are used to construct anti-HIV CARs. CD4, which interacts with gp120 during HIV infection, has naturally high affinity to HIV. The CD4 receptor-based CAR T cell was demonstrated to have the same level of kinetics of lysis and efficiency of inhibition as naturally occurring CTL clones (100). Despite the fact that the CD4 receptor can fully neutralize all HIV isolates, the CD4 receptor-based CARs make the gene-modified T cells vulnerable to HIV infection.
Broadly neutralizing antibodies against HIV are found in ~20% of HIV-infected individuals. These bNAbs target HIV envelope glycoprotein (Env) and have the ability to neutralize most circulating HIV strains (101). Due to their affinity, potency, and breadth of anti-HIV neutralization, it is believed that developing bNAb-CAR for HIV cure would be effective. Hale et al. tested four types of bNAb-based CARs (PGT-128, PGT-145, VRC07-523, and 10E8). Co-culturing with a stably infected HIV-positive T cell line in the presence of ART, primary human T cells engineered with bNAb-based CARs showed specific activation and killing of HIV-infected cells (99). CTL can mediate infected cell lysis with the help of major histocompatibility complex class I (MHC-I) molecules, but HIV could downregulate the surface expression of MHC-I in infected cells to escape this immune response (102, 103). However, CAR T cells could overcome this viral escape mechanism, as the chimeric antigen receptor directly recognizes antigen without MHC I restriction (75, 104). Further, the long-term persistence of CAR T-cell therapy promises prolonged therapeutic benefit (85, 105). Scholler et al. reported that CD4 receptor-based CAR T cells have a decay half-life exceeding 16 years with stable levels of engraftment. As this group measured >500 patient-years of follow-up, their results also emphasized the safety of this therapy (105). One explanation for the long survival time of CAR T cells could be that a portion of these cells persist as functional memory T cells (88), whose life expectancy is much longer than that of effector T cells. Memory CAR T cells also promise to react rapidly and robustly, even if HIV infection reoccurs years later. Additionally, it was observed that CAR T cells persisted at high levels for at least 6 months in the cerebrospinal fluid (CSF) (106), a compartment that contains a significant but hard-to-reach reservoir of HIV (107). A recent study showed that primary T cells transduced with a multi-specific CAR (targeting both the gp120 CD4-binding site and the gp120 co-receptor–binding site) had the ability to potently reduce cellular HIV infection by up to 99% in vitro and >97% in vivo (108).
Two phase II clinical trials of CAR T therapy were carried out in 2000, using the same CAR design but treating two groups of patients with either undetectable viremia or active viral replication. In these trials, both CD4+ and CD8+ T cells were engineered with CD4ζ CAR including a CD28 costimulatory domain. The help provided by CD4-CAR CD4+ T cells was believed to contribute to the prolonged survival of engineered T cells in both studies (97, 109). In the trial with active viral replication, CAR T cells had high persistence in blood for the 8 week observation period (1–3% of peripheral blood mononuclear cells) and survived in 17 of 18 subjects for at least 1 year. In this trial, researchers also reported a >0.5 log mean decrease for at least 14 days in rectal tissue-associated HIV RNA, suggesting antiviral activity of these CAR T cells against this important tissue reservoir of HIV (109). In another trial in 2002, in which the plasma viral loads were <50 copies/ml, infusion of CD4-CAR T cells decreased HIV burden from baseline but caused no differences in the size of the viral reservoir (97). These trials confirmed the safety and feasibility of CD4-CAR T-cell therapy and suggested the necessity of enhancing in vivo expansion of chimeric receptor-modified T cells and optimizing in vivo function. Another two clinical trials of CAR T-cell therapy in HIV-positive patients under cART treatment are ongoing or in recruitment: one (NCT03240328) is testing a bNAb (VRC01)-based CAR, and the other (NCT03617198) is evaluating a CD4-CAR T cell modified by ZFN disruption of its CCR5 for HIV resistance.
Even though it is predicted that anti-HIV CAR T therapy should be effective based on its performance in vitro, early clinical trials showed the safety of CAR T therapy but little efficacy in vivo. Besides, there are several obstacles or limitations as described below.
Several groups infusing expanded modified (110, 111) or even natural (112) CTL specific to HIV all showed that those cells died as a result of immune response or apoptosis in a very short time. Scholler et al. observed that CD4 receptor-based CAR T cells had no evidence of expansion and even persisted in high levels in vivo for decades (105). The problem of CAR T-cell expansion and persistence also occurred when applied in tumor treatment (113, 114). It is still unknown how much cell expansion and persistence contributes to the modest effect of CAR T cell in vivo. However, researchers consider it a possible direction for achieving a better outcome for CAR T therapy (Figure 3A). Sockolosky et al. utilized interleukin-2 (IL-2), a cytokine required for effector T cell expansion, survival, and function to help in CAR T-cell expansion. To avoid universal stimulation of effector T cells by IL-2, they engineered interleukin-2 (IL-2) cytokine-receptor orthogonal (ortho) pairs to interact with one another only, without interacting with their natural counterparts, while still transmitting native IL-2 signals. By introducing this engineered orthoIL-2Rβ into T cells, they selectively exploited the benefit of IL-2 with limited off-target effects and negligible toxicity (115). By using CRISPR/Cas9, Eyquem et al. directed CD19-specific CAR to the T-cell receptor α constant locus to help with CAR T-cell expansion. They reported that CAR expressed uniformly in human peripheral blood T cells, enhanced T-cell potency, performed tonic CAR signaling, and re-expressed following single or repeated exposure to antigen. Their results also showed a delay in effector T-cell differentiation and exhaustion (116). Fraietta et al. knocked down methylcytosine dioxygenase TET2 gene to improve the expansion and efficacy of CAR T cells and observed that, in one subject, 94% of CAR T cells likely originated from a single colony in which the TET2 gene was disrupted by the integrated CAR gene (117). Lack of antigen stimulation due to very low viral replication under cART might also contribute to the short-term persistence of infused CAR T cells. Therefore, in addition to a strong killing effect, the ideal shock and kill strategy using CAR T cells also needs to effectively reactivate latent reservoirs to produce virus, which will not only help CAR T cells to recognize the latently infected CD4+ T cells but will also help them to expand and achieve a better outcome.
