 Yukinori Take
Yukinori Take Shinichi Koizumi
Shinichi Koizumi Atsushi Nagahisa
Atsushi Nagahisa- 1AskAt Inc., Nagoya, Japan
- 2DNA Partners, Fujisawa, Japan
A highly expressed prostaglandin E2 (PGE2) in tumor tissues suppresses antitumor immunity in the tumor microenvironment (TME) and causes tumor immune evasion leading to disease progression. In animal studies, selective inhibition of the prostaglandin E receptor 4 (EP4), one of four PGE2 receptors, suppresses tumor growth, restoring the tumor immune response toward an antitumorigenic condition. This review summarizes PGE2/EP4 signal inhibition in relation to the cancer-immunity cycle (C-IC), which describes fundamental tumor-immune interactions in cancer immunotherapy. PGE2 is suggested to slow down C-IC by inhibiting natural killer cell functions, suppressing the supply of conventional dendritic cell precursors to the TME. This is critical for the tumor-associated antigen priming of CD8+ T cells and their translocation to the tumor tissue from the tumor-draining lymph node. Furthermore, PGE2 activates several key immune-suppressive cells present in tumors and counteracts tumoricidal properties of the effector CD8+ T cells. These effects of PGE2 drive the tumors to non-T-cell-inflamed tumors and cause refractory conditions to cancer immunotherapies, e.g., immune checkpoint inhibitor (ICI) treatment. EP4 antagonist therapy is suggested to inhibit the immune-suppressive and tumorigenic roles of PGE2 in tumors, and it may sensitize the therapeutic effects of ICIs in patients with non-inflamed and C-IC-deficient tumors. This review provides insight into the mechanism of action of EP4 antagonists in cancer immunotherapy and suggests a C-IC modulating opportunity for EP4 antagonist therapy in combination with ICIs and/or other cancer therapies.
Introduction
Increased expressions of PGE2 and cyclooxygenase 2 (COX-2), a key enzyme for PGE2 synthesis, are routinely identified in a variety of tumor tissues in human and animals, and the contributions of PGE2 in tumor initiation, proliferation, and metastasis have been reported (1, 2). Inhibition of the PGE2 signal by non-steroidal anti-inflammatory drugs (NSAIDs) or COX-2 inhibitors has been shown to suppress tumor growth in animal tumor models (1, 3). However, cardiovascular and gastrointestinal safety concerns at high doses may have prevented further development of the drugs in human. To avoid the toxicity and achieve intrinsic efficacy of this mechanism, discovery of drugs that inhibit the downstream signaling of PGE2 has continued.
Tumors directly or indirectly upregulate the expression of PGE2 in tissues, and the highly expressed PGE2 regulates tumors and other cells present in the tumor microenvironment (TME), leading to tumor growth. PGE2 receptor EP4 is a Gs protein-coupled receptor and activates cAMP-ERK and PI3K signaling (4). EP4 receptors are expressed on the surface of tumor cells, fibroblasts, and immune cells in tumor stroma (2). Recent research evidence suggests that the effects of PGE2 on immune cells in the TME actively trigger tumor immune evasion and influence tumor cell growth and patient survival (5). Furthermore, animal experiments using tumor models suggest the involvement of EP4 receptor on immune cells in tumor growth (6–10). The mechanism of action of EP4 signal inhibition in cancer immunotherapy, however, has not yet been clearly demonstrated.
Advances in a therapy using immune checkpoint inhibitors (ICIs) have led to remarkable outcomes in several solid tumors and have established principles of cancer immunity in clinical fields; however, there are still significant limitations with the ICI therapy. The cancer-immunity cycle (C-IC) concept has clearly demonstrated the mechanisms of immune cells to attack and kill tumors through CD8+ T-cell cytotoxicity (11) and represented a basis for understanding the present state of cancer immunotherapy, contributing to drug selection and combination therapies for patients refractory to ICIs and conventional therapies (12, 13). This review summarizes the roles of PGE2 and EP4 signaling in the concept of C-IC and proposes a position and opportunities for EP4 antagonist therapy in cancer immunotherapy, including in combination with ICIs and other therapies.
