 Gaurav Shrivastava1†
Gaurav Shrivastava1† Giovani Visoso-Carvajal1
Giovani Visoso-Carvajal1 Julio Garcia-Cordero1
Julio Garcia-Cordero1 Moisés Leon-Juarez2Bibiana Chavez-Munguia3
Moisés Leon-Juarez2Bibiana Chavez-Munguia3 Tomas Lopez4
Tomas Lopez4 Porfirio Nava5Nicolás Villegas-Sepulveda1
Porfirio Nava5Nicolás Villegas-Sepulveda1 Leticia Cedillo-Barron1*
Leticia Cedillo-Barron1*- 1Departmento de Biomedicina Molecular Centro de Investigación y Estudios Avanzados-Instituto Politécnico Nacional, Mexico City, Mexico
- 2Departamento de Inmunobioquímica, Instituto Nacional de Perinatología, Mexico City, Mexico
- 3Departamento de Infectomica y Biologia Molecular, Centro de Investigación y Estudios Avanzados-Instituto Politécnico Nacional, Mexico City, Mexico
- 4Departamento de Genética del Desarrollo y Fisiología Molecular, Instituto de Biotecnología, UNAM Cuernavaca, Cuernavaca, Mexico
- 5Departamento de Fisiologia, Biofisica y Neurociencias, Cinvestav Zacatenco, Mexico City, Mexico
Dengue is the most prevalent and rapidly transmitted mosquito-borne viral disease of humans. One of the fundamental innate immune responses to viral infections includes the processing and release of pro-inflammatory cytokines such as interleukin (IL-1β and IL-18) through the activation of inflammasome. Dengue virus stimulates the Nod-like receptor (NLRP3-specific inflammasome), however, the specific mechanism(s) by which dengue virus activates the NLRP3 inflammasome is unknown. In this study, we investigated the activation of the NLRP3 inflammasome in endothelial cells (HMEC-1) following dengue virus infection. Our results showed that dengue infection as well as the NS2A and NS2B protein expression increase the NLRP3 inflammasome activation, and further apoptosis-associated speck-like protein containing caspase recruitment domain (ASC) oligomerization, and IL-1β secretion through caspase-1 activation. Specifically, we have demonstrated that NS2A and NS2B, two proteins of dengue virus that behave as putative viroporins, were sufficient to stimulate the NLRP3 inflammasome complex in lipopolysaccharide (LPS)-primed endothelial cells. In summary, our observations provide insight into the dengue-induced inflammatory response mechanism and highlight the importance of DENV-2 NS2A and NS2B proteins in activation of the NLRP3 inflammasome during dengue virus infection.
Introduction
Dengue Virus (DENV) is a positive-sense single-stranded RNA virus from the Flaviviridae family. Dengue is caused by any of the four serotypes DENV-1 to DENV-4. It is transmitted to humans by female mosquitoes of the genus, Aedes (1, 2). The infection results in a broad spectrum of illness ranging from subclinical and mild self-limiting to severe dengue fever (DF) (2, 3). Severe dengue is associated with a secondary infection with a heterologous serotype (1, 3), and is characterized by immune dysfunction that can progress to life-threatening hypovolemic shock due to hemorrhage and leakage of vascular fluid. During severe infection, a massive and aberrant production of cytokines called a “cytokine storm” contributes to its deadly pathology (4). For example, the interleukin IL-1β plays a crucial role in the cytokine storm during dengue infection (5, 6). It is an extremely potent cytokine that is regulated and induced by dengue-infected macrophages and monocytes (6–8). Further, most dengue virus-infected patients present with fever, which is the most common symptom caused by the endogenous pyrogen molecule (EP). Additionally, the pro-inflammatory cytokine IL-1β has been shown to play a crucial role in increasing the deregulation of hemostasis and thrombosis during DENV infection (4, 6, 9). A dual pathway is required for the production of IL-1β, the priming signal, to stimulate the transcription and synthesis of pro-IL-1β, which must undergo post-translational cleavage to mature IL-1β by activated caspase-1. Caspase-1 activation is regulated by a second independent stimulus such as the “inflammasome,” a multi-protein complex assembled upon activation (10).
Pathogen recognition receptors (PPRs) include several nucleotide-binding domain leucine rich repeat-containing proteins (NLRPs) and other innate immune receptors, such as AIM2, which show different specificities toward pathogen- and danger-associated molecular patterns (10). During viral infections, NLRP3, binds to caspase-1 through the adaptor molecule, ASC containing a caspase recruitment domain, to assemble the inflammasome (11). The NLRP3 inflammasome responds to many stimuli from viral components, such as ATP and reactive oxygen species (ROS). Evaluation of macrophages infected with dengue virus has revealed that C-type lectin 5A (CLEC5A) plays a crucial role in dengue virus-induced NLRP3 activation (12).
Increased amounts of IL-1β have been observed in dengue virus patients (4) and several viral proteins have been linked to this process. It is also demonstrated that platelets from dengue-infected patients contribute to increased vascular permeability during infection by the synthesis and release of IL-1β (13). Spleen Tyrosine Kinase (Syk) augments IL-1β induction during antibody-enhanced dengue virus infection in primary human monocytes, however caspase-1 and NLRP3 are required for the maturation of pro-IL-1β during antibody-dependent enhancement (7). Altogether, these observations indicated that inflammasome activation might play a critical role in the pathogenesis of DENV infection. However, the molecular mechanisms through which DENV-2 provides secondary signals that activate the inflammasome are still elusive.
Several reports have confirmed the role of viroporins in inflammasome activation during viral infections (14). These molecules participate in several steps of the life cycle and are usually associated with the pathogenesis of viral infections. Viroporins are small, hydrophobic, transmembrane proteins that cause changes in the cellular permeability by forming hydrophilic pores in host cellular membranes, and disturb balance among the corresponding intracellular ions (e.g., Na+, K+, Ca++, Cl−, and H+). The ion concentration changes induced by those proteins often activates innate immune responses aimed to counter the viral infection (15–17). Interestingly, several viroporins from different viruses have been shown to activate the NLRP3 inflammasome by disrupting the Ca++ balance, including Rotavirus NSP4 (18), HCV p7 (19), EMCV 2B (20), Agnoprotein (21), polio 2BC (22), Influenza PB1-F2 (23), and coxsackievirus 2B (24). Calcium levels are disturbed in two ways; primarily, free cytosolic Ca++ are elevated through the release of Ca++ from the endoplasmic reticulum, Golgi complex, mitochondria, and lysosomes and secondly, influx of extracellular Ca++ via plasma membrane channels or Ca++ pump disturbance (25).
Among the dengue virus non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5), DENV NS2B has been reported to participate in replication, as a co-factor of viral protease and degradation of cGAS (26). In addition, DENV protease complex NS2B3 has been shown to partially cleave mitochondrial fusion proteins i.e., Mfn1 and Mfn2 resulting in the inhibition of mitochondrial fusion. Also, DENV induces cytopathic effects through destabilizing the interferon response and facilitating mitochondrial membrane potential (MMP) disruption (27). In our lab, two non-structural proteins (NSP) DENV-2 NS2B and DENV-2 NS2A have been demonstrated to behave as viroporins. Both molecules permeabilize different membrane models by forming membrane channels, as well as self-oligomerize and participate in a range of viral functions (28, 29). Furthermore, DENV NS2A a 22–25 kDa hydrophobic transmembrane protein plays a critical role in the virus life cycle by modulating the replication, viral assembly, and viral release probably by inhibiting the interferon and JAK-STAT pathways (30–32). DENV NS2A hydrophobic regions have demonstrated to have strong interactions with several Eukariothic model membrane systems (33). Although several studies have provided some clues regarding the role of different molecules of DENV that trigger the inflammatory process (34, 35), the mechanism of NLRP3 activation is not fully understood. In the current study, we have attempted to shed more light on the role of the putative viroporins NS2B and NS2A in the regulation of inflammasome activation. Initially, we demonstrated that macrovascular endothelial cells (HMEC-1) infected with DENV-2 activate the NLRP3 inflammasome, followed by ASC oligomerization, activation of caspase-1 and secretion of IL-1β. We further investigated whether this phenomenon is conserved in other cell lines such HepG2 and THP-1 cells. Finally, we have evaluated whether the viroporin-like proteins, NS2A, and NS2B, which induce changes in membrane permeability (29), are able to trigger the activation of the inflammasome. Thus, we found that the expression of recombinant NS2A-GFP and NS2B-GFP in the ER and mitochondria of HMEC-1 cells, significant increases the expression of NLRP3, ASC oligomerization, caspase-1 activation, and IL-1β secretion in the cell supernatant after priming with LPS. Furthermore, using a genetic knockout strain (ASC gene) and also pharmacological agents (NLRP3 inhibitor or caspase-1 inhibitor), we demonstrated that IL-1β release during DENV infection relays mainly in the assembly of the NLRP3 inflammasome and the activation of Caspase-1. Finally, we suggest that NS2A may be involved in intracellular Ca++ homeostasis and/or mitochondrial disruption, thereby boosting the activation of the NLRP3 inflammasome that leads to the overproduction of IL-1β. In summary, our observations unravel the mechanism by which dengue virus activates the NLRP3 inflammasome and emphasize the activity of viroporins in inducing NLRP3 inflammasome activation.
Materials and Methods
Cell Culture and DENV Serotype 2
HMEC-1 (HMEC line 1; Centers for Disease Control, Atlanta, GA, USA) were grown at 37°C under 5% CO2 in MCDB131 medium (Gibco/Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 1 mg/mL hydrocortisone (Sigma Aldrich, St. Louis, MO, USA), 10 ng/mL epidermal growth factor (Gibco), 100 U penicillin, and 100 mg/mL streptomycin. Cells were detached by treatment with 1,000 U/mL trypsin and 0.5 mM EDTA. Mosquito C6/36 cells derived from Aedes albopictus were grown in MEM supplemented with 10% fetal bovine Serum (FBS) (Gibco Carlsbad, CA) at 34°C. The DENV-2 with high nucleotide sequence homology to the New Guinea strain; (36) was obtained from a clinical isolate from a 1997 DF patient from the Mexican east coast. The stock preparation and titration have been described previously (37). The virus stock was prepared by infecting a C6/36 cell monolayer in 75 cm2 tissue culture flasks at 75–85% confluence. When the infected monolayer showed cytopathic effects, the cells supernatant were homogenized and diluted in a 40% polyethylene glycol solution in 2 M NaCl (Sigma-Aldrich St. Louis, MO) and incubated at 4°C overnight. The suspension was centrifuged at 6,000 rpm for 1 h, and then the virus stock in the bottom was resuspended in 1/15 of the total volume with a glycine buffer (Tris 50 mM, Glycine 200 mM, NaCl 100 mM and EDTA 1 mM) and 1/30 of the total volume of FBS. The virus was homogenized, aliquoted and frozen at −70°C until use.
Antibodies
The Table 1 describe all the antibodies used during this work.