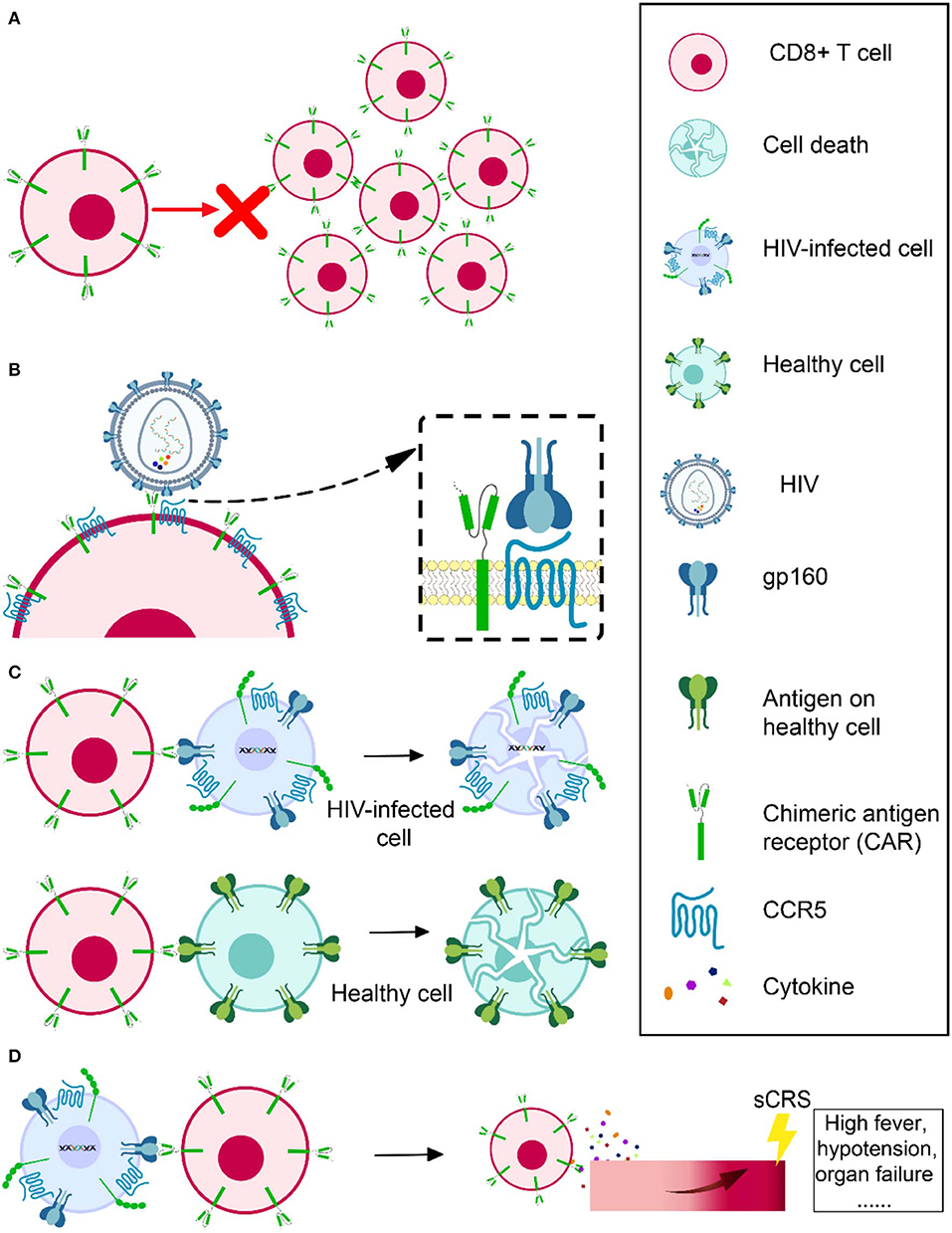
Figure 3. Obstacles in HIV-specific CAR T therapy development. Cell expansion and persistence in vivo (A), susceptibility to HIV infection (B), off-target effects (C), and severe cytokine release syndrome (D).