Suppression of Antitumor Immunity by PGE2-EP4 Signal
Inhibition of Conventional Dendritic Cell (cDC) Recruitment Into Tumor by PGE2-EP4 Signaling
PGE2 Inhibits cDC Recruitment Into Tumor Through a Suppression of NK-DC Crosstalk
Clinical evidence demonstrated that patients with a relatively high number of immature DCs in the tumor had longer overall survival. A decrease in the number of DCs in the tumor significantly correlated with advanced tumor stage in patients with colorectal cancer (14). Recent research results suggest that a conventional DC1 (cDC1, CD103+ DC in mice and CD141+/BDCA3+ DC in human) has been reported as the only professional antigen-presenting DC that can capture tumor-specific antigen on their MHC class I molecule and cross-present the antigen to CD8+ T cells (15–17). The recruitment of cDC1 into the TME is critically supported by natural killer (NK) cells in the tumors by secreting cDC1 chemoattractants XCL1 and CCL5. In patients with cancer, intratumoral XCL1 and CCL5 levels correlate with the presence of NK and cDC1 cells in the tumor and are associated with increased overall survival (18). NK cells also produce formative cytokine for cDC1, Fms-related tyrosine kinase 3 ligand (FLT3L), and stably form conjugates with cDC1. The NK-DC crosstalk positively regulates cDC1 abundance in melanoma. The FLT3L further leads to the expansion of cDC progenitors in bone marrow and promotes the accumulation of immature cDC1 at the tumor site (19). In human melanoma, the numbers of NK cells and cDC1 are demonstrated to correlate with increased overall survival and patient responsiveness to anti-PD-1 immunotherapy (20, 21). Importantly, the tumor-produced PGE2 impairs the viability and chemokine-producing property of NK cells and downregulates chemokine receptor expression in cDC1s in the TME, resulting in cancer immune evasion (18). Thus, the inhibition of NK cell functions by PGE2 and subsequent suppression of the NK-DC axis is suggested as the key trigger to induce the absence and functional failure of cDCs in the TME and the disruption of C-IC (Figure 1).
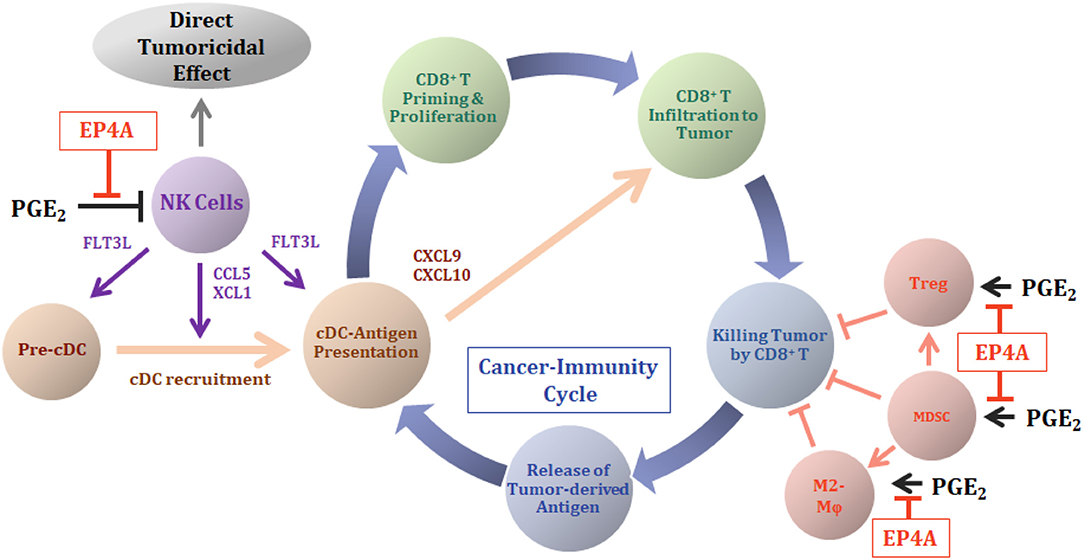
Figure 1. Antitumor mechanism of EP4 antagonist in cancer-immunity cycle (C-IC). The role of the highly expressed PGE2 in tumors and antitumor mechanisms of action of EP4 antagonist (EP4A) therapy are illustrated with the concept of the C-IC. The C-IC includes cyclic steps: a release of tumor-derived antigens (tdAs) to the TME, an antigen presentation of cDCs and a priming of CD8+ T cells and proliferation in tumor-draining lymph node, translocation of effector CD8+ T cells into the tumors, and killing of the tumor cells by the effector CD8+ T cells, and the release of tdAs again. Tumor cell death results in a release of damage-associated molecular patterns (DAMPs), which contains molecules that can be recognized as antigens to specify the tumor cells in the body. Conventional dendritic cells (cDCs) capture and process these tdAs and prime the naïve CD8+ T cells. During the ICI therapy, continuous supply of the pre-cDCs from bone marrow is required for the consecutive continuation of the C-IC result in the success of ICI therapy. NK cells in tumors secrete XCL1 and CCL5 that induce an infiltration of cDCs into the TME. The NK cells further secrete a growth factor FLT3L, which supports survival of cDCs and enhances local cDC differentiation. In TME with high levels of PGE2, the PGE2 shut off supply of cDCs into the TME through the inhibition of NK cell functions, thereby causing disruption of the C-IC and the failure of ICI therapy. The cDCs located in the tumor further secrete chemokines CXCL9/10 that attract CD8+ effector T cells into the tumor. EP4A therapy inhibits the effects of PGE2 on the NK cells and revitalizes the supply of cDCs into the TME. Highly expressed PGE2 in tumors increases populations of immune-suppressive cell species, MDSC, Treg, and M2-like macrophage (M2-Mϕ) in the TME and inhibits cytotoxic ability of effector CD8+ T cells against tumor cells. The tumor-produced PGE2 regulates these immune cells directly or indirectly and produces the non-T-cell-inflamed tumor environment, accelerating tumor immune evasion. EP4A inhibits tumor-mediated PGE2 functions on the immune-suppressive cell species and eliminates the elements causing disruption of an effective C-IC in cancer immunotherapy.