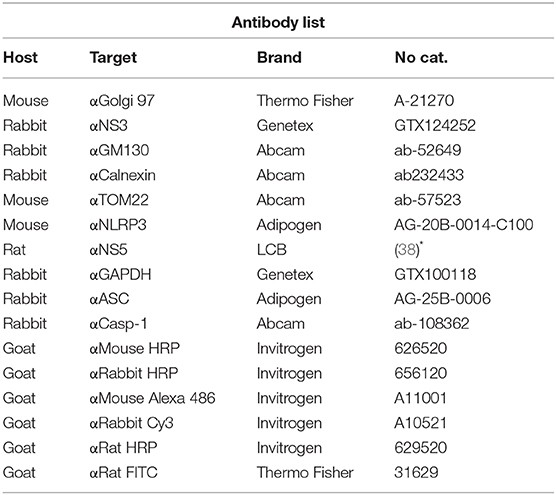
Table 1. List of Antibodies.
Plasmid Construction
The full length cDNAs encoding the NS2A and NS2B proteins of DENV-2 were cloned in the eukaryotic expression vector pEGFPN1 (Clontech). Briefly, the cDNAs encoding NS2A and NS2B were obtained by reverse transcription and PCR (by using specific oligonucleotides) of total RNA extracted from DENV-2 infected C6/36 cells with Trizol (Gibco, USA) according the manufacturer instructions. Both NS2A and NS2B amplicons and the plasmid pEGFPN1 were digested simultaneously with the restriction enzymes XhoI and HindIII (New England Biolabs), and then ligated in frame with GFP. The plasmid constructs pNS2A-GFP and pNS2B-GFP were verified by DNA sequencing. Further, the bulk production of plasmids were obtained by using endotoxin free Maxiprep kit (Qiagen, USA).
SDS-PAGE and Immunoblotting
Protein samples were resolved by SDS-PAGE using 12 or 15 % gels for 80 min at 100 V (Mini-Protean Cell; Amersham Biosciences, Piscataway, NJ, USA) and then electro transferred (120 V for 2 h) onto nitrocellulose membranes (Hybond ECL; GE Healthcare, Little Chalfont, UK). Membranes were blocked and then incubated with the appropriate primary antibody, followed by the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (1:3,000) in PBS-Tween-20. After further washing with PBS-Tween-20, the membranes were developed with western lightning enhanced chemiluminescence reagent (Pearce, Rockford, IL, USA). The membranes were stripped if necessary. All the antibodies used in this work are presented in Table 1.
Mitochondrial Membrane Potential Assay
HMEC-1 cells were seeded at 1 × 105 per well in a 6 well-plate and transiently transfected with plasmids i.e., GFP, NS2A-GFP, NS2B-GFP. After 36 h post-transfection, HMEC-1 cells were treated with 175 mM with Tetramethyl rodhamine Methyl Ester Perchlorate (TMRE Waltham, Massachusetts, USA) in MCDB base medium and incubate for 30 min at 37°C. Then TMRE solution was discarded and the cells were washed. Cells were detached and washed with 2 times with PBS 1X, and cells pellets resuspended in 0.2% BSA in 1X PBS. Mitochondria membrane potential was analyzed in the flow-cytometry at 488 nm.
Quantification of IL-1β Production
LPS-primed HMEC-1 cells were grown in 24 well-plates, transfected as described earlier and incubated at 5% CO2 and 37°C overnight. Further, cell supernatants were harvested at the indicated times post-transfection and analyzed for the presence of IL-1β using an enzyme-linked immunosorbent assay (Human IL-1β ELISA Kit II BD biosciences San Jose CA, USA) according to the manufacturer's instructions, and the concentration of IL-1β of the unknown samples and controls was determined from a standard curve. Samples producing signals higher than that of the highest standard (250 pg/ml) were diluted and re-analyzed. Each sample was run in triplicate and the assay were repeated for at least three times. The absorbance was read at 450 nm within 30 min of stopping reaction.
Transmission Electron Microscopy Analysis
The HMEC-1 cells were grown overnight in a 25 cm2 tissue culture flasks (Corning, New York, USA). Then, cells were mock-infected or infected with DENV-2 at 5 MOIs for 48 h. Further, the samples were fixed with 2.5% of glutaraldehyde in 0.1 M Sodium cacodylate buffer (pH 7.2) for 1 h at room temperature (RT), and post-fixed with 1% osmium tetroxide for 1 h at RT. The samples were dehydrated through an ethanol gradient and propylene oxide, and then were embedded in Polybed epoxy resins and polymerized at 60°C for 24 h. Finally, 70 nm thin sections were stained with uranyl acetate and citrate and then the preparations were analyzed by using a Jeol JEM-1011 transmission electron microscope (JeolLtd., Tokyo, Japan).
Transfection and Immunofluorescence
HMEC-1 cells were trypsinized and resuspended in MCDB131 medium. Cells were then seeded on glass coverslips (1 × 105 cells/ mL). After 24 h, the culture medium was removed, and the monolayers were washed and transfected. Briefly, HMEC-1 cells were grown at 50% confluence in a 6-well-plate, the medium was removed, and the cells were exposed to the transfection complex with different constructed plasmids [1 μg of DNA and 1 μl of Lipofectamine 2000 reagent (Invitrogen Life Technologies) mixed in 100 μl of serum-free Opti-MEM] for 4.5 h at 37°C. Then the cells were cultured in complete medium. At different times post-tranfections, the cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) in PBS for 20 min at room temperature, permeabilized with a solution of PBS supplemented with 0.1% Triton-X100 and blocked with 10% normal goat serum. The cell monolayer was incubated for 60 min with primary antibodies. Further, glass cover slip was washed and the following fluorochrome-conjugated secondary antibodies were added. Irrelevant isotype antibody was used as a negative control. Nuclei were labeled with DAPI (1 μg/ml) in PBS for 10 min, and the slides were mounted with VECTASHIELD (Vector Labs, Burlingame, CA, USA). The images were captured with a confocal microscope (Leica SP2, Barcelona, Spain).
Knockout of ASC by CRISPR-CAS9 Technology
The knockout was performed according to the protocol (37). lentiCRISPR v2 was a gift from Feng Zhang (Addgene plasmid # 52961; http://n2t.net/addgene:52961; RRID:Addgene_52961). Briefly, to clone the specific guide RNA specific to the vector, 5 μg of the lentiviral CRISPR plasmid (lentiCRISPRv2) was digested with BsmBI for 30 min at 37°C. In parallel, guide RNA was phosphorylated and annealed with each pair of oligos. The reaction mixture contained 1 μl Oligo Forward (100 μM), 1 μl Oligo Reverse (100 μM), 1 μl of 10X T4 Ligation Buffer (NEB), 6.5 μl dd H2O, 0.5 μl T4 PNK (NEB M0201S). Briefly, the phosphorylation/annealing reactions of the guide RNA were performed at 37°C for 30 min, 95°C for 5 min and then ramped down to 25°C at the rate of 5°C/min. The annealed oligos were diluted at a 1:200. The ligation reaction was incubated at room temperature for 10 min. Later, 1.5 μl ligated vector DNA was mixed with 20 μl competent Stbl3 bacterial cells, and incubated on ice for 30 min. After incubation, the cells were heat-shocked at 42°C for 45 s and more tubes were placed on ice for 2 min. SOC media (50 μl) was added and the bacteria were cultured at 37°C for 60 min on a shaker. Cultured bacteria were plated on LB-Agar plates and analyzed by colony PCR using the forward primer from the vector plasmid and the reverse primer from the reverse guide RNA (S-1). The amplified DNA was analyzed in 2% agarose gel and the expected band size (125 bp) was observed. Further, HEK 293T (3 × 106) cells were plated and then transfected with 4 μg of pLentiCRISPR v2 encoding sgRNA against GFP (control) or human ASC and packaging plasmids pVSVg and psPAX2 (1 μg each). After 6 h of transfection, 10 mL growth medium (cDMEM with 20% FBS) was replaced. Viral supernatants were collected after 48 and 72 h of transfection, concentrated by centrifugation at 20,000 × g for 2 h and was used to transduce the HMEC-1 cells in the presence of polybrene (2 μg/ml) in the 6-well plate containing HMEC-1 media. The plates were centrifuged for 45 min at 2,000 × g at 37°C, followed incubation at 37°C under 5% CO2 in MCDB131. After 12 h (overnight incubation), cells were tripsinized, washed and transferred to a 75 cm2 tissue culture flask with HMEC-1 media containing puromycin (1 μg/mL). The cells were checked every 3 days and the puromycin-containing medium was changed. Surviving populations derived in this manner were propagated and expanded for 6 weeks before cryopreserving stock cultures. The expression of wild type and ASC KO HMEC-1 was verified by western blotting with Anti-ASC antibody.
Results
Dengue Virus Infection Activates the NLRP3 Inflammasome in HMEC-1 Cells
Considering the major function of endothelial cells during the infection with dengue virus, we wanted to evaluate whether DENV is capable of triggering the immune response mediated by the inflammasome in HMEC-1 cells. We initially characterized the inflammasome activation induced by DENV-2, in different cell lines such as HepG2, THP-1 (Figures S1, S2), and our experimental model HMEC-1 cells. Cell lysates from DENV-2 infected (5 MOI) HMEC-1 and mock-infected (UV treated DENV-2) cells were western blotted for caspase 1. As shown in Figure S1A, DENV-2 infection induced and increased the cleavage of caspase-1, when compared with mock-infected cells. LPS/ATP stimuli was used as a positive control. Moreover, DENV-2 infection was confirmed by western blotting DENV-2 NS3 protein in THP-1 cells (Figure S2A) and by western blotting and imaging (immunofluorescence) DENV-2 NS5 protein in HMEC-1 cells (Figure 1A and Figure S1B).