Another challenge is that CD4 CAR engineered CD8+ T cells are vulnerable to HIV infection (118), since CD8+ T cells have abundant CCR5 coreceptor on the cell surface (119) (Figure 3B). For this reason, Kamata et al. co-expressed two shRNAs, one targeting CCR5 expression and another targeting the HIV LTR, to prevent CD4 receptor-based CAR T cells from becoming HIV infected (120). Their results showed that over time, the number of shRNA-expressing CD4 receptor-based CAR T cells was higher than those without shRNA expression, suggesting that the susceptibility of CAR CD8+ T cell to HIV infection might partly account for its short persistence (120). Liu et al. constructed a novel bispecific CD4-CAR by adding a single-chain variable fragment of the 17b human monoclonal antibody, which can recognize a highly conserved epitope on gp120. Their CD4-17b CAR showed resistance to HIV infection, with higher potency of viral suppression than CD4 receptor-based CAR (18). To improve this bispecific CAR, the same group next substituted 17b with carbohydrate recognition domains (CRD) of a human C-type lectin that had been reported to interact with HIV gp120 mannose-containing glycans, including serum mannose-binding lectin (MBL), langerin, and liver/lymph node-specific intercellular adhesion molecule-3 grabbing non-integrin (L-SIGN). It was reported that the recognition by CRDs was a universal feature that would be hard for HIV to escape. They also emphasized that MBL, a soluble protein present in the circulation, should be safer, as immune reactivity against it should not target normal cellular self-protein. According to their results, this new bispecific CD4-lectin CARs enhanced potency and successfully prevented HIV infection (121). Using bNAb in CAR construction is another approach to prevent the susceptibility of engineered T cells. These bNAbs would not serve as a natural molecule on the cell surface to mediate HIV infection. Researchers using bNAb-based CAR have not reported any CAR T cells infected by HIV (98). Hale et al. even used a gene-editing tool to recombine a bNAb-CAR gene cassette into the CCR5 locus in order to destroy the CCR5 gene and ensure the engineered cells are HIV resistant and confirmed its feasibility and effectiveness (99).
Chimeric antigen receptors (CARs) could be constructed to specifically target HIV latently infected cells to achieve a cure. However, it is possible that CAR could also attack other healthy cells expressing the same or similar target antigen, which is known as an off-target effect (Figure 3C). For example, as CD19 is expressed on both normal and malignant B cells, B cell aplasia is inevitable when using CD19-based CAR T therapy to treat hematologic malignancies (84–86, 88). Fortunately, no evidence showed cytolysis of MHC II (CD4 counterpart)-expressing cell lines in CD4-CAR trials (18, 122). It is even safer to utilize bNAb-based CAR T therapy, because these bNAbs are specifically targeting HIV envelope glycoprotein, while CD4 may have more orthologs in nature. However, the ability of CAR to recognize its specific antigen without MHC I assistance raises another concern that it could recognize soluble antigen (e.g., cell-free or virion-associated HIV protein) and be activated to release cytokines toxic to surrounding tissues instead of specifically targeting HIV-infected cells.
When CAR T cells are infused into a recipient, they will be activated through cognate interaction with their specific antigens. While they could specifically attack the target antigen-expressing cells, they might also result in a progressive systemic inflammatory process, known as severe cytokine release syndrome (sCRS), whose typical manifestations are usually high fever, hypotension, or even organ failure (Figure 3D). After the infusion of CAR T cells, CRS usually occurs within 1–14 days and has a duration of 2–3 weeks to reach full resolution, influenced by the product, clinical trial design, the individual being treated, and the intervention. IL-6 is a signature cytokine of CRS. CAR T cell-mediated clearance of cancer could trigger elevated IL-6 levels. The most important source of IL-6 during CRS is human monocytes. Therefore, getting rid of human monocytes or blocking IL-6 receptor with tocilizumab could prevent CRS. It was reported that tocilizumab rapidly reversed sCRS induced in a critically ill child by CAR T therapy (106). Corticosteroids are also helpful in some cases to control CRS. However, in contrast to tocilizumab, high doses of corticosteroids may impact the antitumor effect of the CAR T cells.
Neurologic events in CAR T therapy include encephalopathy, delirium, aphasia, focal deficits, and seizures. Neurologic toxicity is poorly understood but is considered to be associated with supraphysiologic levels of cytokines and CAR T cells crossing blood–brain barriers. Within the first few weeks of CAR T-cell therapy, neurologic events could occur following CRS or during resolution. Although both neurologic toxicity and CRS are related to high cytokine levels due to CAR T-cell activation, more severe systemic CRS can be a risk factor for neurologic toxicity. However, these two adverse events have independent definitions and need independent management. Tocilizumab was observed to have limited efficacy in resolving neurologic toxicity, possibly because of its poor central nervous system penetration (16, 123, 124). Thus, corticosteroids are used by some centers as first-line therapy for isolated neurologic toxicity (125–127). Optimized treatment algorithms and further research on mechanisms of CAR T-cell therapy-related neurologic toxicity are needed to achieve better management outcomes.
Beside the above-mentioned advantages and potential problems of CAR T therapy, there are some other important developments in HIV CAR T strategies, including improvements of CAR T in tumor therapy that might be useful for HIV cure. Leibman et al. optimized the construction of CD4 receptor-based CAR from the lessons of engineering CARs for hematologic malignancies (128). In terms of capacity to control HIV replication, their re-engineered CAR was at least 50-fold more effective than the original CD4 receptor-based CAR, a TCR-based approach, and bNAb-based CARs. By switching the MMLV-based gamma-retroviral vector to an HIV-based lentiviral vector and by switching the PGK promoter to an EF1α promoter, CAR surface expression was significantly augmented. In addition, the CD8α transmembrane (TM) domain took the place of the CD4 TM domain, which would be downregulated by HIV Vpu. The CD8α TM domain also promoted CAR dimerization, increased variability from the HIV cellular receptor, and enhanced the cytotoxicity of the resultant CAR. The substitution of the transmembrane domain also decreased the susceptibility of CAR CD8+ T cells to HIV infection. Next, different costimulatory molecules, including CD28, 4-1BB, CD28+4-1BB, OX40, ICOS, and CD27, were compared in culture. Experiments showed that ICOS, CD27, and 4-1BB co-stimulation impaired the suppression of HIV duplication. However, when compared with CD28-containing CARs, 4-1BB-containing CARs performed better in controlling early infection in the HIV prevention model and exhibited more durability in the treatment model. The authors concluded that the 4-1BB zeta signaling domain was optimal in HIV cure strategies for (1) acting rapidly to prevent HIV transmission, (2) durably preventing viral rebound, and (3) promoting T-cell survival in the absence of antigen.