EP4 Signal Inhibits NK Cell Functions Following cDC Recruitment Into TME
NK cells have direct cytotoxicity especially to MHC class I molecule-downregulated tumor cells. EP4 signal inhibition is reported to decrease the expression of the MHC class I on mammary tumor cells and stimulates antitumor effects of NK cells (22). PGE2 secreted by thyroid cancer cells promote antitumor immune suppression through the inhibition of NK cell cytotoxicity via EP2 and EP4 receptors (23). EP4 signaling, in addition to the direct antitumor cytotoxicity of NK cells, has demonstrated tumoricidal efficacy through diverse interactions within the immune system. A direct treatment of PGE2 or EP4 agonist to murine splenic NK cells in vitro blocked NK cell functions such as IFNγ and TNFα productions and cancer cell migration (24). The re-activation of NK cell functions in tumor-bearing mice was reported via an ex vivo study using the EP4 antagonist RQ-15986. RQ-15986 was administrated systemically to 66.1 breast tumor-bearing mice for 3 weeks and then splenic NK cells were isolated and tested for their ability to produce IFNγ by IL-2 stimulation. Although NK cells isolated from tumor-bearing mice lost their ability to produce IFNγ almost completely compared with that of normal mice, NK cells from RQ-15986-treated tumor-bearing mice completely recovered IFNγ-producing ability (25). Moreover, in an obesity-associated hepatocellular carcinoma mice model, the daily systemic therapy of the EP4 antagonist AAT-008 for 3 weeks showed significant induction of cDC1 (CD103+ DC) frequency with no change in the frequency of other DC classes (10). Although no evidence of EP4 receptor inhibition on the NK cells is evaluated in this study, the NK-DC crosstalk-mediated mechanism should be included in the evidence observed as changes on cDCs. Taken together, PGE2 is suggested to inhibit cDC1 recruitment into the TME through the suppression of NK cells and the NK-DC crosstalk. The EP4 antagonist restores the cDC1 population in the TME by re-activating NK cells, and then the NK-DC crosstalk, which is suppressed by tumor-induced PGE2 (Figure 1).
cDCs in Tumor Accelerate Effector CD8+ T-Cell Infiltration Into the Tumors Through CXCL9/10 Production
The absence of the effector CD8+ T cells in tumors correlates with a poor prognosis for clinical outcome in both human and animal cancer models (26, 27). The exclusion of CD8+ T cells from the tumor tissue is often observed in many cancers, and they are commonly referred to as non-T-cell-inflamed tumors (28). The non-T-cell-inflamed tumors are difficult to treat with ICIs compared with inflamed tumor therapies in the clinic. A failure in effector CD8+ T cells' traffic into melanoma tissue from tumor-draining lymph nodes in a mice model is mediated by the lack of chemokines CXCL9 and CXCL10 produced by CD103+ DCs. These DCs are generally present in inflamed tumors, whereas these are absent in non-inflamed tumors (29). Thus, the importance of cDCs in tumors is suggested not only for tumor-specific effector CD8+ T-cell production and proliferation but also for the active infiltration of the effector CD8+ T cells into the tumor bed. Production of chemokine CXCL10 in human immature DCs is reported to be negatively regulated by PGE2 and EP2 and/or EP4 receptors are involved in this effect (30). Treatment with EP4 antagonist E7046 demonstrated a significant increase in CD8+ T-cell frequency in tumors in a CT26 colon cancer–bearing mice model (9). The evidence strongly suggests that EP4 antagonist therapy increases the infiltration of effector CD8+ T cells into the tumor through the active production of chemokines CXCL9 and 10 from the DCs in tumors corresponding to the restoration of NK cell functions by EP4 signal inhibition (Figure 1).