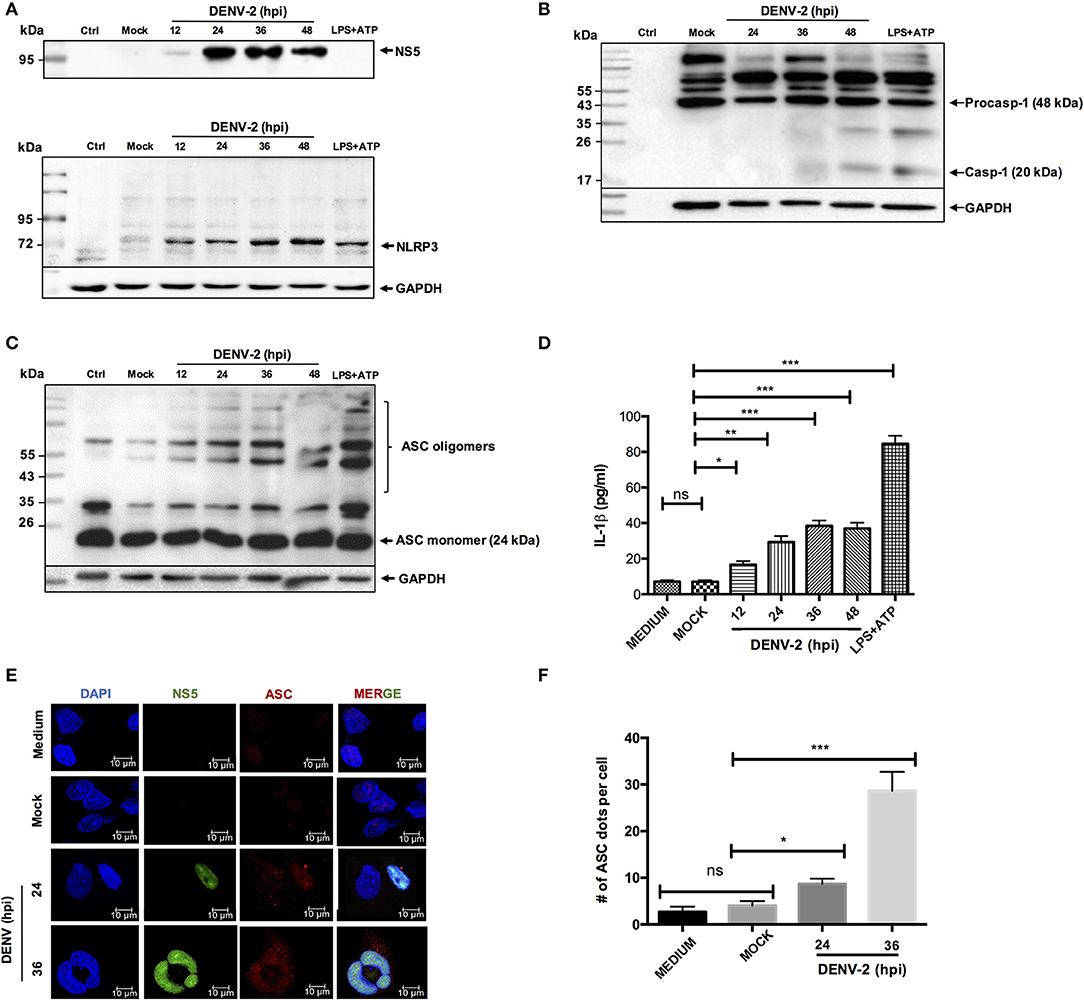
Figure 1. NLRP3 inflammasome activation by DENV-2. HMEC-1 were infected with DENV-2 (5 MOI) at different time points, mock-infected or treated with LPS (1 μg/mL) and ATP (5 mM) as a positive control. Cell lysates were analyzed by western blot using an anti-NLRP3 antibody (1:500) (A), an anti-NS5 antibody (1:100) (B), an anti-caspase-1 antibody (1:1,000) (C), or an anti-ASC antibody (1:1,000) (D). HMEC-1 were infected with DENV-2 (5 MOI) at different time points, mock-infected or treated with LPS (1 μg/mL) for 6 h followed by ATP (5 mM) for 45 min as a positive control, and cell-free supernatants were collected at 12, 24, 36, and 48 h post-infection and analyzed for IL-1β by ELISA (E), HMEC-1 were infected with DENV-2 (5 MOI) at 24 and 36 h or mock-infected. Cells were stained with anti-NS5 (green) and anti-ASC (red) and analyzed by confocal microscopy. Mock-treated cells were also examined. Nuclei were visualized by staining with DAPI (F). The number (#) of ASC puncta per cell was counted by confocal microscopy. ASC puncta was calculated from a total of 20 cells. Data are representative of at least three independent experiments, and indicate the mean ± S.D. (E,F). *P < 0.05, **P < 0.01, and ***P < 0.001. ns, non significant.
Likewise, to determine whether DENV-2 activates the NLRP3 inflammasome, cell lysates from DENV-2 infected HMEC-1 cells were analyzed by western blot at different infection times using specific anti-NLRP3 antibody. During DENV-2 infection. Changes in the NLRP3 expression were observed at the late phase (36 and 48 h) pos-DENV-2 infection, in contrast to HMEC-1 untreated and mock-infected cell controls, shown low expression of NLRP3. In the positive control, the HMEC-1 cells treated with LPS (1 μg/mL) (LPS -O111:B4, sigma Aldrich) (signal 1) for 6 h, and ATP for 45 min (signal 2), to induce the assembly of the inflammasome complex, a high expression of NLRP3 was observed (Figure 1A). Western blot analysis showed the activated caspase 1 (~20 kDa) at 24 h post-infection, and a more prominent band was observed at 48 h post-infection with DENV-2 (Figure 1B). To analyze the oligomerization of the ASC adapter protein, western blot analysis was conducted on the cell lysates infected with DENV-2 at 24, 36, and 48 h post-infection, and cells treated with LPS and ATP. No oligomerization of ASC was observed in mock-infected HMEC-1 cells, however prominent oligomerization of ASC was observed after 12, 24, 36, and 48 h post-infection with DENV-2, along with the positive control (LPS + ATP) (Figure 1C). As expected, a significant increase of IL-1β was detected in the supernatant of DENV-2-infected cells at 36 and 48 h post-infection, in contrast with mock-infected cell supernatant (Figure 1D). Similar to the observed with HMEC-1, DENV-2 triggered caspase-1 activation and IL-1β secretion in HepG2 and THP-1 cells (Figures S2B–E). Furthermore, HMEC-1 cells were DENV-2 or mock-infected at different time points and the presence of ASC punctate structures, which serve as a marker of the “inflammasome complex,” were found in the cells infected with DENV-2 at 36 h, in contrast to mock-infected cells (Figure 1E). The number of ASC puncta structures per cell was calculated for total 20 cells (Figure 1F). Taken together, these results demonstrate that DENV-2 induces activation of the NLRP3 inflammasome in HepG2, THP1, and HMEC-1 cells.
Expression and Localization of NS2A in HMEC-1 Cells
To fully characterize the subcellular localization of DENV-2 NS2A and evaluate its role in inflammasome activation, the NS2A (657 bp) sequence of DENV-2 was cloned as an eGFP-fused protein (eGFPN1 vector). The pNS2A-GFP, pNS2B-GFP, and pGFPN1 plasmids were transfected into HMEC-1 cells and analyzed by western blot and immunofluorescence assays at different times (24 and 48 h) to determine the subcellular localization of DENV-2 NS2A. The expression of NS2A-GFP, was observed in the perinuclear space in HMEC-1, demonstrating its functional expression in the cytoplasmic region (Figure 2A). In addition, NS2A-GFP and NS2B-GFP and GFP transfected cells were lysed, and analyzed by western blot using anti-GFP antibody. Clear bands were observed in the transfected cells with DENV constructs. The molecular weight for NS2A-GFP (48 kDa) and NS2B (43 kDa) were as expected according to the fusion protein (Figure 2B). Additionaly a sharp 27-kDa band was observed in the cells transfected with the NS2A-GFP and NS2B-GFP (in this case, GFP might be produced as a result of leaky translation scanning in the frame fused transcript) as well as in the parental vector GFP (as expected). Using different organelle markers for ER, Golgi apparatus, and mitochondria, we observed that NS2A exhibited significant overlap with calnexin A, which is a transmembrane protein that resides in the ER membrane, indicating that NS2A partially localizes to the ER network. Further, NS2A co-localized with mitochondria (MitoTracker) and Tom22, an outer mitochondrial membrane protein. In contrast, NS2A did not co-localize with the Golgi apparatus marker (GOLGI) (Figure 2C). Hence, our co-localization studies indicated that the NS2A protein localizes to both the ER and mitochondria organelles with Pearson's coefficient values of 0.79 and 0.81, respectively (Figure 2D). The Pearson's coefficients between NS2A and the different markers analyzed were calculated as average values from 20 individual cells. It is important to demonstrate that the NS2A-GFP and NS2B-GFP behave as the viral proteins in the context of DENV infection. Thus, we evaluated the localization of NS2B protein by infecting and transfecting cells simultaneously to corroborate the localization of NS2B. We observed the same localization pattern of NS2B during DENV-2 infection as well as during transfection of NS2B-GFP tag. Thus, GFP tag does not modify the localization of the NS2B (Figure S3).
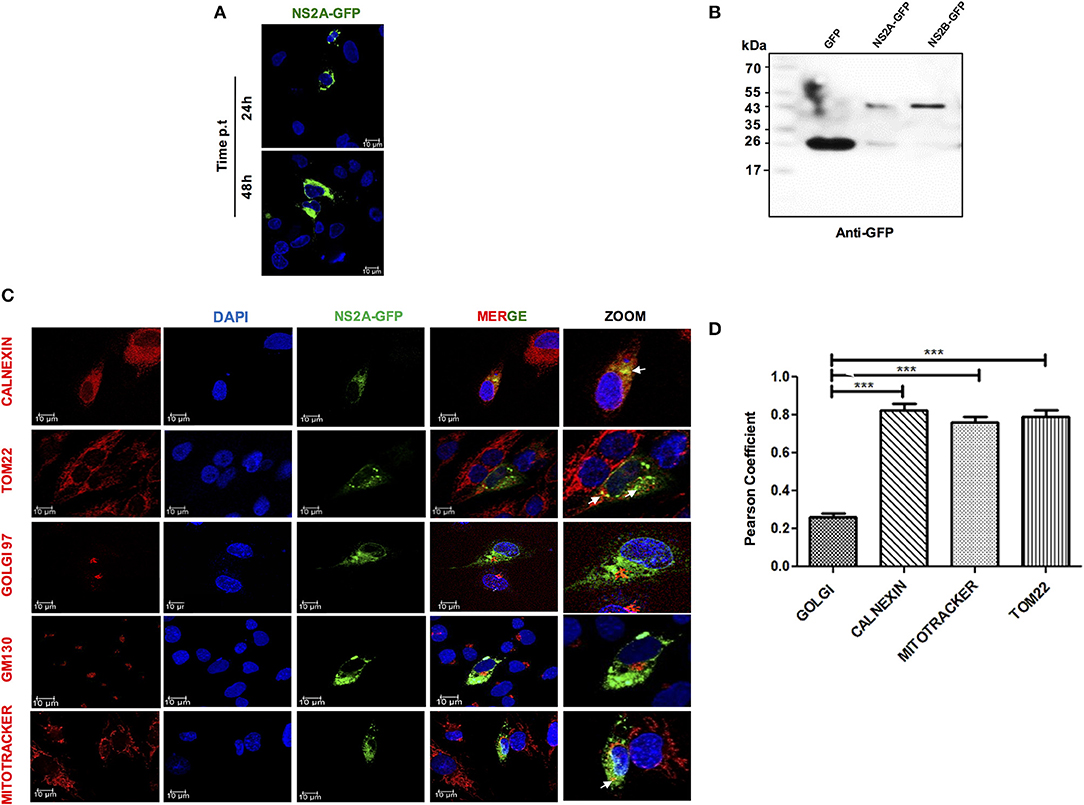
Figure 2. Expression and localization of NS2A protein in HMEC-1 cells. (A) HMEC-1 cells were transiently transfected with plasmids coding for NS2A-GFP and analyzed at 24 and 48 h. Cells were fixed and analyzed by confocal microscopy (B). HMEC-1 cells were transiently transfected with the plasmids coding for GFP and the NS2A-GFP protein and cell lysates were analyzed by western blot using an anti-GFP antibody (1:1,000). Lane 1: molecular weight (MW), Lane 2: pEGFPN1 transfected lysate, Lane 3: pNS2A-GFP transfected cell lysate showing band of GFP (27 kDa) as well as NS2A-GFP (48 kDa) and NS2B-GFP (43 kDa) (C). To evaluate the localization of the NS2A protein, HMEC-1 cells were transfected with a plasmid encoding GFP-tagged DENV-2 NS2A and stained with either anti-calnexin (ER Marker; red), anti-GM (Golgi marker; red), MitoTracker (mitochondrial marker; red) or TOM22 (mitochondrial outer membrane marker, red) and observed with a confocal microscope 24 h after transfection (D). Pearson's Coefficient of the NS2A-GFP localization in different membrane organelles. ***P < 0.001.