It has been reported that, within B cell follicles where T follicular helper (Tfh) cells are located, the number of specific CTLs was too low to stop ongoing viral replication during chronic HIV infection. To solve this problem, Haran et al. co-expressed a potent bispecific anti-SIV CAR (rhCD4–MBL) with the B-cell follicle-homing chemokine receptor CXCR5 to direct CAR T cells. In in vitro migration assay, they showed that the CAR/CXCR5 T cells migrated to CXCL13, the ligand for CXCR5. Meanwhile, CXCR5 co-expression improved the concentration of CAR T cell in the B cell follicles in ex vivo tissues. They also showed that the co-expression of CXCR5 did not compromise the SIV-suppressive activity of the CAR T cell (129).
In addition to successful CD19 CAR T-cell therapy, there are also many other explorations of CARs for various tumor treatments, shedding light on the possible future applications of CAR T treatment for HIV infection. Wu et al. designed “ON-switch” CARs, whose function is controlled by a heterodimerizing small molecule, with the purpose of gaining more precise control over the timing, location, and dosage of T-cell activity, thus decreasing toxicity (130). CAR T strategies in current studies are all using patient-specific cells, the generation of which is prohibitively expensive. Researchers are now trying to develop universal CAR T cells that can adapt to multiple recipients. Torikai et al. took the very first step by using zinc-finger nucleases (ZFN) to eliminate the endogenous αβ-TCR in CAR T cells to avoid graft-vs.-host response. This study suggested that future studies should focus on inhibiting the attack of the recipient's immune system on allogeneic CAR+ T cells (131). Similarly, Hale et al. used homology-directed recombination (HDR) to target the CAR gene to the T cell receptor alpha constant (TRAC) locus, producing TCR-deficient CAR T cells. They then produced CCR5-negative anti-CD19 CAR T cells in the same way, which could be applied in treating HIV-associated B-cell malignancies. The most attractive feature of this HDR-generated CAR T cell was that the CAR gene integrated into a single, definitive target site, diminishing the risk of randomly insertional mutagenesis (132).
Besides CD8+ T cells, researchers have also investigated other immune cell types for alternative CAR therapy. For example, NK cells are considered to be promising candidates, because they do not require prior sensitization, they are not MHC-independent in nature, and they have shown less severe adverse effects since they are tightly controlled by inhibitory receptors. In addition, there are sufficient numbers of NK cells in peripheral blood and functional NK cell lines that can be used in clinical trials (133). Zhen et al. modified HSPCs with CD4zeta-based CAR and successfully differentiated them into functional T cells and NK cells upon transplantation into humanized mice. These modified HSPCs could continuously provide functional antigen-specific cells and suppress HIV replication, with possible resistance to virus infection (134).
In summary, cART cannot eradicate HIV latency; therefore, new strategies for HIV cure are still needed and under development. There are three possible ways to achieve this goal: “block and lock,” “shock and kill,” and gene therapy. It is widely accepted that chimeric antigen receptor (especially bNAb-based CAR) engineered CD8+ T-cell therapy is a promising approach for curing HIV infection that is worth further exploration, even though there are limitations such as CAR T-cell expansion, persistence, off-target effect, and sCRS.
JQ, CD, and YG wrote sections of the manuscript. YG and XJ edited the manuscript.
This study was supported by grants from the Canadian Institutes of Health Research (OVV-152411) and the Anhui Science and Technology Department (201904b11020044).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Rodney P. DeKoter for his help in proofreading and editing the manuscript.
1. Murphy EL, Collier AC, Kalish LA, Assmann SF, Para MF, Flanigan TP, et al. Highly active antiretroviral therapy decreases mortality and morbidity in patients with advanced HIV disease. Ann Intern Med. (2001) 135:17–26. doi: 10.7326/0003-4819-135-1-200107030-00005
2. Sadowski I, Hashemi FB. Strategies to eradicate HIV from infected patients: elimination of latent provirus reservoirs. Cell Mol Life Sci. (2019) 76:3583–600. doi: 10.1007/s00018-019-03156-8
3. Delannoy A, Poirier M, Bell B. Cat and mouse: HIV transcription in latency, immune evasion and cure/remission strategies. Viruses. (2019) 11:269. doi: 10.3390/v11030269
4. Davey RT Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci USA. (1999) 96:15109–14. doi: 10.1073/pnas.96.26.15109
5. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA. (1997) 94:13193–7. doi: 10.1073/pnas.94.24.13193
6. Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. (1997) 278:1295–300. doi: 10.1126/science.278.5341.1295
7. Rong L, Perelson AS. Modeling latently infected cell activation: viral and latent reservoir persistence, and viral blips in HIV-infected patients on potent therapy. PLoS Comput Biol. (2009) 5:e1000533. doi: 10.1371/journal.pcbi.1000533
8. Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. (1999) 5:512–7. doi: 10.1038/8394
9. Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. (2003) 9:727–8. doi: 10.1038/nm880
10. Lorenzo-Redondo R, Fryer HR, Bedford T, Kim EY, Archer J, Pond SLK, et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature. (2016) 530:51–6. doi: 10.1038/nature16933
11. Guaraldi G, Prakash M, Moecklinghoff C, Stellbrink HJ. Morbidity in older HIV-infected patients: impact of long-term antiretroviral use. AIDS Rev. (2014) 16:75–89.