PGE2-EP4 Signal Activates Suppressive Immune Cells and Inhibits Effector CD8+ T Cells
Myeloid-derived suppressor cells (MDSCs), M2-like macrophages in tumors often called tumor-associated macrophages, and regulatory T cells (Tregs) are the major immune-suppressive cell species in the TME. These cells directly or indirectly inhibit cytotoxic activity of effector CD8+ T cells against tumor cells, primarily through the production of immune-suppressive cytokines such as IL-10 and TGFβ. The PGE2-EP4 signal is demonstrated to activate the functions of these immune-suppressive cells and to promote inactivation of antitumor activity in effector CD8+ T cells, leading to tumor immune evasion (Figure 1).
PGE2-EP4 Signal Regulates MDSC Differentiation and Macrophage Polarization
The MDSCs frequently exist in TME and mediate immune suppression through the production of arginase, inducible NOS, TGFβ, IL-10, and COX-2. These molecules directly or indirectly induce Treg activities (31), suppress NK cell functions (32), and promote the polarization of macrophages toward M2-like properties. They cooperatively and synergistically impair the tumoricidal property of effector CD8+ T cells, thereby leading to the evasion of tumor cells from the host's antitumor immunity (33). PGE2 is demonstrated to stimulate MDSC differentiation and proliferation in tumors (32, 34–36), and the EP4 and/or EP2 receptors promote the activation. Albu et al. demonstrated the significant increase in the number of MDSCs in CT-26 colon cancer–bearing mice spleen and the almost complete reversal of the number of MDSCs as a result of continuous treatment with the EP4 receptor antagonist E7046 (9).
Macrophages polarize to functionally different M1- and M2-like phenotypes by various stimulation in tissues with inflammation and tumor. In in vitro experiments, a human peripheral blood mononuclear cell primary culture in the presence of GM-CSF plus IL-4 promotes differentiation to DCs. An addition of PGE2 in this culture suppresses the formation of DCs and skewed the differentiation into the M2-like macrophage. EP4 antagonist E7046 treatment in this culture dose-dependently reversed the effects of PGE2 on the proportion of differentiation to DCs or M2-like macrophages (9). Similarly, in human cervical cancer cell culture, PGE2 hampered monocyte to DC differentiation and skewed their differentiation toward M2-like macrophages. A depletion of PGE2 restored the normal monocyte to DC differentiation (37). In an Apcmin/+ mice, a deletion of the myeloid EP4 receptor led to the loss of the arginase 1-expressing M2 phenotype macrophage population in a histochemical study (38). In K19-Wnt1/C2mE mice, a PGE2 highly producing transgenic gastric cancer mice model, EP4 antagonist RQ-15986 therapy achieved almost complete shrinkage of the tumor. M2-like macrophage polarization was observed in the tumor, and the EP4 signal inhibition by RQ-15986 released this polarization (6). Collectively, the evidence strongly supports the EP4 antagonist's role in the prevention of M2-like macrophage polarization and accumulation in tumors derived by tumor-associated PGE2.
PGE2-EP4 Signal Activates Treg Cells
The frequency of circulating or tumor-infiltrating Tregs is associated with poor survival of patients in many cancers, including colon, breast, melanoma, and lung (39). PGE2 is reported to increase the expression of the Treg-specific marker Foxp3 and to stimulate the functions of Treg cells in vitro and in a mice lung cancer model. The activated Treg cells also induce COX-2 expression and PGE2 production, which then support immune-suppressive functions by themselves. These autocrine and paracrine effects of PGE2 cooperatively activate Treg cells in tumors (40, 41). In Treg cell culture isolated from the spleen of EP2 or EP4 knockout mice, expressions of Foxp3 in the presence of PGE2 were significantly reduced compared with those Treg cells isolated from normal mice. The contribution of EP2 and EP4 receptors in PGE2 signaling in activated Tregs has also been demonstrated using the antagonists AH6809 and AH23848, respectively, by evaluating the reversal of Treg cell-mediated T-cell effector function failure (41). An immunosuppression caused by UV irradiation was mediated through the induction of Treg proliferation, and PGE2-EP4 signaling was also reported to be involved (42).