NS2A and NS2B Proteins Are Sufficient to Trigger NLRP3 Inflammasome
Viroporins from different viruses have been demonstrated to activate the inflammasome (14). Furthermore, we have shown that NS2A and NS2B behave as viroporins, thus, we evaluated the ability of dengue virus viroporin NS2A and NS2B to trigger inflammasome activation. HMEC-1 cells were primed with LPS (O111:B4, sigma Aldrich) (signal 1) followed by transfection with GFP-tagged plasmids expressing dengue NS2A, NS2B, proteins. Western blot analysis demonstrated higher protein levels of NLRP3 expression, ASC oligomerization, and caspase-1 activation in LPS-primed HMEC-1 cells transiently expressing NS2A or NS2B but not in HMEC-1 cells transfected with NS3, and NS2B-NS3 with the parental plasmid (eGFPN1). We also found that NS2A and NS2B expression triggered Caspase-1 cleavage in HepG2 cells (Figure S1C). ATP (5 mM) stimulation was used as a positive control for an NLRP3 inflammasome inducer (Figures 3A–C). Cell free supernatants were collected at 36 h post-transfection and the presence of IL-1β was analyzed using an enzyme-linked immunosorbent assay (ELISA). As expected, significant IL-1β was released from LPS-primed HMEC-1 cells transfected with pNS2A-GFP or pNS2B-GFP vectors when compared with HMEC-1 cells transfected with pNS3-GFP, pNS2B-NS3-GFP or the parental vector (peGFPN1) (Figure 3D). This data strongly suggested a direct involvement of NS2A and NS2B in the activation of the inflammasome complex (NLRP3, ASC, Caspase-1) and the subsequently release of IL-1β. To support the above results, we also examined the oligomerization of ASC as a marker of inflammasome complex activation by NS2A and NS2B. To do this, HMEC-1 cells were primed with LPS for 6 h followed by transfection with peGFPN1-plasmid or GFP tagged NS2A and NS2B plasmid. We have shown that ASC punctate structures were formed in the cytoplasm of HMEC-1 cells transfected with the plasmid expressing the NS2A-GFP and NS2B-GFP, in contrast to the cells transfected with peGFPN1 (Figure 3E). As a positive control, HMEC-1 cells were re-treated with ATP for 45 min to observe the presence of ASC punctate structures. The number of ASC per cell was counted and demonstrated the role of DENV-2 NS2A and NS2B in inflammasome activation (Figure 3F). We also examined the intracellular localization of NLRP3. In agreement with previous reports (39), stimulation of LPS-primed cells with HMEC-1 induced NLRP3 expression in the cytosol. Upon cell transfection with dengue NS2A-GFP after 36 h, NLRP3 was redistributed to the perinuclear region or cytoplasmic granular structures, which are considered a hallmark of NLRP3 activation, in contrast to resting or control cells transfected only with GFP. We also shown that NLRP3 was co-localized with NS2A-GFP in HMEC-1 cells, in contrast to either GFP or NS2B-GFP (not shown). About 60% of cells expressing the NS2A-GFP showed very strong co-localization with NLRP3, very as a 0.8427 (>0.5) Pearson's coefficient was observed (Figures 3G,H). Together, these data provide evidence that the expression of dengue virus viroporins, NS2A, and NS2B are sufficient to activate the NLRP3 inflammasome.
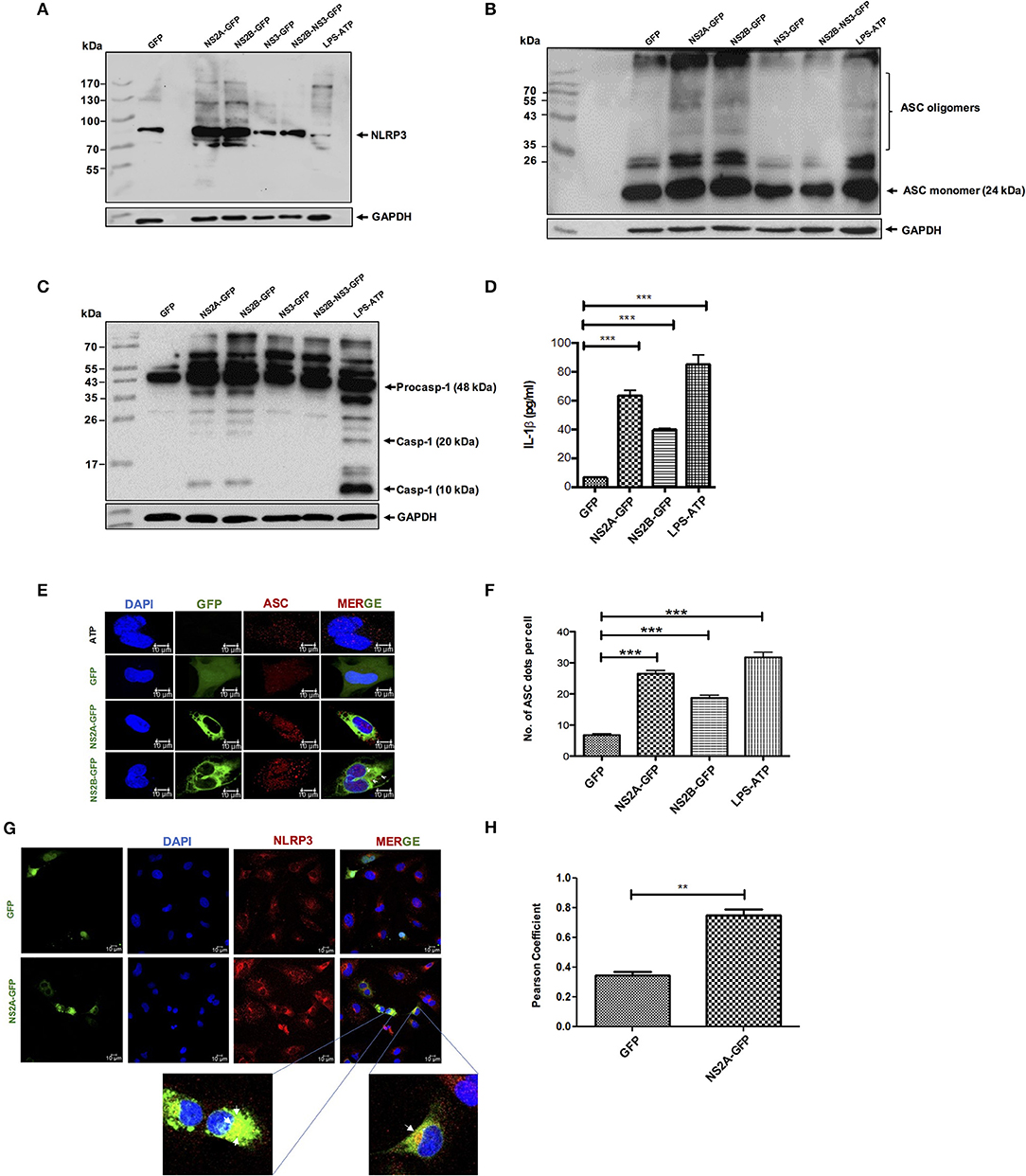
Figure 3. NLRP3 inflammasome activation by DENV-2 viroporin, NS2A, and NS2B. HMEC-1 cells were transfected with the expression plasmid encoding GFP-tagged DENV-2 NS2A, NS2B, or the pEGFPN1 empty vector for 36 h or treated with LPS (1 μg/mL) for 6 h followed by ATP (5 mM) for 45 min as a positive control. Cell lysates were analyzed by western blot using (A) an anti-NLRP3 antibody (1:500), (B) an anti-ASC antibody (1:1,000), and (C) an anti-caspase-1 antibody (1:1,000). (D) HMEC-1 cells were transfected with the expression plasmid encoding GFP-tagged DENV-2 NS2A, NS2B, or the pEGFPN1 empty vector for 36 h or treated with LPS (1 μg/mL) for 6 h followed by ATP (5 mM) for 45 min as a positive control, and the cell free supernatant was analyzed for IL-1β by ELISA. (E) HMEC-1 cells were transfected with expression plasmids encoding GFP-tagged DENV-2 NS2A, NS2B, or the pEGFPN1 parental vector for 36 h or treated with LPS (1 μg/mL) for 6 h followed by ATP (5 mM) for 45 min as a positive control, and the cells were stained with anti-ASC (Red) and analyzed by a confocal microscope. (F) The number (#) of ASC puncta per cell was counted by confocal microscopy. ASC puncta was calculated from a total of 20 cells. (G) HMEC-1 cells were transfected with pNS2A-GFP, or the pEGFPN1 empty vector for 36 h and the cells were stained with anti-NLRP3 (1:200) (Red) and analyzed by a confocal microscope. Nuclei were visualized by staining with DAPI. (H) Pearson's Coefficient of the NS2A-GFP or GFP co-localization with NLRP3. Data are representative of at least three independent experiments, and indicate the mean ± S.D. (D,F,H). **P < 0.01 and ***P < 0.001.