12. Boender TS, Kityo CM, Boerma RS, Hamers RL, Ondoa P, Wellington M, et al. Accumulation of HIV-1 drug resistance after continued virological failure on first-line ART in adults and children in sub-Saharan Africa. J Antimicrob Chemother. (2016) 71:2918–27. doi: 10.1093/jac/dkw218
13. Liu H, Ma Y, Su Y, Smith MK, Liu Y, Jin Y, et al. Emerging trends of HIV drug resistance in Chinese HIV-infected patients receiving first-line highly active antiretroviral therapy: a systematic review and meta-analysis. Clin Infect Dis. (2014) 59:1495–502. doi: 10.1093/cid/ciu590
14. Gupta RK, Abdul-Jawad S, Mccoy LE, Mok HP, Peppa D, Salgado M, et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature. (2019) 568:244–8. doi: 10.1038/s41586-019-1027-4
15. Brudno JN, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for lymphoma. Nat Rev Clin Oncol. (2018) 15:31–46. doi: 10.1038/nrclinonc.2017.128
16. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. (2018) 24:20–8. doi: 10.1038/nm.4441
17. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
18. Liu L, Patel B, Ghanem MH, Bundoc V, Zheng Z, Morgan RA, et al. Novel CD4-based bispecific chimeric antigen receptor designed for enhanced anti-HIV potency and absence of HIV entry receptor activity. J Virol. (2015) 89:6685–94. doi: 10.1128/JVI.00474-15
19. Zhen A, Peterson CW, Carrillo MA, Reddy SS, Youn CS, Lam BB, et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog. (2017) 13:e1006753. doi: 10.1371/journal.ppat.1006753
20. Saleh S, Solomon A, Wightman F, Xhilaga M, Cameron PU, Lewin SR. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood. (2007) 110:4161–4. doi: 10.1182/blood-2007-06-097907
21. Pierson T, Hoffman TL, Blankson J, Finzi D, Chadwick K, Margolick JB, et al. Characterization of chemokine receptor utilization of viruses in the latent reservoir for human immunodeficiency virus type 1. J Virol. (2000) 74:7824–33. doi: 10.1128/JVI.74.17.7824-7833.2000
22. Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci USA. (1997) 94:1925–30. doi: 10.1073/pnas.94.5.1925
23. Gornalusse GG, Mummidi S, Gaitan AA, Jimenez F, Ramsuran V, Picton A, et al. Epigenetic mechanisms, T-cell activation, and CCR5 genetics interact to regulate T-cell expression of CCR5, the major HIV-1 coreceptor. Proc Natl Acad Sci USA. (2015) 112:E4762–71. doi: 10.1073/pnas.1423228112
24. Shan L, Deng K, Gao H, Xing S, Capoferri AA, Durand CM, et al. Transcriptional reprogramming during effector-to-memory transition renders CD4(+) T Cells permissive for latent HIV-1 infection. Immunity. (2017) 47:766–75. doi: 10.1016/j.immuni.2017.09.014
25. Whitney JB, Hill AL, Sanisetty S, Penaloza-MacMaster P, Liu J, Shetty M, et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature. (2014) 512:74–7. doi: 10.1038/nature13594
26. Brenchley JM, Hill BJ, Ambrozak DR, Price DA, Guenaga FJ, Casazza JP, et al. T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: implications for HIV pathogenesis. J Virol. (2004) 78:1160–8. doi: 10.1128/JVI.78.3.1160-1168.2004
27. Bailey JR, Sedaghat AR, Kieffer T, Brennan T, Lee PK, Wind-Rotolo M, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol. (2006) 80:6441–57. doi: 10.1128/JVI.00591-06
28. Tobin NH, Learn GH, Holte SE, Wang Y, Melvin AJ, McKernan JL, et al. Evidence that low-level viremias during effective highly active antiretroviral therapy result from two processes: expression of archival virus and replication of virus. J Virol. (2005) 79:9625–34. doi: 10.1128/JVI.79.15.9625-9634.2005
29. Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. (2002) 110:521–9. doi: 10.1016/S0092-8674(02)00864-4
30. Symons J, Cameron PU, Lewin SR. HIV integration sites and implications for maintenance of the reservoir. Curr Opin HIV AIDS. (2018) 13:152–9. doi: 10.1097/COH.0000000000000438
31. Wang Z, Gurule EE, Brennan TP, Gerold JM, Kwon KJ, Hosmane NN, et al. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc Natl Acad Sci USA. (2018) 115:E2575–84. doi: 10.1073/pnas.1720665115
32. Cohn LB, Silva IT, Oliveira TY, Rosales RA, Parrish EH, Learn GH, et al. HIV-1 integration landscape during latent and active infection. Cell. (2015) 160:420–32. doi: 10.1016/j.cell.2015.01.020
33. Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. (2014) 345:179–83. doi: 10.1126/science.1254194
34. Simonetti FR, Sobolewski MD, Fyne E, Shao W, Spindler J, Hattori J, et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc Natl Acad Sci USA. (2016) 113:1883–8. doi: 10.1073/pnas.1522675113
35. Wagner TA, McLaughlin S, Garg K, Cheung CY, Larsen BB, Styrchak S, et al. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. (2014) 345:570–3. doi: 10.1126/science.1256304
36. Hosmane NN, Kwon KJ, Bruner KM, Capoferri AA, Beg S, Rosenbloom DI, et al. Proliferation of latently infected CD4(+) T cells carrying replication-competent HIV-1: potential role in latent reservoir dynamics. J Exp Med. (2017) 214:959–72. doi: 10.1084/jem.20170193
37. Bui JK, Halvas EK, Fyne E, Sobolewski MD, Koontz D, Shao W, et al. Ex vivo activation of CD4+ T-cells from donors on suppressive ART can lead to sustained production of infectious HIV-1 from a subset of infected cells. PLoS Pathog. (2017) 13:e1006230. doi: 10.1371/journal.ppat.1006230
38. Lorenzi JC, Cohen YZ, Cohn LB, Kreider EF, Barton JP, Learn GH, et al. Paired quantitative and qualitative assessment of the replication-competent HIV-1 reservoir and comparison with integrated proviral DNA. Proc Natl Acad Sci USA. (2016) 113:E7908–16. doi: 10.1073/pnas.1617789113
39. Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. (2013) 3:2510. doi: 10.1038/srep02510
40. Dash PK, Kaminski R, Bella R, Su H, Mathews S, Ahooyi TM, et al. Sequential LASER ART and CRISPR treatments eliminate HIV-1 in a subset of infected humanized mice. Nat Commun. (2019) 10:2753. doi: 10.1038/s41467-019-10366-y
41. Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. (1996) 381:661–6. doi: 10.1038/381661a0
42. Cohen J. Likely HIV cofactor found. Science. (1996) 272:809–10. doi: 10.1126/science.272.5263.809
43. Biti R, Ffrench R, Young J, Bennetts B, Stewart G, Liang T. HIV-1 infection in an individual homozygous for the CCR5 deletion allele. Nat Med. (1997) 3:252–3. doi: 10.1038/nm0397-252
44. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. (1996) 382:722–5. doi: 10.1038/382722a0
45. Scarlatti G, Tresoldi E, Bjorndal A, Fredriksson R, Colognesi C, Deng HK, et al. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat Med. (1997) 3:1259–65. doi: 10.1038/nm1197-1259
46. Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. (2009) 360:692–8. doi: 10.1056/NEJMoa0802905
47. Allers K, Hutter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, et al. Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood. (2011) 117:2791–9. doi: 10.1182/blood-2010-09-309591
48. Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. (2014) 370:901–10. doi: 10.1056/NEJMoa1300662
49. Xu L, Wang J, Liu Y, Xie L, Su B, Mou D, et al. CRISPR-Edited stem cells in a patient with HIV and acute lymphocytic leukemia. N Engl J Med. (2019) 381:1240–7. doi: 10.1056/NEJMoa1817426
50. Wei X, Nielsen R. CCR5-32 is deleterious in the homozygous state in humans. Nat Med. (2019) 25:909–10. doi: 10.1038/s41591-019-0459-6
51. Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. (2004) 2:E234. doi: 10.1371/journal.pbio.0020234
52. Vranckx LS, Demeulemeester J, Saleh S, Boll A, Vansant G, Schrijvers R, et al. LEDGIN-mediated inhibition of integrase-LEDGF/p75 interaction reduces reactivation of residual latent HIV. EBioMedicine. (2016) 8:248–64. doi: 10.1016/j.ebiom.2016.04.039
53. Mousseau G, Clementz MA, Bakeman WN, Nagarsheth N, Cameron M, Shi J, et al. An analog of the natural steroidal alkaloid cortistatin a potently suppresses tat-dependent HIV transcription. Cell Host Microbe. (2012) 12:97–108. doi: 10.1016/j.chom.2012.05.016
54. Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST. The tat Inhibitor didehydro-cortistatin a prevents HIV-1 reactivation from latency. MBio. (2015) 6:e00465. doi: 10.1128/mBio.00465-15
55. Chun TW, Engel D, Mizell SB, Hallahan CW, Fischette M, Park S, et al. Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat Med. (1999) 5:651–5. doi: 10.1038/9498
56. Prins JM, Jurriaans S, van Praag RM, Blaak H, van RR, Schellekens PT, et al. Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS. (1999) 13:2405–10. doi: 10.1097/00002030-199912030-00012
57. Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. (2012) 36:491–501. doi: 10.1016/j.immuni.2012.01.014
58. Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. (2013) 9:e1003834. doi: 10.1371/journal.ppat.1003834
59. Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses. (2009) 25:207–12. doi: 10.1089/aid.2008.0191
60. Bouchat S, Gatot JS, Kabeya K, Cardona C, Colin L, Herbein G, et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS. (2012) 26:1473–82. doi: 10.1097/QAD.0b013e32835535f5
61. Imai K, Togami H, Okamoto T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem. (2010) 285:16538–45. doi: 10.1074/jbc.M110.103531
62. Del Prete GQ, Oswald K, Lara A, Shoemaker R, Smedley J, Macallister R, et al. Elevated plasma viral loads in romidepsin-treated simian immunodeficiency virus-infected rhesus macaques on suppressive combination antiretroviral therapy. Antimicrob Agents Chemother. (2015) 60:1560–72. doi: 10.1128/AAC.02625-15
63. Lucera MB, Tilton CA, Mao H, Dobrowolski C, Tabler CO, Haqqani AA, et al. The histone deacetylase inhibitor vorinostat (SAHA) increases the susceptibility of uninfected CD4+ T cells to HIV by increasing the kinetics and efficiency of postentry viral events. J Virol. (2014) 88:10803–12. doi: 10.1128/JVI.00320-14
64. Shirakawa K, Chavez L, Hakre S, Calvanese V, Verdin E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. (2013) 21:277–85. doi: 10.1016/j.tim.2013.02.005
65. Wei DG, Chiang V, Fyne E, Balakrishnan M, Barnes T, Graupe M, et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog. (2014) 10:e1004071. doi: 10.1371/journal.ppat.1004071
66. Demoustier A, Gubler B, Lambotte O, de Goer MG, Wallon C, Goujard C, et al. In patients on prolonged HAART, a significant pool of HIV infected CD4 T cells are HIV-specific. AIDS. (2002) 16:1749–54. doi: 10.1097/00002030-200209060-00006
67. Douek DC, Betts MR, Brenchley JM, Hill BJ, Ambrozak DR, Ngai KL, et al. A novel approach to the analysis of specificity, clonality, and frequency of HIV-specific T cell responses reveals a potential mechanism for control of viral escape. J Immunol. (2002) 168:3099–104. doi: 10.4049/jimmunol.168.6.3099
68. Pankrac J, Klein K, Mann JFS. Eradication of HIV-1 latent reservoirs through therapeutic vaccination. AIDS Res Ther. (2017) 14:45. doi: 10.1186/s12981-017-0177-4