Activation of Immune-Suppressive Mechanisms by PGE2-EP4 Signal Impairs Effector CD8+ T-Cell Functions
The effector CD8+ T cells infiltrated to tumors are primarily able to fight against the antigen-targeted tumor cells. However, the tumors activate the previously described immune-suppressive cell species to avoid the attack from effector CD8+ T cells. PGE2 is one of the key molecules triggering the immune suppression in TME (34). The EP4 antagonist E7046 therapy increased the population of intratumoral effector CD8+ T cells, inhibited myeloid MDSC activation, and suppressed the polarization of macrophages to M2-like properties in a CT26 mouse colon cancer model. It showed potent antitumor efficacy compared with non-treated controls (9). The aforementioned evidence strongly suggests that tumor-produced, PGE2-activated immune-suppressive cell species in the TME inhibit the tumoricidal function of effector CD8+ T cells in the tumor through EP4 signaling, and that the EP4 antagonist rescues the tumoricidal function of CD8+ T cells (Figure 1).
Mechanism of Action of EP4 Antagonist in the C-IC
Cancer-Immunity Cycle
The concept of the C-IC is proposed by Chen and Mellman and suggests the respective roles of tumor cells, tumor-derived antigens, DCs, and effector CD8+ T cells in a series of stepwise events, which proceed and expand iteratively in cancer immunotherapy (11). For patients with cancer with deficiency in immunological antitumor ability, rescuing single or few defects among these steps is essential for the C-IC to optimally perform. Amplifying the entire C-IC will provide effective antitumor activity; however, drugs such as ICI generally target only a single step in the cycle. Therefore, the most effective approaches will require targeting the rate-limiting steps in a given patient using the appropriate combination of drugs (12). The EP4 antagonist is one of the drug classes that can reactivate antitumor immunity, rescuing the defects of the steps in the C-IC as described above. Understanding the mechanism of action of the EP4 antagonist in cancer immunotherapy in combination with the C-IC concept would be a useful procedure when considering therapeutic strategy, drug regimen combination, or radiotherapy in the clinical environment.
Roles of PGE2 and EP4 in C-IC and Perspectives on EP4 Antagonist in Immunotherapy
As summarized in the previous sections, PGE2 mediates the immunologically deficient tumor environment by regulating several immune cell species that express the EP4 receptor and develop multiple dysfunctions in the C-IC. The EP4 antagonist is suggested to demonstrate antitumor efficacy by restoring the PGE2-mediated dysfunctions in the host's antitumor immune systems. Figure 1 illustrates the roles of PGE2 and the EP4 antagonist in cancer immunotherapy combined with the C-IC concept. Tumor-produced PGE2 inhibits the NK cell function in the TME and suppresses XCL1 and CCL5 production, which promotes the infiltration of cDC precursors (pre-cDCs) into the TME. The suppression of NK cells further mediates the reduction of cDC function in tumors, with FLT3L playing a key role. Moreover, the decrease in the cDC population and function in the TME causes the reduction of CXCL9/10 production by cDCs and suppresses the translocation of effector CD8+ T cells into the tumor from draining lymph nodes. Thus, the PGE2-mediated exhausting of NK cell triggers the failure of sequential recruitment of cDCs and the effector CD8+ T cells, essential elements of the C-IC, into the TME (43). The EP4 antagonist therapy is demonstrated to block the PGE2-mediated dysfunction of NK cells, which enables the continuous infiltration of cDCs into the TME, thereby increasing the population of tumor-specific effector CD8+ T cells in the TME (9, 25). The PGE2 further activates the functions of immune-suppressive cell species, such as MDSC, Tregs, and tumor-associated M2-like macrophages, thereby directly or indirectly wresting the functions of effector CD8+ T cells from the tumor cells. The EP4 antagonist supports the roles of antitumor immune functions by inhibiting the roles of PGE2 in the immune-suppressive cells as noted previously (Figure 1).