Confirmation of a NS2A and NS2B Effect on Inflammasome Activation by CRISPR-CAS9
Next, to confirm the effect of NS2A and NS2B in the activation of NLRP3 inflammasome, the ASC gene was knocked out in HMEC-1 cells using a lenti-CRISPRv2-ASC viral particle. ASC guide RNA was cloned in a lenti-CRISPRv2 vector and the confirmation of cloned lenti-CRISPRv2-ASC was obtained with colony-PCR using appropriate primers that showed the positive clone as 125 bp by agarose gel electrophoresis (Figure S4B). In addition to ASC, NLRP3 and CASP-1 guide RNA were also cloned into the lentiCRISPRv2 plasmid (Figure S4A). To confirm the ASC knockout, HMEC-1 transduced cell lysates were analyzed by western blot; a complete ASC gene knockout was observed, and a lack of ASC was confirmed in HMEC-1 transduced cells (Figure 4A). Then, ASC−/− HMEC-1 cells were selected to analyze inflammasome activation due to NS2A and NS2B. To do this, ASC−/− HMEC-1 cells or WT HMEC-1 cells were primed with 1 μg/mL LPS for 6 h and then transfected with peEGFPN, pNS2A-GFP, and pNS2B-GFP for 36 h. As a positive control, cells were primed with 2 μg/mL LPS for 6 h followed by 5 mM ATP for 45 min. Lysates were analyzed by western blot and demonstrated caspase-1 activation due to the presence of NS2A and ATP in WT HMEC-1 cells in contrast to ASC−/− HMEC-1 cells (Figure 4B). Cell supernatants were also analyzed for IL-1β secretion, which was absent in ASC−/− HMEC-1 cells, in contrast to WT HMEC-1. A similar result was observed for ATP. This result confirmed the effects of NS2A on caspase-1 activation and the subsequent inflammasome assembly (Figure 4C).
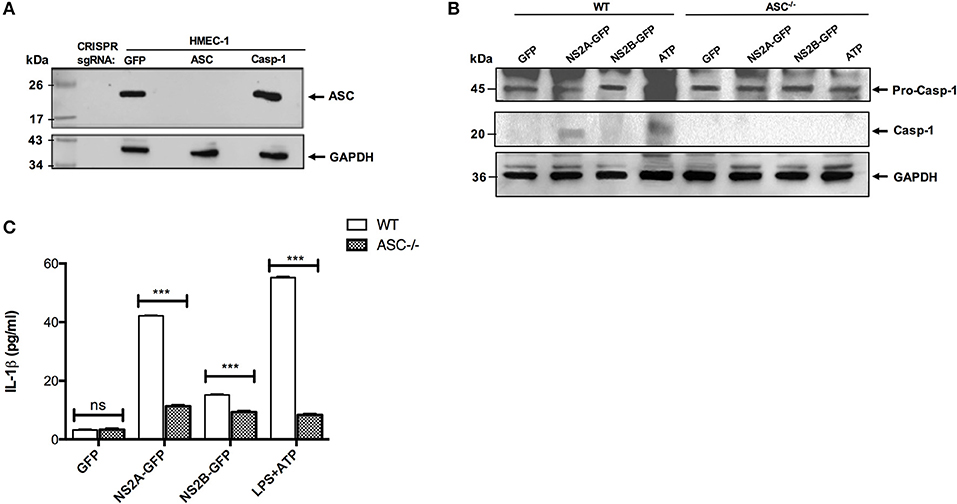
Figure 4. Confirmation of inflammasome activation by viroporin via CRISPR-CAS 9. (A) HMEC-1 lysates expressing lentiCRISPRv2-GFP/Caspase/ASC were resolved by western blot. The primary antibody against ASC was used at a 1:1,000 dilution. HRP-anti rabbit was used as the secondary antibody (1:5,000). (B) ASC−/− HMEC-1 cells and WT HMEC-1 cells were primed with LPS 1 μg/mL for 6 h, followed by transfection with GFP, NS2A-GFP, and NS2B-GFP for 36 h. Positive control cells were primed with LPS 1 μg/mL for 6 h, followed by 5 mM ATP for 45 min. Lysates were analyzed using western blot. Caspase-1 primary antibody was used at a 1:1,000 dilution. HRP-anti rabbit was used as the secondary antibody (1:5,000). (C) HMEC-1 cells (WT or ASC−/−) were transfected with the expression plasmid encoding DENV-2 NS2A-GFP, NS2B-GFP or the pEGFPN1 parental vector for 36 h or treated with LPS (1 μg/mL) for 6 h followed by ATP (5 mM) for 45 min as a positive control, and the cell free supernatant was analyzed for IL-1β by ELISA. ***P < 0.001.
NS2A and NS2B Mediated Inflammasome Activation Was Dependent on NLRP3 and Caspase-1
Further, we examined whether IL-1β release was dependent on the NLRP3 inflammasome activation induced by DENV viral proteins (NS2A, NS2B). We found the absence of caspase-1 activation in the presence of glyburide (NLRP3 inhibitor) in extracts from cells transfected with pNS2B-GFP and a significant reduction in cells transfected with pNS2A-GFP. However, activation of caspase-1 was observed in non-treated cells due to pNS2A-GFP or pNS2B-GFP (Figure 5A). Similarly, IL-1β secretion was reduced in the presence of the NLRP3 inhibitor, suggesting that NS2A and NS2B activate caspase-1 and promote secretion of IL-1β, are dependent on NLRP3 (Figure 5B). Studies also suggested the existence of other pathways through which IL-1β is secreted (40, 41). Therefore, to determine whether secretion of IL-1β was dependent on caspase-1, HMEC-1 cells were treated with YVAD (a caspase-1 inhibitor) for 1 h prior to transfection with the pNS2A-GFP and pNS2B-GFP plasmids. After 36 h post-transfection, a reduced secretion of IL-1β in the presence of the caspase-1 inhibitor (YVAD) was observed, suggesting that NS2A and NS2B-dependent secretion of IL-1β requires caspase-1 activation (Figure 5C). As a positive control, ATP was used; however, the NLRP3 inhibitor had low effect on caspase 1 activation, as observed by western blot, while IL-1β secretion was reduced up to 50 %, as observed by ELISA. However, in the presence of the caspase-1 inhibitor, secretion of IL-1β was reduced by 50% due to ATP. These observations suggested that activation of the inflammasome, and subsequently, secretion of IL-1β in the cell supernatant by dengue virus viroporins NS2A and NS2B were specific to NLRP3 and Caspase-1.
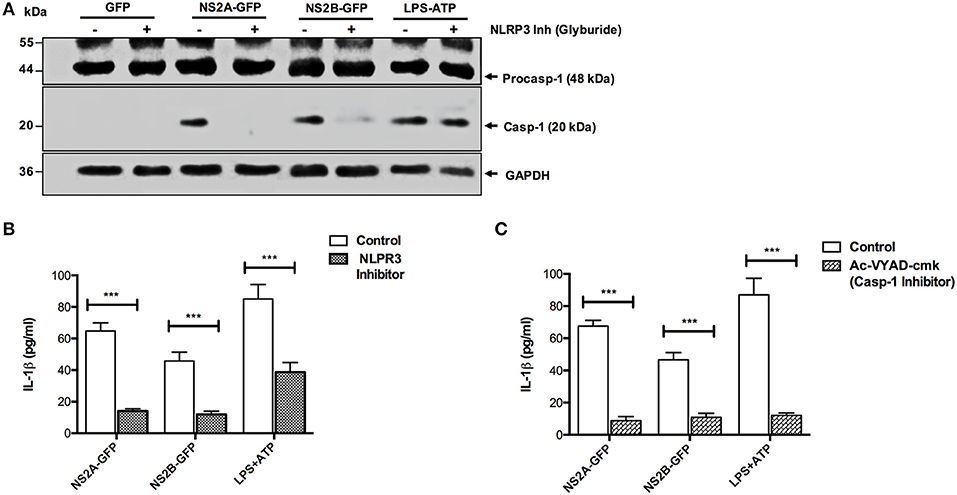
Figure 5. NLRP3 and caspase-1 specific activation of the inflammasome by NS2A and NS2B. HMEC-1 cells were transfected with expression plasmids encoding NS2A-GFP, NS2B-GFP or the pEGFPN1 empty vector for 36 h or treated with LPS (1 μg/mL) for 6 hrs followed by ATP (5 mM) for 1 h as a positive control, in the presence or absence of Glyburide (200 μM). (A) The cell lysates were analyzed by western blot using an anti-Caspase 1 antibody (1:1,000), and (B) the cell free supernatant was analyzed for IL-1β by ELISA. (C) HMEC-1 cells were transfected with expression plasmids encoding NS2A-GFP, NS2B-GFP or the pEGFPN1 empty vector for 36 h or treated with LPS (1 μg/mL) for 6 h followed by ATP (5 mM) for 45 min as a positive control, in the presence or absence of AcVYAD-cmk (50 μM), and the cell free supernatant was analyzed for IL-1β by ELISA. Data are representative of at least three independent experiments and indicate the mean ± S.D. (B,C). ***P < 0.001.
Regulation of Ca++ Might Impair Inflammasome Activation
NS2A and NS2B proteins are mainly localized at the endoplasmic reticulum (ER) (33, 42). We therefore investigated whether the activation of NLRP3 involved the release of Ca++ from organelles. To this end, we analyzed if a cell-permeable Ca++chelator BAPTA-AM inhibit inflammasome activation induced by NS2A-GFP, NS2B-GFP. We found that cells treated with the Ca++ chelator significantly decreased caspase-1 activation due to DENV-2 NS2A, NS2B, and ATP (Figure 6A). Furthermore, treatment of HMEC-1 with BAPTA-AM significantly blocked IL-1β secretion by DENV-2 NS2A (Figure 6B). These results suggest that DENV viroporins may induce Ca++ flux in the cytoplasm from intracellular storages, which activate the NLRP3 inflammasome.
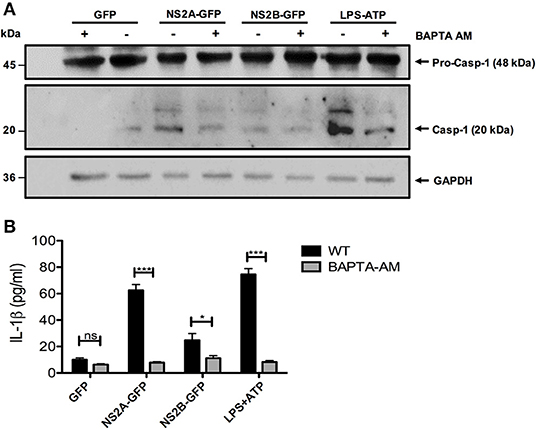
Figure 6. Requirement of increased intracellular Ca++ concentration for NLRP3 inflammasome activation by NS2A and NS2B. HMEC-1 cells were transfected with plasmids encoding NS2A-GFP, NS2B-GFP, or the pEGFPN1 empty vector for 36 h or treated with LPS (1 μg/mL) for 6 hrs followed by ATP (5 mM) for 45 min as a positive control, in the presence or absence of BAPTA-AM (10 μM). (A) The cell lysates were analyzed by western blot using an anti-caspase-1 antibody (1:1,000), and (B) the cell free supernatants were analyzed for IL-1β by ELISA. Data are representative of at least three independent experiments, and indicate the mean ± S.D. (B). *P < 0.05 and ***P < 0.001. ns, non significant.