69. Tsai A, Irrinki A, Kaur J, Cihlar T, Kukolj G, Sloan DD, et al. Toll-like receptor 7 agonist GS-9620 induces HIV expression and HIV-specific immunity in cells from HIV-infected individuals on suppressive antiretroviral therapy. J Virol. (2017) 91:e02166–16. doi: 10.1128/JVI.02166-16
70. Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat Med. (2014) 20:425–9. doi: 10.1038/nm.3489
71. Sogaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. (2015) 11:e1005142. doi: 10.1371/journal.ppat.1005142
72. Narasimhan S, Coumou J, Schuijt TJ, Boder E, Hovius JW, Fikrig E. A tick gut protein with fibronectin III domains aids Borrelia burgdorferi congregation to the gut during transmission. PLoS Pathog. (2014) 10:e1004278. doi: 10.1371/journal.ppat.1004278
73. Waibel M, Christiansen AJ, Hibbs ML, Shortt J, Jones SA, Simpson I, et al. Manipulation of B-cell responses with histone deacetylase inhibitors. Nat Commun. (2015) 6:6838. doi: 10.1038/ncomms7838
74. Barouch DH, Deeks SG. Immunologic strategies for HIV-1 remission and eradication. Science. (2014) 345:169–74. doi: 10.1126/science.1255512
75. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
76. Ramos CA, Dotti G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin Biol Ther. (2011) 11:855–73. doi: 10.1517/14712598.2011.573476
77. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
78. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res. (2017) 5:22. doi: 10.1186/s40364-017-0102-y
79. Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. (1998) 161:2791–7.
80. Krause A, Guo HF, Latouche JB, Tan C, Cheung NK, Sadelain M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med. (1998) 188:619–26. doi: 10.1084/jem.188.4.619
81. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. (2009) 17:1453–64. doi: 10.1038/mt.2009.83
82. Tammana S, Huang X, Wong M, Milone MC, Ma L, Levine BL, et al. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum Gene Ther. (2010) 21:75–86. doi: 10.1089/hum.2009.122
83. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
84. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
85. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
86. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, et al. Chimeric antigen receptor T cells in refractory B-Cell lymphomas. N Engl J Med. (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
87. Newick K, O'Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med. (2017) 68:139–52. doi: 10.1146/annurev-med-062315-120245
88. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
89. Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. (1999) 283:857–60. doi: 10.1126/science.283.5403.857
90. Okulicz JF, Lambotte O. Epidemiology and clinical characteristics of elite controllers. Curr Opin HIV AIDS. (2011) 6:163–8. doi: 10.1097/COH.0b013e328344f35e
91. Loffredo JT, Sidney J, Bean AT, Beal DR, Bardet W, Wahl A, et al. Two MHC class I molecules associated with elite control of immunodeficiency virus replication, Mamu-B*08 and HLA-B*2705, bind peptides with sequence similarity. J Immunol. (2009) 182:7763–75. doi: 10.4049/jimmunol.0900111
92. Saez-Cirion A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, et al. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc Natl Acad Sci USA. (2007) 104:6776–81. doi: 10.1073/pnas.0611244104
93. Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. (1997) 3:205–11. doi: 10.1038/nm0297-205
94. Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. (1994) 68:4650–5. doi: 10.1128/JVI.68.7.4650-4655.1994
95. Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. (2015) 517:381–5. doi: 10.1038/nature14053
96. Mothe B, Llano A, Ibarrondo J, Zamarreno J, Schiaulini M, Miranda C, et al. CTL responses of high functional avidity and broad variant cross-reactivity are associated with HIV control. PLoS ONE. (2012) 7:e29717. doi: 10.1371/journal.pone.0029717
97. Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther. (2002) 5:788–97. doi: 10.1006/mthe.2002.0611
98. Ali A, Kitchen SG, Chen ISY, Ng HL, Zack JA, Yang OO. HIV-1-specific chimeric antigen receptors based on broadly neutralizing antibodies. J Virol. (2016) 90:6999–7006. doi: 10.1128/JVI.00805-16
99. Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, et al. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol Ther. (2017) 25:570–9. doi: 10.1016/j.ymthe.2016.12.023
100. Yang OO, Tran AC, Kalams SA, Johnson RP, Roberts MR, Walker BD. Lysis of HIV-1-infected cells and inhibition of viral replication by universal receptor T cells. Proc Natl Acad Sci USA. (1997) 94:11478–83. doi: 10.1073/pnas.94.21.11478
101. Kwong PD, Mascola JR, Nabel GJ. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat Rev Immunol. (2013) 13:693–701. doi: 10.1038/nri3516
102. Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. (1998) 391:397–401. doi: 10.1038/34929
103. Schwartz O, Marechal V, Le GS, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. (1996) 2:338–42. doi: 10.1038/nm0396-338
104. Lipowska-Bhalla G, Gilham DE, Hawkins RE, Rothwell DG. Targeted immunotherapy of cancer with CAR T cells: achievements and challenges. Cancer Immunol Immunother. (2012) 61:953–62. doi: 10.1007/s00262-012-1254-0
105. Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. (2012) 4:132ra53. doi: 10.1126/scitranslmed.3003761
106. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