Therapy using ICIs in humans led to remarkable antitumor efficacy in several tumors, enabling complete elimination of tumors in some cases. However, the proportion of patients who achieve a response remains generally modest. Absence or functional failure of effector CD8+ T cells in tumors, the non-inflamed tumor, is suggested to be one reason for being refractory to ICI therapy according to current clinical experience. For a portion of ICI-refractory patients, the failure of C-IC in their TME is suggested. As summarized in this review, tumor-mediated PGE2 is one of the key molecules that disrupt the C-IC and promote the non-inflamed tumor environment, resulting in ICI therapy deficiency. When drugs are used concomitantly, EP4 antagonist therapy may sensitize the efficacy of ICI therapy in patients with ICI-refractory by restoring the sequential NK and cDC cell functions. Animal experiments support the combination of the EP4 antagonist E7046 (ER0886046) with anti-CTLA4 in a mouse melanoma B16F10 model and with anti-PD-1 or anti-PD-L1 antibodies in a mouse colon cancer CT26 model. Higher antitumor efficacies were demonstrated in combination, compared with E7046 or the ICI antibody alone (44). Figure 2 illustrates the opportunity to introduce EP4 antagonist therapy with ICIs, which may cooperatively interact leading to an efficient C-IC. An additional opportunity for EP4 antagonist therapy is in combination with the other cancer therapies that induce immunogenic cell death (ICD), such as tumor radiotherapy and some types of cancer chemotherapies. The ICD, by releasing damage-associated molecular patterns (DAMPs) and tumor-derived antigens, has been shown to support the activation of DCs' functions and their traffic to lymph nodes, activating tumor-targeted immune responses (45). The EP4 antagonist E7046 was demonstrated to show improved antitumor efficacy and prolonged survival of mice in combination with radiotherapy, compared with radiotherapy or E7046 alone in a CT26 colon cancer mouse model (46). The combination of radiotherapy and/or chemotherapy with the EP4 antagonist is thought to synergistically accelerate the C-IC. Simultaneous increases in the cDC population by EP4 therapy and the production of DAMPs through therapy-producing ICD in tumors are essentially required for the effective production of tumor-specific effector CD8+ T cells (44) (Figure 2).
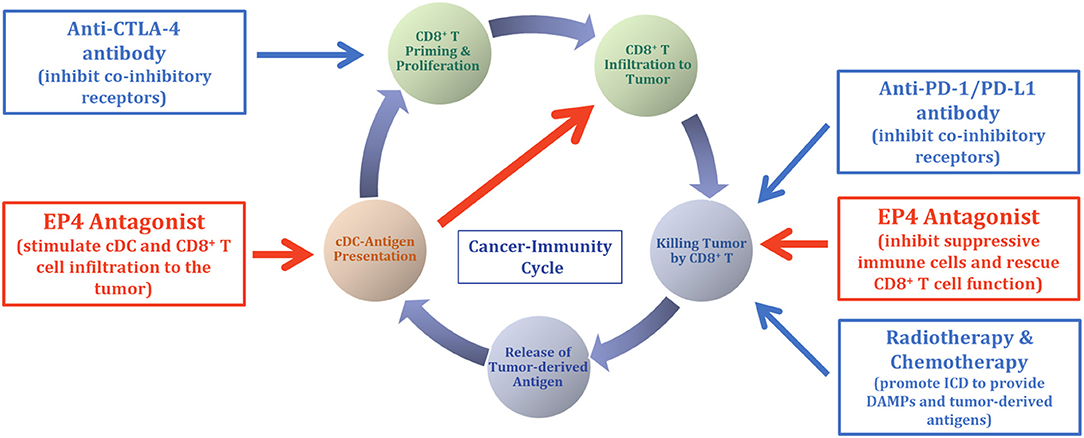
Figure 2. Opportunity for EP4 antagonist therapy in cancer immunotherapy. Based on the mechanisms of action of the EP4 antagonist (Figure 1), opportunities for the EP4 antagonist therapy in cancer immunotherapy are illustrated. Because EP4 antagonist therapy is suggested to change the tumor environment from non-T-cell inflamed to inflamed, concomitant use of EP4 antagonist with ICI will sensitize the efficacy of ICI in some portion of patients whose prior ICI monotherapy was non-responsive. Combination of chemotherapy and radiotherapy with ICI therapy is suggested to show cooperative antitumor efficacy via the production of DAMPs and activation and restoration of tumor-targeted immune responses. This combination is reasonable from the standpoint of C-IC theory, and the addition of EP4 antagonist therapy with ICI, together with chemotherapy and/or radiotherapy, may further accelerate the potential of the C-IC and antitumor therapeutic efficacy.
Conclusion
Several companies are currently conducting clinical trials of EP4 receptor-selective antagonists for cancer therapy (ClinicalTrials ID: NCT03658772, NCT03696212, NCT03152370, NCT03661632) and evaluating EP4 antagonist therapy in anti-PD-1-refractory tumors, microsatellite stable tumors, and in combination with tumor radiotherapy. Enhanced understanding of the mechanism of action of the EP4 antagonist therapy within the C-IC concept enabled us to pursue a new therapeutic approach to EP4 antagonist therapy.
Author Contributions
YT wrote the manuscript. SK and AN contributed to the scientific discussion.
Conflict of Interest
The authors are employees and shareholders of AskAt Inc. AN is also an employee of DNA Partners.
Acknowledgments
We would like to thank Professor Amy Fulton and Andrew M. Saidel for reviewing the manuscript.