ROS Generation Is Required for DENV-2, NS2A Driven NLRP3 Inflammasome
Several viral proteins including viroporins induce membrane permeability, ionic imbalance and can disrupt mitochondria functions (43, 44). Therefore, we investigated if DENV-2 infection affects the mitochondria in HMEC-1 cells. As shown in Figure 7A elongated mitochondria were detected in infected HMEC-1 when compared to uninfected HMEC-1 cells which exhibit a typical mitochondria ultrastructural morphology (Mt) (Figure 7A). In addition, as expected from our previous results NS2A over-expression induced changes in the mitochondrial morphology, those mithocondria appearing as fragmented, longed entities or with perinuclear localization, in contrast to the typical elongated healthy mitochondria (Figure 7B). To further analyze the effects of DENV-2 viroporin on the mitochondria, we evaluated whether DENV viroporins could change the mitochondrial membrane potential (ΔΨm). We found that the flow cytometric distribution of the fluorescence intensity of the ΔΨm indicator in HMEC-1 transfected with pNS2A-GFP decreased membrane potential and fail to sequester TMRE approximately 1.84-fold compared to that in HMEC-1 cells transfected with pEGFPN1 (Figure 7C). NS2B-transfected HMEC-1 cells showed only a minor effect in mitochondrial membrane potential compared to HMEC-1 cells transfected with pEGFPN1 (Figures 7C,D). These data suggest that NS2A induce changes in mitochondrial integrity and polarized the mitochondrial membrane potential (ΔΨm).
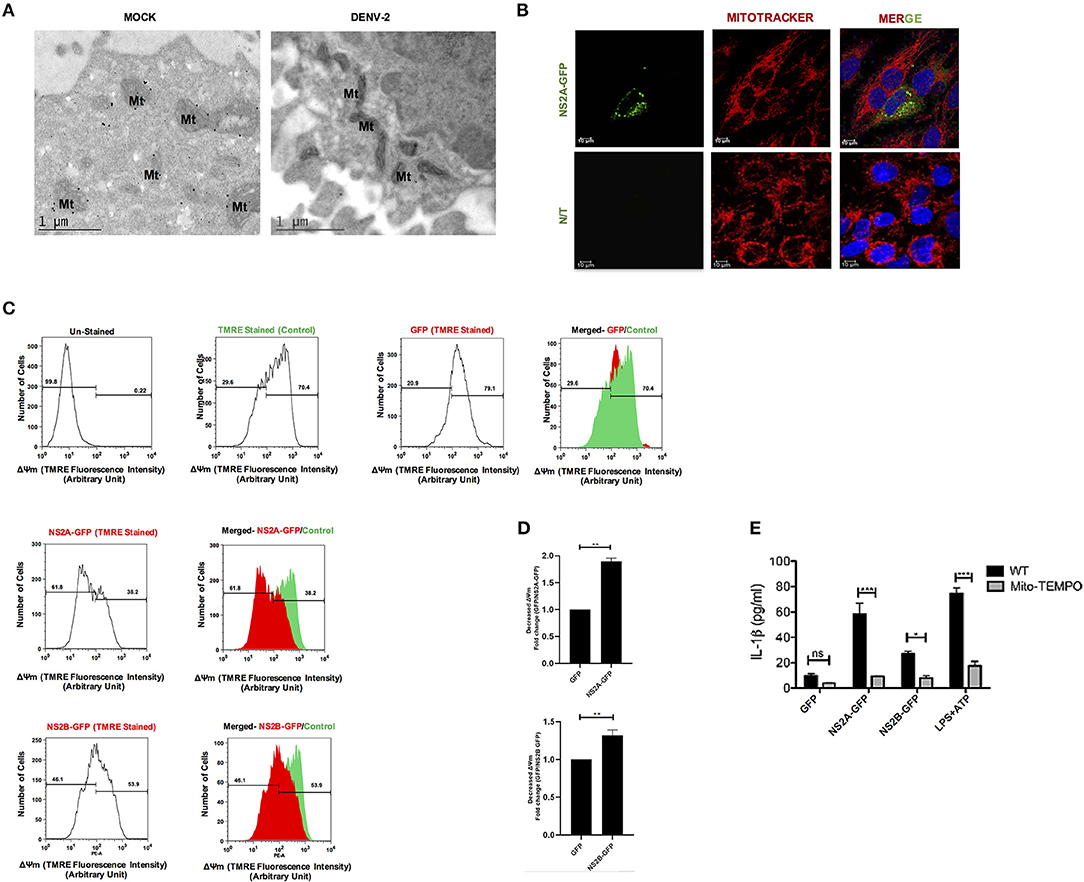
Figure 7. DENV-2 viroporin induce the NLRP3 inflammasome through the mitochondria. (A) Ultra-thing sections (TME) images (70 nm) of resin-embedded HMEC-1 cells, stained with uranyl acetate. Ultrastructural analysis of non-infected and DENV-2 infected HMEC-1 cells show their typical structure of mitochondria (Mt), captured by Jeol JEM-1011 transmission electron microscope (JeolLtd., Tokyo, Japan). (B) To evaluate the localization of the NS2A protein within mitochondria, HMEC-1 cells were transfected with pNS2A-GFP for 36 h, stained with Mito Tracker (mitochondrial marker; red) and analyzed by confocal microscopy. (C,D) Mitochondrial membrane potential was detected in HMEC-1 cells transfected with plasmids encoding NS2A-GFP, NS2B-GFP, or the pEGFPN1 parental plasmid for 36 h and stained with TMRE (175 mM) for 30 min and analyzed using flow cytometry at 488 nm (excitation peak-595 nm, emission-575 nm). (E) HMEC-1 cells were transfected with expression plasmids encoding NS2A-GFP, NS2B-GFP, or the pEGFPN1 empty vector for 36 h, treated with LPS (1 μg/mL) for 6 h followed by ATP (5 mM) for 45 min as a positive control, in the presence or absence of Mito-TEMPO (10 μM), and the cell free supernatant was analyzed for IL-1β by ELISA. Data are representative of at least three independent experiments and indicate the mean ± S.D. *P < 0.05, **P < 0.01, and ***P < 0.001. ns, non significant.
Additionally, mitochondria are reported to play a crucial role in the activation of the NLRP3 inflammasome (45–47). Specifically, mitochondrial ROS has found to be important in fueling NLRP3 inflammasome activation (48, 49). Therefore, we tested the effect of Mito-TEMPO, a scavenger specific for mitochondrial ROS (50, 51), in the activation of the NLRP3 inflammasome due to DENV-2 viroporins. Treatment with the antioxidant Mito-TEMPO had inhibitory effects on the secretion of IL-1β in HMEC-1 transfected with DENV-2 pNS2A-GFP and pNS2B-GFP, as well as on the response to ATP (Figure 7E). Thus, our data strongly suggest that the generation of ROS during DENV-2 viroporin expression might act as a stress signal for inflammasome activation, which in turn is crucial for IL-1β production.
Discussion
The main finding of our present study is that DENV-2, a positive-strand RNA virus, triggers NLRP3 inflammasome–mediated IL-1β production through the expression of the virus-encoded NS2A, NS2B proteins. Our data reveal the physiopathologic relevance of the NLRP3 inflammasome during DENV-2 infection, which may provide therapeutic targets along these pathways for novel anti-DENV-2 treatment.
Endogenous pyrogens (Ex: IL-1β) is induced during dengue virus infection causes fever, a primary symptom of the disease (4). IL-1β processing and release is regulated by caspase-1 through the activation of an inflammasome complex (10). Although dengue virus pathogenesis is not fully elucidated, recent evidence support the central role of pro-inflammatory cytokines in endothelial activation and plasma leakage during DENV-2 infection (4, 6, 52). In this study, we found that DENV-2 triggered NLRP3 activation in endothelial cells (HMEC-1). Microvascular endothelial cells are regarded as the permissive target of DENV-2 (53) and we have demonstrated that DENV-2 is able to activate the inflammasome complex by NLRP3 expression, ASC oligomerization, and caspase-1 activation, following IL-1β secretion in the cell supernatant. These data are in agreement with a recent study that demonstrate NLRP3 inflammasome activation in DENV-2-infected macrophages in cultures and in platelets (12, 13). In addition, a study reported the increased expression of caspase-1 in DENV-2-infected cultured cells (54).
Several studies have reported NLRP3 and RIG-I inflammasome-induced caspase-1 activation during RNA virus infections (55–58). An array of RNA viruses have also been shown to induce IL-1β production through the NLRP3 inflammasome (59). Flaviviruses, including the West Nile virus, swine fever virus (CSFV), Japanese encephalitis virus, and hepatitis C virus, have been shown to assemble the NLRP3 inflammasome and promote IL-1β production during infection (60–63). Our results clearly demonstrated that DENV-2 was able to activate the NLRP3 inflammasome on its own, without the prerequisite for priming the cells with another pathogen associated molecular pattern (PAMP), which was previously shown to be necessary for other viruses, such as encephalomyocarditis virus and stomatitis vesicular virus (VSV) (63). A wide range of stimuli have been reported to activate the NLRP3 inflammasome, including bacterial components, environment irritants, endogenous danger signals from damaged cells, and other viruses (64, 65). Three mechanisms for NLRP3 inflammasome activation have been suggested so far. First, according to the “ion channel model,” high concentrations of extracellular ATP can induce K+ efflux through the P2X7 ATP-gated ion channel, by forming cell membrane pannexin-1 pores, and further facilitating the influx of PAMPs and damage-associated molecular patterns (DAMPs), which trigger NLRP3 activation (66). The second model is the “lysosomal rupture model,” in which large crystals and environmental irritants are phagocytosed and induce the release of cathepsin B by lysosome rupture. Released cathepsin B further activates NLRP3 (67). The third model is “mitochondrial ROS,” in which damaged mitochondria produce a large amount of ROS that stimulates NLRP3 inflammasome activation (68, 69). Although a recent study on macrophages and platelets have shown the importance of the Syk-coupled C-type lectin CLEC5A and RIP kinases during DENV-2-induced NLRP3 inflammasome activation (12, 13), no direct role of dengue proteins in the activation of inflammasomes have been reported.
We recently reported that dengue virus NS2B and NS2A proteins behave as viroporins (28, 29). Therefore, we investigated the mechanism by which the proposed dengue virus like-viroporins (NS2A & NS2B) activate the NLRP3 inflammasome, as several viroporins of RNA viruses have been reported to activate the NLRP3 inflammasome (14). Our results showed a clear activation of the NLRP3 inflammasome by both the DENV-2 NS2B and NS2A proteins.
Several viruses encode viroporins that increase the permeability of host cellular, endoplasmic, and mitochondrial membranes, facilitating virus entry and exit mechanisms. The increased ion concentration helps viral replication and transcription. Increased ion concentration (Na+, K+, Ca++) by viroporins trigger NLRP3 inflammasome activation (14). Using the DENV-2 viroporins, NS2A and NS2B GFP fused-proteins, we have shown that DENV-2 NS2A and NS2B were able to activate the NLRP3 inflammasome complex following IL-1β secretion in the cell supernatant. Furthermore, NS2A was shown to colocalize with NLRP3. Recently, it has been demonstrated that DENV-2 envelope protein domain III and M proteins also trigger NLRP3 activation (34, 35). Furthermore, studies have reported that some RNA viruses (Chikungunya and Zika) activate the AIM2-specific inflammasome (70, 71). Since several DNA viruses have been reported to be sensed by AIM2, the mechanism by which AIM2 senses RNA viruses remains unknown. Here, we demonstrated that IL-1β secretion by NS2A and NS2B was specific to NLRP3 and caspase-1.