107. Thompson KA, Cherry CL, Bell JE, McLean CA. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol. (2011) 179:1623–9. doi: 10.1016/j.ajpath.2011.06.039
108. Anthony-Gonda K, Bardhi A, Ray A, Flerin N, Li M, Chen W, et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci Transl Med. (2019) 11:eaav5685. doi: 10.1126/scitranslmed.aav5685
109. Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus-infected subjects. Blood. (2000) 96:785–93. doi: 10.1182/blood.V96.3.785
110. Brodie SJ, Lewinsohn DA, Patterson BK, Jiyamapa D, Krieger J, Corey L, et al. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nat Med. (1999) 5:34–41. doi: 10.1038/4716
111. Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, Manley SA, et al. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat Med. (1996) 2:216–23. doi: 10.1038/nm0296-216
112. Tan R, Xu X, Ogg GS, Hansasuta P, Dong T, Rostron T, et al. Rapid death of adoptively transferred T cells in acquired immunodeficiency syndrome. Blood. (1999) 93:1506–10. doi: 10.1182/blood.V93.5.1506
113. Jena B, Dotti G, Cooper LJ. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. (2010) 116:1035–44. doi: 10.1182/blood-2010-01-043737
114. Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. (2009) 21:215–23. doi: 10.1016/j.coi.2009.02.009
115. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. (2018) 359:1037–42. doi: 10.1126/science.aar3246
116. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. (2017) 543:113–7. doi: 10.1038/nature21405
117. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. (2018) 558:307–12. doi: 10.1038/s41586-018-0178-z
118. Bitton N, Verrier F, Debre P, Gorochov G. Characterization of T cell-expressed chimeric receptors with antibody-type specificity for the CD4 binding site of HIV-1 gp120. Eur J Immunol. (1998) 28:4177–87. doi: 10.1002/(SICI)1521-4141(199812)28:12<4177::AID-IMMU4177>3.0.CO;2-J
119. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. (2004) 200:749–59. doi: 10.1084/jem.20040874
120. Kamata M, Kim PY, Ng HL, Ringpis GE, Kranz E, Chan J, et al. Ectopic expression of anti-HIV-1 shRNAs protects CD8(+) T cells modified with CD4zeta CAR from HIV-1 infection and alleviates impairment of cell proliferation. Biochem Biophys Res Commun. (2015) 463:216–21. doi: 10.1016/j.bbrc.2015.05.026
121. Ghanem MH, Bolivar-Wagers S, Dey B, Hajduczki A, Vargas-Inchaustegui DA, Danielson DT, et al. Bispecific chimeric antigen receptors targeting the CD4 binding site and high-mannose glycans of gp120 optimized for anti-human immunodeficiency virus potency and breadth with minimal immunogenicity. Cytotherapy. (2018) 20:407–19. doi: 10.1016/j.jcyt.2017.11.001
122. Romeo C, Seed B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell. (1991) 64:1037–46. doi: 10.1016/0092-8674(91)90327-U
123. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4(+): CD8(+) composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
124. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T Cells. Cancer Discov. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
125. Neelapu S, Locke F, Bartlett N, Lekakis L, Miklos D, Jacobson C, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
126. Lee D, Gardner R, Porter D. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2016) 128:1533. doi: 10.1182/blood-2016-07-730689
127. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
128. Leibman RS, Richardson MW, Ellebrecht CT, Maldini CR, Glover JA, Secreto AJ, et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. Plos Pathog. (2017) 13:e1006613. doi: 10.1371/journal.ppat.1006613
129. Haran KP, Hajduczki A, Pampusch MS, Mwakalundwa G, Vargas-Inchaustegui DA, Rakasz EG, et al. Simian immunodeficiency virus (SIV)-specific chimeric antigen receptor-T cell engineered to target B cell follicles and suppress SIV replication. Front Immunol. (2018) 9:492. doi: 10.3389/fimmu.2018.00492
130. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. (2015) 350:aab4077. doi: 10.1126/science.aab4077
131. Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. (2012) 119:5697–705. doi: 10.1182/blood-2012-01-405365
132. Hale M, Lee B, Honaker Y, Leung WH, Grier AE, Jacobs HM, et al. Homology-directed recombination for enhanced engineering of chimeric antigen receptor T cells. Mol Ther Methods Clin Dev. (2017) 4:192–203. doi: 10.1016/j.omtm.2016.12.008
133. Liu D, Tian S, Zhang K, Xiong W, Lubaki NM, Chen Z, et al. Chimeric antigen receptor (CAR)-modified natural killer cell-based immunotherapy and immunological synapse formation in cancer and HIV. Protein Cell. (2017) 8:861–77. doi: 10.1007/s13238-017-0415-5
Keywords: human immunodeficiency virus (HIV), acquired immune deficiency syndrome (AIDS), immunotherapy, chimeric antigen receptor-modified T cell (CAR T), HIV latent reservoir, HIV cure
Citation: Qi J, Ding C, Jiang X and Gao Y (2020) Advances in Developing CAR T-Cell Therapy for HIV Cure. Front. Immunol. 11:361. doi: 10.3389/fimmu.2020.00361
Received: 06 November 2019; Accepted: 14 February 2020;
Published: 10 March 2020.
Edited by:
Ezequiel Ruiz-Mateos, Institute of Biomedicine of Seville (IBIS), SpainReviewed by:
Amarendra Pegu, National Institutes of Health (NIH), United StatesCopyright © 2020 Qi, Ding, Jiang and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xian Jiang, amVubnl4aWFuakAxNjMuY29t; Yong Gao, eWdhbzM4N0B1c3RjLmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.