References
1. Wang D, DuBois RN. Role of prostanoids in gastrointestinal cancer. J Clin Invest. (2018) 128:2732–42. doi: 10.1172/JCI97953
2. Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev. (2013) 65:1010–52. doi: 10.1124/pr.112.007195
3. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. (2010) 10:181–93. doi: 10.1038/nrc2809
4. Konya V, Marsche G, Schuligoi R, Heinemann A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol Ther. (2013) 138:485–502. doi: 10.1016/j.pharmthera.2013.03.006
5. Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. (2015) 162:1257–70. doi: 10.1016/j.cell.2015.08.015
6. Oshima H, Hioki K, Popivanova BK, Oguma K, Van Rooijen N, Ishikawa TO, et al. Prostaglandin E2 2 signaling and bacterial infection recruit tumor-promoting macrophages to mouse gastric tumors. Gastroenterology. (2011) 140:596–607. doi: 10.1053/j.gastro.2010.11.007
7. Fulton AM, Ma X, Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. (2006) 66:9794–7. doi: 10.1158/0008-5472.CAN-06-2067
8. Majumder M, Xin X, Liu L, Girish GV, Lala PK. Prostaglandin E2 receptor EP4 as the common target on cancer cells and macrophages to abolish angiogenesis, lymphangiogenesis, metastasis, and stem-like cell functions. Cancer Sci. (2014) 105:1142–51. doi: 10.1111/cas.12475
9. Albu DI, Wang Z, Huang K-C, Wu J, Twine N, Leacu S, et al. EP4 antagonism by E7046 diminishes myeloid immunosuppression and synergizes with treg-reducing IL-2-Diphtheria toxin fusion protein in restoring anti-tumor immunity. Oncoimmunology. (2017) 6:e1338239. doi: 10.1080/2162402X.2017.1338239
10. Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y, et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov. (2017) 7:522–38. doi: 10.1158/2159-8290.CD-16-0932
11. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
12. Gao D. Compound-therapy based on cancer-immunity cycle: promising prospects for antitumor regimens. Am J Cancer Res. (2019) 9:212–8. Available online at: http://www.ajcr.us/files/ajcr0091499.pdf
13. Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature. (2019) 574:45–56. doi: 10.1038/s41586-019-1593-5
14. Gulubova MV, Ananiev JR, Vlaykova TI, Yovchev Y, Tsoneva V, Manolova IM. Role of dendritic cells in progression and clinical outcome of colon cancer. Int J Colorectal Dis. (2012) 27:159–69. doi: 10.1007/s00384-011-1334-1
15. Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. (2010) 207:1247–60. doi: 10.1084/jem.20092140
16. Bachem A, Güttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med. (2010) 207:1273–81. doi: 10.1084/jem.20100348
17. Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun. (2016) 7:13720. doi: 10.1038/ncomms13720
18. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
19. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. (2016) 44:924–38. doi: 10.1016/j.immuni.2016.03.012
20. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
21. Böttcher JP, Reis E Sousa C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer. (2018) 4:784–92. doi: 10.1016/j.trecan.2018.09.001
22. Kundu N, Ma X, Holt D, Goloubeva O, Ostrand-Rosenberg S, Fulton AM. Antagonism of the prostaglandin E receptor EP4 inhibits metastasis and enhances NK function. Breast Cancer Res Treat. (2009) 117:235–42. doi: 10.1007/s10549-008-0180-5
23. Park A, Lee Y, Kim MS, Kang YJ, Park Y-J, Jung H, et al. Prostaglandin E2 secreted by thyroid cancer cells contributes to immune escape through the suppression of natural killer (NK) cell cytotoxicity and NK cell differentiation. Front Immunol. (2018) 9:1859. doi: 10.3389/fimmu.2018.01859
24. Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E-2 (PGE(2)) suppresses natural killer cell function primarily through the PGE(2) receptor EP4. Cancer Immunol Immunother. (2011) 60:1577–86. doi: 10.1007/s00262-011-1064-9
25. Ma X, Holt D, Kundu N, Reader J, Goloubeva O, Take Y, et al. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. Oncoimmunology. (2013) 2:e22647. doi: 10.4161/onci.22647
26. Lohneis P, Sinn M, Bischoff S, Jühling A, Pelzer U, Wislocka L, et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur J Cancer. (2017) 83:290–301. doi: 10.1016/j.ejca.2017.06.016
27. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci USA. (2018) 115:E4041–50. doi: 10.1073/pnas.1720948115
28. Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349
29. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. (2017) 31:711–23.e4. doi: 10.1016/j.ccell.2017.04.003
30. McIlroy A, Caron G, Blanchard S, Frémaux I, Duluc D, Delneste Y, et al. Histamine and prostaglandin E up-regulate the production of Th2-attracting chemokines (CCL17 and CCL22) and down-regulate IFN-gamma-induced CXCL10 production by immature human dendritic cells. Immunology. (2006) 117:507–16. doi: 10.1111/j.1365-2567.2006.02326.x
31. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. (2015) 125:3356–64. doi: 10.1172/JCI80005
32. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-E2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res. (2014) 20:4096–106. doi: 10.1158/1078-0432.CCR-14-0635
33. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. (2013) 138:105–15. doi: 10.1111/imm.12036
34. Rong Y, Yuan C-H, Qu Z, Zhou H, Guan Q, Yang N, et al. Doxorubicin resistant cancer cells activate myeloid-derived suppressor cells by releasing PGE2. Sci Rep. (2016) 6:23824. doi: 10.1038/srep23824
35. Xu Y, Zhao W, Xu J, Li J, Hong Z, Yin Z, et al. Activated hepatic stellate cells promote liver cancer by induction of myeloid-derived suppressor cells through cyclooxygenase-2. Oncotarget. (2016) 7:8866–78. doi: 10.18632/oncotarget.6839
36. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE2-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. (2011) 71:7463–70. doi: 10.1158/0008-5472.CAN-11-2449
37. Heusinkveld M, de Vos van Steenwijk PJ, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJ, et al. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J Immunol. (2011) 187:1157–65. doi: 10.4049/jimmunol.1100889
38. Chang J, Vacher J, Yao B, Fan X, Zhang B, Harris RC, et al. Prostaglandin E receptor 4 (EP4) promotes colonic tumorigenesis. Oncotarget. (2015) 6:33500–11. doi: 10.18632/oncotarget.5589
39. Ward-Hartstonge KA, Kemp RA. Regulatory T-cell heterogeneity and the cancer immune response. Clin Transl Immunol. (2017) 6:e154. doi: 10.1038/cti.2017.43
40. Sharma S, Yang S-C, Zhu L, Reckamp K, Gardner B, Baratelli F, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+CD25+ T regulatory cell activities in lung cancer. Cancer Res. (2005) 65:5211–20. doi: 10.1158/0008-5472.CAN-05-0141
41. Mahic M, Yaqub S, Johansson CC, Taskén K, Aandahl EM. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J Immunol. (2006) 177:246–54. doi: 10.4049/jimmunol.177.1.246
42. Soontrapa K, Honda T, Sakata D, Yao C, Hirata T, Hori S. Prostaglandin E2-prostoglandin E receptor subtype 4 (EP4) signaling mediates UV irradiation-induced systemic immunosuppression. Proc Natl Acad Sci. USA. (2011) 108:6668–73. doi: 10.1073/pnas.1018625108
43. Fessenden TB, Duong E, Spranger S. A team effort: natural killer cells on the first leg of the tumor immunity relay race. J Immunother Cancer. (2018) 6:67. doi: 10.1186/s40425-018-0380-4
44. van Gulijk M, Dammeijer F, Aerts JGJV, Vroman H. Combination strategies to optimize efficacy of dendritic cell-based immunotherapy. Front Immunol. (2018) 9:2759. doi: 10.3389/fimmu.2018.02759
45. Rapoport BL, Anderson R. Realizing the clinical potential of immunogenic cell death in cancer chemotherapy and radiotherapy. Int J Mol Sci. (2019) 20:959. doi: 10.3390/ijms20040959
46. Bao X, Albu D, Woodall-Jappe M. Combination Therapies for the Treatment of Cancer. Geneva: World Intellectual Property Organization. International Publication Number WO2015/179615A1. Available online at: https://patentimages.storage.googleapis.com/21/34/cb/884c2efafa77ca/WO2015179615A1.pdf
Keywords: EP4 antagonist, PGE2, NK-DC crosstalk, cancer-immunity cycle, cancer immunotherapy, immune checkpoint inhibitor (ICI), inflamed tumor, non-inflamed tumor
Citation: Take Y, Koizumi S and Nagahisa A (2020) Prostaglandin E Receptor 4 Antagonist in Cancer Immunotherapy: Mechanisms of Action. Front. Immunol. 11:324. doi: 10.3389/fimmu.2020.00324
Received: 07 December 2019; Accepted: 10 February 2020;
Published: 10 March 2020.
Edited by:
Giovanna Schiavoni, Higher Institute of Health (ISS), ItalyReviewed by:
Kawaljit Kaur, University of California, Los Angeles, United StatesRodabe N. Amaria, University of Texas MD Anderson Cancer Center, United States
Copyright © 2020 Take, Koizumi and Nagahisa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yukinori Take, eXVraW5vcmkudGFrZSYjeDAwMDQwO2Fza2F0LmNvLmpw