In platelets, inflammasome induction by Dengue virus is dependent on ROS production in the mitochondria (13). We observed that IL-1β production induced by both NS2A and NS2B was dependent on ROS; one hypothesis is that the expression of these proteins in mitochondria could be part of the mechanism. Induction of IL-1β secretion has two components; since the secretion was inhibited by both Mito-TEMPO and BAPTA-AM, these suggest that both Ca++ and ROS production are necessary for IL-1β secretion. Alternatively, one of these events induces the other. In this sense, it was reported that ATP can activate the inflammasome by inducing Ca++ that triggers mitochondrial ROS production (72); it has already been reported that viroporins regulate NLRP3 activation through calcium mobilization (25). These studies support our results.
In summary, the current study shows that DENV-2 infection triggers inflammasome and high level of IL-1β in HMEC-1 as well as in THP-1, HepG2 via NLRP3 inflammasome activation. We also oberved that the maturation and secretion of IL-1β during DENV-2 infection is mediated by NLRP3 inflammasome. DENV-2 NS2A and NS2B protein were found to facilitate the assembly of the NLRP3 inflammasome complex and lead to caspase-1 activation and IL-1β secretion through calcium mobilization or by disrupting mitochondria potential and by inducing ROS production. Further research is required for an in-depth understanding the role of these viroporins in the regulation of innate immunity. These results reveal a novel mechanism for the DENV-2-mediated inflammatory response, which may provide therapeutic targets along these pathways for novel strategies to treat DENV-2 associated disease.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
GS and GV-C performed research, analyzed data, and wrote the paper. JG-C, GV-C, ML-J, and BC-M performed research, analyzed the data, and prepared the images. GS, GV-C, JG-C, BC-M, ML-J, TL, PN, NV-S, and LC-B conceived, designed, conducted the research, interpreted data, and wrote the paper.
Funding
This work was supported by Centro de Investigacion y Estudios Avanzados-IPN. LC-B and in part by funds allocated to NV-S by CINVESTAV and by grant 256261 from CONACyT.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the microscope and cytometry facility team of the CINVESTAV. We thank Raul Bonilla for excellent technical assistance. GS and GV-C are fellow holders from the National Council for Science and Technology (CONACYT) Mexico. Additionally, JG-C, ML-J, PN, TL, NV-S, and LC-B are members of the National System of Researchers, SNI.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00352/full#supplementary-material
Figure S1. DENV-2 and NS2A, NS2B tagged with GFP activate NLRP3 inflammasome in HepG2 cells. Cells were infected with DENV-2 at (5 MOI), mock infected or treated with LPS (1 μg/mL) for 6 hrs followed by ATP (5 mM) for 45 min as a positive control. (A) Cell lysates were analyzed by western blot using an anti-caspase-1 antibody (1:1,000) and anti-NS5 antibody (1:100); (B) HMEC-1 were infected with DENV-2 (5 MOI) for 36 h or treated with medium. Immunofluorescence with double staining was performed in HMEC-1 using anti-NS5 antibody (1:100) and anti-NLRP3 antibody (1:500) (C) HepG2 cells were transfected with either parental plasmid eGFPN1, pNS2A-GFP, pNS2B-GFP, or treated with LPS (1 μg/mL) for 6 h followed by ATP (5 mM) for 1 h as a positive control. At 36 h post-tranfection, cell lysates were analyzed for western blot with anti-caspase-1 antibody (1:1,000). ←Expression of NLRP3.
Figure S2. THP1 cells infected wit DENV 2 trigger the NLRP3 inflammasome activation. THP-1 cells grown in RPMI 1640 medium supplemented with 10% FBS in a 37°C incubator with 5% CO2 were differentiated for 1 days with 100nM phorbol-12-myristate-13-acetate (PMA), followed by DENV-2 infection at 5 MOIs or mock infected. At 24 and 48 h cell lysates and supernatants were obtained. (A) Infection was detected by western blot with anti-NS3 dengue antibody (Genetex USA). (B) Western blot of the same lysates were analyzed with Pro-Caspase 1, and GAPDH. (C) Caspasa and IL-1β. (D) Inflammasome Assay monitor released caspase-1 in culture medium. THP-1 cells grown in RPMI 1640 medium supplemented with 10% FBS in a 37°C incubator with 5% CO2 were differentiated for 1 days with 100 nM phorbol-12-myristate-13-acetate (PMA), followed by infected with either Mock or DENV-2 (5 MOIs for 2 h). After the 24 and 48 h post-infection half of the culture medium (50 μl/well) was transferred to a second plate, 50 μl/well of Caspase-Glo® 1 Reagent or Caspase-Glo® 1 YVAD-CHO Reagent was added and luminescence was recorded using a GloMax® Multi+ Detection System as directed in the GloMax® Multi+ Detection System with Instinct® Software Technical Manual #TM340. For cells, 100 μl/well of reagent was added directly to 100μl/well of cultured cells. (E) THP-1 cells grown in RPMI 1640 medium supplemented with 10% FBS in a 37°C incubator with 5% CO2 were differentiated for 1 days with 100 nM phorbol-12-myristate-13-acetate (PMA), followed by infected with either Mock or DENV-2 (5 MOIs for 2 h). After the 24 and 48 h post-infection, supernatants were collected and IL-1β was checked using R&D IL-1β Elisa kit. ns, non significant, ***P < 0.001.
Figure S3. The expression of NS2B in infected HMEC-1 cells has the same distribution as transfected pNS2B-GFP. (A) HMEC-1 cells were infected with DENV-2 at 5 MOI. 24 and 48 h post-infection, the cells were fixed and stained with anti-NS2B polyclonal antibody (RED) and then analyzed by confocal microscopy (B) To evaluate the distribution of NS2B, HMEC-1 cells were infected with DENV-2 at 5 MOI for 24 h, further the same cells were transiently transfected with plasmid coding for NS2B-GFP and analyzed at 24 h post-transfection. Cells were fixed with 4 % paraformaldehyde and stained with anti-NS2B polyclonal antibody (RED) and analyzed by confocal microscopy.
Figure S4. Confirmation of guide RNA cloning in LentiCRISPR plasmid. (A) Guide RNA specific to NLRP3, ASC and Caspase-1 were cloned in LentiCRISPRv2 plasmid, according to protocol. Clones were transformed in STBL3 bacteria and Colony PCR of transformed clones, specific to lenti-CRISPRv2 (NLRP3, Caspase-1, ASC), was performed. Bands corresponding to 125 bp showed positive clones for the respective Guide RNA. Amplified PCR were resolved using 0.8% agarose gel. (B) Sequence of guide RNA used.
References
1. Schmidt AC. Response to dengue fever–the good, the bad, and the ugly? N Engl J Med. (2010) 363:484–7. doi: 10.1056/NEJMcibr1005904
2. World Health Organization. Dengue: guidelines for diagnosis, treatment, prevention, and control. Spec Program Res Train Trop Dis. World Health Organization (2009) 147. Available online at: https://apps.who.int/iris/
3. Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, et al. Dengue in the early febrile phase: viremia and antibody responses. J Infect Dis. (1997) 176:322–330. doi: 10.1086/514048
4. Bozza FA, Cruz OG, Zagne SMO, Azeredo EL, Nogueira RMR, Assis EF, et al. Multiplex cytokine profile from dengue patients: MIP-1beta and IFN-gamma as predictive factors for severity. BMC Infect Dis. (2008) 8:86. doi: 10.1186/1471-2334-8-86
5. Malavige GN, Ogg GS. Pathogenesis of vascular leak in dengue virus infection. Immunology. (2017) 51:261–9. doi: 10.1111/imm.12748
6. Suharti C, Van Gorp ECM, Setiati TE, Dolmans WMV, Djokomoeljanto RJ, Hack CE, et al. The role of cytokines in activation of coagulation and fibrinolysis in dengue shock syndrome. Thromb Haemost. (2002) 87:42–46. doi: 10.1055/s-0037-1612941
7. Callaway JB, Smith SA, McKinnon KP, de Silva AM, Crowe JE Jr, Ting JP. Spleen Tyrosine Kinase (Syk) mediates IL-1β induction by primary human monocytes during antibody-enhanced dengue virus infection. J Biol Chem. (2015) 290:17306–20. doi: 10.1074/jbc.M115.664136
8. Houghton-Trivino N, Martin K, Giaya K, Rodriguez JA, Bosch I, Castellanos JE. [Comparison of the transcriptional profiles of patients with dengue fever and dengue hemorrhagic fever reveals differences in the immune response and clues in immunopathogenesis. Biomedica. (2010) 30:587–97. doi: 10.7705/biomedica.v30i4.297
9. Jaiyen Y, Masrinoul P, Kalayanarooj S, Pulmanausahakul R, Ubol S. Characteristics of dengue virus-infected peripheral blood mononuclear cell death that correlates with the severity of illness. Microbiol Immunol. (2009) 53:442–50. doi: 10.1111/j.1348-0421.2009.00148.x
10. Davis BK, Wen H, Ting JP-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. (2011) 29:707–35. doi: 10.1146/annurev-immunol-031210-101405
11. Lamkanfi M, Dixit VM. Modulation of inflammasome pathways by bacterial and viral pathogens. J Immunol. (2011) 187:597–602. doi: 10.4049/jimmunol.1100229
12. Wu MF, Chen ST, Yang AH, Lin WW, Lin YL, Chen NJ, et al. CLEC5A is critical for dengue virus-induced inflammasome activation in human macrophages. Blood. (2013) 121:95–106. doi: 10.1182/blood-2012-05-430090
13. Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. (2013) 122:3405–14. doi: 10.1182/blood-2013-05-504449
14. Guo HC, Jin Y, Zhi XY, Yan D, Sun SQ. NLRP3 inflammasome activation by viroporins of animal viruses. Viruses. (2015) 7:3380–91. doi: 10.3390/v7072777
15. Sze CW, Tan YJ. Viral membrane channels: role and function in the virus life cycle. Viruses. (2015) 7:3261–84. doi: 10.3390/v7062771
16. Luis Nieva J, Carrasco L. Viroporins: structures and functions beyond cell membrane permeabilization. Viruses. (2015) 7:5169–71. doi: 10.3390/v7102866
17. Nieto-Torres JL, Verdiá-Báguena C, Castaño-Rodriguez C, Aguilella VM, Enjuanes L. Relevance of viroporin ion channel activity on viral replication and pathogenesis. Viruses. (2015) 7:3552–73. doi: 10.3390/v7072786
18. Hyser JM, Collinson-Pautz MR, Utama B, Estes MK. Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. mBio. (2010) 1:e00265-10. doi: 10.1128/mBio.00265-10
19. Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, et al. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. (2003) 535:34–8. doi: 10.1016/S0014-5793(02)03851-6
20. Ito M, Yanagi Y, Ichinohe T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. (2012) 8:e1002857. doi: 10.1371/journal.ppat.1002857
21. Suzuki T, Orba Y, Okada Y, Sunden Y, Kimura T, Tanaka S, et al. The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Pathog. (2010) 6:e1000801. doi: 10.1371/journal.ppat.1000801
22. Aldabe R, Irurzun A, Carrasco L. Poliovirus protein 2BC increases cytosolic free calcium concentrations. J Virol. (1997) 71:6214–7. doi: 10.1128/JVI.71.8.6214-6217.1997
23. Henkel M, Mitzner D, Henklein P, Meyer-Almes FJ, Moroni A, Difrancesco ML, et al. The proapoptotic influenza A virus protein PB1-F2 forms a nonselective ion channel. PLoS ONE. (2010) 5:e11112. doi: 10.1371/journal.pone.0011112
24. Van Kuppeveld FJM, Hoenderop JGJ, Smeets RLL, Willems PHGM, Dijkman HBPM, Galama JMD, et al. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. (1997) 16:3519–32. doi: 10.1093/emboj/16.12.3519
25. Hyser JM, Estes MK. Pathophysiological Consequences of Calcium-Conducting Viroporins. Annu Rev Virol. (2015) 2:473–96. doi: 10.1146/annurev-virology-100114-054846
26. Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol. (2017) 2:17037. doi: 10.1038/nmicrobiol.2017.37
27. Yu CY, Liang JJ, Li JK, Lee YL, Chang BL, Su CI, et al. Dengue virus impairs mitochondrial fusion by cleaving mitofusins. PLoS Pathog. (2015) 11:e1005350. doi: 10.1371/journal.ppat.1005350
28. León-Juárez M, Martínez-Castillo M, Shrivastava G, García-Cordero J, Villegas-Sepulveda N, Mondragón-Castelán M, et al. Recombinant Dengue virus protein NS2B alters membrane permeability in different membrane models. Virol J. (2016) 13:1. doi: 10.1186/s12985-015-0456-4
29. Shrivastava G, García-Cordero J, León-Juárez M, Oza G, Tapia-Ramírez J, Villegas-Sepulveda N, et al. NS2A comprises a putative viroporin of Dengue virus 2. Virulence. (2017) 8:1450–6. doi: 10.1080/21505594.2017.1356540
30. Leung JY, Pijlman GP, Kondratieva N, Hyde J, Mackenzie JM, Khromykh AA. Role of nonstructural protein NS2A in flavivirus assembly. J Virol. (2008) 82:4731–41. doi: 10.1128/JVI.00002-08
31. Xie X, Gayen S, Kang C, Yuan Z, Shi PY. Membrane topology and function of dengue virus NS2A protein. J Virol. (2013) 87:4609–22. doi: 10.1128/JVI.02424-12
32. Xie X, Zou J, Puttikhunt C, Yuan Z, Shi PY. Two distinct sets of NS2A molecules are responsible for dengue virus RNA synthesis and virion assembly. J Virol. (2015) 89:1298–313. doi: 10.1128/JVI.02882-14
33. Nemésio H, Villalaín J. Membrane interacting regions of Dengue virus NS2A protein. J Phys Chem B. (2014) 118:10142–55. doi: 10.1021/jp504911r
34. Khan RA, Afroz S, Minhas G, Battu S, Khan N. Dengue virus envelope protein domain III induces pro-inflammatory signature and triggers activation of inflammasome. Cytokine. (2019) 123:154780. doi: 10.1016/j.cyto.2019.154780
35. Pan P, Zhang Q, Liu W, Wang W, Lao Z, Zhang W, et al. Dengue virus M protein promotes NLRP3 inflammasome activation to induce vascular leakage in mice. J Virol. (2019) 93:e00996-19. doi: 10.1128/JVI.00996-19
36. Gruenberg A, Woo WS, Biedrzycka A, Wright PJ. Partial nucleotide sequence and deduced amino acid sequence of the structural proteins of dengue virus type 2, New Guinea C and PUO-218 strains. J Gen Virol. (1988) 69:1391–8. doi: 10.1099/0022-1317-69-6-1391
37. García Cordero J, León Juárez M, González-Y-Merchand JA, Cedillo Barrón L, Gutiérrez Castañeda B. Caveolin-1 in lipid rafts interacts with dengue virus NS3 during polyprotein processing and replication in HMEC-1 cells. PLoS ONE. (2014) 9:e90704. doi: 10.1371/journal.pone.0090704
38. García-Cordero J, Carrillo-Halfon S, León-Juárez M, Romero-Ramírez H, Valenzuela-León P, López-González M, et al. Generation and characterization of a rat monoclonal antibody against the RNA polymerase protein from Dengue Virus-2. Immunol Invest. (2014) 43:28–40. doi: 10.3109/08820139.2013.833622
39. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
40. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. (2011) 22:189–95. doi: 10.1016/j.cytogfr.2011.10.001
41. Eder C. Mechanisms of interleukin-1beta release. Immunobiology. (2009) 214:543–53. doi: 10.1016/j.imbio.2008.11.007
42. Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, et al. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. (2009) 5:365–75. doi: 10.1016/j.chom.2009.03.007
43. Madan V, Castelló A, Carrasco L. Viroporins from RNA viruses induce caspase-dependent apoptosis. Cell Microbiol. (2008) 10:437–51. doi: 10.1111/j.1462-5822.2007.01057.x
44. Anand SK, Tikoo SK. Viruses as modulators of mitochondrial functions. Adv Virol. (2013):738–94. doi: 10.1155/2013/738794
45. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. doi: 10.1038/nature09663
46. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNAactivates the NLRP3 inflammasome during apoptosis. Immunity. (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
47. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
48. Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol. (2013) 191:5230–8. doi: 10.4049/jimmunol.1301490
49. Sorbara MT, Girardin SE. Mitochondrial ROS fuel the inflammasome. Cell Res. (2011) 21:558–60. doi: 10.1038/cr.2011.20
50. Trnka J, Blaikie FH, Logan A, Smith RA, Murphy MP. Antioxidant properties of MitoTEMPOL and its hydroxylamine. Free Radic Res. (2009) 43:4–12. doi: 10.1080/10715760802582183
51. Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, et al. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. (2010) 107:106–16. doi: 10.1161/CIRCRESAHA.109.214601
52. Pang T, Cardosa MJ, Guzman MG. Of cascades and perfect storms: the immunopathogenesis of dengue haemorrhagic fever-dengue shock syndrome (DHF/DSS). Immunol Cell Biol. (2007) 85:43–45. doi: 10.1038/sj.icb.7100008
53. Talavera D, Castillo AM, Dominguez MC, Gutierrez AE, Meza I. IL8 release, tight junction and cytoskeleton dynamic reorganization conducive to permeability increase are induced by dengue virus infection of microvascular endothelial monolayers. J Gen Virol. (2004) 85(Pt 7):1801–13. doi: 10.1099/vir.0.19652-0
54. Nasirudeen AM, Liu DX. Gene expression profiling by microarray analysis reveals an important role for caspase-1 in dengue virus-induced p53-mediated apoptosis. J Med Virol. (2009) 81:1069–81. doi: 10.1002/jmv.21486
55. Wang X, Jiang W, Yan Y, Gong T, Han J, Tian Z, et al. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat Immunol. (2014) 15:1126–33. doi: 10.1038/ni.3015
56. Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. (2008) 20:17–22. doi: 10.1016/j.coi.2008.01.002
57. Kell AM, Gale M Jr. RIG-I in RNA virus recognition. Virology. (2015) 479–80:110–21. doi: 10.1016/j.virol.2015.02.017
58. Jacobs SR, Damania B. NLRs, inflammasomes, and viral infection. J Leukoc Biol. (2012) 92:469–77. doi: 10.1189/jlb.0312132
59. Chen IY, Ichinohe T. Response of host inflammasomes to viral infection. Trends Microbiol. (2015) 23:55–63. doi: 10.1016/j.tim.2014.09.007
60. Kaushik DK, Gupta M, Kumawat KL, Basu A. NLRP3 inflammasome: key mediator of neuroinflammation in murine Japanese encephalitis. PLoS ONE. (2012) 7:e32270. doi: 10.1371/journal.pone.0032270
61. Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. (2013) 9:e1003330. doi: 10.1371/journal.ppat.1003330
62. Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, et al. IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. (2012) 8:e1003039. doi: 10.1371/journal.ppat.1003039
63. Fan S, Yuan J, Deng S, Chen Y, Xie B, Wu K, et al. Activation of interleukin-1β release by the classical swine fever virus is dependent on the NLRP3 inflammasome, which affects virus growth in monocytes. Front Cell Infect Microbiol. (2018) 8:225. doi: 10.3389/fcimb.2018.00225
64. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
65. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
66. Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. (2007) 26:433–43. doi: 10.1016/j.immuni.2007.03.008
67. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. (2008) 9:847–56. doi: 10.1038/ni.1631
68. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. (2007) 282:2871–9. doi: 10.1074/jbc.M608083200
69. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. (2010) 11:136–40. doi: 10.1038/ni.1831
70. Ekchariyawat P, Hamel R, Bernard E, Wichit S, Surasombatpattana P, Talignani L, et al. Inflammasome signaling pathways exert antiviral effect against Chikungunya virus in human dermal fibroblasts. Infect Genet Evol. (2015) 32:401–8. doi: 10.1016/j.meegid.2015.03.025
71. Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, et al. Biology of zika virus infection in human skin cells. J Virol. (2015) 89:8880–96. doi: 10.1128/JVI.00354-15
Keywords: dengue, inflammasome, non-structural proteins NS2A and NS2B, viroporins, IL-1β, NLRP3, Caspase-1
Citation: Shrivastava G, Visoso-Carvajal G, Garcia-Cordero J, Leon-Juarez M, Chavez-Munguia B, Lopez T, Nava P, Villegas-Sepulveda N and Cedillo-Barron L (2020) Dengue Virus Serotype 2 and Its Non-Structural Proteins 2A and 2B Activate NLRP3 Inflammasome. Front. Immunol. 11:352. doi: 10.3389/fimmu.2020.00352
Received: 30 April 2019; Accepted: 13 February 2020;
Published: 10 March 2020.
Edited by:
Rosana Pelayo, Mexican Social Security Institute, MexicoReviewed by:
Eva Harris, University of California, Berkeley, United StatesGerardo Santos-López, Biomedical Research Center of East (CIBIOR), Mexico
Philippe Desprès, Université de la Réunion, France
Copyright © 2020 Shrivastava, Visoso-Carvajal, Garcia-Cordero, Leon-Juarez, Chavez-Munguia, Lopez, Nava, Villegas-Sepulveda and Cedillo-Barron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leticia Cedillo-Barron, bGNlZGlsbG9AY2ludmVzdGF2Lm14
†Present address: Gaurav Shrivastava, Laboratory of Malaria and Vector Research, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Rockville, MD, United States